

Abstract
Endometriosis is a common, estrogen-dependent, inflammatory disorder characterized by the growth of endometrial-like tissue at extrauterine locations. Its pathogenesis and mechanisms underlying its pathophysiology are poorly understood, although genetic variation is strongly implicated in these processes. Genetic studies reveal that approximately 50% of risk for endometriosis is due to genetic factors and the other 50% likely owing to environmental factors. As with other complex diseases, genetic variants in the DNA sequence increasing endometriosis risk all have small effects, unlike most single-gene disorders. It is the combinations of these variants adding together that contribute to higher risks for individual women. In addition, recent data on disease lesions demonstrate a high frequency of somatic (likely acquired) mutations, some of which are present in the eutopic endometrium and specifically in the epithelial cell compartment, raising the possibility that abnormal epithelial progenitors in the eutopic endometrium give rise to ectopic disease. Discovery in this field is occurring at a rapid pace, and further definitions of genetic (germline) and environmental (somatic) contributions to the pathogenesis and pathophysiology of this disorder are anticipated soon. These discoveries are expected to increase diagnostic, therapeutic, and preventive strategies to minimize disease and its associated morbidities.
Keywords: Genetic risk factors, Somatic mutations, Disease model
Endometriosis is a common, estrogen-dependent disease in women associated with a high prevalence of pelvic pain and reduced fertility [1,2]. The underlying causes are unclear, and understanding and treating the disease remain a major clinical challenge [2,3]. Endometriosis is characterized by endometrial-like tissue implants outside the uterus, found primarily on the pelvic peritoneum, ovaries, and rectovaginal septum. In rare cases, it can occur at other sites in the body including the lung, pericardium, and brain [1]. Endometriosis affects 7% to 10% of women of reproductive age with significant costs for both affected women and for society, including increased healthcare costs, time off work, reduced productivity, and reduced social and economic participation [2,4].
The disease, similar to many other common noncommunicable conditions, has a complex etiology, influenced by both genetic and environmental factors. Whereas the role of genetic risk factors was once controversial, advances in genetics and genomics applied to the analysis of genetic risk factors for endometriosis have clearly demonstrated a role for genetic factors in disease risk [5–7]. Genomic locations of more than 40 genetic risk factors have been identified [7–14], and their functional consequences and altered gene regulation underlying increased disease risk in some genomic regions are actively being pursued [15–18]. In addition, genomic studies have recently identified a significant burden of somatic mutations in endometriosis lesions, extending the likely role of DNA variation in the pathogenesis of endometriosis. The aim of this review is to summarize the contribution of genetic variation (germline or inherited and somatic or acquired) to endometriosis, the progress in discovery and identification of the specific genetic risk factors, and the role of genomic studies in understanding the etiology of this disease.
Genetic Variation and Disease Risk
A wide spectrum of genetic variants and mutations contribute to human disease. It is useful to consider the characteristics of different categories of genetic variation in interpreting the results of genetic mapping and genomic studies. Rare single-base mutations or deletions in protein-coding sequences can disrupt the function of critical proteins, with major effects on development and function leading to disease. Examples include mutations in genes from the hypothalamic-pituitary-axis such as follicle-stimulating hormone (FSH) receptor (FSHR) causing idiopathic hypogonadotropic hypogonadism and the absence of puberty, or activating mutations in kisspeptin (KISS1) and kisspeptin receptor (KISS1R) resulting in central precocious puberty [19].
In contrast, genetic variation influencing common diseases such as endometriosis generally result from a large number of variants, each with small effects [20]. The most common variation between individuals is differences at single-base positions in the DNA generally referred to as single-nucleotide polymorphisms (SNPs). Many common variants we all carry have no functional consequences. Some are located in regulatory sequences in the DNA, responsible for ensuring the right set of proteins are made in the correct cells and at the right stage of development. Disrupting the function of these regulatory sequences has more subtle effects. It is the combination of altered regulation by this class of variants that predisposes to many common diseases. Consequently, genetic risk for complex diseases results from the combined effects of a large number of genetic variants in these regulatory sequences, each with small effects on disease risk.
Approaches to genetic mapping and success in gene discovery are determined by the size of effects we expect to see from the different classes of genetic variation. Variants disrupting protein function with large effects, such as the FSHR mutations discussed previously, can be followed in families and mapped by linkage and sequencing. It is only in the last 10 years that genome-wide association studies (GWASs) have allowed us to test the effects of millions of SNPs across the genome in many thousands of cases and controls. This has transformed our ability to map genetic risk factors with small effects typical for complex diseases [20]. Large studies can be conducted to discover these variants, and the number of risk factors identified in individual studies shows an almost linear relationship with study size [20]. Mapping genetic risk factors for endometriosis have followed developments in genomics over the last 20 years, with the most robust results coming from the advent of GWAS methods [10,12,14].
Heritability
Before mapping genetic risk factors, it is important to first determine that genetic factors play a role in disease risk. Evidence for genetic effects on endometriosis is supported by higher rates of endometriosis among the relatives (sisters and daughters) of women with endometriosis (cases) than in controls in both hospital [21,22] and population [23] studies. The relative risk for women who have immediate relatives with endometriosis has been estimated at 2.3 in a study of Australian twins and their families [24]. The proportion of risk accounted for by genetic factors can be estimated using classical twin studies comparing the rates of disease of identical twins who inherit exactly the same genetic makeup from their parents with those of non-identical twins who share half their genes (equivalent to brothers and sisters). Studies of Australian twins observed higher rates of endometriosis in identical twin sisters and estimated the genetic contribution to risk, or heritability, of endometriosis at 0.51 [24]. Similar studies of twins from Sweden estimated the heritability of endometriosis at 0.47 [25]. The estimates have relatively wide confidence intervals, but these 2 studies have shown that genetic factors contribute to about half of the variation in endometriosis risk. This estimate of heritability is similar to estimates for genetic contributions to age at menarche and age at menopause and is less than estimates of heritability for polycystic ovarian syndrome and uterine myomas [19].
Genetic Risk Factors for Endometriosis
Endometriosis fits the pattern of a complex disease [5], and GWASs provide strong evidence for the role of many genetic risk factors contributing to endometriosis [7,8,10,12,13,26,27]. For example, the most recently published study included approximately 17 000 cases, analyzed association with 6 979 035 individual SNPs across the genome, and identified 14 genomic regions with 19 independent signals contributing to endometriosis risk [12]. The number of genomic regions associated with endometriosis shows a linear relationship with the number of cases in the studies [5], similar to studies of other traits and diseases. Genomic regions carrying variants influencing endometriosis (Fig. 1) have been generally well replicated in subsequent GWASs and follow-up studies [5]. Genomic regions with variants contributing to endometriosis risk from recent meta-analyses [9,11,12] are summarized in Table 1 with the risk allele, effect size, and nearest gene in each region.
Fig. 1.
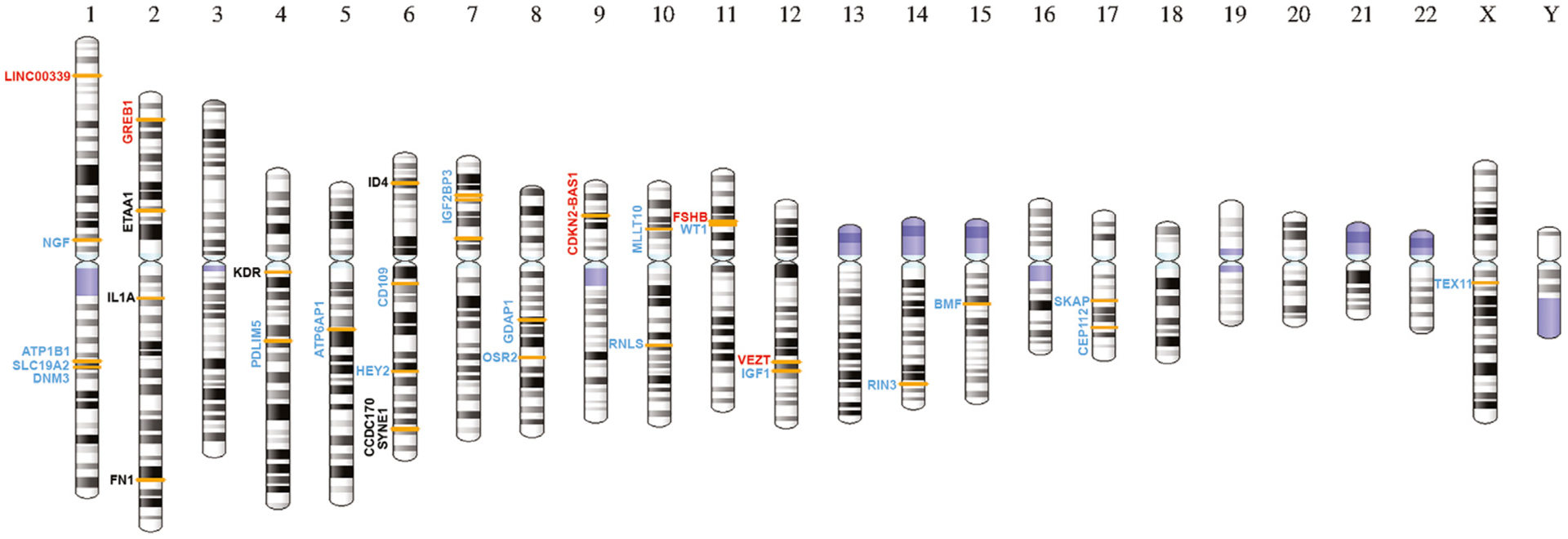
The location of genetic risk factors reported to influence endometriosis risk (7, 8, 9, 10, 11, 12, 13, 14) mapped to an ideogram of human chromosomes. The location of genetic markers showing genome-wide significant evidence of association with endometriosis is indicated by the orange bars on the ideogram. Genes close to the critical variant at each location are shown. Genes in red have evidence for functional relevance, genes in blue are novel regions identified in recent meta-analyses [9,11], and genes in black have been replicated in several studies.
Table 1.
Summary of genome-wide significant loci associated with endometriosis identified in recent GWAS meta-analyses
| Chr | SNP | BP | RA | OR (95% CI) | p value | Associated gene/cytoband |
|---|---|---|---|---|---|---|
| Previously reported loci | ||||||
| 1 | rs12037376 | 22462111 | A | 1.16 (1.12–1.19) | 8.87 × 10−17 | WNT4/1p36.12 |
| 2 | rs11674184 | 11721535 | T | 1.13 (1.10–1.15) | 2.67 × 10−17 | GREB1/2p25.1 |
| 2 | rs10167914 | 113563361 | G | 1.12 (1.08–1.15) | 1.10 × 10−9 | IL1A/2q13 |
| 2 | rs6546324 | 67856490 | A | 1.08 (1.05–1.11) | 3.01 × 10−8 | ETAA1/2p14 |
| 4 | rs1903068 | 56008477 | A | 1.11 (1.07–1.13) | 1.04 × 10−11 | KDR/4q12 |
| 6 | rs760794 | 19790560 | T | 1.09 (1.06–1.12) | 1.79 × 10−10 | ID4/6p22.3 |
| 7 | rs12700667 | 25901639 | A | 1.10 (1.07–1.13) | 9.08 × 10−10 | 7p15.2 |
| 9 | rs1537377 | 22169700 | C | 1.09 (1.06–1.12) | 1.33 × 10−10 | CDKN2B-AS1/9p21.3 |
| 12 | rs4762326 | 95668951 | T | 1.08 (1.05–1.11) | 2.20 × 10−9 | VEZT/12q22 |
| 2 | rs1250241 | 216295312 | T | 1.06 (1.03–1.09) | 6.20 × 10−5 | FN1/2q35 |
| 6 | rs1971256 | 151816011 | C | 1.09 (1.06–1.13) | 3.74 × 10−8 | CCDC170/6q25.1 |
| 6 | rs71575922 | 152554014 | G | 1.11 (1.07–1.15) | 2.02 × 10−8 | SYNE1/6q25.1 |
| 7 | rs74491657 | 46947633 | G | 1.08 (1.03–1.13) | 1.23 × 10−3 | 7p12.3 |
| 11 | rs74485684 | 30242287 | T | 1.11 (1.07–1.15) | 2.00 × 10−8 | FSHB/11p14.1 |
| Novel loci | ||||||
| 1 | rs12030576 | 115817221 | G | 1.07 (1.05–1.09) | 5.2 × 10−13 | NGF/1p13.2 |
| 1 | rs1209731 | 169324793 | C | 1.19 (1.12–1.26) | 2.0 × 10−8 | ATP1B1-F5/1q24.2 |
| 1 | rs1894692 | 169467654 | A | 1.18 (1.13–1.24) | 2.88 × 10−13 | SLC19A2/1q24.2 |
| 1 | rs495590 | 172152202 | G | 1.07 (1.05–1.10) | 6.73 × 10−10 | DNM3/1q24.3 |
| 4 | rs2510770 | 95479372 | A | 1.05 (1.03–1.06) | 8.25 × 10−10 | PDLIM5/4q22.3 |
| 5 | rs13177597 | 82052282 | G | 1.06 (1.04–1.08) | 1.30 × 10−8 | ATP6AP1L/5q14.2 |
| 6 | rs1595344 | 74611632 | G | 1.05 (1.03–1.07) | 1.2 × 10−8 | CD109/6q13 |
| 6 | rs2226158 | 125995467 | G | 1.05 (1.03–1.07) | 2.6 × 10−8 | HEY2/6q22.31 |
| 7 | rs62468795 | 23530051 | G | 1.10 (1.07–1.14) | 8.05 × 10−9 | IGF2BP3/7p15.3 |
| 8 | rs10090060 | 75257608 | A | 1.08 (1.06–1.11) | 5.72 × 10−11 | GDAP1/8q21.11 |
| 8 | rs6468654 | 100062724 | C | 1.06 (1.04–1.08) | 2.5 × 10−8 | OSR2-VPS13B/8q22.2 |
| 10 | rs1802669 | 21827796 | A | 1.07 (1.05–1.10) | 5.52 × 10−9 | MLLT10/10p12.31 |
| 10 | rs796945 | 90150837 | C | 1.07 (1.05–1.10) | 1.78 × 10−9 | RNLS/10q23.31 |
| 11 | rs7924571 | 32350027 | C | 1.06 (1.04–1.08) | 3.5 × 10−8 | WT1/11p13 |
| 12 | rs17727841 | 102809630 | G | 1.06 (1.04–1.08) | 5.33 × 10−11 | IGF1/12q23.2 |
| 14 | rs7151531 | 93113547 | C | 1.07 (1.04–1.10) | 3.80 × 10−8 | RIN3/14q32.12 |
| 15 | rs4923850 | 40352278 | A | 1.05 (1.04–1.06) | 3.07 × 10−13 | BMF/15q15.1 |
| 17 | rs66683298 | 46277748 | C | 1.08 (1.06–1.11) | 1.73 × 10−10 | SKAP1/17q21.32 |
| 17 | rs76731691 | 63960269 | G | 1.08 (1.05–1.11) | 9.27 × 10−9 | CEP112/17q24.1 |
| X | rs13441059 | 70108889 | A | 1.05 (1.03–1.07) | 4.1 × 10−8 | TEX11-SLC7A3/Xq13.1 |
BP = genomic position in base pairs shown relative to GRCh37 (hg19); Chr = chromosome; CI = confidence interval; GWAS = genome-wide association study, OR = odds ratio with respect to RA; p = p value for association; RA = risk allele; SNP = single-nucleotide polymorphism.
Endometriosis is an estrogen-dependent disease. Genomic signals associated with endometriosis include signals close to estrogen receptor 1 gene (ESR1), the predominant receptor for estrogen action in the endometrium. Other candidate regions with genes in the reproductive pathway are signals upstream of FSH beta subunit (FSHB), also associated with increased FSH concentrations, and signals near the estrogen-regulated and early response gene (GREB1) first identified in breast cancer cell lines and tumors [19]. Functional follow-up studies have implicated genes with roles in cell migration, adhesion, and proliferation including cell division cycle 42 (CDC42) [18], the long noncoding RNA cyclin-dependent kinase inhibitor 2B antisense RNA (CDKN2BAS) [17], and vezatin (VEZT) [15,16]. Lower expression of CDKN2BAS in carriers of the risk allele for the causal SNP in a Japanese study has been associated with reduced expression of the cell-cycle inhibitors p16INK4A and p15INK4B and may contribute to survival and proliferation of ectopic endometrial cells and promote the development of endometriosis [17].
A number of candidate gene studies have reported protein-coding variants implicated in endometriosis risk. Many of these results from small studies have not been replicated in the larger GWASs. A specific search, genotyping 240 000 common and low-frequency protein-coding variants in 9004 patients with endometriosis identified only 1 significant result that survived replication. This was SNP rs13394619 in the coding region of GREB1, a gene already implicated from GWASs [10,12]. The SNP may affect RNA splicing and is located approximately 6 kilobases from and strongly correlated with SNP rs11674184, the risk SNP with the strongest association signal from our GWAS results [28]. It is unclear whether this splice acceptor variant plays a direct role in genetic effects on endometriosis.
International mapping efforts are continuing with collaborative projects combining results from many groups around the world. Results from 2 additional studies are reported in manuscripts submitted to the bioRxiv preprint server [9,11], providing strong support for most genomic regions identified in earlier studies and describing a number of novel regions associated with endometriosis (Table 1). One consistent observation from the genetic studies is that many of the variants associated with endometriosis have bigger effects in patients with severe disease [7,10,12]. The overall contribution of all common variants to disease can be calculated from the GWAS results. It is estimated that all common variants account for 26% of variation in endometriosis risk [7,29]. When this is calculated separately for patients with severe disease, the estimate is 34%, which is significantly higher than the 15% reported for patients with minimal or mild disease [7,29].
One limitation of current genetic studies is few data sets included in large studies have surgically confirmed disease, and very few have detailed clinical data. Consistent evidence for stronger genetic effects in severe cases supports the view that there is more to learn about disease subtypes and disease progression. Future large studies with genetic marker data and detailed clinical information will contribute to a greater understanding of the causes of endometriosis.
Somatic Mutations
An important recent development is the high frequency of somatic mutations observed in endometriosis lesions. Somatic mutations are alterations in DNA not inherited from parents. They arise in individual cells throughout life as a result of errors in DNA replication or DNA damage from environmental exposures. Cells with somatic mutations can escape cell death or senescence and/or acquire growth advantages over surrounding cells. As a consequence, somatic mutations are a major contributing factor for many cancers.
Recent studies in endometriosis have identified a high burden of somatic mutations in endometriosis lesions [30–33]. Somatic mutations were identified in 79% of the 24 patients with deep infiltrating endometriosis lesions [30]. Five patients had known cancer driver mutations in ARID1A, PIK3CA, KRAS, or PPP2R1A [30]. Whole exome sequencing of 13 ovarian endometriotic lesions [33] also identified recurrent nonsilent somatic mutations (missense, nonsense, or splice site mutations) in 16 genes. These included mutations in several cancer-associated genes, including KRAS, PIK3CA, FBXW7, PPP2R1A, and PIK3R.
Lesions contain multiple cell types, and the study by Anglesio et al [30] detected KRAS mutations only in the epithelium and not in the stromal cells. A comparison of cancer driver mutations and selection-neutral passenger mutations that are unlikely to result in a growth advantage to cells demonstrated that endometrial epithelium in endometriotic lesions is clonal, whereas stromal cells are not [32]. These results suggest that endometriosis does not result from a single stem or progenitor cell that leads to formation of the lesions. It is more likely that a single endometrial epithelial progenitor cell undergoes expansion to form glandular tissue at the site of the lesion [32]. This view is supported in another recent study sequencing ovarian endometriotic epithelial samples and normal endometrial epithelial samples [33]. The study identified numerous somatic mutations in cancer-associated genes in ovarian endometriotic epithelium cells and showed that genes most frequently mutated in endometriosis-associated ovarian cancers were also frequently mutated in uterine endometrial epithelial samples, including samples of eutopic endometrium of women without endometriosis [33]. This is an important finding in view of the constant shedding and regeneration of uterine endometrium with successive menstrual cycles, and it will be important to determine if different somatic mutations contribute to the different types of endometriotic lesions. This evidence strongly points to an important role for somatic mutations in both endometriosis development and progression.
Discussion
The question posed in this review is “should genetics be considered the pre-eminent etiologic factor in endometriosis?” Do genetic risk factors provide the best current explanation for the origin and progression of the disease? Great progress has been made in the last decade confirming genetic factors are a major contributor to endometriosis risk, identifying more than 40 genomic regions harboring variants contributing to this risk, and beginning to identify the target genes and functional consequences of these variants. The genetic risk constitutes additive effects of a large number of common variants with small effects [12], with little evidence for protein-modifying variants with moderate or large effects [28]. When we add up the effects of all common variants associated with endometriosis, we estimate they contribute to 26% of the variation in endometriosis risk (accounting for about half of the genetic risk) [29]. The gap between the genetic contribution from common variants and the heritability estimated from twin studies could have several explanations including other types of genetic variation not captured by the arrays, possible differences in genetic contributions to different types of endometriosis lesions, variation in the accuracy of diagnosis, and/or the selection of patients recruited. Low-frequency coding variants with large effects were not detected in exome chip studies [28]. DNA sequencing studies in high-risk pedigrees may identify rare variants with large effect, but given the low frequency of these rare variants in the population, we would need to find many of these rare variants to account for the missing heritability. Similarly, rare copy number variants could contribute, although signals from common copy number variants should be picked up by correlated common SNPs typed in the GWAS. The most likely source of missing heritability is many low-frequency variants (minor allele frequencies < 5%) where the signals are not adequately captured in estimates from the GWAS data.
If genetic factors are to explain the etiology of endometriosis, the evidence should identify the origin(s) of cells responsible for initiation of lesions, differences in the types and locations of lesions, and the variable presentation of symptoms among patients. Theories for the origin of cells and initiation of lesions include (1) the deposition of viable cells shed from the endometrium transported to the pelvic cavity through retrograde menstruation, (2) activation of cells left behind from differentiation and migration of the müllerian ducts during development, and (3) metaplasia or transformation of 1 differentiated cell type into another (e.g., transformation of the coelomic epithelium covering the ovary) [3,34]. No 1 theory provides an adequate explanation for all cases of endometriosis or the variable presentation of disease among patients. For example, in some cases, endometriosis occurs in young women before the onset of puberty, and there are rare reports of endometriosis in males. Transport of viable cells from the endometrium through retrograde menstruation could account for many cases, but some must arise from alternate mechanisms [3,34].
Inherited genetic variation could be expressed in all cells. Expression of a number of genes in the endometrium is under genetic regulation, but few of these overlap with key variants in genomic regions associated with endometriosis risk [15,35]. Functional effects of genetic risk factors may be specific to individual cell types from endometrium or in other tissues. Analysis of genetic regulation of gene expression in individual cell types will determine whether some risk factors act only in restricted cell types, and the overlap with endometriosis risk regions may also help identify cellular origins of disease.
Genetic risk factors account for only half of the variation in endometriosis risk. The other half of the variation in disease risk is due to environmental or other factors, and identifying major factors that contribute to this environmental variation has proved elusive. The observation of a high frequency of somatic mutations in endometriotic lesions provides new insights into causes of the disease. Somatic mutations are not inherited as are genetic risk factors. They influence a small number of cells, arise over a women’s lifetime, and are part of the environmental risk. Their contribution to this component of risk has yet to be quantified, but studies suggest 80% of deep infiltrating lesions carry somatic mutations. As in genetic risk factors, the functional consequences of many of these somatic mutations remain to be determined. The published studies have focused on the 20% of somatic mutations that are cancer driver mutations in part because of the documented effects on cell survival and proliferation. However, other somatic mutations may also have a role to play [36]. For example, an in-frame deletion mutation in ID4 has been detected in 1 patient [30]. ID4 is a strong candidate gene in one of the regions associated with endometriosis.
The discovery that somatic mutations are found in epithelial cells and not in stromal cells in lesions provides an important clue. Moreover, evidence shows that somatic cancer driver mutations are present in some epithelial cells in the eutopic endometrium. This suggests that at least 1 cellular origin for the initiation of endometriosis is epithelial cells from eutopic endometrium harboring somatic mutations with cells transported to the peritoneal cavity by retrograde menstruation as first proposed by Sampson in 1927 [37]. If eutopic endometrium is the source of cells for initiation of endometriosis, a difference in the viability of epithelial and/or stromal cells in menstrual fluid could be the explanation why only some women develop endometriosis when retrograde menstruation is common. The presence of cancer driver and other somatic mutations in some women could determine cell survival at menstruation. Current information on functional consequences of genetic risk factors also suggests effects on cell proliferation and cell adhesion.
We propose a model for the development of many cases of endometriosis where genetic risk factors, somatic mutations, and environmental factors all work together in an additive fashion, resulting in the survival and viability of epithelial cells from the eutopic endometrium reaching the peritoneal cavity (Fig. 2). Genetic risk factors alone do not explain the etiology of endometriosis, rather we suggest that the combined effects of genetic variation and somatic mutations comprise the pre-eminent etiologic factor in endometriosis. One prediction from the model is we might expect to see some somatic mutations influencing risk in genes or pathways implicated by germline variation such as the ID4 mutation observed in 1 study [30]. Other environmental risk factors may well act through these mechanisms by inducing DNA damage and somatic mutations or interacting in pathways altered by genetic background. Studies directed to understanding the spectrum of somatic mutations in endometriosis and the functional consequences of genetic risk factors will provide a much greater understanding of pathways to disease.
Fig. 2.
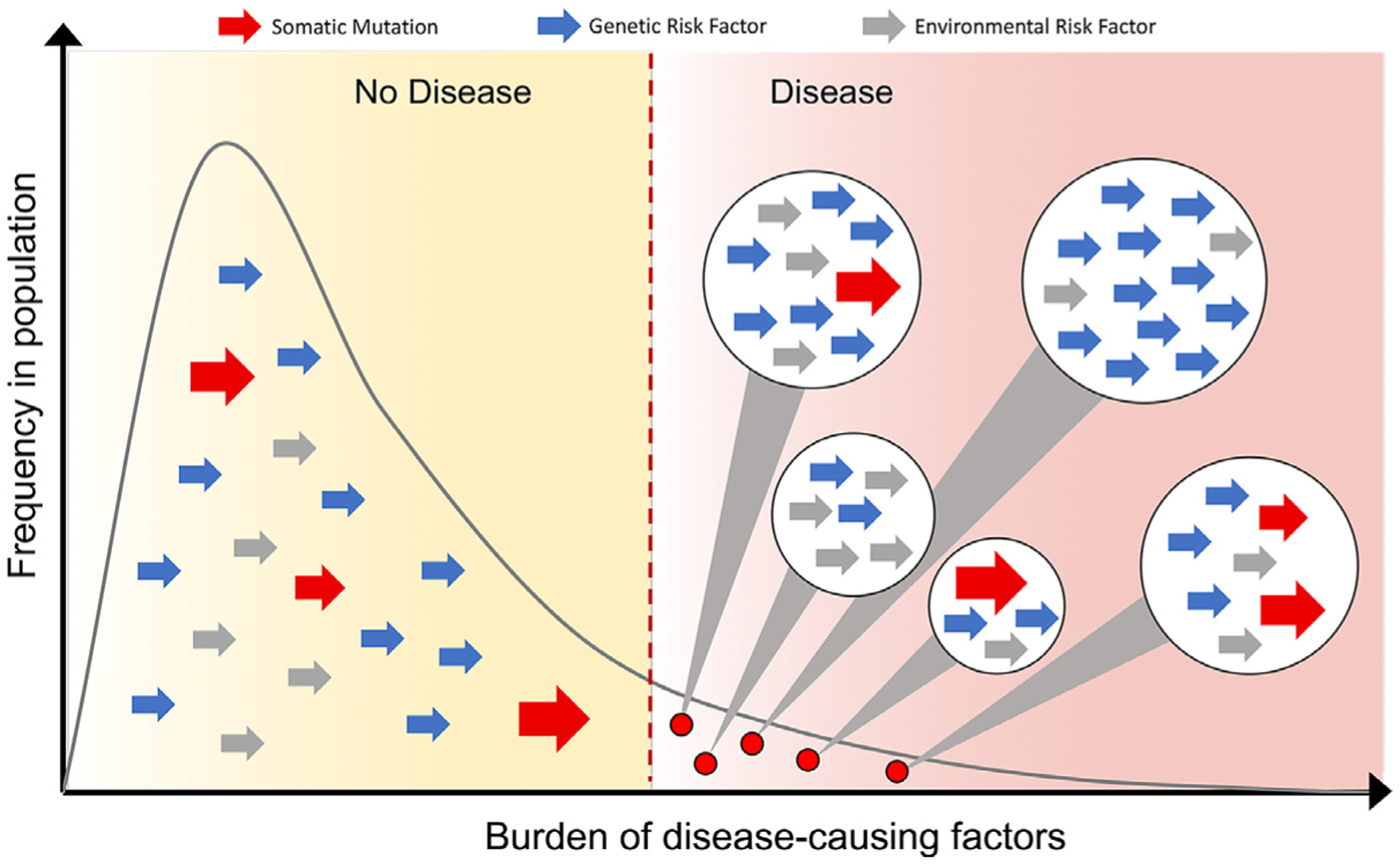
Threshold model for how the additive effects of genetic risk factors, somatic mutations, and environmental risk factors in the population may combine to increase individual risk beyond the threshold for development of the disease. The large red arrows represent somatic mutations, blue arrows represent individual genetic risk factors, and gray arrows represent other environmental risk factors. Women with endometriosis will have a high burden of disease-causing risk factors made up from different combinations of genetic risk factors, somatic mutations in critical cell types, and environmental risk factors that predispose to disease (combinations of risk factors for individual women represented in circles on the right-hand side of the figure).
Acknowledgments
Dr. Montgomery is supported by a National Health and Medical Research Council Australia Fellowship GNT1078399. Supported, in part, by NIH NICHD R01 HD089511.
Footnotes
The authors declare that they have no conflict of interest.
References
- 1.Giudice LC. Clinical practice. Endometriosis. N Engl J Med. 2010;362:2389–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zondervan KT, Becker CM, Koga K, Missmer SA, Taylor RN, Viganò P. Endometriosis. Nat Rev Dis Primers. 2018;4:9. [DOI] [PubMed] [Google Scholar]
- 3.Vercellini P, Viganò P, Somigliana E, Fedele L. Endometriosis: pathogenesis and treatment. Nat Rev Endocrinol. 2014;10:261–275. [DOI] [PubMed] [Google Scholar]
- 4.Soliman AM, Yang H, Du EX, Kelley C, Winkel C. The direct and indirect costs associated with endometriosis: a systematic literature review. Hum Reprod. 2016;31:712–722. [DOI] [PubMed] [Google Scholar]
- 5.Fung JN, Montgomery GW. Genetics of endometriosis: state of the art on genetic risk factors for endometriosis. Best Pract Res Clin Obstet Gynaecol. 2018;50:61–71. [DOI] [PubMed] [Google Scholar]
- 6.Lee SH, Sapkota Y, Fung JN, Montgomery GW. Genetic biomarkers for endometriosis In: D’Hooghe T, editor. Biomarkers for Endometriosis, Cham: Springer; 2017, 83–93. [Google Scholar]
- 7.Painter JN, Anderson CA, Nyholt DR, et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat Genet. 2011;43:51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albertsen HM, Chettier R, Farrington P, Ward K. Genome-wide association study link novel loci to endometriosis. PLoS One. 2013;8:e58257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galarneau G, Fontanillas P, et al. the Celmatix Research Team. Genome-wide association studies on endometriosis and endometriosis-related infertility. bioRxiv. 2018. 10.1101/401448. [DOI] [Google Scholar]
- 10.Nyholt DR, Low SK, Anderson CA, et al. Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat Genet. 2012;44:1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rahmioglu N, Banasik K, Paraskevi C, et al. Large-scale genome-wide association meta-analysis of endometriosis reveals 13 novel loci and genetically-associated comorbidity with other pain conditions. bioRxiv. 2018. 10.1101/406967. [DOI] [Google Scholar]
- 12.Sapkota Y, Steinthorsdottir V, Morris AP, et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat Commun. 2017;8:15539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinthorsdottir V, Thorleifsson G, Aradottir K, et al. Common variants upstream of KDR encoding VEGFR2 and in TTC39B associate with endometriosis. Nat Commun. 2016;7:12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uno S, Zembutsu H, Hirasawa A, et al. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat Genet. 2010;42:707–710. [DOI] [PubMed] [Google Scholar]
- 15.Fung JN, Mortlock S, Girling JE, et al. Genetic regulation of disease risk and endometrial gene expression highlights potential target genes for endometriosis and polycystic ovarian syndrome. Sci Rep. 2018;8:11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mortlock S, Restuadi R, Levien R, et al. Genetic regulation of methylation in human endometrium and blood and gene targets for reproductive diseases. Clin Epigenetics. 2019;11:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakaoka H, Gurumurthy A, Hayano T, et al. Allelic imbalance in regulation of ANRIL through chromatin interaction at 9p21 endometriosis risk locus. PLoS Genet. 2016;12:e1005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Powell JE, Fung JN, Shakhbazov K, et al. Endometriosis risk alleles at 1p36.12 act through inverse regulation of CDC42 and LINC00339. Hum Mol Genet. 2016;25:5046–5058. [DOI] [PubMed] [Google Scholar]
- 19.Gajbhiye R, Fung JN, Montgomery GW. Complex genetics of female fertility. NPJ Genom Med. 2018;3:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kennedy S, Mardon H, Barlow D. Familial endometriosis. J Assist Reprod Genet. 1995;12:32–34. [DOI] [PubMed] [Google Scholar]
- 22.Simpson JL, Bischoff FZ. Heritability and molecular genetic studies of endometriosis. Ann N Y Acad Sci. 2002;955:239–251. discussion 293 −295, 396–406. [DOI] [PubMed] [Google Scholar]
- 23.Stefansson H, Geirsson RT, Steinthorsdottir V, et al. Genetic factors contribute to the risk of developing endometriosis. Hum Reprod. 2002;17:555–559. [DOI] [PubMed] [Google Scholar]
- 24.Treloar SA, O’Connor DT, O’Connor VM, Martin NG. Genetic influences on endometriosis in an Australian twin sample. sueT@qimr.edu.au Fertil Steril. 1999;71:701–710. [DOI] [PubMed] [Google Scholar]
- 25.Saha R, Pettersson HJ, Svedberg P, et al. Heritability of endometriosis. Fertil Steril. 2015;104:947–952. [DOI] [PubMed] [Google Scholar]
- 26.Adachi S, Tajima A, Quan J, et al. Meta-analysis of genome-wide association scans for genetic susceptibility to endometriosis in Japanese population. J Hum Genet. 2010;55:816–821. [DOI] [PubMed] [Google Scholar]
- 27.Sobalska-Kwapis M, Smolarz B, Słomka M, et al. New variants near RHOJ and C2, HLA-DRA region and susceptibility to endometriosis in the Polish population-the genome-wide association study. Eur J Obstet Gynecol Reprod Biol. 2017;217:106–112. [DOI] [PubMed] [Google Scholar]
- 28.Sapkota Y, Vivo I, Steinthorsdottir V, et al. Analysis of potential protein-modifying variants in 9000 endometriosis patients and 150000 controls of European ancestry. Sci Rep. 2017;7:11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SH, Harold D, Nyholt DR, et al. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum Mol Genet. 2013;22:832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anglesio MS, Papadopoulos N, Ayhan A, et al. Cancer-associated mutations in endometriosis without cancer. N Engl J Med. 2017;376: 1835–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anglesio MS, Yong PJ. Endometriosis-associated ovarian cancers. Clin Obstet Gynecol. 2017;60:711–727. [DOI] [PubMed] [Google Scholar]
- 32.Noë M, Ayhan A, Wang TL, Shih IM. Independent development of endometrial epithelium and stroma within the same endometriosis. J Pathol. 2018;245:265–269. [DOI] [PubMed] [Google Scholar]
- 33.Suda K, Nakaoka H, Yoshihara K, et al. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Rep. 2018;24:1777–1789. [DOI] [PubMed] [Google Scholar]
- 34.Gordts S, Koninckx P, Brosens I. Pathogenesis of deep endometriosis. Fertil Steril. 2017;108:872–885.e1. [DOI] [PubMed] [Google Scholar]
- 35.Fung JN, Girling JE, Lukowski SW, et al. The genetic regulation of transcription in human endometrial tissue. Hum Reprod. 2017;32:893–904. [DOI] [PubMed] [Google Scholar]
- 36.Montgomery GW, Giudice LC. New lessons about endometriosis - somatic mutations and disease heterogeneity. N Engl J Med. 2017;376: 1881–1882. [DOI] [PubMed] [Google Scholar]
- 37.Sampson JA. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol. 1927;14:422–469. [Google Scholar]