

Abstract
DNA polymerase from bacteriophage T7 undergoes large, substrate-induced conformational changes that are thought to account for high replication fidelity, but prior studies were adversely affected by mutations required to construct a Cys-lite variant needed for site-specific fluorescence labeling. Here we have optimized the direct incorporation of a fluorescent un-natural amino acid, (7-hydroxy-4-coumarin-yl)-ethylglycine, using orthogonal amber suppression machinery in Escherichia coli. MS methods verify that the unnatural amino acid is only incorporated at one position with minimal background. We show that the single fluorophore provides a signal to detect nucleotide-induced conformational changes through equilibrium and stopped-flow kinetic measurements of correct nucleotide binding and incorporation. Pre-steady-state chemical quench methods show that the kinetics and fidelity of DNA replication catalyzed by the labeled enzyme are largely unaffected by the unnatural amino acid. These advances enable rigorous analysis to establish the kinetic and mechanistic basis for high-fidelity DNA replication.
Keywords: DNA polymerase, fluorescence, enzyme kinetics, protein dynamics, conformational change, unnatural amino acid, bacteriophage, enzyme mechanism, induced-fit, transient kinetics
The role of substrate-induced conformational changes in enzyme specificity has been a controversial topic for decades (1). In particular, the extraordinarily high fidelity of DNA replication by DNA polymerases has been the subject of studies in enzymology for many years (2, 3). Paradoxically, high-fidelity polymerases make fewer mistakes and copy DNA at a much faster rate than low-fidelity DNA polymerases (1, 4). Crystal structures of the T7 DNA polymerase (5), a model system for high-fidelity DNA replication, showed large structural changes in the enzyme upon nucleotide binding, as have other polymerases, raising the question of the contribution of this step to the enzyme's high fidelity. The original T7 DNA polymerase papers were published almost 30 years ago, measuring the correct incorporation (2), misincorporation (3), and exonuclease proofreading reactions (6). These papers provided valuable insights into the kinetic basis for the high DNA replication fidelity of T7 DNA polymerase but were flawed in the analysis of data regarding a prechemistry nucleotide-induced conformational change, based on the interpretation of an observed small thio-elemental effect. The notion that a small observed thio-elemental effect can be interpreted as evidence for the existence of a rate-limiting conformational change preceding chemistry has since been shown to be invalid because the magnitude of the elemental effect is largely dependent on the nature of the transition state and is smaller than previously expected (7, 8). The first direct measurement of a prechemistry conformational change came from studies on the low-fidelity repair enzyme, polymerase β, using a 2-aminopurine fluorescent reporter in the DNA (9–11). These studies showed that the conformational change was faster than chemistry, challenging previous notions that the conformational change had to be rate-limiting to contribute to fidelity. Critics of these studies, however, proposed that the fluorescence signal could be due to changes in the DNA structure and translation, rather than the conformational change in the enzyme. To directly measure the rate of the conformational change for the T7 DNA polymerase, an 8-Cys-lite T7 DNA polymerase variant was labeled with 7-diethylamino-3-((((2-maleimidyl)ethyl)amino)carbonyl)coumarin (MDCC) at a surface-exposed engineered cysteine in the highly mobile fingers domain (4). By labeling the protein instead of the DNA and measuring the relevant rate constants, these studies confirmed that the rate of the conformational change was indeed faster than chemistry for both correct and mismatched nucleotides and discovered that the reverse of the conformational change was much slower than chemistry for the correct nucleotide but much faster than chemistry for the mismatched nucleotide, suggesting a new paradigm for understanding enzyme specificity (4). However, the extensive mutagenesis required to create the 8-Cys-lite variant negatively affected the enzyme. This variant had low solubility and most importantly at least a 50-fold decrease in the apparent Kd for a mismatched nucleotide, reducing the enzyme's fidelity. Subsequent similar studies using HIV reverse transcriptase, where only a single cysteine had to be removed, confirmed the central role of the nucleotide-induced conformational change in DNA replication fidelity (12). However, HIV reverse transcriptase has only moderate fidelity, so it is important to more accurately assess the role of conformational changes in specificity with a high-fidelity enzyme, which is all the more important given the controversies (13).
Because the T7 DNA polymerase is an ideal model system for a fast, high-fidelity enzyme, it was essential to improve upon strategies for labeling the enzyme with a fluorophore to report changes in enzyme structure. Here, we present the results of our optimization of the expression, purification, and basic kinetic characterization of a variant containing the fluorescent unnatural amino acid (7-hydroxy-4-coumarin-yl)-ethylglycine (7-HCou) on the surface of the fingers domain. We show that the fluorescence is sensitive to conformational changes of the protein during nucleotide incorporation (Fig. 1). By comparison to WT enzyme, we show that labeling of the enzyme with the fluorescent amino acid has negligible effects on polymerization kinetics and fidelity. By optimizing expression, the proteins could be purified without the use of added tags which also might alter enzyme activity. The methods in this paper overcome the limitations of the cysteine-maleimide labeling strategy and provide the groundwork for full characterization of the role of conformational dynamics of this enzyme on DNA replication fidelity and proofreading. Our optimized methods can be extended to other enzyme systems.
Figure 1.
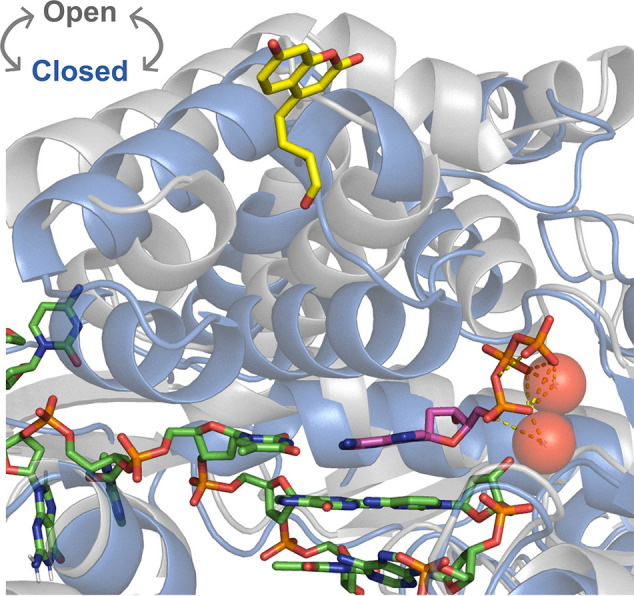
Conformational changes around the active site of T7 DNA polymerase upon nucleotide binding. Large rearrangements are seen in the fingers domain around the polymerase active site going from the open binary E-D complex (PDB: 2AJQ, gray) to the ternary closed E-D-[Mg2+-dATP] complex (PDB: 1T7P, blue). We show DNA bases (green) and the incoming dATP (magenta) with two chelated Mg2+ ions (red) for the closed state with residue 514 shown with the unnatural amino acid 7-HCou (yellow) modeled in place of glutamic acid (ligand ID: DV7).
Results
Unnatural amino acid amber codon suppression strategy
Small fluorescence probes with quantum yields that are sensitive to local environment have significant advantages over larger probes commonly used in biochemistry, such as GFP, Cy3, quantum dots, and dual probes for FRET distance measurements. With a probe sensitive to local environment, there is no need to do traditional FRET experiments that require dual labeling with commensurate higher chances to interfere with enzyme function, because comprehensive global data fitting defines the step being measured by the fluorescence signal (14, 15). The optical properties of 7-hydroxycoumarin in various solvents (16) and at different pH values (pKa ∼ 7.8 (17)) have been well-characterized and show that this fluorophore can be used to probe local environment.
To circumvent the problems associated with the cysteine labeling strategy used for the 8-Cys-lite variant (18), we tested the amber suppression to insert a site-specific unnatural amino acid, requiring only one residue substitution in the protein to directly incorporate a fluorescent unnatural amino acid into the fingers domain of the T7 DNA polymerase (Fig. 1). This unnatural amino acid system repurposes the amber stop codon (TAG) as the signal for insertion of an unnatural amino acid, using an evolved orthogonal Methanocaldococcus jannaschii tyrosyl-tRNA synthetase variant (aminoacyl tRNA synthetase; AARS) and M. jannaschii tyrosyl-tRNA with CUA in the anticodon loop (tRNAMj) expressed in E. coli cells. The amber stop codon is the least used of the three stop codons in E. coli (∼9%) and rarely terminates essential genes (19). The M. jannaschii AARS is amenable to directed evolution to incorporate a wide variety of unnatural amino acids (4). To implement this system with the T7 DNA polymerase, the codon for residue 514 in T7 gene 5 in pcIts-(T7 G5x−, trxA), described below, was mutated to an amber codon by site-directed mutagenesis to provide the codon for unnatural amino acid incorporation; the resulting plasmid was designated pcIts-(T7 G5x− E514TAG, trxA). This position was chosen because it was the amino acid position that gave the best signal using the cysteine-MDCC labeling strategy with the 8-Cys-lite T7 DNA polymerase variant (4).
Bicistronic, heat-induced T7 DNA polymerase expression/purification
We first worked to optimize the expression and purification of WT T7 DNA polymerase. Previous studies showed that coexpression of E. coli thioredoxin and T7 gene product 5 (GP5), the two polypeptides that together constitute the T7 DNA polymerase holoenzyme, greatly enhanced the solubility of a histidine-tagged T7 DNA polymerase variant (20). Al-though thioredoxin was constitutively expressed in E. coli, significant levels of overexpression of T7 GP5 required overexpression of the thioredoxin cofactor as well. We therefore constructed the plasmid pcIts-(T7 G5x−, trxA) (Fig. 2A), containing a bicistronic operon of T7 gene 5 (without a histidine tag) and trxA cloned into the pcIts backbone (21). The bicistronic operon contains a short linker between the genes for thioredoxin and T7 GP5, consisting of a PmeI restriction site followed by a ribosome binding site (Fig. 2B). The pcIts backbone includes a temperature-sensitive λ cIts repressor encoded on the plasmid that binds tightly to the operator sequence at 30 °C, blocking expression, but becomes inactive at temperatures greater than ∼35 °C. Having the repressor encoded by the plasmid allows expression in most E. coli strains, and no chemical inducer is required for high-level expression.
Figure 2.

Temperature-induced coexpression of T7 gene product 5 and thioredoxin from a bicistronic operon. A, T7 GP5/thioredoxin expression plasmid: pcIts-(T7 G5x−, trxA). The main features of this plasmid include a high copy number pUC origin of replication (yellow), gene for TEM-1 b-lactamase enzyme, conferring ampicillin resistance (AmpR) (green), and a λ promoter system (pλ, black) driving expression of a temperature-sensitive λ cIts repressor (brown) that regulates expression of an exonuclease-deficient T7 GP5 variant (T7 G5x−, orange) and thioredoxin (trxA, purple). A strong E. coli rrnB T1T2 terminator (gray) ensures efficient transcription termination of the bicistronic operon. Restriction sites used in cloning are shown on the outside of the plasmid. B, schematic of the linker region between T7 gene 5 and trxA. The 3′ end of T7 gene 5 (orange box) terminates in a TGA stop codon (red letters) and is followed by a PmeI restriction site (gray letters; the cut site is given by the gray line), a ribosome binding site (RBS, dots between strands), a short linker, and a start codon (green letters) for the trxA gene (purple box). C, E. coli lysates before and after heat induction, analyzed by SDS-PAGE. Lanes labeled Pre-Induction contain samples harvested before heat-induction, corresponding to the total cell protein (TCP), soluble protein (Sol.), and insoluble protein (Ins.), from left to right. Lanes labeled Post-Induction contain samples harvested 3 h after heat-induction. The arrow to the right of the gel around 80 kDa marks the position of T7 GP5. D, results of optimized purification protocol. Purified thioredoxin was loaded in the lane labeled Trx at the same molar concentration as the purified T7 GP5/Trx complex in the lane labeled T7 DNAP. Similar intensity of the bands corresponding to thioredoxin in the two samples demonstrates that the two proteins copurify in approximately a 1:1 molar ratio.
We tested the utility of this system for expressing the T7 DNA polymerase in E. coli harboring the plasmid pcIts-(T7 G5x−, trxA). The cells were grown at 30 °C before heat induction at 42 °C for 20 min, followed by expression at 38 °C for 3 h, and were isolated by centrifugation. Cell pellets were later lysed and processed to obtain samples corresponding to the “Total Cell Protein,” “Soluble Protein,” and “Insoluble Protein” fractions at each time point. The gel in Fig. 2C shows soluble and highly overexpressed T7 DNA polymerase, easily seen above the other E. coli proteins. The procedure for purification of T7 DNA polymerase was based on further optimization of previous protocols (2) to improve purity, increase the capacity of the method to accommodate the large increase in protein expression, and reduce the number of time-consuming dialysis steps. The optimized expression/purification protocol routinely yielded 50–100 mg of WT T7 DNA polymerase/liter of media. The purity and stoichiometry of the final product are shown in Fig. 2D, with T7 GP5 and thioredoxin copurifying in approximately a 1:1 ratio.
Plasmid optimizations for amber suppression machinery
To change the specificity of the M. jannaschii tyrosyl-tRNA synthetase (MjYRS) from tyrosine to 7-HCou, eight mutations were made in the AARS gene of the unnatural amino acid machinery plasmid pSUP-(MjYRS-6TRN), as described by Schultz and co-workers (22).The resulting plasmid containing the mutant AARS was designated pSUP-(CouRS-6TRN) (Fig. 3A). Amber suppression efficiency at position 514 with this system was tested with E. coli harboring both pSUP-(CouRS-6TRN) and pcIts-(T7 G5x− E514TAG, trxA) (Fig. 4). As a control, a parallel culture was grown in the absence of 7-HCou to test for background incorporation of naturally occurring amino acids at position 514 in T7 GP5. No band for GP5 was observed in the lanes where 7-HCou was not added to the media (Fig. 4, lane 4), whereas a faint band appeared on the gel for the culture grown in the presence of 7-HCou (Fig. 4, lane 2), indicating low amber suppression efficiency. The literature contains many examples of optimization of various inefficient unnatural amino acid systems (23, 24), so we began testing a number of them to improve the yield of T7 DNA polymerase E514Cou.
Figure 3.

Unnatural amino acid machinery plasmids. A, plasmid map of pSUP-(CouRS-6TRN). Main features include the chloramphenicol resistance gene (CmR, green), p15A origin of replication (yellow), the evolved 7-HCou aminoacyl tRNA synthetase gene (CouRS, red) under the constitutive glnS' promoter (black), and six copies of M. jannaschii tRNAMj (blue) contained in two tricistronic operons under the adjacent E. coli prolyl tRNACGG operon promoters (proK, black) and terminators (gray). B, plasmid map of pSUP-(CouRS-tRNAopt). Components labeled as in (A), but with a single copy of tRNAopt in place of the six copies of tRNAMj.
Figure 4.
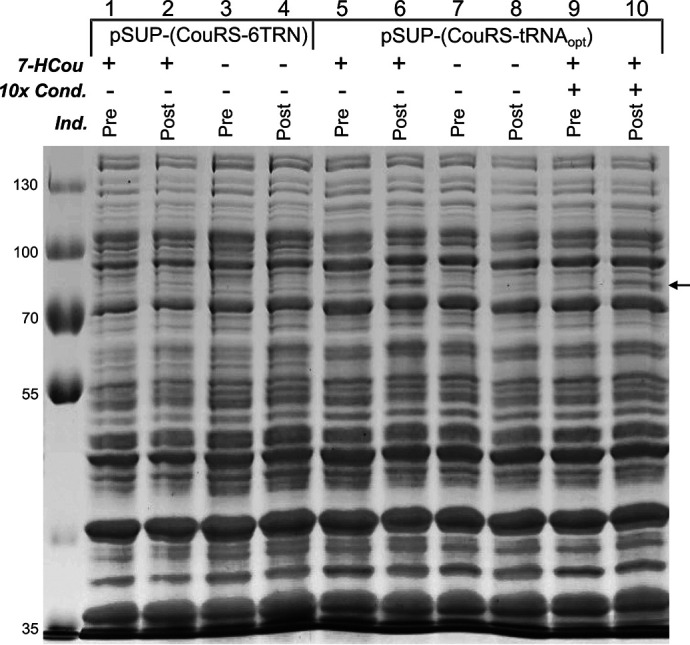
Effect of optimized tRNA and condensed culture strategy on amber suppression efficiency. Lanes 1–4 contain samples from E. coli harboring pcIts-(T7 G5x− E514TAG, trxA) and pSUP-(CouRS-6TRN), analyzed by SDS-PAGE. Lanes 5–10 contain samples from E. coli harboring pcIts-(T7 G5x− E514TAG, trxA) and pSUP-(CouRS-tRNAopt), analyzed by SDS-PAGE. Samples with a + in the row labeled 10 × Cond. were condensed 10× by gentle centrifugation of the cells and resuspension into media with 1 mm 7-HCou before inducing protein expression. The row labeled 7-HCou indicates whether 7-HCou was added to the expression media (+), or not added to the expression media (−) to test for background incorporation of naturally occurring amino acids. The row labeled Ind. indicates whether the cells were collected before inducing protein expression (Pre) or 3 h after inducing protein expression (Post). The arrow to the right of the gel indicates the position of the band for T7 GP5 at ∼80 kDa.
The first plasmid optimization was designed to test the effect of an amber suppressor tRNA variant of tRNAMj evolved to improve unnatural amino acid incorporation efficiency (25). Cells expressing six copies of tRNAMj from pSUP-(CouRS-6TRN) grew very slowly and did not reach the same stationary phase density as cells expressing the WT enzyme. As previously reported by others (23), one of the tricistronic operons was occasionally lost during cloning steps, presumably from homologous recombination because of the adjacent, identical tRNA operons, resulting in a mixture of cells with different numbers of copies of tRNAMj. Substituting one copy of an optimized tRNAMj, designated tRNAopt (25), for six copies of tRNAMj has been successful with other unnatural amino acid systems and is reported to reduce the toxicity (25). The optimized tRNA was inserted in place of the six copies of tRNAMj with ligation-independent cloning (26), and the resulting plasmid containing a single copy of tRNAopt is designated pSUP-(CouRS-tRNAopt) (Fig. 3B). Expression tests of E. coli harboring both pcIts-(T7 G5x-E514TAG, trxA) and pSUP-(CouRS-tRNAopt) show a large improvement in expression over the pSUP-(CouRS-6TRN) plasmid (Fig. 4, lane 2 versus lane 6). Additionally, no significant band was detected for the T7 DNA polymerase in the cultures without 7-HCou added to the media (Fig. 4, lane 4 versus lane 8), indicating that CouRS appears to be inefficient however specific for 7-HCou over endogenous amino acids. Cells harboring pSUP-(CouRS-tRNAopt) grew much faster and to higher densities than cells harboring pSUP-(CouRS-6TRN) and were not prone to loss of one of the operons. Other studies have found that an inducible copy of the AARS gene, combined with the constitutive copy from the mutant promoter for the E. coli glutamine-tRNA ligase gene (glnS') (27), can enhance amber suppression efficiency (23, 28). To test the effect of an inducible copy of CouRS in our system, we added a copy of the gene under control of the λ promoter. Because the T7 DNA polymerase genes are also expressed under this promoter, supplemental expression of CouRS occurs simultaneously upon heat induction. This plasmid, pSUP-(2×CouRS-tRNAopt) (Fig. 5A), contains a single copy of tRNAopt, p15A origin of replication, CmR gene, and two copies of CouRS. This construct was tested for T7 DNA polymerase E514Cou expression, as above. An enhancement in amber suppression efficiency was observed, so that a significant band of the expected molecular mass appeared in the post-induction samples with 7-HCou added to the expression media (Fig. 5B, lane 2 versus lane 7). One final CouRS optimization was attempted by making the D286R mutation in CouRS (not shown). Some unnatural amino acid systems have shown this mutation to further enhance amber suppression (29); however, we found that this mutation did not significantly enhance the efficiency of our system. The plasmid pSUP-(2×CouRS-tRNAopt) was therefore used for all further expressions.
Figure 5.

Effect of inducible copy of CouRS on amber suppression efficiency. A, plasmid containing inducible copy of CouRS: pSUP-(2×CouRS-tRNAopt). Main features include a p15A origin of replication (yellow), CmR gene (green), one copy of tRNAopt (blue) under the proK promoter (black), and two copies of the CouRS gene (red): one inducible copy under the λ promoter (pλ, black), and one constitutive copy under the glnS' promoter (black). B, effect of inducible copy of CouRS on amber suppression efficiency. Samples in lanes 1–5 were from E. coli cultures harboring both pcIts-(T7 G5x− E514TAG, trxA) and pSUP-(2×CouRS-tRNAopt). Samples in lanes 6-9 were from E. coli cultures harboring the plasmids pcIts-(T7 G5x− E514TAG, trxA) and pSUP-(CouRS-tRNAopt). The row labeled 7-HCou indicates whether 7-HCou was added to the growth media (+) or not (−) before inducing expression. Induced cultures without 7-HCou give an indication of the level of background amber suppression with naturally occurring amino acids. The row labeled Ind. indicates whether the sample corresponds to the total cell protein before induction (Pre), the total cell protein after induction (Post), or the soluble protein after induction (Sol.). The arrow to the right of the gel marks the position of T7 GP5 on the gel (∼80 kDa).
Other nonplasmid-based optimizations
Some optimization strategies were tested that did not require any DNA manipulation. Expression tests on the WT enzyme comparing rich media (Terrific Broth) with a supplemented M9 media (see “Experimental procedures”) showed slightly higher specific expression of T7 DNA polymerase with the minimal media (not shown). This has the added advantage of greatly reducing the concentration of naturally occurring amino acids in the media, therefore reducing the likelihood of mis-acylation of the amber suppressor tRNA. The supplemented M9 media was used for all expression tests and large-scale expression of the T7 DNA polymerase E514Cou variant. A high concentration of unnatural amino acid (≥1 mm) is generally added to the media to have a high enough concentration inside the cells for incorporation into the target protein. Generally, only a small fraction is actually translated into proteins and the rest is lost in the media after the cells have been harvested. A strategy commonly used in the NMR and structural biology fields to minimize amounts of probe needed is the use of a condensed culture (24). The condensed culture strategy involves growing large quantities of cells in standard media, gently pelleting them and resuspending in a much smaller volume of media containing the unnatural amino acid before inducing expression. In principle, the concentration is still high enough for efficient incorporation, although the yield of pellet with protein of interest can be increased greatly. When tested in our optimized plasmid system, a band of similar intensity, as compared with the uncondensed culture, appeared in the postinduction sample (Fig. 4, lane 6 versus lane 10), even at high cell densities. We therefore used this strategy in combination with the plasmid-based optimizations for large-scale expression of T7 DNA polymerase E514Cou.
Large-scale expression and purification of T7 DNA polymerase E514Cou
Large-scale protein expression was performed by inoculating six 1-liter cultures of supplemented M9 media with E. coli harboring pSUP-(2×CouRS-tRNAopt) and pcIts-(T7 G5x− E514TAG, trxA). The cultures were grown to an A600 of 0.5, and then the cells were gently pelleted and resuspended in 600 ml of supplemented M9 media containing 1 mm 7-HCou. Protein expression was induced by elevating the culture temperature from 30 to 42 °C, incubating for 30 min, and then dropping the temperature to 38 °C and incubating for 3 h. The T7 DNA polymerase E514Cou variant and the WT enzyme were purified with an optimized purification protocol to increase the capacity and reduce the time required to purify the protein, as described for WT enzyme. The presence of 7-HCou in the protein simplified the chromatography steps of the purification because it provided a convenient optical signature that could be followed by monitoring absorbance at 335 nm on the HPLC. Purification from a pellet from the 600-ml condensed culture yielded 5–10 mg of the enzyme containing 7-HCou.
MS
The 7-HCou–labeled T7 DNA polymerase variant was analyzed by MS performed two ways to address two questions regarding quality control on the purified protein. We reasoned that potential inhomogeneity in our protein preparation due to the unnatural amino acid system could be from either naturally occurring amino acids incorporated at position 514 or, less likely, 7-HCou incorporation at other positions in the protein. Aminoacylation of the orthogonal tRNA with natural amino acids and incorporation into the protein could occur either by promiscuous activity of CouRS or promiscuous activity of endogenous E. coli translation machinery. Alternatively, endogenous aminoacylated tRNAs could wobble base pair with the amber codon for insertion. Either situation would lead to a fraction of purified protein that does not contain 7-HCou. To test whether naturally occurring amino acids were incorporated at residue 514 in a fraction of our purified protein, samples of WT T7 DNA polymerase and the E514Cou variant were separated by SDS-PAGE and the gel bands containing GP5 were excised and trypsinized. The tryptic peptides were then separated by reverse-phase HPLC and analyzed continuously by MS/MS on a Thermo Fisher Scientific Orbitrap Fusion mass spectrometer, and the data were processed as described under “Experimental procedures.” Approximately 1,700 spectra were matched to T7 gene product 5 peptides for each sample. For the E514Cou sample, 59 spectra were obtained showing the Glu → 7-HCou modification at position 514, and a representative spectrum is shown in Fig. 6. Peptides identified for the E514Cou variant are given in the table in the supporting information. We also separately searched the data against databases with T7 gene product 5 containing other amino acid substitutions at position 514 and found three spectra with glutamine substitution at 514 and one spectrum with tryptophan insertion at this position, suggesting that ∼93% of the protein was labeled with 7-HCou at position 514.
Figure 6.

MS/MS spectrum of tryptic peptide containing 7-HCou. Tryptic peptides from gel slices containing T7 DNA polymerase E514Cou were resolved by reverse-phase HPLC and analyzed by MS/MS to confirm successful incorporation of 7-HCou. A representative MS/MS spectrum after collision-induced dissociation of the tryptic peptide corresponding to amino acids 509–518 is shown. Collision-induced dissociation most often produces b ions or y ions, with intact N or C termini, respectively. The subscript number following the letter indicates the number of amino acid residues in the ionized peptide fragment. Peaks identified as b or y ions for the parent peptide 509NQIAAXLPTR518 are shown in red and blue, respectively. The parent peptide (+2H +1) and the b2 ion (−NH3) are shown in green. X denotes the position of 7-HCou in the peptide, with a mass shift of +118 Da relative to glutamic acid in the WT enzyme.
To address whether 7-HCou was incorporated at other positions in the protein, a slightly different mass spectrometry approach was employed. 7-HCou–labeled T7 DNA polymerase was trypsinized in solution, and the peptides were subsequently separated by reverse phase HPLC. Absorbance of 7-HCou was monitored at 335 nm during the separation and two peaks were obtained with an absorbance at this wavelength (not shown). These peaks were analyzed by MALDI-MS, which showed that they were the peptide 509NQIAA[7-HCou]LPTR518 and the missed cleavage peptide 509NQIAA[7-HCou]LPTRDNAK522. Based on these results, there was no significant evidence for incorporation of 7-HCou at any position other than 514 in the labeled protein, and we conservatively estimate the fraction of protein with 7-HCou inserted at position 514 versus natural amino acids to be greater than 90%.
Fluorescence excitation/emission spectra
Next, the fluorescence excitation and emission spectra were collected for various T7 DNA polymerase E514Cou complexes (Fig. 7) to determine whether the fluorescence intensity was sensitive to changes in protein conformation upon nucleotide binding. DNA substrates consist of a 27-nt primer annealed to a 45-nt template and are based on the semi-random but defined sequence used in a prior study of HIV reverse transcriptase (30). A dsDNA region of 27 bases has been shown to be long enough for tight binding to the T7 DNA polymerase (2). To prevent chemistry but still allow nucleotide binding and conformational change, a DNA substrate was used containing a 2′,3′-dideoxy modification on the 3′-terminus of the primer strand (4). The excitation spectrum showed an excitation maximum of 361 nm, with a slightly lower intensity peak around 335 nm. A smaller peak of intensity around 280 nm was also observed, resulting from excitation of tryptophan residues and FRET to the 7-HCou. This result gives us the option of exciting the fluorophore directly (ex: 335 nm, not shown) or exciting tryptophan residues (ex: 295 nm) in the protein and monitoring the FRET signal from 7-HCou emission. T7 DNA polymerase E514Cou also showed a small increase in fluorescence upon DNA binding and a further increase in fluorescence (∼20%) upon addition of the correct nucleotide to the E + Ddd complex, indicating that the T7 DNA polymerase variant could potentially be used to measure the nucleotide-induced conformational change (Fig. 7).
Figure 7.

Fluorescence excitation and emission spectra for various T7 DNA polymerase E514Cou complexes. The excitation spectrum (red) has a maximum at 361 nm but also contains a smaller peak with a local maximum around 280 nm. Emission spectra were collected with excitation of the sample at 295 nm, first for the free enzyme (300 nm) with thioredoxin (6 µm) and Mg2+ (E, black). An excess of DNAdd (400 nm) was added, resulting in a small increase in fluorescence for the binary complex (E + Ddd, blue). dATP (10 µm) was then added and resulted in a further increase in fluorescence for the ternary complex (E + Ddd + dATP, green). The emission maximum was at 447 nm.
Both excitation wavelengths gave a signal change upon nucleotide binding; however, the FRET signal was much larger and less sensitive to solution conditions, so it was used in subsequent kinetic experiments and for the fluorescence emission spectra shown here. We cannot identify which Trp residues contribute to the FRET signal. There are 20 total tryptophan residues in the T7 DNA polymerase, two in thioredoxin and 18 in gene product 5. Tallies for the distances of Trp residues to Glu514 are as follows based on the crystal structure: two at 25-30 Å, seven at 30–40 Å, five at 40–50 Å, four at 50–60 Å, and two at 60–70 Å. Most (seven) are in the palm domain, with three in the fingers domain, two in the thumb domain, five in the exonuclease domain, one in the thioredoxin binding domain, and two in thioredoxin. Because of the large number of tryptophan residues in the protein and the added complication of the FRET efficiency effect of the orientation factor for tryptophan residues with similar distances, we are not able to suggest a single candidate Trp as the donor. Nonetheless, kinetic and equilibrium measurements show that the change in fluorescence intensity is correlated with the formation of the closed enzyme state.
Stopped-flow kinetics
Because of the rapid rate of nucleotide incorporation by the T7 DNA polymerase, a stopped-flow instrument was used to measure the fluorescence signal from 7-HCou upon addition of nucleotide and all experiments were performed at 4 °C. To initially simplify the reaction scheme to only nucleotide binding steps (Scheme 1), the DNAdd substrate was used to measure the nucleotide binding rate at a fixed nucleotide concentration, with the observed rate a function of K1, k2, and k−2. T7 DNA polymerase E514Cou (750 nm), thioredoxin (15 µm), and DNAdd (1 µm) were mixed with dATP (10 µm) and Mg2+ (12.5 mm) to start the reaction (Fig. 8A). A ∼20% increase in fluorescence was observed. The data were best fit using a single exponential function, with an observed decay rate of 135 s−1. To monitor fluorescence changes during the complete nucleotide incorporation reaction, a similar experiment was performed with DNA containing a standard 3′-OH in the primer strand to allow polymerization (Fig. 8B). T7 DNA polymerase E514Cou (750 nm), thioredoxin (15 µm), and DNA 27/45-18T (1 µm) were mixed with dATP (10 µm) and Mg2+ (12.5 mm) to start the reaction. The resulting stopped-flow trace best fit a double exponential equation, with an increase in fluorescence at an exponential decay rate of 136 s−1 (the same rate obtained for the experiment with the DNAdd substrate), followed by a slower decrease in fluorescence back to the original value at a decay rate of 100 s−1. The slower phase corresponds to structural changes in the enzyme occurring after the chemistry step, based on measurements of the rate of the chemical reaction under identical conditions, as described below.
Scheme 1.

Kinetic pathway for nucleotide incorporation.
Figure 8.

Stopped-flow dATP (correct nucleotide) binding and incorporation kinetics at 4 °C. A, stopped-flow dATP binding-rate experiment. T7 DNA polymerase E514Cou (750 nm), thioredoxin (15 µm), and 27dd/45-18T DNA (1 µm) were mixed with dATP (10 µm) and Mg2+ (12.5 mm) in a KinTek SF-300 stopped-flow instrument. The single exponential fit to the data (black line) gives an observed decay rate of 136 ± 1 s−1. B, stopped-flow dATP incorporation experiment. T7 DNA polymerase E514Cou (750 nm), thioredoxin (15 µm), and 27/45-18T DNA (1 µm) were mixed with dATP (10 µm) and Mg2+ (12.5 mm). The fit of the data to a double exponential function is shown as a black line. With a fast phase at 136 s−1, the slower phase showing a decrease in fluorescence has an observed decay rate of 100 ± 2 s−1. For comparison, the exponential phase of the quench-flow burst data at 10 µm dATP gave an observed rate of 92 ± 10 s−1 (Fig. 9).
Experimental strategy to measure fidelity/correct nucleotide burst experiments
The MDCC-labeled 8-Cys-lite T7 DNA polymerase (4, 18) has reduced fidelity because of the extensive mutagenesis required to achieve site-specific labeling at an engineered cysteine residue. We therefore tested whether the 7-HCou substitution altered the fidelity of our T7 DNA polymerase E514Cou variant. To address this question, pre-steady-state kinetic ex-periments were performed at various nucleotide concentrations for both the correct and mismatched nucleotide to estimate the parameters kpol and Kd,app for each nucleotide (30). These parameters are important because the ratio kpol/Kd,app determines enzyme specificity and the ratio of (kpol/Kd,app)Mismatch versus (kpol/Kd,app)Correct defines the fidelity of the enzyme. To measure kpol and Kd,app for the correct nucleotide, rapid-quench pre-steady-state burst experiments were performed (Fig. 9), which allowed reaction times down to 2.5 ms to be accurately measured. The temperature of the reaction was maintained at 4 °C with a circulating water bath. T7 DNA polymerase (60 nm WT and 75 nm E514Cou) with thioredoxin (2 µm), BSA (0.1 mg/ml), and 6-carboxyfluorescein (FAM)-labeled-27/45-18T (with T as the templating base) DNA (125 nm WT and 150 nM E514Cou) were mixed with various concentrations of dATP (2.5–110 µm) and Mg2+ (12.5 mm) to start the reaction. EDTA from the quench syringe was used to stop the reaction, and products were analyzed by denaturing PAGE or capillary electrophoresis. The exponential “burst” phase arises from the fast rate of incorporation of nucleotide by the DNA-bound enzyme, relative to the slow rate of DNA dissociation, before binding a new substrate DNA molecule to allow further nucleotide incorporation. Data were fit using KinTek Explorer with the model in Scheme 2 to extract the parameters kpol and Kd,app. For comparison, the results of equation-based data fitting are shown in Fig. S5. Global data fitting returned values with standard errors based on confidence contour analysis to define kpol = 181 ± 25 s−1 and Kd,app = 28 ± 8 µm for WT, and kpol = 202 ± 14 s−1 and Kd,app = 15 ± 3 µm E514Cou T7 DNA polymerase. A χ2 threshold of 0.96 was used to define confidence limits based on the number of parameters and the number of data points in the fitting (14, 31). Both enzymes have almost identical values of kpol, whereas the apparent Kd for the nucleotide appears slightly lower for the E514Cou variant. Nonetheless, these data indicate that incorporation of 7-HCou has a minimal effect on the kinetics of correct nucleotide incorporation.
Figure 9.

dATP (correct nucleotide) burst kinetics at 4 °C for WT and E514Cou T7 DNA polymerase by rapid quench. A, pre-steady-state burst kinetics for WT T7 DNA polymerase. WT T7 DNA polymerase (60 nm), thioredoxin (1.2 µm), BSA (0.1 mg/ml), and FAM-27/45-18T DNA (125 nm) were mixed with Mg2+ (12.5 mm) and varying concentrations of dATP (5–110 µm) in a KinTek RQF-3 quench-flow instrument, and the reaction was quenched with EDTA from a quench syringe. Data for each dATP concentration are shown in different colors. The solid lines through the data points are the best fit obtained by fitting the data globally in KinTek Explorer using the model in Scheme 2. The best-fit values derived from the fitting are kpol: 181 ± 25 s−1 and Kd,app: 28 ± 8 µm. B, pre-steady-state burst kinetics for T7 DNA polymerase E514Cou. T7 DNA polymerase E514Cou (75 nm), thioredoxin (1.5 µm), BSA (0.1 mg/ml), and FAM-27/45-18T DNA (150 nm) were mixed with Mg2+ (12.5 mm) and varying concentrations of dATP (2.5–110 µm) in the quench-flow to start the reaction and then with EDTA to stop the reaction at various times. Data for each dATP concentration are shown in different colors. The solid lines are the best global fit of the data using KinTek Explorer with the model in Scheme 2 to derive the parameters kpol: 202 ± 14 s−1 and Kd,app: 15 ± 3 µm.
Scheme 2.

Kinetic pathway for nucleotide binding.
Misincorporation reactions
The misincorporation reaction was much slower and could therefore be assayed by handmixing. Reactions were performed using a modified tube rack, designed to sit inside a refrigerated water bath to provide a constant temperature of 4 °C for the reaction for consistency with the correct incorporation reactions. T7 DNA polymerase (225 nm), thioredoxin (4.5 µm), BSA (0.1 mg/ml), and FAM 27/45-18A DNA (75 nm, with an A as the templating base) were mixed with a solution of Mg2+-dATP (0.25–3 mm) and Mg2+ (12.5 mm) to start the reaction. Reactions were quenched by adding EDTA, and products were analyzed by electrophoresis as described above for correct incorporation. The time course of misincorporation at varying nucleotide concentrations for both WT and E514Cou T7 DNA polymerase are shown in Fig. 10. The reaction progress curves are biphasic, even though the experiment was set up with a large excess of enzyme over DNA. This can be explained with the minimal model in Scheme 3, where chemistry (k2) is reversible and pyrophosphate release is slow (k3). Traditional methods of fitting data using equations for biphasic reaction progress curves are highly error prone and difficult to interpret. In contrast, this type of reaction scheme can be easily modeled in KinTek Explorer including statistical analysis to determine how well each parameter is constrained. A good fit to the data was obtained for the model in Scheme 3; however, the individual rate constants were not well-defined. Rather, kpol/Kd,app = k1k2k3/(k2k3+k-1(k-2+k3)) was obtained from fitting the data. Values of kpol/Kd,app of 3.90 ± 0.12 m−1 s−1 and 21.0 ± 0.7 m−1 s−1 from the confidence contour analysis were obtained for the WT and E514Cou variant, respectively, at a χ2 threshold of 0.96. This result is sufficient to define the fidelity of both the WT and E514Cou T7 DNA polymerase variant when combined with the values extracted from the analysis of the pre-steady-state burst experiments for the correct nucleotide.
Figure 10.

Misincorporation kinetics for WT and E514Cou T7 DNA polymerase at 4 °C. A, time course of WT T7 DNA polymerase misincorporation. T7 DNA polymerase (225 nm), thioredoxin (4.5 µm), BSA (0.1 mg/ml), and FAM-27/45-18A DNA (75 nm) were mixed with Mg2+ (12.5 mm) and varying concentrations of Mg2+-dATP (0.25–3 mm), and the reaction was quenched at various time points with EDTA. Data for each dATP concentration are shown in different colors, and the solid lines through the data show the results of global fitting using KinTek Explorer with the model in Scheme 3. For this experiment, kpol and Kd,app were not individually determined because Kd,app is too high to measure; however, the specificity constant, kpol/Kd,app, was accurately determined (3.90 ± 0.12 m−1 s−1) from the fit by simulation. B, time course of T7 DNA polymerase E514Cou misincorporation. T7 DNA polymerase E514Cou (225 nm), thioredoxin (4.5 µm), BSA (0.1 mg/ml), and FAM-27/45-18A DNA (75 nm) were mixed with Mg2+ (12.5 mm) and varying concentrations of dATP (0.25–3 mm). Data for each dATP concentration are shown in different colors, and the solid lines through the data show the results of global data fitting by simulation using KinTek Explorer with the model in Scheme 3. The parameter kpol/Kd,app was determined to be 21.0 ± 0.7 m−1 s−1, as in (A).
Scheme 3.

Kinetic pathway for nucleotide incorporation with slow PPi release.
Fidelity of the WT enzyme versus the E514Cou variant
The parameter kpol/Kd,app is equivalent to kcat/Km, the specificity constant. The ratio of this parameter for the correct versus mismatched nucleotide incorporation reaction defines the fidelity:
defined as the probability the enzyme incorporates a mismatch relative to a correct nucleotide. For the pre-steady-state burst experiment for the correct incorporation, both kpol and Kd,app were individually defined so the ratio gives (kpol/Kd,app)Correct. For the misincorporation reaction, neither parameter was individually defined; however, the specificity constant (kpol/Kd,app)Mismatch was well-defined. These constants are reported in Table 1, along with the calculated base substitution fidelity of the WT enzyme and E514Cou variant. The fidelity of both enzymes is on the order of 1 × 10−6 (see Table 1), differing only by a factor of 2 or 3. This shows that our labeling strategy was effective in providing a fluorescence signal upon nucleotide binding without significantly altering the fidelity of the enzyme.
Table 1.
Parameters governing fidelity for T7 DNA polymerase: WT versus E514Cou
| Enzyme | Nucleotide | kpol (s−1) | Kd,app (µm) | kpol/Kd,app (m−1 s−1) | Fidelity |
|---|---|---|---|---|---|
| WT | Correct (dATP:T) | 181 (154, 221) | 28.1 (20.4, 41.0) | 6.4 × 106 (3.9, 10.8) | 0.61 × 10−6 (1.03, 0.35) |
| Mismatch (dATP:A) | -- | 3.90 (3.78, 4.02) | |||
| E514Cou | Correct (dATP:T) | 202 (188, 225) | 15.0 (12.8, 18.2) | 13.5 × 106 (10.3, 17.6) | 1.56 × 10−6 (2.10, 1.56) |
| Mismatch (dATP:A) | -- | 21.0 (20.3, 21.7) | |||
Parameter (best-fit (lower boundary, upper boundary)).
Discussion
In the spirit of Arthur Kornberg's Fourth Commandment (32), “Do not waste clean thinking on dirty enzymes,” we began by optimizing the expression and purification of the WT enzyme without any tags. Various strategies such as low growth temperature and growth media additives were tested (not shown) to increase the yield of soluble protein. However, we found that coexpression of thioredoxin resulted in a very high level of soluble overexpression of WT enzyme without additional expression strategies.
Alternative failed strategies
In preliminary studies not reported here, inspection of the T7 DNA polymerase crystal structure suggested that most of the cysteines on the protein were not surface-exposed, so we tested whether we could achieve site-specific labeling of the T7 DNA polymerase with MDCC with fewer mutations. After multiple rounds of site-directed mutagenesis, expression, purification, MDCC labeling, and mass spectrometry, we realized that we were chasing a moving target. One cysteine in the WT protein was labeled, but after mutagenesis of this residue, other cysteines became labeled, and the cycle continued until we reached the eight cysteine mutations previously reported (18). To circumvent the problems associated with cysteine-maleimide conjugation for protein labeling, we progressed to the amber suppression system to incorporate unnatural amino acids. We first tested amber suppression efficiency with p-acetyl-phenylalanine in E. coli harboring both pcIts-(T7 G5x− E514TAG, trxA) and pSUP-(pAcPhe-6TRN) (pSUP-CouRS-6TRN with AARS evolved for p-acetyl-phenylalanine), grown in the presence of 1 mm p-acetyl-phenylalanine. This unnatural amino acid has the potential for site-specific labeling of the acetyl group through an oxime ligation reaction (33). Incorporation of p-acetyl-phenylalanine was successful and had high efficiency; however, difficulties arose when attempting the labeling reaction, despite numerous optimization efforts (labeling time, catalysts (34, 35), temperature, etc.).
Labeling with 7-HCou
Based on prior failures with other methods, we opted to directly incorporate 7-HCou into the enzyme, eliminating the labeling step required with the other systems. Others have used this unnatural amino acid to measure folding of myoglobin (22), detect antibody-antigen interactions (36), improve CFP with a eCFP(Cou) variant with a large stokes shift (37), and measure NaK channel preference for K+ by fluorescence lifetime and anisotropy analysis (38), among other uses (39–41). Our initial attempts to incorporate 7-HCou resulted in very low yield of labeled protein, a common issue with some of these unnatural amino acid systems. Various strategies to optimize low-yield systems have worked in a large number of these systems; however, the data were unclear as to how to optimize this particular evolved AARS/tRNA pair. Adding an inducible copy of CouRS to complement a constitutive copy on the plasmid has proven effective in multiple instances (28, 30), and we found that this strategy greatly increased the amber suppression efficiency for CouRS. The tRNA used also has shown to have a major impact on amber suppression efficiency and in multiple unnatural amino acid systems (23, 25). We found that a single copy of optimized tRNA outperformed six copies of the original tRNAMj and eliminated issues of plasmid stability and toxicity that were caused by high expression of foreign tRNA. Finally, the condensed culture strategy (24) greatly enhanced the yield of wet cell pellet and greatly reduced the amount of 7-HCou used per expression, with minimal effects on protein expression level.
It should also be noted that we used heat to induce expression of proteins, but this strategy causes many proteins to form inclusion bodies. However, this can be circumvented with our expression plasmid by using nalidixic acid to induce expression of RecA and allow expression from the λ promoter at lower temperatures. Future work could also optimize the sequence surrounding the amber codon, which in some cases has been shown to have a large effect on amber suppression efficiency (42). Also, future studies could try using a different amber-less E. coli strain and/or a strain with an altered RF1 (43). Our yield for T7 DNA polymerase E514Cou was only ∼10–20% of WT yield; however, this was still sufficient for our extensive kinetic studies because we had first optimized soluble overexpression of the WT enzyme. The reduced expression level of the 7-HCou variant may limit studies with this system on enzymes where the expression level of WT enzyme is low.
We then showed that our T7 DNA polymerase E514Cou variant was sensitive to the nucleotide-induced conformational change through equilibrium fluorescence emission scans and stopped-flow experiments. A roughly 20% change in fluorescence was observed upon nucleotide binding, which was sufficient to measure fast reactions in the stopped-flow instrument. Because the enzyme provides a signal to measure conformational changes in the stopped flow, the other important question is whether the fidelity of the enzyme was affected by 7-HCou incorporation. Although the MDCC-labeled 8-Cys-lite T7 DNA polymerase variant provided valuable insights into the issue of conformational dynamics of DNA replication, the extensive mutagenesis required to site-specifically label the enzyme made the enzyme more difficult to work with (reduced solubility and stability) and reduced its fidelity (4). Whereas kpol stayed the same, the fidelity was lowered by at least one order of magnitude (3 × 105 kpol = 230 s−1) (4). The conformational dynamics of slower, lower-fidelity DNA polymerases have been characterized (12), so our goal was to study truly high-fidelity replication on a more robust system. We therefore performed the important pre-steady-state kinetic experiments for both the correct incorporation and misincorporation to determine whether the fidelity of the enzyme was significantly affected by insertion of 7-HCou and found that the effect of 7-HCou incorporation on fidelity for our T7 DNA polymerase variant was minimal. Overall, the fidelity of the variant is within 3-fold of the WT enzyme, which is the highest fidelity labeled DNA polymerase that we are aware of to date. This work lays the foundation for extensive kinetic analysis to establish the role of enzyme conformational dynamics in specificity and error correction by high-fidelity DNA polymerases and other enzymes. Moreover, our optimized labeling strategies should reduce the effort needed to pursue fluorescent labeling of other enzymes to examine the role of conformational dynamics in enzyme catalysis and specificity.
Experimental procedures
Preparation of reagents
The fluorescent unnatural amino acid 7-HCou was purchased from Sigma-Aldrich and was prepared as a 10 mm stock in H2O and titrated with NaOH to roughly pH 9.5. The stock solution was aliquoted and stored at −80 °C. Lysozyme, PEI, and PMSF were from Sigma-Aldrich. All chromatography resin/pre-packed columns and an AKTA Pure HPLC with multiwavelength detection were from GE Healthcare. Unless otherwise noted, all buffer components, E. coli growth media components, and antibiotics were purchased from either Sigma-Aldrich or Thermo Fisher Scientific. Phusion DNA polymerase, T4 ligase, T4 polynucleotide kinase, BSA, dNTPs, restriction enzymes, and T4 DNA polymerase were purchased from New England Biolabs. SDS-PAGE was performed by the method of Laemmli (44). Plasmid maps were constructed with SnapGene (GSL Biotech, RRID:SCR_015052). All plasmid maps are included in the supporting data files accompanying this article. Protein structures were prepared with the PyMOL Molecular Graphics System, Version 2.0 (Schrödinger, LLC). Sanger sequencing was performed by the University of Texas at Austin's sequencing facility in the Center for Biomedical Research Support at all cloning steps to ensure mutations were not introduced during PCR.
Cloning and mutagenesis
All experiments in this paper using T7 GP5 refer to an exonuclease-deficient variant (D5A, E7A) to allow measurements of the polymerization reaction without complications from the competing exonuclease reaction. The plasmids pUC19-(trxA) (Fig. S1) and pT7-(T7 G5x−) (Fig. S2) containing the trxA gene and exo− T7 gene 5, respectively, were used to make the 6-kb bicistronic expression plasmid pcIts-(T7 G5x−, trxA). This plasmid contains the genes for thioredoxin and T7 GP5, each with an upstream ribosome binding site, arranged in a bicistronic operon between the λ promoter and E. coli rrnB T1T2 terminator. This plasmid backbone was derived from pcIts-ind+-(MKT V649C) (21) (Fig. S3). Site-directed mutagenesis was used to make the E514TAG amber mutation in the gene for T7 GP5 of pcIts-(T7 G5x−, trxA) by PCR with phosphorylated oligos (45) 1 and 2 from Table 2. The resulting plasmid is designated pcIts-(T7 G5x− E514TAG, trxA). The 4.4-kb plasmid pSUP-(MjYRS-6TRN) (27) was a kind gift from Dr. Peter Schultz. Site-directed mutagenesis was used to make the following mutations (22) in the MjYRS gene to change the specificity from tyrosine to 7-HCou: Y32E, L65H, A67G, H70G, F108Y, Q109H, D158G, and L162G. Mutagenesis was performed by PCR using oligos 3–10 in Table 2, either with the QuikChange method (46) or the inverse PCR method with phosphorylated oligos (45). The resulting plasmid with eight mutations in MjYRS was designated pSUP-(CouRS-6TRN). The D286R (29) mutant of CouRS was made with oligos 11 and 12 from Table 2. pSC101Duet-(tacI-MjYRS-proK-Napopt-Gent) was a kind gift from Dr. Ross Thyer and Dr. Andrew Ellington of the University of Texas at Austin and contains a single copy of an optimized variant of tRNAMj, here referred to as tRNAopt (25). Sequences for both tRNAs are given in Table 3. A 570-bp region of the plasmid containing tRNAopt was amplified with oligos 21 and 22 in Table 2 and a 3.3-kb region of pSUP-(CouRS-6TRN) was amplified with oligos 23 and 24 in Table 2, and the two fragments were joined by ligation-independent cloning (26). The resulting 3.9-kb plasmid is designated pSUP-(CouRS-tRNAopt). To add an inducible copy of CouRS, the CouRS gene was amplified from pSUP-(CouRS-6TRN) with oligos 15 and 16 from Table 2, adding NdeI and EcoRI restriction sites at the 5′ and 3′ end of the CouRS gene, respectively. This fragment was digested and cloned into the backbone from pcIts-(T7 G5x−, trxA). The resulting 4.4-kb plasmid was designated pcIts-(CouRS) (Fig. S4). The 2.3-kb portion of pcIts-(CouRS) containing the λ cIts repressor, λ promoter, CouRS gene, and rrnB T1T2 terminator was amplified with oligos 19 and 20 from Table 2, adding BamHI and SpeI restriction sites downstream of the rrnB T1T2 terminator and upstream of the λ cI repressor, respectively. pSUP-(CouRS-tRNAopt) was amplified with oligos 17 and 18 from Table 2, adding BamHI and SpeI restriction sites in the region between the CmR gene and the p15A origin of replication. The PCR products were digested with BamHI and SpeI and ligated to yield the 6.3-kb plasmid pSUP-(2×CouRS-tRNAopt).
Table 2.
Cloning/mutagenesis DNA oligos
| Oligo No. | Description | Sequence (5′ → 3′) | Method* |
|---|---|---|---|
| 1 | pcI(T7x-, bicis.) E514TAG F | GCTACCTACCCGAGATAACGC | IPCR |
| 2 | pcI(T7x-, bicis.) E514TAG R: | TAAGCAGCTATCTGGTTCTTAGTG | IPCR |
| 3 | MjTyrRS Y32E F | GATGAAAAATCTGCTGAGATAGGTTTTGAACCAAGTGG | QC |
| 4 | MjTyrRS Y32E R | CCACTTGGTTCAAAACCTATCTCAGCAGATTTTTCATC | QC |
| 5 | MjTyrRS L65H/A67G/H70G F | GATTTAGGCGCCTATTTAAACCAGAAAGG | IPCR |
| 6 | MjTyrRS L65H/A67G/H70G R | ACCCAAATGTATAATTATATCAAATCCAGCATTTTGTAAATC | IPCR |
| 7 | MjTyrRS F108Y/Q109H F | CATCTTGATAAGGATTATACACTGAATGTCTATAGATTG | IPCR |
| 8 | MjTyrRS F108Y/Q109H R | ATATTCACTTCCATAAACATATTTTGCCTTTAACC | IPCR |
| 9 | MjTyrRS D158G/L162G F | TGGTGGCGTTGATGTTGCAGTTG | IPCR |
| 10 | MjTyrRS D158G/L162G R | TAATGAATGGGATTAACCTGCATTATTGGATAGATAACTTCAG | IPCR |
| 11 | MjTyrRS D286R F | GTTTAAAAAATGCTGTAGCTGAAGAACTTATAAAGATTTTAGAG | IPCR |
| 12 | MjTyrRS D286R R | GCATTGGATGCAATTCCTTATTTTTAAATAAACTCTC | IPCR |
| 13 | pcI EcoRI F | ACGTAGCTTGAATTCATGCAGTAGGGAACTGCCAG | |
| 14 | pcI NdeI R: | TTGCTCGTTGCATATGAACCTCCTTAGTACATGCAACC | |
| 15 | CouRS NdeI F | TTCGCTGTGCATATGGACGAATTTGAAATGATAAAGAG | |
| 16 | CouRS EcoRI R | GGCATGAACCGAATTCAGTTATAATCTCTTTCTAATTGGCTCTAAAATC | |
| 17 | pSUP-CouRS BamHI F | ATGCTAAGAGGATCCCGATAAGCTTGGTACCGAGCTC | |
| 18 | pSUP-CouRS SpeI R | TTGGTCTAGCACTAGTTTACAACTTATATCGTATGGGGCTGAC | |
| 19 | pcI(CouRS) BamHI R | TTTACTAGCTGGATCCCAGGAAACAGCTATGACCATGATTAC | |
| 20 | pcI(CouRS) SpeI F | TTACGCATACTAGTGTGCAATGTAACATCAGAGATTTTG | |
| 21 | LIC tRNAopt F: | GAACCACGGAACACAATCAATTCTTGCGGAGAAC | |
| 22 | LIC tRNAopt R | CGACACGGCAAGACCATTCATGTTGTTGCTC | |
| 23 | LIC pSUP tRNA F: | TGGTCTTGCCGTGTCGACGAATTTCTGCCATTCATCC | |
| 24 | LIC pSUP tRNA R | TTGTGTTCCGTGGTTCATCACACTGCTTCCGGTAGTC | |
*IPCR, inverse PCR, QC, QuikChange PCR.
Table 3.
M. jannaschii amber suppressor tRNA variants
| tRNA | Sequence (5′ → 3′) |
|---|---|
| tRNAMj | CCGGCGGUAGUUCAGCAGGGCAGAACGGCGGACUCUAAAUCCGCAUGGCGCUGGUUCAAAUCCGGCCCGCCGGACCA |
| tRNAopt | CCGGCGGUAGUUCAGCAGGGCAGAACGGCGGACUCUAAAUCCGCAUGGCAGGGGUUCAAAUCCCCUCCGCCGGACCA |
Sequences in green are the bases of the anticodon loop. Bases in red are mutations in the optimized tRNA.
Expression tests to monitor amber suppression efficiency
Starter cultures of LB (1% (w/v) Bacto-Tryptone, 0.5% (w/v) yeast extract, and 1% (w/v) NaCl) plus 100 μg/ml ampicillin and 30 μg/ml chloramphenicol were inoculated with BL21 E. coli (fhuA2 [lon] ompT gal [dcm] ΔhsdS), harboring both pcIts-(T7 G5x− E514TAG, trxA) and various pSUP plasmids, and grown overnight at 30 °C. Starter cultures were inoculated into expression cultures of sterile-filtered supplemented M9 media (48 mm Na2HPO4, 22 mm KH2PO4, 8.6 mm NaCl, 19 mm NH4Cl, 0.126 mg/ml arginine, 0.02 mg/ml glutamine, 10 µm FeSO4, 5 mm MgSO4, 0.2 mm CaCl2, 0.1 mm thiamine-HCl, 1% (v/v) glycerol, and 0.5% (w/v) casamino acids, pH 7.4) with ampicillin and chloramphenicol and incubated at 30 °C with shaking at 250 rpm until the A600 reached 0.5. A “Pre-Induction” sample was taken by pelleting a volume of cells equal to 1 ml at an A600 of 3, aspirating the supernatant, and storing the pellet at −80 °C. Expression cultures were then moved to a water bath set to 42 °C for 15 min to induce expression. Cultures were incubated at 38 °C for 3 h, and then a 1-ml volume of cells at an A600 of 3 was harvested as the Post-Induction sample and stored at −80 °C. Samples were processed for separation by SDS-PAGE as previously described (21). Following electrophoresis, gels were stained with Coomassie Brilliant Blue.
Large-scale expression of T7 DNA polymerase WT and E514Cou
For WT T7 DNA polymerase expression, C2984H E. coli (F′ proA+B+ lacIq ΔlacZM15/fhuA2 Δ(lac-proAB) glnV galK16 galE15 R(zgb-210::Tn10)TetS endA1 thi-1 Δ(hsdS-mcrB)5), harboring pcIts-(T7 G5x−, trxA), were inoculated into a starter culture of LB plus 100 µg/ml ampicillin and incubated overnight with shaking at 30 °C. The following morning, the overnight culture was inoculated into 1 liter of Terrific Broth (2.4% (w/v) yeast extract, 2% (w/v) tryptone, 0.4% (v/v) glycerol, 17 mm KH2PO4, and 72 mm K2HPO4) plus 100 µg/ml ampicillin and incubated with shaking at 30 °C, 250 rpm until the A600 reached 1.5. The culture was then incubated at 42 °C for 30 min with shaking to induce expression, followed by incubation at 38 °C for 3 h with shaking. The cells were harvested by centrifugation at 6,000 × g for 15 min at 4 °C and stored at −80 °C until purification. For expression of T7 DNA polymerase E514Cou, BL21 E. coli harboring pSUP-(2×CouRS-tRNAopt) and pcIts-(T7 G5x− E514TAG, trxA) were inoculated into an overnight starter culture of LB plus 100 µg/ml ampicillin and 25 µg/ml chloramphenicol and incubated with shaking at 30 °C, 250 rpm. The following morning, 20 ml of overnight culture was inoculated into each of 6 × 1-liter of sterile-filtered supplemented M9 media plus 100 µg/ml ampicillin plus 25 µg/ml chloramphenicol in 2.8-liter baffle bottomed flasks. The cultures were incubated at 30 °C with shaking at 200 rpm. When the A600 of the cultures reached 0.5, the cultures were pelleted at 3,500 × g for 12 min at 4 °C. The cells were then resuspended in a total of 600 ml of sterile filtered supplemented M9 plus 1 mm 7-HCou, 100 µg/ml ampicillin, and 25 µg/ml chloramphenicol in a 2.8-liter baffle bottomed flask. The condensed culture was incubated for 30 min with shaking at 30 °C. The incubator was then set to 42 °C with shaking for 30 min to induce expression. The culture was then incubated at 38 °C with shaking for 3 h, and the cells were harvested as described for the WT enzyme. The condensed 600-ml culture generally yielded a pellet of 6–9 g.
Purification of T7 DNA polymerase
WT T7 DNA polymerase and T7 DNA polymerase E514Cou were purified using the same protocol, and all steps were performed at 4 °C unless otherwise noted. The frozen pellet was thawed and resuspended in lysis buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 2.5 mm EDTA, 0.1 mm DTT, and 1 mm β-mercaptoethanol) at 5 ml of buffer/gram of E. coli, and PMSF was added to a final concentration of 10 mm. Lysozyme was added to 0.3 mg/ml, and the mixture was stirred for 15 min at room temperature. The lysate was sonicated on ice for 20 min with constant stirring, followed by addition of sodium deoxycholate (Sigma-Aldrich) to 0.1% (w/v) and NaCl to 500 mm. The lysate was stirred for 30 min, and then cellular debris was pelleted by ultracentrifugation at 100,000 × g for 30 min. The clarified lysate was diluted with dilution buffer (50 mm Tris-HCl, pH 7.5, 0.1 mm EDTA, and 5 mm DTT) to bring the concentration of NaCl to 100 mm. A solution of 10% (v/v) PEI, pH 8, was added to a final concentration of 0.1% (v/v), then stirred for another 15 min. The precipitated protein was collected by centrifugation at 4,000 × g for 15 min, and then protein was extracted and re-extracted in a small volume of PEI extraction buffer (dilution buffer plus 0.5 m NaCl) and the supernatants were pooled. Ammonium sulfate was slowly added to 35% saturation, the mixture was stirred for 30 min, and the sample was then centrifuged at 24,000 × g for 20 min. Ammonium sulfate was slowly added to the supernatant to 70% saturation and stirred for 30 min, and then the precipitated protein was collected as above. The pellet was dissolved in a small volume of resuspension buffer (dilution buffer plus 10% (v/v) glycerol) and dialyzed against two changes of the same buffer using 50,000-Da molecular weight cutoff dialysis tubing (Spectra-Por) for 1 h each. Resuspension buffer was added to bring the conductivity of the sample down to the conductivity of buffer A (resuspension buffer plus 100 mm NaCl). The sample was filtered through a 0.2-µm syringe filter and loaded on a 50-ml Q Sepharose Fast Flow column, monitoring absorbance at both 280 nm and 335 nm. The column was washed with 10% buffer B (buffer A with 400 mm NaCl), then eluted with a linear gradient of 10–70% buffer B over 20 column volumes. Fractions containing T7 DNA polymerase were pooled and loaded on a 5-ml HiTrap heparin column equilibrated in buffer A. The column was washed with 20% buffer C (buffer A with 1 m NaCl), and then the protein was eluted with a linear gradient of 20–70% buffer C over 20 column volumes. Fractions containing T7 DNA polymerase were pooled, diluted 6-fold with resuspension buffer, and loaded on a 5-ml HiTrap SP Sepharose Fast Flow column equilibrated in buffer A. The column was washed with buffer A, and then the protein was eluted with a linear gradient of 0–40% buffer C over 20 column volumes. Fractions containing T7 DNA polymerase were pooled and concentrated using an Ultracel 30,000 centrifugal concentrator (Amicon). The protein was dialyzed into storage buffer (40 mm Tris-HCl, pH 7.5, 50 mm NaCl, 0.1 mm EDTA, 50% (v/v) glycerol, and 1 mm DTT) using 1,000-Da molecular weight cutoff dialysis tubing (Spectra-Por). The concentration of T7 DNA polymerase (T7 GP5 plus thioredoxin) was determined by absorbance at 280 nm using the extinction coefficient ε280: 150,000 m−1 cm−1, calculated based on the amino acid sequence of each polypeptide. The protein was aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C. The final purity of the purified enzyme was >99% as estimated by SDS-PAGE. Purification from 1 liter of cells for the WT enzyme yielded 50–100 mg of purified protein, whereas purification from the 0.6-liter condensed culture for the unnatural amino acid yielded between 5 and 10 mg of purified protein.
Expression and purification of thioredoxin
Thioredoxin was expressed and purified as described previously (47) with two modifications. A heat denaturation step for 10 min at 63 °C before the DEAE column was used and a Superdex 200 column (GE Healthcare) was used for gel filtration. Concentration of purified thioredoxin was determined by absorbance at 280 nm, using an extinction coefficient of 13,980 m−1 cm−1. Purity was determined to be greater than 99% as estimated by SDS-PAGE. Roughly 35 mg of purified thioredoxin was obtained per liter of cells.
MS
MS analyses including protein digest, LC–MS/MS, and database searching were conducted by the University of Texas Austin Center for Biomedical Research Support's Biological MS facility. Analysis of MDCC-labeled Cys mutants was performed as described by Tsai et al. (18). For WT and E514Cou T7 DNA polymerase, purified protein was separated by SDS-PAGE and the band for T7 gene product 5 was excised, alkylated with iodoacetamide, and digested with trypsin. The extracted peptides were analyzed by LC–MS/MS on a Dionex Ultimate 3000 RSLCnano Nanoflow UPLC connected to an Orbitrap Fusion instrument (Thermo Fisher Scientific) with a 60-min gradient. Proteome Discoverer v2.4 with Sequest HT (Thermo Fisher Scientific) was used for data processing by searching against a database consisting of 12 common keratin contaminants and T7 gene product 5. Two missed cleavages by trypsin were allowed. Cysteine carbamidomethylation was used as a fixed modification, and variable methionine oxidation and Glu → 7-HCou (+C8H4O) modifications were used. The mass tolerance for precursor ions was 10 ppm (monoisotopic), and the mass tolerance for fragment ions was 0.8 Da (monoisotopic).
Data analysis and visualization were performed with Scaffold v4.8 (RRID:SCR_019109), which was also used for preparing figures. The peptide threshold used was 99.9%, resulting in 1,723 total spectra for the WT enzyme and 1,762 spectra for the E514Cou variant. Sequence coverage for both samples was 90%. The peptide 509NQIAAXLPTR518 and the missed cleavage peptide 509NQIAAXLPTRDNAK522 (where X is either glutamic acid of 7-HCou at position 514) ionized well and had a high abundance for both the peptide with glutamic acid in the WT sample and the peptide with 7-HCou for the labeled protein sample. To test for 7-HCou incorporation at other positions in T7 DNA polymerase, purified protein was digested in solution with TPCK-trypsin (Promega) and the peptides were separated by reverse phase HPLC on a C18 column with acetonitrile/TFA mobile phase (not shown). The two peaks with an absorbance at 335 nm were analyzed by MALDI-MS in reflectron mode with α-cyano-4-hydroxy-cinnamic acid as a matrix. Data analysis was performed with the program mmass (48) using the same search parameters as above, with a 0.1-Da error tolerance.
Preparation of oligonucleotide substrates
All synthetic oligonucleotides were synthesized by Integrated DNA Technologies with standard desalting, and oligos used for kinetics assays were further purified in-house by denaturing PAGE and had a final purity of >99% full-length oligo. Concentrations of purified oligos for kinetics assays were determined by absorbance at 260 nm, using the extinction coefficients given in Table 4. DsDNA substrates for kinetic assays were prepared by mixing the appropriate 27-nt primer with the 45-nt template oligos (Table 4) at a 1:1.05 ratio in annealing buffer (10 mm Tris-HCl, pH 7.5, 50 mm NaCl, and 1 mm EDTA). Primer/template complexes were then heated to 95 °C, slowly cooled to room temperature over two hours, and stored at −20 °C.
Table 4.
Kinetics DNA oligos
| Name | Sequence (5′ → 3′) | Extinction coefficient, 260 nm (m−1 cm−1) |
|---|---|---|
| 27 | CCGTCGCAGCCGTCCAACCAACTCAAC | 245,700 |
| 27dd* | CCGTCGCAGCCGTCCAACCAACTCAACdd | 245,700 |
| FAM-27 | [6-carboxyfluorescein]-CCGTCGCAGCCGTCCAACCAACTCAAC | 266,660 |
| 45-18T | GGACGGCATTGGATCGATGTTGAGTTGGTTGGACGGCTGCGACGG | 433,700 |
| 45-18A | GGACGGCATTGGATCGAAGTTGAGTTGGTTGGACGGCTGCGACGG | 437,600 |
*Cdd, 2′,3′ dideoxy-CMP.
Kinetic measurements
All experiments were carried out in T7 reaction buffer (2) (40 mm Tris-HCl, pH 7.5, 50 mm NaCl, 12.5 mm MgCl2,1 mm DTT, and 1 mm EDTA). Reactions with 2′,3′ dideoxy-CMP–terminated 27-nt primer (27dd in Table 4) annealed to a 45-nt template (45-18T in Table 4, where the T represents the templating base), designated DNAdd, allowed measurements of nucleotide binding without the chemistry step. In all experiments, a 20-fold molar excess of thioredoxin over T7 DNA polymerase was included. All reactions were performed with 12.5 mm Mg2+ during the reaction. Reactions were set up by preincubating enzyme with DNA in the absence of MgCl2 in one syringe and mixing with a solution of dNTP and MgCl2 from the other syringe to start the reaction. No difference in kinetic data was observed for the reactions where both syringes had Mg2+ versus one syringe with Mg2+ at 2× the final concentration mixed with a syringe containing no Mg2+. Concentrations of reaction components given in the text are concentrations after mixing. Rapid-quench experiments were performed on an RQF-3 rapid-quench Instrument (KinTek Corporation) with a circulating water bath set to 4 °C. All experiments were performed with 0.6 m EDTA in the quench syringe (giving a final concentration of 0.2 m) and reaction buffer without magnesium in the drive syringes. All stopped-flow experiments were performed on an SF-300 instrument with a circulating water bath set to 4 °C, a 150-watt xenon lamp as the light source, and a dead time of 1.3 ms. Stopped-flow data shown in the main text are averages of at least eight individual traces. Stopped-flow experiments were performed on at least three separate occasions to ensure reproducibility, and each replicate yielded similar rates. Fluorescence experiments containing T7 DNA polymerase E514Cou were performed with excitation at 295 nm and emission at 445 nm, observed with a 45-nm bandpass filter (Semrock). Misincorporation reactions were performed by hand-mixing a solution containing the enzyme-DNA complex with a solution containing dATP plus Mg2+ to start the reaction. Reactions were quenched by the addition of EDTA to a final concentration of 0.2 m. Samples collected by hand-mixing and by rapid-quench were analyzed either by denaturing PAGE or by capillary electrophoresis. Both methods rely upon detecting the fluorescence of the FAM label to quantify the concentrations of products and reactants at each time point. A control pre-steady-state burst experiment with 32P-labeled DNA gave the same rates and amplitudes as the 5′-[6-FAM]-labeled DNA (data not shown), so all subsequent experiments were performed with the FAM-DNA for convenience. Samples separated by PAGE were imaged on a Typhoon FLA 9500 Scanner (GE Healthcare) and analyzed with the program ImageQuant (GE Healthcare). Samples separated by capillary electrophoresis were diluted into HiDi formamide (Thermo Fisher Scientific) with a 28-nt Cy3 internal standard oligo added for sizing. Samples were then injected onto a 36-cm capillary array filled with POP-6TM polymer (Thermo Fisher Scientific) on a 3130xl Genetic Analyzer (Applied Biosystems). Samples analyzed by either method gave identical results, and the capillary electrophoresis method will be elaborated in a future paper.
Steady-state fluorescence excitation/emission scans
Fluorescence excitation and emission spectra of various T7 DNA polymerase E514Cou complexes were collected at room temperature (∼25 °C) on a Horiba Fluorolog3 fluorimeter. Final concentrations of reaction components are given in the main text. Excitation scans were performed by monitoring emission at 460 nm (10-nm bandpass) while varying the excitation wavelength from 250–425 nm (bandpass: 5 nm, integration time: 0.5 s). Emission spectra were collected by scanning emission wavelengths (integration time: 0.5 s, bandpass: 5 nm) with a fixed excitation wavelength of either 298 nm or 335 nm (bandpass: 8 nm). Excitation and emission spectra were analyzed with the program a|e v1.2 (FluorTools, RRID:SCR_019108). All spectra shown in the main text have been corrected by subtracting the spectrum for the buffer alone in the cuvette, collected under the same instrument settings and experimental conditions. Emission scans were replicated three times to ensure reproducibility.
Data analysis
Data fitting/analysis was performed with the simulation software KinTek Explorer (49, 50) v9. This software was also used in preparing figures for kinetic data. Stopped-flow traces were fit in the software using the afit function to either a single or double exponential equation. The equation for a single exponential used is , where A0 is the starting fluorescence, A1 is the amplitude of the fluorescence change, b1 is the decay rate, and t is time. The equation for a double exponential used is , where A0 is the starting fluorescence and A1, A2, b1, and b2 are the amplitudes and decay rates for the first and second phases, respectively. After global data fitting (Fig. 9 and Fig. 10), confidence contour analysis was performed for each dataset using the Fitspace (31) feature of KinTek Explorer. Parameter boundaries are reported in Table 1, using a χ2 cutoff of 0.96 in the Fitspace calculation, which was recommended by the software as a reasonable limit based on the number of parameters and number of data points used in the fitting.
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (51) partner repository with the dataset identifier PXD021676. All remaining data are contained within this article and in the supporting information.
Supplementary Material
This article contains supporting information.
Author contributions—T. L. D. and K. A. J. conceptualization; T. L. D. and K. A. J. formal analysis; T. L. D. investigation; T. L. D. methodology; T. L. D. writing-original draft; K. A. J. resources; K. A. J. software; K. A. J. supervision; K. A. J. funding acquisition; K. A. J. project administration; K. A. J. writing-review and editing.
Funding and additional information—This work was supported by NIGMS, National Institutes of Health, Grant 5R01GM114223 (to K. A. J.) and the Welch Foundation Grant F-1604 (to K. A. J.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—K. A. J. is president of KinTek Corporation, which provided the SF300x stopped-flow and RQF-3 rapid quench-flow instruments and KinTek Explorer software used in this study.
- 7-HCou
- (7-hydroxy-4-coumarin-yl)-ethylglycine
- MDCC
- 7-diethylamino-3-((((2-maleimidyl)ethyl)amino)carbonyl)coumarin
- GP5
- gene product 5
- FAM
- carboxyfluorescein
- CmR
- gene for chloramphenicol acetyltransferase enzyme, conferring chloramphenicol resistance
- trxA
- gene for Escherichia coli thioredoxin
- proK
- E. coli prolyl tRNACGG operon
- glnS'
- mutant promoter for the E. coli glutamine-tRNA ligase gene
- tRNAMj
- M. jannaschii tyrosyl-tRNA with CUA anticodon loop
- tRNAopt
- tRNAMj with three mutations in one of the stem loops that enhances amber suppression efficiency
- AARS
- aminoacyl tRNA synthetase
- MjYRS
- M. jannaschii tyrosyl-tRNA synthetase
- CouRS
- MjYRS mutant with specificity for 7-HCou.
References
- 1. Roettger, M. P., Bakhtina, M., and Tsai, M.-D. (2008) Mismatched and matched dNTP incorporation by DNA polymerase β proceed via analogous kinetic pathways. Biochemistry 47, 9718–9727 10.1021/bi800689d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patel, S. S., Wong, I., and Johnson, K. A. (1991) Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 30, 511–525 10.1021/bi00216a029 [DOI] [PubMed] [Google Scholar]
- 3. Wong, I., Patel, S. S., and Johnson, K. A. (1991) An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry 30, 526–537 10.1021/bi00216a030 [DOI] [PubMed] [Google Scholar]
- 4. Tsai, Y.-C., and Johnson, K. A. (2006) A new paradigm for DNA polymerase specificity. Biochemistry 45, 9675–9687 10.1021/bi060993z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doublié, S., Tabor, S., Long, A. M., Richardson, C. C., and Ellenberger, T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature 391, 251–258 10.1038/34593 [DOI] [PubMed] [Google Scholar]
- 6. Donlin, M. J., Patel, S. S., and Johnson, K. A. (1991) Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction. Biochemistry 30, 538–546 10.1021/bi00216a031 [DOI] [PubMed] [Google Scholar]
- 7. Showalter, A. K., and Tsai, M.-D. (2002) A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry 41, 10571–10576 10.1021/bi026021i [DOI] [PubMed] [Google Scholar]
- 8. Wang, H., Huang, N., Dangerfield, T., Johnson, K. A., Gao, J., and Elber, R. (2020) Exploring the reaction mechanism of HIV reverse transcriptase with a nucleotide substrate. J. Phys. Chem. B 124, 4270–4283 10.1021/acs.jpcb.0c02632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhong, X., Patel, S. S., Werneburg, B. G., and Tsai, M. D. (1997) DNA polymerase β: Multiple conformational changes in the mechanism of catalysis. Biochemistry 36, 11891–11900 10.1021/bi963181j [DOI] [PubMed] [Google Scholar]
- 10. Zhong, X., Patel, S., and Tsai, M. D. (1998) DNA polymerase β: 5. Dissecting the functional roles of the two metal ions with Cr(III)dTTP. J. Am. Chem. Soc. 120, 235–236 10.1021/ja973507r [DOI] [Google Scholar]
- 11. Dunlap, C. A., and Tsai, M.-D. (2002) Use of 2-aminopurine and tryptophan fluorescence as probes in kinetic analyses of DNA polymerase β. Biochemistry 41, 11226–11235 10.1021/bi025837g [DOI] [PubMed] [Google Scholar]
- 12. Kellinger, M. W., and Johnson, K. A. (2010) Nucleotide-dependent conformational change governs specificity and analog discrimination by HIV reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 107, 7734–7739 10.1073/pnas.0913946107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ram Prasad, B., Kamerlin, S. C. L., Florián, J., and Warshel, A. (2012) Prechemistry barriers and checkpoints do not contribute to fidelity and catalysis as long as they are not rate limiting. Theor. Chem. Acc. 131, 1288 10.1007/s00214-012-1288-6 [DOI] [Google Scholar]
- 14. Johnson, K. A. (2019) Kinetic analysis for the new enzymology: Using computer simulation to learn kinetics and solve mechanisms, KinTek Corporation, Austin, U. S. A. [Google Scholar]
- 15. Kellinger, M. W., and Johnson, K. A. (2011) Role of induced fit in limiting discrimination against AZT by HIV reverse transcriptase. Biochemistry 50, 5008–5015 10.1021/bi200204m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zinsli, P. E. (1974) Investigation of rate parameters in chemical reactions of excited hydroxycoumarins in different solvents. J. Photochem. 3, 55–69 10.1016/0047-2670(74)80006-7 [DOI] [Google Scholar]
- 17. Fink, D. W., and Koehler, W. R. (1970) pH effects on fluorescence of umbelliferone. Anal. Chem. 42, 990–993 10.1021/ac60291a034 [DOI] [Google Scholar]
- 18. Tsai, Y.-C., Jin, Z., and Johnson, K. A. (2009) Site-specific labeling of T7 DNA polymerase with a conformationally sensitive fluorophore and its use in detecting single-nucleotide polymorphisms. Anal. Biochem. 384, 136–144 10.1016/j.ab.2008.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xie, J., and Schultz, P. G. (2005) An expanding genetic code. Methods 36, 227–238 10.1016/j.ymeth.2005.04.010 [DOI] [PubMed] [Google Scholar]
- 20. Chiu, J., Tillett, D., and March, P. E. (2006) Coexpression of the subunits of T7 DNA polymerase from an artificial operon allows one-step purification of active gp5/Trx complex. Protein Expr. Purif. 47, 264–272 10.1016/j.pep.2005.10.016 [DOI] [PubMed] [Google Scholar]
- 21. Brandis, J. W., and Johnson, K. A. (2009) High-cell density shake-flask expression and rapid purification of the large fragment of Thermus aquaticus DNA polymerase I using a new chemically and temperature inducible expression plasmid in Escherichia coli. Protein Expr. Purif. 63, 120–127 10.1016/j.pep.2008.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Summerer, D., Chen, S., Wu, N., Deiters, A., Chin, J. W., and Schultz, P. G. (2006) A genetically encoded fluorescent amino acid. Proc. Natl. Acad. Sci. U. S. A. 103, 9785–9789 10.1073/pnas.0603965103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Young, T. S., Ahmad, I., Yin, J. A., and Schultz, P. G. (2010) An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374 10.1016/j.jmb.2009.10.030 [DOI] [PubMed] [Google Scholar]
- 24. Liu, J., Castañeda, C. A., Wilkins, B. J., Fushman, D., and Cropp, T. A. (2010) Condensed E. coli cultures for highly efficient production of proteins containing unnatural amino acids. Bioorg. Med. Chem. Lett. 20, 5613–5616 10.1016/j.bmcl.2010.08.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guo, J., Melançon, C. E., Lee, H. S., Groff, D., and Schultz, P. G. (2009) Evolution of amber suppressor tRNAs for efficient bacterial production of proteins containing nonnatural amino acids. Angew. Chem. Int. Ed. Engl. 48, 9148–9151 10.1002/anie.200904035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aslanidis, C., and de Jong, P. J. (1990) Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074 10.1093/nar/18.20.6069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ryu, Y., and Schultz, P. G. (2006) Efficient incorporation of unnatural amino acids into proteins in Escherichia coli. Nat. Methods 3, 263–265 10.1038/nmeth864 [DOI] [PubMed] [Google Scholar]
- 28. Cellitti, S. E., Jones, D. H., Lagpacan, L., Hao, X., Zhang, Q., Hu, H., Brittain, S. M., Brinker, A., Caldwell, J., Bursulaya, B., Spraggon, G., Brock, A., Ryu, Y., Uno, T., Schultz, P. G., et al. (2008) In vivo incorporation of unnatural amino acids to probe structure, dynamics, and ligand binding in a large protein by nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 130, 9268–9281 10.1021/ja801602q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kobayashi, T., Nureki, O., Ishitani, R., Yaremchuk, A., Tukalo, M., Cusack, S., Sakamoto, K., and Yokoyama, S. (2003) Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Biol. 10, 425–432 10.1038/nsb934 [DOI] [PubMed] [Google Scholar]
- 30. Kati, W. M., Johnson, K. A., Jerva, L. F., and Anderson, K. S. (1992) Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267, 25988–25997 [PubMed] [Google Scholar]
- 31. Johnson, K. A., Simpson, Z. B., and Blom, T. (2009) FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 10.1016/j.ab.2008.12.025 [DOI] [PubMed] [Google Scholar]
- 32. Kornberg, A. (2000) Ten commandments: lessons from the enzymology of DNA replication. J. Bacteriol. 182, 3613–3618 10.1128/jb.182.13.3613-3618.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang, L., Zhang, Z., Brock, A., and Schultz, P. G. (2003) Addition of the keto functional group to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 100, 56–61 10.1073/pnas.0234824100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hahn, A., Reschke, S., Leimkühler, S., and Risse, T. (2014) Ketoxime coupling of p-acetylphenylalanine at neutral pH for site-directed spin labeling of human sulfite oxidase. J. Phys. Chem. B 118, 7077–7084 10.1021/jp503471j [DOI] [PubMed] [Google Scholar]
- 35. Wendeler, M., Grinberg, L., Wang, X., Dawson, P., and Baca, M. (2014) Enhanced catalysis of oxime-based bioconjugations by substituted anilines. Bioconjug. Chem. 25, 93–101 10.1021/bc400380f [DOI] [PubMed] [Google Scholar]
- 36. Mills, J. H., Lee, H. S., Liu, C. C., Wang, J., and Schultz, P. G. (2009) A genetically encoded direct sensor of antibody-antigen interactions. Chembiochem 10, 2162–2164 10.1002/cbic.200900254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuhn, S. M., Rubini, M., Müller, M. A., and Skerra, A. (2011) Biosynthesis of a fluorescent protein with extreme pseudo-stokes shift by introducing a genetically encoded non-natural amino acid outside the fluorophore. J. Am. Chem. Soc. 133, 3708–3711 10.1021/ja1099787 [DOI] [PubMed] [Google Scholar]
- 38. Liu, S., Lv, P., Li, D., Guo, X., Zhang, B., Yu, M., Li, D., Xiong, Y., Zhang, L., and Tian, C. (2015) K+ preference at the NaK channel entrance revealed by fluorescence lifetime and anisotropy analysis of site-specifically incorporated (7-hydroxycoumarin-4-yl)ethylglycine. Chem. Commun. 51, 15971–15974 10.1039/c5cc06124e [DOI] [PubMed] [Google Scholar]
- 39. Chen, S., Fahmi, N. E., Wang, L., Bhattacharya, C., Benkovic, S. J., and Hecht, S. M. (2013) Detection of dihydrofolate reductase conformational change by FRET using two fluorescent amino acids. J. Am. Chem. Soc. 135, 12924–12927 10.1021/ja403007r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dean, S. F., Whalen, K. L., and Spies, M. A. (2015) Biosynthesis of a novel glutamate racemase containing a site-specific 7-hydroxycoumarin amino acid: Enzyme–ligand promiscuity revealed at the atomistic level. ACS Cent. Sci. 1, 364–373 10.1021/acscentsci.5b00211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ugwumba, I. N., Ozawa, K., Xu, Z.-Q., Ely, F., Foo, J.-L., Herlt, A. J., Coppin, C., Brown, S., Taylor, M. C., Ollis, D. L., Mander, L. N., Schenk, G., Dixon, N. E., Otting, G., Oakeshott, J. G., et al. (2011) Improving a natural enzyme activity through incorporation of unnatural amino acids. J. Am. Chem. Soc. 133, 326–333 10.1021/ja106416g [DOI] [PubMed] [Google Scholar]
- 42. Schwark, D. G., Schmitt, M. A., and Fisk, J. D. (2018) Dissecting the contribution of release factor interactions to amber stop codon reassignment efficiencies of the Methanocaldococcus jannaschii orthogonal pair. Genes 9, 546 10.3390/genes9110546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lajoie, M. J., Rovner, A. J., Goodman, D. B., Aerni, H.-R., Haimovich, A. D., Kuznetsov, G., Mercer, J. A., Wang, H. H., Carr, P. A., Mosberg, J. A., Rohland, N., Schultz, P. G., Jacobson, J. M., Rinehart, J., Church, G. M., et al. (2013) Genomically recoded organisms expand biological functions. Science 342, 357–360 10.1126/science.1241459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laemmli, U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 45. Ochman, H., Gerber, A. S., and Hartl, D. L. (1988) Genetic applications of an inverse polymerase chain reaction. Genetics 120, 621–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zheng, L., Baumann, U., and Reymond, J.-L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lunn, C. A., Kathju, S., Wallace, B. J., Kushner, S. R., and Pigiet, V. (1984) Amplification and purification of plasmid-encoded thioredoxin from Escherichia coli K12. J. Biol. Chem. 259, 10469–10474 [PubMed] [Google Scholar]
- 48. Strohalm, M., Kavan, D., Novák, P., Volný, M., and Havlíček, V. (2010) mMass 3: A cross-platform software environment for precise analysis of mass spectrometric data. Anal. Chem. 82, 4648–4651 10.1021/ac100818g [DOI] [PubMed] [Google Scholar]
- 49. Johnson, K. A. (2009) Fitting enzyme kinetic data with KinTek global kinetic explorer. Methods Enzymol. 467, 601–626 10.1016/S0076-6879(09)67023-3 [DOI] [PubMed] [Google Scholar]
- 50. Johnson, K. A., Simpson, Z. B., and Blom, T. (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 10.1016/j.ab.2008.12.024 [DOI] [PubMed] [Google Scholar]
- 51. Vizcaíno, J. A., Csordas, A., del-Toro, N., Dianes, J. A., Griss, J., Lavidas, I., Mayer, G., Perez-Riverol, Y., Reisinger, F., Ternent, T., Xu, Q. W., Wang, R., and Hermjakob, H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 10.1093/nar/gkv1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (51) partner repository with the dataset identifier PXD021676. All remaining data are contained within this article and in the supporting information.