

Abstract
The lymph node is a structurally complex organ of the immune system, whose dynamic cellular arrangements are thought to control much of human health. Currently, no methods exist to precisely stimulate substructures within the lymph node or analyze local stimulus-response behaviors, making it difficult to rationally design therapies for inflammatory disease. Here we describe a novel integration of live lymph node slices with a microfluidic system for local stimulation. Slices maintained the cellular organization of the lymph node while making its core experimentally accessible. The 3-layer polydimethylsiloxane device consisted of a perfusion chamber stacked atop stimulation ports fed by underlying microfluidic channels. Fluorescent dextrans similar in size to common proteins, 40- and 70-kDa, were delivered to live lymph node slices with 284 ± 9 μm and 202 ± 15 μm spatial resolution, respectively, after 5 sec, which is sufficient to target functional zones of the lymph node. The spread and quantity of stimulation were controlled by varying the flow rates of delivery; these were predictable using a computational model of isotropic diffusion and convection through the tissue. Delivery to two separate regions simultaneously was demonstrated, to mimic complex intercellular signaling. Delivery of a model therapeutic, glucose-conjugated albumin, to specific regions of the lymph node indicated that retention of the drug was greater in the B-cell zone than in the T-cell zone. Together, this work provides a novel platform, the lymph node slice-on-a-chip, to target and study local events in the lymph node and to inform the development of new immunotherapeutics.
Keywords: microfluidics, slice culture, diffusion, immunotherapy, bioanalytical chemistry
Introduction
This paper describes a novel approach to the study of localized signaling in live lymph node tissue. Events in the lymph node determine how well we fight infections and respond to vaccines, whether a nascent tumor is recognized and destroyed, and whether our own tissues remain safe from autoimmunity. Over a decade of confocal and two-photon live imaging has revealed that cells and signaling molecules in the lymph node are organized in dynamic, intricate arrangements.1,2 It is thought that each cell type is strategically positioned to sense and release molecular signals at just the right location.3,4 Cells called B cells, which produce antibodies once activated, are localized in the outer layer, or cortex, of the lymph node (Figure 1a), where they can rapidly internalize proteins that drain from the periphery.5,6 Cells called T cells are concentrated in the interior, or paracortex, and recently have become a central focus for targeted cancer therapeutics.7 Although these essential substructures are readily imaged, at a few hundred microns in length scale, they are challenging to manipulate experimentally.
Figure 1:

Microfluidic device for local stimulation of lymph-node slices. a) Schematic of key structural features of the lymph node. b) Exploded view of 3-layer PDMS device. Layer 1 contained ten 100 μm x 100 μm microchannels, each with a Y-inlet (Layer 1). Ports in Layer 2 were aligned over the channels, forming the bottom of a perfusion chamber for slice culture (Layer 3). c) Side-view schematic of the device in use. The tissue slice (brown) was perfused with media (blue) above. Stimulus solution (green) was delivered through a microchannel, partially exiting through a port to stimulate a local region of the slice. d) Photograph of assembled device and e) micrograph of the ports aligned over the microchannels. The M-shape design facilitated multiple access points across the lymph node.
Peripheral infection, vaccination, and drug delivery create molecular stimuli that are routed specifically to either the B-cell or T-cell zone8-10, but the role of such local stimulation is difficult to test with current technology. Precisely targeting functional regions in the lymph node, e.g. with cytokines or pharmaceuticals, would elucidate differences in how each region responds to vaccinations or therapeutics. We hypothesize that the ability to probe and monitor local signaling in intact tissue could shift our understanding of how spatial organization drives immune responses. However, current experimental models are either too reduced (cell culture) or too complex (in vivo) to access specific regions precisely. In vitro cell culture is a powerful technique for studying population and single cells, but without the extracellular matrix, structural information about signaling is not available. In vivo experiments maintain the structural integrity, but locally controlled stimulation requires sophisticated systems with transgenic photo-activated receptors11,12 or micromanipulated injections.13 Furthermore, it is challenging to control the concentration and spread of stimuli when bathing cell or tissue samples in vitro or when injecting in vivo. New technologies with intermediate complexity are needed to probe the spatially complex signaling in the lymph node.
We hypothesized that local stimulation of the lymph node was possible by using microfluidics in combination with ex vivo tissue slices. Using viable slices of lymph node tissue, rather than cell cultures or intact explanted lymph nodes, maintains the spatial integrity of the organ while rendering it experimentally accessible. Lymph node14-18 and thymus slices19,20 have been used previously for measurements of T-cell motility, and tissue slices are well established in neurophysiology work and for analysis of tumor tissue.21-24 Here we took advantage of lymph node slices as a novel means to reach specific substructures of the tissue.
Microfluidics, a technology to control fluidic delivery at the 10 – 1000 μm scale,25 provides a unique solution for spatial analysis of living systems.26,27 For example, microfluidic devices have been used to deliver stimuli to slices of brain tissue on chip28-32 and to local regions in cell cultures or even individual cells.33-36 These technologies have provided spatially and temporally precise stimulation by using ports below the tissue,29 laminar flow beneath a culture membrane,34,37 or a mobile probe.31,33,35,36 Micromanipulation has also provided localized stimulation of other tissue samples and whole organisms.23,38-41 If developed for tissues from the immune system, microfluidic analysis could shift our understanding of immunity and aid in developing targeted immunotherapies in the future.
Here we describe the development of a microfluidic platform to provide the first local chemical stimulation of lymph node tissue. After confirming the viability of 300-μm thick lymph node slices, we identified the conditions needed for fluidic delivery with 200-300 μm spatial resolution. Stimulus concentration and timing of delivery were easily controlled via a Y-channel inlet. Multiple stimulations were possible across the slice due to the design of the exit port layer. As a proof-of-principle, we tested local responses to stimulation with a model therapeutic agent in specific regions of the lymph node slice, illustrating the potential to illuminate spatial dynamics of the immune system.
Experimental
Device fabrication:
The top and bottom layers of the microfluidic device were fabricated using standard soft lithography techniques.42 Briefly, transparency masks were drawn in AutoCAD LT 2015 and printed at 20,000 DPI by CAD/Art Services, Inc. (Bandon, OR). Master molds were fabricated using photolithography of SU-8 photoresist (Microchem, Westborough MA, USA) on 3” silicon wafers in a class 1000 cleanroom. Replicas of the device were made using soft lithography of polydimethylsiloxane at a 10:1 ratio of silicon elastomer base to curing agent (PDMS, Elllsworth Adhesives, Germantown WI, USA). The perfusion chamber in Layer 3 was punched with a 12-mm tissue punch; its inlet and outlet were created with a 1.25 mm I.D. tissue punch to accommodate the polyethylene 1.57 O.D. x 1.14 I.D. perfusion tubing (Warner Instruments, Hamden CT, USA). A 0.75 mm I.D. tissue punch was used to punch channel inlets in Layer 3. All tissue punches were obtained from World Precision Instruments, Sarasota FL, USA. The middle layer of the device consisted of a 250 μm-thick PDMS sheet (SSP, Ballston Spa NY, USA) that had been laser etched (Versa Laser 3.5, Universal Laser Systems, Scottsdale AZ, USA) with 80-μm ports and 1000-μm through-holes designed to align with the channel inlets in Layer 3. Layers 2 (middle) and 3 (top) were manually aligned and permanently bonded with air plasma (Tegal Plasmod). These layers were then aligned with Layer 1 (bottom), which was held in place by non-covalent conformal contact.
Determining the flow rate through the port:
Each channel in the device had two potential outlets for fluid flow: the port and waste outlet at the terminus of the channel. To estimate the flow rate passing through the port, a slice of agarose was placed over the port and immersed in PBS. The channel was pre-filled with PBS. PBS was flowed through the channel for 30 min at 0.4 μL/min and collected in a centrifuge tube at the waste outlet. The theoretical volume (12 μL) was compared to the actual volume collected (8.32 ± 0.02 μL, n = 3 channels). Therefore, the flow rate through the port in this experiment was estimated as 3.67 ± 0.02 μL in 30 min, or 0.12 μL/min.
Lymph node slice preparation:
Lymph node slices were prepared similarly to rodent brain slices43,44, and based on published protocols for lymph node and thymus.14,16 All animal work was approved by the Animal Care and Use Committee at the University of Virginia. Male and female C57BL/6 mice were purchased from Jackson Laboratory or Taconic (USA) and used while 6-12 weeks old. Mice were housed in a vivarium and given food and water ab libitum. On the day of the experiment, mice were anesthetized with isoflurane and immediately euthanized by cervical dislocation. The six inguinal, brachial, and axillary lymph nodes were immediately removed and placed in ice-cold DPBS without calcium or magnesium (Lonza, Walkersville MD USA, #17-512F), supplemented with 2 % heat-inactivated FBS (Gibco, Fisher Scientific), for 2 min for recovery. The lymph nodes were then embedded in 6 % low melting point agarose (Lonza, Walkersville MD, USA) prepared in PBS and chilled on ice. Tissue was sliced 300 μm thick in ice-cold PBS using a vibratome (Leica VT1000S, Bannockburn, IL, USA) set to a speed of 90 and frequency of 3. Slices were immediately immersed in RPMI supplemented with 10 % FBS, 1x L-glutamine, and 1x Pen/Strep (Fisher Scientific) and kept in a humidified sterile incubator at 37°C with 5% CO2 for at least one hour before the experiment. Slices of agarose without tissue were prepared in the same manner. See supplemental methods for methods used to test slice viability.
Viability of lymph node slices:
To assess the viability of cultured lymph node slices, slices were collected as described in the Methods section and placed in an incubator at 37 °C with 5 % CO2 for an initial 1 hr recovery period after surgery. Slices remained immersed in media, without media exchange, for the entire culture period. At each time point (0, 2, 6, or 24 hours after the recovery period), pools of 20 slices were crushed through a 70-μm filter, centrifuged (Sorvall ST40R Centrifuge, Fisher Scientific) at 400G for 5 minutes, and resuspended in PBS. Cell suspensions were immediately stained with 2 μM Calcein AM (Life Technologies) and 0.5 μg/mL 7-AAD (Biolegend, San Diego CA, USA). A subset of slices were killed at time 0 with 70% ethanol for 15 minutes before staining, as a negative control. Cell viability was analyzed using a FACS Calibur flow cytometer (BD, USA).
To assess on-chip viability, lymph node slices were cultured in PBS on the microfluidic chip with and without perfusion for 1 hr and 4 hr. Perfusion was achieved using a peristaltic pump (Watson Marlow 120S/DM2, Wilmington MA, USA) with Manifold Solva orange pump tubing (1.D. 0.89 mm, Fisher Scientific) connected to Polyethylene 1.57 O.D. x 1.14 I.D. PE-160/100 tubing (Warner Instruments) at a rate of 0.15 mL/min. A subset of slices were killed with 70 % ethanol for 15 minutes to serve as a negative control. Another subset of slices were cultured in a well plate in media (37°C with 5 % CO2) to serve as a positive control. After 1 hr or 4 hr, slices were stained for 20 minutes with a LIVE/DEAD assay consisting of Calcein AM (Life Technologies) to stain live cells and propidium iodide (Fisher Scientific) to stain dead cells. Slices were analyzed on a ClarioSTAR plate reader (BMG LABTECH, Ortenberg, Germany). The ratio of LIVE/DEAD fluorescent intensities from the plate reader was calculated to quantitate the viability of the tissue after each condition.
Microfluidic delivery of dextrans to lymph node slices and agarose:
For delivery experiments, a single slice of tissue or agarose was placed in the perfusion chamber of the device and submerged with PBS with 2% FBS. The slice was weighted down by placing a small stainless steel washer (10 mm O.D. and 5.3 mm I.D., Grainger USA) over top the agarose. Chemyx syringe pumps (Houston TX, USA) with nonshrinkable PTFE TT-30 tubing with 0.012” I.D. and 0.009” wall thickness (Weico Wire, Edgewood NY, USA) were used for delivery (100-μL Hamilton Model 1710 RN syringe with 26s gauge, large hub needle). Solutions of FITC-dextran of varying molecular weight (3, 10, 40, 70 kDa; all at 0.2 mg/mL; Life Technologies) in PBS were delivered to the slice at room temperature and in room air for varying lengths of time from 3 to 60 sec (“pulse time”). Each delivery was performed as follows: Separate syringes loaded with PBS or FITC-dextran were connected to the two inlets of a Y-channel. The channel was primed with PBS, which was kept at a constant flow rate throughout the experiment. A pulse of dextran was delivered by starting and stopping the flow of dextran solution at designated time intervals; the PBS was left on to continually flush the dextran out of the tissue. This procedure allowed multiple pulse times to be tested in a single slice. Perfusion over the top of the slice did not significantly affect the spread of dextrans in the tissue (Figure S4). Therefore, for ease of experimentation, all characterization was done in the absence of perfusion. The flow rate for the dextran and PBS delivery was 0.2 μL/min each (total flow rate of 0.4 μL/min; final dextran concentration 0.1 mg/mL) unless otherwise noted. At this flow rate, the dextran and PBS streams mixed within the first few millimeters, providing a uniform concentration distribution at the port. See supplemental methods for estimation of actual flow rate through the port.
For the flow rate experiment, the dextran and PBS flow rates were varied while being held in a constant ratio, to keep the concentration delivered constant at 0.1 mg/mL. For the concentration experiment, the flow rate ratio was varied while holding the total flow rate constant at 0.4 μL/min. For the dual delivery experiment, FITC-conjugated 70-kDa Dextran and Texas Red-conjugated 10-kDa Dextran (Life Technologies) were delivered through separate ports.
Fluorescent microscopy and image analysis:
All delivery experiments were imaged in real time on a Zeiss AxioZoom macroscope (Carl Zeiss Microscopy, Germany) with an Axiocam 506 Mono camera (for glucose-BSA, delivery to agarose, flow rate and concentration experiments, and dual-delivery experiments) and Axiocam 506 color camera (for delivery of dextran and fluorescein to tissue). Filter cubes used were GFP (Zeiss filter set #38), Cy5 (#50), Texas Red (#64), Rhodamine (#43), and DAPI (#49). Time lapse images (14 bit) were collected for the duration of delivery at 500-ms intervals for dextran/fluorescein delivery and 1-s intervals for dual-delivery. Images were analyzed using Zen software (Zeiss) and Image J. Linescans were drawn across the delivery region once the dextran reached maximum intensity in the slice, immediately before PBS flushed it out. Background fluorescence from the slice was measured before each delivery pulse and subtracted from the linescan data. Each linescan was fit with a Gaussian distribution, and the area under each linescan curve was calculated and plotted versus pulse time. Pulse width was calculated at 61 % of the maximum intensity; this gives a width equal to 2 standard deviations of the Gaussian curve.
Microfluidic delivery of Glucose-BSA to lymph node slices:
Bovine serum albumin conjugated to glucose (Glucose-BSA) was purchased from Vector Laboratories (Burlingame CA, USA) and fluorescently labelled with Alexa Fluor 647-succinimidyl ester (Life Technologies) according to manufacturer recommendations. Live lymph node slices were stained with 10 μg/mL FITC anti-mouse CD45R/B220 antibody (clone RA3-6B2) for 60 minutes (Biolegend, San Diego CA, USA) and 1 μg/mL Hoescht 33342 trihydrochloride trihydride for 10 minutes in the incubator before the experiment (Life Technologies). Slices were glucose-starved in PBS + 2% FBS for 1 hour to facilitate enhanced uptake, then transferred to the microfluidic perfusion chamber. 400 μg/mL Alexa Fluor 647-Glucose-BSA in PBS was locally delivered to the T-cell and/or B-cell zones of the slice at a flow rate of 0.4 μL/min for 5 s and was not flushed out with PBS. After local delivery, slices were immersed in media in a 24-well plate and transferred to an incubator at 37 °C and 5% CO2. Images were collected at 5, 15, 30, and 60 min after delivery. For comparison, a different set of slices were soaked in 400 μg/mL Alexa Fluor 647-Glucose-BSA in PBS for one hour in an incubator (global delivery), then washed 3x with PBS and immediately imaged. Images were analyzed in Zeiss Zen software. To quantify spatial distribution, the integrated fluorescence intensity of glucose-BSA was calculated in the slice. Next, the B-cell and T-cell zones were manually outlined according to the intensity of the B220 staining, and the integrated fluorescent intensity of glucose-BSA was calculated in each zone. The percent of signal in each zone was obtained from the ratio of fluorescent intensity in the zone to the total fluorescent signal. See supplemental methods for glucose-BSA uptake analysis by flow cytometry.
Glucose-BSA uptake analysis:
To confirm cellular uptake of glucose-BSA, splenocytes were harvested from naïve mice and incubated in 400 μg/mL Alexa Fluor 647-Glucose-BSA or PBS (control) for 1 hr in an incubator (37 °C and 5 % CO2). The cells were washed, immunostained with 10 μg/mL B220/CD45R-Pacific blue (clone RA-6B2), 10 μg/mL CD4-APC-Cy7 (clone GK1.5), 2.5 μg/mL CD11c-PE (clone N418), and 0.5 μg/mL 7-AAD (all from Biolegend, San Diego CA, USA) analyzed using flow cytometry. Gating was set using single-color and FMO controls.
Statistics:
All values are reported as mean ± SEM unless otherwise noted. All statistics were performed in GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA USA) and considered significant at the 95% confidence interval (p<0.05).
Results and Discussion
Device design for the lymph node slice-on-a-chip:
We designed an integrated microfluidic chip that would provide localized chemical targeting of lymph node slices while permitting live fluorescent imaging (Figure 1b-e). The three-layer microfluidic device was comprised of an on-chip perfusion chamber aligned atop a set of ten parallel 100 μm x 100 μm microchannels, each feeding a single port 80 μm in diameter. Channels beneath the tissue slice provide the ability to discretely deliver stimulant solutions to local regions of tissue.37,45 The channel spacing (200 μm center-to-center distance) and the port diameter were chosen to provide spatially resolved delivery to lymph node substructures such as the B cell and T cell zones, which are typically 200-300 μm in length scale. A Y-channel inlet for each microchannel enabled straightforward control of the concentration and timing of delivery, as well as the ability to rapidly flush out the stimulus solution from the tissue without having to reload syringes or pumps. We used an M-shaped layout for the ports, rather than a straight line, to facilitate multiple access points across the lymph node slice.
Viability of lymph node slices off-chip and on-chip:
Culture of live lymph node slices is a relatively new technique14-16, so we first sought to verify the viability of the tissue over the timescale of the experiments. To collect slices, the brachial, inguinal, and axillary lymph nodes from mice were removed, embedded in 6 % agarose, sliced on a vibratome, and allowed to recover for 1 hr while immersed in media in a cell culture incubator (5 % CO2) at 37 °C. Slice viability during culture was first tested using flow cytometry (Figure S1). Under these conditions, slices remained viable for at least 6 hours (<7 % membrane-permeable cells) with a drop in viability by 24 hours, similar to cultures of splenocytes46 and comparable to acute brain slice culture.43 Further optimization of lymph node slice viability will be required for study of longer term immunological responses, such as antigen recognition (24 or 48 hr), proliferation of T cells (3-5 days) or induction of germinal centers (6 days).47
Next, lymph node slice viability was validated on the microfluidic device (Figure S2). There was no significant difference in viability between slices cultured in an incubator or cultured on chip, after 1 or 4 hr. This result indicates that slice viability was not altered by the chip on this timescale. Furthermore, there was no difference in viability with or without perfusion of media on the chip in this time period, similar to results in other microtissues23, although we note that perfusion or media exchange may be needed when studying function in future experiments.
Local delivery to agarose and lymph node tissue:
To study how cells in specific regions respond to stimuli, migrate before and after inflammatory triggers, or how local signals initiate structural changes in the lymph node, it is essential to control the quantity and resolution of delivery. To characterize the spatial resolution of our device, we delivered peptide and protein-sized molecules into slices by using FITC-labelled dextrans of varying molecular weight (3 to 70 kDa) while taking time lapse images every 500 ms (Movie S1). Repeated deliveries were made possible by flushing the dextran out of the tissue with PBS immediately after each delivery. Delivery into tissue slices (Figure 2) was compared to delivery into 2 % agarose (Figure S3) in order to isolate any effects of the tissue structure. Delivery profiles were well fit by a Gaussian distribution in both the agarose (Figure S3a,b) and the tissue (Figure 2a,b). The quantity delivered was assessed by the area under the Gaussian curve (AUC), and the width of the peak, or pulse width, was assessed at 61% of maximum intensity. AUC increased linearly at small pulse times in both agarose and tissue (Figure S3c and Figure 2c, respectively), then continued to increase linearly with a shallower slope at longer times (> 20 s; Figure S3e and Figure S5). This suggested that for longer delivery times, the tissue may approach a saturation point or the dextran may diffuse out the top of the tissue.
Figure 2:

Delivery of FITC-Dextran to 300 μm-thick murine lymph node tissue slices. a) Representative bright field (top) and fluorescent (bottom) images of 0.1 mg/mL 40 kDa-FITC dextran delivery to a tissue slice. In the lower image, the slice is outlined in white and the direction of flow through the active channel is shown by yellow arrow. A line scan was taken (green dotted line) to analyze quantity and resolution of delivery. b) Representative line scan data for a 5-s delivery of 40 kDa-FITC Dextran. Curve was fit with a Gaussian distribution (black trace), which was used to calculate the area under the curve (AUC) and the width of the delivered pulse, taken at 61 % of the max (2 standard deviations). c) AUC increased linearly with increasing pulse time to 15 s (r2=0.96-0.98 for all curves). d) Pulse width increased linearly at small pulse times (r2=0.80-0.99). n = 5, mean ± SEM.
Pulse width also increased linearly at small pulse times (3 - 15 s), providing predictable local stimulation (Figure S3d, Figure 2d). Delivery times longer than 20 s did not provide local stimulation under these conditions, and the width of delivery began to reach saturation. On average, a 317 ± 16 μm region of agarose (n = 5 slices) and a 284 ± 9 μm region of tissue (n = 5 slices) were stimulated by a 5-s delivery of 40-kDa FITC dextran. Consistent with a diffusion-controlled process, there was a significant dependency of pulse width on molecular weight (Figure S3g, two-way ANOVA, F(4,31) = 14.4, p < 0.0001), with larger molecules travelling shorter distances. We note that both diffusion and convection from the port contribute to the observed lateral spread in these experiments, due to continuous flow of dextran solution and PBS into the tissue during delivery. Overall, the device provides sufficient resolution to deliver proteins such as cytokines, which typically have molecular weights of 15 - 50 kDa, to specific sub-structures in the lymph node.
Experimental modeling of mass transport in brain tissue is often done in dilute (0.2 – 1 %) agarose due its similarity in permeability.48 It is not known how molecular diffusion in lymph node tissue compares to brain. Interestingly, we observed that pulse width was not dependent on whether the probes were delivered to 2 % agarose or lymph node tissue (Figure S3g, two-way ANOVA, F(4,31) = 0.295, p = 0.88). The similarity between agarose and tissue suggests that 2 % agarose may be a suitable matrix for modeling mass transport in lymph node tissue.
Microfluidic control over removal of stimuli:
Precise temporal control of stimulation is an advantage of using microfluidics. Here, the Y-channel design provided a means to rapidly remove the stimulus from the tissue after timed delivery. During experiments dextran was cleared from the tissue after each pulse by continued flow of PBS (Movie S1). We observed that the FITC-dextran signal was cleared from the tissue in 30 - 80 s, depending on the molecular weight. In a 2-dimensional computational model of the experiment (Comsol Multiphysics, see supplemental methods), which incorporated convection and isotropic diffusion, this removal was predicted to take approximately 60 - 120 s at a flow rate of 0.4 μL/min (Figure 3a and Movie S2). The shorter experimental clearance times may be due to lack of sensitivity to trace quantities of fluorophore in the experimental system, or to differences in fluid flow and mass transport through the tissue compared to the simulated matrix. Nonetheless, these results show that protein-sized molecules were removed by continued flushing of saline to below the limit of detection in seconds to minutes after delivery.
Figure 3:
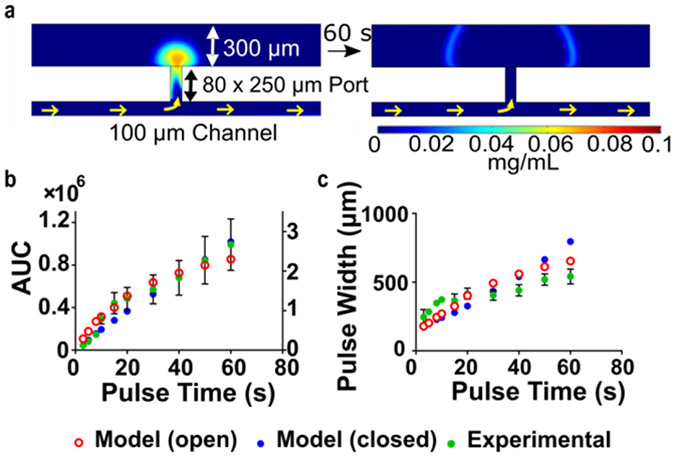
Diffusion out the top of the tissue is predicted to effect lateral spread. a) Concentration distribution plot (left) after a simulated 5-s delivery of 0.1 mg/mL FITC 40 kDa-Dextran, and (right) after 60 s of continued buffer flow. The tissue was modeled as a porous matrix, in this case with an open boundary so that mass transport could occur out the top. Color scale below shows concentration. Predicted AUC (right Y-axis in b) and pulse width (c) vs. pulse time for tissue with an open (red open circles) or closed (blue circles) top boundary. Experimental data (green circles) is overlaid and was most comparable to the open-boundary model.
We hypothesized that the spatial resolution of delivery was improved by diffusion out the top of the tissue. Simulations of the experiment supported this hypothesis at long but not short delivery times (Figure 3 b-c). By modeling the tissue with an open or closed top boundary, it was confirmed that loss out the top slowed the accumulation and lateral spread of the dextran at longer delivery times (>40-60 s). At shorter times, the open-top model actually had increased influx (AUC) of dextran, because of the increased vertical convection passing through the slice. Experimental data for AUC and pulse width followed similar trends as the open-boundary model, which suggests that diffusion does occur out the top of the tissue and impacts the quantity and resolution of delivery.
On-demand microfluidic control over quantity delivered and spatial resolution:
When delivering chemical stimuli to tissue, it is desirable to be able to control the quantity and spatial resolution of delivery separately. We hypothesized that increasing the concentration of stimulus at the port would increase the quantity delivered, independent of the pulse width. To test this hypothesis experimentally in lymph node slices, the total flow rate during delivery was held constant (0.4 μL/min), while the individual flow rates of 40-kDa dextran and saline at the Y-channel inlets were varied (Figure 4a; raw data in Figure S6a). The experiment was replicated in the computational model by varying the concentration of dextran at the channel inlet. The total quantity delivered (area under the curve) increased linearly with increasing concentration in both the experiment and the model (Figure 4c). In contrast, pulse width was not significantly dependent on delivery concentration in the experiment or the model (Figure 4e; one-way ANOVA, p>0.05, n = 5). Thus, quantity delivered could be increased with minimal effect on the pulse width, simply by varying the flow rates at the Y-channel inlet.
Figure 4:

Varying pulse width and quantity delivered on the microfluidic chip. a-b) Schematics of experiments. In lymph node slices, area under the curve (AUC) increased linearly with c) increasing concentration and d) increasing flow rate. AUC represents integrated fluorescent units and integrated concentration in the experiment and model, respectively. Because of differing units, only trends, not values, can be compared. e) Pulse width was not significantly dependent on delivery concentration (one-way ANOVA, P>0.05) but f) increased linearly with increasing flow rate. n = 5, mean ± SEM. Experiments were simulated using COMSOL Multiphysics (red and right Y-axis for d-e).
The flow through the port drives convection through the lymph node matrix that is expected to contribute to the entry and spread of chemical stimuli. The estimated flow rate through the port when delivering at a total flow rate of 0.4 μL/min was 0.12 μL/min, approximately 1/3 of the total flow rate (see Methods). To test the contribution of convection through the lymph node tissue on the chip, total flow rate was varied by increasing the flow rate in each channel equally (Figure 4b,d,f; raw data in Figure S6b). The experiment was simulated by varying the flow velocity at the channel inlet. In both the experiment and the model, AUC and pulse width were significantly dependent on flow rate (one-way ANOVA, p<0.05 for each). The amount delivered was less sensitive to flow rate than to concentration: doubling the concentration or the flow rate increased the experimental AUC by 3.5-fold or 1.7-fold, respectively. This property may be useful when aiming to deliver stimuli at physiologically relevant flow rates. Alternatively, to mitigate the contribution of flow, a porous membrane could be incorporated as a barrier beneath the tissue.37
Dual stimulation of lymph node slices
To recreate the complex intercellular signaling that occurs in the node, both the location and timing of delivery must be controlled, for molecules of varying size and molecular weight. We modelled this situation by delivering FITC-conjugated 70-kDa dextran and Texas Red-conjugated 10-kDa Dextran through different ports into a single tissue slice (Figure 5 and Movie S2). Ports were chosen to deliver the reagents to non-overlapping regions, and the initiation of flow and PBS flush were staggered in time. FITC-70-kDa Dextran was delivered first (Figure 5b) for 5 seconds, followed by a subsequent flush with PBS, and Texas red-10-kDa Dextran (Figure 5d) was delivered in between. Next, Texas red-10-kDa Dextran was flushed out (Figure 5f-g), and within a few seconds both dextrans were removed from the tissue by PBS (flushing flow rate increased to 5 μL/min, Figure 5h). The flushing caused an increase in lateral spread through the tissue. To minimize this, future versions of the device could include additional sources of buffer flow to minimize the lateral spread during delivery and flushing.37 This experiment demonstrates the potential for delivery of multiple drugs or stimuli in parallel in a single microfluidic experiment.
Figure 5:

Selected time lapse images from a dual delivery of 70 kDa FITC-Dextran (green) and 10 kDa Texas Red-Dextran (red) to a lymph node tissue slice. In (a), the slice is outlined in white dashed lines, and the active ports are traced in white dotted lines. Images are shown from each stage of the experiment, according to the timeline above.
Local activation of immune cells on-chip:
To test the hypothesis that local stimulation of lymph node tissue can reveal how specific regions respond to stimuli, we delivered a model therapeutic to specific regions of the lymph node and monitored the response (Figure 6). Glucose-conjugates have been utilized as drug delivery systems to target cancer cells through receptor-mediated endocytosis49,50; conjugates with glucose or other metabolites may also provide a means to preferentially reach glycolitically active lymphocytes7. Targeting T-cells with glucose conjugates could potentially prevent T-cell dysfunction in hostile environments during cancer7, while targeting B-cells could potentially be used to deactivate B-cell receptor signaling to improve tumor viability and resistance to therapy.51
Figure 6:

Local stimulation of the T-cell and B-cell regions of lymph node slices on-chip. Live slices were stained with FITC-anti-mouse B220 (green) and Hoechst (blue) to identify B-cell zones. a) Representative delivery of Alexa Fluor-647-Glucose-BSA (red) to the T-cell zone. Representative images collected 5 min and 15 min after delivery to the T-cell zone (b, c), B-cell zone (e, f), and the intersection of the two zones (h, i). All images shown with equal scaling of intensities. Graphs show the average percent of total glucose-BSA signal that was located in each zone, 5 and 15 minutes after delivery. d) Percent signal in the T cell zone dropped significantly by 15 min after delivery there (paired t-test, p<0.05, n = 2). g) Percent signal in the B-cell zone remained high after delivery there (n = 3). j) After delivery to the T/B interface, percent signal in the T cell zone dropped significantly by 15 min (paired t-test, p<0.05, n = 3), but not in the B cell zone. Values are mean ± SEM.
We conjugated glucose-BSA to AlexaFluor 647, then verified uptake by incubating splenocytes with the conjugate and analyzing by flow cytometry (Figure S7). To test whether local targeting was in fact needed, lymph node slices were soaked and incubated with 400 μg/mL of Alexa Fluor 647-glucose-BSA for 1 hr. Both the quantity and the location of uptake differed between slices (Figure S8). Some slices took up glucose-BSA only in the sinus area or B cell zones (Figure S9a,c), while others showed uptake across the tissue (Figure S9b). We hypothesized that local stimulation could circumvent this unpredictability by specifically targeting the region of interest.
Based on its molecular weight, BSA (66 kDa) conjugated to glucose was expected to remain localized within a 300-μm region after short delivery pulses on the microfluidic chip without flushing. This made it a good candidate to test local delivery to the T-cell and B-cell zones within the lymph node (Figure 6). To enable targeting of specific regions, live lymph node slices were immunostained prior to the experiment with FITC-anti-mouse B220, to identify the B-cell zones, and counterstained with Hoechst (blue). For simplicity, the T-cell zone was defined as any region outside the B-cell zone. The device was able to deliver glucose-BSA to these localized regions. To promote retention of the protein in the tissue, all flow was stopped immediately after delivery instead of flushing it out with PBS. When imaged 5 min after delivery to the T-cell (Figure 6a-d) or B-cell (Figure 6e-g) zones, glucose-BSA signal was found predominantly in the targeted region (69 ± 5 % in the T cell zone and 77 ± 5 % in the B cell zone, respectively).
Interestingly, the response to stimulation with Alexa Fluor 647-glucose-BSA differed between the regions. By 15 min after delivery to the T cell zone, glucose-BSA signal was completely cleared from the T-cell zone (0 % of remaining signal), while remaining unchanged in the B-cell zone (Figure 6c,d). In contrast, after delivery to the B-cell zone, the glucose-BSA remained concentrated in that zone (93 ± 3 % of remaining signal, Figure 6f,g). These data suggest that glucose-BSA in the T-cell zone either diffused rapidly out of the tissue slice or, possibly, was transported to the B-cell zone.
To further test these observations, we delivered glucose-BSA to the intersection of the T-cell and B-cell zones for simultaneous stimulation (Figure 6h-i). Five minutes after delivery, the glucose-BSA signal was equally distributed between the T-cell and B-cell zones (49 ± 11 % and 48 ± 13 %, respectively; Figure 6j). However, by 15 minutes, only 1.3 ± 0.4 % was in the T-cell zone and 93 ± 7 % was in the B-cell zone. This data supported the observation that glucose-BSA does not remain in the T-cell zone after short deliveries. The differential response in the T-cell and B-cell zones illustrates the need for analysis of local behaviors. Future experiments with higher resolution microscopy will allow tracking of cell migration and molecular diffusion after delivery, to determine the extent to which glucose-BSA is transported between regions.
Recently, glucose-conjugates were proposed as a potential strategy for targeted drug-delivery to T-cells.7 Interestingly, our initial results suggest that such conjugates may be preferentially captured in the B cell zone rather than the T cell zone of the lymph node, although these experiments cannot distinguish cellular endocytosis from retention in the extracellular matrix. It is possible that longer deliveries to the T-cell zone would facilitate more uptake, although flow cytometry data suggested that, on average, B-cells picked up more glucose-BSA than T-cells did after 1 hr incubation in vitro (Figure S8). If upheld with other conjugates, then targeting the B-cell zone may provide a useful method to facilitate active transport into cells for drug delivery. These findings suggest that local delivery of potential therapeutics with real-time monitoring of the response may become a useful platform for drug-screening and may inform the development of spatially targeted therapies.
Microfluidic delivery is complementary to existing methods for local stimulation of intact lymphoid tissue. For instance, light-induced stimulation of photo-activated receptors11,12,52 and release of caged molecules53,54 offer potentially higher spatial and temporal resolution than fluidic delivery, but require genetically modified animals 55 or specifically designed cage systems for each molecule of interest. Microinjection is another well-established approach for local stimulation in brain13,56, although it may increase the risk of local tissue damage around the insertion site.57 In small organs such as murine lymph nodes, a 30 or 31-gauge needle (310 or 260 μm outer diameter) is sufficient to deliver to an intact node and elicit powerful immune responses58,59, but may cause damage not suitable for studying the effects of spatial organization.60 In contrast, the damage from slicing tissue is localized to the outer 10 – 100 μm of the slice15,61,62, leaving the inner layers intact for microfluidic delivery to specifically immunostained sub-structures. In addition, microfluidics provides the ability to remove or flush out the stimuli from the tissue. Flushing is advantageous because it provides temporal control over the stimulation and enables repeated measurements. Here, we showed removal of stimuli within 30 - 80 s, even at the slow flow rates used here (0.2 μL/min), potentially enabling temporally resolved stimulations on the order of once per minute. Future work to optimize the device for more frequent stimulations could provide rapid pulsing of stimuli in discrete regions of the tissue.
Applications of microfluidic devices to immunology have primarily focused on studying chemotaxis63, purifying specific cell types64,65, quantitating cell secretion at the single cell level66-68, and studying single cell interactions.66,69,70 These techniques illuminate how single cells migrate and communicate, but provide limited spatial information. In complementary work, researchers have used a “bottom-up” approach to recreate complex immunological structures on-chip, starting from cell cultures.71-74 Microfluidics has been used to build 3D cellular constructs for paracrine signaling75 and leukocyte extravasation74, and to mimic lymphatic drainage71,76 and immune cell reorganization during cancer.77 Tissue engineering strategies have created sophisticated in vitro models of lymphoid tissue from cells and extracellular matrix components.78 Here, we took an alternative “top-down” approach to retain the spatial organization of intact tissue while rendering it experimentally accessible.
Conclusions
This paper presents the first experimental system for local chemical stimulation of lymph node tissue on a microfluidic device with 200 – 300 μm spatial resolution. This spatial resolution was sufficient to target discrete regions of a murine lymph node slice, including the B cell and T cell zones. The device provides real-time control over the pulse width and quantity delivered and a unique array of ports for multi-focal activation of tissue. Local stimulation with glucose-BSA revealed that T-cell and B-cell zones responded differently, supporting the need for a system that can tease apart the effects of spatial structure by targeting specific regions within the tissue. Future iterations of the device to improve spatial resolution and incorporate ex vivo detection assays may improve its utility for detecting immune responses in small clusters of cells. In the future, the lymph node slice-on-a-chip may become a powerful platform to study the local responses that drive immunity, with potential applications in fundamental biomedical research and in the development of new immunotherapies.
Supplementary Material
Acknowledgements:
This work was funded by an Individual Biomedical Research Award from The Hartwell Foundation, the Starter Grant Award from the Society for Analytical Chemists of Pittsburgh, and the Department of Chemistry at the University of Virginia. A.E.R. was supported through The American Association of Immunologists Careers in Immunology Fellowship Program. Access to the Versa Laser system was kindly provided by Dr. James Landers. We thank Ben Groff for assistance with immunofluorescence staining.
References:
- 1.Miller MJ, Safrina O, Parker I and Cahalan MD, Imaging the Single Cell Dynamics of CD4+ T Cell Activation by Dendritic Cells in Lymph Nodes. J. Exp. Med, 2004, 200, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang J, van Panhuys N, Kastenmüller W and Germain RN, The Future of Immuno-imaging – Deeper, Bigger, More Precise, and Definitively More Colorful. Eur. J. Immunol, 2013, 43, 1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kastenmüller W, Torabi-Parizi P, Subramanian N, Lämmermann T and Germain RN, A Spatially-Organized Multicellular Innate Immune Response in Lymph Nodes Limits Systemic Pathogen Spread. Cell, 2012, 150, 1235–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Subramanian N, Torabi-Parizi P, Gottschalk RA, Germain RN and Dutta B, Network representations of immune system complexity. Wiley Interdiscip. Rev. Syst. Biol. Med, 2015, 7, 13–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Victora GD and Nussenzweig MC, Germinal centers. Annu. Rev. Immunol, 2012, 30, 429–457. [DOI] [PubMed] [Google Scholar]
- 6.Willard-Mack CL, Normal Structure, Function, and Histology of Lymph Nodes. Toxicol. Pathol, 2006, 34, 409–424. [DOI] [PubMed] [Google Scholar]
- 7.O’Sullivan D and Pearce EL, Targeting T cell metabolism for therapy. Trends Immunol., 2015, 36, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, von Andrian UH and Carroll MC, Conduits Mediate Transport of Low-Molecular-Weight Antigen to Lymph Node Follicles. Immunity, 2009, 30, 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, Pabst R, Lutz MB and Sorokin L, The Conduit System Transports Soluble Antigens from the Afferent Lymph to Resident Dendritic Cells in the T Cell Area of the Lymph Node. Immunity, 2005, 22, 19–29. [DOI] [PubMed] [Google Scholar]
- 10.Girard J-P, Moussion C and Förster R, HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol, 2012, 12, 762–773. [DOI] [PubMed] [Google Scholar]
- 11.Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P and Bamberg E, Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. PNAS, 2003, 100, 13940–13945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chambers JJ and Kramer RH, Light-Activated Ion Channels for Remote Control of Neural Activity. Methods Cell Biol., 2008, 90, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lohman R-J, Liu L, Morris M and O’Brien TJ, Validation of a method for localised microinjection of drugs into thalamic subregions in rats for epilepsy pharmacological studies. J. Neurosci. Methods, 2005, 146, 191–197. [DOI] [PubMed] [Google Scholar]
- 14.Dzhagalov IL, Melichar HJ, Ross JO, Herzmark P and Robey EA, Two-photon imaging of the immune system Curr. Protoc. Cytom. Editor. Board J Paul Robinson Manag. Ed. Al, 2012, Chapter 12, Unit12.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asperti-Boursin F, Real E, Bismuth G, Trautmann A and Donnadieu E, CCR7 ligands control basal T cell motility within lymph node slices in a phosphoinositide 3–kinase– independent manner. J. Exp. Med, 2007, 204, 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salmon H, Rivas-Caicedo A, Asperti-Boursin F, Lebugle C, Bourdoncle P and Donnadieu E, Ex vivo Imaging of T Cells in Murine Lymph Node Slices with Widefield and Confocal Microscopes. J. Vis. Exp. JoVE, 2011, e3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katakai T, Kondo N, Ueda Y and Kinashi T, Autotaxin Produced by Stromal Cells Promotes LFA-1–Independent and Rho-Dependent Interstitial T Cell Motility in the Lymph Node Paracortex. J. Immunol, 2014, 193, 617–626. [DOI] [PubMed] [Google Scholar]
- 18.Katakai T, Habiro K and Kinashi T, Dendritic Cells Regulate High-Speed Interstitial T Cell Migration in the Lymph Node via LFA-1/ICAM-1. J. Immunol, 2013, 191, 1188–1199. [DOI] [PubMed] [Google Scholar]
- 19.Ehrlich LIR, Oh DY, Weissman IL and Lewis RS, Differential Contribution of Chemotaxis and Substrate Restriction to Segregation of Immature and Mature Thymocytes. Immunity, 2009, 31, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhakta NR, Oh DY and Lewis RS, Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat. Immunol, 2005, 6, 143–151. [DOI] [PubMed] [Google Scholar]
- 21.Dunwiddie TV and Diao L, Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J PharmacolExpTher, 1994, 268, 537–545. [PubMed] [Google Scholar]
- 22.Joseph JD, Wang YM, Miles PR, Budygin EA, Picetti R, Gainetdinov RR, Caron MG and Wightman RM, Dopamine autoreceptor regulation of release and uptake in mouse brain slices in the absence of D(3) receptors. Neuroscience, 2002, 112, 39–49. [DOI] [PubMed] [Google Scholar]
- 23.Astolfi M, Péant B, Lateef MA, Rousset N, Kendall-Dupont J, Carmona E, Monet F, Saad F, Provencher D, Mes-Masson A-M and Gervais T, Micro-dissected tumor tissues on chip: an ex vivo method for drug testing and personalized therapy. Lab. Chip, 2016, 16, 312–325. [DOI] [PubMed] [Google Scholar]
- 24.Hattersley SM, Dyer CE, Greenman J and Haswell SJ, Development of a microfluidic device for the maintenance and interrogation of viable tissue biopsies. Lab. Chip, 2008, 8, 1842–1846. [DOI] [PubMed] [Google Scholar]
- 25.Whitesides GM, The origins and the future of microfluidics. Nature, 2006, 442, 368–373. [DOI] [PubMed] [Google Scholar]
- 26.Young EWK and Beebe DJ, Fundamentals of microfluidic cell culture in controlled microenvironments. Chem. Soc. Rev, 2010, 39, 1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Velve-Casquillas G, Le Berre M, Piel M and Tran PT, Microfluidic tools for cell biological research. Nano Today, 2010, 5, 28–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mauleon G, Fall CP and Eddington DT, Precise Spatial and Temporal Control of Oxygen within In Vitro Brain Slices via Microfluidic Gas Channels. PLoS ONE, 2012, 7, e43309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mohammed JS, Caicedo HH, Fall CP and Eddington DT, Microfluidic add-on for standard electrophysiology chambers. Lab. Chip, 2008, 8, 1048–1055. [DOI] [PubMed] [Google Scholar]
- 30.Blake AJ, Rodgers FC, Bassuener A, Hippensteel JA, Pearce TM, Pearce TR, Zarnowska ED, Pearce RA and Williams JC, A microfluidic brain slice perfusion chamber for multisite recording using penetrating electrodes. J. Neurosci. Methods, 2010, 189, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Queval A, Ghattamaneni NR, Perrault CM, Gill R, Mirzaei M, McKinney RA and Juncker D, Chamber and microfluidic probe for microperfusion of organotypic brain slices. Lab. Chip, 2010, 10, 326–334. [DOI] [PubMed] [Google Scholar]
- 32.Huang Y, Williams JC and Johnson SM, Brain slice on a chip: opportunities and challenges of applying microfluidic technology to intact tissues. Lab. Chip, 2012, 12, 2103–2117. [DOI] [PubMed] [Google Scholar]
- 33.Chen D, Du W, Liu Y, Liu W, Kuznetsov A, Mendez FE, Philipson LH and Ismagilov RF, The chemistrode: A droplet-based microfluidic device for stimulation and recording with high temporal, spatial, and chemical resolution. Proc. Natl. Acad. Sci, 2008, 105, 16843–16848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peterman MC, Noolandi J, Blumenkranz MS and Fishman HA, Localized chemical release from an artificial synapse chip. Proc. Natl. Acad. Sci. U. S. A, 2004, 101, 9951–9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Safavieh M, Qasaimeh MA, Vakil A, Juncker D and Gervais T, Two-Aperture Microfluidic Probes as Flow Dipole: Theory and Applications. Sci. Rep, 2015, 5, 11943–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Juncker D, Schmid H and Delamarche E, Multipurpose microfluidic probe. Nat. Mater, 2005, 4, 622–628. [DOI] [PubMed] [Google Scholar]
- 37.Chang TC, Mikheev AM, Huynh W, Monnat RJ, Rostomily RC and Folch A, Parallel microfluidic chemosensitivity testing on individual slice cultures. Lab Chip, 2014, 14, 4540–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucchetta EM, Lee JH, Fu LA, Patel NH and Ismagilov RF, Dynamics of Drosophila embryonic patterning network perturbed in space and time using microfluidics. Nature, 2005, 434, 1134–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lovchik RD, Kaigala GV, Georgiadis M and Delamarche E, Micro-immunohistochemistry using a microfluidic probe. Lab. Chip, 2012, 12, 1040–1043. [DOI] [PubMed] [Google Scholar]
- 40.Candelier R, Sriti Murmu M, Alejo Romano S, Jouary A, Debregeas G and Sumbre G, A microfluidic device to study neuronal and motor responses to acute chemical stimuli in zebrafish. Sci. Rep, 2015, 5, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Feng X, Du W and Liu B, Microfluidic worm-chip for in vivo analysis of neuronal activity upon dynamic chemical stimulations. Anal. Chim. Acta, 2011, 701, 23–28. [DOI] [PubMed] [Google Scholar]
- 42.Qin D, Xia Y and Whitesides GM, Soft lithography for micro- and nanoscale patterning. Nat. Protoc, 2010, 5, 491–502. [DOI] [PubMed] [Google Scholar]
- 43.Ankri L, Yarom Y and Uusisaari MY, Slice It Hot: Acute Adult Brain Slicing in Physiological Temperature. JoVE J. Vis. Exp, 2014, e52068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross AE and Venton BJ, Adenosine transiently modulates stimulated dopamine release in the caudate-putamen via A1 receptors. J. Neurochem, 2015, 132, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaikh Mohammed J, Caicedo H, Fall CP and Eddington DT, Brain Slice Stimulation Using a Microfluidic Network and Standard Perfusion Chamber. J. Vis. Exp. JoVE, 2007, e302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klein AB, Witonsky SG, Ahmed SA, Holladay SD, Gogal RM, Link L and Reilly CM, Impact of different cell isolation techniques on lymphocyte viability and function. J. Immunoassay Immunochem, 2006, 27, 61–76. [DOI] [PubMed] [Google Scholar]
- 47.Murphy K, Travers P, Walport M and Janeway C, Janeway’s immunobiology, Garland Science, New York, 8th ed., 2012. [Google Scholar]
- 48.Tao L and Nicholson C, Diffusion of albumins in rat cortical slices and relevance to volume transmission. Neuroscience, 1996, 75, 839–847. [DOI] [PubMed] [Google Scholar]
- 49.Calvaresi EC and Hergenrother PJ, Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. R. Soc. Chem. 2010, 2013, 4, 2319–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Ma F-K, Dang Q-F, Liang X-G and Chen X-G, Glucose-conjugated chitosan nanoparticles for targeted drug delivery and their specific interaction with tumor cells. Front. Mater. Sci, 2014, 8, 363–372. [Google Scholar]
- 51.Fowler N and Davis E, Targeting B-cell receptor signaling: changing the paradigm. Hematol. Educ. Program Am. Soc. Hematol. Am. Soc. Hematol. Educ. Program, 2013, 2013, 553–560. [DOI] [PubMed] [Google Scholar]
- 52.Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML and Nussenzweig MC, Germinal Center Dynamics Revealed by Multiphoton Microscopy with a Photoactivatable Fluorescent Reporter. Cell, 2010, 143, 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tischer D and Weiner OD, Illuminating cell signalling with optogenetic tools. Nat. Rev. Mol. Cell Biol, 2014, 15, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stutts L and Esser-Kahn AP, A Light-Controlled TLR4 Agonist and Selectable Activation of Cell Subpopulations. ChemBioChem, 2015, 16, 1744–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A and Kanagawa O, Monitoring cellular movement in vivo with photoconvertible fluorescence protein ‘Kaede’ transgenic mice. PNAS, 2008, 105, 10871–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gonzalez-Perez O, Guerrero-Cazares H and Quiñones-Hinojosa A, Targeting of Deep Brain Structures with Microinjections for Delivery of Drugs, Viral Vectors, or Cell Transplants. J. Vis. Exp. JoVE, 2010, e2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bjornsson CS, Oh SJ, Al-Kofahi YA, Lim YJ, Smith KL, Turner JN, De S, Roysam B, Shain W and Kim SJ, Effects of insertion conditions on tissue strain and vascular damage during neuroprosthetic device insertion. J. Neural Eng, 2006, 3, 196. [DOI] [PubMed] [Google Scholar]
- 58.Jewell CM, López SCB and Irvine DJ, In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc. Natl. Acad. Sci, 2011, 108, 15745–15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andorko JI, Tostanoski LH, Solano E, Mukhamedova M and Jewell CM, Intra-lymph node injection of biodegradable polymer particles. J. Vis. Exp. JoVE, 2014, e50984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johansen P and Kündig TM, Intralymphatic immunotherapy and vaccination in mice. J. Vis. Exp. JoVE, 2014, e51031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Germain RN, Bajénoff M, Castellino F, Chieppa M, Egen JG, Huang AYC, Ishii M, Koo LY and Qi H, Making friends in out-of-the-way places: how cells of the immune system get together and how they conduct their business as revealed by intravital imaging. Immunol. Rev, 2008, 221, 163–181. [DOI] [PubMed] [Google Scholar]
- 62.Toga AW and Mazziotta JC, Brain Mapping: The Methods, Academic Press, 2002. [Google Scholar]
- 63.Wu J, Wu X and Lin F, Recent developments in microfluidics-based chemotaxis studies. Lab. Chip, 2013, 13, 2484–2499. [DOI] [PubMed] [Google Scholar]
- 64.Hulspas R, Villa-Komaroff L, Koksal E, Etienne K, Rogers P, Tuttle M, Korsgren O, Sharpe JC and Berglund D, Purification of regulatory T cells with the use of a fully enclosed high-speed microfluidic system. Cytotherapy, 2014, 16, 1384–1389. [DOI] [PubMed] [Google Scholar]
- 65.Lin L, Ma C, Wei B, Aziz N, Rajalingam R, Yusung S, Erlich HA, Trachtenberg EA, Targan SR, McGovern DPB, Heath JR and Braun J, Human NK cells licensed by killer Ig receptor genes have an altered cytokine program that modifies CD4+ T cell function. J. Immunol, 2014, 193, 940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ogunniyi AO, Thomas BA, Politano TJ, Varadarajan N, Landais E, Poignard P, Walker BD, Kwon DS and Love JC, Profiling human antibody responses by integrated single-cell analysis. Vaccine, 2014, 32, 2866–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Torres AJ, Hill AS and Love JC, Nanowell-Based Immunoassays for Measuring Single-Cell Secretion: Characterization of Transport and Surface Binding. Anal. Chem, 2014, 86, 11562–11569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chokkalingam V, Tel J, Wimmers F, Liu X, Semenov S, Thiele J, Figdor CG and Huck WTS, Probing cellular heterogeneity in cytokine-secreting immune cells using droplet-based microfluidics. Lab. Chip, 2013, 13, 4740–4744. [DOI] [PubMed] [Google Scholar]
- 69.Kim SK, Moon WK, Park JY and Jung H, Inflammatory mimetic microfluidic chip by immobilization of cell adhesion molecules for T cell adhesion. The Analyst, 2012, 137, 4062–4068. [DOI] [PubMed] [Google Scholar]
- 70.Faley S, Seale K, Hughey J, Schaffer DK, VanCompernolle S, McKinney B, Baudenbacher F, Unutmaz D and Wikswo JP, Microfluidic platform for real-time signaling analysis of multiple single T cells in parallel. Lab. Chip, 2008, 8, 1700–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wong KHK, Truslow JG, Khankhel AH, Chan KLS and Tien J, Artificial lymphatic drainage systems for vascularized microfluidic scaffolds. J. Biomed. Mater. Res. A, 2013, 101A, 2181–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gopalakrishnan N, Hannam R, Casoni GP, Barriet D, Ribe JM, Haug M and Halaas Ø, Infection and immunity on a chip: a compartmentalised microfluidic platform to monitor immune cell behaviour in real time. Lab. Chip, 2015, 15, 1481–1487. [DOI] [PubMed] [Google Scholar]
- 73.Giese C, Demmler CD, Ammer R, Hartmann S, Lubitz A, Miller L, Müller R and Marx U, A human lymph node in vitro--challenges and progress. Artif. Organs, 2006, 30, 803–808. [DOI] [PubMed] [Google Scholar]
- 74.Molteni R, Bianchi E, Patete P, Fabbri M, Baroni G, Dubini G and Pardi R, A novel device to concurrently assess leukocyte extravasation and interstitial migration within a defined 3D environment. Lab. Chip, 2015, 15, 195–207. [DOI] [PubMed] [Google Scholar]
- 75.Byrne MB, Trump L, Desai AV, Schook LB, Gaskins HR and Kenis PJA, Microfluidic platform for the study of intercellular communication via soluble factor-cell and cell-cell paracrine signaling. Biomicrofluidics, 2014, 8, 44104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bonvin C, Overney J, Shieh AC, Dixon JB and Swartz MA, A multichamber fluidic device for 3D cultures under interstitial flow with live imaging: development, characterization, and applications. Biotechnol. Bioeng, 2010, 105, 982–991. [DOI] [PubMed] [Google Scholar]
- 77.Agliari E, Biselli E, De Ninno A, Schiavoni G, Gabriele L, Gerardino A, Mattei F, Barra A and Businaro L, Cancer-driven dynamics of immune cells in a microfluidic environment. Sci. Rep, 2014, 4, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Purwada A, Jaiswal MK, Ahn H, Nojima T, Kitamura D, Gaharwar AK, Cerchietti L and Singh A, Ex vivo engineered immune organoids for controlled germinal center reactions. Biomaterials, 2015, 63, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.