

Abstract
In eukaryotic DNA replication, DNA polymerase ε (Polε) is responsible for leading strand synthesis, whereas DNA polymerases α and δ synthesize the lagging strand. The human Polε (hPolε) holoenzyme is comprised of the catalytic p261 subunit and the noncatalytic p59, p17, and p12 small subunits. So far, the contribution of the noncatalytic subunits to hPolε function is not well understood. Using pre-steady-state kinetic methods, we established a minimal kinetic mechanism for DNA polymerization and editing catalyzed by the hPolε holoenzyme. Compared with the 140-kDa N-terminal catalytic fragment of p261 (p261N), which we kinetically characterized in our earlier studies, the presence of the p261 C-terminal domain (p261C) and the three small subunits increased the DNA binding affinity and the base substitution fidelity. Although the small subunits enhanced correct nucleotide incorporation efficiency, there was a wide range of rate constants when incorporating a correct nucleotide over a single-base mismatch. Surprisingly, the 3′→5′ exonuclease activity of the hPolε holoenzyme was significantly slower than that of p261N when editing both matched and mismatched DNA substrates. This suggests that the presence of p261C and the three small subunits regulates the 3′→5′ exonuclease activity of the hPolε holoenzyme. Together, the 3′→5′ exonuclease activity and the variable mismatch extension activity modulate the overall fidelity of the hPolε holoenzyme by up to 3 orders of magnitude. Thus, the presence of p261C and the three noncatalytic subunits optimizes the dual enzymatic activities of the catalytic p261 subunit and makes the hPolε holoenzyme an efficient and faithful replicative DNA polymerase.
Keywords: pre-steady-state kinetics, human DNA polymerase epsilon, eukaryotic DNA polymerase, exonuclease, DNA replication, DNA binding, polymerase fidelity, nucleotide incorporation kinetics, function regulation, accessory subunits, DNA polymerase, pre-steady-state kinetics, enzyme mechanism, substrate specificity, the 3′→5′, exonuclease
Three B-family DNA polymerases are responsible for replicating the majority of the eukaryotic nuclear genome: DNA polymerases α (Polα), δ (Polδ), and ε (Polε) (1). DNA replication is initiated on both the leading and lagging strands by the Polα-primase complex. Primase synthesizes short primers of 7-12 ribonucleotides (rNTPs) that are extended by Polα with an additional 20-25 deoxyribonucleotides (dNTPs) (2). Following primer synthesis, processive DNA synthesis is taken over by Polε and Polδ on the leading and lagging strands, respectively (3–6).
The eukaryotic replicative DNA polymerases are present as multisubunit complexes in vivo (1). Each of the replicative DNA polymerases shares a common core consisting of a single large catalytic subunit and a smaller B-subunit, e.g. Polα p180-p70, Polδ p125-p50, and Polε p261-p59 in humans. In addition to this core complex, each replicative DNA polymerase associates with a set of accessory subunits to form its heterotetrameric holoenzyme. For example, human Polδ (hPolδ) core complex associates with p66 (sometimes called p68) and p12 accessory subunits (7), whereas human Polε (hPolε) core complex interacts with p17 and p12 (distinct from hPolδ p12) accessory subunits (8). In the case of human Polα (hPolα), however, the p180-p70 core complex forms a heterotetrameric complex with the primase heterodimer, which is comprised of the p49 catalytic subunit and the p58 regulatory subunit (9).
Structures of Saccharomyces cerevisiae Polα (10), Polδ (11, 12), and Polε (13) as well as human Polδ (14) illustrate that all three replicative DNA polymerases are characterized by a globular catalytic core and an extended structure, with the latter element largely composed of the catalytic subunit C-terminal domain as well as the B- and accessory subunits. The structures further reveal that the extended regions play a role in modulating the coordination and dynamics of the replicative DNA polymerases. For example, the crystal structure of the hPolα-primase complex reveals that the catalytic subunits of Polα and primase are linked to each other through the primase p58 regulatory subunit to allow for efficient coordination of the polymerase and primase activities during primer synthesis (15). Additionally, in the recent high-resolution cryo-EM structure of human Polδ in complex with human proliferating cell nuclear antigen (PCNA), DNA, and dNTP, hPolδ is tethered to one of the three PCNA monomers through the extended C-terminal domain of the p125 catalytic subunit, whereas the p50, p66, and p12 small subunits extend laterally away from the DNA- and PCNA-binding sites (14). In this configuration, p12 likely stabilizes p125, while providing a scaffold for the p50 and p66 subunits to contact p125 and regulate its polymerization activity (14).
In the absence of the three small subunits, the catalytic subunit of each of the replicative DNA polymerases is capable of catalyzing template-dependent DNA synthesis in vitro. However, the B- and accessory subunits either regulate or enhance the DNA polymerization activities of the replicative DNA polymerase holoenzymes. For example, the hPolα p70 B-subunit limits the processivity of RNA primer extension by the p180 catalytic subunit (16). Furthermore, the polymerase activity of the trimeric hPolδ complex (p125-p50-p66) is enhanced by 4.6-fold in the presence of the p12 accessory subunit, whereas its 3ʹ→5ʹ exonuclease activity is reduced by as much as 5-fold on a DNA substrate containing a single-base mismatch at the primer 3ʹ terminus (17). Additionally, the polymerization processivity of S. cerevisiae Polε is enhanced in the presence of all three of its small subunits (13, 18). Thus, to accurately characterize the activities of the eukaryotic replicative DNA polymerases in vitro, it is imperative that kinetic studies are carried out with the full, intact DNA polymerase complexes.
Previously, we kinetically characterized the N-terminal fragment of the hPolε p261 catalytic subunit (residues 1−1189, p261N) and established a minimal mechanism for DNA polymerization catalyzed by p261N (19). However, detailed analysis of the hPolε heterotetrameric complex has been hampered by the difficulty of preparing sufficient quantities of pure complex. Initial studies of hPolε heterotetramer overexpressed in insect cells revealed that the three small subunits do not enhance the activity of the catalytic subunit alone (20). However, these studies were performed with excess enzyme on a large M13 DNA plasmid template (20), which does not allow for a quantitative comparison of DNA binding and nucleotide binding and incorporation kinetics between p261N and the hPolε heterotetramer. Furthermore, these studies did not investigate the impact of the small subunits on the 3ʹ→5ʹ exonuclease activity of hPolε. To determine whether the small subunits affect the DNA binding and polymerization kinetics of hPolε, we overexpressed and purified fully-assembled hPolε heterotetramer in insect cells and performed a pre-steady-state kinetic analysis of the heterotetramer. We determined that the p261 C-terminal domain (p261C) and the three small subunits enhance the DNA binding affinity of hPolε by reducing the DNA dissociation rate constant (19). Although p261C and the small subunits moderately affect base substitution fidelity, the contribution of 3ʹ→5ʹ exonuclease activity to hPolε fidelity is surprisingly reduced compared with that of p261N, primarily due to a nearly 10-fold attenuation of the excision rate constant on a DNA substrate containing a single-base mismatch (19, 21). Together, these observations suggest that p261C and the small subunits drive hPolε toward processive DNA synthesis to ensure rapid synthesis of the leading strand in vivo.
Results
Previously, we carried out the kinetic characterization of p261N, the N-terminal fragment (residues 1-1189) of the hPolε p261 catalytic subunit. Our initial characterization of p261N alone was motivated by the relative ease of its overexpression and purification from Escherichia coli in suitable quantities for pre-steady-state kinetic studies. Importantly, the p261N fragment contains all of the conserved polymerase and 3′→5′ exonuclease motifs (22). From our pre-steady-state kinetic analysis, we defined a minimal kinetic mechanism of nucleotide incorporation, a simplified version of which is shown in Scheme 1A (19). We also established a kinetic basis for the high fidelity of DNA polymerization catalyzed by p261N (21). In a study that followed, we examined WT hPolε holoenzyme prepared from baculovirus-infected insect cells and performed burst assays with both hPolε and p261N under identical conditions. We observed that both exhibited a fast burst phase and a slow linear phase of product formation (23). The multiphasic kinetics of correct nucleotide incorporation suggests that hPolε likely utilizes a similar minimal kinetic mechanism as p261N (Scheme 1A) for DNA polymerization. To provide more evidence for the kinetic mechanism and to further kinetically characterize the hPolε holoenzyme, we overexpressed and purified an exonuclease-deficient variant (D275A/E277A/D368A, hPolε exo−) and carried out the following kinetic analysis at 20 °C:
Scheme 1.

A, minimal and B, revised kinetic mechanism of correct nucleotide incorporation catalyzed by the human DNA polymerase ε holoenzyme.
Measurement of burst kinetics, the steady-state rate constant of correct nucleotide incorporation, and the dissociation rate constant of the E•DNA complex
If the mechanism in Scheme 1A can be applied to hPolε, then it must display multiphasic burst kinetics during single-nucleotide incorporation under enzyme-limiting conditions. To confirm that hPolε exo− displays burst kinetics as previously observed for p261N (19, 23) and the WT hPolε holoenzyme (23), we performed a burst assay in which a preincubated solution of hPolε exo− (20 nm, UV concentration) and a 4-fold excess of D-1 DNA substrate (80 nm, Table 1) was rapidly mixed with dTTP (100 μm) for various time points. The time course of product formation showed a clear burst of nucleotide incorporation followed by a slow linear phase (Fig. 1A), indicating that hPolε follows a similar mechanism of single-nucleotide incorporation as p261N. The data were best fit to Equation 1 in which the initial burst phase is described by two exponential rate constants, kfast and kslow, and the second linear phase is described by klinear. The rate constants for kfast and kslow were 139 ± 27 s−1 and 1.3 ± 0.5 s−1, respectively, whereas klinear was 0.0044 ± 0.0008 s−1. Although Scheme 1A does not explain why two exponential phases are present in the initial burst of product formation, we address this observation in more detail in the analysis of our pre-steady-state nucleotide incorporation assays (see below).
Table 1.
Sequences of DNA substrates
| D-1 | 5′-CGCAGCCGTCCAACCAACTCA-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTAGCAGCTAGGTTACGGCAGG-5′ | |
| M-1 | 5′-CGCAGCCGTCCAACCAACTCAC-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTAGCAGCTAGGTTACGGCAGG-5′ | |
| M-8 | 5′-CGCAGCCGTCCAACCAACTCAC-3′ |
| 3′-GCGTCGGCAGGTTGGTTGAGTCGCAGCTAGGTTACGGCAGG-5′ |
Figure 1.

Pre-steady-state and steady-state kinetics of correct dTTP incorporation by hPolε exo−. A, the burst kinetics of dTTP incorporation were measured by mixing a preincubated solution of hPolε exo− (20 nm, UV concentration) and 5′-radiolabeled D-1 DNA substrate (80 nm) with 100 μm dTTP and Mg2+ and then quenching at various time intervals with addition of EDTA. The data were fit to Equation 1 to yield a fast exponential rate constant of 139 ± 27 s−1, a slow exponential rate constant of 1.3 ± 0.5 s−1, and a linear phase rate constant of 0.0044 ± 0.0008 s−1. B, the steady-state DNA polymerization rate constant was measured by mixing a preincubated solution of hPolε exo− (1 nm, active site concentration) and 5′-radiolabeled D-1 DNA substrate (400 nm) with dTTP (100 μm) and Mg2+ for varying reaction times and then quenching the reaction with the addition of EDTA. Product concentration was plotted against time and the data were fit to Equation 2 to yield a kss of 0.0070 ± 0.0003 s−1. C, to directly measure the DNA dissociation rate constant, k-1, a preincubated solution of hPolε exo− (50 nm, UV concentration) and 5′-radiolabeled D-1 DNA substrate (100 nm) was mixed with unlabeled D-1 DNA substrate (2.5 μm) for varying incubation times before the reaction was initiated with the addition of dTTP (100 μm) and Mg2+. The reaction was allowed to proceed for 15 s and then quenched with the addition of EDTA. Product concentration was plotted against time and the data were fit to Equation 3 to yield a k-1 of 0.0058 ± 0.0007 s−1.
Typically, the linear phase of the burst time course is equal to the steady-state rate constant of nucleotide incorporation. To verify this assumption, we directly measured the steady-state DNA polymerization rate constant (kss) by mixing hPolε exo− (1 nm, active site concentration) and a large excess of D-1 DNA substrate (400 nm) with a solution of dTTP (100 μm) and Mg2+ (see “Experimental procedures”). The time course of product formation was fit to Equation 2 and the kss was determined to be 0.0070 ± 0.0003 s−1 (Fig. 1B), which is in good agreement with the linear phase rate constant measured in the burst assay (Fig. 1A).
Finally, for hPolε to follow a similar mechanism as in Scheme 1A, the dissociation of hPolε from the E•DNA complex must be the slowest step in a multiple turnover reaction and its rate must be equal to the steady-state rate constant of nucleotide incorporation (24–27). Subsequently, we directly measured the rate constant of dissociation of hPolε exo− from DNA (k−1, Scheme 1A). A preincubated solution of hPolε exo− (50 nm, UV concentration) and 5′-radiolabeled D-1 DNA substrate (100 nm) was mixed with a large excess of unlabeled D-1 DNA trap (2.5 μm) for varying incubation times. During the incubation period, any hPolε exo− that dissociated from the labeled D-1 would rebind to the unlabeled D-1, which was present in a 25-fold molar excess over the labeled D-1. Thus, when the reaction was initiated with the addition of dTTP (100 μm) and Mg2+, only hPolε exo− that was still bound to labeled D-1 would catalyze observable product formation. Product concentration was plotted against DNA trap incubation time and the data were fit to Equation 3, resulting in a k-1 of 0.0058 ± 0.0007 s−1 (Fig. 1C). The k-1 value is in good agreement with the measured kss value and confirms that multiple enzyme turnovers are limited by the rate constant of DNA dissociation as observed for p261N and that hPolε follows a similar mechanism as in Scheme 1A. Thus, the presence of the triple mutations and C-terminal residues 1190-2257 of p261 as well as the p59, p17, and p12 subunits did not change the kinetic mechanism and the rate-limiting step of multiple turnovers of DNA polymerization.
Active site titration
To determine whether the triple mutations (D275A/E277A/D368A), p261C, and the presence of the small subunits (p59, p17, and p12) affect the binding affinity of hPolε to a DNA substrate, we measured the apparent equilibrium dissociation constant of hPolε exo− binding to DNA to form the E•DNA binary complex () using an active site titration assay. Because correct nucleotide incorporation during the first turnover was previously shown to be much faster than the equilibration of hPolε binding to DNA (E + DNA ↔ E•DNA) (23), a titration of the polymerase active site with DNA can be used to measure by examining the DNA concentration dependence of the first-turnover amplitude (24–27). Specifically, a fixed concentration of hPolε exo− (50 nm, UV concentration) was preincubated with varying concentrations of D-1 DNA substrate (10-125 nm) to allow the E•DNA complex to form prior to initiation of the reaction with the addition of correct dTTP (100 μm) and Mg2+. The concentration of the product formed during the first turnover was measured by quenching each reaction after 50 ms, which allowed adequate time for the dTTP incorporation to reach the maximum first-turnover amplitude with a negligible contribution of multiple turnovers (23). Importantly, the amplitude of product formation from the first turnover was a direct measurement of the amount of productive E•DNA complex that formed during the preincubation period. Therefore, the maximum amplitude corresponds to the active enzyme concentration. As observed previously for p261N (19) and WT hPolε (23), the active hPolε exo− concentration is low (14.7 nm, 29%), suggesting that the majority of hPolε may not bind to DNA or binds to DNA in a nonproductive conformation. Notably, we have observed that hPolε is prone to forming aggregates, even after purification by size-exclusion chromatography. Such hPolε aggregates certainly contribute to the low active enzyme concentration. Therefore, the determined by active site titration mostly reflects the hPolε population that is bound to DNA in a productive, polymerization-ready state (see “Discussion”).
The concentration of E•DNA complex was plotted against the concentration of total D-1 DNA substrate and the data were fit to a quadratic equation (Equation 4) to give a of 22 ± 4 nm (Fig. 2). Notably, the measured here is comparable to the of 33 nm that we measured previously for WT hPolε (23), indicating that the triple mutations to inactivate the 3′→5′ exonuclease activity of hPolε did not impact the ability of hPolε to bind to a DNA substrate. Previously, a of 79 nm was measured using the same assay to monitor the binding of p261N to the same D-1 DNA substrate (19). Thus, the C-terminal 1,068 residues of p261 and the three small subunits (p59, p17, and p12) collectively increase the binding affinity of hPolε by 3.6-fold (Table 2). Furthermore, the second-order rate constant of DNA and hPolε association (k1 = k-1/, k-1 = 0.0058 s−1, = 22 nm) was calculated to be 2.6 × 105 m−1 s−1, which is nearly identical to the k1 value of 2.7 × 105 m−1 s−1 measured for p261N (19). Consequently, the 3.6-fold binding affinity difference between hPolε and p261N is almost completely contributed by the 3.6-fold difference in their k-1 values (Table 2).
Figure 2.

Active site titration assay to measure the for hPolε exo− binding to DNA. A preincubated solution of hPolε exo− (50 nm, UV concentration) and increasing concentrations of 5′-radiolabeled D-1 DNA substrate (10-125 nm) was rapidly mixed with dTTP (100 μm) and Mg2+ for 50 ms and then quenched with the addition of EDTA. All measurements were performed in triplicate and the average concentration of E•DNA complex that formed during the preincubation period, given by product concentration, was plotted against total D-1 DNA concentration and the data were fit to Equation 4 to yield a of 22 ± 4 nm and an active enzyme concentration (E0) of 14.7 ± 0.8 nm. Error bars represent the S.D. from the calculated average product concentration.
Table 2.
Comparison of kinetic parameters determined for p261N and hPolε at 20 °C
Pre-steady-state kinetics of correct nucleotide incorporation
We then sought to determine whether the presence of p261C, the B-subunit, and the accessory subunits (p17 and p12) affect the kinetics of nucleotide binding and incorporation. To explore this possibility, we measured the equilibrium dissociation constant for correct nucleotide binding () and the maximum nucleotide incorporation rate constant (kmax) by monitoring the dependence of the observed rate constant of nucleotide incorporation on nucleotide concentration under single-turnover kinetic conditions. Briefly, a preincubated solution of hPolε exo− (100 nm, UV concentration) and D-1 DNA substrate (20 nm) was rapidly mixed with increasing concentrations of correct dTTP in the presence of Mg2+. After varying reaction times, the reaction was quenched with the addition of 0.37 m EDTA. The time course of product formation at each nucleotide concentration was fit to Equation 5 to yield a fast (kfast) and a slow (kslow) observed rate constant for nucleotide incorporation (Fig. 3A), similar to the double-exponential kinetics in the burst assay. The fast observed rate constants were plotted against dTTP concentration and the data were fit to Equation 7 (Fig. 3B). The resulting kmax and values were 411 ± 26 s−1 and 11 ± 2 μm, respectively, and the substrate specificity (kmax/) was calculated to be 37 μm−1 s−1.
Figure 3.
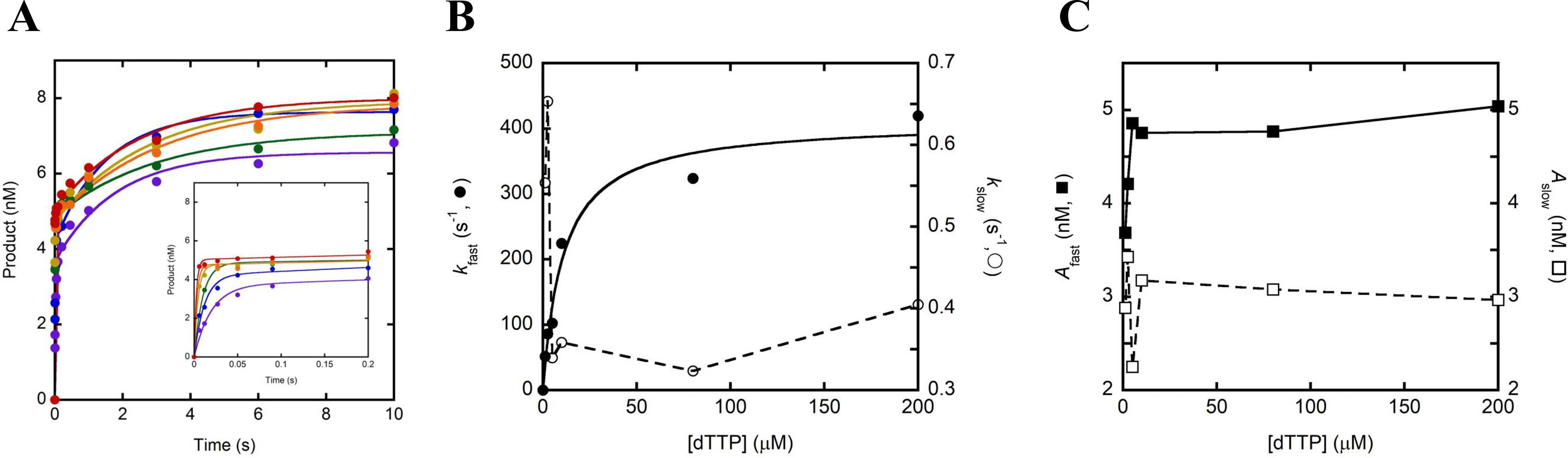
Pre-steady-state kinetics of correct nucleotide incorporation. A, a preincubated solution of hPolε exo− (100 nm, UV concentration) and 5′-radiolabeled D-1 DNA substrate (20 nm) was rapidly mixed with 1.25 (purple), 2.5 (blue), 5 (green), 10 (yellow), 80 (orange), or 200 μm (red) dTTP and Mg2+ for varying incubation times before the reaction was quenched with the addition of EDTA. Product concentration was plotted against time and the data were fit to Equation 5. The inset shows the first 0.2 s of each time course. B, the kfast and kslow values for each dTTP concentration were plotted against their respective dTTP concentration. The plot of kfast versus [dTTP] was fit to Equation 7 to yield a kmax of 411 ± 26 s−1 and a of 11 ± 2 μm. The plot of kslow versus [dTTP] was fit with a smoothed line. C, the Afast and Aslow values for each dTTP concentration were plotted against their respective dTTP concentration, and the data were fit with a smoothed line.
The dependence of kfast on dNTP concentration indicates that kfast reports on the rate constant for nucleotide incorporation onto DNA that is productively bound to the polymerase active site (EP•DNA), and the maximal rate (kmax) therefore reflects the rate-limiting pre-chemistry conformational change (k3) preceding the chemistry step (kchem) (Scheme 1A). In contrast, the observed slow rate constants of product formation, which ranged from 0.32 to 0.65 s−1, showed only a weakly negative correlation with nucleotide concentration (Fig. 3B), which is suggestive of an additional slow step prior to rapid nucleotide binding and incorporation that is exhibited by a significant population of hPolε bound to DNA. A similar result was observed for single-turnover nucleotide incorporation assays performed with S. cerevisiae Polε heterotetramer and the slow phase was attributed either to switching of the DNA primer terminus from the 3′→5′ exonuclease active site to the polymerase active site or to slow binding of Polε to DNA (18). Although plausible, the former case is unlikely given that strand transfer from the 3′→5′ exonuclease active site to the polymerase active site was shown to be very fast (> 700 s−1) for the replicative DNA polymerase from T7 bacteriophage (28). Additionally, we believe that the latter situation is unlikely because a 10-fold molar excess of S. cerevisiae Polε was preincubated with DNA before adding a correct dNTP to initiate nucleotide incorporation (18). Kinetically, hPolε (Fig. 3A) likely bound to DNA in a productive (EP•DNA) and a nonproductive state (EN•DNA) at the polymerase active site, leading to the observed fast and slow nucleotide incorporation phases, respectively (29–34). To illustrate these two possible binding modes, we have proposed a revised Scheme 1B, which shows that hPolε may bind directly to DNA in a productive conformation (E + DNA ↔ EP•DNA) or in a nonproductive conformation (E + DNA ↔ EN•DNA). hPolε that is bound to DNA in a productive, polymerization-ready state will catalyze rapid nucleotide incorporation with a rate constant kP, which is equal to kmax. On the other hand, hPolε that is bound to DNA in a nonproductive state must first transition to the productive state at a rate constant kN→P, which is equivalent to kslow (Scheme 1B). This isomerization is intrinsic and its rate does not depend on nucleotide concentration (28). Thus, the slow phase in Fig. 3A was likely limited by isomerization and thus, its rate constant was not significantly affected by dNTP concentration. Importantly, inclusion of an excess of an unlabeled DNA trap in the reaction mixture does not eliminate the slow phase or alter its rate constant (Fig. S2), supporting our conclusion that the slow phase results from the transition of DNA-bound hPolε from a nonproductive to a productive state. Finally, we note that the amplitude of the fast phase (Afast) shows a weak positive correlation with nucleotide concentration, whereas the amplitude of the slow phase (Aslow) is relatively unaffected (Fig. 3C). The relationship between the amplitudes of the two phases and nucleotide concentration is unclear and is not further explored in the present study.
Elemental effect on correct nucleotide incorporation
To further determine whether p261C, the B-subunit, and the accessory subunits affect the rate-limiting step of correct nucleotide incorporation by hPolε exo−, we compared the incorporation rate constants of dTTP and α-thiophosphate-substituted dTTP (Sp-dTTPαS). A preincubated solution of hPolε exo− (100 nm, UV concentration) and D-1 DNA substrate (20 nm) was rapidly mixed with dTTP (or Sp-dTTPαS) (5 μm) and Mg2+ for varying incubation times. The reaction was quenched with the addition of 0.37 m EDTA. The time courses of product formation were fit to Equation 5 to yield fast observed incorporation rate constants of 127 ± 15 s−1 and 118 ± 21 s−1 for dTTP and Sp-dTTPαS, respectively (Fig. 4A). The elemental effect () was calculated to be 1.1 for hPolε exo−. Previously, an elemental effect of 4−11 was used as suggestive evidence for a rate-limiting chemistry step during nucleotide incorporation (19, 24–26, 35). Thus, the protein conformational change (EP•DNAn•dNTP → EP′•DNAn•dNTP), rather than phosphodiester bond formation (EP′•DNAn•dNTP → EP′•DNAn+1•PPi), is likely rate-limiting for each round of correct nucleotide incorporation catalyzed by hPolε exo− (Scheme 1B). Notably, a similarly small elemental effect (0.9) was observed for correct nucleotide incorporation catalyzed by p261N (19). Thus, both hPolε and p261N share the same rate-limiting step during correct nucleotide incorporation (Scheme 1). Finally, the slow phases for incorporation of dTTP and Sp-dTTPαS yielded an elemental effect of 1.2, indicating that, as expected, the slow phase of nucleotide incorporation, limited by the intrinsic isomerization rate kN→P (Scheme 1B), was not affected by the α-thiophosphate substitution in Sp-dTTPαS.
Figure 4.

Elemental effect on correct and incorrect nucleotide incorporation. A, a preincubated solution of hPolε exo− (100 nm, UV concentration) and 5′-radiolabeled D-1 DNA substrate (20 nm) was rapidly mixed with 5 μm dTTP (●) or Sp-dTTPαS (○) and Mg2+ for varying incubation times before quenching with the addition of EDTA. Product concentration was plotted against time and the data were fit to Equation 5 to yield fast rate constants of 127 ± 15 s−1 and 118 ± 21 s−1 for dTTP and Sp-dTTPαS, respectively, resulting in an elemental effect of 1.1. B, the elemental effect on incorrect dATP incorporation was tested under identical conditions, except the reaction was initiated with the addition of 500 μm dATP (●) or Sp-dATPαS (○) and Mg2+. Product concentration was plotted against time and the data for dATP incorporation were fit to Equation 6, whereas the data for Sp-dATPαS were fit to a line. The incorporation rate constants were 0.073 ± 0.007 s−1 for dATP and 0.0016 s−1 for Sp-dATPαS, giving an elemental effect of 46.
Pre-steady-state kinetics of incorrect nucleotide incorporation
The p261N fragment of hPolε exhibits high base substitution fidelity (10−4–10−7) resulting from decreases in both kmax and the ground-state binding affinity (1/) for incorrect relative to correct nucleotides (21). To probe whether p261C, the B-subunit, and the accessory subunits affect the base substitution fidelity of hPolε, we measured the pre-steady-state kinetic parameters for the incorporation of all three incorrect nucleotides with D-1 as described above. The kmax and values for all four nucleotides are listed in Table 3. As for correct dTTP incorporation, we calculated the substrate specificities for each of the incorrect nucleotides and subsequently determined the base substitution fidelity (Fpol) of hPolε exo−, which was 10−5–10−7 (Table 3). We also measured the elemental effect for incorrect nucleotide incorporation onto D-1 by comparing the incorporation rate constant of dATP and Sp-dATPαS (500 μm). The observed incorporation rate constants were 0.073 ± 0.007 s−1 for dATP and 0.0016 s−1 for Sp-dATPαS (Fig. 4B), yielding an elemental effect () of 46, which indicates that phosphodiester bond formation is at least partially rate-limiting for incorrect nucleotide incorporation (see above). This is similar to the large elemental effect (167) on incorrect nucleotide incorporation observed with p261N (19).
Table 3.
Kinetic parameters for correct and incorrect nucleotide incorporation onto the D-1 DNA substrate catalyzed by hPolε exo− at 20 °C
Interestingly, only a single phase of product formation was observed for incorporation of dATP and Sp-dATPαS (Fig. 4B). Likewise, misincorporation of dCTP lacked a second phase (data not shown). Unlike correct nucleotide incorporation, the rate of intrinsic conversion from the nonproductive to productive bound state for DNA binding to hPolε exo− is faster than or comparable to the rate of misincorporation, and the overall observed rate constant of nucleotide incorporation was either limited by slow incorrect nucleotide incorporation or was a function of both kN→P (Scheme 1B) and slow nucleotide incorporation. In the case of dGTP misincorporation, a fast phase of incorporation was observed for a very small population of the total DNA concentration (< 5%), and the reaction time course was dominated by very slow nucleotide incorporation (data not shown). The significance of the small population of more rapidly misincorporated dGTP is unclear and requires further studies.
Pre-steady-state kinetics of mismatch extension
Previously, we determined that p261N exo− very poorly extends single-base mismatches due to large decreases in both kmax and 1/for the next correct nucleotide (21). To determine whether the presence of p261C, the B-subunit, and the accessory subunits affect the ability of hPolε exo− to extend a single-base mismatch, we measured the kinetic parameters for incorporation of the next correct nucleotide, dCTP, onto two different mismatched substrates containing either a C:A mismatch (M-1, Table 1) or a C:C mismatch (M-8, Table 1) at the primer 3′ terminus. For extension of both the C:A and the C:C mismatched DNA substrates, the kmax of correct nucleotide incorporation was reduced by 100- and 63,000-fold (Table 4), respectively, relative to extension of a correctly-matched A:T base pair in D-1 (Tables 1 and 3). Furthermore, the binding affinity for the correct nucleotide was reduced by 22-fold when extending from the C:A base pair, and 64-fold when extending from the C:C base pair. Overall, the substrate specificity of the correct nucleotide was reduced by 2.2 × 103- and 4.0 × 106-fold with the C:A and C:C mismatched DNA substrates (Tables 3 and 4), respectively, indicating that hPolε exo− very poorly extended mismatched DNA substrates compared to matched substrates as observed with p261N exo− (21). However, both hPolε exo− and p261N exo− demonstrate a marked variability in their abilities to extend different mismatched base pairs. Based on the current study, hPolε exhibits an 1,800-fold preference for extension of a C:A mismatch versus a C:C mismatch (Table 4), whereas p261N only displayed a 28-fold preference for C:A versus C:C (21). The reason for this difference in mismatch type discrimination remains to be determined. We further tested the ability of hPolε exo− to extend the C:A mismatch with an incorrect nucleotide, dGTP. From the measured kinetic parameters, we calculated a substrate specificity of 1.6 × 10−7 μm−1 s−1 for dGTP (Table 4), which is similar to the substrate specificity values determined for extension of a mismatched base pair with an incorrect nucleotide by p261N exo− (21). Thus, both hPolε exo− and p261N exo− are similarly unlikely to bury a mismatched base pair with an additional mismatch. Based on both a 20,000-fold decrease in kmax and a 5.4-fold decrease in binding affinity relative to extension of the same mismatch with correct dCTP, we determined the base substitution fidelity during mismatch extension (Fext) to be 9.4 × 10−6 (Table 4), demonstrating that hPolε exo− remains highly selective for the correct nucleotide even when extending from a mismatch.
Table 4.
Kinetic parameters for mismatch extension and excision catalyzed by hPolε exo− and hPolε exo+ at 20 °C
| dNTP | kmax | kmax/ | Fexta | b | kX | Fexoc | |
|---|---|---|---|---|---|---|---|
| s−1 | μm | μm−1 s−1 | s−1 | s−1 | |||
| C:A mismatch (M-1) | |||||||
| dCTP | 4.1 ± 0.3 | (2.4 ± 0.5) × 102 | 1.7 × 10−2 | - | 1.2 | - | |
| dGTP | (2.1 ± 0.2) × 10−4 | (1.3 ± 0.2) × 103 | 1.6 × 10−7 | 9.4 × 10−6 | 1.5 × 10−5 | - | |
| - | - | - | - | - | - | 0.4 ± 0.1 | 0.3 |
| C:C mismatch (M-8) | |||||||
| dCTP | (6.5 ± 0.5) × 10−3 | (7 ± 1) × 102 | 9.3 × 10−6 | - | 8.1 × 10−4 | - | |
| - | - | - | - | - | - | 0.5 ± 0.1 | 617 |
a Calculated as (kmax/)incorrect/[(kmax/)correct + (kmax/)incorrect].
b Calculated as kmax[dNTP]/( + [dNTP]) during extension from a mismatched primer terminus at an intracellular dCTP concentration of 100 μm.
c Calculated as kX/.
Pre-steady-state kinetics of matched and mismatched base pair excision
Previous pre-steady-state kinetic analysis of mismatch extension and excision by p261N revealed that the 3′→5′ exonuclease activity of p261N increased its fidelity of DNA synthesis by 3.5 × 102- to 1.2 × 104-fold due to a strong preference for excision of a mismatched DNA substrate coupled with poor efficiency of extension (21). Here, we used a similar approach to measure the excision rate constants for a matched A:T base pair in D-1 and a mismatched C:A base pair in M-1 (Table 1) by WT hPolε heterotetramer. A preincubated solution of WT hPolε (100 nm, UV concentration) and a 5′-radiolabeled DNA substrate (20 nm) was rapidly mixed with Mg2+ in the absence of nucleotides. The concentration of remaining DNA substrate was plotted against time (Fig. 5). The data for excision of the matched D-1 DNA substrate were fit to Equation 9 to yield an excision rate constant of 0.015 ± 0.003 s−1. In contrast to the matched D-1 DNA substrate, the mismatched M-1 DNA substrate displayed two clear phases of excision (Fig. 5). Accordingly, the data were fit Equation 10 to yield a fast excision rate constant of 0.4 ± 0.1 s−1 and a slow excision rate constant of 0.0099 ± 0.0006 s−1. Thus, in the presence of a single mismatch, the excision rate constant increased by 27-fold. This is comparable with the 15-fold 3′→5′ exonuclease activity enhancement measured previously for p261N exo+ in the presence of a single-base mismatch (Table 2). Similarly, for the M-8 substrate, which contains a C:C mismatch, we measured a fast excision rate constant of 0.5 ± 0.1 s−1 and a slow mismatch excision rate constant of 0.0041 ± 0.0002 s−1 (data not shown), indicating that the identity of the mismatched base pair does not significantly affect the excision rate constant of hPolε as was observed with p261N (21).
Figure 5.

Pre-steady-state kinetics of matched and mismatched base pair excision. A preincubated solution of WT hPolε (100 nm, UV concentration) and 5′-radiolabeled D-1 (●) or M-1 (○) DNA substrate (20 nm) was rapidly mixed with Mg2+ for varying incubation times before quenching with the addition of EDTA. Remaining substrate concentration was plotted against time. The data for excision of the matched D-1 substrate were fit to Equation 9 to yield an excision rate constant of 0.015 ± 0.003 s−1. The data for excision of the mismatched M-1 DNA substrate were fit to Equation 10 to yield a fast excision rate constant of 0.4 ± 0.1 s−1 and a slow excision rate constant of 0.0099 ± 0.0006 s−1.
The single excision rate constant for the matched DNA substrate and the slow excision rate constants for the mismatched DNA substrates are comparable to each other and likely represent a slow transition of the DNA substrate from the polymerase active site to the 3′→5′ exonuclease active site (28). Accordingly, we have added this strand transfer step (EP•DNA ↔ EX•DNA) to Scheme 1B, and the rate of this strand transfer, EP→X, is equal to the rate of the slow phase of mismatch excision. The appearance of an initial fast phase for the mismatch-containing DNA substrates likely corresponds to direct binding of the mismatched primer terminus to the 3′→5′ exonuclease active site of hPolε (E + DNA ↔ EX•DNA) as was observed previously for hPolγ (36). For the M-1 and M-8 substrates, the amplitudes of their fast phases accounted for 12–17% of the total DNA concentration, indicating that a significant percentage of the DNA is bound at the 3′→5′ exonuclease site of hPolε during preincubation with a mismatch-containing DNA substrate. Accordingly, we have added a third possible configuration for hPolε binding to DNA in Scheme 1B. Mismatched DNA that is pre-bound to the 3′→5′ exonuclease active site of hPolε is cleaved at a rate constant kX, which is equal to the fast phase of excision. Notably, the rate constants for excision of both matched and mismatched DNA substrates by hPolε are reduced by 11- and 5.8-fold, respectively, compared with the excision rate constants measured for p261N (Table 2). Thus, the presence of p261C, the B-subunit, and the accessory subunits appears to modulate the 3′→5′ exonuclease activity of hPolε.
Discussion
Kinetic mechanism for DNA polymerization catalyzed by the hPolε holoenzyme
Using pre-steady-state kinetic methods, we previously determined that p261N, the N-terminal 140-kDa fragment of the p261 catalytic subunit, catalyzes correct nucleotide incorporation via an induced-fit mechanism (19) that is generally followed by all DNA polymerases (Scheme 1) and is characterized by a rate-limiting pre-chemistry conformational change following nucleotide binding (37, 38). In the present study, as well as in our previous study (23), we demonstrated that the hPolε holoenzyme, like p261N, incorporated correct nucleotides by following multiphasic kinetics (Fig. 1A). We then determined that the multiple-turnover (or steady-state) rate (0.007 s−1) of nucleotide incorporation was governed by the hPolε•DNA complex dissociation (0.0058 s−1) (Fig. 1). Through an active site titration assay, the apparent binding affinity of D-1 DNA substrate to hPolε (1/, = 22 nm) was measured (Fig. 2), indicating that the first turnover of nucleotide incorporation was much faster than the equilibration of hPolε and DNA binding (E + DNA ↔ E•DNA) (23).
Based on the results of our pre-steady-state kinetic assays (discussed below), hPolε likely binds to DNA in three different modes during preincubation with a duplex DNA substrate: a productive polymerization-ready state (EP•DNA), a nonproductive state (EN•DNA), and with the DNA bound at the 3′→5′ exonuclease active site (EX•DNA). Thus, the measured by the active site titration assay (Fig. 2) is a function of , and (Scheme 1B). However, is expected to be a good approximation of for the following reasons. First, the reaction is quenched after 50 ms, which is too short to observe any additional product formation resulting from the slow conversion of EN•DNA to EP•DNA (EN→P; 0.32-0.65 s−1; t1/2 ∼1.1-2.2 s). Second, excision of a matched DNA substrate is dominated by the rate of strand transfer from the polymerase active site to the 3′→5′ exonuclease active site (EP→X). The lack of a fast phase of excision indicates that no appreciable population of DNA is bound to the 3′→5′ exonuclease active site of hPolε (EX•DNA). Thus, is expected to represent a good approximation of the equilibrium dissociation constant for productive binding of DNA to the polymerase active site of hPolε (EP•DNA).
Through the single-turnover kinetic assay, we determined the maximum correct dTTP incorporation rate (kmax = 411 s−1) and ground-state binding affinity (1/, = 11 μm) of correct dTTP with hPolε (Fig. 3). Furthermore, we measured the elemental effect (1.1) of dTTP incorporation onto D-1 by hPolε (Fig. 4A), suggesting that phosphodiester bond formation did not limit the single-turnover rate of correct nucleotide incorporation (19, 24–26, 35). Together, these kinetic assays demonstrate that hPolε and p261N possess similar kinetic behaviors and likely follow the similar minimal kinetic mechanism for productive nucleotide incorporation (compare Scheme 1, A and B) involving a rate-limiting conformational change (k3 in Scheme 1A or kP in Scheme 1B) that is triggered by correct nucleotide binding ( = k-2/{k2[dNTP]}) and precedes phosphodiester bond formation (kchem) (Scheme 1).
The presence of two distinct phases of correct nucleotide incorporation by hPolε provides evidence for hPolε binding to DNA in at least two modes: a productive, polymerization-ready state (EP•DNA) and a nonproductive state (EN•DNA). Although our pre-steady-state exonuclease assays demonstrate that DNA may also bind directly to the 3′→5′ exonuclease active site of hPolε (EX•DNA), we argue that the slow phase of nucleotide incorporation is not rate-limited by strand transfer from the 3′→5′ exonuclease active site to the polymerase active site (kX→P) as the rate constant for this strand transfer was previously measured to be very fast (>700 s−1) (28). Moreover, the absence of a fast excision phase in our exonuclease assays with D-1 (Fig. 5) shows that almost no matched DNA substrate is bound to the 3′→5′ exonuclease active site after the pre-incubation phase. Therefore, the slow phase of polymerization most likely results from DNA-bound enzyme isomerizing from the nonproductive to the productive state (kN→P). In contrast, the strong dependence of the fast phase of nucleotide incorporation on nucleotide concentration indicates that the maximal rate of the fast phase (kmax) represents the rate-limiting pre-chemistry conformational change (kP). Accordingly, Scheme 1B illustrates the interconversion between nonproductive and productive polymerization states, the latter of which binds nucleotide to catalyze rapid correct nucleotide incorporation.
Effect of the C-terminal domain of p261 and the three small subunits on nucleotide incorporation kinetics with hPolε
Previously, we determined that p261N rapidly incorporates correct nucleotides with an average kmax of 252 s−1 at 20 °C and an average of 23 μm (Table 2), and exhibits large decreases in both kmax and ground-state nucleotide binding affinity (1/) for incorrect nucleotides thereby achieving the high base substitution fidelity (10−4–10−7) (21). In this study, we determined that hPolε catalyzed correct dTTP incorporation onto D-1 at 20 °C with a slightly higher kmax (411 s−1, 1.6-fold), a slightly lower (11 μm, 2.1-fold), and a moderately higher kmax/ (37 μm−1 s−1, 3.4-fold) (Tables 2 and 3). Furthermore, we measured the kinetic parameters for all three misincorporations onto D-1 and calculated the base substitution fidelity of hPolε to be 10−5–10−7, which is 8- to 20-fold higher than that of p261N (Table 3). The high base substitution fidelity of hPolε is a hallmark of replicative DNA polymerases (39). Taken together, the presence of p261C and the three small subunits favorably affect the nucleotide incorporation kinetics and fidelity of Polε. In comparison, the pre-steady-state kinetic parameters determined for correct nucleotide incorporation by S. cerevisiae Polε are nearly identical to those measured for the C-terminal truncated Pol2 catalytic subunit, indicating that the small subunits do not significantly affect nucleotide incorporation kinetics with S. cerevisiae Polε (18). This conclusion is supported by the positioning of the small subunits away from the globular Pol2 catalytic domain in the low-resolution cryo-EM structure of S. cerevisiae Polε (13). Other than the solution structure of an N-terminal fragment of the p59 subunit (40), there are currently no structures of the hPolε heterotetramer. Although in vitro transcription/translation and immunoprecipitation studies have assigned the binding sites for the three small subunits within the p261C domain (20), the structure of the polymerase active site within p261N is likely affected by the association of the small subunits in the hPolε holoenzyme.
Interestingly, hPolε catalyzes correct dCTP incorporation during extension from a single-base mismatch with a kmax of 4.1 s−1 for a C:A mismatch and 0.0065 s−1 for a C:C mismatch (Table 4). This wide kmax range (10−3–10° s−1) contrasts significantly from the values determined for p261N, which generally catalyzes single-base mismatch extension with a kmax on the order of 10−2 s−1 regardless of mismatch identity (Table 2). The wide kmax range also affects the contribution of the 3′→5′ exonuclease to the overall fidelity of hPolε, which was calculated to be 0.3- or 617-fold under a typical intracellular nucleotide concentration of 100 μm (Table 4). Consequently, the wide-ranging contribution to fidelity by the 3′→5′ exonuclease activity of hPolε is likely one of the factors to cause sequence-dependent DNA replication errors in vivo (41, 42).
Enhanced DNA binding affinity of hPolε by the C-terminal domain of p261 and three small subunits
Earlier structural and biochemical studies of S. cerevisiae Polε implicated a direct role for the three small subunits in modulating the polymerase processivity of Polε. A low-resolution cryo-EM structure of S. cerevisiae Polε heterotetramer revealed an extended tail-like structure that is hypothesized to encircle dsDNA that trails Polε as it synthesizes the leading strand, thereby tethering Polε to DNA and enhancing processivity (13). This hypothesis is supported by the observation that S. cerevisiae Polε heterotetramer is able to synthesize longer products than the Pol2 catalytic subunit alone under single-hit conditions but only when the length of dsDNA is at least 40 nucleotides long (43). Consistently, the Dpb3/Dpb4 (corresponding to p12/p17 in humans) heterodimer forms a stable complex with dsDNA in the absence of Pol2/Dpb2 (corresponding to p261/p59 in humans) (44) and significantly enhances the polymerase and 3′→5′ exonuclease processivities of the Pol2/Dpb2 heterodimer (43), suggesting that the increased processivity results from additional interactions between S. cerevisiae Polε and DNA afforded by the small subunits. In the presence of p261C and the three small subunits, the hPolε heterotetramer binds to D-1 DNA substrate with a of 22 nm (Fig. 2), which is 3.6-fold lower than the previously measured for p261N binding to the same D-1 DNA (Tables 1 and 2). Additionally, the D-1 dissociation rate constant of 0.0058 s−1 was determined using unlabeled DNA trap (Fig. 1C). Notably, this DNA dissociation rate constant is 3.6-fold slower than that measured for p261N (Table 2) and essentially accounts for the 3.6-fold difference in DNA binding affinity, indicating that the C-terminal residues 1190-2257 of p261 and the p59, p17, and p12 subunits collectively enable hPolε to form a tighter, more stable complex with its DNA substrate. Surprisingly, our previous data show that the processivities of p261N and hPolε heterotetramer are indistinguishable when the polymerases are in excess over a DNA substrate (23). In addition, p261N and hPolε are comparably active on a primed M13 DNA template and their activities are similarly enhanced in the presence of PCNA, replication factor C, and replication protein A (20). Thus, the increased DNA binding affinity does not appear to affect the polymerization processivity of hPolε, contrasting sharply with the effect of the small subunits on the polymerase processivity of S. cerevisiae Polε. The difference will be answered by future high-resolution structures of hPolε and S. cerevisiae Polε in complex with DNA and an incoming dNTP. Surprisingly, the recent high-resolution cryo-EM structure of the complex of hPolδ, PCNA, DNA, and dNTP shows that the C-terminal domain of hPolδ and the three subunits do not directly interact with DNA but may indirectly facilitate DNA binding through stabilizing the complex by interactions between the subunits and with one of the three PCNA monomers (14). Thus, the contributions of p261C and the three small subunits to DNA binding by hPolε may be different in the presence and absence of PCNA.
Modulation of hPolε proofreading activity by the C-terminal domain of p261 and the three small subunits
WT hPolε excised a matched A:T in D-1 with a single-turnover excision rate constant of 0.015 s−1. In contrast, the C:A mismatch in M-1 and the C:C mismatch in M-8 were excised with fast rate constants of 0.4 s−1 and 0.5 s−1 and slow rate constants of 0.0099 s−1 and 0.0041 s−1, respectively (Tables 1, 2, and 4). Thus, the excision rate constant is enhanced roughly 27- to 33-fold in the presence of a single-base mismatch compared with a matched base pair.
The presence of two phases of excision for a mismatched DNA substrate suggests that some population of mismatched DNA substrate may bind directly to the 3′→5′ exonuclease active site of hPolε. Although the fast phase excision rate constant (0.4-0.5 s−1) is much faster than the single rate constant for excision of a matched DNA substrate (0.015 s−1), the slow phase excision rate constants for the two mismatched DNA substrates tested here (0.0041-0.0099 s−1) are more comparable with the rate constant for matched DNA excision. Therefore, these rate constants are likely representative of the rate constant for strand transfer from the polymerase active site to the 3′→5′ exonuclease active site (kP→X), whereas the fast phase excision rate constants represent cleavage of DNA pre-bound to the 3′→5′ exonuclease active site of hPolε. Accordingly, Scheme 1B shows a third DNA binding mode in which DNA directly binds to the 3′→5′ exonuclease active site of hPolε or transfers from the polymerase active site to the 3′→5′ exonuclease active site. Once bound to the 3′→5′exonuclease site, the DNA substrate is cleaved with a rate constant kX.
Surprisingly, the excision rate constants measured with WT hPolε are significantly lower than those measured for p261N exo+ and are reduced by 11-fold for excision of a matched DNA substrate and 5.8-fold for excision of a mismatched DNA substrate (Table 2). Furthermore, excision of mismatched substrates by p261N exo+ was dominated by a single fast phase, whereas hPolε displayed a low-population fast phase of excision in the presence of a mismatch. Thus, the presence of p261C and the three small subunits appears to modulate the 3′→5′ exonuclease activity of hPolε by limiting both strand transfer from the polymerase active site to the 3′→5′ exonuclease active site (kP→X) as well as direct binding of the DNA to the 3′→5′ exonuclease active site. Interestingly, this result contrasts sharply with the pre-steady-state kinetic analysis of S. cerevisiae Polε, which demonstrated that the excision rate constants for matched, mismatched, and ssDNA substrates were largely unaffected by the presence or absence of the Pol2 C-terminal domain and the three small subunits (18).
Previously, pre-steady-state kinetic methods have been used to examine the 3′→5′ exonuclease activities of hPolδ, the major lagging strand replicative DNA polymerase, and hPolγ, the mitochondrial replicative DNA polymerase, as well as their oligomeric assemblies. Notably, kinetic comparison of a heterotrimeric hPolδ assembly (p125-p50-p66), and the fully assembled heterotetramer (p125-p50-p66-p12), revealed that addition of p12 to the hPolδ heterotrimer results in a 4.6-fold increase in polymerase activity, but 8.3- and 4.8-fold decreases in the excision rates of matched and single-base mismatched DNA substrates, respectively (17). The high-resolution cryo-EM structure of the complex of hPolδ, PCNA, DNA, and dNTP shows that p12 bridges the exonuclease and C-terminal domains of p125 as well as the oligonucleotide-binding domain of p50 and stabilizes hPolδ (14). The stabilization likely enhances hPolδ polymerase activity but restricts the transfer of a mismatched primer from the polymerase to the 3′→5′ exonuclease active site for editing. Consequently, processive polymerization by hPolδ heterotetramer is less likely to be interrupted by the slow switching of the DNA primer from the polymerase to the 3′→5′ exonuclease active site. Consistent with the enhanced polymerase activity and attenuated exonuclease activity, hPolδ heterotetramer exhibits increased tolerance for translesion synthesis and mismatch incorporation and extension in the presence of p12 (45). Interestingly, it has been shown that treatment of human cells with UV, methyl methanesulfonate, and other DNA damaging agents results in degradation of p12 (46). Taken together, damage-dependent p12 degradation resulting in a more proofreading active form of hPolδ is a potential mechanism that human cells have evolved for minimizing mutagenic DNA synthesis while maintaining the high rate of DNA synthesis required for timely genome replication under normal cellular conditions. Similarly, hPolγ catalyzes nucleotide incorporation 5-fold faster and is 7.8-fold more processive in the presence of its p55 accessory subunit (47), but exhibits a 2-fold decrease in exonuclease activity and greater selectivity against excision of correctly matched dsDNA (36). However, unlike hPolδ, a similar mechanism of proofreading activity control has not been identified for hPolγ in vivo. Finally, whereas the polymerization rate we have measured for hPolε heterotetramer increases slightly from that of p261N (411 s−1 versus 252 s−1, 1.6-fold; Table 2), the >5-fold decrease in exonuclease-activity is consistent with the regulatory effects exerted upon hPolδ and hPolγ by their small subunits. As with hPolγ, the significance of such control is unknown. Overall, the proofreading activities of these three replicative DNA polymerases are comparably modulated in the presence of their respective small subunits, although the physiological role for proofreading control may not be common between these DNA polymerases.
Experimental procedures
Materials
Reagents used for the experiments described were purchased from the following sources: [γ-32P]ATP from PerkinElmer Life Sciences (Boston, MA); Optikinase from U.S. Biochemicals (Cleveland, OH); and dNTPs from Bioline (Taunton, MA).
Preparation of the human DNA polymerase ε heterotetramer
The 11A/hPolε1exo− vector, which encodes an exonuclease-deficient (D275A/E277A/D368A) variant (19) of the full-length p261 catalytic subunit, and the 11A/hPolε234 vector, which encodes the p59 B-subunit as well as the p17 and p12 accessory subunits, were kindly provided by Drs. Yoshihiro Matsumoto and Alan Tomkinson at the University of New Mexico (Albuquerque, NM). The exo− p261 sequence contains C-terminal PKA, FLAG, and His6 tags. The p59 sequence contains an N-terminal StrepII tag, a thrombin cleavage site, a PKA tag, and a hemagglutinin tag. The p17 and p12 sequences are WT and contain no tags. Recombinant baculoviruses were generated using the Bac-to-Bac expression system (Life Technologies) and amplified to a high titer according to the manufacturer's instructions. The hPolε exo− heterotetramer was overexpressed by simultaneously infecting 1.8 × 109 Sf9 cells in a 1-liter shaking culture with p261 exo− virus and virus encoding all three small subunits. The infected insect cells were incubated at 25 °C for 60 h, and then harvested and lysed by Dounce homogenization. The lysate was clarified by ultracentrifugation at 20,000 × g and hPolε exo− heterotetramer was purified from the cleared lysate essentially as described (23). The WT hPolε heterotetramer was also prepared as described (23).
DNA substrates
The DNA substrates listed in Table 1 were purchased from Integrated DNA Technologies, Inc. (Coralville, IA) and purified by denaturing PAGE as described (48). All primers were radiolabeled by incubation with [γ-32P]ATP and OptiKinase at 37 °C for 3 h. Following the labeling reaction, the kinase was inactivated by heating the reaction mixture at 95 °C for 5 min, and unreacted [γ-32P]ATP was removed by passing the reaction mixture through a Bio-Spin 6 column (Bio-Rad). The 5′-radiolabeled primers were annealed to the template oligonucleotides in Table 1 by incubating the primer with a 1.15-fold molar excess of template at 95 °C for 5 min and then slowly cooling the mixture to room temperature over several hours.
Pre-steady-state kinetic assays
All assays were performed at 20 °C in reaction buffer containing 50 mm Tris-OAc (pH 7.4), 8 mm Mg(OAc)2, 1 mm DTT, 10% glycerol, 0.1 mg/ml BSA, and 0.1 mm EDTA. Fast reactions were performed using a rapid chemical quench-flow apparatus (KinTek). All reported concentrations are final after mixing. All reactions were quenched with the addition of EDTA. Use of 1 m HCl as a quenching solution decreases the observed rate constants by ∼2-fold (Fig. S3). However, there are still distinct fast and slow phases with acid quench as with EDTA quench (Fig. S3), indicating that the slow phase did not result from slow incorporation by the E•DNA•dNTP complex that cannot be stripped of Mg2+ by the EDTA quench. Thus, the 2-fold rate constant decrease observed with acid quench relative to EDTA quench does not change our proposed kinetic mechanism (Scheme 1B). Accordingly, we chose to use an EDTA quench to avoid strong acid damage to our rapid chemical quench-flow apparatus. Unless otherwise specified, most experimental data were collected from only single trials due to the insufficient quantities of purified hPolε holoenzyme to repeat all assays in triplicate.
Active site titration assay
A preincubated solution of hPolε exo− (50 nm, UV concentration) and 5′-radiolabeled D-1 DNA substrate (10-125 nm) was rapidly mixed with a solution containing dTTP (100 μm). The reaction was allowed to proceed for 50 ms and then quenched with the addition of 0.37 m EDTA. Each data point for the active site titration assay was measured in triplicate.
Measurement of the E•DNA complex dissociation rate constant
A preincubated solution of hPolε exo− (50 nm, UV concentration) and 5′-radiolabeled D-1 DNA substrate (100 nm) was mixed with an excess of unlabeled D-1 DNA substrate (2.5 μm). After varying incubation times, the reaction mixture was supplemented with dTTP (100 μm). The nucleotide incorporation reaction was allowed to proceed for an additional 15 s and then was quenched with addition of 0.37 m EDTA.
Measurement of the steady-state rate constant of correct nucleotide incorporation
A preincubated solution of hPolε exo− (1 nm, active site concentration) and 5′-radiolabeled D-1 DNA (400 nm) was mixed with a solution containing dTTP (100 μm). After varying incubation times, an aliquot of the reaction mixture was quenched in 0.37 m EDTA.
Measurement of the elemental effect on nucleotide incorporation
A preincubated solution of hPolε exo− (100 nm, UV concentration) and 5′-radiolabeled D-1 DNA substrate (20 nm) was rapidly mixed with a solution containing either dTTP or Sp-dTTPαS (5 μm) for varying reaction times and quenched with the addition of 0.37 m EDTA. The elemental effect on incorrect nucleotide incorporation was measured under identical assay conditions, but with the addition of incorrect dATP or Sp-dATPαS (500 μm).
Single-turnover exonuclease assays
A preincubated solution of WT hPolε (100 nm, UV concentration) and 5′-radiolabeled DNA substrate (20 nm) was rapidly mixed with a solution containing 8 mm Mg(OAc)2 to initiate the 3′→5′ exonuclease reaction. After varying reaction times, the reaction was quenched with the addition of 0.37 m EDTA.
Product analysis
Reaction substrates and products were separated by denaturing PAGE (17% acrylamide, 8 m urea, and 1× TBE running buffer) and quantified using a Typhoon TRIO (GE Healthcare) and ImageQuant (Molecular Dynamics).
Data analysis
Data were fit by nonlinear regression using Kaleidagraph (Synergy Software). Data from the burst assay were fit to Equation 1,
| (Eq. 1) |
where Afast and Aslow are the amplitudes of product formation of the fast and slow phases, kfast and kslow are the observed rate constants of the fast and slow phases, and klinear is the observed steady-state rate constant.
Data from the steady-state nucleotide incorporation assay were fit to Equation 2,
| (Eq. 2) |
where kss is the steady-state rate constant of nucleotide incorporation at the initial enzyme concentration of E0.
Data from the DNA dissociation assay were fit to Equation 3,
| (Eq. 3) |
where A is the product concentration in the absence of the DNA trap and k-1 is the DNA dissociation rate constant.
Data from the active site titration assay were fit to Equation 4,
| (Eq. 4) |
where is the apparent equilibrium dissociation constant for hPolε binding to DNA to form the E•DNA binary complex, E0 is the enzyme concentration, and D0 is the DNA concentration.
Data from single-turnover polymerization assays were fit to Equation 5,
| (Eq. 5) |
where Afast and Aslow are the amplitudes of product formation of the fast and slow phases and kfast and kslow are the observed rate constants of the fast and slow phases. For some misincorporations, only a single exponential phase of product formation was observed, or the first fast phase was populated by less than 5% of the total DNA substrate. Accordingly, these data were fit to Equation 6,
| (Eq. 6) |
where A is the amplitude of product formation and kobs is the rate constant of nucleotide incorporation.
The kfast (or kobs) values were plotted against nucleotide concentration and the data were fit to Equation 7,
| (Eq. 7) |
where kmax is the maximum rate constant of nucleotide incorporation and is the equilibrium dissociation constant for dNTP binding. When was very large, the data were fit to Equation 8,
| (Eq. 8) |
to yield the substrate specificity constant, kmax/.
Data from single-turnover exonuclease assays with a matched DNA duplex were fit to Equation 9,
| (Eq. 9) |
where Aexo is the amplitude of DNA substrate excision and kexo is the observed excision rate constant.
Data from single-turnover exonuclease assays with a mismatched DNA duplex were fit to Equation 10,
| (Eq. 10) |
where Aexo and Aexo2 are the amplitudes of DNA substrate excision of the fast and slow phases and kexo and kexo2 are the observed excision rate constants of the fast and slow phases.
All reported errors were generated by fitting the data to the above equations in Kaleidagraph.
Data availability
All data are in the manuscript and supporting information, or are available from the authors Walter J. Zahurancik (zahurancik.2@osu.edu) and Zucai Suo (zucai.suo@med.fsu.edu) upon request.
Supplementary Material
This article contains supporting information.
Author contributions—W. J. Z. data curation; W. J. Z. formal analysis; W. J. Z. and Z. S. funding acquisition; W. J. Z. and Z. S. validation; W. J. Z. and Z. S. methodology; W. J. Z. and Z. S. writing-review and editing; Z. S. conceptualization; Z. S. resources; Z. S. supervision; Z. S. investigation; Z. S. visualization; Z. S. writing-original draft; Z. S. project administration.
Funding and additional information—This work was supported by National Institutes of Health Grant R01ES028271 and the Florida State University startup fund (to Z. S.). W. J. Z. was supported by a Pelotonia Graduate Fellowship from The Ohio State University Comprehensive Cancer Center and a Presidential Fellowship from The Ohio State University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare no conflicts of interest in regards to this manuscript.
- hPolα
- human DNA polymerase α heterodimer
- hPolε
- human DNA polymerase ε heterotetramer
- p261N
- the N-terminal fragment (residues 1−1189) of p261
- p261C
- the C-terminal domain (residues 1190-2257) of p261
- hPolδ
- human DNA polymerase δ heterotetramer
- cryo-EM
- cryo-electron microscopy
- PCNA
- proliferating cell nuclear antigen
- PKA
- protein kinase A
- Sp-dTTPαS
- Sp diastereoisomer of deoxythymidine α-thiotriphosphate
- Sp-dATPαS
- Sp diastereoisomer of deoxyadenosine α-thiotriphosphate
- TBE
- Tris-boric acid-EDTA
- hPolγ
- human DNA polymerase γ.
References
- 1. Doublié, S., and Zahn, K. E. (2014) Structural insights into eukaryotic DNA replication. Front. Microbiol. 5, 444 10.3389/fmicb.2014.00444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pellegrini, L. (2012) The Polα-primase complex. Subcell. Biochem. 62, 157–169 10.1007/978-94-007-4572-8_9 [DOI] [PubMed] [Google Scholar]
- 3. McElhinny, S. A. N., Gordenin, D. A., Stith, C. M., Burgers, P. M. J., and Kunkel, T. A. (2008) Division of labor at the eukaryotic replication fork. Mol. Cell 30, 137–144 10.1016/j.molcel.2008.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miyabe, I., Kunkel, T. A., and Carr, A. M. (2011) The major roles of DNA polymerases ε and δ at the eukaryotic replication fork are evolutionarily conserved. PLoS Genet. 7, e1002407 10.1371/journal.pgen.1002407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pursell, Z. F., Isoz, I., Lundstrom, E. B., Johansson, E., and Kunkel, T. A. (2007) Yeast DNA polymerase ε participates in leading-strand DNA replication. Science 317, 127–130 10.1126/science.1144067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shinbrot, E., Henninger, E. E., Weinhold, N., Covington, K. R., Göksenin, A. Y., Schultz, N., Chao, H., Doddapaneni, H., Muzny, D. M., Gibbs, R. A., Sander, C., Pursell, Z. F., and Wheeler, D. A. (2014) Exonuclease mutations in DNA polymerase ε reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 24, 1740–1750 10.1101/gr.174789.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xie, B., Mazloum, N., Liu, L., Rahmeh, A., Li, H., and Lee, M. Y. (2002) Reconstitution and characterization of the human DNA polymerase δ four-subunit holoenzyme. Biochemistry 41, 13133–13142 10.1021/bi0262707 [DOI] [PubMed] [Google Scholar]
- 8. Li, Y., Pursell, Z. F., and Linn, S. (2000) Identification and cloning of two histone fold motif-containing subunits of HeLa DNA polymerase ε. J. Biol. Chem. 275, 23247–23252 10.1074/jbc.M002548200 [DOI] [PubMed] [Google Scholar]
- 9. Muzi-Falconi, M., Giannattasio, M., Foiani, M., and Plevani, P. (2003) The DNA polymerase α-primase complex: multiple functions and interactions. ScientificWorld J. 3, 21–33 10.1100/tsw.2003.05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nunez-Ramirez, R., Klinge, S., Sauguet, L., Melero, R., Recuero-Checa, M. A., Kilkenny, M., Perera, R. L., Garcia-Alvarez, B., Hall, R. J., Nogales, E., Pellegrini, L., and Llorca, O. (2011) Flexible tethering of primase and DNA Polα in the eukaryotic primosome. Nucleic Acids Res. 39, 8187–8199 10.1093/nar/gkr534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jain, R., Hammel, M., Johnson, R. E., Prakash, L., Prakash, S., and Aggarwal, A. K. (2009) Structural insights into yeast DNA polymerase δ by small angle X-ray scattering. J. Mol. Biol. 394, 377–382 10.1016/j.jmb.2009.09.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jain, R., Rice, W. J., Malik, R., Johnson, R. E., Prakash, L., Prakash, S., Ubarretxena-Belandia, I., and Aggarwal, A. K. (2019) Cryo-EM structure and dynamics of eukaryotic DNA polymerase δ holoenzyme. Nat. Struct. Mol. Biol. 26, 955–962 10.1038/s41594-019-0305-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Asturias, F. J., Cheung, I. K., Sabouri, N., Chilkova, O., Wepplo, D., and Johansson, E. (2006) Structure of Saccharomyces cerevisiae DNA polymerase ε by cryo-electron microscopy. Nat. Struct. Mol. Biol. 13, 35–43 10.1038/nsmb1040 [DOI] [PubMed] [Google Scholar]
- 14. Lancey, C., Tehseen, M., Raducanu, V. S., Rashid, F., Merino, N., Ragan, T. J., Savva, C. G., Zaher, M. S., Shirbini, A., Blanco, F. J., Hamdan, S. M., and De Biasio, A. (2020) Structure of the processive human Polδ holoenzyme. Nat. Commun. 11, 1109 10.1038/s41467-020-14898-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baranovskiy, A. G., Babayeva, N. D., Zhang, Y., Gu, J., Suwa, Y., Pavlov, Y. I., and Tahirov, T. H. (2016) Mechanism of concerted RNA-DNA primer synthesis by the human primosome. J. Biol. Chem. 291, 10006–10020 10.1074/jbc.M116.717405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang, Y., Baranovskiy, A. G., Tahirov, T. H., and Pavlov, Y. I. (2014) The C-terminal domain of the DNA polymerase catalytic subunit regulates the primase and polymerase activities of the human DNA polymerase α-primase complex. J. Biol. Chem. 289, 22021–22034 10.1074/jbc.M114.570333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meng, X., Zhou, Y., Lee, E. Y., Lee, M. Y., and Frick, D. N. (2010) The p12 subunit of human polymerase delta modulates the rate and fidelity of DNA synthesis. Biochemistry 49, 3545–3554 10.1021/bi100042b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ganai, R. A., Osterman, P., and Johansson, E. (2015) Yeast DNA polymerase ε catalytic core and holoenzyme have comparable catalytic rates. J. Biol. Chem. 290, 3825–3835 10.1074/jbc.M114.615278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zahurancik, W. J., Klein, S. J., and Suo, Z. (2013) Kinetic mechanism of DNA polymerization catalyzed by human DNA polymerase ε. Biochemistry 52, 7041–7049 10.1021/bi400803v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bermudez, V. P., Farina, A., Raghavan, V., Tappin, I., and Hurwitz, J. (2011) Studies on human DNA polymerase ε and GINS complex and their role in DNA replication. J. Biol. Chem. 286, 28963–28977 10.1074/jbc.M111.256289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zahurancik, W. J., Klein, S. J., and Suo, Z. (2014) Significant contribution of the 3'–>5' exonuclease activity to the high fidelity of nucleotide incorporation catalyzed by human DNA polymerase. Nucleic Acids Res. 42, 13853–13860 10.1093/nar/gku1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goksenin, A. Y., Zahurancik, W., Lecompte, K. G., Taggart, D. J., Suo, Z., and Pursell, Z. F. (2012) Human DNA polymerase ε is able to efficiently extend from multiple consecutive ribonucleotides. J. Biol. Chem. 287, 42675–42684 10.1074/jbc.M112.422733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zahurancik, W. J., Baranovskiy, A. G., Tahirov, T. H., and Suo, Z. (2015) Comparison of the kinetic parameters of the truncated catalytic subunit and holoenzyme of human DNA polymerase ε. DNA Repair 29, 16–22 10.1016/j.dnarep.2015.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fiala, K. A., Sherrer, S. M., Brown, J. A., and Suo, Z. (2008) Mechanistic consequences of temperature on DNA polymerization catalyzed by a Y-family DNA polymerase. Nucleic Acids Res. 36, 1990–2001 10.1093/nar/gkn004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fiala, K. A., and Suo, Z. (2004) Mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry 43, 2116–2125 10.1021/bi035746z [DOI] [PubMed] [Google Scholar]
- 26. Brown, J. A., and Suo, Z. (2009) Elucidating the kinetic mechanism of DNA polymerization catalyzed by Sulfolobus solfataricus P2 DNA polymerase B1. Biochemistry 48, 7502–7511 10.1021/bi9005336 [DOI] [PubMed] [Google Scholar]
- 27. Patel, S. S., Wong, I., and Johnson, K. A. (1991) Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 30, 511–525 10.1021/bi00216a029 [DOI] [PubMed] [Google Scholar]
- 28. Donlin, M. J., Patel, S. S., and Johnson, K. A. (1991) Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry 30, 538–546 10.1021/bi00216a031 [DOI] [PubMed] [Google Scholar]
- 29. Sherrer, S. M., Sanman, L. E., Xia, C. X., Bolin, E. R., Malik, C. K., Efthimiopoulos, G., Basu, A. K., and Suo, Z. (2012) Kinetic analysis of the bypass of a bulky DNA lesion catalyzed by human Y-family DNA polymerases. Chem. Res. Toxicol. 25, 730–740 10.1021/tx200531y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tokarsky, E. J., Gadkari, V. V., Zahurancik, W. J., Malik, C. K., Basu, A. K., and Suo, Z. (2016) Pre-steady-state kinetic investigation of bypass of a bulky guanine lesion by human Y-family DNA polymerases. DNA Repair 46, 20–28 10.1016/j.dnarep.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vyas, R., Efthimiopoulos, G., Tokarsky, E. J., Malik, C. K., Basu, A. K., and Suo, Z. (2015) Mechanistic basis for the bypass of a bulky DNA adduct catalyzed by a Y-family DNA polymerase. J. Am. Chem. Soc. 137, 12131–12142 10.1021/jacs.5b08027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gadkari, V. V., Tokarsky, E. J., Malik, C. K., Basu, A. K., and Suo, Z. (2014) Mechanistic investigation of the bypass of a bulky aromatic DNA adduct catalyzed by a Y-family DNA polymerase. DNA Repair 21, 65–77 10.1016/j.dnarep.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brown, J. A., Newmister, S. A., Fiala, K. A., and Suo, Z. (2008) Mechanism of double-base lesion bypass catalyzed by a Y-family DNA polymerase. Nucleic Acids Res. 36, 3867–3878 10.1093/nar/gkn309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fiala, K. A., Hypes, C. D., and Suo, Z. (2007) Mechanism of abasic lesion bypass catalyzed by a Y-family DNA polymerase. J. Biol. Chem. 282, 8188–8198 10.1074/jbc.M610718200 [DOI] [PubMed] [Google Scholar]
- 35. Herschlag, D., Piccirilli, J. A., and Cech, T. R. (1991) Ribozyme-catalyzed and nonenzymatic reactions of phosphate diesters: rate effects upon substitution of sulfur for a nonbridging phosphoryl oxygen atom. Biochemistry 30, 4844–4854 10.1021/bi00234a003 [DOI] [PubMed] [Google Scholar]
- 36. Johnson, A. A., and Johnson, K. A. (2001) Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 276, 38097–38107 10.1074/jbc.M106743200 [DOI] [PubMed] [Google Scholar]
- 37. Johnson, K. A. (2010) The kinetic and chemical mechanism of high-fidelity DNA polymerases. Biochim. Biophys. Acta 1804, 1041–1048 10.1016/j.bbapap.2010.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raper, A. T., Reed, A. J., and Suo, Z. (2018) Kinetic mechanism of DNA polymerases: contributions of conformational dynamics and a third divalent metal ion. Chem. Rev. 118, 6000–6025 10.1021/acs.chemrev.7b00685 [DOI] [PubMed] [Google Scholar]
- 39. Ganai, R. A., and Johansson, E. (2016) DNA replication: a matter of fidelity. Mol. Cell 62, 745–755 10.1016/j.molcel.2016.05.003 [DOI] [PubMed] [Google Scholar]
- 40. Nuutinen, T., Tossavainen, H., Fredriksson, K., Pirilä, P., Permi, P., Pospiech, H., and Syvaoja, J. E. (2008) The solution structure of the amino-terminal domain of human DNA polymerase ε subunit B is homologous to C-domains of AAA+ proteins. Nucleic Acids Res. 36, 5102–5110 10.1093/nar/gkn497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Park, V. S., and Pursell, Z. F. (2019) POLE proofreading defects: contributions to mutagenesis and cancer. DNA Repair (Amst.) 76, 50–59 10.1016/j.dnarep.2019.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Haradhvala, N. J., Kim, J., Maruvka, Y. E., Polak, P., Rosebrock, D., Livitz, D., Hess, J. M., Leshchiner, I., Kamburov, A., Mouw, K. W., Lawrence, M. S., and Getz, G. (2018) Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun. 9, 1746 10.1038/s41467-018-04002-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Aksenova, A., Volkov, K., Maceluch, J., Pursell, Z. F., Rogozin, I. B., Kunkel, T. A., Pavlov, Y. I., and Johansson, E. (2010) Mismatch repair-independent increase in spontaneous mutagenesis in yeast lacking non-essential subunits of DNA polymeraseε. PLoS Genet. 6, e1001209 10.1371/journal.pgen.1001209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsubota, T., Maki, S., Kubota, H., Sugino, A., and Maki, H. (2003) Double-stranded DNA binding properties of Saccharomyces cerevisiae DNA polymerase ε and of the Dpb3p-Dpb4p subassembly. Genes Cells 8, 873–888 10.1046/j.1365-2443.2003.00683.x [DOI] [PubMed] [Google Scholar]
- 45. Meng, X., Zhou, Y., Zhang, S., Lee, E. Y., Frick, D. N., and Lee, M. Y. (2009) DNA damage alters DNA polymerase delta to a form that exhibits increased discrimination against modified template bases and mismatched primers. Nucleic Acids Res. 37, 647–657 10.1093/nar/gkn1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang, S., Zhou, Y., Trusa, S., Meng, X., Lee, E. Y., and Lee, M. Y. (2007) A novel DNA damage response: rapid degradation of the p12 subunit of DNA polymerase δ. J. Biol. Chem. 282, 15330–15340 10.1074/jbc.M610356200 [DOI] [PubMed] [Google Scholar]
- 47. Johnson, A. A., Tsai, Y., Graves, S. W., and Johnson, K. A. (2000) Human mitochondrial DNA polymerase holoenzyme: reconstitution and characterization. Biochemistry 39, 1702–1708 10.1021/bi992104w [DOI] [PubMed] [Google Scholar]
- 48. Fiala, K. A., and Suo, Z. (2004) Pre-steady-state kinetic studies of the fidelity of Sulfolobus solfataricus P2 DNA polymerase IV. Biochemistry 43, 2106–2115 10.1021/bi0357457 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are in the manuscript and supporting information, or are available from the authors Walter J. Zahurancik (zahurancik.2@osu.edu) and Zucai Suo (zucai.suo@med.fsu.edu) upon request.