

Abstract
HIV Type 1 (HIV-1) and simian immunodeficiency virus (SIV) display differential replication kinetics in macrophages. This is because high expression levels of the active host deoxynucleotide triphosphohydrolase sterile α motif domain and histidine-aspartate domain–containing protein 1 (SAMHD1) deplete intracellular dNTPs, which restrict HIV-1 reverse transcription, and result in a restrictive infection in this myeloid cell type. Some SIVs overcome SAMHD1 restriction using viral protein X (Vpx), a viral accessory protein that induces proteasomal degradation of SAMHD1, increasing cellular dNTP concentrations and enabling efficient proviral DNA synthesis. We previously reported that SAMHD1-noncounteracting lentiviruses may have evolved to harbor RT proteins that efficiently polymerize DNA, even at low dNTP concentrations, to circumvent SAMHD1 restriction. Here we investigated whether RTs from SIVmac239 virus lacking a Vpx protein evolve during in vivo infection to more efficiently synthesize DNA at the low dNTP concentrations found in macrophages. Sequence analysis of RTs cloned from Vpx (+) and Vpx (−) SIVmac239–infected animals revealed that Vpx (−) RTs contained more extensive mutations than Vpx (+) RTs. Although the amino acid substitutions were dispersed indiscriminately across the protein, steady-state and pre-steady-state analysis demonstrated that selected SIVmac239 Vpx (−) RTs are characterized by higher catalytic efficiency and incorporation efficiency values than RTs cloned from SIVmac239 Vpx (+) infections. Overall, this study supports the possibility that the loss of Vpx may generate in vivo SIVmac239 RT variants that can counteract the limited availability of dNTP substrate in macrophages.
Keywords: lentiviruses, reverse transcriptase, enzyme kinetics, Vpx, macrophages, dNTPs, lentivirus, nucleotide, sterile α motif domain and histidine-aspartate domain–containing protein 1 (SAMHD1)
During their pathogenesis, lentiviruses such as HIV type 1 (HIV-1), HIV-2, and simian immunodeficiency virus (SIV) infect both dividing CD4+ T cells and terminally differentiated/nondividing myeloid cells such as macrophages and microglia (1–4). Although sharing a selective cellular tropism, cell-dependent replication kinetics differ among the viruses because HIV-1 replication kinetics are delayed in nondividing cell populations (5). Slowed replication kinetics in macrophage and microglial populations, particularly in the brain, support the persistent production of HIV-1 at low levels (6). This is in stark contrast to the robust replication of HIV-1 in CD4+ T cells, which leads to rapid cell death. Whereas dividing cells, like activated CD4+ T cells, undergo dNTP biosynthesis in S phase, terminally differentiated cells like macrophages have no necessity to support chromosomal DNA replication or mitotic division and are thus characterized by lower dNTP pools (7, 8). Indeed, cellular dNTP concentrations in human primary monocyte–derived macrophages (20–40 nm) are 100–250× lower than those found in activated/dividing CD4+ T cells (2–5 μm) (8). Recent studies have revealed that host deoxynucleotide triphosphohydrolase (dNTPase) sterile α motif (SAM) domain and histidine-aspartate (HD) domain–containing protein 1 (SAMHD1), which is active and highly expressed in macrophages, is responsible for the depletion of cellular dNTPs within this nondividing target cell type (9–12). Although macrophages are not refractory to HIV-1 infection, the SAMHD1-mediated limited substrate availability for RT during viral replication kinetically restricts HIV-1 proviral DNA synthesis in macrophages (13, 14). Conversely, HIV-2 and some SIV strains replicate rapidly in this nondividing myeloid cell type through the implementation of a virally encoded accessory protein called viral protein X (Vpx) (11, 15, 16). Lentiviral Vpx targets host SAMHD1 for proteasomal degradation through the E3 ubiquitination pathway, robustly reducing SAMHD1 protein levels and increasing intracellular dNTP concentrations in infected macrophages (17–19). In the absence of SAMHD1, abundant dNTP substrate accelerates reverse transcription during the viral replication cycle and enables rapid proviral DNA synthesis of SAMHD1-counteracting viruses (20). Because Vpx arose from a gene duplication event of accessory protein viral protein R (Vpr) (21, 22), Vpr proteins of some SIV strains lacking Vpx (e.g. SIVagm677, SIVagm9648, SIVdeb, and SIVmus1) also possess the ability to target SAMHD1 for proteasomal degradation through the same pathway hijacked by Vpx (23–25). However, the Vpr proteins of SAMHD1-noncounteracting strains such as HIV-1 and SIVcpz are unable to induce degradation of their host SAMHD1 proteins (25, 26).
Unlike SAMHD1-counteracting viruses, including SIVmac239 and SIVagm677, which replicate under abundant cellular dNTP concentrations even in macrophages (20), SAMHD1-noncounteracting viruses such as HIV-1 and SIVcpz replicate within limited dNTP pools during the infection of this nondividing target cell type. Interestingly, recent studies have suggested that SAMHD1-noncounteracting lentiviral RTs have been evolutionarily honed to complete proviral DNA synthesis even at the low dNTP concentrations found in macrophages. We previously observed that RTs from SAMHD1-noncounteracting lentiviruses (e.g. HIV-1) were characterized by lower steady-state Km values and displayed faster pre-steady-state rates of dNTP incorporation (kpol) when compared with RTs from SAMHD1-counteracting lentiviruses (e.g. SIVmac239) (27, 28). Further pre-steady-state kinetic analysis revealed that SAMHD1-noncounteracting lentiviruses overcome low dNTP concentrations in macrophages through the use of RT proteins that execute a faster conformational change during the incorporation of an incoming nucleotide substrate (29). Although less effective than Vpx/Vpr-induced degradation of SAMHD1, this RT-mediated mechanism enables the slow, but complete, reverse transcription of the viral genome during infection of macrophages by SAMHD1-noncounteracting lentiviruses like HIV-1.
Given the influence of Vpx on viral reverse transcription and replication kinetics in macrophages, Westmoreland et al. (30) previously sought to characterize the cellular and tissue targets of a Vpx-deleted mutant of SIVmac239 (Vpx (−)) in infected Rhesus macaques. Apart from the 101-base deletion of the vpx gene, Vpx (−) mutant virus was identical to WT SIVmac239 (Vpx (+)) virus in this study (31, 32). Although macaques infected with Vpx (−) virus eventually developed AIDS with opportunistic infections and AIDS-defining lesions, Vpx (−)-infected animals (mean survival: 935.4 dpi) lived roughly 2.5× longer than those infected with Vpx (+) WT virus (mean survival: 364.3 dpi). Additionally, the group observed that viral replication in myeloid cells was drastically impaired in the absence of Vpx (30). Previous studies have shown that HIV-1 disease progression is associated with the appearance of viral variants that utilize an array of different coreceptors that enable the virus to infect a wider range of host cell types (33). These viral variants are speculated to arise from selective pressure exerted by the host immune system that drives HIV-1 evolution during infection. Similarly, steady-state and pre-steady-state studies of SAMHD1-noncounteracting RTs (HIV-1 RTs) have demonstrated that SAMHD1-noncounteracting primate lentiviruses might have evolved over time to harbor RTs that can more efficiently incorporate nucleotides at the low dNTP concentrations found in macrophages, which allows these lentiviruses to circumvent the antiviral selective pressure from dNTPase SAMHD1 (27–29).
In this study, we sought to investigate whether SIVmac239 RT, which is kinetically less efficient than RTs from SAMHD1-noncounteracting HIV-1 strains (27), undergoes enzymatic improvement in the absence of Vpx during pathogenesis to circumvent the SAMHD1 restriction in myeloid cells. For this, we biochemically characterized SIVmac239 RT variants cloned from adult Rhesus macaques infected with either Vpx (+) or Vpx (−) SIVmac239 virus and observed that Vpx (−) RT variants, encoding frequently identified amino acid mutations, displayed elevated steady-state catalytic efficiency (kcat/Km) and pre-steady-state incorporation efficiency (kpol/Kd) values when compared with Vpx (+) RTs. Overall, our sequence and kinetic analyses support the idea that RTs of SAMHD1-noncounteracting lentiviruses may evolve in vivo to better support proviral DNA synthesis in the SAMHD1-mediated low dNTP pools of target macrophage cell populations.
Results
Isolation and sequencing of SIVmac239 RT variants from Rhesus macaques infected with Vpx (+) WT and Vpx (−) mutant SIVmac239 viruses
We previously observed that RT of SAMHD1-counteracting SIVmac239 synthesizes DNA with low efficiency at macrophage-like dNTP concentrations, displaying lower steady-state catalytic efficiency (kcat/Km) values when compared with RTs of SAMHD1-noncounteracting HIV-1 strains (27). We predicted that the lower enzymatic efficiency of RTs originating from SAMHD1-counteracting lentiviruses (e.g. SIVmac239) is an evolutionary consequence of natural viral replication occurring within high cellular dNTP concentrations, even in nondividing myeloid target cells, because of the virus-induced SAMHD1 proteasomal degradation. Therefore, here, we reasoned that if Vpx (−) mutant SIVmac239 replicates in animals, the viral RTs may be selectively honed during in vivo infections to contain mutations that can improve viral DNA synthesis efficiency, even in the low dNTP pools found in macrophages that result from the presence of dNTPase SAMHD1 and the absence of Vpx, much like HIV-1 RT (34).
To test this, we utilized samples previously collected from Rhesus macaques infected with either Vpx (+) WT or Vpx (−) mutant SIVmac239. We chose samples collected upon the development of AIDS characteristics in these infected animals (e.g. AIDS-defining lesions with opportunistic infections). To begin, we extracted RNAs from two samples originating from animals infected with Vpx (+) WT virus (27 and 29 weeks postinfection) and two samples from animals infected with Vpx (−) mutant virus (both 36 months postinfection (30)). Because these time points immediately precede the development of AIDS in the infected animals, the initially inoculated viruses underwent the entirety of in vivo evolution and host pathogenesis. In this near-terminal stage, viral titers were considerably elevated (30), even in animals infected with Vpx (−) mutant SIVmac239, which was convenient for our collection of the abundant viral RNA samples. Next, we conducted RT-PCR for the full-length reverse transcriptase gene and cloned the resulting amplicons to obtain plasmids containing Vpx (+) and Vpx (−) RT variants. The cloned RT plasmids were sequenced and compared with WT RT SIVmac239 nucleotide and amino acid sequences (GeneBank: AY588946.1). A few minor RT clones containing premature stop codons or frameshift mutations were discarded, and a total of 40 complete RT sequences were compiled for each animal sample: 80 clones for Vpx (−) infections and 80 clones for Vpx (−) infections. We first observed that nucleotide and amino acid mutations in both Vpx (+) and Vpx (−) RTs are indiscriminately distributed throughout the five RT functional and structural subdomains (i.e. the fingers, palm, thumb, connection, and RNase H domains) (Table 1). Interestingly, when analyzing the number of amino acid mutations present per cloned RT, we found that Vpx (−) RTs, on average, harbored about 2.6× more amino acid mutations than Vpx (+) RTs (Fig. 1A). In fact, when observing the distribution of the number of amino acid mutations present in each clone, the predominant number of mutations shifts from 1 in Vpx (+) RTs to 3–4 in Vpx (−) RTs (Fig. 1B). Consistent with this observation, we determined that more than 20 of 80 RT clones remained unmutated (WT) in the Vpx (+)-infected samples whereas only three of 80 RT clones showed WT amino acid sequences in the Vpx (−)-infected samples (Fig. 1B). The maximum number of amino acid mutations present in a single clone was eight for both groups, whereas the maximum number of nucleic acid mutations was 11 and 16 for Vpx (+) and Vpx (−) RTs, respectively. The data shown in Fig. 1 suggest that more mutations per cloned RT were found in Vpx (−) samples compared with Vpx (+) samples, which may simply result from Vpx (−)-infected animals experiencing infection periods ∼6× longer than Vpx (+)-infected animals (Vpx (−): 36 months; Vpx (+): 27–29 weeks).
Table 1.
Redundant amino acid mutations found in Vpx (+) and Vpx (−) RTs
| Mutation | RT subdomains | Number of clones (of 40) with noted mutation |
|||
|---|---|---|---|---|---|
| A1 Vpx (−) | A2 Vpx (−) | A3 Vpx (+) | A4 Vpx (+) | ||
| I2L | Fingers | 2 | - | - | - |
| V32I | Fingers | 1 | 21 | - | 1 |
| V32A | Fingers | - | - | 2 | 1 |
| R82K | Fingers | 1 | - | 1 | - |
| L100I | Palm | - | 2 | - | - |
| E122K | Fingers | - | - | 2 | 1 |
| I145M | Fingers | - | - | 1 | 2 |
| M164L | Palm | 1 | 38 | - | - |
| L196P | Palm | - | - | 1 | 1 |
| S211N | Palm | 24 | 9 | - | - |
| I212L | Palm | 2 | - | - | - |
| E218D | Palm | 23 | 2 | - | - |
| Q228R | Palm | - | - | 1 | 1 |
| M230I | Palm | - | - | 1 | 1 |
| R281K | Thumb | 5 | - | - | - |
| G285E | Thumb | - | 1 | 1 | - |
| G322D | Connection | - | 2 | - | - |
| P324L | Connection | - | 16 | - | - |
| I330V | Connection | - | 15 | - | - |
| V385I | Connection | 1 | - | 1 | - |
| K394R | Connection | 2 | 37 | - | 1 |
| E412G | Connection | 1 | - | 1 | - |
| S417P | Connection | - | - | 1 | 1 |
| S449T | RNaseH | - | 2 | - | - |
| T486I | RNaseH | 2 | 1 | - | - |
| V465I | RNaseH | - | - | 1 | 2 |
| A492V | RNaseH | 2 | - | 2 | - |
| C508R | RNaseH | 1 | - | 1 | 1 |
| E522K | RNaseH | 1 | 2 | 1 | - |
| E523K | RNaseH | 1 | 1 | - | - |
| I525S | RNaseH | 1 | 1 | - | - |
Figure 1.

Summary of amino acid mutations found in Vpx (+) and Vpx (−) RT variants. A, the average number of amino acid mutations per cloned RT was plotted for Vpx (+) (black) and Vpx (−) (blue) RTs, with error bars representing standard deviation. Statistical significance from an unpaired two-tailed student's t test is indicated as **** p < 0.0001. B, RT clones were grouped by the number of mutations present in the amino acid sequence. The distribution of the number of amino acid mutations present in Vpx (+) and Vpx (−) RTs are shown using black and blue lines, respectively.
Interestingly, whereas amino acid mutations M164L, S211N, and E218D are found in multiple clones from both Vpx (−) animals, a number of mutations, including K394R, A492V, and E522K, are seen in both Vpx (−) and Vpx (+) RT proteins (Table 1). Even though a greater redundancy in amino acid mutations is seen in Vpx (−) RTs, there are surprisingly six mutations exclusive to Vpx (+) RTs (Table 1). Overall, we found that if RT clones were identical in amino acid mutation profiles, differing mutations were observed at the gene levels (e.g. different codons for the same amino acid), thus supporting that redundancy in amino acid mutations is less likely the result of amplification during RT-PCR. Overall, these data reveal that although mutations are not isolated to one region of the enzyme, Vpx (−) RTs contain more mutations than Vpx (+) RTs at both the nucleic acid and amino acid levels.
Steady-state kinetic analysis of Vpx (+) and Vpx (−) RTs
Previous studies have shown that there are kinetic variations between RTs originating from lentiviruses with and without the ability to counteract SAMHD1 (27, 28). To broadly characterize the differential steady-state multinucleotide incorporation kinetics in Vpx (+)- and Vpx (−)-infected animals, we conducted a steady-state kinetic assay. Starting with the 40 RT clones that were collected and sequenced per animal (Table 1), we systematically chose 10 RT clones per animal with commonly identified mutations, proper protein expression in Esherichia coli, and close-to-WT RT purification yields to conduct multiple-nucleotide incorporation kinetic analysis in steady-state conditions. Ultimately, 20 Vpx (+) and 20 Vpx (−) RT variant proteins were chosen for enzymatic analysis. To determine the steady-state substrate efficiency (Vmax/Km) for each RT, we examined the RNA-dependent DNA polymerization activity of these purified RT proteins using a 39mer RNA template annealed to a 5′-32P-labeled 17mer DNA primer and varying concentrations of dNTPs (25 nm to 50 μm). RTs displaying substrate efficiency values lower than 0.1 × 10−5 s−1 were considered dead enzymes and were omitted from final quantifications. As summarized in Fig. 2, Vpx (−) RTs synthesized DNA from an RNA template with substrate efficiencies approximately 2-fold greater than Vpx (+) RTs, indicating that SIVmac239 RT might have improved its steady-state kinetic DNA synthesis efficiency during infection in the absence of Vpx.
Figure 2.

Steady-state kinetic measurement of Vpx (+) and Vpx (−) RT variants. Substrate efficiency (Vmax/Km) for Vpx (+) (black) and Vpx (−) (blue) RTs are represented using scatter dot plots with the gray lines signifying mean and standard deviation values. Statistical significance from an unpaired two-tailed student's t test is indicated as * p = 0.0182.
Steady-state single-nucleotide incorporation kinetic activity of two Vpx (−) RT variants
Interestingly, the initial kinetic analysis enabled the identification of two Vpx (−) RT variants with substrate efficiencies much higher than the mean substrate efficiency of Vpx (+) RTs (1.831 × 10−5 s−1): RTs 2G7 (6.781 × 10−5 s−1) and 2N0 (8.135 × 10−5 s−1). Relative to WT SIVmac239 RT, 2G7 and 2N0 variants both contain four amino acid mutations, two of which they share (M164L and K394R); however, they differ in the number of nucleic acid mutations present in their sequences (Table 2). Here, we conducted a more extensive kinetic characterization of these two SIVmac239 RT variants identified from Vpx (−) infections. First, considering that these two RTs contain multiple mutations, we tested whether these mutations alter their observed protein thermostability, which is indicative of overall protein stability (35, 36). As shown in Table 3, both RT variant proteins possess melting temperatures (Tm values) similar to that of WT SIVmac239 RT protein, indicating that the amino acid mutations present in 2G7 and 2N0 RT variants cause no significant abnormalities in protein folding or structure.
Table 2.
Amino acid mutations present in Vpx (−) clones 2G7, 2N0, and 1M6
| Clone | NTa | AAb | Mut 1 | Mut 2 | Mut 3 | Mut 4 | Mut 5 | Mut 6 | Mut 7 | Mut 8 |
|---|---|---|---|---|---|---|---|---|---|---|
| 2G7 | 10 | 4 | V32A | M164L | G322D | K394R | - | - | - | - |
| 2N0 | 8 | 4 | M164L | P324L | I340V | K394R | - | - | - | - |
| 1M6 | 10 | 8 | A158S | S211N | E250K | K357R | A368T | S514N | E522K | E523K |
a Number of nucleic acid substitutions.
b Number of amino acid substitutions.
Table 3.
Melting temperatures (Tm) of various Vpx (+) and Vpx (−) RTs compared with that of WT SIVmac239 RT
| Clonea | Tm (°C) |
|---|---|
| SIVmac239 | 54.65 |
| 1M6 | 51.24 |
| 2N0 | 53.78 |
| 2G7 | 54.26 |
a Prefix of 1 or 2 indicates that the cloned RT originated from Vpx (−) animals 1 and 2, respectively.
To better understand the kinetic activities of 2G7 and 2N0 RT variants during first- and second-strand synthesis, we conducted steady-state single-nucleotide incorporation kinetic analysis to observe their RNA- and DNA-dependent DNA polymerization efficiency. Consistent with previous studies, we found that the Km associated with dTTP incorporation by WT SIVmac239 RT from a DNA template (100 nm) was two times greater than that observed when using an RNA template (50 nm) (Table 4) (37, 38). This was not the case for 2G7 and 2N0 RT variants, which displayed slightly reduced Km values when polymerizing from a DNA template (2G7: 36 nm, 2N0: 34 nm) compared with those observed when using an RNA template (2G7: 50 nm, 2N0: 48 nm). However, RT variants 2G7 and 2N0 were found to turn over substrate faster than WT SIVmac239 RT regardless of template type, as demonstrated by significantly larger kcat values (Table 4). As expected, we found that WT SIVmac239 RT incorporated dTTP using an RNA template (Fig. 3A, 22,000 s−1 μm−1) with almost twice the catalytic efficiency (kcat/Km) than when polymerizing on a DNA template (Fig. 3B, 11,900 s−1 μm−1). Conversely, 2G7 RT catalyzed dTTP incorporation from RNA and DNA templates with similar kinetics, whereas 2N0 RT more efficiently incorporated dTTP when utilizing a DNA template (Fig. 3). Despite sharing similar Km values when polymerizing from an RNA template, the large template-independent kcat values of 2G7 and 2N0 enable the Vpx (−) RTs to be more catalytically efficient than SIVmac239 RT during DNA synthesis from both RNA and DNA templates (Table 4 and Fig. 3).
Table 4.
Steady-state dTTP incorporation by WT SIVmac239 RT and Vpx (−) RTs 2G7 and 2N0 from RNA and DNA templates
| Clone | RNA template | DNA template |
|---|---|---|
| Km (nm) | ||
| WT | 50 ± 30 | 100 ± 10 |
| 2G7 | 50 ± 20 | 36 ± 5 |
| 2N0 | 48 ± 6 | 34 ± 7 |
| kcat (s−1) | ||
| WT | 9.0 × 10−4 ± 0.6 × 10−4 | 11.7 × 10−4 ± 1.6 × 10−4 |
| 2G7 | 16.4 × 10−4 ± 1.4 × 10−4 | 15.1 × 10−4 ± 0.6 × 10−4 |
| 2N0 | 24 × 10−4 ± 1.0 × 10−4 | 32.2 × 10−4 ± 1.9 × 10−4 |
| kcat/Km (s−1m−1) | ||
| WT | 22,000 ± 9000 | 11,900 ± 200 |
| 2G7 | 41,000 ± 17,000 | 43,000 ± 7000 |
| 2N0 | 50,000 ± 5000 | 98,000 ± 18,000 |
Figure 3.
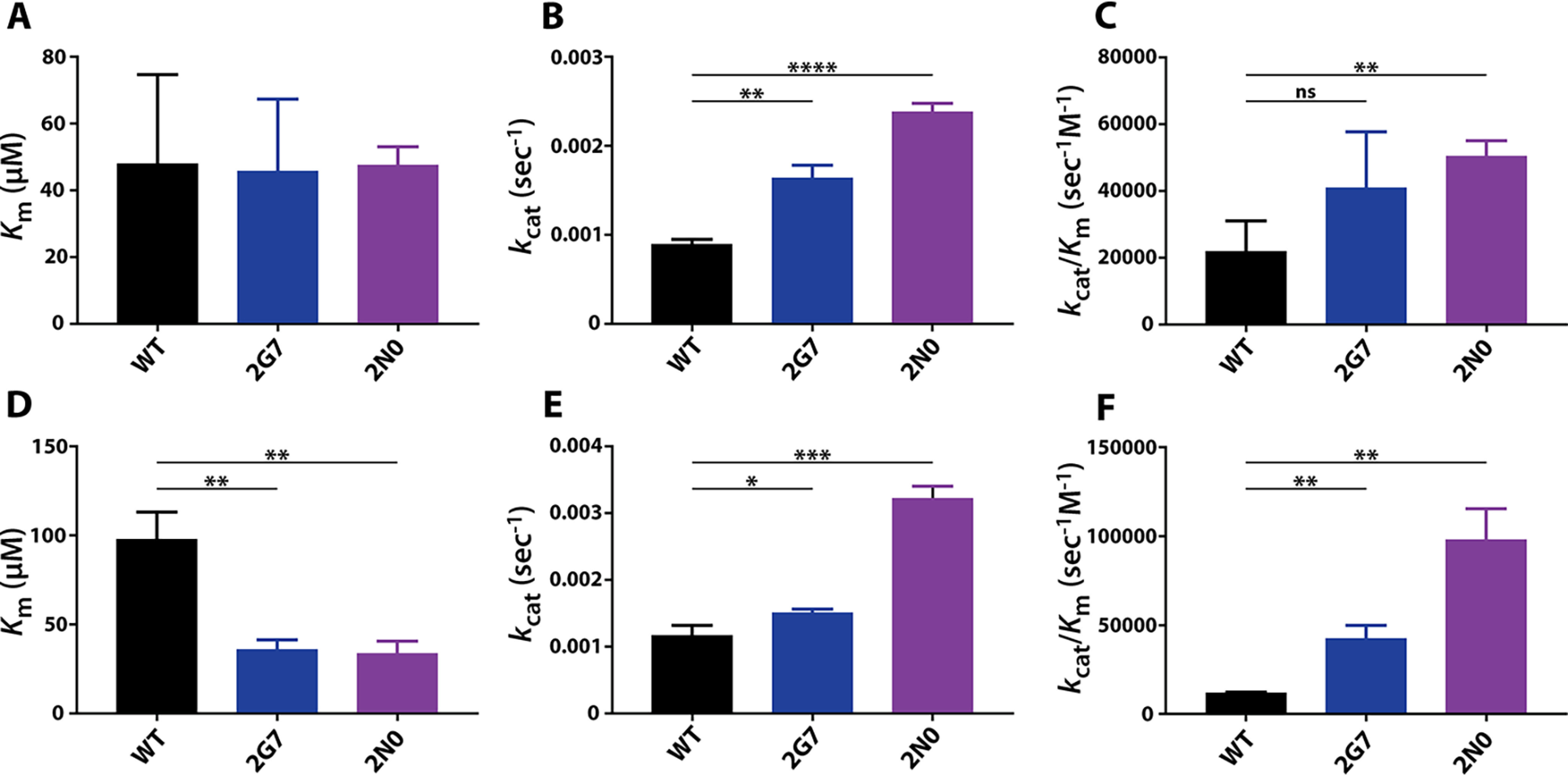
Steady-state kinetic activity of Vpx (−) 2G7 and 2N0 RT variants compared with that of WT SIVmac239 RT. Steady-state kinetic parameters were determined for WT SIVmac239 RT and Vpx (−) RTs 2G7 and 2N0 with dTTP using RNA (A–C) and DNA (D–F) templates (Table 3) as described under “Experimental procedures.” The assays were conducted in triplicate before Km (A and D), kcat (B and E), and catalytic efficiency (kcat/Km) (C and F) values of Vpx (−) RTs were compared to those of WT RT. Statistical significance from unpaired two-tailed student's t tests is indicated as ns, not significant (p < 0.1234); * p < 0.0332; ** p < 0.0021; *** p < 0.002; and **** p < 0.0001.
Pre-steady-state kinetic analysis of two Vpx (−) RT variants
Previous studies have detailed the pre-steady-state kinetic differences between SAMHD1-counteracting and -noncounteracting lentiviral RTs. Although characterized by similar dNTP dissociation constants (Kd), SAMHD1-noncounteracting RTs (e.g. HIV-1 RTs) have demonstrated higher rates of polymerization (kpol) and elevated incorporation efficiency values (kpol/Kd) relative to SAMHD1-counteracting lentiviral RTs (27, 29). Because it has been shown that SAMHD1-noncounteracting lentiviral RTs can more efficiently incorporate an incoming nucleotide, pre-steady-state single-turnover experiments were conducted using normalized concentrations of active 2G7, 2N0, and WT SIVmac239 RT proteins to assess pre-steady-state kinetic activity.
First, we determined the active site concentrations of these purified SIVmac239 RT proteins by conducting pre-steady-state burst experiments using a 5′-32P-labeled 17mer DNA primer annealed to a 39mer DNA template that enables incorporation of a dATP molecule (A-T/P). We measured the 18mer product formation resulting from the mixture of a solution containing RT protein (100 nm RT proteins, see below) prebound to A-T/P (300 nm, excess T/P) with a solution containing 300 μm dATP and 10 mm MgCl2 for various durations ranging from 0.05–3 s. As shown in Fig. 4, there was an initial burst of product formation by WT (Fig. 4A), 2G7 (Fig. 4B), and 2N0 (Fig. 4C) RTs because of dATP incorporation onto the prebound RT·T/P complex (pre-steady-state kinetics), which is followed by a slower, linear phase of product formation corresponding to the steady-state kinetics associated with multiple rounds of DNA polymerization. By fitting these results to the burst equation (Eq. 1), we observed that 12–30% the SIVmac239 RT variant proteins are active.
Figure 4.
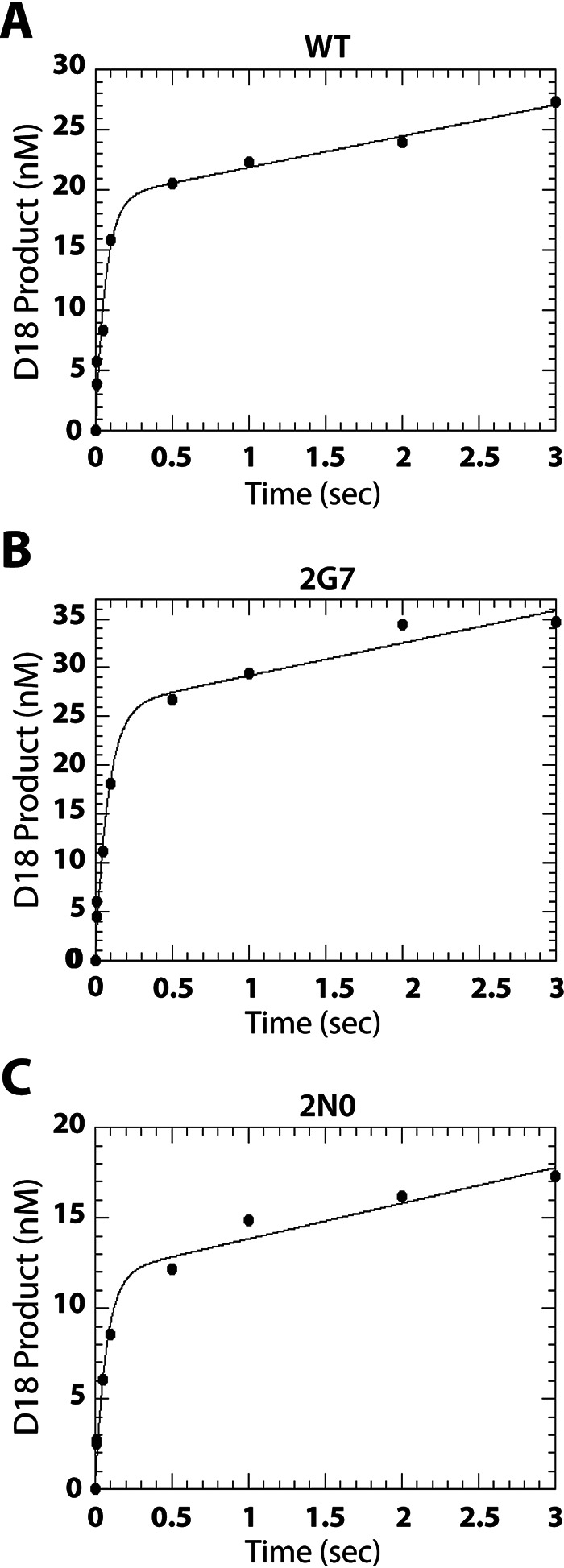
Active site determination for WT SIVmac239 RT and Vpx (−) 2G7 and 2N0 RT variants. Pre-steady-state burst kinetics of (A) WT, (B) 2G7, and (C) 2N0 SIVmac239 RTs detailing the incorporation of a single dATP molecule onto A-T/P in excess T/P conditions (“Experimental procedures,” 3 T/P: 1 active RT) was used to determine the active site concentration of these proteins. The solid line represents the fit of the data to a burst equation (Eq. 1). Burst experiments were conducted in triplicate for each enzyme. These RT proteins display 15–30% active enzyme.
Next, using concentrations of dTTP ranging from 1.6–100 μm, 250 nm active RT, and a 5′-32P-labeled 22mer T primer annealed to the same 39mer DNA template used for the active site determination (T-T/P), we determined the rate of pre-steady-state single-nucleotide incorporation at each dNTP concentration (Eq. 2). The resulting rates were then plotted as a function of substrate concentration and fit to a nonlinear regression curve equation (Eq. 3) to obtain kpol and Kd values. As summarized in Table 5 and Fig. 5A, 2G7 displays faster rates of nucleotide incorporation (35.3 s−1) compared with 2N0 (21 s−1) and WT SIVmac239 RT (20.3 s−1). Whereas previous studies have shown that SAMHD1-counteracting and -noncounteracting lentiviral RTs are characterized by similar Kd values (27, 29), both Vpx (−) RTs displayed lower Kd values (2G7: 16 μm, 2N0: 17 μm) compared with WT SIVmac239 RT (33 μm) (Table 5 and Fig. 5B). Overall, incorporation efficiency for 2G7 (2.3 s−1 μm−1) and 2N0 (1.3 s−1 μm−1) is 2–3-fold greater than that of WT RT (0.61 s−1 μm−1) (Table 5 and Fig. 5C). Because the increased pre-steady-state kinetic activity of 2G7 and 2N0 could be due to their shared amino acid mutations, Vpx (−) RT mutant 1M6 was also kinetically assessed. Like 2G7 and 2N0, Tm measurements revealed that 1M6 is not significantly less stable than WT SIVmac239 RT, suggesting that it is not structurally impaired (Table 2). 1M6 contains 10 nucleic acid mutations and eight amino acid mutations, none of which are shared by 2G7 or 2N0 (Table 3). Vpx (−) RT 1M6 displayed faster kpol (31.5 s−1) and lower Kd (22 μm) values than WT SIVmac239 RT, resulting in a significantly larger incorporation efficiency (1.4 s−1 μm−1) (Table 5 and Fig. 5). This result suggests that Vpx (−) RTs can achieve increased enzyme kinetics through numerous differential amino acid mutations.
Table 5.
Pre-steady-state dTTP incorporation by WT SIVmac239 RT and Vpx (−) RT variants 2G7, 2N0, and 1M6 from DNA template
| RT | Kd (μm) | Kpol (s−1) | Kpol/Kd (s−1 μm−1) |
|---|---|---|---|
| WT | 33 ± 2 | 20.3 ± 1.1 | 0.61 ± 0.01 |
| 2G7 | 16 ± 5 | 35.3 ± 0.4 | 2.3 ± 0.6 |
| 2N0 | 17 ± 3 | 21 ± 3 | 1.3 ± 0.4 |
| 1M6 | 22 ± 4 | 31.5 ± 1.8 | 1.4 ± 0.3 |
Figure 5.

Pre-steady-state kinetic activity of Vpx (−) 2G7, 2N0 RT, and 1M6 RT variants compared with that of WT SIVmac239 RT. Pre-steady-state kinetic values were determined for WT SIVmac239 RT and Vpx (−) RTs 2G7, 2N0, and 1M6 with dTTP using a DNA template (Table 4) as described under “Experimental procedures.” The assays were conducted in triplicate before kpol (A), Kd (B), and incorporation efficiency (kpol/Kd) (C) values of Vpx (−) RTs were compared with those of WT RT. Statistical significance from unpaired two-tailed student's t tests is indicated as: ns, not significant (p < 0.1234); * p < 0.0332; ** p < 0.0021; *** p < 0.002; and **** p < 0.0001.
Discussion
It was previously demonstrated that the limited intracellular dNTP pools found in nondividing myeloid cells restrict lentiviral reverse transcription and that this limited dNTP availability is a biochemical restriction factor against HIV-1 RT in nondividing myeloid target cells (8). Later studies revealed that host dNTPase SAMHD1 is responsible for the low dNTP pools found in these nondividing target cells (16, 39, 40) and that, for SAMHD1-noncounteracting lentiviruses (beyond and including HIV-1), the SAMHD1-mediated low dNTP pools in macrophages serve as a kinetic hurdle that delays complete reverse transcription and slows proviral DNA synthesis kinetics during viral replication. Indeed, SAMHD1 is a strong myeloid-specific HIV-1 restriction factor that can suppress three distinct intracellular dNTP-dependent steps during a single HIV-1 lifecycle: 1) reverse transcription, 2) DNA gap filling, and 3) endogenous reverse transcription (41). Conversely, SAMHD1-counteracting lentiviruses, like SIVmac239, employ viral Vpx proteins to target host dNTPase SAMHD1 for proteasomal degradation, resulting in increased cellular dNTPs and the alleviation of kinetic restriction.
Studies have shown that RTs from lentiviruses without the ability to counteract SAMHD1 have evolved over time to execute a faster enzyme conformational change during dNTP incorporation. This enables SAMHD1-noncounteracting lentiviruses to circumvent SAMHD1 restriction and complete reverse transcription even in the low dNTP concentrations found in nondividing myeloid cells. Because kinetic similarities are seen among RTs originating from various lentiviral phylogroups depending on their ability or inability to counteract SAMHD1, the evolution of SAMHD1-noncounteracting lentiviral RTs is presumed to have occurred over many years and by viral passage through many hosts (42, 43). In this study, we reasoned that SIVmac239 RT, an enzyme known to synthesize DNA with low efficiency within the limited dNTP pools of the macrophage (27, 28), can improve its enzyme kinetics to resemble enzymatically efficient HIV-1 RT when SIVmac239 virus replicates within animals in the absence of Vpx (like HIV-1), an in vivo environment likely to create a strong antiviral selective pressure in myeloid cells because of the presence of host SAMHD1.
SIVmac239 RT proteins assayed in this study originated from single infected animal sources with survival durations of 27–29 weeks and 3 years postinfection for Vpx (+) and Vpx (−) infections, respectively. We found that RTs originating from Vpx (−) infections, on average, contained more amino acid mutations and displayed faster steady-state and pre-steady-state kinetics than WT SIVmac239 RT and RTs cloned from Vpx (+) infections. However, because Vpx (−) animals lived roughly six times longer than the Vpx (+) animals used in this study, we cannot exclude the possibility that the mutations we observed in Vpx (−) RTs are the result of randomly accumulated viral mutagenesis rather than mutations that have been selected for because of the evolutionary pressure exerted by the absence of Vpx (44–46). It is also possible that some of the commonly observed mutations are escape products resulting from certain types of immune selections. Lastly, because vpx is the result of a gene duplication event of vpr, both proteins have been found to have the potential to counteract SAMHD1, depending on the species origins of the viral Vpx/Vpr and host SAMHD1 proteins. Although SIVmac239 Vpr does not possess SAMHD1 counteraction activity, it is possible that Vpx (−) viruses evolved a Vpr protein that can induce the degradation of host dNTPase SAMHD1. Vpx protein was cloned and sequenced from Vpx (−)-infected samples in a manner identical to the RTs in this study, revealing truncated vpx genes that presumably resulted in no protein product (data not shown). However, neither the anti-SAMHD1 activity of Vpr nor the SAMHD1 protein expression in Vpx (−)-infected cells was verified in this study. Therefore, we cannot negate the possibility that Vpx (−) viruses evolved SAMHD1-counteraction ability through the evolution of Vpr.
As shown in our data, the selected SIVmac239 RT variants from Vpx (−) infections improved their steady-state DNA synthesis abilities by having lower Km values and higher dNTP binding affinity (Kd) compared with WT SIVmac239 RT. Whereas both 2G7 and 1M6 displayed faster kpol values, 2N0 exhibited no significant difference from WT SIVmac239 RT. These kinetic changes are different from the uniformity we observed between SAMHD1-noncounteracting HIV-1 RTs and SAMHD1-counteracting SIV RTs: HIV-1 RT is characterized by lower steady-state Km values, similar Kd values, and faster pre-steady-state kpol values when compared with SIV RT (27, 28). To improve their enzyme kinetic efficiency, it appears that the two SIVmac239 RT variants (2G7 and 2N0), originating from a single Vpx (−) infection, employ differential mechanistic pathways from what has been previously observed for HIV-1 RTs. Multiple-nucleotide incorporation experiments revealed that Vpx (−) RTs, on average, display substrate efficiency values 2-fold greater than those observed for Vpx (+) RTs. Although a 2-fold difference seems minute, a 2-fold kinetic increase in vivo translates into a 6-h replication period rather than 12-h, thus drastically decreasing the time required for viral replication. Conversely, a 2-fold decrease in kinetics would result in replication taking 24 h rather than 12 h, exposing the viral material to cellular defenses for longer durations of time and potentially negatively affecting viral replication.
With no solved structure for the SIV RT heterodimer, a structure of heterodimeric HIV-1 RT bound to nucleic acid (PBD ID: 1RTD) was used to highlight the location of the equivalent residues mutated in Vpx (−) RT clones 2G7 (top panels), 2N0 (middle panels), and 1M6 (bottom panels) (Fig. 6). This structure of HIV-1 RT shows a large portion of the p66 subunit participating in T/P binding. Interestingly, all three clones contain mutations of residues near the T/P binding cleft, providing the possibility that these mutations may affect not only the formation of the binary complex but also polymerase T/P binding and processivity during steady-state polymerization. Whereas many mutations are surface exposed (i.e. 3 of 4 residues in 2G7, 2 of 4 in 2N0, and 4 of 8 in 1M6), residue Met-164 lies just outside of the active site and is not only substituted in both 2G7 and 2N0 RTs but has also been adopted by 38 of the 40 RT variants cloned from one Vpx (−)-infected animal (Table 1). Additionally, 1M6 contains substitutions of three residues near the heterodimeric interface: Ala-158, Lys-357, and Ala-368. Nevertheless, because Vpx (−) RT variants contain substitutions at residues that are not close to any known residues involved in dNTP binding or catalysis in HIV-1 RT, it is difficult to explain the mechanistic and structural impact made by the various combinations of the observed mutations. Possibly, some, or all, of the mutations found in the three Vpx (−) RTs indirectly coordinate local structural changes to improve dNTP binding affinity or enhance the rate of enzyme conformational change in these polymerases. Interestingly, studies have shown that HIV-1 RT specificity is determined by two factors: the rate of nucleotide substrate binding (including the subsequent isomerization of the substrate within the closed enzyme complex) and the rate of the enzyme conformational change (47, 48). These studies revealed that whereas kcat is determined by the rate of chemistry, kcat/Km is a function of both Kd for nucleotide binding to the open state and the rate of conformational change. With this new understanding of the interplay between polymerase efficiency and fidelity, future experiments could also include comparison of mismatch incorporation and extension events between WT and Vpx (−) RT variants.
Figure 6.

Location of mutated residues in 2G7, 2N0, and 1M6 Vpx (−) RT variants. The crystal structure of heterodimeric HIV-1 RT bound to nucleic acid (PBD ID: 1RTD) was used to map the location of the equivalent mutated residues (blue spheres) in 2G7 (first row), 2N0 (second row), and 1M6 (third row) SIVmac239 Vpx (−) RT variants. Front and back views of the polymerase are displayed in the first and second column, respectively. The p66 fingers (pink), palm (orange), thumb (green), connection (yellow), and RNaseH (cyan) subdomains and p51 (gray) subunits are colored accordingly.
Finally, these biochemical findings can be further verified through virological investigations that compare the viral infectivity of infectious SIVmac239ΔVpx mutant virus harboring the two Vpx (−) RT variants to that of SIVmac239ΔVpx encoding for WT RT. These experiments seek to characterize the differential impact of these mutated RTs during single-round infection in activated CD4+ T cells and macrophages. Further studies could also identify the specific RT amino acid mutations contributing to the altered enzyme kinetics in Vpx (−) RTs and explore the associated mechanisms. Collectively, these sequencing and kinetic studies support the idea that the absence of Vpx in SIVmac239 infections results in RT enzymes with more numerous amino acid mutations and enhanced kinetics that enable more efficient polymerization from RNA and DNA templates.
Experimental procedures
Animal samples
Serum samples originating from two Rhesus macaques infected with Vpx-deleted SIVmac239 (animals 1 and 2) were obtained from the Westmoreland et al. study (30), and PBMCs from two Rhesus macaques infected with WT SIVmac239 (animals 3 and 4) were provided by Dr. Guido Silvestri. Vpx-deleted SIVmac239 samples were collected 3 years postinfection, whereas WT SIVmac239 samples for animals 3 and 4 were collected 27 and 29 weeks postinfection, respectively.
Cloning and sequencing Vpx+ and Vpx− RTs
RNA was extracted from infected samples using the RNeasy Mini Kit (Qiagen). SIVmac239 RT sequences were amplified by RT-PCR using SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Invitrogen) with 5NdeIF (5′-AAAAAAACATATGCCCATAGCTAAAGTAGAGCC-3′; resulting amplicon has 5′ NdeI site) and 3XhoIR (5′-AAAAAACTCGAGTTATTGACTAACTAG-3′; resulting amplicon has 3′ XhoI site) for 40 cycles with primer annealing at 45 °C for 30 s and primer extension at 68 °C for 3 min. PCR fragments were isolated using the QIAquick PCR Purification Kit (Qiagen) and subsequently cloned directly into the pCR4-TOPO vector (TOPO-RT) using the TOPO TA Cloning Kit (Invitrogen). TOPO-RT vectors were sequenced using M13 Forward (−20) and M13 Reverse primers. Viral RT nucleotide and amino acid sequences were analyzed and compared with parental viral clone SIVmac239 RT (30–32). Ultimately, 40 complete RT clones were identified from each infected sample, and alterations in nucleotide and amino acid sequences were noted. The first number within the name of the cloned RT signifies the originating animal: animals 1 and 2 are Vpx (−)-infected animals, whereas animals 3 and 4 were infected by WT or Vpx (+) SIVmac239 virus. The subsequent letter and number following the animal distinction are in reference to sequencing records.
RT protein expression and purification
N-terminal His-tagged RT proteins were expressed from cloned pET28a-RT expression plasmids in E. coli BL21 Rosetta 2 DE3 (Millipore), and the p66/p66 homodimers were purified as described previously (49) with the following changes. For large-scale purifications, clear lysate obtained through sonication of 1-liter cultures was applied to charged His•Bind resin (Millipore) equilibrated with a binding buffer containing 40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, 20 mm imidazole, and 10% glycerol. The column was washed with 15 column volumes of binding buffer prior to being eluted in 1-ml fractions by a solution containing 40 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm MgCl2, 240 mm imidazole, and 10% glycerol. Fractions containing the His-tagged p66/p66 were pooled and dialyzed for 16 h in a storage buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm KCl, 0.25 mm EDTA, 1 mm β-mercaptoethanol, and 20% glycerol. To examine the purity of the proteins, the dialyzed RTs were run on a 4–15% SDS-PAGE gel (Bio-Rad). All RTs were determined to have at least 95% purity and were flash-frozen in liquid nitrogen prior to being stored at −80 °C for future use. Small-scale protein preparations used in the steady-state multiple-nucleotide incorporation kinetic screen were obtained from 250-ml cultures using His SpinTrap (GE Healthcare) columns and the same buffers utilized in large-scale purifications.
Steady-state multiple-nucleotide incorporation assay
The previously described primer extension assay (8, 27) was slightly modified for this study. Briefly, a 32P-labeled template/primer (T/P) was prepared by annealing a 5′-32P 17mer DNA primer (17D: 5′-CGCGCCGAATTCCCGCT-3′, Integrated DNA Technologies) to a 3-fold excess of 39mer RNA (39R: 5′-AGCUUGGCUGCAGAAUAUUGCUAGCGGGAAUUCGGCGCG-3′, Integrated DNA Technologies). Assay mixtures (20 μl) contained 10 nm T/P, RT, and varying dNTP concentrations (50, 25, 10, 5, 1, 0.5, 0.25, 0.1, 0.05, and 0.025 μm). RT activity was normalized for 50% extension at the highest dNTP concentration (50 μm), and with all dNTPs supplied, this reaction allows multiple rounds of primer extension. Reaction mixtures were incubated at 37 °C for 5 min, quenched with 40 mm EDTA in 99% formamide, and placed at 95 °C for 2 min to further inactivate RT. Reaction products were separated on a 14% polyacrylamide/8-m urea gel, visualized using a PharosFX (Bio-Rad) phosphoimager, and quantified with Image Laboratory Software (Bio-Rad). Data were fit to a nonlinear regression curve to obtain Michealis-Menten kinetic parameters describing the maximal reaction velocity (Vmax) and the amount of substrate required to reach half-Vmax (Km).
Steady-state single-nucleotide incorporation assay
Single-nucleotide incorporation experiments were conducted using a 5′-32P-labeled 22mer DNA primer (5′-CGCGCCGAATTCCCGCTAGCAA-3′, Integrated DNA Technologies) annealed to a 39mer RNA (39R) or DNA (39D: 5′-AGCTTGGCTGCAGAATATTGCTAGCGGGAATTCGGCGCG-3′, Integrated DNA Technologies) template using the same methods described above with the following changes. Instead of adding all four dNTPs to the reaction mixture, the incoming nucleotide (dTTP) was supplied in various concentrations ranging from 25 nm to 50 μm. Reactions were conducted and quantified as previously described. Per Michaelis-Menten kinetics, Vmax values were converted to catalytic turnover (kcat) values using total enzyme concentrations. Steady-state experiments were conducted in triplicate for each RT.
Pre-steady-state single-turnover experiments
The pre-steady-state kinetic parameters kpol and Kd were determined as previously described (27, 29, 50). Briefly, the active site concentrations of WT SIVmac239 RT and Vpx (−) RT clones 2G7, 2N0, and 1M6 were determined using pre-steady-state burst experiments conducted with an RFQ-3 rapid quench-flow apparatus (KinTek Corporation) and a 17mer 32P-labeled DNA primer (17D) annealed to a 39mer DNA template (39D) supplied in 3-fold excess to RT. The reactions were quenched with EDTA at the following time points: 0, 0.005, 0.01, 0.05, 0.1, 0.5, 1, 2, and 3 s. Active site concentrations were determined by fitting product formation to the burst equation (Eq. 1):
| (Equation 1) |
in which A is the amplitude of the burst and reflects the concentration of enzyme that is in an active form, kobs represents the observed first-order burst rate for dNTP incorporation, and kss is the linear steady-state rate constant (27, 51, 52). Active site titrations were performed in triplicate for each RT.
RT active site concentrations were used to ensure that subsequent single-turnover experiments contained 250 nm active RT enzyme, a 5-fold excess to the 50 nm T/P (a 5′-32P-labeled 22mer DNA primer annealed to a 39mer DNA template that enables incorporation of a dTTP molecule, i.e. T-T/P) present in the reactions. In single-turnover experiments, excess RT prebound to T-T/P was rapidly mixed with a solution containing 10 mm MgCl2 and varying concentrations of dTTP (1.6–100 μm). The reactions were quenched with EDTA at various time points ranging from 0.01 to 2 s and visualized using the same methods as above. The data were then fit to a single exponential equation (Eq. 2) to obtain the observed pre-steady-state rate for dNTP incorporation (kobs) at every dTTP substrate concentration tested.
| (Equation 2) |
In this equation, A is the amplitude of product formation, kobs is the observed pre-steady-state rate for dNTP incorporation, and t is time.
Next, kobs was plotted as a function of substrate concentration and fit to a nonlinear regression curve (Eq. 3) to obtain kinetic parameters kpol and Kd for each characterized RT.
| (Equation 3) |
in which kpol is the maximum rate of dNTP incorporation and Kd is the equilibrium dissociation constant for the dNTP substrate (53). Pre-steady-state single-turnover experiments were conducted in triplicate for each RT.
Thermostability shift assay
Protein mixtures (40 μl) containing 2.5 μm purified RT protein and 5 mm MgCl2 in RT storage buffer (50 mm Tris-HCl, pH 7.5, 150 mm KCl, 0.25 mm EDTA, 1 mm β-mercaptoethanol, and 20% glycerol) were added to a 96-well plate (Lightcycler 480 Multiwell Plate 96 white, Roche) in triplicate for each RT. Wells containing either no enzyme or no dye were performed in triplicate as negative controls. Sypro Orange Protein Gel Stain (Sigma-Aldrich) was diluted 1:20 in RT storage buffer and 1 μl of the dilution was added to the protein mixture in each well. Reactions mixtures were heated from 32 to 99 °C at the rate of 0.02 °C/s by a real-time PCR device (LightCycler 480 II, Roche) that monitored protein unfolding signified by changes in fluorescence of the Sypro Orange fluorophore. The resulting fluorescence intensities were plotted against temperature for each sample well and fit to the Boltzmann equation using Spyder Software (Anaconda). The midpoint of each transition (Tm), or melting temperature of the enzyme, was calculated for each well, and the average Tm of each RT was calculated by averaging the results of the triplicate wells.
Data availability
All data are contained within this article.
Author contributions—S. A. C. data curation; S. A. C. formal analysis; S. A. C. validation; S. A. C. and B. K. investigation; S. A. C., D.-H. K., and R. F. S. methodology; S. A. C. writing-original draft; D.-H. K., R. F. S., R. C. D., and B. K. conceptualization; D.-H. K., R. F. S., R. C. D., and B. K. writing-review and editing; R. F. S. and B. K. funding acquisition; R. C. D. resources; B. K. supervision.
Funding and additional information—This work was supported by NIAID National Institutes of Health Grants AI136581 (to B. K.), AI150451 (B. K.), and MH116695 (R. F. S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- SIV
- simian immunodeficiency virus
- SAM
- sterile α motif
- HD
- histidine-aspartate
- SAMHD1
- SAM domain and HD domain–containing protein 1
- Vpr
- viral protein R
- Vpx
- viral protein X
- dNTP
- deoxyribonucleoside triphosphate
- A-T/P
- 5′-32P-labeled 17-mer A primer annealed to a 39mer DNA template.
References
- 1. Mori, K., Ringler, D. J., Kodama, T., and Desrosiers, R. C. (1992) Complex determinants of macrophage tropism in env of simian immunodeficiency virus. J. Virol. 66, 2067–2075 10.1128/JVI.66.4.2067-2075.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jamburuthugoda, V. K., Chugh, P., and Kim, B. (2006) Modification of human immunodeficiency virus type 1 reverse transcriptase to target cells with elevated cellular dNTP concentrations. J. Biol. Chem. 281, 13388–13395 10.1074/jbc.M600291200 [DOI] [PubMed] [Google Scholar]
- 3. Weissman, D., Rabin, R. L., Arthos, J., Rubbert, A., Dybul, M., Swofford, R., Venkatesan, S., Farber, J. M., and Fauci, A. S. (1997) Macrophage-tropic HIV and SIV envelope proteins induce a signal through the CCR5 chemokine receptor. Nature 389, 981–985 10.1038/40173 [DOI] [PubMed] [Google Scholar]
- 4. Swanstrom, A. E., Del Prete, G. Q., Deleage, C., Elser, S. E., Lackner, A. A., and Hoxie, J. A. (2018) The SIV envelope glycoprotein, viral tropism, and pathogenesis: novel insights from nonhuman primate models of AIDS. Curr. HIV Res. 16, 29–40 10.2174/1570162X15666171124123116 [DOI] [PubMed] [Google Scholar]
- 5. Bejarano, D. A., Puertas, M. C., Börner, K., Martinez-Picado, J., Müller, B., and Kräusslich, H. G. (2018) Detailed characterization of early HIV-1 replication dynamics in primary human macrophages. Viruses 10, 620 10.3390/v10110620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Williams, K. C., and Hickey, W. F. (2002) Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu. Rev. Neurosci. 25, 537–562 10.1146/annurev.neuro.25.112701.142822 [DOI] [PubMed] [Google Scholar]
- 7. Traut, T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 140, 1–22 10.1007/BF00928361 [DOI] [PubMed] [Google Scholar]
- 8. Diamond, T. L., Roshal, M., Jamburuthugoda, V. K., Reynolds, H. M., Merriam, A. R., Lee, K. Y., Balakrishnan, M., Bambara, R. A., Planelles, V., Dewhurst, S., and Kim, B. (2004) Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 279, 51545–51553 10.1074/jbc.M408573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmidt, S., Schenkova, K., Adam, T., Erikson, E., Lehmann-Koch, J., Sertel, S., Verhasselt, B., Fackler, O. T., Lasitschka, F., and Keppler, O. T. (2015) SAMHD1's protein expression profile in humans. J. Leukoc. Biol. 98, 5–14 10.1189/jlb.4HI0714-338RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin, C., Peng, X., Liu, F., Cheng, L., Lu, X., Yao, H., Wu, H., and Wu, N. (2014) MicroRNA-181 expression regulates specific post-transcriptional level of SAMHD1 expression in vitro. Biochem. Biophys. Res. Commun. 452, 760–767 10.1016/j.bbrc.2014.08.151 [DOI] [PubMed] [Google Scholar]
- 11. Hollenbaugh, J. A., Tao, S., Lenzi, G. M., Ryu, S., Kim, D. H., Diaz-Griffero, F., Schinazi, R. F., and Kim, B. (2014) dNTP pool modulation dynamics by SAMHD1 protein in monocyte-derived macrophages. Retrovirology 11, 63 10.1186/s12977-014-0063-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Franzolin, E., Pontarin, G., Rampazzo, C., Miazzi, C., Ferraro, P., Palumbo, E., Reichard, P., and Bianchi, V. (2013) The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 110, 14272–14277 10.1073/pnas.1312033110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Plitnik, T., Sharkey, M. E., Mahboubi, B., Kim, B., and Stevenson, M. (2018) Incomplete suppression of HIV-1 by SAMHD1 permits efficient macrophage infection. Pathog. Immun. 3, 197–223 10.20411/pai.v3i2.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim, B., Nguyen, L. A., Daddacha, W., and Hollenbaugh, J. A. (2012) Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J. Biol. Chem. 287, 21570–21574 10.1074/jbc.C112.374843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hrecka, K., Hao, C., Gierszewska, M., Swanson, S. K., Kesik-Brodacka, M., Srivastava, S., Florens, L., Washburn, M. P., and Skowronski, J. (2011) Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661 10.1038/nature10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Laguette, N., Sobhian, B., Casartelli, N., Ringeard, M., Chable-Bessia, C., Ségéral, E., Yatim, A., Emiliani, S., Schwartz, O., and Benkirane, M. (2011) SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657 10.1038/nature10117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hrecka, K., Gierszewska, M., Srivastava, S., Kozaczkiewicz, L., Swanson, S. K., Florens, L., Washburn, M. P., and Skowronski, J. (2007) Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. U. S. A. 104, 11778–11783 10.1073/pnas.0702102104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahn, J., Hao, C., Yan, J., DeLucia, M., Mehrens, J., Wang, C., Gronenborn, A. M., and Skowronski, J. (2012) HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 287, 12550–12558 10.1074/jbc.M112.340711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Srivastava, S., Swanson, S. K., Manel, N., Florens, L., Washburn, M. P., and Skowronski, J. (2008) Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 4, e1000059 10.1371/journal.ppat.1000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lahouassa, H., Daddacha, W., Hofmann, H., Ayinde, D., Logue, E. C., Dragin, L., Bloch, N., Maudet, C., Bertrand, M., Gramberg, T., Pancino, G., Priet, S., Canard, B., Laguette, N., Benkirane, M., et al. (2012) SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13, 223–228 10.1038/ni.2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Etienne, L., Hahn, B. H., Sharp, P. M., Matsen, F. A., and Emerman, M. (2013) Gene loss and adaptation to hominids underlie the ancient origin of HIV-1. Cell Host Microbe 14, 85–92 10.1016/j.chom.2013.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sharp, P. M., Bailes, E., Stevenson, M., Emerman, M., and Hahn, B. H. (1996) Gene acquisition in HIV and SIV. Nature 383, 586–587 10.1038/383586a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou, X., DeLucia, M., Hao, C., Hrecka, K., Monnie, C., Skowronski, J., and Ahn, J. (2017) HIV-1 Vpr protein directly loads helicase-like transcription factor (HLTF) onto the CRL4-DCAF1 E3 ubiquitin ligase. J. Biol. Chem. 292, 21117–21127 10.1074/jbc.M117.798801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Belzile, J. P., Duisit, G., Rougeau, N., Mercier, J., Finzi, A., and Cohen, E. A. (2007) HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 3, e85 10.1371/journal.ppat.0030085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lim, E. S., Fregoso, O. I., McCoy, C. O., Matsen, F. A., Malik, H. S., and Emerman, M. (2012) The Ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe 11, 194–204 10.1016/j.chom.2012.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fregoso, O. I., Ahn, J., Wang, C., Mehrens, J., Skowronski, J., and Emerman, M. (2013) Evolutionary toggling of Vpx/Vpr specificity results in divergent recognition of the restriction factor SAMHD1. PLoS Pathog. 9, e1003496 10.1371/journal.ppat.1003496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lenzi, G. M., Domaoal, R. A., Kim, D. H., Schinazi, R. F., and Kim, B. (2015) Mechanistic and kinetic differences between reverse transcriptases of Vpx coding and non-coding lentiviruses. J. Biol. Chem. 290, 30078–30086 10.1074/jbc.M115.691576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lenzi, G. M., Domaoal, R. A., Kim, D. H., Schinazi, R. F., and Kim, B. (2014) Kinetic variations between reverse transcriptases of viral protein X coding and noncoding lentiviruses. Retrovirology 11, 111 10.1186/s12977-014-0111-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coggins, S. A., Holler, J. M., Kimata, J. T., Kim, D. H., Schinazi, R. F., and Kim, B. (2019) Efficient pre-catalytic conformational change of reverse transcriptases from SAMHD1 non-counteracting primate lentiviruses during dNTP incorporation. Virology 537, 36–44 10.1016/j.virol.2019.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Westmoreland, S. V., Converse, A. P., Hrecka, K., Hurley, M., Knight, H., Piatak, M., Lifson, J., Mansfield, K. G., Skowronski, J., and Desrosiers, R. C. (2014) SIV vpx is essential for macrophage infection but not for development of AIDS. PLoS ONE 9, e84463 10.1371/journal.pone.0084463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gibbs, J. S., Regier, D. A., and Desrosiers, R. C. (1994) Construction and in vitro properties of SIVmac mutants with deletions in “nonessential” genes. AIDS Res. Hum. Retroviruses 10, 333–342 10.1089/aid.1994.10.333 [DOI] [PubMed] [Google Scholar]
- 32. Desrosiers, R. C., Lifson, J. D., Gibbs, J. S., Czajak, S. C., Howe, A. Y., Arthur, L. O., and Johnson, R. P. (1998) Identification of highly attenuated mutants of simian immunodeficiency virus. J. Virol. 72, 1431–1437 10.1128/JVI.72.2.1431-1437.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Connor, R. I., Sheridan, K. E., Ceradini, D., Choe, S., and Landau, N. R. (1997) Change in coreceptor use correlates with disease progression in HIV-1–infected individuals. J. Exp. Med. 185, 621–628 10.1084/jem.185.4.621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hollenbaugh, J. A., Montero, C., Schinazi, R. F., Munger, J., and Kim, B. (2016) Metabolic profiling during HIV-1 and HIV-2 infection of primary human monocyte-derived macrophages. Virology 491, 106–114 10.1016/j.virol.2016.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yutani, K., Ogasahara, K., and Sugino, Y. (1985) Effect of amino acid substitutions on conformational stability of a protein. Adv. Biophys. 20, 13–29 10.1016/0065-227X(85)90028-0 [DOI] [PubMed] [Google Scholar]
- 36. Miyazawa, S., and Jernigan, R. L. (1994) Protein stability for single substitution mutants and the extent of local compactness in the denatured state. Protein Eng. 7, 1209–1220 10.1093/protein/7.10.1209 [DOI] [PubMed] [Google Scholar]
- 37. Parker, W. B., White, E. L., Shaddix, S. C., Ross, L. J., Buckheit, R. W. Jr, Germany, J. M., Secrist, J. A. III, Vince, R., and Shannon, W. M. (1991) Mechanism of inhibition of human immunodeficiency virus type 1 reverse transcriptase and human DNA polymerases α, β, and γ by the 5′-triphosphates of carbovir, 3′-azido-3′-deoxythymidine, 2′,3′-dideoxyguanosine and 3′-deoxythymidine. A novel RNA template for the evaluation of antiretroviral drugs. J. Biol. Chem. 266, 1754–1762 [PubMed] [Google Scholar]
- 38. Reardon, J. E., and Miller, W. H. (1990) Human immunodeficiency virus reverse transcriptase. substrate and inhibitor kinetics with thymidine 5′-triphosphate and 3′-azido-3′-deoxythymidine 5′-triphosphate. J. Biol. Chem. 265, 20302–20307 [PubMed] [Google Scholar]
- 39. Coggins, S. A. A., Mahboubi, B., Schinazi, R. F., and Kim, B. (2020) SAMHD1 functions and human diseases. Viruses 12, 382 10.3390/v12040382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goldstone, D. C., Ennis-Adeniran, V., Hedden, J. J., Groom, H. C., Rice, G. I., Christodoulou, E., Walker, P. A., Kelly, G., Haire, L. F., Yap, M. W., de Carvalho, L. P., Stoye, J. P., Crow, Y. J., Taylor, I. A., and Webb, M. (2011) HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382 10.1038/nature10623 [DOI] [PubMed] [Google Scholar]
- 41. Mahboubi, B., Gavegnano, C., Kim, D. H., Schinazi, R. F., and Kim, B. (2018) Host SAMHD1 protein restricts endogenous reverse transcription of HIV-1 in nondividing macrophages. Retrovirology 15, 69 10.1186/s12977-018-0452-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yokoyama, S., Chung, L., and Gojobori, T. (1988) Molecular evolution of the human immunodeficiency and related viruses. Mol. Biol. Evol. 5, 237–251 10.1093/oxfordjournals.molbev.a040495 [DOI] [PubMed] [Google Scholar]
- 43. Yokoyama, S. (1988) Molecular evolution of the human and simian immunodeficiency viruses. Mol. Biol. Evol. 5, 645–659 10.1093/oxfordjournals.molbev.a040520 [DOI] [PubMed] [Google Scholar]
- 44. Mansky, L. M. (1996) Forward mutation rate of human immunodeficiency virus type 1 in a T lymphoid cell line. AIDS Res. Hum. Retroviruses 12, 307–314 10.1089/aid.1996.12.307 [DOI] [PubMed] [Google Scholar]
- 45. Coffin, J. M. (1995) HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267, 483–489 10.1126/science.7824947 [DOI] [PubMed] [Google Scholar]
- 46. Mansky, L. M. (1998) Retrovirus mutation rates and their role in genetic variation. J. Gen. Virol. 79, 1337–1345 10.1099/0022-1317-79-6-1337 [DOI] [PubMed] [Google Scholar]
- 47. Kellinger, M. W., and Johnson, K. A. (2010) Nucleotide-dependent conformational change governs specificity and analog discrimination by HIV reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 107, 7734–7739 10.1073/pnas.0913946107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kirmizialtin, S., Nguyen, V., Johnson, K. A., and Elber, R. (2012) How conformational dynamics of DNA polymerase select correct substrates: experiments and simulations. Structure 20, 618–627 10.1016/j.str.2012.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim, B. (1997) Genetic selection in Escherichia coli for active human immunodeficiency virus reverse transcriptase mutants. Methods 12, 318–324 10.1006/meth.1997.0485 [DOI] [PubMed] [Google Scholar]
- 50. Skasko, M., Weiss, K. K., Reynolds, H. M., Jamburuthugoda, V., Lee, K., and Kim, B. (2005) Mechanistic differences in RNA-dependent DNA polymerization and fidelity between murine leukemia virus and HIV-1 reverse transcriptases. J. Biol. Chem. 280, 12190–12200 10.1074/jbc.M412859200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kati, W. M., Johnson, K. A., Jerva, L. F., and Anderson, K. S. (1992) Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267, 25988–25997 [PubMed] [Google Scholar]
- 52. Reardon, J. E. (1992) Human immunodeficiency virus reverse transcriptase: steady-state and pre-steady-state kinetics of nucleotide incorporation. Biochemistry 31, 4473–4479 10.1021/bi00133a013 [DOI] [PubMed] [Google Scholar]
- 53. Johnson, K. A. (1995) Rapid quench kinetic analysis of polymerases, adenosinetriphosphatases, and enzyme intermediates. Methods Enzymol. 249, 38–61 10.1016/0076-6879(95)49030-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are contained within this article.