

1. Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive genetic defects in cortisol synthesis. The altered negative feedback of cortisol to the hypothalamus and the pituitary gland prompts corticotropin-releasing hormone (CRH) and adrenocorticotrophic hormone (ACTH) elevations. Increased ACTH, in turn, has two downstream effects: 1) it overstimulates adrenal steroidogenesis, resulting in an accumulation of steroids above the enzymatic blockage; and 2) when sustained, ACTH elevation promotes adrenal gland enlargement (hence, the term CAH). A variety of mutations in one or more genes encoding enzymes essential for cortisol synthesis (Figure 1) lead to a spectrum of disorders and disease severity. In general, complete or nearly complete enzymatic defects result in overt adrenal insufficiency, and are conventionally referred to as “classic” CAH. In milder forms of the disease, also termed “late-onset” or “non-classic” CAH (NCCAH), partial enzymatic defects are overcome by ACTH elevations. These patients have compensated cortisol and aldosterone production. Defects in the gene encoding 21-hydroxylase (CYP21A2) account for more than 95% of all cases of CAH1 and, unless otherwise specified, the term CAH will refer to 21-hydroxylase deficiency (21OHD) throughout this article. A brief overview of rare forms of CAH is presented in Table 1.
Figure 1 : Steroidogenic pathway.
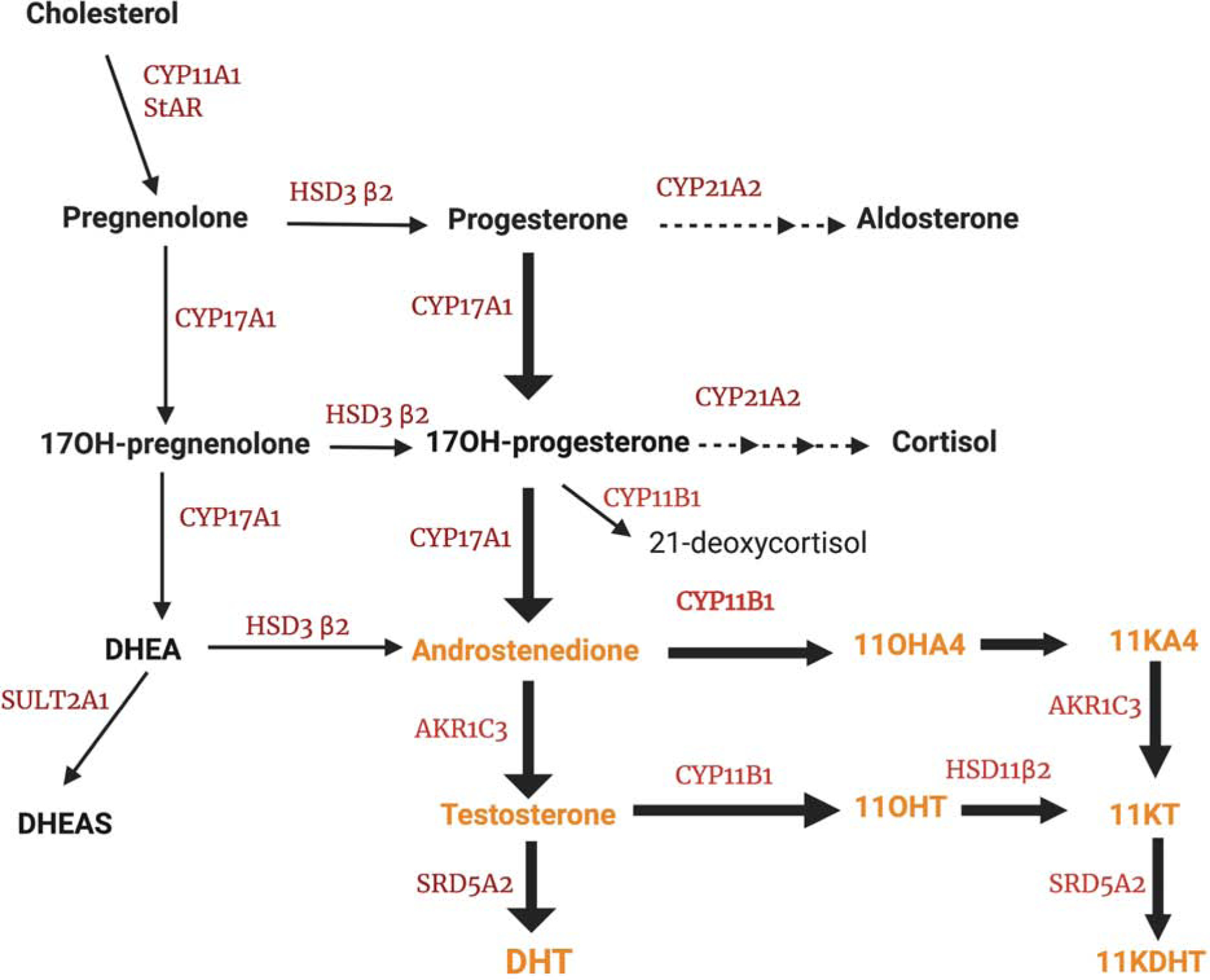
Genetic defects in 21-hydroxylase result in accumulation of 17α-hydroxyprogesterone, which is diverted towards androgens, including: androstenedione, testosterone, and 11-oxygenated androgens (11OHA4 and 11OHT); the latter two are oxidized to 11KA4 and 11KT, respectively, in the kidneys and other tissues.
Abbreviations: StAR: steroidogenic acute regulatory protein; CYP11A1, cholesterol side-chain cleavage; HSD3β2, 3β-hydroxysteroid dehydrogenase type 2; CYP17A1, 17α-hydroxylase/17,20-lyase; CYB5A, cytochrome b5 type A; CYP11B1, 11β-hydroxylase; AKR1C3, 17β-hydroxysteroid dehydrogenase type 5; HSD11B2, 11β-hydroxysteroid dehydrogenase, type 2; SULT2A1, sulfotransferase 2A1; SRD5A2, steroid 5α-reductase type 2; DHEA: dehydroepiandrosterone; DHEAS: dehydroepiandrosterone sulfate; 11OHA4: 11β-hydroxyandrostenedione; 11OHT: 11β-hydroxytestosterone; 11KA4: 11-ketoandrostenedione; 11KT: 11-ketotestosterone;.
Table 1:
Uncommon Causes of Congenital Adrenal Hyperplasia (CAH)
| Side chain cleavage (SCC) deficiency: | |
| Prevalence: |
|
| Presentation: |
|
| Diagnosis: |
|
| Non-classic form: |
|
| Management: |
|
| StAR deficiency: Lipoid CAH | |
| Prevalence: |
|
| Presentation: |
|
| Diagnosis: |
|
| Non-classic form: |
|
| Management: |
|
| HSD3B2 (Δ5- Δ4 Isomerase) deficiency | |
| Prevalence: |
|
| Presentation: |
|
| Diagnosis: |
|
| Non-classic form: |
|
| Management: |
|
| 11β-hydroxylase deficiency | |
| Prevalence: |
|
| Presentation: |
|
| Diagnosis: |
|
| Non-classic form: |
|
| Management: |
|
| CYP17A1 deficiency (both 17α-hydroxylase deficiency and 17,20 lyase deficiency) | |
| Prevalence: |
|
| Presentation: |
|
| Diagnosis: |
|
| Non-classic form: |
|
| Management: |
|
| Isolated 17,20-lyase activity | |
| Prevalence: |
|
| Presentation: |
|
| Diagnosis: |
|
| Non-classic form: |
|
| Management: |
|
| P450 oxidoreductase (POR) deficiency | |
| Prevalence: | |
| Presentation: |
|
| Diagnosis: |
|
| Non-classic form: |
|
| Management: |
|
Patients with classic CAH are typically diagnosed at birth or early in life, and their transition of care to adult endocrinology occurs with the diagnosis previoulsly established. Conversely, adult endocrinologists must know when to suspect NCCAH, which has a considerably higher prevalence. In this review, we will focus primarily on NCCAH.
2. Epidemiology
The worldwide incidence of classic CAH is roughly 1:14,000 to 1:18,000 births2. In contrast, NCCAH, is relatively common, with an overall prevalence of 1:200 in the Caucasian US population,2,3 and a higher frequency among Ashkenazi Jews, Hispanics, Mediteraneans, Middle Eastearns, and Eskimos4. Among women presenting with symptoms of androgen excess, the overall prevalence of NCCAH is approximately 4%5.
3. Genetics of CAH
The CYP21A2 gene is located on the long arm of chromosome 6, within the human leukocyte antigen (HLA) locus and adjacent to the genes for the fourth component of complement6–8, a region within which genetic recombinations occur frequently9. A highly homologous non-functional pseudogene (CYP21A1P), which encodes a truncated, inactive enzyme, also resides in the vicinity10. Most patients with NCCAH are compound heterozygotes with different mutations in the two alleles5. Mutations that fully annul 21-hydroxylase activity, such as complete deletions, large gene conversions, and non-sense or frame-shift mutations, lead to salt-wasting CAH, in which both glucocorticoid and mineralocorticoid production is lacking. Mutations that render even minimum residual enzyme activity lead to the so-called “simple virilizing” CAH, where sufficient aldosterone synthesis occurs. In NNCAH, 20–60% of the enzyme activity is preserved, which ensures cortisol concentrations equal to those of unaffected individuals. NCCAH patients might harbor one classic and one non-classic allele or two non-classic alleles. Parents with NCCAH have a 1.5–2.5% risk of having a child with classic 21OHD11,12. Thus, CYP21A2 genotyping in patients with 21OHD and their partners can refine the risk of having an affected offspring2.
Although a strong genotype-phenotype correlation exists for the most severe and the mildest forms of CAH, the clinical manifestations of moderate CAH forms are quite variable13,14. Certain mutations, such as P30L, I172N or I2G, and combinations of mutations (homozygous vs. heterozygous) can yield different phenotypes14. Characterizing the predominant phenotype for a given genotype can assist in genetic counseling of parents at risk of having a child with CAH.
4. Clinical features of patients with CAH
The clinical manifestations of CAH can result from cortisol and/or aldosterone deficiency when present (in classic CAH), and from excessive synthesis of bioactive steroids, which is prompted by ACTH and facilitated by the enzymatic blockade. In the case of 21OHD, variable degrees of androgen excess occur (Figure 1). In patients with classic 21OHD, in-utero exposure to profound androgen excess leads to virilization of the genitalia in girls, which is easily identified at birth. In contrast, affected boys have minimal or no physical findings, putting them at risk for incorrect or missed diagnosis when newborn screening is not pursued. The implementation of newborn screening across all USA and several other countries has significantly decreased the death rates among infants with salt wasting 21OHD2 and debunked the preponderance of disease in females.
Patients with NCCAH may present during childhood with premature pubarche, which has been described as early as 6 months of age5. In contrast, females with NCCAH often present as adolescents or young adults with acne, hirsutism, menstrual abnormalities, or infertility, features that overlap greatly with those of PCOS. Males with NCCAH often remain undiagnosed, or only identified during genetic screening performed for pre-conception counselling or after the birth of an affected offspring5.
Female patients with non-classic 3β-hydroxysteroid dehydrogenase type 2 (HSD3B2) or 11β-hydroxylase (CYP11B1) deficiency (11OHD) present with similar clinical features with those with 21OHD and PCOS, but both are extremely rare forms of CAH15,16. The distinction between these types of non-classic CAH and PCOS is unreliable based on clinical presentation alone17. Hirsutism (59% in NCCAH and 60–70% in PCOS) and acne (33% in NCCAH and 14–25% in PCOS) occur at comparable rates in both disorders18. In constrast, menstrual irregularity (10–17% in women with NCCAH vs 75–90% in PCOS)18,19 and infertility (approximately 13% in NCCAH vs 25–50% with PCOS) tend to occur more frequently among women with PCOS18. Similarly, polycystic ovarian morphology, although more common in PCOS, has also been reported in 30–40% patients with NCCAH18,19. Metabolic features typically associated with PCOS, including obesity, insulin resistance, and dyslipidemia, have also been reported in up to 40% of patients with NCCAH18,19. Although the prevalence of PCOS is about 40–50 times higher than that of non-classic 21OHD among women with hyperandrogenemia18, testing for non-classic 21OHD should be pursued in all such patients, as the correct diagnosis has implications for treatment and family planning.
5. Biochemistry of CAH - implication for diagnosis and disease monitoring
5.1. Diagnosis of CAH
All enzymatic defects within the steroidogenic pathway generate a large precursor to product ratio (Figure 1), which is further enhanced by ACTH stimulation. The prominent elevation of the main substrate for the defective enzyme forms the basis of CAH diagnosis. For 21OHD, the diagnosis relies on17-hydroxyprogesterone (17OHP) elevations, which span a gradient reflective of the spectrum of enzymatic defects. Both neonatal and clinically-prompted screening for 21OHD consists in 17OHP measurement, and values >200 ng/dL (6 nmol/L) are suggestive of the diagnosis. Patients with classic 21OHD typically have 17OHP concentrations above 10,000 ng/dL. In patients with modest 17OHP baseline elevations, of 200–1000 ng/dL (6–30 nmol/ L), a cosyntropin stimulation test is the current standard-of-care, and 17OHP values >1000 ng/dL establish the diagnosis2. Because 17OHP follows closely the circadian rhythm of ACTH, blood should be obtained in early morning. After puberty, 17OHP should be measured during the follicular phase to detect NCCAH, as ovarian surges of 17OHP occur during the luteal phase of the menstrual cycle.
A caveat to CAH testing is that these diagnostic thresholds should be considered in the context of the assay methodology, population, and individual background. Some patients with NCCAH could be missed based on a 17OHP cutoff >200 ng/dL (> 6nmol/L),20 and lower thresholds have been proposed, particularly when using mass spectrometry assays17,20,21. False positive 17OHP screening results are even more common, particularly when immunoassays are used, and when the blood sample is obtained in the luteal phase. Modestly elevated 17OHP is reported in 25% women with PCOS19,22, likely because the high rates of irregular menses and amenorrhea make follicular phase testing impractical. Other tests suggestive of PCOS, such as elevated LH/ FSH ratio, have poor sensitivity and specificity and can also be seen in approximately 10% women with NCCAH19. Simultaneous elevation of 21-deoxycortisol, and lower corticosterone, further support the diagnosis of CAH17,23. Such multistoried panels could circumvent the need for cosyntropin-stimulated testing in the future.
17OHP is typically elevated in patients with classic HSD3B2 deficiency or 11OHD1, although normal levels may be seen in non-classic forms16. The diagnosis of non-classic HSD3B2 deficiency is made when 17-hydroxypregnenolone exceeds 3,000 ng/dL, along with a cortisol >18 μg/dL after cosyntropin stimulation, and a 17-hydroxypregnenolone/cortisol ratio >10 standard deviations above normal24. Non-classic 11OHD is diagnosed when 11-deoxycortisol reaches concentrations >1,800 ng/dL and cortisol is >18 μg/dL after cosyntropin25. Modestly abnormal baseline and even cosyntropin-stimulated steroid ratios can be deceiving, especially when immunoassays are used, and diagnostic confirmation with genetic testing is preferred for these very rare forms of CAH.
5.2. Androgen excess in CAH
Aside from its diagnostic significance, the excess 17OHP serves as substrate for fully functional enzymes, resulting in overproduction of adrenal androgens and precursors (Figure 1). This includes androstenedione (A4) and testosterone (T), as well as their 11-oxygenated metabolites, also called 11-oxyandrogens26. T and A4 are produced both by the gonads and adrenal glands, which might explain their poor correlation with clinical evidence of androgen excess in CAH patients27,28. In contrast, the major source of 11-oxyandrogens is the adrenal gland29, which expresses CYP11B1 abundantly30. In fact, 11-hydroxyandrostenedione (11OHA4) is the most abundant unconjugated C19 steroid produced by the healthy human adrenal gland31, and its synthesis is further enhanced in patients with classic32 and non-classic 21OHD23. The downstream metabolites of 11OHA4, 11-ketoandrostenedione (11KA4) and 11-ketotestosterone (11KT), are produced primarily in periphery.
In vitro studies have shown that 11KT is a bioactive androgen, with maximum androgen potency comparable with that of T33,34. Several lines of evidence also support the androgenic bioactivity of 11KT in vivo. 11KT has been associated with physiological and premature adrenarche, and its concentrations exceed those of T prior to puberty33. 11-oxyandrogens are 3- to 4-fold higher in patients with classic CAH relative to age and sex-matched controls32. Higher concentrations of 11-oxyandrogens have been associated with higher adrenal volume, presence of testicular adrenal rest tumors (TARTs), and menstrual irregularities in patients with classic CAH35. In addition, while in women with classic CAH, 11KT and T correlate directly, suggesting their common adrenal source, T correlates inversely with 11KT in sexually mature males with classic 21OHD,32 pointing towards hypothalamic-pituitary-gonadal axis suppression by excessive adrenal androgens in uncontrolled patients.
In a recent study of 86 patients undergoing testing for NCCAH, 11-oxyandrogens were disproportionately elevated relative to conventional androgens in patients with confirmed NCCAH vs. those without CAH, 50% of whom had PCOS36. In contrast, A4 and T were similar between the two groups, reinforcing that the clinical utility of these traditional androgens in assessing the source of hyperandrogenism is limited Generally, a high A4/T ratio is suggestive of adrenal androgen excess.37 To further complicate things, however, women with CAH often secondarily develop PCOS38, and, conversely, a large subset of women with PCOS have increased adrenal androgen production.39,40
6. Management
The goal of treatment in patients with NCCAH is to suppress the excess adrenal androgen synthesis and the associated complications. Unlike patients with classic CAH, patients with NCCAH do not have adrenal insufficiency and do not need hormonal replacement with glucocorticoids. In children with precocious puberty or accelerated growth velocity, glucocorticoids may be used to suppress the ACTH-driven adrenal androgen excess2. Consideration for glucocorticoid discontinuation should be given once these children have attained their final adult height or two to three years post-menarche for girls2.
For adolescents and adult women presenting with signs of hyperandrogenism, such as acne or hirsutism, oral contraceptive therapy (OCs) with estrogen-progestin preparations are the treatment of choice.41 Treatment with OCs reduces hyperandrogenemia by a) suppression of LH and subsequently of ovarian androgen production; b) stimulation of hepatic synthesis of sex hormone-binding globulin, with resultant reduction of free androgen concentration; c) slight reduction of adrenal androgen synthesis; d) decreased binding of androgens to their receptors; e) increased clearance of testosterone; and f) mild inhibition in the pilosebaceous unit of 5α-reductase, the enzyme catalyzing conversion of testosterone to the most potent androgen, dihydrotestosterone (DHT)41. OCs containing progestins with low androgenicity like norgestimate, desogestrel, and gestodene might have an advantage over levonorgestrel over metabolic risk factors41. While norgestimate is the only available progestin with low androgeneicity that does not enhace the risk of venous thromboembolism over earlier generation progestins, evidence to support higher effectiveness of a particular OC for treating hirsutism is lacking. Women who present with hirsutism should be counselled about the expected time frame for improvement, which may take as long as 6–12 months,41 and the potential need for adjunct direct hair removal methods, like photoepilation or electrolysis.
Addition of anti-androgens can be considered if results are unsatisfactory with OCs alone. These therapies should never be used in reproductive age women without reliable contraception methods, due to their potential effects on the developing male genitalia in utero. Spironolactone, primarily a mineralocorticoid antagonist, also has anti-androgenic effects.42 Other therapies for hirsutism include finasteride, a 5α-reductase inhibitor; or cyproterone acetate (CPA), a progesterone derivative that competes with DHT for binding to androgen receptor (not available in the US). Flutamide, a non-steroidal androgen receptor blocker is not recommended due to hepatotoxicity. Both spironolactone and finasteride have similar efficacy in improving hirsutism, with a pooled weighted mean difference of −7.02 [95% CI (−11.51 to −2.52)] Ferriman-Gallway units in comparison to placebo41,43 and their effect is additive to that of OCs, further reducing hirsutism pooled weighted Ferriman-Gallway units by −1.73 [95% CI (−3.32 to −0.13)]43. Glucocorticoids are the mainstay of androgen suppression therapy only in classic CAH. Although glucocorticoids were shown to be more effective than OCs or antiandrogens for suppressing serum adrenal androgen concentrations in women with NCCAH, they were less effective in improving hirsutism44,45, with an increased risk of toxicity Thus, glucocorticoids are only used for managing hirsutism in female patients with NCCAH when intolerance to OCs and/or anti-androgens has been established41.
Monitoring treatment of patients with NCCAH relies mostly on clinical evaluation, as reliable tests have been lacking. When elevated, T and A4 should be normalized, while 17OHP elevations should be permissible. Normalization of 17OHP typically indicates overtreatment with glucocorticoids. Interestingly, elevations of 11-oxyandrogens have been reported in a subset of women with PCOS relative to healthy controls46,47. Nevertheless, limited data suggests that these steroids might be higher in patients with NCCAH48. Although some commercial laboratories offer 11KT measurement, further studies are needed to clarify the clinical utility of 11-oxyandrogens as biomarkers of adrenal vs. gonadal over-activity. Standardized protocols, with testing that accounts for circadian rhythmicity and that directly compares women with NCCAH and PCOS will be essential to promulgate 11-oxyandrogens into future practical applications.
Pregnancy in women with NCCAH
For women with NCCAH with irregular and or anovulatory cycles who are interested in conceiving, glucocorticoids are the first-line therapy49. Ovulation induction with clomiphene citrate, followed by other reproductive endocrinology interventions are recommended when pregnancy is not achieved during treatment with glucocorticoids50.
Limited evidence suggests that treatment with glucocorticoids might be reduce the time to conception and the risk of miscarriages in women with NCCAH, which occur at higher rates in this population as compared with healthy women2,11,12,51. Hydrocortisone, prednisone, or prednisolone are all safe to use in women attempting pregnancy, while dexamethasone, which is not inactivated by the placenta, should be avoided, due to risk of fetal hypothalamic-pituitary-adrenal axis and growth suppression2. Women with NCCAH who become pregnant spontaneously, without treatment with glucocorticoids, do not need to be treated with glucocorticoids.
Maternal 17OHP and androstenedione are elevated during pregnancy and cannot be used as biomarkers of CAH control. Hence, pregnant women should be followed clinically. Guidelines are lacking in regards to the optimal management of women with NCCAH during pregnancy. Some practices continue low doses of glucocorticoids thoughout pregnancy if used pre-conception, while others stop glucocorticoids once pregnancy is confirmed or after the first trimester.
Summary and conclusions
NCCAH is a relatively common disease, and it should be suspected and excluded in all women with PCOS-like phenotype, including hirsutism, acne, and menstrual abnormalities. Other non-classic forms of CAH that might mimic PCOS, such as HSD3B or CYP11B1 deficiencies, are extremely rare. Screening with 17OHP, followed by a confirmatory cosyntropin stimulation test if needed, establishes the diagnosis of 21OHD. Because hormonal cutoffs can be ambiguous, in part due to the low numbers, along with immunoassay artefacts, genetic testing is often indicated to confirm the very rare HSD3B or CYP11B1 deficiency when suspected based on clinical and hormonal abnormalities. Although the prevalence of PCOS far exceeds that of CAH, and although overlap in management exists (particularly for hirsutism and acne), treatment of infertility and pre-conception considerations warrant accurate diagnosis. Biomarkers indicative of adrenal as opposed to gonadal androgen excess, such as 11-oxyandrogens, might facilitate the management of patients with hyperandrogenism; studies directly comparing patients with CAH and PCOS are lacking, and will be key to translating pathophysiology knowledge into clinical applications.
Key Points.
Non-classic congenital adrenal hyperplasia (NCCAH) has an overall prevalence of 1:200 in the US population and up to 1:30 in certain ethnic groups, particularly Mediterraneans and Ashkenazi Jews.
21-hydroxylase deficiency accounts for 95% of CAH cases.
Patients with NCCAH do not have adrenal insufficiency and do not need hormonal replacement.
Glucocorticoids can be used in children with NCCAH who present with precocious puberty in order to suppress the adrenal androgen excess; the need for therapy should be reassessed after final height is attained.
Hormonal screening for NCCAH with 17-hydroxyprogesterone is indicated in all patients with polycystic ovarian syndrome (PCOS)-like phenotype, as the two cannot be distinguished clinically.
The treatment of women with NCCAH who present with signs of hyperandrogenism and who are not planning pregnancy is similar to those with PCOS, and includes oral contraceptives +/− anti-androgen therapy. Treatment of infertility, however, starts with glucocorticoids in non-classic CAH women.
Research suggests that steroid biomarkers of primarily adrenal origin, such as 11-hydroxyandrostenedione and its downstream bioactive metabolite 11-ketotestosterone, may offer guidance regarding disease control and management in non-classic CAH.
Synopsis.
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive genetic defects in cortisol synthesis. The altered negative feedback of cortisol to the hypothalamus and the pituitary gland prompts corticotropin-releasing hormone (CRH) and adrenocorticotrophic hormone (ACTH) elevations. Increased ACTH, in turn, has two downstream effects: 1) it overstimulates adrenal steroidogenesis, resulting in an accumulation of steroids above the enzymatic blockage; and 2) when sustained, ACTH elevation promotes adrenal gland enlargement (hence, the term CAH).
Acknowledgement
Figure 1 was created using Biorender.com.
Sources of Funding: This work was supported by the NIH Intramural Research Program and by NIDDK (grant 1K08DK109116 awarded to AFT) and University of Michigan M-Cubed (grant U064177 awarded to AFT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinology and metabolism clinics of North America. 2015;44(2):275–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Speiser PW, Arlt W, Auchus RJ, et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hannah-Shmouni F, Morissette R, Sinaii N, et al. Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians. Genet Med. 2017;19(11):1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. High frequency of nonclassical steroid 21-hydroxylase deficiency. American journal of human genetics. 1985;37(4):650–667. [PMC free article] [PubMed] [Google Scholar]
- 5.Carmina E, Dewailly D, Escobar-Morreale HF, et al. Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Hum Reprod Update. 2017;23(5):580–599. [DOI] [PubMed] [Google Scholar]
- 6.Carroll MC, Campbell RD, Porter RR. Mapping of steroid 21-hydroxylase genes adjacent to complement component C4 genes in HLA, the major histocompatibility complex in man. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(2):521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White PC, Grossberger D, Onufer BJ, et al. Two genes encoding steroid 21-hydroxylase are located near the genes encoding the fourth component of complement in man. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(4):1089–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White PC, New MI, Dupont B. Structure of human steroid 21-hydroxylase genes. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(14):5111–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller WL, Merke DP. Tenascin-X, Congenital Adrenal Hyperplasia, and the CAH-X Syndrome. Hormone research in paediatrics. 2018;89(5):352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21(3):245–291. [DOI] [PubMed] [Google Scholar]
- 11.Moran C, Azziz R, Weintrob N, et al. Reproductive outcome of women with 21-hydroxylase-deficient nonclassic adrenal hyperplasia. J Clin Endocrinol Metab. 2006;91(9):3451–3456. [DOI] [PubMed] [Google Scholar]
- 12.Bidet M, Bellanne-Chantelot C, Galand-Portier MB, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95(3):1182–1190. [DOI] [PubMed] [Google Scholar]
- 13.New MI, Abraham M, Gonzalez B, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 2013;110(7):2611–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finkielstain GP, Chen W, Mehta SP, et al. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011;96(1):E161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32(1):81–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reisch N, Hogler W, Parajes S, et al. A diagnosis not to be missed: nonclassic steroid 11beta-hydroxylase deficiency presenting with premature adrenarche and hirsutism. J Clin Endocrinol Metab. 2013;98(10):E1620–1625. [DOI] [PubMed] [Google Scholar]
- 17.Oriolo C, Fanelli F, Castelli S, et al. Steroid biomarkers for identifying non-classic adrenal hyperplasia due to 21-hydroxylase deficiency in a population of PCOS with suspicious levels of 17OH-progesterone. J Endocrinol Invest. 2020. [DOI] [PubMed] [Google Scholar]
- 18.Papadakis G, Kandaraki EA, Tseniklidi E, Papalou O, Diamanti-Kandarakis E. Polycystic Ovary Syndrome and NC-CAH: Distinct Characteristics and Common Findings. A Systematic Review. Front Endocrinol (Lausanne). 2019;10:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pall M, Azziz R, Beires J, Pignatelli D. The phenotype of hirsute women: a comparison of polycystic ovary syndrome and 21-hydroxylase-deficient nonclassic adrenal hyperplasia. Fertil Steril. 2010;94(2):684–689. [DOI] [PubMed] [Google Scholar]
- 20.Maffazioli GDN, Bachega T, Hayashida SAY, et al. Steroid Screening Tools Differentiating Nonclassical Congenital Adrenal Hyperplasia and Polycystic Ovary Syndrome. J Clin Endocrinol Metab. 2020;105(8). [DOI] [PubMed] [Google Scholar]
- 21.Escobar-Morreale HF, Sanchon R, San Millan JL. A prospective study of the prevalence of nonclassical congenital adrenal hyperplasia among women presenting with hyperandrogenic symptoms and signs. J Clin Endocrinol Metab. 2008;93(2):527–533. [DOI] [PubMed] [Google Scholar]
- 22.Rudnicka E, Kunicki M, Radowicki S. [Androgen and 17-hydroxyprogesterone concentrations in blood serum versus menstrual patterns in women with polycystic ovary syndrome (PCOS)]. Ginekologia polska. 2010;81(10):745–749. [PubMed] [Google Scholar]
- 23.Turcu AF, El-Maouche D, Zhao L, et al. Androgen excess and diagnostic steroid biomarkers for nonclassic 21-hydroxylase deficiency without cosyntropin stimulation. European journal of endocrinology / European Federation of Endocrine Societies. 2020;183(1):63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mermejo LM, Elias LL, Marui S, Moreira AC, Mendonca BB, de Castro M. Refining hormonal diagnosis of type II 3β-hydroxysteroid dehydrogenase deficiency in patients with premature pubarche and hirsutism based on HSD3B2 genotyping. The Journal of clinical endocrinology and metabolism. 2005;90(3):1287–1293. [DOI] [PubMed] [Google Scholar]
- 25.White PC, Curnow KM, Pascoe L. Disorders of steroid 11β-hydroxylase isozymes. Endocrine reviews. 1994;15(4):421–438. [DOI] [PubMed] [Google Scholar]
- 26.Turcu AF, Rege J, Auchus RJ, Rainey WE. 11-Oxygenated androgens in health and disease. Nat Rev Endocrinol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Speiser PW, Dupont J, Zhu D, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. The Journal of clinical investigation. 1992;90(2):584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. The Journal of clinical endocrinology and metabolism. 2000;85(3):1059–1065. [DOI] [PubMed] [Google Scholar]
- 29.Turcu AF, Rege J, Auchus RJ, Rainey WE. 11-Oxygenated androgens in health and disease. Nat Rev Endocrinol. 2020;16(5):284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rege J, Nakamura Y, Wang T, Merchen TD, Sasano H, Rainey WE. Transcriptome profiling reveals differentially expressed transcripts between the human adrenal zona fasciculata and zona reticularis. The Journal of clinical endocrinology and metabolism. 2014;99(3):E518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rege J, Nakamura Y, Satoh F, et al. Liquid chromatography-tandem mass spectrometry analysis of human adrenal vein 19-carbon steroids before and after ACTH stimulation. The Journal of clinical endocrinology and metabolism. 2013;98(3):1182–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turcu AF, Nanba AT, Chomic R, et al. Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency. European journal of endocrinology / European Federation of Endocrine Societies. 2016;174(5):601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rege J, Turcu AF, Kasa-Vubu JZ, et al. 11-Ketotestosterone Is the Dominant Circulating Bioactive Androgen During Normal and Premature Adrenarche. The Journal of clinical endocrinology and metabolism. 2018;103(12):4589–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pretorius E, Africander DJ, Vlok M, Perkins MS, Quanson J, Storbeck KH. 11-Ketotestosterone and 11-Ketodihydrotestosterone in Castration Resistant Prostate Cancer: Potent Androgens Which Can No Longer Be Ignored. PloS one. 2016;11(7):e0159867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turcu AF, Mallappa A, Elman MS, et al. 11-Oxygenated Androgens Are Biomarkers of Adrenal Volume and Testicular Adrenal Rest Tumors in 21-Hydroxylase Deficiency. The Journal of clinical endocrinology and metabolism. 2017;102(8):2701–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turcu AF, El-Maouche D, Zhao L, et al. Androgen excess and diagnostic steroid biomarkers for nonclassic 21-hydroxylase deficiency without cosyntropin stimulation. European Journal of Endocrinology. 2020;183(1):63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Auchus RJ, Arlt W. Approach to the patient: the adult with congenital adrenal hyperplasia. The Journal of clinical endocrinology and metabolism. 2013;98(7):2645–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. The Journal of clinical endocrinology and metabolism. 1994;79(5):1328–1333. [DOI] [PubMed] [Google Scholar]
- 39.Azziz R, Black V, Hines GA, Fox LM, Boots LR. Adrenal androgen excess in the polycystic ovary syndrome: sensitivity and responsivity of the hypothalamic-pituitary-adrenal axis. The Journal of clinical endocrinology and metabolism. 1998;83(7):2317–2323. [DOI] [PubMed] [Google Scholar]
- 40.Puurunen J, Piltonen T, Jaakkola P, Ruokonen A, Morin-Papunen L, Tapanainen JS. Adrenal androgen production capacity remains high up to menopause in women with polycystic ovary syndrome. The Journal of clinical endocrinology and metabolism. 2009;94(6):1973–1978. [DOI] [PubMed] [Google Scholar]
- 41.Martin KA, Anderson RR, Chang RJ, et al. Evaluation and Treatment of Hirsutism in Premenopausal Women: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103(4):1233–1257. [DOI] [PubMed] [Google Scholar]
- 42.Corvol P, Michaud A, Menard J, Freifeld M, Mahoudeau J. Antiandrogenic effect of spirolactones: mechanism of action. Endocrinology. 1975;97(1):52–58. [DOI] [PubMed] [Google Scholar]
- 43.Swiglo BA, Cosma M, Flynn DN, et al. Clinical review: Antiandrogens for the treatment of hirsutism: a systematic review and metaanalyses of randomized controlled trials. J Clin Endocrinol Metab. 2008;93(4):1153–1160. [DOI] [PubMed] [Google Scholar]
- 44.Frank-Raue K, Junga G, Raue F, Vecsei P, Ziegler R. [Therapy of hirsutism in females with adrenal enzyme defects of steroid hormone biosynthesis: comparison of dexamethasone with cyproterone acetate]. Klin Wochenschr. 1990;68(12):597–601. [DOI] [PubMed] [Google Scholar]
- 45.Spritzer P, Billaud L, Thalabard JC, et al. Cyproterone acetate versus hydrocortisone treatment in late-onset adrenal hyperplasia. The Journal of clinical endocrinology and metabolism. 1990;70(3):642–646. [DOI] [PubMed] [Google Scholar]
- 46.O’Reilly MW, Kempegowda P, Jenkinson C, et al. 11-oxygenated C19 steroids are the predominant androgens in polycystic ovary syndrome. The Journal of clinical endocrinology and metabolism. 2016:jc20163285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida T, Matsuzaki T, Miyado M, et al. 11-oxygenated C19 steroids as circulating androgens in women with polycystic ovary syndrome. Endocr J. 2018;65(10):979–990. [DOI] [PubMed] [Google Scholar]
- 48.Turcu AF, Wannachalee T, Tsodikov A, et al. Comprehensive Analysis of Steroid Biomarkers for Guiding Primary Aldosteronism Subtyping. Hypertension. 2020;75(1):183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Birnbaum MD, Rose LI. The partial adrenocortical hydroxylase deficiency syndrome in infertile women. Fertility and sterility. 1979;32(5):536–541. [DOI] [PubMed] [Google Scholar]
- 50.Lo JC, Grumbach MM. Pregnancy outcomes in women with congenital virilizing adrenal hyperplasia. Endocrinology and metabolism clinics of North America. 2001;30(1):207–229. [DOI] [PubMed] [Google Scholar]
- 51.Eyal O, Ayalon-Dangur I, Segev-Becker A, Schachter-Davidov A, Israel S, Weintrob N. Pregnancy in women with nonclassic congenital adrenal hyperplasia: Time to conceive and outcome. Clin Endocrinol (Oxf). 2017;87(5):552–556. [DOI] [PubMed] [Google Scholar]
- 52.Miller WL. MECHANISMS IN ENDOCRINOLOGY: Rare defects in adrenal steroidogenesis. Eur J Endocrinol. 2018;179(3):R125–R141. [DOI] [PubMed] [Google Scholar]
- 53.Al Alawi AM, Nordenstrom A, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase type 2 deficiency. Endocrine. 2019;63(3):407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khattab A, Haider S, Kumar A, et al. Clinical, genetic, and structural basis of congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. Proc Natl Acad Sci U S A. 2017;114(10):E1933–E1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Costa-Santos M, Kater CE, Auchus RJ, Brazilian Congenital Adrenal Hyperplasia Multicenter Study G. Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab. 2004;89(1):49–60. [DOI] [PubMed] [Google Scholar]
- 56.Sousa Paredes SC, Marques O, Alves M. Partial deficiency of 17alpha-hydroxylase: a rare cause of congenital adrenal hyperplasia. BMJ Case Rep. 2019;12(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arlt W, Walker EA, Draper N, et al. Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet. 2004;363(9427):2128–2135. [DOI] [PubMed] [Google Scholar]
- 58.Krone N, Reisch N, Idkowiak J, et al. Genotype-phenotype analysis in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2012;97(2):E257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]