

Abstract
The bacterial enhancer-binding protein (bEBP) FlrC, controls motility and colonization of Vibrio cholerae by regulating the transcription of class-III flagellar genes in σ54-dependent manner. However, the mechanism by which FlrC regulates transcription is not fully elucidated. Although, most bEBPs require nucleotides to stimulate the oligomerization necessary for function, our previous study showed that the central domain of FlrC (FlrCC) forms heptamer in a nucleotide-independent manner. Furthermore, heptameric FlrCC binds ATP in “cis-mediated” style without any contribution from sensor I motif 285REDXXYR291 of the trans protomer. This atypical ATP binding raises the question of whether heptamerization of FlrC is solely required for transcription regulation, or if it is also critical for ATPase activity. ATPase assays and size exclusion chromatography of the trans-variants FlrCC-Y290A and FlrCC-R291A showed destabilization of heptameric assembly with concomitant abrogation of ATPase activity. Crystal structures showed that in the cis-variant FlrCC-R349A drastic shift of Walker A encroached ATP-binding site, whereas the site remained occupied by ADP in FlrCC-Y290A. We postulated that FlrCC heptamerizes through concentration-dependent cooperativity for maximal ATPase activity and upon heptamerization, packing of trans-acting Tyr290 against cis-acting Arg349 compels Arg349 to maintain proper conformation of Walker A. Finally, a Trp quenching study revealed binding of cyclic-di-GMP with FlrCC. Excess cyclic-di-GMP repressed ATPase activity of FlrCC through destabilization of heptameric assembly, especially at low concentration of protein. Systematic phylogenetic analysis allowed us to propose similar regulatory mechanisms for FlrCs of several Vibrio species and a set of monotrichous Gram-negative bacteria.
Keywords: Flagellar synthesis, Vibrio cholera, bacterial enhancer binding protein, σ54-dependent transcription, oligomeric assembly, ATPase activity, mutagenesis, crystal structures, sequence analysis, bacterial transcription, ATPases associated with diverse cellular activities (AAA), site-directed mutagenesis, oligomerization, cyclic di-GMP (c-di-GMP)
Vibrio cholerae, the facultative human pathogen that causes diarrheal disease cholera, is highly motile by means of a single, polar sheathed flagellum. V. cholerae enters inside the human host through ingestion of contaminated food or water, adheres to the apical surface of the intestinal epithelial cell, and expresses virulence factors (1, 2). Motility and colonization of V. cholerae are prerequisites of producing the virulence factors and immune-resistant biofilms which, in turn, are governed by flagellar synthesis (3, 4).
Expression of the proteins required to synthesize the functional flagellum of V. cholerae is regulated by a four-tiered transcriptional hierarchy (4–7). The class-I gene product and bacterial enhancer-binding protein (bEBP), FlrA activates σ54-dependent transcription of the class-II genes flrBC, which encode another bEBP, FlrC and its cognate kinase FlrB (4, 5). Transcription of class-III flagellar genes, which encode important flagellar components like basal body hook and the flagellin FlaA are regulated by FlrC (4, 8). The anti-σ factor FlgM is secreted through the basal body-hook to allow σ28-dependent transcription of class-IV genes, which encode four additional flagellins and some of the motor components (4, 6).
Motility and biofilm formation of V. cholerae are further regulated by ubiquitous second messenger cyclic di-guanosine monophosphate (c-di-GMP) at the transcriptional level. Although the precise molecular mechanisms by which c-di-GMP affects motility in V. cholerae are less well understood, a high c-di-GMP level was found to inhibit the production and function of V. cholerae's single polar flagellum (9–12). Available evidences suggest that V. cholerae responds to an elevated level of c-di-GMP by increasing the transcription of the vps, eps, and msh genes and decreasing that of flagellar genes (12). This clearly indicates a distinctive mode of interactions of c-di-GMP with the bEBPs involved in exopolysaccaride production and flagellar synthesis.
FlrC of V. cholerae is made of N-terminal response regulator (R) domain, central AAA+ ATPase domain, and C-terminal DNA-binding domain. Phosphorylation occurs in the R domain of FlrC by cognate kinase FlrB (8). Previous studies by Klose and colleagues (6) delineated that a V. cholerae strain containing a deletion of flrC is nonmotile and also displays a modest colonization defect, whereas a strain expressing a hyperactive form of FlrC has an altered cell morphology (6, 8, 13). They further showed that both inactive and constitutively active mutants of FlrC cause more severe colonization defects than a strain lacking FlrC entirely, which implies that both unphosphorylated and phosphorylated forms of FlrC are required for the colonization, and locking FlrC into either an active or an inactive state would send incorrect stimuli into this stepwise colonization process (6, 8, 13).
Although domain organization portrays FlrC as NtrC-type bEBP, this is atypical in many respects. The σ54-dependent activators usually bind to the enhancer elements located upstream of the RNAP-σ54 binding site and contact this complex at the promoter by DNA looping mechanism (14). In contrast, FlrC binds enhancer elements located downstream of the σ54-binding and transcriptional start sites of the flaA and flgK promoters (8). The same feature was observed in FleQ of Pseudomonas aeruginosa for flhA, fliE, and fliL genes where the authors argued for direct interaction between RNAP-σ54 and the activator without DNA looping (15). However, the mechanistic details of binding RNAP-σ54 at the promoter is yet to be deciphered for FleQ or FlrC.
Usually, oligomerization of bEBPs takes place through the AAA+ domain. Despite that, bEBPs display remarkable diversity in terms of oligomerization, ATP binding, and hydrolysis, mediated by different motifs within the AAA+ domain, which possibly play roles in directing the proteins toward specific functions (16–22). Generally, in the NtrC class of proteins, oligomerization is guided by Nt-dependent subunit remodeling from the inactive dimer to active hexa/heptamer (17, 21, 22). We have previously determined the crystal structure of the AAA+ domain of FlrC (FlrCC) in Nt-free (Fig. 1a) and AMP-PNP–bound (Fig. 1b) states (20). In both cases FlrCC was found in the heptameric state demonstrating that, unlike the NtrC class of bEBPs, FlrC spontaneously forms heptamer without Nt-dependent subunit remodeling (PDB codes 4QHS and 4QHT) (20). The major presence of the heptameric species of FlrCC was also established in solution by size exclusion chromatography and dynamic light scattering (20). Although asymmetric split-ring hexamer was observed for NtrC1 to contact the RNAP-σ54 complex at promoter DNA (18), heptameric FlrCC with a much wider central pore was presumed to be adequate for the same purpose without any serious ring distortion (20).
Figure 1.
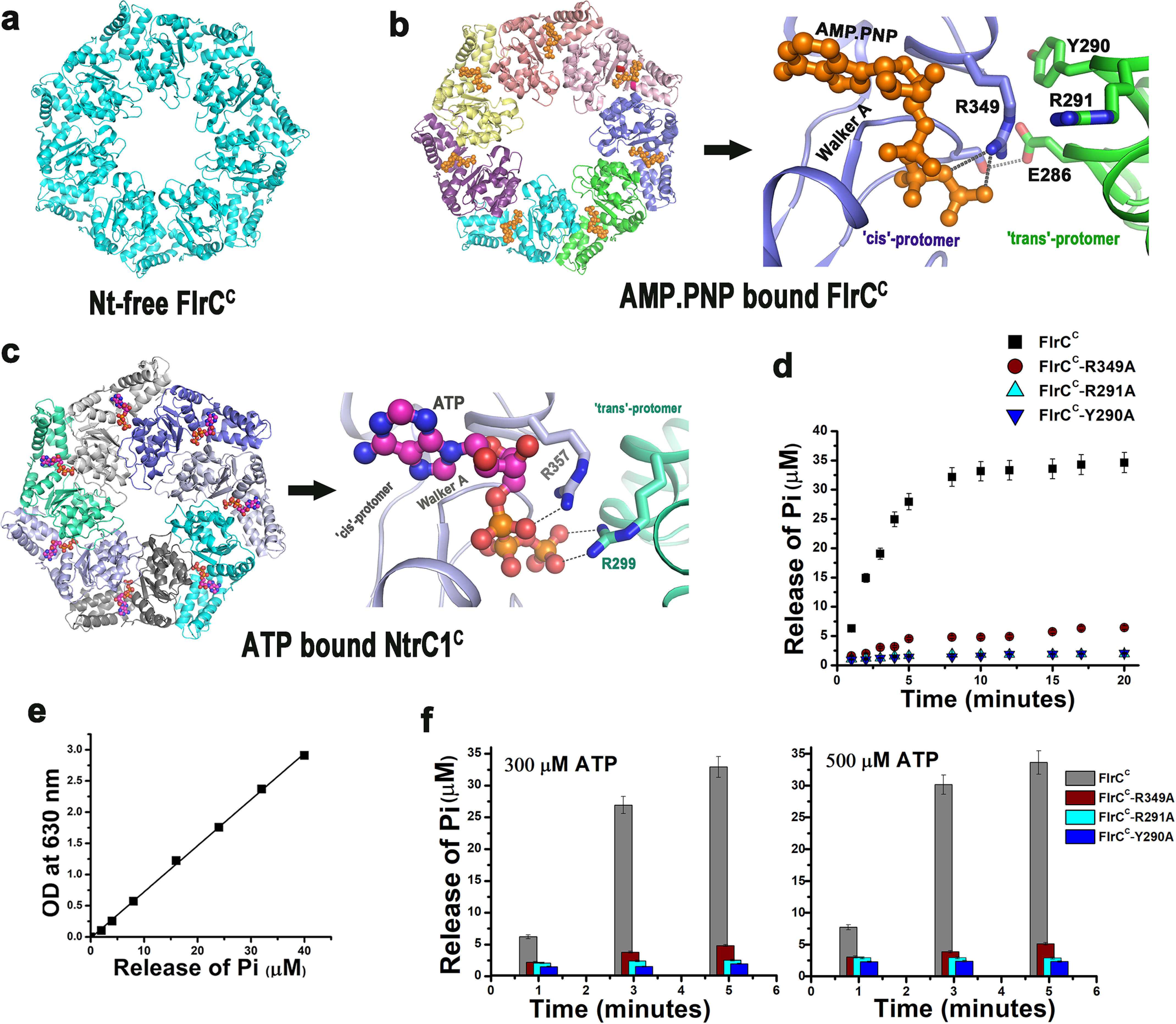
cis-Mediated ATP binding in FlrCC and loss-of-function variants. a, structure of Nt-free heptameric FlrCC (PDB code 4QHS). b, at left is the AMP-PNP–bound heptameric structure of FlrCC (PDB code 4QHT) where bound AMP-PNP molecules are shown in orange spheres; at the right is the zoomed view of cis-mediated AMP-PNP binding. c, at the left is the ATP-bound heptameric structure of NtrCC (PDB code 3M0E) where bound ATP molecules are shown in magenta spheres; at the right is the zoomed view of ATP binding showing contribution of trans-acting Arg finger (R299). In b and c, cis- and trans-protomers are labeled with respective chain colors. d, time course ATPase activities of FlrCC, the cis-mutant FlrCC-R349A, and the trans-mutants FlrCC-Y290A and FlrCC-R291A with 100 μm ATP by Malachite green assay showed that the mutants are nonfunctional; e, release of Pi was estimated against the standard curve of KH2PO4. f, nonfunctional nature of the mutants is evident from ATPase activities measured at elevated (300 and 500 μm) ATP concentrations at three time points. For d and f, error bars are mean ± S.D. values from three replicates.
In the NtrC class of bEBPs, such as NtrC1 and PspF, ATP binds in the protomer interface having predominant contacts with Walker A of the cis-protomer. Trans-acting arginine(s) belonging to the conserved “RXDXXXR” motif of sensor I acts as “R-finger” to stabilize the γ-phosphate of ATP (Fig. 1c) (17). Conversely, a novel and solely cis-mediated ATP binding and hydrolysis occurs in the heptameric FlrCC, where γ-phosphate of ATP is stabilized by cis-acting Arg349 of sensor II (Fig. 1b). Although the 285REDXXYR291 motif of sensor I was conserved in FlrC and adjacent to the ATP-binding site in trans, no residue of this motif participates in binding ATP (Fig. 1b) (20). This atypical “cis-mediated” mode of ATP binding in FlrC necessitates further investigations on mechanistic relationships between heptamerization and ATPase activity. Because no residues of trans-protomer interact with ATP, FlrC should be capable of binding and hydrolyzing ATP even in its monomeric state. Our current study, therefore, investigated if the heptameric structure of FlrC is required for binding the RNAP-σ54 complex at promoter, or it is decisive for ATPase activity as well.
To address this issue, we have used three variants where cis-acting Arg349 of sensor II and two trans-acting residues Tyr290 and Arg291 (belonging to 285REDXXYR291 motif) of sensor I were replaced by Ala. Interestingly, all three variants, FlrCC-R349A, FlrCC-Y290A, and FlrCC-R291A showed impaired ATPase activity with destabilization of the heptameric state in varying degrees. Through the crystal structures of FlrCC-R349A and FlrCC-Y290A, we have addressed the molecular basis of the aforesaid structural and functional changes. Our observations suggest that heptamerization coupled with optimal conformation of Walker A is essential for efficient ATP binding and hydrolysis by FlrC. FlrCC forms heptamer through concentration-dependent positive cooperativity leading to maximal ATPase activity.
Of the two key bEBPs involved in flagellar synthesis of V. cholerae, c-di-GMP abrogates interactions of FlrA with the promoters of the flrBC operon, leading to reduced expression of the downstream flagellar genes (11). However, the role of c-di-GMP in regulation of FlrC was as yet unexplored. Our study has revealed for the first time that high concentrations of c-di-GMP repress ATPase activity of FlrCC by destabilizing heptameric assembly. Based on database and phylogenetic analyses we have further envisaged existence of such mechanisms in several other Vibrio species and a set of monotrichous Gram-negative bacteria.
Results
ATP hydrolysis was impaired in all three FlrCC variants
FlrCC and the variants were purified as His6-tagged proteins and tested for ATP hydrolysis using Malachite green assay as per the protocol described earlier (20, 23, 24). Time course ATPase assays were performed where reaction mixtures containing 2.5 μm FlrCC (or variants) and 0.1 mm ATP (Sigma Aldrich) were incubated at 298 K for different time periods from 1 to 20 min (Fig. 1d). The release of Pi was measured at 630 nm upon incubation with Malachite green. Released Pi from each reaction was quantified by comparing with a Pi standard curve prepared using KH2PO4 (Fig. 1e). Time course experiments showed that the amount of Pi produced by FlrCC as a result of ATP hydrolysis increased approximately linearly with time for the first 5 min before it slowed down (Fig. 1d). However, ATPase activity of all three variants was drastically low throughout the time course (Fig. 1d). Concentrations of ATP inside the bacterial host may elevate up to 1 mm under certain conditions (25, 26). Therefore, we measured ATPase activities of FlrCC and variants upon elevating ATP concentrations to 0.3 and 0.5 mm as well (Fig. 1f). Considering the linearity pattern of the time course graph in Fig. 1d, release of Pi by the proteins were measured after 1, 3, and 5 min incubation (Fig. 1f). Negligible Pi release by the variants compared with FlrCC further established inertness of the variants in terms of ATPase activity (Fig. 1f). Because trans-acting Tyr290 and Arg291 of sensor I have no direct interaction with ATP (Fig. 1b), drastic reduction in ATP hydrolysis upon mutation of these residues to Ala was truly thought provoking. These results indicated that Tyr290 and neighboring residue Arg291 of trans protomer have indirect yet definite roles in the ATPase activity of FlrC.
Heptameric state is impaired upon mutation of Try290, Arg291, and Arg349
The oligomeric states of FlrCC and its variants were compared by size exclusion chromatography (SEC) using Superdex 200 increase column 10/300) (Fig. 2, a–e). Peak II of Fig. 2a having an elution volume of 12.13 ml was indicative of the heptamer of FlrCC where peak I denoted a higher molecular weight aggregate. In contrast, FlrCC-Y290A eluted at 15.75 ml exclusively as a monomeric species (Fig. 2, b and e). During SEC experiments, 100 μl of each protein was loaded in the column. Concentrations of FlrCC and FlrCC-R291A were 360 μm. Although FlrCC-Y290A was initially 360 μm, its exclusive existence as monomeric species intended us to elevate its concentration to 500 μm, which produced the same result (Fig. 2b). Exclusive existence as a monomeric species of FlrCC-Y290A in SEC was further validated through dynamic light scattering experiments (Fig. 2f).
Figure 2.
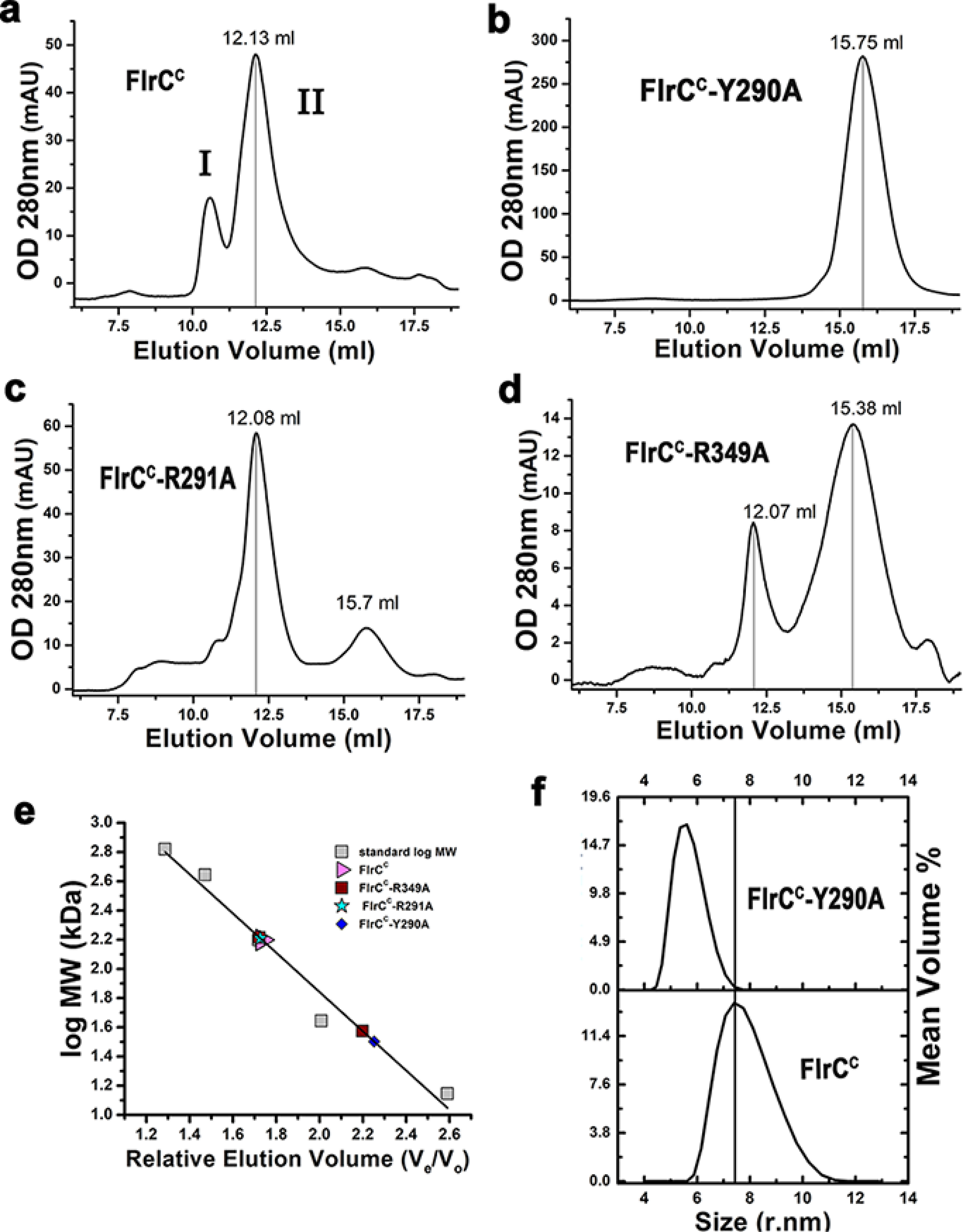
Heptameric state was disassembled in the variants. Size exclusion chromatography elution profiles of (a) FlrCC, (b) FlrCC-Y290A, (c) FlrCC-R291A, (d) FlrCC-R349A. e, the molecular weight of the peaks was determined from the calibration curve prepared using molecular weight standards. Elution volumes suggest predominant heptameric state of FlrCC, monomeric state of FlrCC-Y290A, and mixed hepta and monomeric states of FlrCC-R291A and FlrCC-R349A. f, existence of FlrCC-Y290A as monomer in solution was reconfirmed by dynamic light scattering.
FlrCC-R291A and FlrCC-R349A, however, eluted as a mixture of heptameric and monomeric species (Fig. 2, c–e). Although the heptameric state was prevalent for FlrCC-R291A, a majority of FlrCC-R349A was found as a monomeric species (Fig. 2, c–e). It should be noted that due to an aggregation problem, the concentration of FlrCC-R349A could not be increased beyond 158 μm (which was less than that of FlrCC-R291A). Prevalence of the monomeric species of FlrCC-R349A probably occurred at that concentration after ∼60-fold dilution during SEC (Fig. 2, d and e). Because of a higher concentration, FlrCC-R291A could maintain more heptameric species, even after similar dilution (Fig. 2, c and e).
Crystal structure determination of FlrCC-R349A and FlrCC-Y290A
Crystal structures of FlrCC-R349A and FlrCC-Y290A were determined up to 3.1 and 3.45 Å, respectively. Very similar unit cell dimensions, space group (P212121), and Mathew's coefficient of the variants to that of FlrCC (apo or AMP-PNP bound) suggested similar assembly. We, therefore, calculated the initial electron density map of the respective variant using the apo coordinates of FlrCC (PDB code 4QHS) after rigid body refinement. Because FlrCC-Y290A and FlrCC-R349A showed complete/major existence as monomeric species in SEC, the heptameric species in their crystal structures suggest that these variant proteins were forced to form heptamer at supersaturation. Interestingly, the crystals of FlrCC-Y290A were short-lived and disappeared in 10-16 h. Nonetheless, the diffraction data were satisfactory and the structures of FlrCC-R349A and FlrCC-Y290A were refined up to Rwork of 19.03% (Rfree = 25.08%) and 18.77% (Rfree = 23.41%), respectively. Data collection and refinement statistics are given in Table 1.
Table 1.
Data collection and refinement statistics
| FlrCC-Y290A | FlrCC-R349A | |
|---|---|---|
| Data collection | ||
| Beamline | PX-Bl-21, RRCAT, Indore, India | PX-Bl-21, RRCAT, Indore, India |
| Wavelength (Å) | 0.979 | 0.979 |
| Space group | P212121 | P212121 |
| Unit cell parameters (Å/°) | 80.93, 154.13, 194.06,90, 90, 90 | 80.75 154.63 194.6090, 90, 90 |
| Resolution range (Å) | 48.01-3.45 (3.64- 3.45) | 48.65-3.10 (3.21-3.10) |
| Number of unique reflections | 32,784 (4,707) | 45,041 (4,337) |
| Completeness (%) | 100 (100) | 99.9 (100) |
| Multiplicity | 7.2 (7.3) | 5.5 (5.3) |
| Averaged I/σI | 9.9 (3.1) | 11.1 (2.5) |
| Rmerge (%) | 23.5 (85.4) | 16.3 (69.4) |
| CC1/2 | 99.0 (81.1) | 99.2 (73.8) |
| Refinement | ||
| Rwork/Rfree (%) | 18.77 (23.41) | 19.03/25.08 |
| No. of atoms | ||
| Protein | 13,424 | 13,482 |
| Solvent | 0 | 61 |
| Ligand | 181 | 0 |
| Average B-factor (all atoms) (Å2) | 56 | 40 |
| RMSD | ||
| Bond lengths (Å) | 0.003 | 0.003 |
| Angles (°) | 0.918 | 0.573 |
| Ramachandran plot | ||
| Favored (%) | 95.68 | 94.87 |
| Allowed (%) | 3.97 | 4.78 |
| Disallowed (%) | 0.35 | 0.35 |
Overall superposition of FlrCC-Y290A heptamer on apo and AMP-PNP–bound FlrCC structures produced RMSD of 0.43 and 0.71 Å. Corresponding RMSD values for FlrCC-R349A were of 0.33 and 0.84 Å, respectively. Superposition of the monomers of the variants produced RMSD ranging from 0.4 to 0.6 Å. This implies that the overall heptameric structure of both variants were similar to the functional WT protein. Except for the ones that were influenced by the mutations, most of the other interactions at inter-protomeric interfaces were retained in these loss-of-function variants.
Drastic change in conformation of Walker A upon mutation of cis-acting R349A
In the case of FlrCC-R349A, reduction in ATPase activity was expected, because the side chain of Arg349 was absent to stabilize the γ-phosphate of ATP. But from the structure of FlrCC-R349A we have identified additional factors responsible for compromised ATP binding.
Although no noticeable change in the overall heptameric structure of FlrCC-R349A was observed, Walker A motif was found in an altered conformation in all seven chains. Fig. 3a shows defined electron density around Walker A of FlrCC-R349A. Flexibility is the characteristic feature of the Walker A motif. Previously we noticed a “closed to open” conformational shift of Walker A in FlrCC to facilitate binding of ATP (20). In the “closed” conformation, which occurs in Nt-free FlrCC, Walker A was in a position to exert steric hindrance to the β- and γ-phosphates of ATP. A closed to open movement of Walker A relieved that hindrance by flipping Ser161 (Fig. 3b). However, the drastic shift of Walker A, observed in the absence of Arg349, was significantly different from Nt-free and AMP-PNP–bound FlrCC (shown in red in Fig. 3b). In FlrCC-R349A, the region between Gly162 to Lys165 experienced a major conformational change where the Cα atom of Ser163 has shifted about 5.85 Å (Fig. 3, b and c). Superposition of FlrCC-R349A on the AMP-PNP–bound FlrCC structure further revealed that altered conformation of Walker A in the variant practically encroached on the ATP-binding site (Fig. 3d).
Figure 3.

ATP-binding site of FlrCC-R349A was obscured by Walker A. a, 2Fo − Fc electron density map, contoured at 1σ, around Walker A motif of FlrCC-R349A. b, superposition of Walker A regions of Nt-free and AMP-PNP–bound FlrCC on that of FlrCC-R349A. The mutation site is shown as R349A. Hydrophobic packing between cis-acting Arg349 (gray) with trans-acting Tyr290 (magenta) is shown in Nt-free FlrCC. Both protomers of AMP-PNP–bound FlrCC are shown in sea green. c, superposition of FlrCC-R349A (brown) on Nt-free FlrCC structure (gray) depicted the conformational difference in Walker A (shown by arrow). d, superposition of FlrCC-R349A on AMP-PNP–bound FlrCC (sea green) has shown encroachment of ATP-binding site by Walker A of the variant. Bound ATP is shown as green ball and stick. In b–d, Walker A of FlrCC-R349A is shown in red for clarity.
Trans-mutant FlrCC-Y290A contains bound ADP at the ATP-binding pocket
All seven ATP-binding sites of FlrCC-Y290A were occupied by ADP molecules (Fig. 4, a and b). Because no nucleotide (ADP or ATP) was added externally during protein preparation and/or crystallization, bound ADP was indicative of the fact that ADP was acquired by FlrCC-Y290A from bacterial host cells after expression. This is further established from the fact that FlrCC, when overexpressed and purified by the same procedure, did not contain any bound nucleotide, as evident from the heptameric structure of Nt-free FlrCC (Fig. 1a) (PDB code 4QHS) (20). Notably, bound ADP was previously observed in the structure of the inactive dimer of Aquifex aeolicus NtrC1RC where ATP was added during protein purification (PDB code 1NY5) (27). Because we have not used ATP/ADP during protein preparation, ADP binding to FlrCC-Y290A was envisaged as the outcome of the mutation of trans-acting Tyr290 to Ala.
Figure 4.

ADP remained bound to ATP-binding site of FlrCC-Y290A. a, 2Fo − Fc electron density map contoured at 1σ around ADP molecules, bound to all seven chains of the heptamer. b, zoomed view of 2Fo − Fc electron density map around ADP and the surrounding residues, contoured at 1σ. c, overlay of the FlrCC-Y290A structure (magenta and orange) on that bound to AMP-PNP (cyan) showed that ADP is bound to the cognate site of ATP binding. AMP-PNP is shown as yellow stick. Site of mutation is designated as Y290A. trans-Protomer of FlrCC-Y290A is shown in orange. d, electrostatic surface of ADP-bound FlrCC-Y290A. Interactions of ADP with neighboring residues are also shown here. e, superposition of all seven chains of FlrCC-Y290A showing ADP molecules and disposition of the side chains around that.
Structural superposition of FlrCC-Y290A on AMP-PNP–bound FlrCC showed that ADP binds exclusively in cis-mediated manner, similar to AMP-PNP binding (Fig. 4c). The base was hydrophobically packed with Val132, Val167, Trp299, and Leu312 (Fig. 4, c and d). The ribose sugar was hydrogen bonded with Arg319 (Fig. 4d). Two phosphates of ADP were stabilized through polar interactions with the backbone NH groups of Walker A residues Gly162, Gly164, and Glu166 (Fig. 4, c and d). However, Lys165, which was observed to stabilize β-phosphate of AMP-PNP, remained indifferent to the ADP binding (Fig. 4, c and e). The side chain of cis-acting Arg349 was seen to be away from the ATP-binding site (Fig. 4, c and e). Rather, it was found to interact with trans-acting Glu286 (Fig. 4c). Another conformation of Arg349 was observed in low contour (0.8 σ) heading further away from the ATP-binding site to form a hydrogen bond with cis-acting Asn353 (Fig. S1). But considering the resolution of the structure and low occupancy of that conformation, we have not assigned any alternate conformation to Arg349 during refinement. Conformation of Walker A of FlrCC-Y290A was similar to that of Nt-free FlrCC. When seven protomers of FlrCC-Y290A were superposed, side chain of Ser161 was found in two different conformations (Fig. 4e). In most of the cases Ser161 was hydrogen bonded with cis-acting “Asn switch” Asn272, like it was in Nt-free state of FlrCC (to stabilize the closed conformation of Walker A).
FlrCC heptamerizes through concentration dependent cooperativity
To inspect probable effect of FlrCC concentration on its ATPase activity, we have measured specific activity of FlrCC with gradually increasing concentrations of protein (300 nm to 1.4 μm) through time course experiments up to 5 min upon incubation with 0.1 mm ATP. Specific activity of FlrCC, ±S.D. (derived from three replicates), was plotted against protein concentration (Fig. 5a). A sharp rise in specific activity occurred above protein concentrations of 600 nm, which was maximized and saturated after 1 μm (Fig. 5a). Saturation in activity was confirmed by continuing the experiments with increased concentration of protein up to 1.4 μm (Fig. 5a). Sigmoidal nature of the curve suggested that an increase in protein concentration encourages oligomerization through positive cooperativity that leads to increased ATPase activity. Elevation in activity is saturated above the concentration where the oligomeric state is maximally accomplished.
Figure 5.
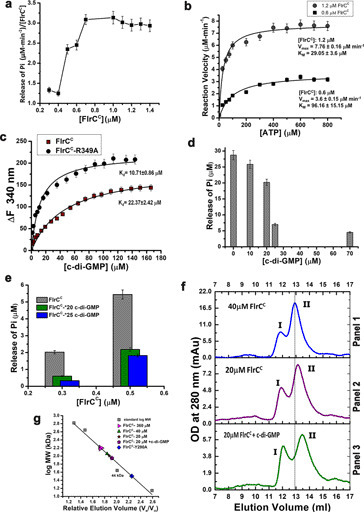
Cooperative heptamerization and effect of c-di-GMP. a, specific ATPase activity of FlrCC, determined through time course experiments, has been plotted against protein concentration. ATP concentration was 100 μm during experiments. b, ATPase activities, measured at two different FlrCC concentrations, 1.2 and 0.6 μm, were plotted against ATP concentrations as per the Michaelis-Menten equation: y = Vmax × x^n/(k^n + x^n) with one site-specific binding model. Adjusted R2 for the two plots were 0.94 and 0.95, respectively. The plots depicted influence of oligomerization on the reaction velocity. c, change in fluorescence (ΔF) of FlrCC and FlrCC-R349A were measured in the presence of c-di-GMP. Nonlinear plots of ΔF as per equation: ΔF = ΔFmax × X^n/(Kd^n + X^n), against c-di-GMP concentration (X), considering one site-specific binding model produced ΔFmax values of 143.8 ± 6.88 (±S.D.) and 210.6 ± 6.22 (±S.D.) for FlrCC and FlrCC-R349A, respectively. Apparent Kd values obtained from the plots indicated stronger c-di-GMP binding with FlrCC-R349A compared with FlrCC. Adjusted R2 values of the nonlinear fit were 0.93 and 0.96, respectively. d, ATPase activity of FlrCC was measured after incubation of the protein with c-di-GMP of different concentrations. Drastic reduction in ATP hydrolysis occurred upon 25 m excess of c-di-GMP. e, effect of c-di-GMP was compared at FlrCC concentrations of 0.3 and 0.5 μm. Significant reductions are observed in the presence of 20- and 25-fold c-di-GMP. f, SEC elution profiles showed right shift of the heptameric peak of FlrCC upon dilution and addition of c-di-GMP. g, the molecular weight of the peaks obtained upon dilution and addition of c-di-GMP were marked in the calibration curve prepared using molecular weight standards. Error bars in a–e are mean ± S.D. values obtained from at least three replicates.
Although we observed previously that FlrCC acquires a heptameric state in a Nt-independent manner, specific activity measurement (Fig. 5a) further intended us to investigate any probable effect of ATP on restoration of the heptameric state of FlrCC specifically at low concentrations where the protein showed relatively low ATPase activity. As described before, we have loaded 100 μl of FlrCC (of concentration 20 μm) in the SEC column in free state or upon incubation with 10-fold molar excess of AMP-PNP (for 30 min on ice). Considering the dilution during experiment, the final concentration of FlrCC was expected to be ∼650 nm (Fig. S2). However, no shift in peak position was observed, further confirming that ATP or AMP-PNP has no effect on the oligomeric state of FlrCC (Fig. S2).
Measurements of Vmax and Km at different FlrCC concentrations confirmed requirement of oligomeric state for optimal ATPase activity
Furthermore, to enquire protein concentration dependence of the kinetic parameters, we have determined maximal velocity (Vmax) and Michaelis constant (Km) at two different FlrCC concentrations through time course ATPase assays. Because Fig. 5a indicated that FlrCC remains maximally active above 1 μm, two sets of experiments were carried out, one with protein concentration of 1.2 μm and the other with 0.6 μm. Considering linearity of Pi production during time course experiments (Fig. 1d), the reaction velocity was measured upon incubation of FlrCC with increasing ATP concentrations (up to 800 μm) through a time scan up to 4 min. Reaction velocity (V0) in terms of Pi release was then plotted against ATP concentrations (Fig. 5b). Fitting of ATPase activities in the Michaelis-Menten equation resulted in Vmax of 7.76 ± 0.16 μm min−1 and Km of 29.05 ± 3.6 μm for FlrCC of 1.2 μm, whereas the corresponding values were 3.6 ± 0.15 μm min−1 and 96.16 ± 15.15 μm for FlrCC of 0.6 μm (Fig. 5b). Noticeable increase in Km value upon dilution of the protein from 1.2 μm to 600 nm pointed toward inefficient ATP binding upon dilution of protein. Presumably, decrease in protein concentration caused instability in the heptameric structure leading to reduction in ATPase activity. This observation was further supported by the fact that measurement of Vmax and Km with an even lesser FlrCC concentration (250 nm) was unsuccessful, since no change in Pi release per min was observed at that concentration.
Interaction of c-di-GMP with FlrCC and the variants
Previous studies on FleQ of P. aeruginosa (which is an ortholog of FlrA of V. cholerae) suggested that c-di-GMP binds near the Walker A motif of its AAA+ ATPase domain, although the binding site is distinct from the ATP-binding site (28). Furthermore, mutational analysis deciphered that c-di-GMP binds near Walker A of the flagella export AAA+ ATPase FliI of P. fluorescens (29). The above mentioned information intended us to assume that c-di-GMP may bind near the Walker A motif of FlrCC. FlrCC and variants contain three tryptophan residues Trp299, Trp344, and Trp379. Among these residues, Trp299 is within the Forster distance of Walker A in FlrCC, which is also near the inter-protomeric region (Fig. 4, c and d). The other two tryptophans are beyond Forster distance from Walker A. Probable interactions of c-di-GMP with FlrCC and its variants were, therefore, monitored where the intrinsic fluorescence of Trp299 was expected to change upon c-di-GMP binding. FlrCC showed substantial quenching upon addition of c-di-GMP with an apparent Kd value of 22.37 ± 2.42 μm (Fig. S3a, Fig. 5c). Interestingly, the variant FlrCC-R349A interacted with c-di-GMP even more efficiently, as reflected from the apparent Kd value of 10.71 ± 0.86 μm (Fig. S3b, Fig. 5c). Although quenching was observed in case of FlrCC-Y290A, the curve did not reach saturation, which essentially points toward low affinity or weak binding (Fig. S3, c and d).
Increased concentrations of c-di-GMP abrogated ATP hydrolysis of FlrCC
We have investigated the effect of c-di-GMP on the ATPase activity of FlrCC (Fig. 5d). The variants were not tested because they hydrolyze ATP poorly (Fig. 1, d and f). 1 μm FlrCC was incubated with varying concentrations of c-di-GMP (as mentioned in Fig. 5d) for 30 min. ATP of 0.1 mm was added and incubated for another 25 min. Release of Pi was monitored by a Malachite green assay and plotted against the c-di-GMP concentration (Fig. 5d). The results showed that an increase in c-di-GMP concentration reduces ATPase activity of FlrCC. Although 30% reduction in ATPase activity was witnessed up to 20 μm c-di-GMP, a sudden reduction of 70% activity was observed at c-di-GMP of 25 μm (Fig. 5d). No significant reduction was witnessed with a further increase in the c-di-GMP concentration. Reduction in activity increased to 85% after that with a c-di-GMP concentration as high as 70 μm. Previous results demonstrated that significant reduction in ATPase activity of FlrCC is caused in the presence of 25-fold molar excess of c-di-GMP. FlrC plays a crucial role in flagellar synthesis and our results were corroborated with a previous finding that suggested inhibition of flagellar synthesis of V. cholerae by a high concentration of c-di-GMP (9).
c-di-GMP attenuates ATPase activity at low FlrCC concentration
Prompted by the results of reduction in ATPase activity with increased c-di-GMP concentrations (Fig. 5d) and concentration-dependent oligomerization of FlrCC (Fig. 5a), we felt it judicious to check the effect of c-di-GMP on ATPase activity at lower concentrations of the protein. Based on the observations of Fig. 5a, 500 and 300 nm FlrCC were treated with 20- and 25-fold molar excess of c-di-GMP separately. Measurement of ATPase activity revealed that whereas 20-fold molar excess of c-di-GMP caused only 30% reduction in activity of 1 μm FlrCC (Fig. 5d), the extent of reduction increased to 55 and 60% with FlrCC concentrations of 500 and 300 nm, respectively (Fig. 5e). c-di-GMP of 25-fold molar excess, however, caused 70% reduction in activity at protein concentrations of 500 nm, whereas reduction was of 85% with 300 nm protein. This observation certainly pointed to the fact that impact of c-di-GMP on reduction of ATPase activity of FlrCC is more pronounced at lower concentrations of the protein.
Heptameric assembly of FlrCC is destabilized upon dilution and/or c-di-GMP binding
In view of previous observations, we carried out qualitative experiments to investigate the impact of dilution and influence of c-di-GMP on the stability of heptameric assembly. SEC experiments were performed with three sets of samples as indicated in Fig. 5f. A right shift of peak II was evident upon dilution when panel 1 of Fig. 5f (FlrCC concentration, 40 μm) was compared with Fig. 2a (FlrCC concentration, 360 μm). In this study, dilution during SEC was the basis of correlation between concentration-dependent ATPase activity (Fig. 5a) and dilution-dependent destabilization of heptameric assembly (Fig. 5f). ∼40-Fold dilution of a protein of 40 μm reduced the effective concentration to 1 μm (or even lesser, considering soluble aggregate, peak I), which was of ∼9 μm in the previous run (Fig. 2a). Right shift of peak II (Figs. 2, b, and 5, f and g), indicate that destabilization of heptameric assembly is manifested at ∼1 μm. This observation is in accordance with the previous results, where we have seen saturation in specific activity at or above protein concentration of 1 μm (Fig. 5a). Further right shift of peak II was observed with further half-dilution, which was increased upon addition of c-di-GMP (panels 2 and 3 of Fig. 5, f and g). These observations underscored destabilization of heptameric assembly at low concentrations of FlrCC, which was facilitated by excess c-di-GMP (Fig. 5, f and g). Abrogation of the functional oligomeric state was observed for FleQ of P. aeruginosa, where the protein was locked in an altered loss-of-function trimeric state in the presence of c-di-GMP (28). Our observations (Fig. 5, a, e, and f), therefore, intended us to hypothesize that high concentrations of c-di-GMP negatively regulate FlrC by destabilizing the heptameric assembly and this effect is more pronounced when the protein is present in low concentrations.
Sequence analysis of FlrC in Vibrio species and monotrichous Gram-negative bacteria
Vibrio species were searched for FlrC and/or similar regulatory proteins. Apart from V. cholerae, we have identified 19 other monotrichous Vibrio species namely, Vibrio mimicus, Vibrio diazotrophicus, Vibrio alginolyticus, Vibrio campbelii, Vibrio harveyi, Vibrio nereis, Vibrio rotiferianus, Vibrio nigripulchritudo, Vibrio anguillarum, Vibrio caribbeanicus, Vibrio tubiashii, Vibrio fluvialis, Vibrio gazogenes, Vibrio mediterranei, Vibrio tapetis, Vibrio aerogenes, Vibrio azureus, Vibrio galatheae, and Vibrio xiamenesis, which possess a two-component system of FlrBC for flagellar synthesis. Multiple sequence alignment depicted strict sequence conservation of different functional residues and motifs such as Walker A, sensor I, sensor II, and RNAP-σ54/promoter-binding region of Loop 1 in FlrCs of these Vibrio species (Fig. S4). Moreover, the amino acids involved in inter-protomeric contacts are all highly conserved in these Vibrio species (Fig. S4). The extent of sequence conservation intended us to envisage a similar mechanism of oligomerization of FlrC to regulate ATPase activity, presumably in a cis-mediated fashion, and of interactions with the RNAP-σ54-promoter complex in case of these Vibrio species.
The two-component systems of FlrB/FlrC (observed in V. cholerae) or FleS/FleR (observed in P. aeruginosa) are necessary for σ54-dependent activation of class-III flagellar genes (5, 6, 8). Hence, we have searched for different organisms possessing any one of such two-component systems. We have identified 18 other monotrichous Gram-negative bacteria that possess flagellar regulatory proteins FlrB/FlrC or FleS/FleR for flagellar synthesis. Multiple sequence alignment with FlrC and FleR of all 19 organisms showed that important functional residues of the AAA+ domain are mostly conserved (Fig. 6a, Fig. S5). Arg349 of sensor II is strictly conserved (Fig. 6a). Although Arg291 of sensor I is also conserved, Tyr290 is replaced by Phe in a couple of organisms (Fig. 6a). Nonetheless, hydrophobic packing with the conserved Arg corresponding to Arg349 should be unaffected upon Tyr to Phe mutation (validated in silico) at this position of sensor I. A cladogram generated with these 19 sequences clustered FlrC and FleR separately. FlrCs of Colwellia psychrerythraea and Shewanella oneidensis were identified as close relatives of V. cholerae (Fig. 6b).
Figure 6.

Sequence analysis and cladogram of monotrichous Gram-negative bacteria. a, selected functional regions of consensus sequence logo generated (using Skylign by followed by ClustalW alignment) from FlrC and similar flagellar regulatory protein sequences of 19 monotrichous Gram-negative bacteria are shown here. Residue numbering is as per FlrC of V. cholerae. b, the cladogram of the same set of protein sequences mentioned in a is generated by MUSCLE (32) that uses Neighbor-joining method after ClustalW alignment.
Discussion
FlrCC, the bEBP that controls transcription of class-III flagellar genes, forms heptamer in Nt-independent manner and exhibits unique cis-mediated ATP binding (20). These atypical features of FlrC stimulated a question whether heptamerization is solely required for interaction with the RNAP-σ54 complex or has any definite role on ATPase activity as well. Our current study that dealt with the effect of crucial mutations on oligomerization, ATPase activity, and structural perturbation, ascertained that heptamerization of FlrCC occurs through concentration-dependent cooperativity to accomplish full ATPase activity. Excess second messenger c-di-GMP, on the other hand, represses the activity by destabilizing the assembly structure (Fig. 7).
Figure 7.

Schematic representation of proposed mechanism. Hydrophobic packing of trans-acting Tyr290 with cis-acting Arg349 is pivotal in maintaining the heptameric state, which is required for ATPase activity. In the presence of c-di-GMP, FlrC loses heptameric state with abrogation of ATPase activity.
All three mutants, the cis-mutant FlrCC-R349A and two trans-mutants FlrCC-Y290A and FlrCC-R291A, showed partial or full destabilization of the heptameric state in solution (Fig. 2). Although FlrCC-Y290A favors a monomeric state (Fig. 2b), FlrCC-R349A and FlrCC-R291A display both heptameric and monomeric states in solution (Fig. 2, c–e), presumably in a concentration-dependent manner. In fact, disruption of the oligomeric state upon dilution is common in some oligomeric AAA+ motors, especially when they coexist in multiple states. Although as low as 2.5 μm FlrCC is sufficient to display full ATPase activities (at different ATP concentrations), all three variants are activity deficient under similar conditions (Fig. 1, d–f), seemingly because of the prevalence of monomeric state. We, therefore, propose that the heptameric state of FlrCC is essential for its ATPase activity and the protein cannot retain its activity in monomeric form.
As observed in the crystal structure of AMP-PNP–bound FlrCC, cis-acting Arg349 of sensor II stabilizes γ-phosphate of ATP (Fig. 1b) (20). Our current study delineated that Arg349 plays multifaceted roles through (i) hydrophobic packing with trans-acting Tyr290, (ii) maintaining the architecture of Walker A, and (iii) stabilizing γ-phosphate of ATP. In the absence of the Arg349 side chain, Walker A experienced a drastic conformational shift pertaining to encroachment of the ATP-binding pocket in FlrCC-R349A (Fig. 3, b and d). Because of the loss of hydrophobic packing between cis-acting Arg349 and trans-acting Tyr290, FlrCC-R349A predominantly turned to monomeric species, especially in a low concentration (Fig. 2d). Furthermore, in the absence of Arg349, Walker A was not compelled to be in place to execute a restrained closed to open conformational shift, optimum for efficient ATP binding. The heptamer structure of FlrCC-R349A was likely formed through hydrophobic packing between trans-acting Tyr290 with Ala349 at supersaturation. Despite that, an unusual shift of Walker A was manifested, which implies that the Arg349 side chain plays the key role to keep Walker A in place (Fig. 3d).
Although the trans-acting motif 285REDXXYR291 had no direct interaction with bound AMP-PNP in FlrCC (20), our current study demonstrated that Tyr290 of this motif plays pivotal role in heptamerization. In the absence of Tyr290, the variant turned monomeric where cis-acting Arg349 was not compelled to orient toward the ATP-binding pocket. As a result, FlrCC-Y290A lacked affinity for the fresh ATP molecule and was found to hold the product ADP at the ATP-binding site (Fig. 4). The variant FlrCC-R291A, on the other hand, loses ATPase activity in the diluted state. Absence of Arg291 side chain presumably provides extra conformational freedom to neighboring Tyr290. If Arg349 is not oriented properly to the ATP-binding pocket in the absence of trans-acting Tyr290 (Fig. 4c, Fig. S1), or due to altered conformation and/or flexibity of Tyr290 (which may be the case in FlrCC-R291A), the protein experiences no compulsion to bind ATP, resulting in impaired ATP hydrolysis, which is evident from time course ATPase assays (Fig. 1, d–f). Absence or flexibility of Tyr290 is, therefore, detrimental for spontaneous oligomerization of FlrCC. In the absence of the heptameric structure, recognition of ATP and synergic contribution of the switches (20), which are the crucial factors for cis-mediated ATP binding and hydrolysis, are compromised severely.
Apart from showing the essentiality of the heptameric structure of FlrCC, our results unveiled the mechanism of heptamerization of this bEBP. The sigmoidal pattern of the specific activity plot of FlrCC pointed toward a concentration-dependent oligomerization of FlrCC (Fig. 5a). Furthermore, a significant increase in the Km value upon dilution of FlrCC from 1.2 μm to 600 nm (Fig. 5b) suggested that destabilization of the heptameric state upon dilution resulted in weak ATP binding and hydrolysis. Dependence of oligomerization on protein concentration was previously observed in NtrC1 (17). But in that case ATP was found to play another lead role, in subunit remodeling. The mechanism of cooperative oligomerization of FlrC, which is independent of nucleotide, has been articulated for the first time. Fig. 5a indicates that elevation of the FlrCC concentration above a certain threshold indulges formation of heptameric assembly in a cooperative manner (promoting highest ATPase activity above 1 μm). Results of SEC experiments further corroborated this hypothesis because destabilization of heptameric assembly occurs (right shift of peak II in Fig. 5f) upon dilution of the protein below 1 μm concentration.
For the first time, we have shown significant reduction in ATPase activity of FlrCC by excess c-di-GMP and the effect is more pronounced at low concentrations of the protein (Fig. 5, d and e). This result naturally generates a question whether c-di-GMP inhibits ATPase activity through competitive inhibition or the effect is indirect, through destabilization of the essential heptameric structure. In the case of FleQ of P. aeruginosa, the c-di-GMP–binding site is not identical to the ATP-binding site (although both sites are near Walker A) and c-di-GMP traps the AAA+ domain of FleQ in an alternative nonfunctional trimeric state, leading to ATPase inhibition (28). A close inspection of the FlrCC structure suggested that in the heptameric form this bEBP has insufficient space to accommodate c-di-GMP at the ATP-binding site. Notably, FlrCC-R349A, which was predominantly observed as monomer in solution, showed stronger binding of c-di-GMP compared with FlrCC (Fig. 5c). Tighter binding of c-di-GMP to FlrCC-R349A compared with FlrCC might be attributed to the ease of transformation to the monomeric state that provides more space to accommodate c-di-GMP near Walker A. In other words, monomeric conformation of FlrCC is more suitable for c-di-GMP binding and therefore, the presence of excess c-di-GMP triggers destabilization of the heptameric structure especially at the low concentration of this bEBP (Fig. 5f). Despite that FlrCC-Y290A primarily acquires a monomeric state in solution, bound ADP turns Walker A rigid and makes it less susceptible to c-di-GMP binding. In that case, a significantly high concentration of c-di-GMP might be required to outcompete ADP, as indicated by Fig. S3.
V. cholerae elevates c-di-GMP expression to down-regulate transcription of flagellar genes during biofilm formation (10). Binding of c-di-GMP to FlrA abrogates its interactions with the promoter of the flrBC operon thus down-regulating expression of flrBC as well as downstream flagellar genes (11). In such a situation, production of FlrC would be compromised and effectively the concentration of FlrC will be diminished in the cell. We propose that c-di-GMP at an elevated concentration will be able to inactivate residual FlrC, present in low concentrations in the Vibrio cells.
In NtrC, ATP-dependent conversion from an inactive dimer to active hexa/heptamer acts as regulator of function. In contrast, FlrC is intrinsically heptameric without Nt-dependent remodeling. We, therefore, hypothesize that the rate of production of FlrC may act as a regulatory factor for downstream flagellar gene expression. Repression of FlrC by c-di-GMP is the second line of action in transcription inhibition of class-III flagellar genes. In brief, whereas spontaneous heptamerization of FlrC escalates quick expression of class-III genes during flagellar synthesis, disassembling of heptamer at low concentrations and in the presence of excess c-di-GMP inhibit transcription of flagellar genes during biofilm formation. Based on sequence and phylogenetic analysis, we have proposed similar mechanisms in several other Vibrio species and some monotrichous Gram-negative bacteria that possess a FlrBC system. Further investigations in this direction will enrich knowledge on the uniqueness of the bEBPs involved in flagellar synthesis of pathogenic bacteria.
Experimental procedures
Cloning, overexpression, and purification
FlrCC-Y290A and FlrCC-R291A were prepared by two-step PCR amplification using FlrCC (aa 132-381 of accession code: A0A0H3AHP1) as template and cloned in pET28a+ vector (Novagen) using NdeI and BamHI as restriction sites. Sequences of the variants were verified by commercial sequencing. For overexpression, a single colony was transferred into 10 ml of LB broth and grown overnight at 310 K. 1 liter of LB broth was inoculated overnight with a 10-ml culture and the culture was grown at 310 K until the OD600 reached 0.6. The cells were induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside and grown at 310 K for 3 h. The cells were then harvested at 4500 × g for 20 min at 277 K and the pellet was resuspended in 10 ml of ice-cold lysis buffer-L (of composition 50 mm Tris–HCl, pH 8.0, 300 mm NaCl, 5 mm MgCl2, 10% (v/v) glycerol). Phenylmethylsulfonyl fluoride (1 mm) and lysozyme (1 mg/ml) were added to the resuspended solution, and the solution was lysed by sonication on ice. The cell lysate was then centrifuged at (12,000 rpm for 60 min) at 277 K. The collected supernatant was applied onto a nickel–nitrilotriacetic acid affinity chromatography media (Qiagen) that was previously equilibrated with buffer-L. The His6-tagged recombinant proteins were eluted with the buffer containing 70-200 mm imidazole. Imidazole was removed through buffer exchange using Amicon centrifugation units. FlrCC-Y290A and FlrCC-R291A were concentrated up to 506 and 451 μm, respectively. FlrCC-R349A was purified with the same protocol and concentrated up to 158 μm. FlrCC was also purified up to a concentration of 652 μm. Protein concentrations were measured by Braford assay using BSA as standard. For all constructs, concentration was reported for monomer. The homogeneity of the purified proteins was checked using SDS-PAGE with 12% polyacrylamide concentration.
ATPase assay
ATPase activities of FlrCC and the variants were determined spectrophotometrically by measuring the release of Pi by the Malachite green assay, as described in Dey et al. (20). Reaction mixtures having 2.5 μm protein and buffer containing 50 mm Tris-HCl (pH 8.0), 300 mm NaCl, and 5 mm MgCl2 were incubated with ATP (Sigma Aldrich) ranging from 0.1 to 0.5 mm at 298 K for different time periods in a reaction volume of 1 ml. Freshly prepared colored reagent Malachite green that contains 10 ml of 0.44 g of Malachite green dissolved in 0.3 m H2SO4, 2.5 ml of 7.5% ammonium molybdate, and 0.2 ml of 11% Tween 20 was added to the reaction mixture, and absorbance was measured at 630 nm within 5 min of adding the coloring reagent. Pi standard curve was prepared by using KH2PO4 and plotting OD630 against release of Pi using OriginPro 8.0 software. Each protein was tested with Malachite green without ATP to measure the contaminant Pi if any, and the negligible absorbance thus obtained at 630 nm was subtracted from the absorbance produced by that protein upon hydrolysis of the added ATP. Similarly, the ATP hydrolysis without protein was judged to nullify the effect of the contaminating Pi with ATP. To check the effect of c-di-GMP on ATPase activity of FlrCC, we followed the same method where the protein concentration was 1 μm. All the experiments were minimally performed in triplicate.
Gel filtration assay
Size exclusion chromatography experiments were performed in a Superdex 200 increase column 10/300 attached with an AKTA purifier (GE Healthcare Life Sciences).
The peak fractions were collected for SDS-PAGE analysis. The column was calibrated with standard molecular weight calibration kit (GE Healthcare) of blue dextran, thyroglobulin (660 kDa), ferritin (440 kDa), aldolase (158 kDa), ovalbumin (43 kDa), and lysozyme (14.3 kDa). The standard graph was prepared against relative elution volume (Ve/Vo) in the x axis (where Ve is the elution volume and Vo is the void volume) and the log molecular weight in the y axis. For analysis of FlrCC and the variants, in each run, 100 μl of protein was injected to the SEC column pre-equilibrated with buffer containing 50 mm Tris-HCl (pH 8.0), 300 mm NaCl, and 5 mm MgCl2 and run at a flow rate of 0.5 ml/min. Protein concentrations were as follows: 360 μm for FlrCC and FlrCC-R291A, 500 μm for FlrCC-Y290A, and 158 μm for FlrCC-R349A. The peak fractions were collected and analyzed in SDS-PAGE.
Dynamic light scattering
Dynamic light scattering measurements with FlrCC and FlrCC-variant were performed by Malvern Zetasizer Nano S instrument (Malvern, Herremberg, Germany) equipped with a 633 nm He–Ne laser and operating at an angle of 173°. Data were collected for a 1-ml sample volume using a quartz cuvette of 1-cm path length at 298 K, with protein concentrations of 30 μm in the buffer containing 50 mm Tris-HCl (pH 8.0), 300 mm NaCl, and 5 mm MgCl2. Protein samples were centrifuged at 10,000 rpm for 2 min before the experiment. The dynamic light scattering results are expressed in terms of size distribution by volume and plotted against hydrodynamic radius. The graph was plotted in OriginPro 8.0 software.
Crystallization, diffraction data collection, and refinement
Initial crystallization trials were performed by the hanging drop vapor diffusion methods in 24-well crystallization trays using PEG 6000 Grid Screen of Hampton Research, USA. Optimal diffraction quality crystals of FlrCC-R349A were obtained by vapor diffusion with precipitant containing 10% (w/v) PEG6000, in 0.1 m HEPES (pH 7.0) and 3% glycerol after 48 h incubation. Diffraction quality crystals of FlrCC-Y290A were obtained with 5% (w/v) PEG6000 after overnight incubation. All the crystals were obtained at 293 K. Crystals were soaked in the cryo-protecting solution made of 40% (v/v) ethylene glycol in mother liquor and flash-frozen in liquid nitrogen.
Crystals were tested for diffraction at Elettra, Italy, and RRCAT, India. X-ray diffraction data up to 3.1 and 3.45 Å resolutions, collected for FlrCC-R349A and FlrCC-Y290A, respectively, at RRCAT, India, have been reported here.
Coordinates of Nt-free FlrCC (PDB code 4QHS) without water and other solvents were used as starting model for refinement. Interestingly, both variants showed heptameric assembly in the crystal structure. Rigid body refinement, followed by positional and TLS refinements, and water picking (in case of FlrCC-R349A) were performed with PHENIX (30). Few cycles of model fitting were performed in between using Coot (31). Seven-fold NCS was used at the initial level of both the refinements. Coordinates of the refined models of FlrCC-R349A and FlrCC-Y290A were submitted to the Protein Data Bank with PDB codes 6LUA and 6LUF, respectively.
Fluorescence quenching study
Fluorescence measurements were carried out using a spectrofluorometer, Hitachi F-7000 using quartz cuvettes of 1-cm path length. Changes in tryptophan fluorescence were measured at a fixed excitation wavelength of 295 nm, and the emission spectra were recorded between 300 and 400 nm with 5-nm band pass slits for both excitation and emission channels at 298 K. For all proteins, the final concentrations were 5 μm in buffer-L, and the changes in fluorescence emission intensity were measured in the presence fluorescence of increasing concentrations of c-di-GMP as ligand. Maximum fluorescence (ΔFmax) value and apparent dissociation constant (Kd) for protein ligand interactions were determined using nonlinear regression with “one site-specifc binding” model (ΔF = ΔFmax × X/(Kd + X), where Y is the change in fluorescence intensity (ΔF), X is the ligand concentration, ΔFmax is the saturating value for maximum fluorescence intensity change upon binding saturation with ligand and Kd is the apparent dissociation constant. Fluorescence changes (ΔF) from three independent experiments were analyzed and fitted according to above equation using the OriginPro 8.0 software. Errors were measured as standard deviations.
Sequence analysis and generation of cladogram
Sequence analysis were carried out using the programs MULTALIN and ClustalW. The cladogram with the FlrC/FleR sequences of monotrichous Gram-negative bacteria were generated by MUSCLE (32) that uses Neighbor-joining method after ClustalW.
Data availability
Coordinates and structure factor files of the crystal structures of FlrCC-R349A and FlrCC-Y290A have been deposited in the Protein Data Bank with the data set identifiers 6LUA and 6LUF, respectively. All other data are contained within the manuscript.
Supplementary Material
Acknowledgments
We thank Prof. Udayaditya Sen of SINP, Kolkata, India, for support and advice at various stages of this work. We also thank BL-21 beamline scientists Dr. Ravindra Makde and Dr. Ashwani Kumar for providing support during data collection at PX-BL21 beamline (BARC) at Indus-2 Synchrotron, India. We thank Prof. B. Gopal of IISc, Bangalore, India, for his cordial support in sending crystals to Elettra. We are also thankful to Dr. Babu A. Manjasetty for his support during crystal screening at Elettra. We thank Kamalendu Pal of SINP, Kolkata, for help during gel filtration experiments. We are also thankful to Sritapa Basu Mallick for assistance in sequence alignment and phylogenetic analysis. We are grateful to Rev. Dr. Dominic Savio, SJ, Principal, St. Xavier's College, Kolkata, for his constant support and encouragement.
This article contains supporting information.
Author contributions—S. C. and B. G. data curation; S. C. and S. A. formal analysis; S. C. and B. G. validation; S. C., M. B., S. D., S. A., and T. C. investigation; S. C., M. B., S. D., and T. C. methodology; B. G. software; J. D. conceptualization; J. D. supervision; J. D. funding acquisition; J. D. writing-original draft; J. D. project administration.
Funding and additional information—This work was supported by DBT Grant BT/03/IYBA/2010 of Govt. of India, DST Grant SR/FST/COLLEGE-014/2010(C), and WBDBT BOOST Grant 335/WBBDC/1P-2/2013 for infrastructural support.
Conflict of interest—The authors declare that they have no conflicts of interest related to the contents of this article.
- bEBP
- bacterial enhancer binding protein
- AAA+
- ATPases associated with various cellular activities
- Nt
- nucleotide
- PDB
- Protein Data Bank
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- c-di-GMP
- cyclic di-guanosine monophosphate
- SEC
- size exclusion chromatography
- R
- regulator
- RMSD
- root mean square deviation.
References
- 1. Holmgren, J., and Svennerholm, A.-M. (1977) Mechanisms of disease and immunity in cholera: a review. J. Infect. Dis. 136, S105–S112 10.1093/infdis/136.Supplement.S105 [DOI] [PubMed] [Google Scholar]
- 2. Taylor, R. K., Miller, V. L., Furlong, D. B., and Mekalanos, J. J. (1987) Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. U.S.A. 84, 2833–2837 10.1073/pnas.84.9.2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yildiz, F. H., and Visick, K. L. (2009) Vibrio biofilms: so much the same yet so different. Trends Microbiol. 17, 109–118 10.1016/j.tim.2008.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Syed, K. A., Beyhan, S., Correa, N., Queen, J., Liu, J., Peng, F., Satchell, K. J. F., Yildiz, F., and Klose, K. E. (2009) The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J. Bacteriol. 191, 6555–6570 10.1128/JB.00949-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prouty, M. G., Correa, N. E., and Klose, K. E. (2001) The novel σ54- and σ28-dependent flagellar gene transcription hierarchy of Vibrio cholerae. Mol. Microbiol. 39, 1595–1609 10.1046/j.1365-2958.2001.02348.x [DOI] [PubMed] [Google Scholar]
- 6. Klose, K. E., and Mekalanos, J. J. (1998) Distinct roles of an alternative sigma factor during both free-swimming and colonizing phases of the Vibrio cholerae pathogenic cycle. Mol. Microbiol. 28, 501–520 10.1046/j.1365-2958.1998.00809.x [DOI] [PubMed] [Google Scholar]
- 7. Echazarreta, M. A., and Klose, K. E. (2019) Vibrio flagellar synthesis. Front. Cell. Infect. Microbiol. 9, 131 10.3389/fcimb.2019.00131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Correa, N. E., and Klose, K. E. (2005) Characterization of enhancer binding by the Vibrio cholerae flagellar regulatory protein FlrC. J. Bacteriol. 187, 3158–3170 10.1128/JB.187.9.3158-3170.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conner, J. G., Zamorano-Sánchez, D., Park, J. H., Sondermann, H., and Yildiz, F. H. (2017) The ins and outs of cyclic di-GMP signaling in Vibrio cholerae. Curr. Opin. Microbiol. 36, 20–29 10.1016/j.mib.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beyhan, S., Tischler, A. D., Camilli, A., and Yildiz, F. H. (2006) Transcriptome and phenotypic responses of Vibrio cholerae to increased cyclic di-GMP level. J. Bacteriol. 188, 3600–3613 10.1128/JB.188.10.3600-3613.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Srivastava, D., Hsieh, M. L., Khataokar, A., Neiditch, M. B., and Waters, C. M. (2013) Cyclic di-GMP inhibits Vibrio cholerae motility by repressing induction of transcription and inducing extracellular polysaccharide production. Mol. Microbiol. 90, 1262–1276 10.1111/mmi.12432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shikuma, N. J., Fong, J. C. N., and Yildiz, F. H. (2012) Cellular levels and binding of c-di-GMP control subcellular localization and activity of the Vibrio cholerae transcriptional regulator VpsT. PLoS Pathog. 8, e1002719 10.1371/journal.ppat.1002719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Correa, N. E., Lauriano, C. M., McGee, R., and Klose, K. E. (2000) Phosphorylation of the flagellar regulatory protein FlrC is necessary for Vibrio cholerae motility and enhanced colonization. Mol. Microbiol. 35, 743–755 10.1046/j.1365-2958.2000.01745.x [DOI] [PubMed] [Google Scholar]
- 14. Xu, H., and Hoover, T. R. (2001) Transcriptional regulation at a distance in bacteria. Curr. Opin. Microbiol. 4, 138–144 10.1016/S1369-5274(00)00179-X [DOI] [PubMed] [Google Scholar]
- 15. Jyot, J., Dasgupta, N., and Ramphal, R. (2002) FleQ, the major flagellar gene regulator in Pseudomonas aeruginosa, binds to enhancer sites located either upstream or atypically downstream of the RpoN binding site. J. Bacteriol. 184, 5251–5260 10.1128/jb.184.19.5251-5260.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bush, M., and Dixon, R. (2012) The role of bacterial enhancer binding proteins as specialized activators of σ54-dependent transcription. Microbiol. Mol. Biol. Rev. 76, 497–529 10.1128/MMBR.00006-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen, B., Sysoeva, T. A., Chowdhury, S., Guo, L., De Carlo, S., Hanson, J. A., Yang, H., and Nixon, B. T. (2010) Engagement of arginine finger to ATP triggers large conformational changes in NtrC1 AAA+ ATPase for remodeling bacterial RNA polymerase. Structure 18, 1420–1430 10.1016/j.str.2010.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sysoeva, T. A., Chowdhury, S., Guo, L., and Tracy Nixon, B. (2013) Nucleotide-induced asymmetry within ATPase activator ring drives σ54–RNAP interaction and ATP hydrolysis. Genes Dev. 27, 2500–2511 10.1101/gad.229385.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Joly, N., Zhang, N., and Buck, M. (2012) ATPase site architecture is required for self-assembly and remodeling activity of a hexameric AAA+ transcriptional activator. Mol. Cell 47, 484–490 10.1016/j.molcel.2012.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dey, S., Biswas, M., Sen, U., and Dasgupta, J. (2015) Unique ATPase site architecture triggers cis-mediated synchronized ATP binding in heptameric AAA+-ATPase domain of flagellar regulatory protein FlrC. J. Biol. Chem. 290, 8734–8747 10.1074/jbc.M114.611434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joly, N., and Buck, M. (2010) Engineered interfaces of an AAA+-ATPase reveal a new nucleotide-dependent coordination mechanism. J. Biol. Chem. 285, 15178–15186 10.1074/jbc.M110.103150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rappas, M., Schumacher, J., Niwa, H., Buck, M., and Zhang, X. (2006) Structural basis of the nucleotide driven conformational changes in the AAA+ domain of transcription activator PspF. J. Mol. Biol. 357, 481–492 10.1016/j.jmb.2005.12.052 [DOI] [PubMed] [Google Scholar]
- 23. Dey, S., and Dasgupta, J. (2013) Purification, crystallization and preliminary X-ray analysis of the AAA+ σ54 activator domain of FlrC from Vibrio cholerae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 69, 800–803 10.1107/S1744309113015613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geladopoulos, T. P., Sotiroudis, T. G., and Evangelopoulos, A. E. (1991) A malachite green colorimetric assay for protein phosphatase activity. Anal. Biochem. 192, 112–116 10.1016/0003-2697(91)90194-X [DOI] [PubMed] [Google Scholar]
- 25. Buckstein, M. H., He, J., and Rubin, H. (2008) Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J. Bacteriol. 190, 718–726 10.1128/JB.01020-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bhate, M. A. P., Molnar, K. A. S., Goulian, M., and Degrado, W. F. (2015) Signal transduction in histidine kinases: insights from new structures. Structure 23, 981–994 10.1016/j.str.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee, S. Y., De La Torre, A., Yan, D., Kustu, S., Nixon, B. T., and Wemmer, D. E. (2003) Regulation of the transcriptional activator NtrC1: structural studies of the regulatory and AAA+ ATPase domains. Genes Dev. 17, 2552–2563 10.1101/gad.1125603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsuyama, B. Y., Krasteva, P. V., Baraquet, C., Harwood, C. S., Sondermann, H., and Navarro, M. V. A. S. (2016) Mechanistic insights into c-di-GMP-dependent control of the biofilm regulator FleQ from Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 113, E209–E218 10.1073/pnas.1523148113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trampari, E., Stevenson, C. E. M., Little, R. H., Wilhelm, T., Lawson, D. M., and Malone, J. G. (2015) Bacterial rotary export ATPases are allosterically regulated by the nucleotide second messenger cyclic-di-GMP. J. Biol. Chem. 290, 24470–24483 10.1074/jbc.M115.661439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams, P. D., Afonine, P. V., Bunkóczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L. W., Kapral, G. J., Grosse-Kunstleve, R. W., McCoy, A. J., Moriarty, N. W., Oeffner, R., Read, R. J., Richardson, D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emsley, P., and Cowtan, K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 32. Edgar, R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factor files of the crystal structures of FlrCC-R349A and FlrCC-Y290A have been deposited in the Protein Data Bank with the data set identifiers 6LUA and 6LUF, respectively. All other data are contained within the manuscript.