

Abstract
Leukocidin ED (LukED) is a pore-forming toxin produced by Staphylococcus aureus, which lyses host cells and promotes virulence of the bacteria. LukED enables S. aureus to acquire iron by lysing erythrocytes, which depends on targeting the host receptor Duffy antigen receptor for chemokines (DARC). The toxin also targets DARC on the endothelium, contributing to the lethality observed during bloodstream infection in mice. LukED is comprised of two monomers: LukE and LukD. LukE binds to DARC and facilitates hemolysis, but the closely related Panton–Valentine leukocidin S (LukS-PV) does not bind to DARC and is not hemolytic. The interaction of LukE with DARC and the role this plays in hemolysis are incompletely characterized. To determine the domain(s) of LukE that are critical for DARC binding, we studied the hemolytic function of LukE–LukS-PV chimeras, in which areas of sequence divergence (divergence regions, or DRs) were swapped between the toxins. We found that two regions of LukE's rim domain contribute to hemolysis, namely residues 57–75 (DR1) and residues 182–196 (DR4). Interestingly, LukE DR1 is sufficient to render LukS-PV capable of DARC binding and hemolysis. Further, LukE, by binding DARC through DR1, promotes the recruitment of LukD to erythrocytes, likely by facilitating LukED oligomer formation. Finally, we show that LukE targets murine Darc through DR1 in vivo to cause host lethality. These findings expand our biochemical understanding of the LukE–DARC interaction and the role that this toxin-receptor pair plays in S. aureus pathophysiology.
Keywords: ACKR1, bacterial toxin, cell surface receptor, DARC, Duffy, erythrocyte, hemoglobin, hemolysis, iron, leukocidin ED, nutritional immunity, Staphylococcus aureus (S. aureus), bacterial pathogenesis, receptor
Staphylococcus aureus is a Gram-positive pathobiont responsible for significant human disease (1). S. aureus virulence is supported through a wide array of virulence factors (2), one class of which is the bicomponent leukocidins. The leukocidins are a class of secreted pore-forming toxins each made up of two soluble monomers that together can form pores in host-cell membranes and cause a range of host-cell responses including cell death (3).
The leukocidin family produced by human-associated S. aureus isolates has five members: LukSF-PV (or PVL), LukED, HlgAB, HlgCB, and LukAB (or LukHG) (4, 5). Each toxin is made of one S subunit, which recognizes target host cells by proteinaceous receptors, and one F subunit, which binds to the S subunit. The components then oligomerize with each other to form a prepore octamer of alternating subunits, which finally matures into a pore spanning the cell membrane (4). The S subunits are LukS-PV, LukE, HlgA, HlgC, and LukA, and the F subunits are LukF-PV, LukD, HlgB, and LukB (Fig. 1A). With the exception of LukA, the S subunits all bind to seven-transmembrane domain G protein–coupled receptors (GPCRs). For example, LukS-PV binds the C5a receptors C5aR and C5L2 (6), whereas LukE binds CCR5, CXCR1 and 2, and DARC (7–9) (Fig. 1A).
Figure 1.
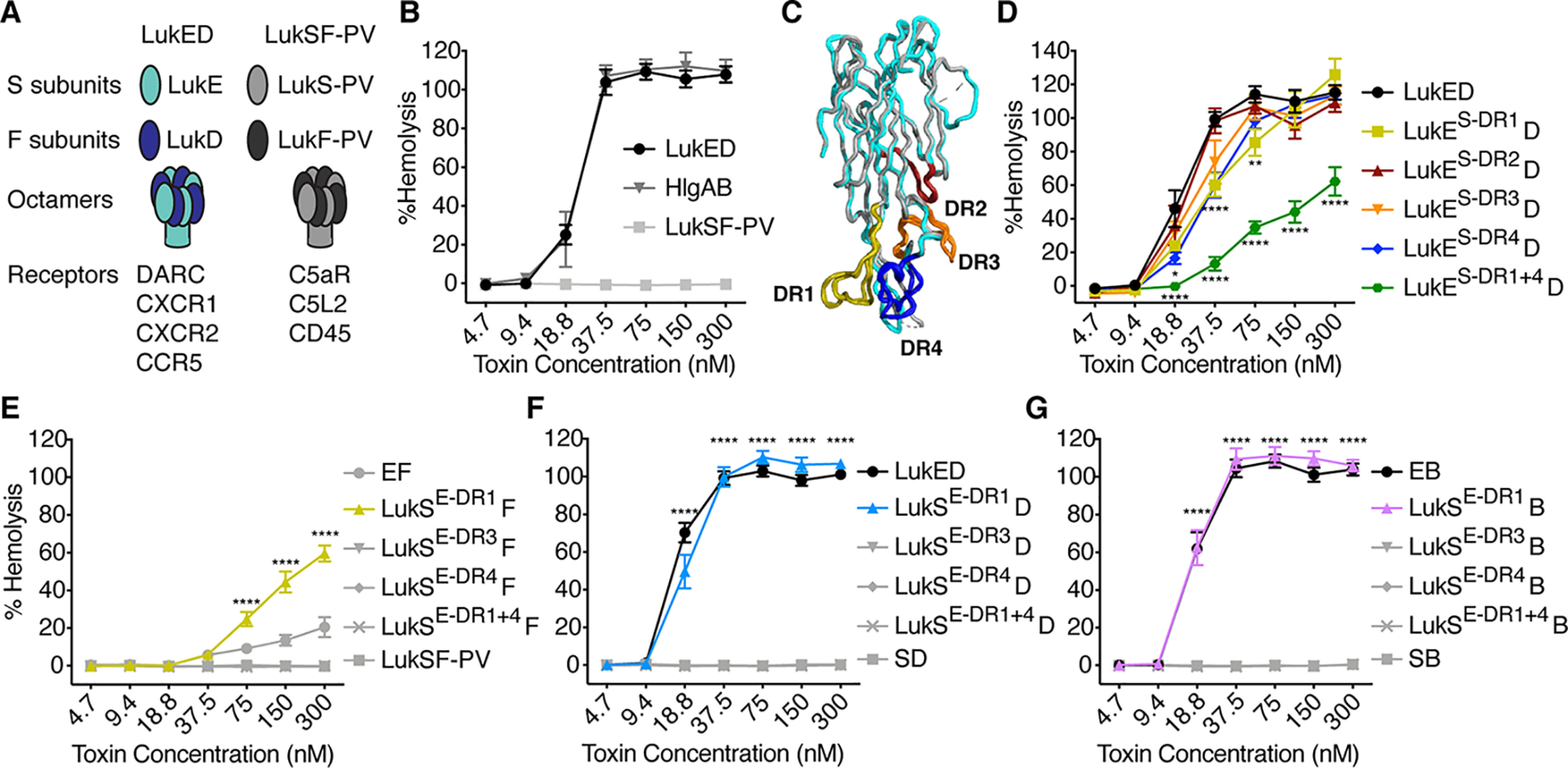
LukE DR1 and DR4 both contribute to hemolysis, although DR1 is sufficient to confer erythrocyte specificity. A, cartoon of LukED and LukSF-PV subunits and receptors. B, hemolysis of primary human erythrocytes treated with WT S. aureus bi-component leukocidins (n = 6 donors). C, structural alignment of LukE (Protein Data Bank code 3ROH, shown in light blue) and LukS-PV (Protein Data Bank code 1T5R, shown in gray) as described (8). Regions of low amino acid identity between the two toxins (referred to as divergence regions or DRs) are highlighted as follows: DR1 in yellow (residues 57–75), DR2 in red (residues 140–150), DR3 in orange (residues 164–178), and DR4 in blue (residues 182–196). See Fig. S1 for amino acid sequences and Fig. S2 (A and B) for cartoon and gels of S subunit chimeras. D–G, hemolysis of primary human erythrocytes treated with LukE and LukS DR chimeras and indicated F subunits (n = 5–17 donors). LukES-DR1 indicates LukE with the DR1 from LukS-PV, LukSE-DR1 indicates LukS-PV with the DR1 from LukE, etc. D indicates LukD, F indicates LukF-PV, and B indicates HlgB. Noncanonical toxin pairs are denoted as EB for LukE + HlgB, etc. The data in B and D–F are pooled from 3–6 independent experiments and represent means ± S.E. ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; *, p < 0.05. D–G, two-way ANOVA with Sidak's correction, compared with WT LukE (D), or WT LukSF-PV (E–G). D, upper asterisks at 18.8 and 37.5 nm reference LukES-DR1 and LukES-DR4, but the upper asterisks at 75 nm only reference LukES-DR1.
Each subunit has three major domains: the cap, or β-sandwich domain, which allows association of the two subunits; the rim domain, which allows recognition of host-cell receptors; and the stem domain, which, upon octamer formation, inserts into the host-cell membrane to form a β-barrel pore together with the stem domains of seven other subunits (10, 11). Although the four S subunits that target GPCRs share 65–80% amino acid identity (3), their rim domains are the most divergent, enabling tropism for different receptors. Previous studies have investigated the S subunit rim domain sequences necessary for receptor targeting. For example, LukS-PV targets C5aR using several residues including Arg73, Tyr184, and Thr244 (12). LukE targets CXCR1 and CXCR2 using residues 182–196 (8) and CCR5 using residues 57–75 (13). Interestingly, multiple regions of the HlgA rim and cap domains are involved in targeting erythrocytes through DARC (14–16).
Host organisms employ nutritional immunity to prevent the acquisition of iron and other metals by pathogens, and thus successful pathogens must devise strategies to acquire these essential nutrients (17, 18). S. aureus accomplishes this by secreting LukED and HlgAB to lyse erythrocytes and access heme iron (9, 19). LukE and HlgA both recognize erythrocytes by binding the DARC receptor (Duffy antigen receptor for chemokines or atypical chemokine receptor 1 (ACKR1)) (9). Interestingly, the F subunits LukD and HlgB bind to erythrocytes even without LukE or HlgA and thus are involved in defining erythrocyte tropism in a different way than other cell types (20–22).
DARC is a member of the atypical chemokine receptor subclass. Like other members of this subclass, DARC binds promiscuously to both CC and CXC chemokines but lacks a G protein–signaling motif (23). DARC is expressed on erythrocytes, where it is the receptor for several plasmodium parasites (24, 25), as well as endothelial cells (26). DARC is thought to regulate chemokine concentrations and their functions in the circulation (27, 28).
Here we investigated the sequence determinants of LukE for targeting DARC on erythrocytes. We report that although multiple regions of the LukE rim domain are involved in DARC targeting, residues 57–75 (designated DR1) are sufficient to confer DARC specificity and hemolytic activity. We explored the interplay between LukE and LukD in erythrocyte targeting and found that LukE DR1, through DARC targeting, enhances LukD binding. Finally, we demonstrate the in vivo relevance of the LukE DR1–DARC interaction using a murine toxin challenge model.
Results
Determination of the critical regions of LukE for hemolysis
LukED and HlgAB are hemolytic, but LukSF-PV is not (Fig. 1B) (9). The LukE and LukS-PV proteins share 70% amino acid identity. To determine regions of LukE critical for hemolysis, we made chimeric toxins where we swapped regions of sequence divergence (divergence regions, or DRs) between LukE and LukS-PV (Fig. 1C and Figs. S1A and S2A), because this approach has been successful for studying the interaction of LukE with other receptors (8, 13). We produced loss-of-function chimeras, in which we took LukE and swapped out a DR for the analogous region of LukS (LukES-DR#), and gain-of-function chimeras, in which we took LukS and swapped out a DR for the analogous region of LukE (LukSE-DR#) (Fig. S2, A and B).
The loss-of-function chimeras were all hemolytic when combined with LukD. LukES-DR1 and LukES-DR4 exhibited slightly lower hemolytic activity than WT LukE, and LukES-DR2 and LukES-DR3 chimeras showed almost WT levels of hemolytic activity (Fig. 1D). We next made a combined chimera where both the DR1 and the DR4 of LukS were swapped into LukE (LukES-DR1+4). This chimera exhibited a greater loss of function but still had some hemolytic activity (Fig. 1D). Thus, several regions of the LukE rim domain, including DR1 and DR4, contribute to erythrocyte targeting.
We next assessed the hemolytic activity of the gain-of-function chimeras when combined with LukF-PV. We found that LukSE-DR1 exhibited partial hemolytic activity, whereas the other chimeras had none (Fig. 1E). We also made LukSE-DR1+4, but surprisingly this chimera had no hemolytic activity (Fig. 1E).
Because LukD and HlgB are critical for targeting erythrocytes (16, 20, 22), and S and F subunits can form active pores in noncanonical pairs (29, 30), we next combined the gain-of-function chimeras with LukD and HlgB. Remarkably, we found that LukSE-DR1 exhibited full hemolytic activity when combined with LukD or HlgB, indistinguishable from WT LukED (Fig. 1, F and G). Thus, the DR1 of LukE is sufficient to render LukS-PV fully hemolytic when combined with LukD or HlgB.
Because LukED can target multiple cell types through multiple receptors, we wanted to assess the overall functionality of our chimeras by testing their ability to kill different cells. We previously found that the loss-of-function chimeras studied here are all cytotoxic to human polymorphonuclear leukocytes (hPMNs) except for LukES-DR4 but that this chimera is still cytotoxic to CCR5+ cells (8). These data confirm that the loss-of-function chimeras are all active toxins. Thus, we tested the gain-of-function chimeras on hPMNs. We found that LukSE-DR1, LukSE-DR3, and LukSE-DR4 were all cytotoxic to hPMNs when combined with LukF-PV, at levels similar to WT LukED and LukSF-PV and noncanonical toxin pairs (Fig. 2, A and B). Further, LukSE-DR1, the gain-of-function chimera with hemolytic activity (Fig. 1, E–G) was cytotoxic to hPMNs in combination with LukF, LukD, or HlgB (Fig. 2C). These data increased our confidence in the specificity of the LukE DR1 for targeting erythrocytes and CCR5+ cells (13), as opposed to being necessary for global toxin function. However, LukSE-DR1+4, which lacked hemolytic activity (Fig. 1, E–G), also lacked cytotoxic activity toward hPMNs (Fig. 2D). These data show that the LukSE-DR1+4 chimera is more globally impaired; thus, we cannot draw conclusions about the additive importance of the DR1 and DR4 regions for hemolysis from this chimera.
Figure 2.

LukSE-DR chimeras are active cytolysins with the exception of LukSE-DR1+4. A–D, viability of primary human PMNs treated with LukE and LukS DR chimeras and indicated F subunits (n = 3–11 donors). LukSE-DR1 indicates LukS-PV with the DR1 from LukE, etc. D indicates LukD, F indicates LukF-PV, and B indicates HlgB. Noncanonical toxin pairs are denoted as EB for LukE + HlgB, etc. The data are pooled from at least two independent experiments and represent means ± S.E. ****, p < 0.0001. D, two-way ANOVA with Sidak's correction, compared with WT LukSF-PV.
Role of loops 1 and 4 within the rim domain in LukE-mediated hemolysis
While conducting the experiments described herein, Peng et al. (16) published a report also studying the sequence determinants of LukE involved in hemolysis. They used a similar approach, making LukE–LukS-PV chimeras but swapped smaller loops at the tip of the rim domain. This work found, as we did, that multiple regions of the LukE rim domain contribute to erythrocyte targeting. However, Peng et al. described loop 4 as the critical determinant of erythrocyte binding and hemolysis, finding both that the LukES-loop4 chimera showed the most pronounced loss of hemolytic function and that the LukSE-loop4 chimera showed some gain of hemolytic function at the high concentration of 1430 nm. Of note, loop 4 is not analogous to our DR4. Rather, it is comprised of residues 245–249, part of our previously defined DR5 (8), which exhibited a global cytotoxicity defect (Fig. S1A). In contrast, the loop 1 is comprised of residues 65–70, part of our DR1 (Fig. S1A). Loop 1 of LukE contributed to hemolysis but more minimally than the larger DR1 (16). Of note, Peng et al. (16) used a different nomenclature for the leukocidins than we do here, in which HlgB is referred to as LukF and HlgA is referred to as Hlg2 (see Ref. 4 for clarification).
Because our findings were different from those of Peng et al. (16), we next generated loop 1 and loop 4 loss- and gain-of-function chimeras and assayed the hemolytic activity of the resulting toxins (Fig. S2, A and C). We were able to recapitulate the finding that LukES-loop4 exhibited no hemolytic activity and that LukES-loop1 has partially decreased hemolytic activity when combined with LukD (Fig. 3A). However, in our hands LukSE-loop4 did not show gain of hemolytic function with any F subunit (Fig. 3D). We repeated these experiments using the same buffer conditions that Peng et al. (16) used, namely PBS instead of saline and a higher concentration of toxin for the gain-of-function chimera. We again produced similar loss of function data but observed no gain of function activity (Fig. 3, G and H).
Figure 3.

Amino acids 249–253 of LukE are not sufficient for erythrocyte targeting. A and D, hemolysis of primary human erythrocytes treated with LukE and LukS loop chimeras and indicated F subunits (n = 6 donors for erythrocytes). B and E, viability of CCR5 expressing SupT1 cells (SupT1-CCR5) treated with LukE and LukS loop chimeras and indicated F subunits (n = 3–4 experiments). C and F, viability of primary human PMNs treated with LukE and LukS loop chimeras and indicated F subunits (n = 6 donors). G and H, hemolysis of primary human erythrocytes treated with LukE and LukS loop chimeras, in PBS as in Ref. 16 (n = 6 donors). In H, a concentration of 1430 nm per subunit was used, as in Ref. 16. The legend in B applies to A and B, the legend in E applies to D and E. LukES-loop1 indicates LukE with residues 65–70 of LukS-PV, LukES-loop4 indicates LukE with residues 249–253 of LukS-PV, LukSE-1oop1 indicates LukS-PV with residues 65–70 of LukE, and LukSE-loop4 indicates LukS-PV with residues 237–271 of LukE. D indicates LukD, F indicates LukF-PV, and B indicates HlgB. Noncanonical toxin pairs are denoted as EB for LukE + HlgB, etc. See Fig. S1 for amino acid sequences and Fig. S2 (A and C) for cartoon and gel of S subunit loop chimeras. The data are pooled from at least two independent experiments and represent means ± S.E., mean ± S.D. for H. ****, p < 0.0001; ns, not significant. A–G, two-way ANOVA with Sidak's correction, compared with WT LukE (A–C and G) or WT LukSF-PV (D–F). H, one-way ANOVA with Tukey's correction, compared with WT LukSF-PV.
To further characterize the loop 1 and loop 4 chimeras, we assayed their cytotoxicity against SupT1-CCR5 cells, a human T cell line that expresses CCR5 (12), and hPMNs, which express CXCR1 and 2. We found that LukES-loop1 had decreased cytotoxic activity toward SupT1-CCR5 cells, consistent with our published data (13), and was cytotoxic to hPMNs at the same level as WT LukED (Fig. 3, B and C). However, LukES-loop4 showed no cytotoxic activity toward SupT1-CCR5 or hPMNs (Fig. 3, B and C). These data reveal that LukES-loop4 has a global cytotoxicity defect rather than a specific defect in targeting erythrocytes. Further, LukSE-loop4 showed little to no cytotoxic activity toward either cell type with any F subunit (Fig. 3, E and F) and thus also has a global cytotoxicity defect. Taken together, our data demonstrate that although several regions of the LukE rim domain can participate in erythrocyte targeting, DR1, rather than loop 4, is sufficient to confer specificity toward erythrocytes.
LukE DR1 interacts with DARC to enhance LukD binding to erythrocytes
We have previously shown that DARC is required for LukED-mediated hemolysis and that LukE targets erythrocytes through binding DARC (9). We further supported these findings by showing that purified recombinant DARC can inhibit LukED-mediated hemolysis (Fig. 4A). We next studied the interaction of LukE, LukS-PV, and DARC by surface plasmon resonance (SPR). We found that WT LukE bound to DARC with a dissociation equilibrium constant (KD) of 58.0 (±3.46) nm, whereas WT LukS-PV did not associate with DARC up to a concentration of 50 μg/ml (1.5 mm) (Fig. 4B). The DR1 of LukE greatly enhances the affinity of LukS-PV for recombinant DARC (KD of 3.13 (±0.48) μm). The DR1 of LukS-PV expressed in LukE decreased the affinity of LukE by 11.5-fold for DARC (KD = 666.7 (±93.9) nm) (Fig. 4B). These data show that LukE DR1 promotes direct binding to DARC.
Figure 4.
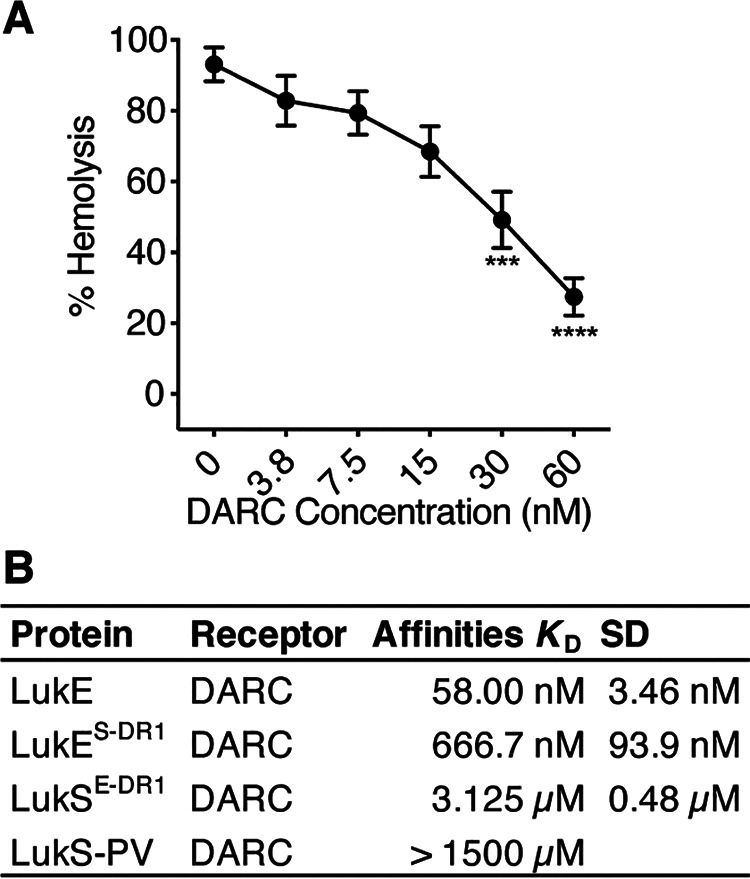
DR1 of LukE binds to DARC. A, hemolysis of primary human erythrocytes treated with 15 nm LukED and increasing concentrations of purified recombinant DARC (n = 4 donors). B, SPR data of the indicated toxins and mutants binding to DARC. LukES-DR1 indicates LukE with the DR1 from LukS-PV, LukSE-DR1 indicates LukS-PV with the DR1 from LukE, etc. The data in A are pooled from two independent experiments and represent means ± S.E. ****, p < 0.0001; ***, p < 0.001. A, one-way ANOVA with Tukey's correction, compared with 0 nm DARC.
LukD and HlgB can bind to erythrocytes independently of LukE and HlgA and can recruit the S subunits by oligomerizing with them (16, 20, 22). In the context of LukED-mediated hemolysis, the observed LukD binding has resulted in a model that on erythrocytes, LukD binds to the plasma membrane first, followed by binding of LukE to LukD to form oligomers (22). However, the interaction between LukE and DARC is necessary for hemolysis (9). It is thus unclear what role DARC plays in facilitating hemolysis. To explore the interplay between DARC, LukE, and LukD in erythrocyte binding, we used flow cytometry to measure toxin binding to erythrocytes on a single-cell level. We fluorescently labeled LukD and LukDΔ130-4 (LukDmut) (Fig. 5, A and B), a stem domain mutant that oligomerizes with LukE but is unable to form pores (31). This mutant allows the measurement of LukD binding in the presence of LukE without lysing the erythrocytes. We found that binding of LukD to erythrocytes is significantly enhanced by the presence of LukE in a DARC-dependent manner (Fig. 5C). In the presence of DARC, the binding of LukD was enhanced, on average, by 9.7-fold by the addition of LukE (Fig. 5D). Finally, we found that the DR1 of LukE, within a LukS backbone, is sufficient to enhance LukD binding (Fig. 5E). Taken together, these data show that not only does LukD recruit LukE to erythrocyte membranes (22), but that LukE, through its interaction with DARC via DR1, further recruits LukD onto the plasma membrane of erythrocytes.
Figure 5.

LukE promotes LukD binding to erythrocytes through DR1. A, left panel, Coomassie Blue–stained gel of DyLight 488–labeled toxins. Right panel, fluorescent image of gel. B, flow cytometry gating strategy of human erythrocytes used in C–E. C and D, binding of LukDmut-488 in the presence or absence of LukE with or without DARC. C, MFI. D, ratio of MFI relative to binding of LukDmut-488 alone after subtracting the background fluorescence of unstained cells (n = 5 donors). E, binding of LukDmut-488 in the presence of the indicated S subunits (n = 5 donors). LukDmut indicates LukDΔ130-4 (pore-formation incompetent), 488 indicates DyLight 488, and LukSE-DR1 indicates LukS-PV with the DR1 from LukE. The data are pooled from five (C and D) or two (E) independent experiments, and means ± S.E. are shown. ****, p < 0.0001; ***, p < 0.001; ns, not significant. C, two-way ANOVA with Sidak's correction. D, Student's t test. E, one-way ANOVA with Tukey's correction.
LukE DR1–DARC interaction in vivo
In addition to facilitating hemolysis, LukED targeting of Darc (the murine homolog of DARC) also contributes to lethality during S. aureus bloodstream infections in mice. In fact, when LukED is injected intravenously, the toxin targets murine Darc on endothelial cells to cause rapid lethality through derangements in the distribution of vascular fluid (32). To further investigate the role of the LukE DR1 in targeting Darc in vivo, we employed this toxin challenge model using Swiss Webster mice. We found that the DR1 of LukE is sufficient to render LukS-PV lethal in this model when injected together with either LukD or HlgB (LukSE-DR1 D and LukSE-DR1 B; Fig. 6A). We next investigated the role of Darc using C57BL/6J mice. Although the WT mice were as susceptible as the Swiss Webster mice, the Darc−/− mice were fully resistant (Fig. 6, B and C). Thus, the LukE DR1–Darc interaction is also important for LukED targeting of endothelial cells in vivo.
Figure 6.

LukE DR1 is sufficient for Darc targeting in vivo. A, survival curve of Swiss Webster mice (females, 4–6 weeks old) challenged with IV purified toxin (n = 3–5 mice/group). B and C, survival curve of B6 WT (B) and Darc−/− (C) mice (mixed males and females, 7–10 weeks old) injected with purified toxin via retro-orbital injection (n = 4–6 mice/group). LukSE-DR1 indicates LukS-PV with the DR1 from LukE. SD indicates LukS-PV + LukD. The data are pooled from 2–3 independent experiments. **, p < 0.01; *, p < 0.05. A and B, Mantel–Cox test.
Discussion
S. aureus deploys a collection of bicomponent pore-forming leukocidins to target host cells and promote its survival within the mammalian host. These toxins, although very similar at the amino acid and structural level, exhibit tremendous specificity toward their cellular surface receptors. This specificity dictates the breadth of cells targeted by the toxins. The molecular means by which leukocidins target their different cellular receptors is incompletely understood. Here we studied the interaction between LukED and DARC on erythrocytes. We found that, rather than one region of the LukE rim domain being necessary for hemolysis, DR1 and DR4 both contribute to erythrocyte targeting. We have previously shown that LukE DR1 targets CCR5 (13) and LukE DR4 targets CXCR1/2 (8). DARC is a promiscuous chemokine receptor, capable of binding many chemokines including CCL5, which binds CCR5, and several ligands of CXCR1 and 2, including CXCL8 (33). Thus, perhaps it is not surprising that LukE binds DARC using both chemokine receptor targeting regions. However, we found that the LukE DR1 is sufficient for binding and hemolysis in the context of a LukS-PV backbone and a hemolytic F subunit (i.e. LukD or HlgB).
Our conclusions conflict with those previously published by Peng et al. (16), which found that loop 4 of LukE is the most critical region for hemolysis. We found that LukES-loop4 indeed lacks hemolytic activity, but this mutant also lacks cytotoxic activity toward CXCR1/2-expressing hPMNs and CCR5-expressing cells. Thus, we conclude that this chimeric mutant is globally defective rather than being specifically deficient in targeting of DARC and erythrocytes. In terms of LukSE-loop4, however, we observed no gain of hemolytic function, in contrast to Peng et al. (16), even when using the same buffer and concentration (Fig. 3H). The most notable remaining difference in our methods is that we used toxins produced by S. aureus, whereas Peng et al. (16) used toxins produced by Escherichia coli, which may explain the discrepancy in our data. We propose two possible explanations as to why mutating loop 4 of LukE abrogates cytotoxic activity globally. One likely possibility is that this mutation leads to changes in the structure of the rim domain loops, making them dysfunctional. Peng et al. (16) interrogated the secondary structure of their chimeras by CD analysis to exclude chimeras that greatly altered the structure of the proteins. However, the rim domain loops of the leukocidins are unstructured in X-ray crystallographic studies, and thus changes in their structure are unlikely to be evident from this analysis. Indeed, a single-residue mutation in loop 4 of LukS-PV has been shown to alter the structure of both loop 4 and DR4 (loop 3), leading to reduced loop flexibility and impaired receptor binding (12). Thus, this region of the protein is highly sensitive to manipulation.
Directly upstream of loop 4 in LukS-PV is the “Z region,” a five-residue stretch similar to HlgC, which includes a threonine and basic residues. This region has been shown to be crucial for receptor binding and cytotoxicity of LukS-PV (12) and HlgC (34, 35). The Z region is absent from LukE and HlgA (Fig. S1B). However, we observed that the residues in loop 4 of LukE and HlgA also contain a threonine in the context of basic residues, somewhat analogous to the Z region of LukS-PV and HlgC. We realigned the S-subunit sequences in this region to highlight this similarity (Fig. S1C). This alignment suggests an alternative explanation as to why LukES-loop4 lacks cytotoxic activity globally. The threonine in the context of basic residues may be important for receptor binding on a more global level, perhaps for binding to a conserved region within GPCRs. If so, we would expect LukES-loop4 to be unable to bind any GPCRs, because it is lacking these crucial residues. This region merits further study, because it may represent a key to inhibiting all of the leukocidins that target GPCRs.
We also addressed herein the role of DARC in facilitating hemolysis. In Fig. 5, we showed that LukE DR1, through its interaction with DARC, recruits additional LukD molecules to the surface of erythrocytes. It is likely that the recruitment we observe represents LukE and LukD oligomerization events, suggesting that DARC enables this. A potential mechanism for this may relate to the homodimerization of DARC (36). Two DARC molecules, each bound to a LukE–LukD dimer, could boost oligomerization of these subunits because of their close proximity. Because the prepore structure of LukED consists of four subunits of LukE and four subunits of LukD, we might expect that LukE and DARC would increase the binding of LukD by a factor of 4. However, we found that the enhancement of LukD binding in the presence of LukE was over 9-fold (Fig. 5D). Thus, we propose the model shown in Fig. 7. In this model, 1) LukD binds to erythrocytes, and LukE binds to DARC dimers. 2) LukD and LukE dimerize, 3) DARC facilitates the recruitment and further oligomerization of LukD and LukE to form a prepore octamer, and 4) the prepore disassociates from DARC and matures into a pore, and DARC is free to bind another LukE and begin the process again. Thus, one dimer of DARC may be capable of facilitating the formation of multiple octamers of LukE and LukD, explaining why the addition of LukE enhances LukD binding by more than 4-fold. An alternative hypothesis is that higher avidity of LukE for DARC compared with LukD for its binding partner results in more octamers associating with the erythrocyte in the presence of LukE than LukD monomers associate in the absence of LukE.
Figure 7.

Model of DARC recruiting LukED into oligomers on the erythrocyte membrane.
A related question is, what is the binding partner of LukD on erythrocytes? F subunits bind to lipids including phosphatidylcholine (37–39), yet this cannot explain the differential binding patterns of the F subunits to erythrocytes. Recently, it was established that LukF-PV binds to CD45 and that this interaction is critical for LukSF-PV toxicity (40), making it likely that other F subunits also have proteinaceous receptors. To put these observations together, there may be a receptor for LukD that allows initial association of the toxin to the cell, and DARC may act as a co-receptor, which by binding LukE facilitates oligomerization of the toxin subunits into prepore octamers (Fig. 7).
Finally, we established the role of LukE DR1 in vivo using a toxin challenge model of endothelial targeting. These findings raise the question of whether the other biochemical observations regarding erythrocyte targeting described here and elsewhere also apply to endothelial targeting. Moreover, the identification of the LukE DR1 as critical for toxin activity pinpoints a domain that could be used to elicit protective immune responses in the form of neutralizing antibodies (41). These are interesting questions for further research.
S. aureus leukocidins play many roles in facilitating pathogenesis (7, 9, 32, 42–45). Further, the leukocidins are promising vaccine targets (41, 46). Thus, understanding the biochemistry of leukocidin interactions with host receptors and cells is an important area of research to continue because these studies may facilitate the development of novel S. aureus therapeutics and vaccines.
Experimental procedures
Ethics statement
Blood was obtained from healthy, deidentified, consenting adult donors as buffy coats from the New York Blood Center. All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of New York University and were performed according to guidelines from the National Institutes of Health, the Animal Welfare Act, and United States Federal Law.
Leukocidin generation and purification from S. aureus
The LukES-DR1+4 chimera and the LukSE-DR chimeras were generated by overlapping extension PCR, as described (31). The primer sequences are listed in Table S1. LukES-DR1+4 was made using LukES-DR1 plasmid (VJT34.08) as a template, with primers VJT629, VJT903, VJT914, and VJT1114, and final PCR was performed with VJT629 and VJT1114. LukSE-DR chimeras were made using LAC genomic DNA as a template, with the following primers: LukSE-DR1: VJT1208, VJT1186, VJT1189, and VJT1117; final PCR: VJT1208, VJT1117, VJT1187, and VJT1188; LukSE-DR2: VJT1208, VJT1190, VJT1193, and VJT1117; final PCR, VJT1208, VJT1117, VJT1191, and VJT1192; LukSE-DR3: VJT1208, VJT1194, VJT1197, and VJT1117; final PCR, VJT1208, VJT1117, VJT1195, and VJT1196; and LukSE-DR4: VJT1208, VJT1198, VJT1201, and VJT1117; final PCR: VJT1208, VJT1117, VJT1199, and VJT1200.
The LukSE-DR1+4 chimera and the loop chimeras described previously (16) were synthesized as gBlocks (Integrated DNA Technologies) and amplified by PCR. LukS mutants were amplified with primers VJT1208 and VJT1117. LukE mutants were amplified with VJT629 and VJT1114.
The final PCR products were cloned into the pOS1-PlukAB-lukAss-His6 plasmid using BamHI and PstI restriction sites, transformed into E. coli DH5α, screened, sequenced, electroporated into S. aureus RN4220, and finally electroporated into S. aureus Newman ΔΔΔΔ (ΔlukED hlgACB::tet lukAB::spec hla::ermC) as described (31). All other mutants were described previously (8).
WT and mutant toxins were purified from supernatants of S. aureus Newman as detailed elsewhere (8, 13, 31). Briefly, culture filtrates were harvested from 5-h subcultures of S. aureus Newman ΔΔΔΔ strains harboring plasmids with the toxin sequences with His tags grown in tryptic soy broth supplemented with chloramphenicol grown at 37°C with 180-rpm shaking. The cultures were centrifuged and filter-sterilized, and the His-tagged proteins were purified on nickel–nitrilotriacetic acid–agarose resin (Qiagen) columns. Protein concentrations were quantified by measuring the absorbance at 280 nm using an ND-1000 Spectrophotometer (NanoDrop) and then multiplying by the molecular weight and dividing by the extinction coefficient. All holotoxin concentrations are represented as per subunit unless otherwise indicated.
Hemolysis assays
Human erythrocytes were isolated from buffy coats obtained from the New York Blood Center. Erythrocytes were sedimented with dextran as described (22) and then washed three times with 0.9% saline. 4 × 106 cells were incubated with purified toxin in a V-bottom plate for a total volume of 100 μl for 30 min at 37 °C + 5% CO2 and then centrifuged at 2,000 rpm for 10 min. 50 μl of supernatant were transferred to a flat-bottomed plate, and the absorbance was measured at 405 nm to quantify hemoglobin release. Each condition was run in duplicate, and the two wells were averaged. Hemolysis was normalized to Triton X-100 lysed control (set to 100%).
To test the hemolytic activity of the loop mutants designed by Peng et al. (16) under the conditions analogous to those described by the authors, the cells were washed three times with PBS, and 108 cells were added to purified toxin in a V-bottom plate for a total volume of 250 μl for 1 h at 37 °C + 5% CO2 and then centrifuged at 2,000 rpm for 10 min. Hemolysis was quantified from supernatant as above.
Cytotoxicity assays with human neutrophils
Primary human PMNs were isolated from buffy coats obtained from the New York Blood Center as described (8). A total of 2 × 105 cells in RPMI supplemented with 10% FBS were incubated with purified toxin in a flat-bottomed plate for a total volume of 100 μl for 1 h at 37 °C + 5% CO2. 10 μl of CellTiter 96 Aqueous One Solution cell proliferation assay dye (Promega) was added to the cells and incubated for an additional 2 h at 37 °C + 5% CO2. Absorbance was measured at 492 nm to quantify cell viability. Each condition was run in duplicate, and the two wells were averaged. Cell death was normalized to Triton X-100 lysed control (set to 100%).
Cytotoxicity assays with SupT1-CCR5 cells
SupT1 cells (ATCC CRL-1942) transduced with CCR5 were cultured as described (13). Briefly, the cells were grown in RPMI supplemented with 10% FBS, 1% penicillin/streptomycin, and 1 μg/ml puromycin at 37 °C + 5% CO2.
To assay cytotoxicity, a total of 1 × 105 cells in RPMI supplemented with 10% FBS were incubated with purified toxin in a flat-bottomed plate for a total volume of 100 μl for 2 h at 37 °C + 5% CO2. Cell viability was measured with CellTiter assay dye as above.
Inhibition of hemolysis by DARC
Purified LukED at a concentration of 15 nm was added to a titration of purified recombinant DARC (OriGene) and incubated at room temperature for 10 min. Human erythrocytes were washed, added to the mixture, and intoxicated as above.
SPR analysis of LukE/LukS binding to DARC
SPR was performed using the Biacore S200 system (Cytiva) as previously described (9) with some minor modifications. Briefly, purified recombinant Duffy/DARC purified from human HEK293T cells (OriGene) were immobilized using the NHS capture kit onto flow cells 2–4 of a series S sensor chip CM5 (Cytiva) with flow cell 1 acting as the blank reference control. Recombinant toxins were flowed over DARC at concentrations ranging from 0.016 to 50 μg·ml−1 using single cycle kinetics. At least duplicate data were obtained from all three flow cells for each interaction, with 1× PBS at pH 7.2 used as the buffer only zero concentration control. A flow rate of 20 μl·min−1 was used for the analysis with a contact time of 60 s, and a final disassociation step of 10 min was performed. The affinities were calculated using the Biacore S200 evaluation software.
Fluorescent toxin labeling of toxins
Purified LukD and LukDΔ130-4 were labeled with Thermo Scientific Dylight 488 NHS-Ester Amine reactive dye kit according to the manufacturer's protocol. 2 μg of labeled toxin was run on an SDS-PAGE gel and imaged using the ChemiDoc Touch imaging system to confirm labeling. The gel was then stained with Coomassie Blue and imaged.
Cell binding assays
Human erythrocytes were prepared from buffy coats as above and then washed three times in 0.9% saline supplemented with 0.55% BSA. 4 × 106 cells were incubated with 180 nm toxin in a V-bottom plate in a total volume of 100 μl for 10 min at 37 °C + 5% CO2, centrifuged at 2000 rpm for 2 min. The cells were then washed twice with FACS buffer (2% FBS, 0.05% sodium azide in PBS), resuspended in 200 μl of FACS buffer, and analyzed by flow cytometry (CytoFLEX S). The data are shown as the geometric mean fluorescence intensity (MFI) of live single cells.
Murine toxin challenge
Swiss Webster mice were purchased from Envigo. C57BL/6J Darc−/− mice were provided by Dr. G. Scott Worthen (University of Pennsylvania), rederived, and bred in-house at New York University School of Medicine as described previously (32). C57BL/6J controls were purchased from The Jackson Laboratory and bred in-house. The mice were maintained under specific pathogen-free conditions and used age-matched at 4–6 weeks of age for Swiss Webster mice and 7–11 weeks of age for C57BL/6J WT and Darc−/− mice. Swiss Webster experiments were performed with all female mice, and C57BL/6J experiments were performed with female and male (sex-matched) mice. The mice were randomly mixed within their genotypes and sex and then assigned to groups. No sex difference was observed.
The mice were challenged with leukocidins as described previously (31). The mice were anesthetized by inhalation of isoflurane gas (2%), and the toxins were administered systemically at the indicated doses via retro-orbital injection in 100 μl of PBS. The time to acute intoxication was recorded after sacrifice of mice displaying signs of morbidity, including ruffled fur, hunched posture, paralysis, inability to walk, inability to consume food or water, or marked difficulty breathing.
Protein sequence alignments
The LukE (strain Newman) and LukS-PV (strain FPR3757) amino acid sequences were aligned with DNASTAR MegAlign software with ClustalW, as described (8). The alignment of the S subunits shown in Fig. S1B was reproduced from Peng et al. (16). To generate Fig. S1C, these sequences were manually realigned to highlight the similarities between the Z region sequences of LukS-PV and HlgC and loop 4 for LukE and HlgA. The sequence alignment figures were prepared using T-Coffee and BoxShade online tools. The structural alignment figure of LukE (Protein Data Bank code 3ROH) and LukS-PV (Protein Data Bank code 1T5R) was prepared using PyMOL.
Graphical and statistical analyses
Statistical details (n numbers, tests used, definitions of the error bars) are described in the figure legends. Analyses of flow cytometric data were performed using FlowJo. Statistical analyses were performed using GraphPad Prism 8, with Student's t test, two-way ANOVA with Sidak's post hoc test for multiple comparisons, one-way ANOVA with Tukey's post hoc test for multiple comparisons, or log-rank (Mantle–Cox) test as indicated.
Data availability
All data are contained within the article.
Supplementary Material
Acknowledgments
We thank current and former members of the Torres laboratory for fruitful discussions.
This article contains supporting information.
Author contributions—M. T. V., A. L., and V. J. T. conceptualization; M. T. V., C. J. D., and M. P. J. data curation; M. T. V., A. L., T. R.-R., C. J. D., and M. P. J. formal analysis; M. T. V., A. L., T. R.-R., K. A. L., and V. J. T. funding acquisition; M. T. V., A. L., T. R.-R., K. A. L., and C. J. D. investigation; M. T. V., T. R.-R., C. J. D., and M. P. J. methodology; M. T. V., A. L., T. R.-R., C. J. D., K. A. L., M. P. J., and V. J. T. writing-review and editing; A. L. and V. J. T. supervision; A. L. and V. J. T. writing-original draft; M. P. J. resources; V. J. T. project administration.
Funding and additional information—This work was supported in part by National Institute of Health Grants R01 AI105129-04S1 (to M. T. V.); T32 GM007308, T32 AI007180, and F30 AI124606 (to A. L.); T32 AI007180 and F31 AI112290 (to T. R.-R.); and R01 AI105129 and HHSN272201400019C (to V. J. T). This work was also supported in part by a postdoctoral fellowship from the Cystic Fibrosis Foundation (to K. A. L.) and National Health and Medical Research Council Principal Research Fellowship 1138466 (to M. P. J.). V. J. T. is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases. Flow cytometry technologies were provided by New York University Langone's Cytometry and Cell Sorting Laboratory, which is supported in part by NCI, National Institutes of Health Grant P30CA016087. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—V. J. T. is an inventor on patents and patent applications filed by New York University, which are currently under the commercial license to Janssen Biotech Inc.
- DARC
- Duffy antigen receptor for chemokines
- LukED
- leukocidin ED
- LukS-PV
- Panton–Valentine leukocidin S
- DR
- divergence region
- GPCR
- G protein–coupled receptor
- hPMN
- human polymorphonuclear leukocyte
- SPR
- surface plasmon resonance
- MFI
- mean fluorescence intensity
- ANOVA
- analysis of variance.
References
- 1. Tong, S. Y., Davis, J. S., Eichenberger, E., Holland, T. L., and Fowler, V. G., Jr. (2015) Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 28, 603–661 10.1128/CMR.00134-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thammavongsa, V., Kim, H. K., Missiakas, D., and Schneewind, O. (2015) Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 13, 529–543 10.1038/nrmicro3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yoong, P., and Torres, V. J. (2013) The effects of Staphylococcus aureus leukotoxins on the host: cell lysis and beyond. Curr. Opin. Microbiol. 16, 63–69 10.1016/j.mib.2013.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alonzo, F., 3rd, and Torres, V. J. (2014) The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol. Mol. Biol. Rev. 78, 199–230 10.1128/MMBR.00055-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spaan, A. N., van Strijp, J. A. G., and Torres, V. J. (2017) Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 15, 435–447 10.1038/nrmicro.2017.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Spaan, A. N., Henry, T., van Rooijen, W. J., Perret, M., Badiou, C., Aerts, P. C., Kemmink, J., de Haas, C. J., van Kessel, K. P., Vandenesch, F., Lina, G., and van Strijp, J. A. (2013) The staphylococcal toxin Panton–Valentine Leukocidin targets human C5a receptors. Cell Host Microbe 13, 584–594 10.1016/j.chom.2013.04.006 [DOI] [PubMed] [Google Scholar]
- 7. Alonzo, F., 3rd, Kozhaya, L., Rawlings, S. A., Reyes-Robles, T., DuMont, A. L., Myszka, D. G., Landau, N. R., Unutmaz, D., and Torres, V. J. (2013) CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 493, 51–55 10.1038/nature11724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reyes-Robles, T., Alonzo, F., 3rd, Kozhaya, L., Lacy, D. B., Unutmaz, D., and Torres, V. J. (2013) Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 14, 453–459 10.1016/j.chom.2013.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spaan, A. N., Reyes-Robles, T., Badiou, C., Cochet, S., Boguslawski, K. M., Yoong, P., Day, C. J., de Haas, C. J., van Kessel, K. P., Vandenesch, F., Jennings, M. P., Le Van Kim, C., Colin, Y., van Strijp, J. A., Henry, T., et al. (2015) Staphylococcus aureus targets the Duffy antigen receptor for chemokines (DARC) to lyse erythrocytes. Cell Host Microbe 18, 363–370 10.1016/j.chom.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olson, R., Nariya, H., Yokota, K., Kamio, Y., and Gouaux, E. (1999) Crystal structure of staphylococcal LukF delineates conformational changes accompanying formation of a transmembrane channel. Nat. Struct. Biol. 6, 134–140 10.1038/5821 [DOI] [PubMed] [Google Scholar]
- 11. Yamashita, K., Kawai, Y., Tanaka, Y., Hirano, N., Kaneko, J., Tomita, N., Ohta, M., Kamio, Y., Yao, M., and Tanaka, I. (2011) Crystal structure of the octameric pore of staphylococcal γ-hemolysin reveals the β-barrel pore formation mechanism by two components. Proc. Natl. Acad. Sci. U.S.A. 108, 17314–17319 10.1073/pnas.1110402108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laventie, B. J., Guerin, F., Mourey, L., Tawk, M. Y., Jover, E., Maveyraud, L., and Prevost, G. (2014) Residues essential for Panton–Valentine leukocidin S component binding to its cell receptor suggest both plasticity and adaptability in its interaction surface. PLoS One 9, e92094 10.1371/journal.pone.0092094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tam, K., Schultz, M., Reyes-Robles, T., Vanwalscappel, B., Horton, J., Alonzo, F., 3rd, Wu, B., Landau, N. R., and Torres, V. J. (2016) Staphylococcus aureus leukocidin LukED and HIV-1 gp120 target different sequence determinants on CCR5. mBio 7, e02024 10.1128/mBio.02024-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nariya, H., and Kamio, Y. (1997) Identification of the minimum segment essential for the Hγ II-specific function of staphylococcal γ-hemolysin. Biosci. Biotechnol. Biochem. 61, 1786–1788 10.1271/bbb.61.1786 [DOI] [PubMed] [Google Scholar]
- 15. Yokota, K., Sugawara, N., Nariya, H., Kaneko, J., Tomita, T., and Kamio, Y. (1998) Further study on the two pivotal parts of Hlg2 for the full hemolytic activity of staphylococcal γ-hemolysin. Biosci. Biotechnol. Biochem. 62, 1745–1750 10.1271/bbb.62.1745 [DOI] [PubMed] [Google Scholar]
- 16. Peng, Z., Takeshita, M., Shibata, N., Tada, H., Tanaka, Y., and Kaneko, J. (2018) Rim domain loops of staphylococcal β-pore forming bi-component toxin S-components recognize target human erythrocytes in a coordinated manner. J. Biochem. 164, 93–102 10.1093/jb/mvy030 [DOI] [PubMed] [Google Scholar]
- 17. Weinberg, E. D. (1975) Nutritional immunity: host's attempt to withold iron from microbial invaders. JAMA 231, 39–41 [DOI] [PubMed] [Google Scholar]
- 18. Hood, M. I., and Skaar, E. P. (2012) Nutritional immunity: transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 10, 525–537 10.1038/nrmicro2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Skaar, E. P., Humayun, M., Bae, T., DeBord, K. L., and Schneewind, O. (2004) Iron-source preference of Staphylococcus aureus infections. Science 305, 1626–1628 10.1126/science.1099930 [DOI] [PubMed] [Google Scholar]
- 20. Ozawa, T., Kaneko, J., and Kamio, Y. (1995) Essential binding of LukF of staphylococcal γ-hemolysin followed by the binding of Hγ II for the hemolysis of human erythrocytes. Biosci. Biotechnol. Biochem. 59, 1181–1183 10.1271/bbb.59.1181 [DOI] [PubMed] [Google Scholar]
- 21. Yokota, K., and Kamio, Y. (2000) Tyrosine72 residue at the bottom of rim domain in LukF crucial for the sequential binding of the staphylococcal γ-hemolysin to human erythrocytes. Biosci. Biotechnol. Biochem. 64, 2744–2747 10.1271/bbb.64.2744 [DOI] [PubMed] [Google Scholar]
- 22. Yoong, P., and Torres, V. J. (2015) Counter inhibition between leukotoxins attenuates Staphylococcus aureus virulence. Nat. Commun. 6, 8125 10.1038/ncomms9125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horuk, R. (2015) The Duffy antigen receptor for chemokines DARC/ACKR1. Front. Immunol. 6, 279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller, L. H., Mason, S. J., Dvorak, J. A., McGinniss, M. H., and Rothman, I. K. (1975) Erythrocyte receptors for (Plasmodium knowlesi) malaria: Duffy blood group determinants. Science 189, 561–563 10.1126/science.1145213 [DOI] [PubMed] [Google Scholar]
- 25. Horuk, R., Chitnis, C. E., Darbonne, W. C., Colby, T. J., Rybicki, A., Hadley, T. J., and Miller, L. H. (1993) A receptor for the malarial parasite Plasmodium vivax: the erythrocyte chemokine receptor. Science 261, 1182–1184 10.1126/science.7689250 [DOI] [PubMed] [Google Scholar]
- 26. Hadley, T. J., Lu, Z. H., Wasniowska, K., Martin, A. W., Peiper, S. C., Hesselgesser, J., and Horuk, R. (1994) Postcapillary venule endothelial cells in kidney express a multispecific chemokine receptor that is structurally and functionally identical to the erythroid isoform, which is the Duffy blood group antigen. J. Clin. Invest. 94, 985–991 10.1172/JCI117465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dawson, T. C., Lentsch, A. B., Wang, Z., Cowhig, J. E., Rot, A., Maeda, N., and Peiper, S. C. (2000) Exaggerated response to endotoxin in mice lacking the Duffy antigen/receptor for chemokines (DARC). Blood 96, 1681–1684 10.1182/blood.V96.5.1681.h8001681a_1681_1684 [DOI] [PubMed] [Google Scholar]
- 28. Luo, H., Chaudhuri, A., Zbrzezna, V., He, Y., and Pogo, A. O. (2000) Deletion of the murine Duffy gene (Dfy) reveals that the Duffy receptor is functionally redundant. Mol. Cell Biol. 20, 3097–3101 10.1128/mcb.20.9.3097-3101.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gravet, A., Colin, D. A., Keller, D., Girardot, R., Monteil, H., and Prévost, G. (1998) Characterization of a novel structural member, LukE–LukD, of the bi-component staphylococcal leucotoxins family. FEBS Lett. 436, 202–208 10.1016/S0014-5793(98)01130-2 [DOI] [PubMed] [Google Scholar]
- 30. Morinaga, N., Kaihou, Y., and Noda, M. (2003) Purification, cloning and characterization of variant LukE–LukD with strong leukocidal activity of staphylococcal bi-component leukotoxin family. Microbiol. Immunol. 47, 81–90 10.1111/j.1348-0421.2003.tb02789.x [DOI] [PubMed] [Google Scholar]
- 31. Reyes-Robles, T., Lubkin, A., Alonzo, F., 3rd, Lacy, D. B., and Torres, V. J. (2016) Exploiting dominant-negative toxins to combat Staphylococcus aureus pathogenesis. EMBO Rep. 17, 428–440 10.15252/embr.201540994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lubkin, A., Lee, W. L., Alonzo, F., Wang, C., Aligo, J., Keller, M., Girgis, N. M., Reyes-Robles, T., Chan, R., O'Malley, A., Buckley, P., Vozhilla, N., Vasquez, M. T., Su, J., Sugiyama, M., et al. (2019) Staphylococcus aureus leukocidins target endothelial DARC to cause lethality in mice. Cell Host Microbe 25, 463–470 10.1016/j.chom.2019.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hansell, C. A., Hurson, C. E., and Nibbs, R. J. (2011) DARC and D6: silent partners in chemokine regulation? Immunol. Cell Biol. 89, 197–206 10.1038/icb.2010.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nariya, H., Nishiyama, A., and Kamio, Y. (1997) Identification of the minimum segment in which the threonine246 residue is a potential phosphorylated site by protein kinase A for the LukS-specific function of staphylococcal leukocidin. FEBS Lett. 415, 96–100 10.1016/S0014-5793(97)01100-9 [DOI] [PubMed] [Google Scholar]
- 35. Nishiyama, A., Nariya, H., and Kamio, Y. (1998) Phosphorylation of LukS by protein kinase A is crucial for the LukS-specific function of the staphylococcal leukocidin on human polymorphonuclear leukocytes. Biosci. Biotechnol. Biochem. 62, 1834–1838 10.1271/bbb.62.1834 [DOI] [PubMed] [Google Scholar]
- 36. Chakera, A., Seeber, R. M., John, A. E., Eidne, K. A., and Greaves, D. R. (2008) The Duffy antigen/receptor for chemokines exists in an oligomeric form in living cells and functionally antagonizes CCR5 signaling through hetero-oligomerization. Mol. Pharmacol. 73, 1362–1370 10.1124/mol.107.040915 [DOI] [PubMed] [Google Scholar]
- 37. Noda, M., Kato, I., Hirayama, T., and Matsuda, F. (1980) Fixation and inactivation of staphylococcal leukocidin by phosphatidylcholine and ganglioside GM1 in rabbit polymorphonuclear leukocytes. Infect. Immun. 29, 678–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Monma, N., Nguyen, V. T., Kaneko, J., Higuchi, H., and Kamio, Y. (2004) Essential residues, W177 and R198, of LukF for phosphatidylcholine-binding and pore-formation by staphylococcal γ-hemolysin on human erythrocyte membranes. J. Biochem. 136, 427–431 10.1093/jb/mvh140 [DOI] [PubMed] [Google Scholar]
- 39. Liu, J., Kozhaya, L., Torres, V. J., Unutmaz, D., and Lu, M. (2020) Structure-based discovery of a small-molecule inhibitor of methicillin-resistant Staphylococcus aureus virulence. J. Biol. Chem. 295, 5944–5959 10.1074/jbc.RA120.012697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tromp, A. T., Van Gent, M., Abrial, P., Martin, A., Jansen, J. P., De Haas, C. J. C., Van Kessel, K. P. M., Bardoel, B. W., Kruse, E., Bourdonnay, E., Boettcher, M., McManus, M. T., Day, C. J., Jennings, M. P., Lina, G., et al. (2018) Human CD45 is an F-component-specific receptor for the staphylococcal toxin Panton–Valentine leukocidin. Nat. Microbiol. 3, 708–717 10.1038/s41564-018-0159-x [DOI] [PubMed] [Google Scholar]
- 41. Tam, K., Lacey, K. A., Devlin, J. C., Coffre, M., Sommerfield, A., Chan, R., O'Malley, A., Koralov, S. B., Loke, P., and Torres, V. J. (2020) Targeting leukocidin-mediated immune evasion protects mice from Staphylococcus aureus bacteremia. J. Exp. Med. 217, e20190541 10.1084/jem.20190541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Labandeira-Rey, M., Couzon, F., Boisset, S., Brown, E. L., Bes, M., Benito, Y., Barbu, E. M., Vazquez, V., Hook, M., Etienne, J., Vandenesch, F., and Bowden, M. G. (2007) Staphylococcus aureus Panton–Valentine leukocidin causes necrotizing pneumonia. Science 315, 1130–1133 10.1126/science.1137165 [DOI] [PubMed] [Google Scholar]
- 43. Alonzo, F., 3rd, Benson, M. A., Chen, J., Novick, R. P., Shopsin, B., and Torres, V. J. (2012) Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol. Microbiol. 83, 423–435 10.1111/j.1365-2958.2011.07942.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. DuMont, A. L., Yoong, P., Day, C. J., Alonzo, F., 3rd, McDonald, W. H., Jennings, M. P., and Torres, V. J. (2013) Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc. Natl. Acad. Sci. U.S.A. 110, 10794–10799 10.1073/pnas.1305121110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berends, E. T. M., Zheng, X., Zwack, E. E., Ménager, M. M., Cammer, M., Shopsin, B., and Torres, V. J. (2019) Staphylococcus aureus impairs the function of and kills human dendritic cells via the LukAB toxin. MBio 10, e01918–18 10.1128/mBio.01918-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomsen, I. P., Dumont, A. L., James, D. B., Yoong, P., Saville, B. R., Soper, N., Torres, V. J., and Creech, C. B. (2014) Children with invasive Staphylococcus aureus disease exhibit a potently neutralizing antibody response to the cytotoxin LukAB. Infect. Immun. 82, 1234–1242 10.1128/IAI.01558-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the article.