

Abstract
Kinases are critical components of intracellular signaling pathways and have been extensively investigated with regard to their roles in cancer. p21-activated kinase-1 (PAK1) is a serine/threonine kinase that has been previously implicated in numerous biological processes, such as cell migration, cell cycle progression, cell motility, invasion, and angiogenesis, in glioma and other cancers. However, the signaling network linked to PAK1 is not fully defined. We previously reported a large-scale yeast genetic interaction screen using toxicity as a readout to identify candidate PAK1 genetic interactions. En masse transformation of the PAK1 gene into 4,653 homozygous diploid Saccharomyces cerevisiae yeast deletion mutants identified ∼400 candidates that suppressed yeast toxicity. Here we selected 19 candidate PAK1 genetic interactions that had human orthologs and were expressed in glioma for further examination in mammalian cells, brain slice cultures, and orthotopic glioma models. RNAi and pharmacological inhibition of potential PAK1 interactors confirmed that DPP4, KIF11, mTOR, PKM2, SGPP1, TTK, and YWHAE regulate PAK1-induced cell migration and revealed the importance of genes related to the mitotic spindle, proteolysis, autophagy, and metabolism in PAK1-mediated glioma cell migration, drug resistance, and proliferation. AKT1 was further identified as a downstream mediator of the PAK1-TTK genetic interaction. Taken together, these data provide a global view of PAK1-mediated signal transduction pathways and point to potential new drug targets for glioma therapy.
Keywords: kinase, glioma, genetic interaction, PAK1, molecular cell biology, cell migration, drug resistance, cell proliferation, signal transduction
Gliomas are tumors that occur in the brain and spinal cord. Glioma, which begins in the glial cells that surround neurons and help them function, is one of the most common types of malignant primary brain tumors. Treatment for glioma depends on the cell type, size, grade of malignancy, and location of the tumor. Treatment is usually a combination of surgery, radiation therapy, chemotherapy, and immunotherapy. Targeted drug therapy is preferred over conventional cytotoxic chemotherapy, as the targeted drug treatments focus on specific abnormalities present within cancer cells (1, 2). By blocking these abnormalities, targeted drug treatments can cause cancer cells to die without affecting noncancer cells (3). However, for the targeted drug therapy to be successfully developed and used in the clinic, one needs to better understand glioma cell behaviors (such as glioma cell migration/invasion, growth/proliferation, death/survival, and drug resistance) and related signaling pathways.
The signaling component “p21-activated kinase-1” (PAK1) is a serine/threonine kinase regulated by small GTP-binding proteins Cdc42 and Rac (4–7). This kinase affects a wide variety of cellular processes, such as cell motility, invasion, metastasis, growth, cell cycle progression, and angiogenesis. It has been previously implicated in a wide range of biological processes and the development of multiple types of cancer (8–14). More specifically, PAK1 has been previously implicated in cell death/survival, cell migration, and glioma (15–19). The expression level of PAK1 has been associated with the invasiveness of glioblastoma and survival time in patients with glioblastoma. Our own previous RNAi selection study indicated that the PAK kinase family regulates cell migration (20).
In this study, to better understand PAK1 kinase signaling pathways and to uncover new drug targets related to these pathways, we sought to exploit a model eukaryote which is amenable to rapid genome-scale experimentation to identify related functions. More specifically, we performed a genetic interaction screen for yeast deletions that can relieve the toxic effects of PAK1 expression in yeast, thereby identifying orthologous human genes as candidate genetic interaction partners. This is based on our previous study, in which overexpression of human PAK1 was toxic in yeast (21). Subsequent mammalian cell- and tissue-based validation experiments of these candidates identified multiple determinants of glioma cell migration, drug resistance, and proliferation, helping to elucidate the molecular mechanisms of PAK1-mediated carcinogenesis and other cellular behaviors.
Results
Identification of PAK1 genetic interactions
Functional analysis of kinase signaling pathways can be achieved by mapping genetic interaction networks for kinase genes (22–24). Here we sought genes for which perturbation modifies the activity of PAK1 kinase. Because Saccharomyces cerevisiae has been a faithful model for many conserved aspects of eukaryotic cell biology (25), and because genetic interaction screens can be efficiently carried out in yeast, we used a pool of yeast deletion mutants that may modify the PAK1 toxicity phenotype. To identify PAK1 genetic interactions, we previously performed a genome-wide pooled screen to identify genetic interactions on the basis of toxicity modification (21). In that screen, the PAK1 gene was first introduced into a pool of 4,653 homozygous diploid yeast deletion mutants such that each mutant harbors unique barcode sequences flanking the deletion locus (26). Second, PAK1 gene expression was induced in yeast cells grown on galactose media. Third, yeast barcodes were amplified from deletion pool cultures. Finally, yeast barcode abundances were quantified using next-generation sequencing (Bar-Seq) to identify fitness values of PAK1 genetic interactions (27). The relative abundance of each yeast barcode is a proxy for differential growth of the corresponding deletion strain, which allowed us to detect modulation of PAK1 toxicity in the absence of a specific yeast gene (28, 29). For each of 4,653 yeast deletions, we identified the relative abundance of each deletion strain after selection in the presence of PAK1. Genetic interactions were identified on the basis of Z-scores: Z-scores >1.96 were identified as toxicity suppressors. Z-score of 1.96 corresponds to confidence level 95% and p value <0.05, which is commonly used for statistical significance (21, 30). Among the 402 yeast suppressors of PAK1 toxicity, 131 yeast genes had human orthologs in our previous study (21). Human orthologs of the yeast toxicity suppressors were identified using the Karolinska Institute's InParanoid Database (31). Of these 131 genes, 19 genes (15%) that had human orthologs and were expressed in glioma cells were selected for further evaluation in the current study (Table 1 and Fig. S1).
Table 1.
List of PAK1 genetic interactions expressed in glioma cells
| No. | Yeast gene names | UniProt ID | Description | Human orthologs | |
|---|---|---|---|---|---|
| 1 | YMR246W | FAA4 | P47912 | Long-chain fatty acid–CoA ligase 4, EC 6.2.1.3 (fatty acid activator 4) (long-chain acyl-CoA synthetase 4) | ACSL4 |
| 2 | YHR028C | DAP2 | P18962 | Dipeptidyl aminopeptidase B, DPAP B, EC 3.4.14.– (YSCV) | DPP4 |
| 3 | YCL011C | GBP2 | P25555 | Single-strand telomeric DNA–binding protein GBP2, G-strand–binding protein 2 (RAP1 localization factor 6) | ELAVL1 |
| 4 | YBL003C | HTA2 | P04912 | Histone H2A.2 | H2AFX |
| 5 | YBR010W | HHT1 | P61830 | Histone H3 | H3F3A |
| 6 | YAL005C | SSA1 | P10591 | Heat shock protein SSA1 (heat shock protein YG100) | HSPA8 |
| 7 | YEL009C | GCN4 | P03069 | General control protein GCN4 (amino acid biosynthesis regulatory protein) | JUNB |
| 8 | YBL063W | KIP1 | P28742 | Kinesin-like protein KIP1 (chromosome instability protein 9) | KIF11 |
| 9 | YER007C-A | TMA20 | P89886 | Translation machinery–associated protein 20 | MCTS1 |
| 10 | YBL091C | MAP2 | P38174 | Methionine aminopeptidase 2, MAP 2, MetAP 2, EC 3.4.11.18 (peptidase M) | METAP2 |
| 11 | YJR066W | TOR1 | P35169 | Serine/threonine-protein kinase TOR1, EC 2.7.11.1 (dominant rapamycin resistance protein 1) (phosphatidylinositol kinase homolog TOR1) (target of rapamycin kinase 1) | mTOR |
| 12 | YPL006W | NCR1 | Q12200 | NPC intracellular cholesterol transporter 1–related protein 1 (Niemann–Pick type C–related protein 1) | NPC1 |
| 13 | YBL024W | NCL1 | P38205 | Multisite-specific tRNA:(cytosine-C(5))-methyltransferase, EC 2.1.1.202 (multisite-specific tRNA:m5C-methyltransferase) (tRNA (cytosine-5-)-methyltransferase NCL1) (tRNA methyltransferase 4) | NSUN2 |
| 14 | YOR347C | PYK2 | P52489 | Pyruvate kinase 2, PK 2, EC 2.7.1.40 | PKM1, PKM2 |
| 15 | YJR100C | AIM25 | P47140 | Altered inheritance rate of mitochondria protein 25 | PLSCR1 |
| 16 | YJL134W | LCB3 | P47013 | Dihydrosphingosine-1-phosphate phosphatase LCB3, EC 3.1.3.- (long-chain base protein 3) (sphingolipid resistance protein 2) | SGPP1 |
| 17 | YHR114W | BZZ1 | P38822 | Protein BZZ1 (LAS17-binding protein 7) | TRIP10 |
| 18 | YCR008W | SAT4 | P25333 | Serine/threonine-protein kinase HAL4/SAT4, EC 2.7.11.1 (halotolerance protein 4) | TTK |
| 19 | YER177W | BMH1 | P29311 | Protein BMH1 | YWHAE |
Genetic interactions of PAK1 in mammalian cells
For functional evaluation of the PAK1 genetic interactions, siRNA-mediated knockdown in glioma cells was performed for these 20 orthologs of yeast interaction partners (note that, for yeast PYK2, we tested two orthologs, PKM1 and PKM2). Five siRNAs were designed for each PAK1 interaction partner (see Table S1 for siRNA sequences and Fig. S1 for knockdown efficiency). In cases where all five siRNAs were ineffective in knocking down the expression of the corresponding gene, an additional siRNA was designed and synthesized. We tested whether the expression knockdown of candidate genes could modify PAK1-induced cell migration and death/survival (see Fig. S2 for knockdown efficiency of siRNAs in PAK1 transfectants). Cell migration was first assessed using GL26 mouse glioma cells stably overexpressing PAK1 (Fig. S3). Because cells expressing either WT or a constitutively active mutant PAK1 cDNA showed similar PAK1 phosphorylation levels (Fig. S4) and enhanced cell migration phenotypes (Fig. S5), further experiments were performed only with WT PAK1-expressing GL26 cells (32). We could not determine exactly whether exogenous PAK1 expression affected endogenous PAK1 activity. Overexpression of WT kinases often induces or enhances their own activity through autophosphorylation, etc. as a result of a potential “artifact” of overexpression (33–35). Thus, under the overexpression conditions, WT and a constitutively active mutant form of PAK1 might have exhibited similar levels of activation. We used two stable cell clones, GL26-PAK1-1 and GL26-PAK1-2, that each showed PAK1 overexpression and activation as indicated by phosphorylation. Overexpression of kinases is commonly used to determine the role of kinases in cancer-related signaling (33). Among the 5–6 siRNAs for each interaction partner, the one with the best knockdown efficiency was introduced into PAK1-transfected GL26 cells, and cell migration was assessed by a wound-healing assay. Among the 20 PAK1 interaction partners, knockdown of seven genes (DPP4, KIF11, mTOR, PKM2, SGPP1, TTK, and YWHAE) attenuated PAK1-induced cell migration, indicating that these genes related to proteases, mitotic spindle, autophagy, and metabolism of glucose and sphingolipids genetically interact with PAK1 to affect cell migration (Table 2 and Fig. S6). The cell migration assays were performed under the condition of no significant cytotoxicity (Fig. S7).
Table 2.
Evaluation of candidate PAK1 human genetic interactions with respect to cell migration using siRNAs
| Serial no. | Gene name | siRNA no. | Migration assaya |
|
|---|---|---|---|---|
| GL26-PAK1-1 | GL26-PAK1-2 | |||
| 1 | ACSL4 | 5 | − | − |
| 2 | DPP4 | 1 | + | + |
| 3 | ELAVL1 | 5 | − | − |
| 4 | H2AFX | 1 | − | − |
| 5 | H3F3A | 1 | − | − |
| 6 | HSPA8 | 1 | − | − |
| 7 | JUNB | 2 | − | − |
| 8 | KIF11 | 5 | + | + |
| 9 | MCTS1 | 1 | − | − |
| 10 | METAP2 | 1 | − | − |
| 11 | mTOR | 1 | + | + |
| 12 | NPC1 | 1 | − | − |
| 13 | NSUN2 | 3 | − | − |
| 14 | PKM1 | 4 | − | − |
| 15 | PKM2 | 6 | + | + |
| 16 | PLSCR1 | 4 | − | − |
| 17 | SGPP1 | 1 | + | + |
| 18 | TRIP10 | 5 | − | − |
| 19 | TTK | 1 | + | + |
| 20 | YWHAE | 5 | + | + |
a Genetic interaction with p value tested by wound-healing assay in two independent clones of PAK1 stable transfectants (GL26-PAK1-1 and GL26-PAK1-2) (+, p < 0.05; −, not significant).
Pharmacological inhibitors were next used to further investigate the yeast-orthologous human genetic modifiers of PAK1 identified in the large-scale screen. Among the 20 PAK1 interaction partners tested by siRNA-mediated knockdown, pharmacological inhibitors were commercially available for six candidates. These six pharmacological inhibitors were used in wound-healing assays in a manner similar to that used for the siRNA-mediated knockdown experiments. Among the six pharmacological inhibitors, four compounds (monastrol, KIF11 inhibitor; MPS1-IN-1, TTK inhibitor; rapamycin, mTOR inhibitor; vildagliptin, DPP4 inhibitor) reduced PAK1-induced GL26 glioma cell migration, whereas two compounds (fumagillin, METAP2 inhibitor; P-M2tide, PKM2 inhibitor) had no significant effects (Table 3 and Fig. S8). All inhibitors were used at optimal concentrations without apparent cytotoxicity based on the IC50 as reported in previous studies (36–39) and as determined by a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) cell viability assay (Figs. S9 and S10). The results of the pharmacological inhibitor experiments were consistent with those of the siRNA-mediated genetic knockdown experiments except for PKM2. PKM2 inhibitor P-M2tide did not significantly influence PAK1-induced cell migration, whereas siRNA-mediated knockdown of PKM2 gene expression had significant effects. This discrepancy may be due to the complex role of PKM2 expression and activity in regulating cell behaviors (40–42).
Table 3.
Evaluation of candidate PAK1 genetic interactions with respect to cell migration using pharmacological inhibitors
| Serial no. | Gene name | Inhibitor name | Migration assaya |
|
|---|---|---|---|---|
| GL26-PAK1-1 | GL26-PAK1-2 | |||
| 1 | DPP4 | Vildagliptin | + | + |
| 2 | KIF11 | Monastrol | + | + |
| 3 | METAP2 | Fumagillin | − | − |
| 4 | mTOR | Rapamycin | + | + |
| 5 | PKM2 | P-M2tide | − | − |
| 6 | TTK | MPS1-IN-1 | + | + |
aGenetic interaction with p value tested by wound-healing assay in two independent clones of PAK1 stable transfectants (GL26-PAK1-1 and GL26-PAK1-2) (+, p < 0.05; −, not significant).
For the functional evaluation of PAK1 genetic interactions with respect to cell death/survival phenotypes, glioma cells were used for anticancer drug toxicity testing and cell proliferation assays. PAK1-overexpressing GL26 glioma cells were transfected with siRNAs and then exposed to bis-chloroethylnitrosourea (BCNU), an anticancer drug that is similar to temozolomide, which is commonly used in the treatment of glioma patients. Glioma cells stably expressing PAK1 clearly showed increased drug resistance compared with that of parental cells. Knockdown of PAK1 interaction partner expression using siRNAs modulated glioma cell death (PAK1-induced drug resistance), as evaluated by lactate dehydrogenase (LDH) and MTT assays (Table 4 and Figs. S11 and S12), indicating genetic interactions between PAK1 and the knocked down genes (DPP4, mTOR, SGPP1, TTK, and YWHAE). A BCNU concentration of 200 μm was used for these experiments, based on dose-dependent toxicity test results (Fig. S13), and the mode of cell death was primarily apoptosis, as determined by a caspase-3 activity assay (Fig. S14). Differences between controls and PAK1-overexpressing cells were modest in some of the results. However, the difference was statistically significant in the key comparisons.
Table 4.
Evaluation of candidate PAK1 genetic interactions with respect to drug resistance using siRNAs
| Serial no. | Gene name | siRNA no. | LDH assaya |
MTT assayb |
||
|---|---|---|---|---|---|---|
| GL26-PAK1-1 | GL26-PAK1-2 | GL26-PAK1-1 | GL26-PAK1-2 | |||
| 1 | ACSL4 | 5 | − | − | − | − |
| 2 | DPP4 | 1 | + | + | + | ++ |
| 3 | ELAVL1 | 5 | − | − | − | − |
| 4 | H2AFX | 1 | − | − | − | − |
| 5 | H3F3A | 1 | − | − | − | − |
| 6 | HSPA8 | 1 | − | − | − | − |
| 7 | JUNB | 2 | − | − | − | − |
| 8 | KIF11 | 5 | − | − | − | − |
| 9 | MCTS1 | 1 | − | − | − | − |
| 10 | METAP2 | 1 | − | − | − | − |
| 11 | mTOR | 1 | + | ++ | + | + |
| 12 | NPC1 | 1 | − | − | − | − |
| 13 | NSUN2 | 3 | − | − | − | − |
| 14 | PKM1 | 4 | − | − | − | − |
| 15 | PKM2 | 6 | − | − | − | − |
| 16 | PLSCR1 | 4 | − | − | − | − |
| 17 | SGPP1 | 1 | + | ++ | + | ++ |
| 18 | TRIP10 | 5 | − | − | − | − |
| 19 | TTK | 1 | ++ | + | ++ | + |
| 20 | YWHAE | 5 | ++ | + | + | + |
a Genetic interaction with p value tested by LDH assay in two independent clones of PAK1 stable transfectants (GL26-PAK1-1 and GL26-PAK1-2) (+, p < 0.05; ++, p < 0.01; −, not significant).
b Genetic interaction with p value tested by MTT assay in two independent clones of PAK1 stable transfectants (GL26-PAK1-1 and GL26-PAK1-2) (+, p < 0.05; ++, p < 0.01; −, not significant).
Functional evaluation of PAK1 genetic interactions with respect to glioma cell proliferation was performed using the six pharmacological inhibitors of the interaction partners. The proliferation rate of PAK1-expressing GL26 glioma cells was measured by real-time imaging of cell confluence using IncuCyte (43), and the results are presented as the cell doubling time. Inhibition of DPP4, METAP2, mTOR, PKM2, and TTK attenuated PAK1-induced effects on glioma cell proliferation, indicating genetic interactions between PAK1 and these five genes in terms of cell proliferation (Table 5 and Fig. S15). Taken together, these results indicate that PAK1 exhibited similar genetic interactions with respect to cell migration, cell death/survival, and proliferation phenotypes in the cultured glioma cell model. These results suggest that the PAK1 genetic interactions identified in the yeast-based screen may be relevant to glioma growth, invasion, and chemoresistance in vivo (see below).
Table 5.
Evaluation of PAK1 genetic interactions with respect to cell proliferation using pharmacological inhibitors
| Serial no. | Gene name | Inhibitor name | Proliferation assaya |
|
|---|---|---|---|---|
| GL26-PAK1-1 | GL26-PAK1-2 | |||
| 1 | DPP4 | Vildagliptin | + | ++ |
| 2 | KIF11 | Monastrol | − | − |
| 3 | METAP2 | Fumagillin | ++ | + |
| 4 | mTOR | Rapamycin | + | + |
| 5 | PKM2 | P-M2tide | ++ | + |
| 6 | TTK | MPS1-IN-1 | + | ++ |
a Genetic interaction with p value tested by cell proliferation assay in two independent clones of PAK1 stable transfectants (GL26-PAK1-1 and GL26-PAK1-2) (+, p < 0.05; ++, p < 0.01; −, not significant).
We next tested the impact of the candidate modifiers on the expression of the PAK1 gene. For none of the eight major interaction partners did siRNA-mediated knockdown significantly affect the expression levels of PAK1 in glioma cells (Fig. S16), indicating that the modulation of PAK1-mediated effects on cell phenotypes (cell migration, cell death/survival, and proliferation) by the genetic interactors was not due to the down-regulation of PAK1 gene expression.
Evaluation of genetic interactions of PAK1 in brain slice cultures and orthotopic glioma models
Among the 20 genetic interactors of PAK1 tested in mammalian cells, DPP4, KIF11, METAP2, mTOR, and TTK were subjected to subsequent evaluation using brain slice cultures (Fig. 1). In this experiment, the invasive capacity of glioma cells transplanted onto organotypic cultures of brain slices was evaluated. To examine the genetic interactions between PAK1 and its interacting partners, GL26 glioma cells overexpressing PAK1 were transplanted onto brain slices, and the degree of glioma cell invasion was monitored in the presence of specific inhibitors of the five interacting partners. Compared with empty vector–transfected control cells, PAK1-transfected glioma cells (GL26-PAK1-1 and GL26-PAK1-2) showed higher degrees of cell invasion when transplanted onto brain slice cultures. The PAK1-induced enhancement of glioma cell invasion was diminished by MPS1-IN-1 (TTK inhibitor), rapamycin (mTOR inhibitor), and vildagliptin (DPP4 inhibitor) but not by fumagillin (METAP2 inhibitor) or monastrol (KIF11 inhibitor) (Fig. 1). The discrepancy between these results and the wound-healing assay performed in a cultured cell line (Table 3) may reflect differences in the migration and invasion capacity of glioma cells in the distinct microenvironments of the two assay systems. Nevertheless, PAK1 genetic interactions with DPP4, mTOR, and TTK were consistent in both assays.
Figure 1.
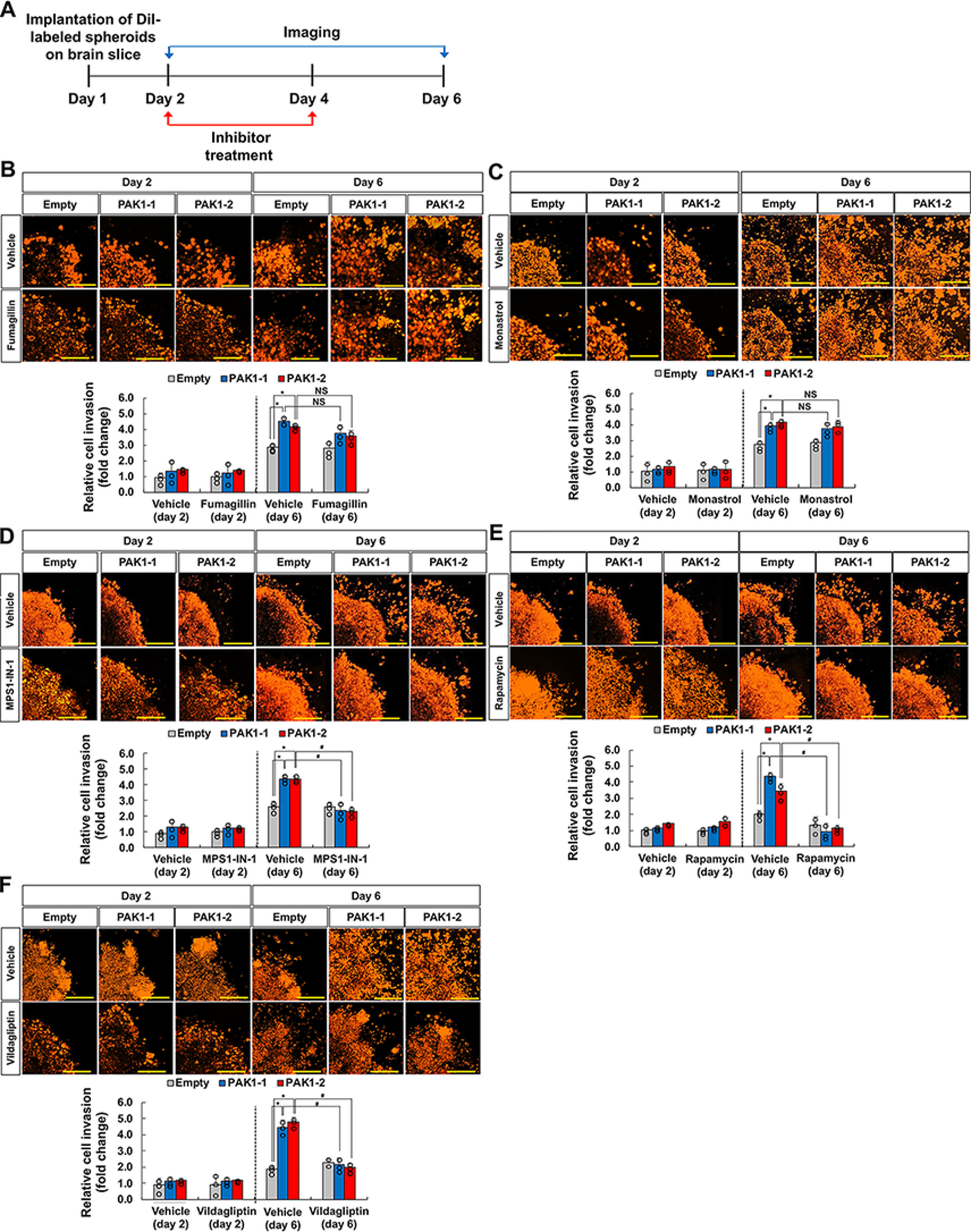
Evaluation of PAK1 genetic interactions in organotypic brain slices. A, experimental timeline. DiI-labeled spheroids were implanted onto brain slice cultures on day 1. Confocal imaging and inhibitor treatment were performed at the given time points. B–F, GL26 glioma spheroids stably expressing empty vector or PAK1 (GL26-PAK1-1 and GL26-PAK1-2) were implanted onto the slice cultures followed by treatment with vehicle or pharmacological inhibitors of potential PAK1 interaction partners: fumagillin, 1 nm (B); monastrol, 1 μm (C); MPS1-IN-1, 100 nm (D); rapamycin, 100 nm (E); vildagliptin, 250 nm (F). The Z-stack images were superimposed into a single image representing the entire spheroid. Invasive cell density was quantified as shown in the graphs. Scale bar, 200 μm. *, p < 0.05 (Student's t test comparing empty versus PAK1 groups, n = 3); #, p < 0.05 (Student's t test comparing vehicle versus inhibitor groups, n = 3); NS, not significant. Error bars, S.D.
Next, an orthotopic mouse model of glioma was used to test the genetic interactions between PAK1 and DPP4, mTOR, and TTK. These interaction partners were selected for genetic interaction evaluation in vivo based on the brain slice culture experiments. PAK1-expressing GL26 glioma cells (GL26-PAK1-1) were injected into the striatum of the mouse brain, and glioma behaviors were assessed by bioluminescence live imaging and histological analyses. Glioma growth was continuously measured up to 16 days after intracranial implantation of glioma cells using real-time bioluminescence imaging to detect GL26 glioma cells transfected with both PAK1 and luciferase. Pharmacological inhibition of the interaction partners by specific inhibitors reduced PAK1-induced increases in glioma growth (Fig. 2), volume, and invasion in the mouse brain (Fig. 3 and Figs. S17 and S18). At the end of the live imaging analysis, animals were sacrificed for histological analyses. Glioma volume was measured by quantitative digital image analysis of serial brain sections containing tumors (Fig. 3, A and B). Glioma invasion was quantified by particle analysis of threshold images (Fig. 3C and Fig. S17) or invasive finger analysis (Fig. S18) of tumor-bearing brain sections. The results indicated strong genetic interactions between PAK1 and these partners (DPP4, mTOR, and TTK) with respect to glioma growth and invasion in vivo.
Figure 2.
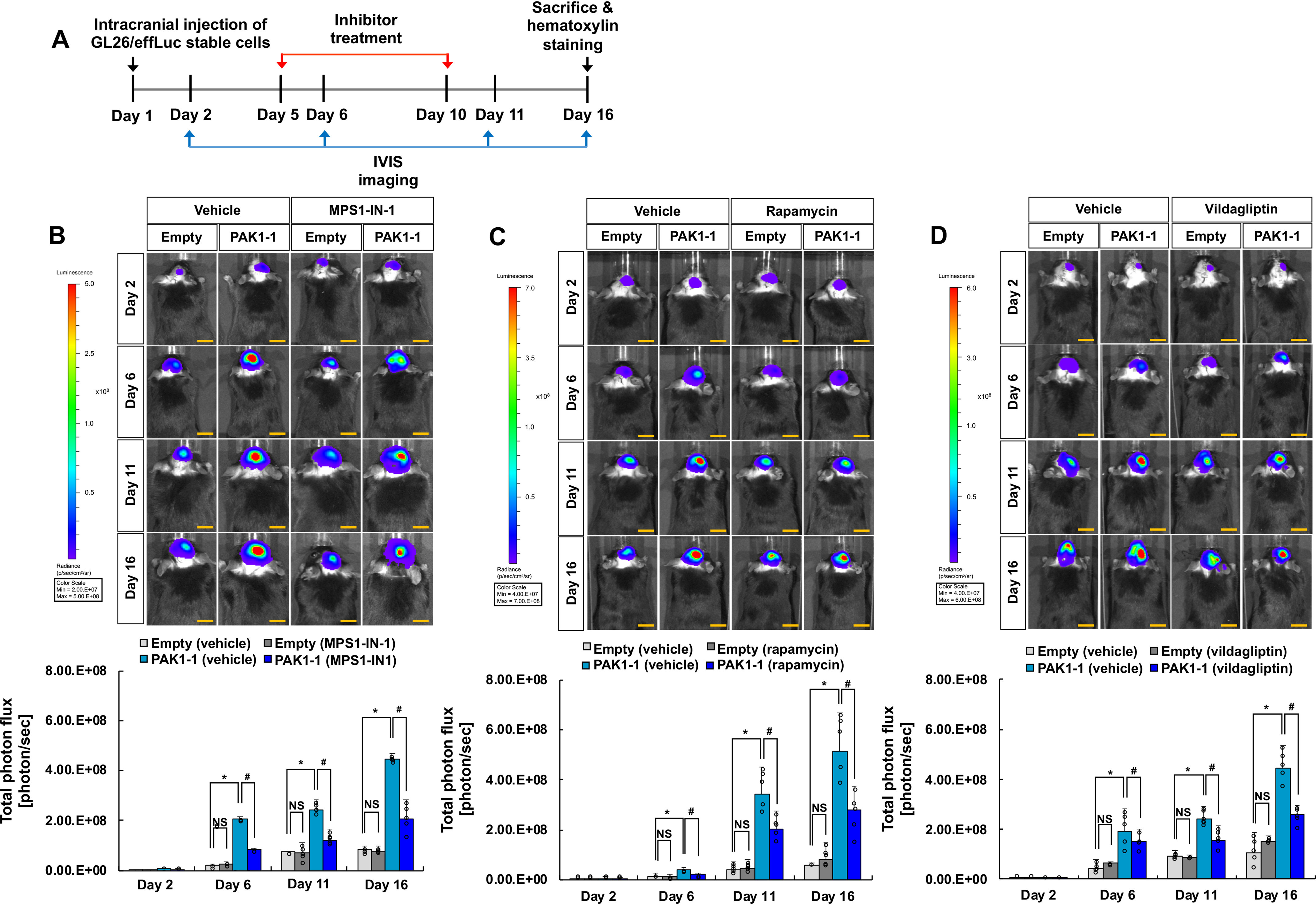
Evaluation of PAK1 genetic interactions in an orthotopic model of glioma. A, experimental timeline. C57BL/6 mice were injected with 9 × 105 empty vector– or PAK1–expressing GL26-effLuc cells in the striatum. Glioma-implanted mice were injected with inhibitors (MPS1-IN-1, rapamycin, or vildagliptin) or vehicle, and IVIS images were obtained at the indicated time points to assess glioma growth. B–D, IVIS images were collected on days 2, 6, 11, and 16, and glioma growth in the presence of vehicle or inhibitors was quantified in the graphs: MPS1-IN-1, 100 nm, 5 µl (B); rapamycin, 100 nm, 5 µl (C); vildagliptin, 250 nm, 5 µl (D). Photon intensity of the images is directly related to tumor size. Scale bar, 5 mm. *, p < 0.05 (Student's t test comparing empty versus PAK1-1 group, n = 5); #, p < 0.05 (Student's t test comparing vehicle versus inhibitor groups, n = 5); NS, not significant. Error bars, S.D.
Figure 3.
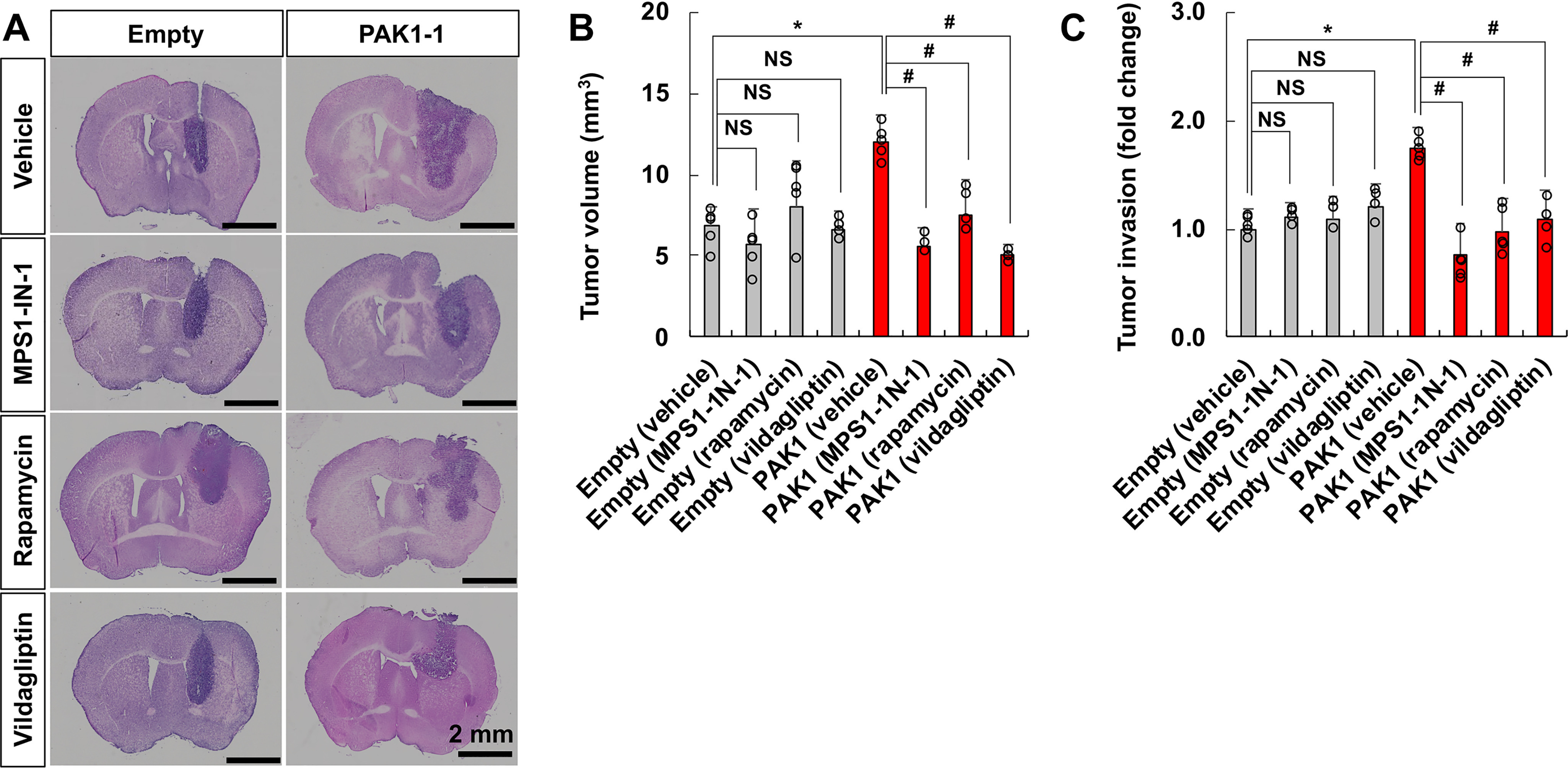
Assessment of tumor volume and invasiveness in an orthotopic model of glioma. After IVIS imaging (as in Fig. 2), mice (n = 5) from each group (empty vector– or PAK1–transfected GL26 glioma cells implanted; vehicle or inhibitors injected) were euthanized to obtain brain tissues, and the tumor volume (A and B) and invasiveness (C) were evaluated. A, representative hematoxylin-stained images from serial sections confirm viable regions of tumor. B, tumor volume was calculated by quantitative digital image analysis of the serial sections containing tumor. C, overall tumor invasion was quantified by particle analysis of threshold images prepared from tumor-bearing sections of engrafted animals. Tumor invasion in each group of animals was assessed by counting the number of tumor particles invading the brain parenchyma per tumor rim length. Scale bar, 2 mm. *, p < 0.05 (Student's t test, n = 5); #, p < 0.05 (Student's t test, n = 5); NS, not significant. Error bars, S.D.
AKT1 as a common downstream mediator of the PAK1-TTK genetic interaction
Among the genetic interactions confirmed in brain slice cultures and orthotopic glioma models, a subsequent mechanistic study focused on the genetic interaction between PAK1 and TTK. Because AKT1 has been previously implicated in cell migration and is known to act downstream of the PAK1 signaling pathway (44–48), the potential role of AKT1 in the genetic interaction between PAK1 and TTK was investigated. AKT1 was identified as a common downstream component of PAK1- and TTK-mediated cell migration signaling pathways (Fig. 4). Transient overexpression of either PAK1 or TTK induced AKT1 phosphorylation in NIH3T3 cells (Fig. 4B). Inhibition of TTK using MPS1-IN-1 reduced PAK1-induced AKT1 phosphorylation; however, PAK1 inhibition using IPA3 did not significantly affect TTK-induced AKT1 phosphorylation (Fig. 4B). Similarly, TTK inhibition decreased PAK1-mediated cell migration, whereas PAK1 inhibition did not significantly influence TTK-mediated cell migration (Fig. 4C), indicating that PAK1 acts upstream of TTK in the cell migration signaling pathway (Fig. 4D). Pharmacological inhibitors were used at concentrations that did not exert cytotoxic effects (Fig. S19). Moreover, PAK1 overexpression increased TTK expression at both the mRNA and protein levels (Fig. S20), further suggesting that PAK1 acts upstream of TTK. Taken together, these results suggest that the genetic interaction between PAK1 and TTK mediating multiple behaviors of glioma cells may involve (i) activation of the common downstream mediator AKT1 and (ii) PAK1 up-regulation of TTK expression. The molecular mechanisms of the genetic interactions between PAK1 and DPP4 or mTOR, which were confirmed in the in vivo glioma model, need to be further investigated; however, the PAK1-mTOR genetic interaction could be explained by a pathway similar to that of the PAK1-TTK interaction (Fig. 4D), as mTOR has been previously shown to activate AKT1 (49, 50).
Figure 4.

Role of AKT1 in the genetic interaction between PAK1 and TTK. A, experimental timeline. NIH3T3 cells were transiently transfected with empty vector, PAK1, or TTK cDNAs and treated with inhibitors (TTK inhibitor, MPS1-IN-1; PAK1 inhibitor, IPA3), and signaling pathway or cell migration was analyzed by Western blotting or a wound-healing assay, respectively. B, phospho-AKT (p-AKT), total AKT (t-AKT), or β-actin protein levels were measured by Western blotting. Quantification of the band intensities is presented in the adjacent graphs. *, p < 0.05 (Student's t test, n = 3); #, p < 0.05 (Student's t test, n = 3). C, quantification of cell migration based on the wound-healing assays. *, p < 0.05 (Student's t test comparing empty vector–transfected versus TTK- or PAK1-transfected group, n = 5); #, p < 0.05 (Student's t test, n = 5). Error bars, S.D. D, schematic representation of the PAK1, TTK, and AKT pathways. The PAK1-TTK-AKT1 axis appears to play an important role in glioma migration/invasion, drug resistance, and proliferation. PAK1-mTOR or PAK1-DPP4 may play a similar role through either AKT1-dependent or -independent pathways.
Validation of the genetic interactions via overexpression of PAK1 interactors
To further investigate the genetic interactions between PAK1 and its interactors, we tested whether the overexpression of some of the PAK1 genetic interactors rescues cell migration, proliferation, drug resistance, and glioma invasion under PAK1-inhibited conditions. For this, mTOR, PKM2, TTK, and AKT1 were either activated or overexpressed in GL26 glioma cells, which were treated with a PAK1 inhibitor (IPA3). Rheb cDNA overexpression was used to activate the mTOR pathway (51), DASA-58 was used as a PKM2 activator, TTK cDNA was overexpressed, SC-79 was used as an AKT1 activator, or Myr-AKT1 cDNA (52) was overexpressed. All compounds were used at the concentrations of no significant cytotoxicity (Fig. S21), and the overexpression of the PAK1 interactors was confirmed by Western blotting or fluorescence imaging (Fig. S22). In the subsequent experiments, migration (Figs. S23–S25), proliferation (Fig. S26), drug resistance (Fig. S27), and glioma invasion (Fig. S28) were all rescued by either activation or overexpression of mTOR, PKM2, TTK, or AKT1 in PAK1-inhibited glioma cells, further supporting the genetic interactions between PAK1 and its effector molecules.
Discussion
Analyses of PAK1 genetic interactions identified many new potential determinants of glioma cell migration, drug resistance, and growth. Among the 131 human genetic interaction partners of PAK1, many were previously associated with cell motility and death/survival. Interestingly, PAK1 is also known to be involved in glioma cell migration, death/survival, and proliferation (15–19). Therefore, several interaction partners of PAK1 were chosen for further investigation using cultured glioma cells, organotypic brain slice cultures, and an orthotopic in vivo glioma model. Genetic interactions between PAK1 and the candidate genes were evaluated in these experimental models with respect to various glioma cell behaviors, such as cell migration, invasion, drug resistance, growth, and proliferation. During this process, a few candidates (DPP4, mTOR, and TTK) were selected for further evaluation of their consistent genetic interactions with PAK1 in an animal model. Finally, one of the genetic interaction partners, TTK, which is itself a protein kinase and has been previously implicated in mitotic spindle regulation (53, 54), was subjected to mechanistic study. After demonstrating the important role of TTK in PAK1-induced glioma cell migration, drug resistance, and growth in vitro and in vivo, we further found that AKT1 is a point of convergence in the signaling pathways of the two kinases PAK1 and TTK.
Overexpression of either PAK1 or TTK induced AKT1 activation and enhanced cell migration. PAK1 was found to be upstream of TTK in the cell migration pathway on the basis of signaling pathway analyses using kinase inhibitors and transient overexpression of the kinases (Fig. 4). However, no protein-protein interaction between PAK1 and TTK was observed in the co-immunoprecipitation assay (Fig. S29). PAK1 and TTK seem to share a common signaling pathway that leads to AKT1 activation and cell migration (Fig. 4D). This relationship among the three kinases (PAK1, TTK, and AKT1) may be important in view of glioma chemotherapy. Inhibition of the common AKT1 pathway may have therapeutic potential for either PAK1- or TTK-mediated oncogenic events. Our results also provide a mechanistic basis for mono- or combination chemotherapy of malignant glioma in the future.
In the evaluation of PAK1 genetic interactions using mammalian cells, we only tested 20 genetic interaction partners among the 131 candidates. The other 111 interaction partners with human orthologs may also be relevant in the context of PAK1 and glioma. In fact, the 131 candidates were associated with many molecular functions and biological processes (Tables S2 and S3), which can be further explored to better understand glioma pathogenesis. However, it remains to be determined to what extent genetic interaction discovered in yeast based on the fitness/toxicity of PAK1 would be relevant in human glioma. Among the 20 PAK1 genetic interactions, not all leads from the yeast toxicity screen were relevant in mammalian cell phenotypes, such as mouse glioma cell migration, drug resistance, and growth. This might be due to differences in the mechanisms underlying the modification of yeast toxicity and mammalian cell phenotypes. For example, the mechanism of PAK1 toxicity suppression by deletion of DAP2, KIP1, MAP2, TOR1, or SAT4 in yeast may be different from that of modification of glioma cell phenotypes by knockdown of their orthologs (DPP4, KIF11, METAP2, mTOR, or TTK) in mammalian cells (see Table 1 for the list of PAK1 genetic interactions in yeast and their human orthologs). Despite these limitations, current results provided testable and subsequently validated hypotheses relating to biological functions of PAK1 in glioma. Moreover, our analyses newly identified numerous determinants of glioma cell migration, drug resistance, and growth as potential drug targets for glioma treatment.
Materials and methods
Yeast strains, media, and plasmids
BY4742 (Mat α; his3Δ1; leu2Δ0, lys2Δ0; ura3Δ0) was used as a WT yeast strain in this study. The Homozygous Diploid Complete Set of Yeast Deletion Clones and Homozygous Diploid Yeast Deletion Pools were purchased from Invitrogen (Carlsbad, CA, USA). Yeast cells were grown in rich medium (YPD) or synthetic medium lacking leucine and containing 2% glucose (SD−Leu), raffinose (SRaf−Leu), or galactose (SGal−Leu). WT and constitutively active mutant (T423E) PAK1 cDNAs were kindly provided by Prof. Eung-Gook Kim at Chungbuk National University (Cheongju, South Korea) (55–57). The Gateway LR reaction was used to shuttle PAK1 cDNA into pAG425GAL-ccdB (Addgene, Cambridge, MA, USA) (58) for yeast expression. All plasmids were 2-μm-based and expressed under the control of the GAL1 promoter. All constructs were verified by Sanger sequencing. PAK1 cDNA in pAG425GAL (yeast destination vector) was transformed into BY4742 or homozygous diploid deletion strains. All yeast strains were grown at 30 °C according to the standard protocol. We used the LiAc/SS carrier DNA/PEG method to transform yeast with plasmid DNA as described previously (59). For functional studies in mammalian cells or mice, the Gateway LR reaction was used to shuttle PAK1 cDNA into pDS-GFP-XB (Invitrogen) destination vectors.
PAK1 genetic interaction screen
The screen for genetic modifiers of PAK1 in S. cerevisiae was carried out as described (Fig. S30) (21). Briefly, PAK1 cDNAs were transformed into homozygous diploid yeast deletion pools containing 4,653 individual deletion clones. Transformants were incubated in SD−Leu medium for 16 h. The cells were washed twice with PBS and then incubated in SGal−Leu medium for 2 days. Cells remaining in glucose-containing SD−Leu medium were used as a control. Genomic DNA was isolated from cells harvested after pooled growth. Each 20-mer UPTAG barcode was amplified and sequenced using a Genome Analyzer (Illumina, San Diego, CA, USA) according to the manufacturer's protocols.
Cell cultures and reagents
NIH3T3 mouse fibroblasts were purchased from ATCC, and GL26 mouse glioma cells were kindly provided by Prof. Eun Jung Park (Cancer Immunology Branch, National Cancer Center, Goyang, Gyeonggi, South Korea). Both cell lines were authenticated, tested negative for Mycoplasma, and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 100 units/ml penicillin at 37 °C. For the stable transfection of GL26 cells, cells were transfected with pDS-GFP-XB containing WT or mutant (T423E) human PAK1 cDNA using Fugene HD transfection reagent (Invitrogen), according to the manufacturer's instructions. Stable transfectants were established in the presence of 1 mg/ml G418 (Sigma–Aldrich), and their GFP expression was observed under fluorescence microscopy (BX50; Olympus, Tokyo, Japan). Desalted and preannealed siRNA duplexes were purchased from Genolution Pharmaceuticals (Seoul, South Korea). The siRNAs were designed using a proprietary algorithm devised by Genolution Pharmaceuticals. The siRNAs were transfected into cells using RNAi MAX reagent (Invitrogen). Transient transfection of NIH3T3 and GL26 cells with human PAK1, TTK, or AKT cDNA was similarly performed using Fugene HD or Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA). Rapamycin (catalog no. 553210) was purchased from Calbiochem. Monastrol (catalog no. M8515) was purchased from Sigma–Aldrich. MPS1-IN-1 (catalog no. 5142) and IPA3 (catalog no. 3622) were purchased from Tocris Bioscience (Bristol, UK). Fumagillin (catalog no. ab141527) was purchased from Abcam (Cambridge, MA, USA). P-M2tide (catalog no. BML-P239) was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Vildagliptin (catalog no. S3033) and DASA-58 (catalog no. S7928) were purchased from Selleckchem (Houston, TX, USA). SC-79 (catalog no. 4635) was purchased from Tocris Bioscience (Bristol, UK). A summary of pharmacological agents used in this study is shown in Table S4.
RT-PCR
Isolated cells were homogenized using TRIzol reagent (Invitrogen) to obtain total RNA. cDNA was then synthesized using Superscript II (Invitrogen) and oligo(dT) primer. Traditional PCR amplification was conducted using specific primer sets (Table S5). Electrophoresis was conducted to analyze PCRs using 1.2% agarose gels.
Wound-healing assay
In vitro wound-healing assay was performed using the IncuCyte ZOOM Live-Cell Imaging system (Essen Bioscience, Ann Arbor, MI, USA). Cells were seeded at a density of 2.5 × 104 cells/well in IncuCyte® ImageLock plates. Cells were then treated with mitomycin C (5 μg/ml) for 2 h before introduction of a wound to inhibit cell proliferation. Wounds were made with an IncuCyte WoundMaker™, and plates were automatically analyzed for wound closure with the IncuCyte ZOOM Live-Cell Imaging system. siRNA transfection was performed 48 h before wound introduction. Alternatively, inhibitor treatment was performed immediately after wound introduction. Real-time images were acquired every 2–3 h for 48 h. Cell confluence was quantified using time-lapse curves generated by IncuCyte ZOOM software and converted to wound closure velocity (μm2/h).
Manual scratch wound-healing assays were performed as described previously (60). In brief, scratch wounds were created using a 10-μl pipette tip on confluent cell monolayers in 24-well culture plates containing DMEM with 10% FBS and 100 units/ml penicillin. Cells were incubated at 37 °C under 5% CO2 during migration of the monolayer into the cleared wound area. The wound area was observed by microscopy (×100 magnification). Relative cell migration distance was determined by measuring the wound width and subtracting this from the initial value: Cell migration distance = initial wound width at day 0 – wound width on the day of measurement. Three non overlapping fields were selected and examined in each well (three wells per experimental group). The results are presented as the -fold increase in migration distance (wound closure).
Boyden chamber migration assay
Cell migration was also determined using a 48-well Boyden chamber (Neuroprobe, Gaithersburg, MD, USA), according to the manufacturer's instructions. DMEM with 10% FBS was placed into the base wells and separated from the top wells by polycarbonate filters (8-μm pore size; 25 × 80 mm, polyvinylpyrrolidone-free; Neuroprobe). Cells were harvested by trypsinization, washed once with serum-free DMEM containing 0.5 mg/ml soybean trypsin inhibitor (Sigma–Aldrich), and washed twice with serum-free DMEM. Cells were resuspended in serum-free medium and added to the upper chamber at 8 × 103 cells/well. Cells were incubated for 16 h at 37 °C under 5% CO2. At the end of the experiment, cells were fixed with methanol for 10 min and stained with Mayer's hematoxylin (Dakocytomation, Glostrup, Denmark) for 20 min. Cells on the upper slide of the membrane were then removed using a cotton swab. Photomicrographs of five randomly chosen fields were taken (×100 magnification), and cells were enumerated to calculate the average number of cells that had migrated.
Cell viability assays
For the MTT assay, cells were treated with various reagents for the designated time period. After treatment, MTT (0.5 mg/ml; Sigma-Aldrich) was added to the cells and incubated at 37 °C in a 5% CO2 incubator. After 2 h, DMSO was added to dissolve the insoluble crystals, and absorbance was measured using a microplate reader (Anthos Labtec Instruments, Wals, Austria) at 570 nm. For the LDH assay, cells were seeded at a density of 2 × 104 cells/well in 96-well plates. Cells were transfected with siRNAs and treated with BCNU at 48 h after cell transfection. After 24 h, culture media were transferred to a new 96-well plate, and LDH activity was measured using the Promega Cytotoxicity kit (Madison, WI, USA) according to the manufacturer's instructions. LDH activity is presented as the percentage of LDH in the culture medium following BCNU or vehicle treatment relative to the maximum LDH release following treatment with cell lysis solution.
Measurement of caspase-3 activity
Caspase-3 activities were assessed using a caspase-3 colorimetric assay kit (Abcam) according to the manufacturer's protocol. Briefly, cells were collected and resuspended in lysis buffer containing 50 mm HEPES (pH 7.4), 0.1% CHAPS, 1 mm DTT, 0.1 mm EDTA, and 0.1% Triton X-100. Following incubation for 30 min on ice, cell lysate was centrifuged at 11,000 × g for 10 min at 4 °C, and the protein concentration in the supernatant was measured using the Bradford dye method. The supernatants were incubated with reaction buffer containing 2 mm Ac-DEVD-AFC and 10 mm DTT at 37 °C for 2 h. Caspase-3 activity was determined by measuring the absorbance at 405 nm.
Cell proliferation assay
The IncuCyte ZOOM Live-Cell Imaging system (Essen Bioscience) was used for monitoring cell proliferation. Cells were seeded at a density of 1 × 103 cells/well in IncuCyte® ImageLock plates. After 24 h, cells were treated with various compounds and analyzed with the IncuCyte ZOOM Live-Cell Imaging system. Real-time images of cell confluence were acquired every 2–3 h for 72 h. Cell proliferation was quantified based on time-lapse curves generated by IncuCyte ZOOM software and is presented as cell doubling time.
Western blotting analysis
Cultured cells were lysed in triple-detergent lysis buffer (50 mm Tris-HCl (pH 8), 150 mm NaCl, 0.02% sodium azide, 0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 1 mm phenylmethylsulfonyl fluoride). Protein concentrations in cell lysates were determined using a protein assay kit (Bio-Rad). Each protein sample was separated by 8 or 12% SDS-PAGE, blotted, and incubated overnight at 4 °C using the following primary antibodies: anti-PAK1 (Cell Signaling Technology (Danvers, MA, USA), catalog no. 2602), anti-phospho-PAK1/2 (Ser-144/Ser-141, Cell Signaling Technology, catalog no. 2606), anti-TTK (Origene (Rockville, MD, USA), TA314651), anti-phospho-AKT (Ser-473, Cell Signaling Technology, catalog no. 9271), anti-AKT (Cell Signaling Technology, catalog no. 9272), anti-p-p70S6K (Thr-389, Cell Signaling Technology, catalog no. 9234), anti-p70S6K (Cell Signaling Technology, catalog no. 2708), anti-GFP (Santa Cruz Biotechnology, Inc. (Dallas, TX, USA), sc-9996), anti-FLAG (Sigma–Aldrich, F7425), anti-α-tubulin (Sigma–Aldrich, T5168), and anti-β-actin (Thermo Fisher Scientific, MAS-15739). Afterward, samples were incubated with horseradish peroxidase–conjugated secondary antibodies and detected using ECL solution.
Mice and animal care procedures
Female C57BL/6 mice (8 weeks old) were obtained from Samtaco (Osan, South Korea). All animal experiments were performed in accordance with animal protocols and guidelines approved by the Animal Care Committee at Kyungpook National University (approval KNU 2018-0084).
Generation of GL26 glioma spheroids
GL25 glioma cell suspensions (GL26-Empty, GL26-PAK1-1, and GL26-PAK1-2) were dispensed into ULA 96-well round-bottomed plates (Corning Inc., Corning, NY, USA) using a multichannel pipette at optimized densities (5 × 104 cells/well). Plates were incubated for 4 days at 37 °C, 5% CO2, and 95% humidity for spheroid formation.
Glioma invasion assay in organotypic brain slice cultures
Organotypic brain slice cultures were prepared by the interface method and grown for the first 3 days in serum-based culture medium as described previously (61–65). Glioma spheroids (200–400 μm) were labeled with 25 μg/ml 1,1′-dioctadecyl-3,3,3′,3′ tetramethylindocarbocyanine perchlorate (DiI) solution (Molecular Probes, Invitrogen) for 24 h, washed, and implanted onto brain slice cultures in the area between the cortex and striatum close to the corpus callosum. To enable capture and correct placement onto brain slice cultures, only spheroids of 200–400 μm were used. Glioma cell invasion was visualized using confocal microscopy (Nikon ECLIPSE TE2000-E inverted microscope). Confocal Z-stacks with 20-μm steps were recorded and superimposed into a single image representing the entire spheroid. This procedure was repeated on days 2 and 6. In the superimposed images, the area of each spheroid was measured using the software Visiomorph (Visiopharm, Hørsholm, Denmark). The spheroids were outlined at the spheroid border, identifying the beginning of the invasion zone. The area of the invasive cells was also measured using a classifier identifying the area of DiI staining, representing only the invasive cells and not the spheroids.
Surgery for mouse orthotopic model of glioma
Glioma cell transplantation surgery was performed as described previously (66, 67) with minor modifications. Mice were anesthetized initially using 2–4% isoflurane and placed on a minivent (Harvard Apparatus, Cambridge, MA, USA). Anesthesia was maintained at 1.5% throughout the surgery. To visualize the transplanted glioma cells, GL26 cells were infected with retroviruses carrying the enhanced firefly luciferase gene (effLuc) and a Thy1.1 (CD90.1) marker. The infected cells were isolated by magnetic-activated cell sorting (Miltenyi Biotech Ltd., Bisley, Surrey, UK) using monoclonal anti-CD90.1 antibody conjugated to magnetic microbeads. The effLuc-expressing GL26 cells were stably transfected with either empty vector or PAK1 constructs to generate GL26-empty/effLuc or GL26-PAK1-1/effLuc cells, respectively. These cells were delivered into the right striatum using a 25-gauge Hamilton syringe and stereotactic frame (Kopf, Tujunga, CA, USA) at an injection rate of 1 μl/min (9 × 105 cells in 2.5 μl of PBS). After tumor implantation, mice received an intratumoral injection of inhibitors (MPS1-IN-1, rapamycin, and vildagliptin) using a 26-gauge Hamilton syringe attached to an automated microinjector. Infusion proceeded at a rate of 0.5 μl/min for 10 min. At the end of the injection, the needle was left in place for an additional 10 min before being retracted slowly.
Bioluminescence imaging of intracranial glioma growth
Intracranial tumor growth was assessed by bioluminescence imaging using the Xenogen IVIS 200 small-animal imaging system (Xenogen Corp., Alameda, CA, USA). The IVIS 200 cooled CCD camera system was used for emitted light acquisition, and Living Image software (Xenogen Corp.) was used for data analysis as an overlay on IGOR software (Wavemetrics Corp., Lake Oswego, OR, USA). Animals received a 200-μl intraperitoneal injection of d-luciferin (Xenogen Corp.) at a concentration of 15 mg/ml under 1–2% inhaled isoflurane anesthesia. Identical illumination settings (lamp voltage, filters, f/stop, field of views, binning, excitation filter block, and emission filter open) were used for acquiring all images. Data were analyzed using total photon flux emission (photons/s) in a region of interest covering the entire brain. After euthanasia with pentobarbital (100 mg/kg, intraperitoneally) followed by extensive perfusion with 4% paraformaldehyde, brains were removed, incubated in 30% sucrose (w/v in PBS) for 18 h, and frozen over liquid nitrogen.
Analysis of tumor volume and invasion in the mouse model of glioma
For serial sectioning and visualization of tumors, fixed mouse brains were processed routinely, embedded in OCT (optimal cutting temperature) compound for cryosectioning, and serially sectioned at 9 μm. Coronal sections were stained with Mayer's modified hematoxylin (100%, 2 min). Serial sections at 150-μm intervals were used for tumor volume and invasion measurements.
For analysis of tumor volume, sections containing tumor were used for quantitative digital image analysis (68). Each section was imaged in its entirety using an Axiovert 35 light microscope (Zeiss, Chester, VA, USA) equipped with a digital camera (Sensys, Roper Scientific, Trenton, NJ, USA) and RGB filter (Microcolor GmbH, Straufhein, Germany). Areas occupied by tumor were outlined in a blinded fashion using a preselected cutoff, and images were acquired and digitized via Metamorph (Universal Imaging Corp., West Chester, PA, USA). The sum of all tumor areas was recorded (68).
For analysis of tumor invasion, the tumor-bearing hemisphere was recorded in a series of ×50 magnification images using Spot Advanced software (Diagnostic Instruments, Sterling Heights, MI, USA). Photomerge automation for Photoshop CS4 (Adobe, San Jose, CA, USA) was then used to reconstruct the entire series into a single image. Inversion of the image into a black-on-white image was used to isolate the tumor, and the threshold was adjusted to distinguish the tumor from any nonspecific background staining. Finally, the ImageJ Analyze Particles (National Institutes of Health, Bethesda, MD, USA) function was used to quantify the total number of discrete particles. Alternatively, tumor invasion was quantified based on the number of invasive fingers, as described previously (69–71). Finger-like tumor projections into the adjacent normal brain tissue at the interface between the tumor and normal tissue were counted in images at ×20 magnification to assess tumor invasion in this area.
Statistical analysis
All data are presented as the mean ± S.D. from three or more independent experiments, unless otherwise stated. Different treatments were compared with Student's t test or one-way analysis of variance with Dunnett's multiple-comparison test using SPSS software (version 18.0; SPSS Inc., Chicago, IL, USA). Differences with a p value of <0.05 were considered statistically significant. The sample size for experiments was chosen to ensure adequate statistical power on the basis of G*power 3.1 software (72).
Data availability
All data pertinent to this work are contained within this article or available upon request. For requests, please contact Kyoungho Suk (ksuk@knu.ac.kr).
Supplementary Material
This article contains supporting information.
Author contributions—J.-H. K., Y. S., M. J., H. J., Q. Z., M. V., and F. P. R. data curation; J.-H. K. and H. J. formal analysis; J.-H. K., Y. S., M. J., H. J., Y.-S. K., E.-J. K., D. S., W.-H. L., N. Y., and Q. Z. validation; J.-H. K., Y. S., M. J., H. J., Y.-S. K., E.-J. K., D. S., W.-H. L., N. Y., and M. V. investigation; J.-H. K., M. J., N. Y., Q. Z., and M. V. methodology; J.-H. K., M. J., N. Y., Q. Z., M. V., F. P. R., and K. S. writing-original draft; J.-H. K., Y. S., M. J., H. J., Y.-S. K., E.-J. K., D. S., W.-H. L., N. Y., Q. Z., M. V., F. P. R., and K. S. writing-review and editing; Y. S., M. J., H. J., Y.-S. K., E.-J. K., D. S., W.-H. L., N. Y., and Q. Z. visualization; S. R. K. and Q. Z. resources; N. Y., Q. Z., F. P. R., and K. S. conceptualization; F. P. R. and K. S. funding acquisition, project administration, and supervision.
Funding and additional information—This work was supported by National Research Foundation of Korea (NRF) Grant 2017R1A5A2015391 and 2018R1A2A1A05077118 funded by the Korean government. F. P. R. was supported by National Institutes of Health Grants HG001715 and HG004233, by the Canada Excellence Research Chairs Program, and by the Krembil and Avon Foundations. N. Y. was supported by a Japan Society for the Promotion of Science (JSPS) fellowship (Research Abroad), a Banting Postdoctoral Fellowship, the National Sciences and Engineering Research Council of Canada, and a PRESTO research grant by the Japan Science and Technology Agency (JST). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- MTT
- 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- BCNU
- bis-chloroethylnitrosourea
- LDH
- lactate dehydrogenase
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- DiI
- 1,1′-dioctadecyl-3,3,3′,3′ tetramethylindocarbocyanine perchlorate.
References
- 1. Le Rhun, E., Preusser, M., Roth, P., Reardon, D. A., van den Bent, M., Wen, P., Reifenberger, G., and Weller, M. (2019) Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 80, 101896 10.1016/j.ctrv.2019.101896 [DOI] [PubMed] [Google Scholar]
- 2. Miller, J. J., and Wen, P. Y. (2016) Emerging targeted therapies for glioma. Expert Opin. Emerg. Drugs 21, 441–452 10.1080/14728214.2016.1257609 [DOI] [PubMed] [Google Scholar]
- 3. Mooney, J., Bernstock, J. D., Ilyas, A., Ibrahim, A., Yamashita, D., Markert, J. M., and Nakano, I. (2019) Current approaches and challenges in the molecular therapeutic targeting of glioblastoma. World Neurosurg. 129, 90–100 10.1016/j.wneu.2019.05.205 [DOI] [PubMed] [Google Scholar]
- 4. Zhao, Z. S., and Manser, E. (2012) PAK family kinases: physiological roles and regulation. Cell Logist. 2, 59–68 10.4161/cl.21912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teramoto, H., Crespo, P., Coso, O. A., Igishi, T., Xu, N., and Gutkind, J. S. (1996) The small GTP-binding protein Rho activates c-Jun N-terminal kinases/stress-activated protein kinases in human kidney 293T cells: evidence for a Pak-independent signaling pathway. J. Biol. Chem. 271, 25731–25734 10.1074/jbc.271.42.25731 [DOI] [PubMed] [Google Scholar]
- 6. Ong, C. C., Jubb, A. M., Zhou, W., Haverty, P. M., Harris, A. L., Belvin, M., Friedman, L. S., Koeppen, H., and Hoeflich, K. P. (2011) p21-activated kinase 1: PAK'ed with potential. Oncotarget 2, 491–496 10.18632/oncotarget.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taglieri, D. M., Ushio-Fukai, M., and Monasky, M. M. (2014) P21-activated kinase in inflammatory and cardiovascular disease. Cell. Signal. 26, 2060–2069 10.1016/j.cellsig.2014.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Zijl, F., Krupitza, G., and Mikulits, W. (2011) Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat. Res. 728, 23–34 10.1016/j.mrrev.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gupta, S. C., Kim, J. H., Prasad, S., and Aggarwal, B. B. (2010) Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 29, 405–434 10.1007/s10555-010-9235-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kichina, J. V., Goc, A., Al-Husein, B., Somanath, P. R., and Kandel, E. S. (2010) PAK1 as a therapeutic target. Expert Opin. Ther. Targets 14, 703–725 10.1517/14728222.2010.492779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bright, M. D., Garner, A. P., and Ridley, A. J. (2009) PAK1 and PAK2 have different roles in HGF-induced morphological responses. Cell. Signal. 21, 1738–1747 10.1016/j.cellsig.2009.07.005 [DOI] [PubMed] [Google Scholar]
- 12. Coniglio, S. J., Zavarella, S., and Symons, M. H. (2008) Pak1 and Pak2 mediate tumor cell invasion through distinct signaling mechanisms. Mol. Cell Biol. 28, 4162–4172 10.1128/MCB.01532-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rane, C. K., and Minden, A. (2018) P21 activated kinase signaling in cancer. Semin. Cancer Biol. 54, 40–49 10.1016/j.semcancer.2018.01.006 [DOI] [PubMed] [Google Scholar]
- 14. Semenova, G., and Chernoff, J. (2017) Targeting PAK1. Biochem. Soc. Trans. 45, 79–88 10.1042/BST20160134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parvathy, M., Sreeja, S., Kumar, R., and Pillai, M. R. (2016) Potential role of p21 activated kinase 1 (PAK1) in the invasion and motility of oral cancer cells. BMC Cancer 16, 293 10.1186/s12885-016-2263-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang, G., Zhang, Q., Song, Y., Wang, X., Guo, Q., Zhang, J., Li, J., Han, Y., Miao, Z., and Li, F. (2015) PAK1 regulates RUFY3-mediated gastric cancer cell migration and invasion. Cell Death Dis. 6, e1682 10.1038/cddis.2015.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ong, C. C., Jubb, A. M., Haverty, P. M., Zhou, W., Tran, V., Truong, T., Turley, H., O'Brien, T., Vucic, D., Harris, A. L., Belvin, M., Friedman, L. S., Blackwood, E. M., Koeppen, H., and Hoeflich, K. P. (2011) Targeting p21-activated kinase 1 (PAK1) to induce apoptosis of tumor cells. Proc. Natl. Acad. Sci. U. S. A. 108, 7177–7182 10.1073/pnas.1103350108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yuan, Z. Q., Kim, D., Kaneko, S., Sussman, M., Bokoch, G. M., Kruh, G. D., Nicosia, S. V., Testa, J. R., and Cheng, J. Q. (2005) ArgBP2γ interacts with Akt and p21-activated kinase-1 and promotes cell survival. J. Biol. Chem. 280, 21483–21490 10.1074/jbc.M500097200 [DOI] [PubMed] [Google Scholar]
- 19. Aoki, H., Yokoyama, T., Fujiwara, K., Tari, A. M., Sawaya, R., Suki, D., Hess, K. R., Aldape, K. D., Kondo, S., Kumar, R., and Kondo, Y. (2007) Phosphorylated Pak1 level in the cytoplasm correlates with shorter survival time in patients with glioblastoma. Clin. Cancer Res. 13, 6603–6609 10.1158/1078-0432.CCR-07-0145 [DOI] [PubMed] [Google Scholar]
- 20. Seo, M., Lee, S., Kim, J. H., Lee, W. H., Hu, G., Elledge, S. J., and Suk, K. (2014) RNAi-based functional selection identifies novel cell migration determinants dependent on PI3K and AKT pathways. Nat. Commun. 5, 5217 10.1038/ncomms6217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim, J. H., Seo, Y., Jo, M., Jeon, H., Lee, W. H., Yachie, N., Zhong, Q., Vidal, M., Roth, F. P., and Suk, K. (2020) Yeast-based genetic interaction analysis of human kinome. Cells 9, 1156 10.3390/cells9051156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Youn, J. Y., Friesen, H., Nguyen Ba, A. N., Liang, W., Messier, V., Cox, M. J., Moses, A. M., and Andrews, B. (2017) Functional analysis of kinases and transcription factors in Saccharomyces cerevisiae using an integrated overexpression library. G3 (Bethesda) 7, 911–921 10.1534/g3.116.038471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee, K. T., So, Y. S., Yang, D. H., Jung, K. W., Choi, J., Lee, D. G., Kwon, H., Jang, J., Wang, L. L., Cha, S., Meyers, G. L., Jeong, E., Jin, J. H., Lee, Y., Hong, J., et al. (2016) Systematic functional analysis of kinases in the fungal pathogen Cryptococcus neoformans. Nat. Commun. 7, 12766 10.1038/ncomms12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharifpoor, S., van Dyk, D., Costanzo, M., Baryshnikova, A., Friesen, H., Douglas, A. C., Youn, J. Y., VanderSluis, B., Myers, C. L., Papp, B., Boone, C., and Andrews, B. J. (2012) Functional wiring of the yeast kinome revealed by global analysis of genetic network motifs. Genome Res. 22, 791–801 10.1101/gr.129213.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Botstein, D., and Fink, G. R. (2011) Yeast: an experimental organism for 21st century biology. Genetics 189, 695–704 10.1534/genetics.111.130765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giaever, G., Shoemaker, D. D., Jones, T. W., Liang, H., Winzeler, E. A., Astromoff, A., and Davis, R. W. (1999) Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat. Genet. 21, 278–283 10.1038/6791 [DOI] [PubMed] [Google Scholar]
- 27. Smith, A. M., Durbic, T., Kittanakom, S., Giaever, G., and Nislow, C. (2012) Barcode sequencing for understanding drug-gene interactions. Methods Mol. Biol. 910, 55–69 10.1007/978-1-61779-965-5_4 [DOI] [PubMed] [Google Scholar]
- 28. Smith, A. M., Heisler, L. E., Mellor, J., Kaper, F., Thompson, M. J., Chee, M., Roth, F. P., Giaever, G., and Nislow, C. (2009) Quantitative phenotyping via deep barcode sequencing. Genome Res. 19, 1836–1842 10.1101/gr.093955.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith, A. M., Heisler, L. E., St Onge, R. P., Farias-Hesson, E., Wallace, I. M., Bodeau, J., Harris, A. N., Perry, K. M., Giaever, G., Pourmand, N., and Nislow, C. (2010) Highly-multiplexed barcode sequencing: an efficient method for parallel analysis of pooled samples. Nucleic Acids Res. 38, e142 10.1093/nar/gkq368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jo, M., Chung, A. Y., Yachie, N., Seo, M., Jeon, H., Nam, Y., Seo, Y., Kim, E., Zhong, Q., Vidal, M., Park, H. C., Roth, F. P., and Suk, K. (2017) Yeast genetic interaction screen of human genes associated with amyotrophic lateral sclerosis: identification of MAP2K5 kinase as a potential drug target. Genome Res. 27, 1487–1500 10.1101/gr.211649.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Brien, K. P., Remm, M., and Sonnhammer, E. L. (2005) Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 33, D476–D480 10.1093/nar/gki107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim, H., Oh, J. Y., Choi, S. L., Nam, Y. J., Jo, A., Kwon, A., Shin, E. Y., Kim, E. G., and Kim, H. K. (2016) Down-regulation of p21-activated serine/threonine kinase 1 is involved in loss of mesencephalic dopamine neurons. Mol. Brain 9, 45 10.1186/s13041-016-0230-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lun, X. K., Szklarczyk, D., Gabor, A., Dobberstein, N., Zanotelli, V. R. T., Saez-Rodriguez, J., von Mering, C., and Bodenmiller, B. (2019) Analysis of the human kinome and phosphatome by mass cytometry reveals overexpression-induced effects on cancer-related signaling. Mol. Cell 74, 1086–1102.e5 10.1016/j.molcel.2019.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ishikawa, E., Kosako, H., Yasuda, T., Ohmuraya, M., Araki, K., Kurosaki, T., Saito, T., and Yamasaki, S. (2016) Protein kinase D regulates positive selection of CD4+ thymocytes through phosphorylation of SHP-1. Nat. Commun. 7, 12756 10.1038/ncomms12756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Camurdanoglu, B. Z., Hrovat, C., Dürnberger, G., Madalinski, M., Mechtler, K., and Herbst, R. (2016) MuSK kinase activity is modulated by a serine phosphorylation site in the kinase loop. Sci. Rep. 6, 38271 10.1038/srep38271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koch, A., Maia, A., Janssen, A., and Medema, R. H. (2016) Molecular basis underlying resistance to Mps1/TTK inhibitors. Oncogene 35, 2518–2528 10.1038/onc.2015.319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang, Y. D., Su, Y. J., Li, J. Y., Yao, X. C., and Liang, G. J. (2015) Rapamycin, a mTOR inhibitor, induced growth inhibition in retinoblastoma Y79 cell via down-regulation of Bmi-1. Int. J. Clin. Exp. Pathol. 8, 5182–5188 [PMC free article] [PubMed] [Google Scholar]
- 38. Jiang, P., Mukthavaram, R., Mukthavavam, R., Chao, Y., Bharati, I. S., Fogal, V., Pastorino, S., Cong, X., Nomura, N., Gallagher, M., Abbasi, T., Vali, S., Pingle, S. C., Makale, M., and Kesari, S. (2014) Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs. J. Transl. Med. 12, 13 10.1186/1479-5876-12-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garcia-Saez, I., DeBonis, S., Lopez, R., Trucco, F., Rousseau, B., Thuéry, P., and Kozielski, F. (2007) Structure of human Eg5 in complex with a new monastrol-based inhibitor bound in the R configuration. J. Biol. Chem. 282, 9740–9747 10.1074/jbc.M608883200 [DOI] [PubMed] [Google Scholar]
- 40. Wiese, E. K., and Hitosugi, T. (2018) Tyrosine kinase signaling in cancer metabolism: PKM2 paradox in the Warburg effect. Front. Cell Dev. Biol. 6, 79 10.3389/fcell.2018.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu, Z., and Hunter, T. (2018) Metabolic kinases moonlighting as protein kinases. Trends Biochem. Sci. 43, 301–310 10.1016/j.tibs.2018.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dayton, T. L., Jacks, T., and Vander Heiden, M. G. (2016) PKM2, cancer metabolism, and the road ahead. EMBO Rep. 17, 1721–1730 10.15252/embr.201643300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johnston, S. T., Shah, E. T., Chopin, L. K., Sean McElwain, D. L., and Simpson, M. J. (2015) Estimating cell diffusivity and cell proliferation rate by interpreting IncuCyte ZOOM assay data using the Fisher-Kolmogorov model. BMC Syst. Biol. 9, 38 10.1186/s12918-015-0182-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Aslan, J. E., Baker, S. M., Loren, C. P., Haley, K. M., Itakura, A., Pang, J., Greenberg, D. L., David, L. L., Manser, E., Chernoff, J., and McCarty, O. J. (2013) The PAK system links Rho GTPase signaling to thrombin-mediated platelet activation. Am. J. Physiol. Cell Physiol. 305, C519–C528 10.1152/ajpcell.00418.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang, Y., Zhou, H., Chen, A., Pittman, R. N., and Field, J. (2000) The Akt proto-oncogene links Ras to Pak and cell survival signals. J. Biol. Chem. 275, 9106–9109 10.1074/jbc.275.13.9106 [DOI] [PubMed] [Google Scholar]
- 46. Thillai, K., Lam, H., Sarker, D., and Wells, C. M. (2017) Deciphering the link between PI3K and PAK: an opportunity to target key pathways in pancreatic cancer? Oncotarget 8, 14173–14191 10.18632/oncotarget.13309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xue, G., and Hemmings, B. A. (2013) PKB/Akt-dependent regulation of cell motility. J. Natl. Cancer Inst. 105, 393–404 10.1093/jnci/djs648 [DOI] [PubMed] [Google Scholar]
- 48. Higuchi, M., Onishi, K., Kikuchi, C., and Gotoh, Y. (2008) Scaffolding function of PAK in the PDK1-Akt pathway. Nat. Cell Biol. 10, 1356–1364 10.1038/ncb1795 [DOI] [PubMed] [Google Scholar]
- 49. Taga, M., Hirooka, E., and Ouchi, T. (2009) Essential roles of mTOR/Akt pathway in Aurora-A cell transformation. Int. J. Biol. Sci. 5, 444–450 10.7150/ijbs.5.444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sarbassov, D. D., Guertin, D. A., Ali, S. M., and Sabatini, D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 10.1126/science.1106148 [DOI] [PubMed] [Google Scholar]
- 51. Kim, S. R., Kareva, T., Yarygina, O., Kholodilov, N., and Burke, R. E. (2012) AAV transduction of dopamine neurons with constitutively active Rheb protects from neurodegeneration and mediates axon regrowth. Mol. Ther. 20, 275–286 10.1038/mt.2011.213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ries, V., Henchcliffe, C., Kareva, T., Rzhetskaya, M., Bland, R., During, M. J., Kholodilov, N., and Burke, R. E. (2006) Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 103, 18757–18762 10.1073/pnas.0606401103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alimova, I., Ng, J., Harris, P., Birks, D., Donson, A., Taylor, M. D., Foreman, N. K., Venkataraman, S., and Vibhakar, R. (2016) MPS1 kinase as a potential therapeutic target in medulloblastoma. Oncol. Rep. 36, 2633–2640 10.3892/or.2016.5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wei, J. H., Chou, Y. F., Ou, Y. H., Yeh, Y. H., Tyan, S. W., Sun, T. P., Shen, C. Y., and Shieh, S. Y. (2005) TTK/hMps1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. J. Biol. Chem. 280, 7748–7757 10.1074/jbc.M410152200 [DOI] [PubMed] [Google Scholar]
- 55. Be Tu, P. T., Nguyen, B. C., Tawata, S., Yun, C. Y., Kim, E. G., and Maruta, H. (2017) The serum/PDGF-dependent “melanogenic” role of the minute level of the oncogenic kinase PAK1 in melanoma cells proven by the highly sensitive kinase assay. Drug Discov. Ther. 10, 314–322 10.5582/ddt.2016.01062 [DOI] [PubMed] [Google Scholar]
- 56. Rahman, M. H., Jha, M. K., Kim, J. H., Nam, Y., Lee, M. G., Go, Y., Harris, R. A., Park, D. H., Kook, H., Lee, I. K., and Suk, K. (2016) Pyruvate dehydrogenase kinase-mediated glycolytic metabolic shift in the dorsal root ganglion drives painful diabetic neuropathy. J. Biol. Chem. 291, 6011–6025 10.1074/jbc.M115.699215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shin, K. S., Shin, E. Y., Lee, C. S., Quan, S. H., Woo, K. N., Soung, N. K., Kwak, S. J., Kim, S. R., and Kim, E. G. (2002) Basic fibroblast growth factor-induced translocation of p21-activated kinase to the membrane is independent of phospholipase C-γ1 in the differentiation of PC12 cells. Exp. Mol. Med. 34, 172–176 10.1038/emm.2002.25 [DOI] [PubMed] [Google Scholar]
- 58. Alberti, S., Gitler, A. D., and Lindquist, S. (2007) A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 24, 913–919 10.1002/yea.1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gietz, R. D., and Schiestl, R. H. (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 10.1038/nprot.2007.13 [DOI] [PubMed] [Google Scholar]
- 60. Liang, C. C., Park, A. Y., and Guan, J. L. (2007) In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333 10.1038/nprot.2007.30 [DOI] [PubMed] [Google Scholar]
- 61. Aaberg-Jessen, C., Norregaard, A., Christensen, K., Pedersen, C. B., Andersen, C., and Kristensen, B. W. (2013) Invasion of primary glioma- and cell line-derived spheroids implanted into corticostriatal slice cultures. Int. J. Clin. Exp. Pathol. 6, 546–560 [PMC free article] [PubMed] [Google Scholar]
- 62. Nørregaard, A., Jensen, S. S., Kolenda, J., Aaberg-Jessen, C., Christensen, K. G., Jensen, P. H., Schrøder, H. D., and Kristensen, B. W. (2012) Effects of chemotherapeutics on organotypic corticostriatal slice cultures identified by a panel of fluorescent and immunohistochemical markers. Neurotox. Res. 22, 43–58 10.1007/s12640-011-9300-9 [DOI] [PubMed] [Google Scholar]
- 63. Stoppini, L., Buchs, P. A., and Muller, D. (1991) A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182 10.1016/0165-0270(91)90128-M [DOI] [PubMed] [Google Scholar]
- 64. Eisemann, T., Costa, B., Strelau, J., Mittelbronn, M., Angel, P., and Peterziel, H. (2018) An advanced glioma cell invasion assay based on organotypic brain slice cultures. BMC Cancer 18, 103 10.1186/s12885-018-4007-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Depner, C., Zum Buttel, H., Böğürcü, N., Cuesta, A. M., Aburto, M. R., Seidel, S., Finkelmeier, F., Foss, F., Hofmann, J., Kaulich, K., Barbus, S., Segarra, M., Reifenberger, G., Garvalov, B. K., Acker, T., et al. (2016) EphrinB2 repression through ZEB2 mediates tumour invasion and anti-angiogenic resistance. Nat. Commun. 7, 12329 10.1038/ncomms12329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yadav, V. N., Zamler, D., Baker, G. J., Kadiyala, P., Erdreich-Epstein, A., DeCarvalho, A. C., Mikkelsen, T., Castro, M. G., and Lowenstein, P. R. (2016) CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: a genetic knockdown study. Oncotarget 7, 83701–83719 10.18632/oncotarget.13295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Candolfi, M., Curtin, J. F., Nichols, W. S., Muhammad, A. G., King, G. D., Pluhar, G. E., McNiel, E. A., Ohlfest, J. R., Freese, A. B., Moore, P. F., Lerner, J., Lowenstein, P. R., and Castro, M. G. (2007) Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J. Neurooncol. 85, 133–148 10.1007/s11060-007-9400-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lampson, L. A., Lampson, M. A., and Dunne, A. D. (1993) Exploiting the lacZ reporter gene for quantitative analysis of disseminated tumor growth within the brain: use of the lacZ gene product as a tumor antigen, for evaluation of antigenic modulation, and to facilitate image analysis of tumor growth in situ. Cancer Res. 53, 176–182 [PubMed] [Google Scholar]
- 69. Cortes-Santiago, N., Hossain, M. B., Gabrusiewicz, K., Fan, X., Gumin, J., Marini, F. C., Alonso, M. M., Lang, F., Yung, W. K., Fueyo, J., and Gomez-Manzano, C. (2016) Soluble Tie2 overrides the heightened invasion induced by anti-angiogenesis therapies in gliomas. Oncotarget 7, 16146–16157 10.18632/oncotarget.7550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Feng, H., Hu, B., Vuori, K., Sarkaria, J. N., Furnari, F. B., Cavenee, W. K., and Cheng, S. Y. (2014) EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene 33, 2504–2512 10.1038/onc.2013.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Feng, H., Hu, B., Liu, K. W., Li, Y., Lu, X., Cheng, T., Yiin, J. J., Lu, S., Keezer, S., Fenton, T., Furnari, F. B., Hamilton, R. L., Vuori, K., Sarkaria, J. N., Nagane, M., et al. (2011) Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRα-stimulated glioma tumorigenesis in mice and humans. J. Clin. Invest. 121, 4670–4684 10.1172/JCI58559 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72. Charan, J., and Kantharia, N. D. (2013) How to calculate sample size in animal studies? J. Pharmacol. Pharmacother. 4, 303–306 10.4103/0976-500X.119726 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data pertinent to this work are contained within this article or available upon request. For requests, please contact Kyoungho Suk (ksuk@knu.ac.kr).