

Abstract
The adipocyte-derived hormone leptin increases trafficking of KATP and Kv2.1 channels to the pancreatic β-cell surface, resulting in membrane hyperpolarization and suppression of insulin secretion. We have previously shown that this effect of leptin is mediated by the NMDA subtype of glutamate receptors (NMDARs). It does so by potentiating NMDAR activity, thus enhancing Ca2+ influx and the ensuing downstream signaling events that drive channel trafficking to the cell surface. However, the molecular mechanism by which leptin potentiates NMDARs in β-cells remains unknown. Here, we report that leptin augments NMDAR function via Src kinase–mediated phosphorylation of the GluN2A subunit. Leptin-induced membrane hyperpolarization diminished upon pharmacological inhibition of GluN2A but not GluN2B, indicating involvement of GluN2A-containing NMDARs. GluN2A harbors tyrosine residues that, when phosphorylated by Src family kinases, potentiate NMDAR activity. We found that leptin increases phosphorylation of Tyr-418 in Src, an indicator of kinase activation. Pharmacological inhibition of Src or overexpression of a kinase-dead Src mutant prevented the effect of leptin, whereas a Src kinase activator peptide mimicked it. Using mutant GluN2A overexpression, we show that Tyr-1292 and Tyr-1387 but not Tyr-1325 are responsible for the effect of leptin. Importantly, β-cells from db/db mice, a type 2 diabetes mouse model lacking functional leptin receptors, or from obese diabetic human donors failed to respond to leptin but hyperpolarized in response to NMDA. Our study reveals a signaling pathway wherein leptin modulates NMDARs via Src to regulate β-cell excitability and suggests NMDARs as a potential target to overcome leptin resistance.
Keywords: Src kinase, β-cells, KATP channel, GluN2A, phosphorylation, glutamate receptor, type 2 diabetes, Src, beta cell (B-cell)
Leptin is an adipocyte-produced hormone that plays a key role in body weight regulation. The physiological actions and signaling mechanisms of leptin in the central nervous system have been extensively studied. Less well-understood is the function and mechanism of leptin signaling in peripheral tissues. In pancreatic islets, leptin was reported to down-regulate glucose-stimulated insulin secretion more than 2 decades ago (1–6). Recent studies find that leptin stimulates potassium channel trafficking to the cell surface to reduce β-cell excitability (7–9). Specifically, leptin causes a transient increase in the number of ATP-sensitive potassium (KATP) channels and Kv2.1 channels in the β-cell plasma membrane (9). KATP channels control β-cell resting membrane potential and couple blood glucose with insulin secretion, whereas Kv2.1 channels play a prominent role in action potential repolarization to stop insulin secretion (10, 11). The increased surface abundance of these channels would reduce β-cell excitability and thus explain how leptin inhibits glucose-stimulated insulin secretion.
Leptin signaling is complex, and multiple signal transduction pathways have been described (12, 13). The best-characterized is JAK2-dependent phosphorylation of STAT3 following activation of the ObRb receptor, but phosphatidylinositol 3-kinase/Akt, mitogen-activated protein kinase, AMPK, and Src family kinases (SFKs) are also possible downstream effectors (13, 14). In addition, transactivation of other cytokine receptors via leptin signaling has also been implicated (13). Studies into the mechanisms by which leptin regulates KATP and Kv2.1 channel trafficking so far have identified several key molecular players. These include the NMDA subtype glutamate receptors (NMDARs), calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ), AMPK, and PKA (15). Evidence that has emerged reveals a novel signaling pathway wherein leptin potentiates NMDAR function to increase Ca2+ influx, resulting in activation of CaMKKβ, which phosphorylates and activates AMPK; AMPK in turn causes PKA-dependent actin depolymerization, culminating in increased trafficking of KATP and Kv2.1 channels to the β-cell surface. A key question of how leptin potentiates NMDAR activity, however, has yet to be addressed.
NMDARs are calcium-permeant ionotropic glutamate receptors that are highly expressed in the brain and are important for learning and memory (16). Although they are extensively studied in the central nervous system, there is growing evidence that they are expressed in pancreatic β-cells and play a role in regulating insulin secretion (17–19). Functional NMDARs generally form as heterotetramers of two obligatory glycine-binding GluN1 subunits (also known as NR1) and two glutamate-binding GluN2 (GluN2A-D or NR2A-D) subunits (20). NMDARs containing GluN2A and -2B subunits are highly sensitive to blockade by extracellular Mg2+ under negative membrane potential and require membrane depolarization to remove Mg2+ block for activity, whereas those with GluN2C and -2D subunits are significantly less sensitive to external Mg2+ (16, 20, 21). Additionally, NMDAR activity can be modulated by post-translational modifications. In particular, GluN2A and GluN2B have long cytoplasmic tails that are known to be phosphorylated at serine/threonine and/or tyrosine residues by a variety of kinases, including CDK5, PKA, PKC, CaMKII, casein kinase II, and protein tyrosine kinases (22). In neurons, these phosphorylation modifications have been linked to regulation of NMDAR trafficking, localization, and function (reviewed in Ref. 23). For example, phosphorylation of several tyrosine residues in GluN2A by SFKs have been shown to enhance NMDAR activity (24, 25). Of note, Src activation is one of the many downstream signaling events that have been reported following stimulation of the ObRb leptin receptor by leptin (14, 26, 27), raising the possibility that leptin may signal through SFKs to modulate NMDAR activity in pancreatic β-cells.
Here, we present evidence that leptin activates Src to phosphorylate GluN2A-containing NMDARs in pancreatic β-cells, which results in potentiation of NMDAR currents and increased trafficking of KATP and Kv2.1 channels to the cell surface to hyperpolarize β-cell membrane potential. Importantly, we show that leptin fails to induce membrane hyperpolarization in β-cells from the leptin-resistant db/db mice and obese diabetic human donors. Interestingly, direct activation of NMDARs by NMDA was able to mimic the effect of leptin. As leptin resistance is frequently associated with obesity-related diabetes, NMDARs may be a potential target to overcome leptin resistance in diabetic β-cells.
Results
Leptin hyperpolarizes pancreatic β-cells through GluN2A-containing NMDARs
In rodent pancreatic β-cell lines as well as primary human β-cells, leptin induces membrane hyperpolarization at glucose concentrations that depolarize β-cell membrane potential (3, 7–9, 19). Recently, we showed that, both in rat insulinoma INS-1 832/13 cells and human β-cells, this effect is mediated by NMDARs (19). Leptin stimulation increased NMDA currents, leading to increased Ca2+ influx and increased surface density of KATP and Kv2.1 channels (19). To determine the mechanism by which leptin increases NMDA currents, we began by characterizing the NMDARs that are expressed in β-cells. Functional NMDARs that are Ca2+-permeant are tetramers of two GluN1 subunits and two of four different GluN2 subunits, GluN2A, -2B, -2C, and -2D (20). The GluN1 subunit is obligatory and is encoded by a single gene, Grin1. In contrast, GluN2 subunits are encoded by four different genes: Grin2a, Grin2b, Grin2c, and Grin2d for GluN2A–D (20). We first examined the expression of these subunits at the mRNA level in INS-1 832/13 cells by RT-PCR. Transcripts for Grin1, Grin2a, Grin2b, and Grin2d were clearly detected, but not Grin2c (Fig. 1A), suggesting that these cells express GluN1, GluN2A, GluN2B, and GluN2D, but not GluN2C.
Figure 1.

Leptin induces membrane hyperpolarization through GluN2A-containing NMDARs. A, RT-PCR detection of mRNA for NMDAR subunits, GluN1 (Grin1), GluN2A (Grin2a), GluN2B (Grin2b), GluN2C (Grin2c), and GluN2D (Grin2d), in rat brain (Br) and INS-1 832/13 cells (INS). β-Actin was included as a control. B, representative traces of NMDAR currents in the absence (top) or presence (bottom) of 100 μm Mg2+. C, sensitivity of NMDA currents (elicited by puff application of 1 mm NMDA) to Mg2+ block. Averaged current amplitudes before (control) and after 100 μm Mg2+ for each cell are connected by a straight line. Mean ± S.E. values for each group are shown next to individual data points. *, p < 0.05 by paired t test. D, Western blots showing protein expression of GluN1, GluN2A, and GluN2B from INS-1 832/13 cell membrane fraction (Memb; 30 µg) and total cell lysate (Lysate; 30 µg). E, individual cell-attached membrane recordings from INS-1 832/13 cells treated with leptin (10 nm) alone (top), with leptin plus the GluN2B inhibitor Ro 25-6981 (middle; Ro25, 1 μm), or with leptin plus the GluN2A inhibitor TCN201 (bottom; 50 μm). F, group data showing the degree of membrane hyperpolarization in mV for leptin alone (n = 11 cells) or leptin co-applied with Ro 25-6981 (n = 14 cells) or TCN201 (n = 16 cells). Here and in subsequent figures, individual cells are represented by symbols and means are indicated by a black line. Error bars, S.E. ***, p < 0.0005 by unpaired t test as compared with leptin.
NMDARs containing GluN2A or -2B are much more sensitive to external Mg2+ block than those containing GluN2C or -2D (20, 28). We thus tested Mg2+ sensitivity of NMDA currents using whole-cell recording as an indicator of the GluN2 subunit composition. Puff application of NMDA (1 mm) at a holding potential of −70 mV in Mg2+-free external solution elicited NMDA currents that were reduced by >80% upon the addition of MgCl2 (100 μm) to the external solution (from 11.3 ± 2.6 to 1.3 ± 0.5 pA, n = 4; p < 0.05 by paired t test) (Fig. 1, B and C). The strong Mg2+ block observed is characteristic of NMDARs containing GluN2A and/or GluN2B subunits (21, 28), suggesting that NMDARs at the surface of these cells are largely made up of GluN1 and GluN2A and/or GluN2B subunits. Consistently, in Western blotting experiments, GluN1 and GluN2A were readily detectable in total cell lysate and further enriched in the membrane fraction, whereas GluN2B was barely detectable in total cell lysate but clearly seen in the membrane fraction (Fig. 1D).
Because both GluN2A and -2B could potentially mediate the leptin response, we sought to determine their relative contributions using subunit-specific inhibitors: TCN-201 for GluN2A and Ro 25-6981 for GluN2B (20, 29, 30). Hyperpolarization of membrane potential in Tyrode's solution containing 11 mm glucose was used as a readout for leptin response. Because GluN2A inhibition by TCN-201 depends on glycine, which at higher concentrations renders the drug less effective (30), we reduced the glycine supplement in Tyrode's solution from 100 to 50 μm. These experiments were performed using cell-attached current-clamp recording, which provides a robust assessment of changes in membrane potential while maintaining cell integrity and preventing dialysis of soluble factors that may be important for intracellular signaling (31, 32). Bath application of leptin alone (10 nm) induced a mean membrane hyperpolarization of −46.8 ± 8.1 mV, similar to that reported previously (9, 19). However, co-application of the GluN2A-selective antagonist TCN-201 (50 μm) with leptin only induced a mean hyperpolarization of −14.4 ± 3.9 mV, significantly less than that observed in cells treated with leptin alone (Fig. 1, E and F). By contrast, co-application of the GluN2B-selective antagonist Ro 25-6981 (33) had little effect on the ability of leptin to hyperpolarize INS-1 832/13 cells (Ro 25-6981 at 1 μm, ΔVm = −35.8 ± 3.8 mV; Fig. 1, E and F). These results suggest that GluN2A is largely responsible for mediating the effect of leptin. Note that there is evidence that GluN2A can form diheteromeric GluN1/GluN2A or triheteromeric GluN1/GluN2A/GluN2B complexes (20). Because the potency and efficacy of TCN-201 and Ro 25-6981 have been shown to be reduced for triheteromeric GluN1/GluN2A/GluN2B channels, especially for Ro 25-6981 (29, 34), the lack of effect by Ro 25-6981 in our experiment does not allow us to exclude the involvement of GluN1/GluN2A/GluN2B heteromers, as both GluN2A and GluN2B (albeit less abundant compared with GluN2A based on Western blotting of total cell lysate) were detected in membrane fractions prepared from INS-1 832/13 cells by Western blotting (Fig. 1D).
Leptin regulates NMDAR activity via Src family kinases
In contrast to GluN1, which has a relatively small cytoplasmic domain of ∼100 amino acids, GluN2A has a long cytoplasmic tail of ∼600 amino acids that contains potential phosphorylation sites for a number of protein kinases, including PKA, PKC, CDK5, and the SFKs (22). Phosphorylation by PKC (35), CDK5 (36), and Src (25, 37) in particular has been reported to potentiate NMDAR currents. We therefore monitored changes in INS-1 832/13 cell membrane potentials in response to leptin in the absence or presence of inhibitors for the various kinases. Neither roscovitine (Ros; 10 μm), an inhibitor of CDK5 kinase, nor Go 6983 (Go; 10 μm), a broad-spectrum PKC kinase inhibitor, had significant effects on leptin-induced hyperpolarization (ΔVm = −32.4 ± 4.1 mV for leptin alone; ΔVm = −39.3 ± 7.0 mV for roscovitine plus leptin; ΔVm = −33.7 ± 5.5 mV for Go 6983 plus leptin; Fig. 2, A and B). By contrast, AZD0530 (AZD; 10 μm), also known as saracatinib (38), a broad-spectrum inhibitor of SFKs, markedly reduced the ability of leptin to induce membrane hyperpolarization (ΔVm = −4.8 ± 3.1 mV, p < 0.0005 compared with leptin alone; Fig. 2, A and B) but had no effect on membrane hyperpolarization triggered by direct activation of NMDARs via NMDA (NMDA: ΔVm = −35.3 ± 8.9 mV, n = 7; NMDA plus AZD0530: ΔVm = −30.4 ± 9.9 mV, n = 7). Another tyrosine kinase inhibitor, dasatinib (Das; 25 μm) (38) also abrogated the effect of leptin, resulting in only a small hyperpolarization (ΔVm = −7.0 ± 2.4 mV, p < 0.0005 compared with leptin alone; Fig. 2B). These results indicate that SFKs but not CDK5 nor PKC are likely involved in the leptin-induced membrane hyperpolarization observed in INS-1 832/13 cells.
Figure 2.

Leptin regulates NMDAR activity via Src family kinases. A, representative cell-attached current-clamp recordings from INS-1 832/13 cells treated with leptin (10 nm) alone (top left) or leptin with the PKC inhibitor Go 6983 (bottom left; Go, 10 μm), the CDK5 inhibitor roscovitine (top right; Ros, 10 μm), or the SFK inhibitor AZD (bottom right; 10 μm). Inhibitors were applied for 10 min prior to the application of leptin and remained in the solution throughout the recording. B, group data showing extent of membrane hyperpolarization for leptin alone (n = 40 cells), and leptin co-applied with roscovitine (Ros, n = 14), Go 6983 (n = 18), AZD (n = 17), or dasatinib (Das, 25 μm; n = 15). ***, p < 0.0005 by unpaired t test as compared with leptin. C, representative whole-cell current-clamp recordings of INS-1 832/13 cells without (top trace) or with (bottom trace) the Src kinase–activating peptide YEEI (1 μm) in the pipette solution. D, group data showing the degree of hyperpolarization for control (n = 6) and YEEI peptide (n = 7). *, p < 0.05, unpaired t test as compared with control.
Next, we determined whether activation of SFKs via an activating phosphopeptide EPQpYEEIPIYL (referred to as YEEI), which binds to the Src homology 2 domain to relieve kinase autoinhibition (39, 40), could mimic the effects of leptin and induce membrane hyperpolarization. For these experiments, whole-cell current-clamp recordings of INS-1 832/13 cells were carried out to apply YEEI intracellularly through the patch pipette. Under whole-cell conditions with 5 mm ATP in the pipette solution (estimated intracellular [ATP] with 11 mm glucose in the bath solution), inclusion of YEEI phosphopeptide induced significant membrane hyperpolarization (ΔVm = −21.1 ± 4.1 mV from break-in to steady state, n = 7) compared with the control without the peptide (ΔVm = −4.9 ± 4.0 mV from break-in to steady state, n = 6) (Fig. 2, C and D). The result supports the notion that direct activation of SFKs is sufficient to mimic the effect of leptin and cause β-cell membrane hyperpolarization.
To directly test whether SFKs underlie the potentiation of NMDAR currents by leptin, we performed whole-cell recording of NMDAR currents and monitored how current amplitudes were affected by leptin and the SFK inhibitor AZD0530. In the absence of leptin, repeated puff applications of 1 mm NMDA at 1-min intervals elicited NMDAR currents of similar amplitudes. Bath application of 10 nm leptin potentiated NMDA-evoked currents within 5 min of leptin treatment from a baseline of 14.8 ± 8.0 pA to 25.8 ± 13.3 pA (p < 0.05 by Friedman's test; Fig. 3), as we reported previously (19). Subsequent co-application of 10 μm AZD0530 with leptin abolished the effect of leptin, with averaged NMDA currents of 15.5 ± 8.7 pA comparable with baseline values (Fig. 3). These results provide direct evidence that leptin-mediated potentiation of NMDAR currents requires SFKs.
Figure 3.
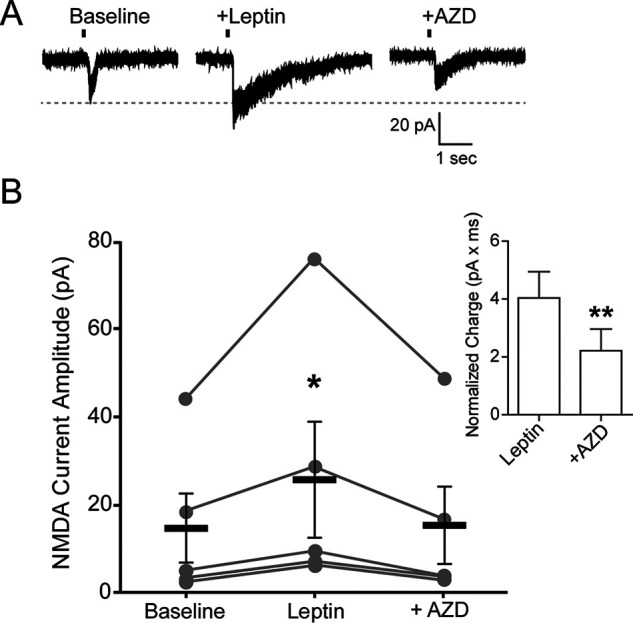
Leptin potentiation of NMDAR currents is prevented by inhibition of Src kinases. A, example of whole-cell currents from a single INS-1 832/13 cell induced by puff application of NMDA (1 mm; vertical line indicates time of puff) during baseline, 5 min after leptin, and following wash-in of AZD (10 μm AZD0350) in the presence of leptin for 5 min. B, within-cell group data showing amplitude of NMDAR currents for each condition (n = 5 cells). Means for each condition are depicted as thick black lines ± S.E. (error bars). Statistical analysis was conducted using Friedman's test (p = 0.0239) followed by a post hoc Dunn's multiple-comparison test with significance set to p < 0.05 (*), as compared with baseline. Inset, comparison of total charge (pA × ms) observed for each NMDA puff between leptin and leptin + AZD treatments normalized to baseline. **, p < 0.005, paired Student's t test as compared with leptin.
Leptin modulation of NMDAR activity requires phosphorylation of GluN2A at Tyr-1292 and Tyr-1387
SFKs have been reported to potentiate NMDAR currents by phosphorylating GluN2 (23, 41, 42). Specifically, three tyrosine residues in GluN2A (Tyr-1292, Tyr-1325, and Tyr-1387) have been identified as targets of SFK-mediated phosphorylation to enhance NMDAR function in different experimental systems (24, 25, 41, 43). To determine whether these sites play a role in leptin-induced membrane hyperpolarization, we mutated the three tyrosine residues in GluN2A to phenylalanines together GluN2AY1292F,Y1325F,Y1387F as well as individually (GluN2AY1292F, GluN2AY1325F, and GluN2AY1387F). To facilitate visualization of expression, a GluN2A construct fused to GFP at its extracellular N terminus was used (44). INS-1 832/13 cells were then transfected to express WT or phosphomutants. To confirm that exogenously expressed GFP-GluN2A co-assembles with endogenous GluN1, we performed co-immunoprecipitation experiments using an anti-GFP nanobody. Both GluN1 and GFP-GluN2A were present in the immunoprecipitate as detected by anti-GFP, anti-GluN2A, and anti-GluN1 antibodies in Western blots (Fig. 4A), indicating association of transfected GluN2A with endogenous GluN1. Surface staining of the GFP tag further demonstrates that transfected WT and mutant GluN2A were expressed in the plasma membrane (Fig. 4B).
Figure 4.

Leptin-induced membrane hyperpolarization requires phosphorylation of GluN2A Tyr-1292 and Tyr-1387 but not Tyr-1325. A, immunoprecipitation of GFP-GluN2A with anti-GFP followed by immunoblotting using anti-GFP, anti-GluN2A, or anti-GluN1 antibodies showing association of endogenous GluN2A and GluN1 with transfected GFP-GluN2A. Arrowheads next to the blots indicate protein bands corresponding to GFP-GluN2AWT (top blot), GFP-GluN2AWT (top arrowhead) and endogenous GluN2A (bottom arrowhead, middle blot), and GluN1 (bottom blot). B, surface staining using anti-GFP antibody showing plasma membrane expression of GFP-GluN2AWT and GFP-GluN2AY1292F,Y1325F,Y1387F. C, representative INS-1 832/13 cell-attached current-clamp recordings from an untransfected cell (unt) or a cell transfected with GluN2AWT treated with leptin (10 nm) or cells transfected with GluN2AY1292F,Y1325F,Y1387F and treated with leptin followed by the KATP channel activator diazoxide (200 μm) or with NMDA (50 μm). All GluN2A constructs in this and subsequent panels contain an N-terminal GFP tag. D, group data showing the degree of hyperpolarization induced by leptin in untransfected cells (n = 10), cells transfected with GluN2AWT (n = 13), and cells transfected with GluN2AY1292F,Y1325F,Y1387F (n = 18) with subsequent exposure to diazoxide (200 μm; n = 14) or treated with NMDA alone (50 μm; n = 11). E, representative cell-attached recordings from an untransfected cell and three different GluN2A phosphorylation mutants (Y1292F, Y1325F, or Y1387F) treated with 10 nm leptin. F, group data showing the amount of hyperpolarization induced by leptin for an untransfected cell (n = 26) and for single GluN2A phosphorylation mutants (GluN2AY1292F, n = 19; GluN2AY1325F, n = 16; GluN2AY1387F, n = 15). ***, p < 0.0005 by unpaired t test as compared with untransfected cells. Error bars, S.E.
Changes in membrane potential following leptin treatment were again monitored using cell-attached current-clamp recording. We first compared cells expressing GFP-GluN2AWT with those expressing the GFP-GluN2AY1292F,Y1325F,Y1387F triple phenylalanine mutant. GFP-negative cells on the same coverslip were also examined as untransfected controls. Representative membrane potential traces from untransfected cells and cells expressing GFP-GluN2AWT or triple phosphomutant GFP-GluN2AY1292F,Y1325F,Y1387F are shown in Fig. 4C. Leptin application to untransfected cells induced a mean hyperpolarization of −51.1 ± 5.6 mV (n = 10) that was not statistically significantly different from leptin responses in cells expressing GluN2AWT (−48.0 ± 6.5 mV, n = 13). By contrast, leptin had little hyperpolarizing effect in cells expressing the triple phosphomutant (Fig. 4, C and D; GluN2AY1292F,Y1325F,Y1387F ΔVm = −12.0 ± 1.7 mV; n = 18; p < 0.0005 by unpaired t test compared with untransfected cells). The diminished response is not due to disruption of KATP channel gating or expression as the KATP activator diazoxide (200 μm) was fully able to hyperpolarize cells expressing GluN2AY1292F,Y1325F,Y1387F (ΔVm = −52.7 ± 4.9 mV; n = 14) (Fig. 4D). Moreover, we tested the response of cells expressing GluN2AY1292F,Y1325F,Y1387F to NMDA, which we have shown hyperpolarizes cells in the absence of leptin (19). We found that NMDA hyperpolarized INS-1 832/13 cells by an average of −38.8 ± 7.0 mV (n = 11), suggesting that the GluN2A phosphomutant also did not disrupt NMDAR expression or function (Fig. 4D). To determine the relative contributions of the three tyrosine residues, we next tested the membrane potential response to leptin in cells expressing GluN2AY1292F, GluN2AY1325F, or GluN2AY1387F. In this cohort of cells, leptin application caused a mean hyperpolarization of −47 ± 3.8 mV in untransfected cells (n = 26) (Fig. 4, E and F). Interestingly, leptin still hyperpolarized cells transfected with GluN2AY1325F (ΔVm = −41.7 ± 4.1 mV; n = 16), not significantly different from untransfected cells. By contrast, in cells expressing GluN2AY1292F or GluN2AY1387F, the response to leptin was significantly reduced (Fig. 4, E and F; ΔVm for GluN2AY1292F = −17.4 ± 4.1 mV, n = 19, and ΔVm for GluN2AY1387F = −24.3 ± 4.7 mV, n = 15; p < 0.0005 by unpaired t test). Taken together, these results identify the tyrosine phosphorylation sites Tyr-1292 and Tyr-1387 in GluN2A as responsible for mediating the leptin effect.
Leptin activates Src kinase in β-cells
The above results suggest that leptin likely activates SFKs to modulate NMDAR currents. Studies of GluN2A phosphorylation using in vitro or the HEK293 cell heterologous expression systems have found that both Src and Fyn can phosphorylate GluN2A (24). Src, when activated, autophosphorylates a highly conserved tyrosine residue, Tyr-418, in the catalytic domain of Src kinase, which is required for its full catalytic activity (45, 46). Increased phosphorylation of Tyr-418 is therefore indicative of kinase activation. We used an antibody raised against a peptide containing Src-pY418 to monitor Src phosphorylation to directly test whether leptin activates Src. Immunocytochemistry experiments were carried out on INS-1 832/13 cells treated with or without leptin (10 nm for 10 min) in the presence or absence of the tyrosine kinase inhibitor dasatinib (50 μm). We found that leptin induced a significant increase in Src-pY418 staining as compared with matched controls with prominent staining at the cell periphery (Fig. 5, A and B) that was inhibited by co-application of dasatinib (Fig. 5, A and B). Further biochemical experiments using similar treatment conditions were carried out. In good agreement with the immunostaining results, leptin increased the ratio of phosphorylated to total Src by 176.0 ± 30.0% as compared with controls (p < 0.05 by unpaired t test), which was reduced to 102.1 ± 40.4% of controls when co-applied with dasatinib (Fig. 5, C and D). Because the Src-pY418 antibody also recognizes conserved corresponding phosphopeptide in other SFKs, including Fyn, and because both Src and Fyn are expressed in β-cells (47), we further tested the requirement of Src activity in leptin-induced response by expressing a dominant-negative kinase-dead Src mutant (48) (see “Experimental procedures”) in INS-1 832/13 cells. For this, we monitored leptin-induced increase of KATP channel surface expression by surface biotinylation, as described in our previous studies (7, 9). As expected, leptin caused an increase in surface biotinylated SUR1, the regulatory subunit of the β-cell KATP channel, in control cells. However, in cells transfected with the kinase-dead Src mutant leptin failed to show an increase in surface biotinylated SUR1 (Fig. 5, E and F). This result lends further support to the requisite role of Src activity in mediating the effect of leptin on KATP channel trafficking.
Figure 5.
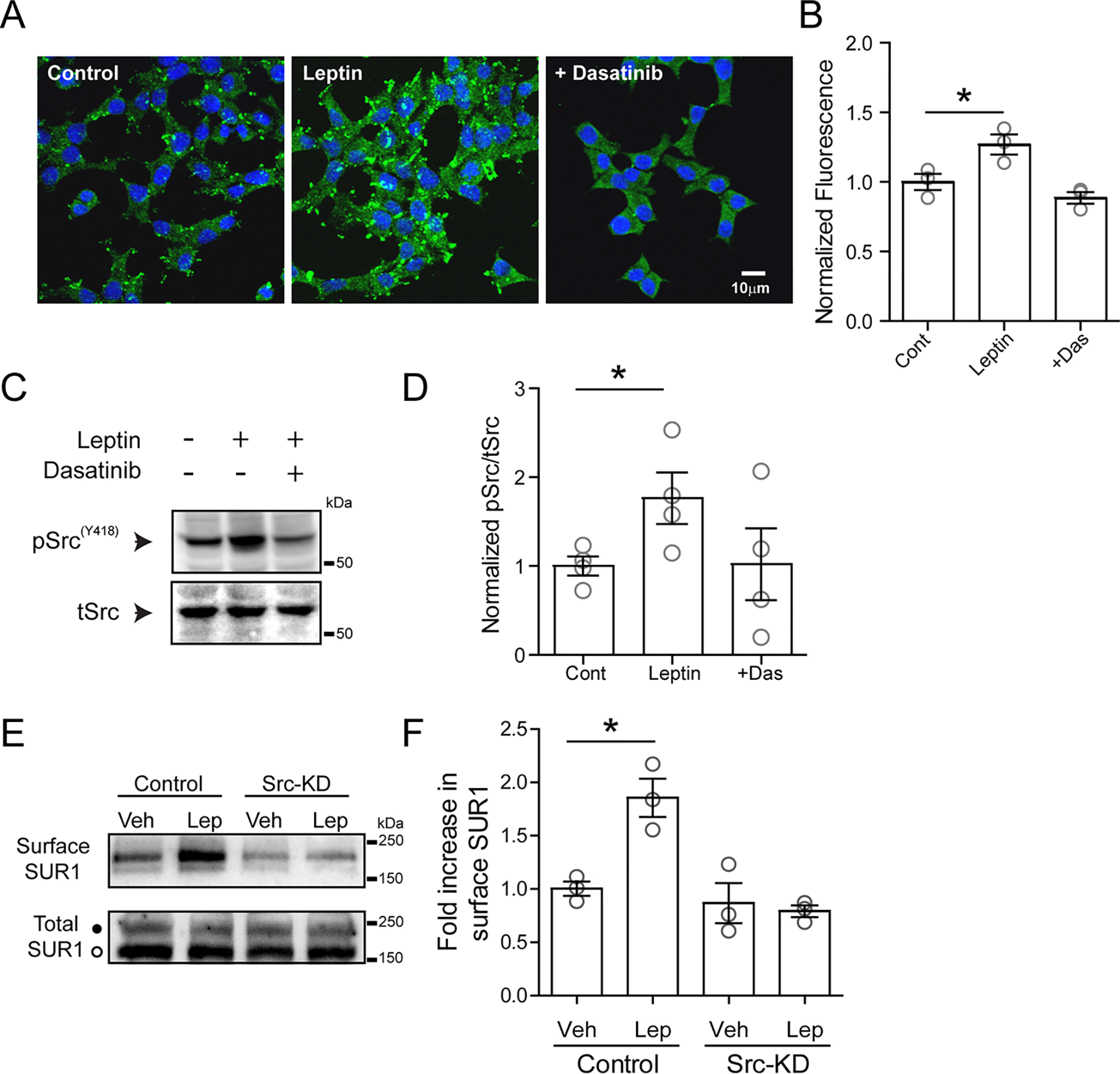
Leptin induces activation of Src kinase. A, immunofluorescence images of cultured INS-1 831/13 cells stained with 4′,6-diamidino-2-phenylindole (blue) and phosphorylated SrcY418 (green) for conditions indicated. Cells were treated with vehicle (control), leptin (10 nm, 10 min), or leptin with dasatinib (25 μm, pretreated for 30 min before leptin was applied). B, group data for phosphorylated SrcY418 fluorescence for each condition normalized to the control (n = 100 cells/condition for each experiment, and the experiment was repeated three times). *, p < 0.05, unpaired Student's t test. C, representative Western blotting of phosphorylated SrcY418 from cell lysates prepared from INS-1 832/13 cells treated with vehicle, leptin, or leptin co-applied with dasatinib. D, group data from four independent experiments as indicated in C. *, p < 0.05, unpaired Student's t test. E, top, Western blotting showing surface SUR1 in response to vehicle or leptin using control cells and cells transfected with an Src kinase–dead mutant (Src-KD). Bottom, total SUR1 showing both the complex glycosylated form (top band, solid circle) that can traffic to the cell surface and the core-glycosylated form (bottom band, open circle) that is in the endoplasmic reticulum/early Golgi. F, quantitation of three independent surface SUR1 experiments. Data were analyzed by one-way analysis of variance (p = 0.0019, F = 12.95) followed by a post hoc Dunnett's multiple-comparison test with significance set to p < 0.05 (*). Error bars, S.E.
Leptin signaling through Src-mediated phosphorylation of GluN2A is conserved in human β-cells
We have previously shown that leptin induces hyperpolarization that depends on NMDARs in human β-cells as in INS-1 832/13 cells (19). Having found that leptin potentiates NMDARs via Src-mediated phosphorylation of GluN2A in INS-1 832/13 cells, we next tested whether the same mechanism applies to human β-cells. Cell-attached current-clamp recordings were made in human β-cells dissociated from islets of three different nondiabetic donors obtained through the Integrated Islets Distribution Program (IIDP). All donor information can be found in Table 1. Bath application of leptin alone induced a mean membrane hyperpolarization of −43.7 ± 8.9 mV (n = 9 cells from three donors) (Fig. 6, A and B). The GluN2A-selective inhibitor TCN-201 largely eliminated the leptin response (ΔVm = −9.1 ± 4.2 mV, n = 12 cells from 3 donors; p < 0.05, unpaired t test) (Fig. 6, A and B), suggesting that GluN2A-containing NMDARs underlie leptin signaling in human β-cells. We next tested whether leptin modulation of NMDARs and membrane potential in human β-cells requires Src. As shown in the example traces in Fig. 6C, the SFK inhibitor AZD0530 nearly abolished the ability of leptin to hyperpolarize human β-cells (ΔVm = −0.4 ± 1.2 mV; n = 6 cells from two nondiabetic donors), whereas another SFK inhibitor, dasatinib, also diminished leptin-induced hyperpolarization, albeit to a lesser extent (ΔVm = −13.5 ± 2.8 mV, n = 7 cells from three donors) compared with human β-cells treated with leptin alone (ΔVm = −33.5 ± 6.0 mV; n = 17 cells from five nondiabetic donors; p < 0.0005 between AZD0530 and control, and p < 0.05 between dasatinib and control, Welch's t test) (Fig. 6D). Taken together, results from both INS-1 832/13 and human β-cells establish a mechanistic link between leptin receptor signaling and modulation of GluN2A-containing NMDARs via Src kinase.
Table 1.
Human islet donor information
| Donor no. (date received)a | T2Db | Age | Gender | BMIc | Cause of death | Islet viability (%) | Islet purity (%) |
|---|---|---|---|---|---|---|---|
| 1 (11/21/17) | N | 62 | M | 28.9 | Stroke | 97 | 80 |
| 2 (01/10/18) | N | 32 | M | 26.2 | Anoxia | 98 | 85 |
| 3 (10/09/18) | N | 47 | F | 24.1 | Stroke | 90 | 90 |
| 4 (10/19/18) | N | 55 | F | 35.7 | Stroke | 98 | 94 |
| 5 (04/02/18) | N | 32 | M | 32.3 | Anoxia | 95 | 85 |
| 6 (11/07/18) | N | 48 | M | 24.4 | Head trauma | 90 | 90 |
| 7 (10/27/15) | Y | 41 | F | 43.1 | Stroke | 95 | 95 |
| 8 (08/09/16) | Y | 52 | F | 39.9 | Anoxia | 95 | 85 |
| 9 (08/29/16) | Y | 57 | M | 34.6 | Head trauma | 98 | 80 |
| 10 (11/07/16) | Y | 55 | M | 32.5 | Anoxia | 98 | 75 |
| 11 (04/03/17) | Y | 65 | F | 42.6 | Anoxia | 90 | 95 |
| 12 (06/01/17) | Y | 42 | F | 37.0 | Head trauma | 89 | 90 |
| 13 (12/13/17) | Y | 37 | F | 38.1 | Stroke | 94 | 85 |
aDonor information for specific experiments is as follows: Fig. 6B: donors 4, 5, 6; Fig. 6C, D: donors 1, 2, 3, 4, 6; Fig. 8A: donor 7, 8, 9; Fig. 8B, C, D: donors 10, 11; total NMDAR currents from normal β-cells: donors 1, 2, 3, 4, 5; total NMDAR currents from T2D β-cells: donors 12, 13.
bT2D: type 2 diabetes.
cBMI: body mass index.
Figure 6.

Leptin signaling through GluN2A-containing NMDARs in human β-cells is mediated by Src family kinases. A, individual cell-attached membrane potential recordings from human β-cells isolated from nondiabetic islets treated with leptin (10 nm) alone (top) or leptin together with TCN-201 (bottom; 50 μm). B, summary data from three nondiabetic donors showing the amount of hyperpolarization induced by leptin alone (n = 2–5 cells/donor) or when leptin was co-applied with TCN-201 (n = 3–4 cells/donor). Inset, normalized responses to leptin across donors in the absence or presence of TCN-201. C, representative traces of cell-attached current-clamp recordings from human β-cells isolated from nondiabetic islets treated with leptin in the absence or presence of AZD (10 μm). D, summary data from five nondiabetic donors showing the extent of membrane hyperpolarization in response to leptin alone (n = 19 cells; 3–6 cells/donor) or when leptin was co-applied with dasatinib (Das; 25 μm; n = 7 cells; 2–3 cells/donor) or AZD (n = 6 cells; 3 cells/donor). Inset, normalized responses to leptin across donors in the absence or presence of dasatinib or AZD. *, p < 0.05; ***, p < 0.0005, Welch's t test compared with leptin alone group. Error bars, S.E.
Leptin signaling through NMDARs is disrupted in β-cells from db/db mice and from human type 2 diabetic donors
Leptin resistance is a common pathological feature of type 2 diabetes associated with obesity (49). To test how the leptin-signaling pathway pertinent to this study is affected in diabetic β-cells, we examined β-cells from leptin-insensitive db/db mice, which harbors a mutation in the leptin receptor gene that renders ObRb signaling-defective (50), as well as from human obese type 2 diabetic donors. Cell-attached recordings of membrane potential in β-cells isolated from C57BL/6J (WT) mice showed a clear response to leptin with a ΔVm of −33.5 ± 5.2 mV (n = 8), as expected. By contrast, β-cells isolated from db/db mice failed to respond to leptin (10 nm leptin for 15 min, ΔVm = −0.4 ± 0.4 mV, n = 6, p < 0.0001), although subsequent application of the KATP channel opener diazoxide (250 μm) evoked large hyperpolarizations (ΔVm = −44.7 ± 8.0 mV, n = 6) (Fig. 7, A and B), suggesting that KATP channels are functionally expressed in these mice. These results also provide clear evidence that leptin-induced hyperpolarization requires functional leptin receptors. Next, we tested whether direct activation of NMDARs using the receptor agonist NMDA can bypass leptin receptor deficiency and cause membrane hyperpolarization in the absence of leptin. Control WT cells hyperpolarized in response to bath application of 50 μm NMDA (ΔVm = −37.9 ± 5.5 mV, n = 9). Strikingly, β-cells from db/db mice also showed robust responses to NMDA (ΔVm = −44.3 ± 6.7 mV, n = 9; Fig. 7, C and D), indicating that the signaling events downstream of NMDAR activation that lead to increased KATP and Kv2.1 channel trafficking and membrane hyperpolarization remain intact despite harboring nonfunctional leptin receptors.
Figure 7.

Direct NMDAR activation causes membrane hyperpolarization in mice deficient in leptin receptor signaling. A, representative cell-attached membrane potential recordings from β-cells isolated from C57BL/6J (WT) mice treated with leptin (top trace) or β-cells isolated from db/db mice treated with leptin (10 nm) followed by diazoxide (250 μm; bottom trace). B, group data showing the degree of membrane hyperpolarization in mV for β-cells isolated from C57BL/6J (WT, n = 9) or db/db (n = 7) mice for the indicated conditions. Gray and black circles indicate different animals. ***, p < 0.0001, unpaired Student's t test compared with WT leptin–treated group. C, as described for A except β-cells were treated with NMDA (50 μm). D, group data showing the degree of membrane hyperpolarization in mV for β-cells isolated from C57BL/6J (n = 9) or db/db (n = 9) mice following NMDA treatment. Error bars, S.E.
To test whether findings from the monogenic leptin-resistant mouse β-cells are also reproduced in human diabetic β-cells, we examined β-cells from obese (body mass index > 30) diabetic donors with presumably different genetic background through the IIDP (see Table 1 for donor information). We first tested whether these cells were resistant to leptin. Whole-cell recordings were performed to assess KATP and Kv2.1 current density with or without leptin treatment (10 nm for 30 min) as described previously from β-cells taken from normal donors (7, 9). In contrast to normal human β-cells (7, 9), leptin did not increase the current density of KATP channels (107.4 ± 23.2 for control versus 75.4 ± 11.7 pA/pF for leptin-treated, n = 6) or Kv2.1 channels (683.3 ± 146.3 for control versus 720.3 ± 158.0 pA/pF for leptin-treated, n = 10) (Fig. 8A). Moreover, leptin failed to potentiate NMDAR currents in β-cells from diabetic donors (12.4 ± 2.3 pA for control versus 11.3 ± 1.8 pA following leptin treatment, n = 5) (Fig. 8B), in contrast to β-cells from normal donors we reported previously (19). Consistent with these results, leptin did not induce membrane hyperpolarization in diabetic human β-cells (ΔVm = 1.3 ± 2.1 mV for leptin, n = 8) (Fig. 8, C and D). Conversely, application of 50 μm NMDA in the bath solution to directly activate NMDARs in β-cells from the same donors used to test leptin response did induce significant hyperpolarization (ΔVm = −22.0 ± 8.6 mV, n = 9) (Fig. 8, C and D), indicating that NMDAR function and downstream signaling events were not compromised as was observed in β-cells from db/db mice. These results show that β-cells from a sample of obese diabetic donors were leptin-resistant but retained the ability to hyperpolarize in response to direct NMDAR activation.
Figure 8.

β-Cells from obese type 2 diabetic human donors failed to respond to leptin but retained response to NMDA. A, current density of KATP channels (left) and Kv2.1 channels (right) in β-cells from human diabetic donors treated with vehicle or 10 nm leptin (30 min). Different symbols are used to denote different donors. There is no statistically significant difference between leptin-treated and control groups. B, whole-cell NMDA currents in β-cells from diabetic donors before leptin application (control) or after 5-min incubation in 10 nm leptin (leptin). The mean (filled circles) and individual cell currents (before and after leptin treatment connected by a solid line) are shown. The open and filled gray diamonds represent different donors. C, representative cell-attached current-clamp recordings from human diabetic β-cells treated with 10 nm leptin (top) or 50 μm NMDA (bottom). Note the spike in the top trace near the end of the recording is a solution suction artifact. D, group data showing membrane potential response to leptin or NMDA in β-cells isolated from human diabetic donors. *, p < 0.05, unpaired Student's t test compared with the leptin-treated group. Error bars, S.E.
Discussion
The results presented in this study elucidate the mechanism by which leptin potentiates NMDAR currents to regulate pancreatic β-cell excitability. Multiple lines of evidence support our conclusion that leptin modulation of β-cell membrane potential requires the phosphorylation of GluN2A-containing NMDARs by Src kinase. First, application of TCN-201, a potent and highly selective inhibitor for GluN2A-containing NMDARs, diminished leptin-induced membrane hyperpolarization in both INS-1 832/13 and human β-cells. Second, inhibition of SFKs blocked the ability of leptin to potentiate NMDAR currents and hyperpolarize β-cells. Third, leptin increased phosphorylation of Src at Tyr-418, a site that is required for its catalytic activity (45, 46), and dominant-negative suppression of Src activity via a kinase-dead mutant prevented the ability of leptin to increase surface KATP channels. Finally, mutation of known Src kinase phosphorylation sites, specifically Tyr-1292 and Tyr-1387, located in the C terminus of GluN2A prevented leptin-induced hyperpolarization. It is worth noting that Src has many downstream targets. For example, Src has been shown to regulate Kv2.1 activity (51) and actin dynamics (52, 53). Therefore, results from experiments involving pharmacological or molecular manipulation of Src activity could be due to Src regulation of other targets in addition to phosphorylation of NMDARs and downstream trafficking of KATP and Kv2.1 channels. Nonetheless, the collective evidence presented in this study together with our previously published studies (7, 9, 19) points to a signaling pathway in which leptin activates Src kinase to phosphorylate GluN2A of NMDARs, which leads to potentiation of NMDAR currents and increased Ca2+ influx. The increased Ca2+ influx then activates CaMKKβ and AMPK, resulting in PKA-dependent actin depolymerization and trafficking of KATP and Kv2.1 channels to the cell surface, which culminates in hyperpolarization of β-cell membrane potential and reduced insulin secretion. Given Ca2+ influx is normally associated with β-cell depolarization and insulin secretion, it is likely that the Ca2+ influx through NMDARs triggered by leptin is localized and specifically coupled to downstream events that regulate potassium channel trafficking and hyperpolarize the membrane. Importantly, we demonstrate in β-cells from mice deficient in leptin signaling and obese diabetic human donors that direct activation of NMDARs by NMDA can bypass leptin resistance and induce membrane hyperpolarization, suggesting that the signaling pathway downstream of Src kinase modulation of NMDARs remains operational and may be targeted to modulate insulin secretion.
Mechanism of NMDAR potentiation by leptin
NMDARs are allosterically modulated by a variety of endogenous extracellular ions. For example, Mg2+ can bind within the pore region of NMDAR and cause a voltage-dependent block (20). In addition, Zn2+ can promote voltage-dependent and voltage-independent inhibition of NMDAR activity (54–56). Voltage-dependent inhibition, like that observed with Mg2+, occurs at a low-affinity GluN1 Zn2+-binding site located within the channel pore (56), whereas voltage-independent inhibition appears to be mediated by a high-affinity Zn2+-binding site located outside the channel pore and is associated with GluN2 subunits (57). Of the GluN2 subunits, GluN2A is 200-fold more sensitive to Zn2+ than GluN2B (58, 59). The binding of Zn2+ to these subunits is thought to trap the receptor in a low open probability state, reducing their activity (54, 55). Zn2+ has been shown to be co-released with several transmitters, including glutamate, and at some glutamatergic synapses within the brain, vesicular Zn2+ is thought to diminish NMDAR activity (60). Interestingly, β-cells contain and release high levels of Zn2+ due to its role in the biosynthesis and packaging of insulin (61). We speculate that NMDARs residing on the β-cell membrane would be subjected to high concentrations of Zn2+ inhibition during bouts of insulin secretion. Of note, phosphorylation of GluN2A by Src has been shown to potentiate NMDAR currents by reducing tonic inhibition of the receptor by Zn2+ (23, 25). A possible scenario is that under high glucose concentrations when β-cells are depolarized to relieve external Mg2+ block, NMDAR activity is still limited by Zn2+ co-released with insulin. However, inhibition by Zn2+ could be reduced by phosphorylating GluN2A via leptin-induced Src activation to regulate KATP and Kv2.1 surface expression and tune insulin secretion. In this way, GluN2A phosphorylation affords a mechanism to allow modulation of β-cell response to glucose stimulation by additional inputs such as leptin. Interestingly, in cells overexpressing GluN2A phosphomutants that failed to respond to leptin, bath application of 50 μm NMDA still induced membrane hyperpolarization. This suggests that NMDAR currents activated by endogenously released ligands and potentiated by leptin represent a fraction of total NMDAR currents that were activated by 50 μm NMDA under our experimental conditions.
Mechanism of Src activation by leptin
Expression of ObRb mRNAs in β-cells has been well-documented (1, 6, 62–65). It is assumed that leptin binds to ObRb expressed by pancreatic β-cells to suppress glucose-stimulated insulin secretion. However, leptin has also been reported to transactivate other cytokine receptor signaling molecules, including epidermal growth factor receptor, type 1 insulin-like growth factor receptor, low-density lipoprotein receptor–related protein, and vascular endothelial growth factor receptor (13). Our finding that db/db β-cells lack leptin-induced hyperpolarization lends strong support to the requirement of ObRb for leptin regulation of K+ channel trafficking. The mechanism by which ObRb activation leads to Src activation in β-cells awaits further investigation. The enzymatic activity of Src is tightly regulated by tyrosine phosphorylation at Tyr-418 in the catalytic domain and Tyr-529 at its C-terminal tail. When Tyr-529 is phosphorylated, the C-terminal tail acts as an autoinhibitory peptide to block kinase activity, and its dephosphorylation relieves autoinhibition, leading to autophosphorylation of Tyr-418, which further activates the kinase (38). Activation of ObRb is known to activate several tyrosine phosphatases, such as Shp2, which could dephosphorylate pY529 to activate the kinase (13, 38). Alternatively, direct recruitment of Src via the kinase's Src homology domains to phosphotyrosine in the activated leptin receptor complex or regulation by other kinases activated by leptin are also possibilities (38).
Tyrosine phosphorylation sites in GluN2A and GluN2B have been extensively investigated in recombinant expression systems and in neurons. The three GluN2A tyrosine residues examined in our study have all been shown to contribute to NMDAR current modulation (24, 25, 43). It is interesting that our data suggest that phosphorylation of Tyr-1292 and Y1387F but not Tyr-1325 are important for leptin response. This is in contrast to previous findings in the striatum neurons that Tyr-1325 is critical for Src-induced increase of NMDAR activity to regulate depression-related behavior (43). Very recently, Bland et al. (66) showed that leptin controls glutamatergic synaptogenesis and NMDAR trafficking via GluN2B phosphorylation by Fyn. Thus, the precise mechanisms and consequences of NMDAR modulation by leptin and SFKs are likely to be cell context–dependent.
Implications for leptin resistance and type 2 diabetes
We find that β-cells from obese type 2 diabetic human donors no longer hyperpolarize in response to leptin, as was also seen in β-cells from leptin-resistant, obese diabetic db/db mice. Examination of KATP and Kv2.1 currents confirms that the lack of hyperpolarization is due to the lack of increase in trafficking of these channels to the cell surface, indicating that leptin signaling was disrupted. Interestingly, we found that NMDAR current density was similar in β-cells from normal and obese diabetic donors with detectable currents (12.26 ± 4.12 pA/pF from normal donors, n = 16 (total of 35 cells from five donors (Table 1); 19 cells had no detectable currents) versus 11.60 ± 2.17 pA/pF from obese diabetic donors, n = 20 (total of 26 cells from two donors (Table 1); 6 cells had no detectable currents). Moreover, direct activation of NMDAR by NMDA triggered membrane hyperpolarization in both db/db β-cells and β-cells from human diabetic donors. Thus, despite leptin resistance, signaling mechanisms downstream of NMDARs (i.e. activation of CaMKKβ, AMPK, and PKA-dependent actin depolymerization we reported previously (7, 9, 19)) remain functional to promote KATP and Kv2.1 channel trafficking to modulate β-cell excitability. The ability of leptin to temper glucose-stimulated insulin secretion has been proposed as part of an adipoinsular feedback loop between adipocytes and β-cells to prevent excessive secretion of insulin, an adipogenic hormone, thereby limiting fat mass (67). Disruption of leptin signaling in β-cells has been shown to disrupt glucose homeostasis in some animal models (64, 65), although others argue against a major role of β-cell leptin signaling in glucose homeostasis (68). Whether the controversial findings were due to different mouse strains and genetic models used (69) remains to be resolved. Nonetheless, it is tempting to speculate on the potential of exploiting the leptin signaling pathway we have identified for the prevention and treatment of obesity-associated type 2 diabetes. In obese individuals, hyperleptinemia may result in leptin resistance (49, 70), which may lead to excessive insulin secretion. Hyperinsulinemia can then cause insulin resistance and hyperglycemia, further fueling obesity, leading to a loss of glucose control and eventually β-cell failure as seen in type 2 diabetes (71). Based on our finding that obese diabetic human β-cells, despite not being able to respond to leptin, retain response to NMDAR activation, stimulating NMDAR in leptin-resistant obese, prediabetic individuals may help to prevent excessive insulin secretion and thereby prevent or slow the development of type 2 diabetes (71). On the other hand, inhibition of NMDARs in individuals who have already developed type 2 diabetes may stimulate insulin secretion and alleviate hyperglycemia, as has been reported by Marquard et al. (17).
In summary, the current study identifies a novel leptin-signaling mechanism in β-cells wherein leptin activates Src kinase to phosphorylate GluN2A-containing NMDARs and potentiate NMDAR activity, thereby reducing β-cell excitability and insulin secretion. The findings build on our previously identified signaling pathway and provide a molecular explanation for the action of leptin. Moreover, they raise the therapeutic potential of targeting the pathway for the prevention and treatment of type 2 diabetes.
Experimental procedures
Chemicals
Leptin was purchased from Sigma. NMDA, diazoxide, roscovitine, Ro 25-6981, and TCN201 were purchased from Tocris Bioscience (Bristol, UK). AZD0530, GO6983, and dasatinib were from Selleckchem (Houston, TX, USA). Src kinase activator peptide (YEEI) was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
INS-1 832/13 cell culture and transfection
INS-1 832/13 cells were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, 10 mm HEPES, 2 mm glutamine, 1 mm sodium pyruvate, and 50 μm β-mercaptoethanol. Only cells within passage number 75 were used for experiments. For experiments involving transfection, cells were prepared as follows. INS-1 832/13 cells were grown on coverslips placed within 35-mm dishes and were transfected with WT (Addgene plasmid 45445), single GluN2A phosphorylation mutants (GluN2AY1292F, GluN2AY1325F, and GluN2AY1387F) or a triple phosphorylation mutant (GluN2AY1292F,Y1325F,Y1387F) using Lipofectamine 2000 (Invitrogen). Plasmids also encoded enhanced GFP at the N terminus to visually discern transfected from nontransfected cells at time of recording 48–72 h after transfection. The kinase-dead Src mutant (pLNCX chick src K295R, a gift from Joan Brugge, Addgene plasmid 13659) transfection was carried out in 10-cm plates (cell confluence, ∼50–60%) using Lipofectamine 2000 (20 μg of DNA and 40 μl of lipofectamine 2000) 48 h prior to experiments.
Dissociation of human pancreatic β-cells
Human β-cells were dissociated from human islets obtained through the Integrated Islets Distribution Program, as described previously (7, 9, 19). Briefly, human islets were cultured in RPMI 1640 medium with 10% FBS and 1% l-glutamine. For recording, islets were dissociated into single cells by trituration in a solution containing 116 mm NaCl, 5.5 mm d-glucose, 3 mm EGTA, and 0.1% BSA, pH 7.4. Dissociated cells were then plated on 0.1% gelatin-coated coverslips placed in 35-mm culture dishes (Falcon 35-3002). For electrophysiological experiments, β-cells were initially identified using the high autofluorescence signature of β-cells to 488-nm excitation, as these cells have high concentrations of unbound flavin adenine dinucleotide (72, 73). Dithizone (Sigma–Aldrich) staining was then used to further confirm β-cell identity at the end of each recording (74). Donor information for specific experiments is provided in Table 1.
Dissociation of mouse pancreatic β-cells
Mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA): control (C57BL/6J) and db/db [B6.BKS(D)-Leprdb/J]. Pancreatic islets were isolated from the mice as described previously (75) and were performed in compliance with institutional guidelines and approved by the Oregon Health and Science University Animal Care and Use Committee. Following isolation, islets were cultured overnight in FBS-supplemented RPMI 1640 medium at 37 °C and 5% CO2. Pancreatic β-cells were dissociated from islets using Spinners/EGTA solution (116 mm NaCl, 5.37 mm KCl, 0.8 mm MgSO4, 26 mm NaHCO3, 11.67 mm NaH2PO4, 5.5 mm d-glucose, 3 mm EGTA, 1% BSA, 1% phenol red (pH 7.4)). Islets were incubated twice in Spinners/EGTA for 10 min with slight agitation every 5 min. In between incubations with Spinners/EGTA, the islets were washed with FBS-supplemented RPMI 1640 medium. Dissociated cells were plated on glass coverslips coated with 1% gelatin (Sigma–Aldrich) and cultured overnight. The identity of individual β-cells was confirmed by dithizone staining at the end of each experiment (74).
RT-PCR
Total RNA was isolated from mouse brain and INS-1 832/13 cells using an RNeasy minikit (Qiagen, Hilden, Germany), and RT-PCR (from 1 μg of mouse brain RNA and 1.8 μg of INS-1 832/13 RNA) was performed using the Superscript RT-PCR system (Invitrogen) to examine the mRNA expression of NMDA receptor subunits, Grin1 and Grin2a–2d. β-Actin mRNA was used as a standard reference housekeeping gene. The primers used were as follows: Grin1, 5′-GGTTTG-AGATGATGCGAGTCTAC (forward) and 5′-CAGC-AGAGCCGTCACATTCTTGG (reverse); Grin2a, 5′-CAATCTGACTGGATCACAGAGC (forward) and 5′-CTG-TCCTTCCCTTGAAAGGATC (reverse); Grin2b, 5′-CAATAACCCACCCTGTGAGG (forward) and 5′-GGTGGGTT-GTCACAGTCATAG (reverse); Grin2c, 5′-ACATGAAGTATCCAGTATGG (forward) and 5′-GTTCTGGTTGTAGCTGACAG (reverse); Grin2d, 5′-AGGTGTTCTATCAGCGTG (forward) and 5′-TGTAGCTGTGCATGTCAG (reverse); and β-actin, 5′-CGTAAAGACCTCTATGCCAA (forward) and 5′-AGCCATGCCAAATGTGTCAT (reverse).
Immunoblotting
INS-1 832/13 cells were lysed in lysis buffer (50 mm Tris-HCl, 2 mm EDTA, 2 mm EGTA, 100 mm NaCl, 1% Triton X-100, pH 7.4, with cOmplete EDTA-free protease inhibitor mixture (Roche, Basel, Switzerland) and Halt-phosphatase inhibitor mixture (Thermo Scientific) for 30 min at 4 °C, and cell lysates were cleared by centrifugation (MIKRO 200R, Helmer Scientific, Noblesville, IN, USA) at 14,000 rpm for 10 min at 4 °C. In some experiments, lysis buffer was replaced with ice-cold PBS to aid in the isolation of membrane proteins. PBS cell lysates were briefly spun at 3000 rpm for 5 min at 4 °C, and the membrane pellet was homogenized in a hypotonic solution containing 15 mm KCl, 10 mm HEPES, 1.5 mm MgCl2 with phosphatase and protease inhibitors by passing 30 times through a 27½ gauge needle. Spun at 6,000 rpm at 4 °C for 2 min to remove nuclei and cellular debris. Supernatant was collected and transferred to 1.5-ml Beckman centrifuge tubes and spun at 54,000 rpm at 4 °C for 60 min. The resulting membrane pellet was resuspended in solubilization buffer containing 0.2 m NaCl, 0.1 m KCl, 0.05 m HEPES, and proteins were separated by SDS-PAGE (7.5% polyacrylamide gel) and transferred onto polyvinylidene difluoride membranes (Millipore, Burlington, MA, USA). Membranes were incubated overnight at 4 °C with primary antibody diluted in TBST (TBS plus 0.1% Tween 20) followed by incubation with horseradish peroxidase–conjugated secondary antibodies in TBST for 1 h at room temperature. Primary antibodies used were anti-phosphorylated SrcY418 (human Src numbering; rabbit monoclonal; Abcam, ab133460; 1:1000 dilution), anti-c-Src; Abcam, ab16885; 1:100 dilution), anti-GluN1 (NeuroMab, 75-272; 1:1000 dilution), anti-GluN2A (NeuroMab, 75-288; 1:1000 dilution), and anti-GluN2B (NeuroMab, 75-101; 1:1000 dilution). Blots were developed using Super Signal West Femto (Pierce) and imaged with FluorChemE (ProteinSimple, San Jose, CA, USA), protein bands were quantified using ImageJ (National Institutes of Health) and normalized to the corresponding controls.
Immunocytochemistry
INS-1 832/13 cells were fixed in 2% paraformaldehyde in PBS for 10 min at room temperature, permeabilized with 0.2% Triton X-100 in 1% BSA/PBS, and blocked for 60 min with 1% BSA in PBST (PBS + 0.1% Tween 20) before being incubated overnight at 4 °C with primary rabbit polyclonal antibodies directed against phospho-SrcY418 (Abcam, ab4816). Proteins were visualized using Alexa 488–conjugated secondary antibodies. Fluorescent images were acquired using a Zeiss LSM780 confocal microscope equipped with a ×63 oil immersion objective. Images were processed and analyzed using NIH ImageJ software (76). For surface staining of GFP-GluN2AWT and GFP-GluN2AY1292F,Y1325F,Y1387F, cells were incubated in cold DPBS containing anti-GFP antibody (1:100; ThermoScientific, G10362) at 4 °C for 30 min. Cells were then fixed as described above, and surface GFP was visualized using a Cy3-conjugated secondary antibody.
Immunoprecipitation
48 h post-transfection of GFP-GluN2AWT, cells were collected in DPBS. A membrane pellet was obtained following the protocol described above. The pellet was then solubilized in co-immunoprecipitation buffer (0.5 mm Tris-HCl, 150 mm NaCl, 0.5% Igepal, pH 7.2) and incubated with 25 μl of GFP-Trap Dynabeads (Chromotek) overnight at 4 °C. The beads were washed twice with co-immunoprecipitation buffer. Proteins bound to the beads were then eluted in 2× protein loading buffer for 10 min at 95 °C. Samples were analyzed via immunoblotting for GFP (1:100; ThermoScientific, G10362), GluN2A (1:1000; NeuroMab, 75-288), and GluN1 (1:1000; NeruoMab, 75-272) and simultaneously imaged using near-IR secondary antibodies (LI-COR IRDye).
Surface biotinylation
INS-1 832/13 cells were incubated in RPMI 1640 for 1 h at 37 °C prior to a 30-min treatment with vehicle or leptin (10 nm). Cells were then washed four times with cold DPBS and incubated with 1 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Pierce) in DPBS with vehicle or leptin for 30 min at 4 °C. The reaction was terminated by incubating cells twice for 5 min with DPBS containing 50 mm glycine followed by two washes with cold DPBS. Cells were then lysed in 300 μl of lysis buffer as described above, and 500 μg of total lysate was incubated with 50 μl of an 50% slurry of NeutraAvidin-agarose beads (Pierce) overnight at 4 °C. Biotinylated proteins were eluted with 2× protein loading buffer for 15 min at 37 °C. Both eluent and input samples (50 μg of total cell lysate) were analyzed by immunoblotting using anti-SUR1 antibody (raised against a hamster SUR1 C-terminal peptide KDSVFASFVRADK) as described previously (7).
Electrophysiology
Electrophysiological recordings were conducted using an Axon 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) with Clampex 9.2 (pCLAMP) software. Signals were acquired at 20 kHz and filtered at 2 kHz. Recording electrodes (tip resistances ranged between 3 and 6 megaohms) were pulled from nonheparinized Kimble glass (Thermo Fisher Scientific, Waltham, MA, USA) using a P-97 micropipette puller (Sutter Instruments). For whole-cell recording of NMDA currents, external Tyrode's solution contained 137 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 0.5 mm MgCl2, 5 mm Na-HEPES, 3 mm NaHCO3, and 0.16 mm NaH2PO4, (pH 7.2). The external solution was supplemented with 0.1 mm glycine and 11 mm glucose, and in some experiments Mg2+ was omitted as specified in the figure legends. The internal pipette solution contained 140 mm potassium gluconate, 6 mm EGTA, 10 mm HEPES, 5 mm K2ATP, 1 mm CaCl2 (pH 7.2). To induce NMDA-mediated currents, cells were held at −70 mV, and NMDA (1 mm) was puffed (3–5 p.s.i. for 0.5 s) with carbogen using 1–0.5-megaohm micropipettes connected to a Multi-function Microforge Controller DMF1000 (World Precision Instruments, Sarasota, FL, USA) equipped with a pressure regulator. In experiments measuring NMDAR current response to Mg2+ block (Fig. 1B), leptin (Figs. 3 and 8B), and leptin plus AZD (Fig. 3), 1 mm NMDA was puffed repeatedly at 1-min intervals 3–5 times to establish control baseline. Only cells that showed reproducible currents to repeated NMDA puffs during control baseline measurements were subsequently treated with 100 μm Mg2+, 10 nm leptin, or leptin plus AZD for 5 min, after which 1 mm NMDA was again puffed at 1-min intervals 3–5 times. The currents before and after treatments were then averaged, and the averaged values are shown as individual data points in the figures.
Whole-cell KATP and Kv2.1 current recordings (Fig. 8A) were conducted as described previously (7, 9). For KATP currents, cells were held at −70 mV, and KATP currents were recorded at two voltage steps (−50 and −90 mV) applied every 2 s. Pipette solution contained 140 mm KCl, 10 mm K-HEPES, 1 mm K-EGTA, pH 7.3. The bath solution contained 137 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 0.5 mm MgCl2, 5 mm Na-HEPES, 3 mm NHCO3, 0.16 mm NaH2PO4, pH 7.2. Diazoxide (200 μm) was applied to the bath solution immediately after break-in to maximally stimulate KATP channels. After the current had plateaued, 300 μm tolbutamide (a KATP channel inhibitor) was applied, and residual currents were subtracted from maximal currents observed in diazoxide and divided by cell capacitance to obtain KATP current density. For Kv2.1, micropipettes were filled with an internal solution containing 140 mm KCl, 1 mm CaCl2, 2 mm MgCl2, 5 mm EGTA, 5 mm ATP, 10 mm glucose, and 10 mm HEPES, pH 7.3. The bath solution contained the following: 140 mm NaCl, 5 mm KCl, 4 mm MgCl2, 11 mm glucose, 10 mm HEPES, pH 7.3. Ca2+ was excluded from the bath solution to eliminate calcium channel currents. A 30-ms prepulse to −10 mV was used to inactivate transient potassium channel currents and voltage-dependent Na+ currents. The sustained current at +80 mV, after subtracting currents remaining in a 10 mm concentration of the potassium current blocker tetraethylammonium, was divided by cell capacitance for Kv2.1 current density calculation.
For cell-attached recording to monitor membrane potentials, micropipettes were filled with 140 mm NaCl. The bath solution contained 137 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 0.5 mm MgCl2, 5 mm Na-HEPES, 3 mm NHCO3, 0.16 mm NaH2PO4, 11 mm glucose, pH 7.2. The liquid junction potential was measured to be −10 mV and corrected from the recorded Vm. Seal resistances between the recording pipette and the cell membrane ranged between 4 and 11 gigaohms, and membrane potentials were monitored in current-clamp mode (I = 0). After correction for the liquid junction potential, the average starting Vm was around −4 mV. Note that the Vm estimated in this configuration depends on the ratio of the seal resistance and the combined patch and cell resistance, where the ratio of recorded Vm and true membrane potential = (Rseal/Rpatch + Rcell)/(1 + (Rseal/Rpatch + Rcell)) (32). As such, the recorded Vm is an underestimate of the actual membrane potential (i.e. more depolarized than the actual Vm) (31, 32). Because the recorded Vm under this configuration is only a proxy of the true membrane potential, we only used it to track changes in membrane potential (31, 32). Seal resistance was monitored before and after the recording, and only cells that showed stable baseline membrane potential prior to leptin/drug application and that maintained good seal resistance were included for analysis. In addition, the KATP channel opener diazoxide or inhibitor tolbutamide was used when applicable to ensure cell and patch integrity as we reported previously (19) and in the current study (see Figs. 4 and 7). For whole-cell current-clamp recordings of membrane potential (Fig. 2, C and D), pipette solution contained 130 mm potassium gluconate, 10 mm KCl, 10 mm HEPES, 6 mm EGTA, 0.1 mm CaCl2, 1 mm MgCl2, and 5 mm ATP, with or without the SF activator YEEI phosphopeptide (EPQpYEEIPIYL at 1 μm). All recordings were analyzed using Clampfit 9.2 (pCLAMP).
Statistical analysis
Results are expressed as mean ± S.E. One-way analysis of variance with a post hoc Dunnett's test (Fig. 5E) or Friedman's test (Fig. 3B) was used for multiple comparisons where different experimental conditions were carried out side by side. For most experiments, difference between control and treated groups was tested using a paired t test or unpaired t tests (for comparable sample sizes) or Welch's t test (for significantly unequal sample sizes in Fig. 6D) as detailed in the figure legends. The level of statistical significance was set at p < 0.05.
Data availability
All data are contained in the article.
Acknowledgments
We thank Dr. Christopher Newgard for the rat insulinoma INS-1 clone 832/13 cells.
Author contributions—V. A. C., Y. W., D. A. F., and S.-L. S. conceptualization; V. A. C., Y. W., D. A. F., and S.-L. S. data curation; V. A. C., Y. W., D. A. F., and S.-L. S. formal analysis; V. A. C., Y. W., Z. Y., A. E., J. D., and D. A. F. investigation; V. A. C., A. E., P. K., D. A. F., and S.-L. S. writing-review and editing; Y. W., D. A. F., and S.-L. S. writing-original draft; S.-L. S. funding acquisition; S.-L. S. project administration.
Funding and additional information—This work was supported by National Institutes of Health Grants R01DK057699 and 3R01DK057699-14S1 (to S.-L. S.). P. K. was supported by National Institutes of Health Grant P51 OD011092 (to the Oregon National Primate Research Center). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- AMPK
- AMP-activated protein kinase
- SFK
- Src family kinase
- NMDA
- N-methyl-d-aspartate
- NMDAR
- NMDA receptor
- CaMKKβ
- calcium/calmodulin-dependent protein kinase kinase β
- PKA
- protein kinase A
- PKC
- protein kinase C
- AZD
- AZD0530
- IIDP
- Integrated Islets Distribution Program
- pF
- picofarads
- FBS
- fetal bovine serum
- DPBS
- Dulbecco's PBS.
References
- 1. Emilsson, V., Liu, Y. L., Cawthorne, M. A., Morton, N. M., and Davenport, M. (1997) Expression of the functional leptin receptor mRNA in pancreatic islets and direct inhibitory action of leptin on insulin secretion. Diabetes 46, 313–316 10.2337/diab.46.2.313 [DOI] [PubMed] [Google Scholar]
- 2. Fehmann, H. C., Peiser, C., Bode, H. P., Stamm, M., Staats, P., Hedetoft, C., Lang, R. E., and Göke, B. (1997) Leptin: a potent inhibitor of insulin secretion. Peptides 18, 1267–1273 10.1016/S0196-9781(97)00135-6 [DOI] [PubMed] [Google Scholar]
- 3. Kieffer, T. J., Heller, R. S., Leech, C. A., Holz, G. G., and Habener, J. F. (1997) Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes 46, 1087–1093 10.2337/diabetes.46.6.1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kulkarni, R. N., Wang, Z. L., Wang, R. M., Hurley, J. D., Smith, D. M., Ghatei, M. A., Withers, D. J., Gardiner, J. V., Bailey, C. J., and Bloom, S. R. (1997) Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J. Clin. Invest. 100, 2729–2736 10.1172/JCI119818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ookuma, M., Ookuma, K., and York, D. A. (1998) Effects of leptin on insulin secretion from isolated rat pancreatic islets. Diabetes 47, 219–223 10.2337/diabetes.47.2.219 [DOI] [PubMed] [Google Scholar]
- 6. Poitout, V., Rouault, C., Guerre-Millo, M., Briaud, I., and Reach, G. (1998) Inhibition of insulin secretion by leptin in normal rodent islets of Langerhans. Endocrinology 139, 822–826 10.1210/endo.139.3.5812 [DOI] [PubMed] [Google Scholar]
- 7. Chen, P. C., Kryukova, Y. N., and Shyng, S. L. (2013) Leptin regulates KATP channel trafficking in pancreatic β-cells by a signaling mechanism involving AMP-activated protein kinase (AMPK) and cAMP-dependent protein kinase (PKA). J. Biol. Chem. 288, 34098–34109 10.1074/jbc.M113.516880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park, S. H., Ryu, S. Y., Yu, W. J., Han, Y. E., Ji, Y. S., Oh, K., Sohn, J. W., Lim, A., Jeon, J. P., Lee, H., Lee, K. H., Lee, S. H., Berggren, P. O., Jeon, J. H., and Ho, W. K. (2013) Leptin promotes KATP channel trafficking by AMPK signaling in pancreatic β-cells. Proc. Natl. Acad. Sci. U. S. A. 110, 12673–12678 10.1073/pnas.1216351110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu, Y., Shyng, S. L., and Chen, P. C. (2015) Concerted trafficking regulation of Kv2.1 and KATP channels by leptin in pancreatic β-cells. J. Biol. Chem. 290, 29676–29690 10.1074/jbc.M115.670877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jacobson, D. A., and Shyng, S. L. (2020) Ion channels of the islets in type 2 diabetes. J. Mol. Biol. 432, 1326–1346 10.1016/j.jmb.2019.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rorsman, P., and Ashcroft, F. M. (2018) Pancreatic β-cell electrical activity and insulin secretion: of mice and men. Physiol. Rev. 98, 117–214 10.1152/physrev.00008.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friedman, J. (2016) The long road to leptin. J. Clin. Invest. 126, 4727–4734 10.1172/JCI91578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wauman, J., Zabeau, L., and Tavernier, J. (2017) The leptin receptor complex: heavier than expected? Front. Endocrinol. (Lausanne) 8, 30 10.3389/fendo.2017.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang, L., Li, Z., and Rui, L. (2008) Leptin stimulates both JAK2-dependent and JAK2-independent signaling pathways. J. Biol. Chem. 283, 28066–28073 10.1074/jbc.M805545200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cochrane, V., and Shyng, S. L. (2019) Leptin-induced trafficking of KATP channels: a mechanism to regulate pancreatic beta-cell excitability and insulin secretion. Int. J. Mol. Sci. 20, 2660 10.3390/ijms20112660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paoletti, P., Bellone, C., and Zhou, Q. (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400 10.1038/nrn3504 [DOI] [PubMed] [Google Scholar]
- 17. Marquard, J., Otter, S., Welters, A., Stirban, A., Fischer, A., Eglinger, J., Herebian, D., Kletke, O., Klemen, M. S., Stožer, A., Wnendt, S., Piemonti, L., Köhler, M., Ferrer, J., Thorens, B., et al. (2015) Characterization of pancreatic NMDA receptors as possible drug targets for diabetes treatment. Nat. Med. 21, 363–372 10.1038/nm.3822 [DOI] [PubMed] [Google Scholar]
- 18. Otter, S., and Lammert, E. (2016) Exciting times for pancreatic islets: glutamate signaling in endocrine cells. Trends Endocrinol. Metab. 27, 177–188 10.1016/j.tem.2015.12.004 [DOI] [PubMed] [Google Scholar]
- 19. Wu, Y., Fortin, D. A., Cochrane, V. A., Chen, P. C., and Shyng, S. L. (2017) NMDA receptors mediate leptin signaling and regulate potassium channel trafficking in pancreatic β-cells. J. Biol. Chem. 292, 15512–15524 10.1074/jbc.M117.802249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hansen, K. B., Yi, F., Perszyk, R. E., Furukawa, H., Wollmuth, L. P., Gibb, A. J., and Traynelis, S. F. (2018) Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 150, 1081–1105 10.1085/jgp.201812032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wrighton, D. C., Baker, E. J., Chen, P. E., and Wyllie, D. J. (2008) Mg2+ and memantine block of rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J. Physiol. 586, 211–225 10.1113/jphysiol.2007.143164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen, B. S., and Roche, K. W. (2007) Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53, 362–368 10.1016/j.neuropharm.2007.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salter, M. W., and Kalia, L. V. (2004) Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 5, 317–328 10.1038/nrn1368 [DOI] [PubMed] [Google Scholar]
- 24. Yang, M., and Leonard, J. P. (2001) Identification of mouse NMDA receptor subunit NR2A C-terminal tyrosine sites phosphorylated by coexpression with v-Src. J. Neurochem. 77, 580–588 10.1046/j.1471-4159.2001.00255.x [DOI] [PubMed] [Google Scholar]
- 25. Zheng, F., Gingrich, M. B., Traynelis, S. F., and Conn, P. J. (1998) Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nat. Neurosci. 1, 185–191 10.1038/634 [DOI] [PubMed] [Google Scholar]
- 26. Heida, N. M., Leifheit-Nestler, M., Schroeter, M. R., Müller, J. P., Cheng, I. F., Henkel, S., Limbourg, A., Limbourg, F. P., Alves, F., Quigley, J. P., Ruggeri, Z. M., Hasenfuss, G., Konstantinides, S., and Schäfer, K. (2010) Leptin enhances the potency of circulating angiogenic cells via Src kinase and integrin αvβ5: implications for angiogenesis in human obesity. Arterioscler. Thromb. Vasc. Biol. 30, 200–206 10.1161/ATVBAHA.109.192807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shanley, L. J., Irving, A. J., and Harvey, J. (2001) Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 21, RC186 10.1523/JNEUROSCI.21-24-j0001.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuner, T., and Schoepfer, R. (1996) Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. J. Neurosci. 16, 3549–3558 10.1523/JNEUROSCI.16-11-03549.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stroebel, D., Casado, M., and Paoletti, P. (2018) Triheteromeric NMDA receptors: from structure to synaptic physiology. Curr. Opin. Physiol. 2, 1–12 10.1016/j.cophys.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Edman, S., McKay, S., Macdonald, L. J., Samadi, M., Livesey, M. R., Hardingham, G. E., and Wyllie, D. J. (2012) TCN 201 selectively blocks GluN2A-containing NMDARs in a GluN1 co-agonist dependent but non-competitive manner. Neuropharmacology 63, 441–449 10.1016/j.neuropharm.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mason, M. J., Simpson, A. K., Mahaut-Smith, M. P., and Robinson, H. P. (2005) The interpretation of current-clamp recordings in the cell-attached patch-clamp configuration. Biophys. J. 88, 739–750 10.1529/biophysj.104.049866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perkins, K. L. (2006) Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J. Neurosci. Methods 154, 1–18 10.1016/j.jneumeth.2006.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fischer, G., Mutel, V., Trube, G., Malherbe, P., Kew, J. N., Mohacsi, E., Heitz, M. P., and Kemp, J. A. (1997) Ro 25-6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J. Pharmacol. Exp. Ther. 283, 1285–1292 [PubMed] [Google Scholar]
- 34. Yi, F., Mou, T. C., Dorsett, K. N., Volkmann, R. A., Menniti, F. S., Sprang, S. R., and Hansen, K. B. (2016) Structural basis for negative allosteric modulation of GluN2A-containing NMDA receptors. Neuron 91, 1316–1329 10.1016/j.neuron.2016.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grant, E. R., Guttmann, R. P., Seifert, K. M., and Lynch, D. R. (2001) A region of the rat N-methyl-d-aspartate receptor 2A subunit that is sufficient for potentiation by phorbol esters. Neurosci. Lett. 310, 9–12 10.1016/S0304-3940(01)02085-7 [DOI] [PubMed] [Google Scholar]
- 36. Li, B. S., Sun, M. K., Zhang, L., Takahashi, S., Ma, W., Vinade, L., Kulkarni, A. B., Brady, R. O., and Pant, H. C. (2001) Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc. Natl. Acad. Sci. U. S. A. 98, 12742–12747 10.1073/pnas.211428098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang, K., Trepanier, C., Sidhu, B., Xie, Y. F., Li, H., Lei, G., Salter, M. W., Orser, B. A., Nakazawa, T., Yamamoto, T., Jackson, M. F., and Macdonald, J. F. (2012) Metaplasticity gated through differential regulation of GluN2A versus GluN2B receptors by Src family kinases. EMBO J. 31, 805–816 10.1038/emboj.2011.453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roskoski, R., Jr (2015) Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 94, 9–25 10.1016/j.phrs.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 39. Boggon, T. J., and Eck, M. J. (2004) Structure and regulation of Src family kinases. Oncogene 23, 7918–7927 10.1038/sj.onc.1208081 [DOI] [PubMed] [Google Scholar]
- 40. Liu, X., Brodeur, S. R., Gish, G., Songyang, Z., Cantley, L. C., Laudano, A. P., and Pawson, T. (1993) Regulation of c-Src tyrosine kinase activity by the Src SH2 domain. Oncogene 8, 1119–1126 [PubMed] [Google Scholar]
- 41. Köhr, G., and Seeburg, P. H. (1996) Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the Src family. J. Physiol. 492, 445–452 10.1113/jphysiol.1996.sp021320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang, W. S., Chen, Z. G., Liu, W. T., Chi, Z. Q., He, L., and Liu, J. G. (2015) Dorsal hippocampal NMDA receptor blockade impairs extinction of naloxone-precipitated conditioned place aversion in acute morphine-treated rats by suppressing ERK and CREB phosphorylation in the basolateral amygdala. Br. J. Pharmacol. 172, 482–491 10.1111/bph.12671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taniguchi, S., Nakazawa, T., Tanimura, A., Kiyama, Y., Tezuka, T., Watabe, A. M., Katayama, N., Yokoyama, K., Inoue, T., Izumi-Nakaseko, H., Kakuta, S., Sudo, K., Iwakura, Y., Umemori, H., Inoue, T., et al. (2009) Involvement of NMDAR2A tyrosine phosphorylation in depression-related behaviour. EMBO J. 28, 3717–3729 10.1038/emboj.2009.300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barria, A., and Malinow, R. (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron 35, 345–353 10.1016/s0896-6273(02)00776-6 [DOI] [PubMed] [Google Scholar]
- 45. Roskoski, R., Jr. (2005) Src kinase regulation by phosphorylation and dephosphorylation. Biochem. Biophys. Res. Commun. 331, 1–14 10.1016/j.bbrc.2005.03.012 [DOI] [PubMed] [Google Scholar]
- 46. Pucheta-Martinez, E., Saladino, G., Morando, M. A., Martinez-Torrecuadrada, J., Lelli, M., Sutto, L., D'Amelio, N., and Gervasio, F. L. (2016) An allosteric cross-talk between the activation loop and the ATP binding site regulates the activation of Src kinase. Sci. Rep. 6, 24235 10.1038/srep24235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yoder, S. M., Dineen, S. L., Wang, Z., and Thurmond, D. C. (2014) YES, a Src family kinase, is a proximal glucose-specific activator of cell division cycle control protein 42 (Cdc42) in pancreatic islet β cells. J. Biol. Chem. 289, 11476–11487 10.1074/jbc.M114.559328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Socodato, R., Santiago, F. N., Portugal, C. C., Domith, I., Encarnação, T. G., Loiola, E. C., Ventura, A. L., Cossenza, M., Relvas, J. B., Castro, N. G., and Paes-de-Carvalho, R. (2017) Dopamine promotes NMDA receptor hypofunction in the retina through D1 receptor-mediated Csk activation, Src inhibition and decrease of GluN2B phosphorylation. Sci. Rep. 7, 40912 10.1038/srep40912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Konner, A. C., and Bruning, J. C. (2012) Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 16, 144–152 10.1016/j.cmet.2012.07.004 [DOI] [PubMed] [Google Scholar]
- 50. Chen, H., Charlat, O., Tartaglia, L. A., Woolf, E. A., Weng, X., Ellis, S. J., Lakey, N. D., Culpepper, J., Moore, K. J., Breitbart, R. E., Duyk, G. M., Tepper, R. I., and Morgenstern, J. P. (1996) Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84, 491–495 10.1016/S0092-8674(00)81294-5 [DOI] [PubMed] [Google Scholar]
- 51. Song, M. Y., Hong, C., Bae, S. H., So, I., and Park, K. S. (2012) Dynamic modulation of the kv2.1 channel by SRC-dependent tyrosine phosphorylation. J. Proteome Res. 11, 1018–1026 10.1021/pr200770v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Destaing, O., Sanjay, A., Itzstein, C., Horne, W. C., Toomre, D., De Camilli, P., and Baron, R. (2008) The tyrosine kinase activity of c-Src regulates actin dynamics and organization of podosomes in osteoclasts. Mol. Biol. Cell 19, 394–404 10.1091/mbc.e07-03-0227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Olivares, M. J., González-Jamett, A. M., Guerra, M. J., Baez-Matus, X., Haro-Acuña, V., Martínez-Quiles, N., and Cardeñas, A. M. (2014) Src kinases regulate de novo actin polymerization during exocytosis in neuroendocrine chromaffin cells. PLoS ONE 9, e99001 10.1371/journal.pone.0099001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Amico-Ruvio, S. A., Murthy, S. E., Smith, T. P., and Popescu, G. K. (2011) Zinc effects on NMDA receptor gating kinetics. Biophys. J. 100, 1910–1918 10.1016/j.bpj.2011.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dolino, D. M., Chatterjee, S., MacLean, D. M., Flatebo, C., Bishop, L. D. C., Shaikh, S. A., Landes, C. F., and Jayaraman, V. (2017) The structure-energy landscape of NMDA receptor gating. Nat. Chem. Biol. 13, 1232–1238 10.1038/nchembio.2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jalali-Yazdi, F., Chowdhury, S., Yoshioka, C., and Gouaux, E. (2018) Mechanisms for zinc and proton inhibition of the GluN1/GluN2A NMDA receptor. Cell 175, 1520–1532.e15 10.1016/j.cell.2018.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fayyazuddin, A., Villarroel, A., Le Goff, A., Lerma, J., and Neyton, J. (2000) Four residues of the extracellular N-terminal domain of the NR2A subunit control high-affinity Zn2+ binding to NMDA receptors. Neuron 25, 683–694 10.1016/S0896-6273(00)81070-3 [DOI] [PubMed] [Google Scholar]
- 58. Paoletti, P., Ascher, P., and Neyton, J. (1997) High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 17, 5711–5725 10.1523/JNEUROSCI.17-15-05711.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Traynelis, S. F., Burgess, M. F., Zheng, F., Lyuboslavsky, P., and Powers, J. L. (1998) Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J. Neurosci. 18, 6163–6175 10.1523/JNEUROSCI.18-16-06163.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vogt, K., Mellor, J., Tong, G., and Nicoll, R. (2000) The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 26, 187–196 10.1016/S0896-6273(00)81149-6 [DOI] [PubMed] [Google Scholar]
- 61. Dodson, G., and Steiner, D. (1998) The role of assembly in insulin's biosynthesis. Curr. Opin. Struct. Biol. 8, 189–194 10.1016/S0959-440X(98)80037-7 [DOI] [PubMed] [Google Scholar]
- 62. Kieffer, T. J., Heller, R. S., and Habener, J. F. (1996) Leptin receptors expressed on pancreatic beta-cells. Biochem. Biophys. Res. Commun. 224, 522–527 10.1006/bbrc.1996.1059 [DOI] [PubMed] [Google Scholar]
- 63. Seufert, J., Kieffer, T. J., Leech, C. A., Holz, G. G., Moritz, W., Ricordi, C., and Habener, J. F. (1999) Leptin suppression of insulin secretion and gene expression in human pancreatic islets: implications for the development of adipogenic diabetes mellitus. J. Clin. Endocrinol. Metab. 84, 670–676 10.1210/jcem.84.2.5460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Covey, S. D., Wideman, R. D., McDonald, C., Unniappan, S., Huynh, F., Asadi, A., Speck, M., Webber, T., Chua, S. C., and Kieffer, T. J. (2006) The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell Metab. 4, 291–302 10.1016/j.cmet.2006.09.005 [DOI] [PubMed] [Google Scholar]
- 65. Morioka, T., Asilmaz, E., Hu, J., Dishinger, J. F., Kurpad, A. J., Elias, C. F., Li, H., Elmquist, J. K., Kennedy, R. T., and Kulkarni, R. N. (2007) Disruption of leptin receptor expression in the pancreas directly affects beta cell growth and function in mice. J. Clin. Invest. 117, 2860–2868 10.1172/JCI30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bland, T., Zhu, M., Dillon, C., Sahin, G. S., Rodriguez-Llamas, J. L., Appleyard, S. M., and Wayman, G. A. (2020) Leptin controls glutamatergic synaptogenesis and NMDA-receptor trafficking via Fyn kinase regulation of NR2B. Endocrinology 161, bqz030 10.1210/endocr/bqz030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kieffer, T. J., and Habener, J. F. (2000) The adipoinsular axis: effects of leptin on pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 278, E1–E14 10.1152/ajpendo.2000.278.1.E1 [DOI] [PubMed] [Google Scholar]
- 68. Soedling, H., Hodson, D. J., Adrianssens, A. E., Gribble, F. M., Reimann, F., Trapp, S., and Rutter, G. A. (2015) Limited impact on glucose homeostasis of leptin receptor deletion from insulin- or proglucagon-expressing cells. Mol. Metab. 4, 619–630 10.1016/j.molmet.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fontaine, D. A., and Davis, D. B. (2016) Attention to background strain is essential for metabolic research: C57BL/6 and the international knockout mouse consortium. Diabetes 65, 25–33 10.2337/db15-0982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhao, S., Kusminski, C. M., Elmquist, J. K., and Scherer, P. E. (2020) Leptin: less is more. Diabetes 69, 823–829 10.2337/dbi19-0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Erion, K., and Corkey, B. E. (2018) β-Cell failure or β-cell abuse? Front. Endocrinol. (Lausanne) 9, 532 10.3389/fendo.2018.00532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Smelt, M. J., Faas, M. M., de Haan, B. J., and de Vos, P. (2008) Pancreatic beta-cell purification by altering FAD and NAD(P)H metabolism. Exp. Diabetes Res. 2008, 165360 10.1155/2008/165360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Van De Winkel, M., and Pipeleers, D. (1983) Autofluorescence-activated cell sorting of pancreatic islet cells: purification of insulin-containing B-cells according to glucose-induced changes in cellular redox state. Biochem. Biophys. Res. Commun. 114, 835–842 10.1016/0006-291X(83)90857-4 [DOI] [PubMed] [Google Scholar]
- 74. Redick, S. D., Leehy, L., Rittenhouse, A. R., Blodgett, D. M., Derr, A. G., Kucukural, A., Garber, M. G., Shultz, L. D., Greiner, D. L., Wang, J. P., Harlan, D. M., Bortell, R., and Jurczyk, A. (2020) Recovery of viable endocrine-specific cells and transcriptomes from human pancreatic islet-engrafted mice. FASEB J. 34, 1901–1911 10.1096/fj.201901022RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tennant, K. G., Lindsley, S. R., Kirigiti, M. A., True, C., and Kievit, P. (2019) Central and peripheral administration of fibroblast growth factor 1 improves pancreatic islet insulin secretion in diabetic mouse models. Diabetes 68, 1462–1472 10.2337/db18-1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P., et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are contained in the article.