

Abstract
Mitochondria are required for cell survival and are best known for their role in energy production. These organelles also participate in many other biological processes that are critical for cellular function and, thus, play a central role in cellular life and death decisions. In a majority of cell types, mitochondria form highly dynamic, reticular networks. Maintaining the shape of these complex, ever-changing networks is critical for mitochondrial and cellular function and requires the conserved activities of mitochondrial fission and fusion. Great advances in our knowledge about the molecular machines that mediate these dynamic activities have been made over the past two decades. These advances have been driven by the use of highly complementary in vitro and in vivo approaches that have proven extremely powerful for studying the complex membrane remodeling processes that drive fission and fusion of the organelle. In this chapter, we detail current methods used to examine the mechanisms and regulation of mitochondrial fission and fusion in vitro and in vivo.
Keywords: mitochondrial dynamics, mitochondrial fusion, mitochondrial fission, mitochondrial division
Methods
| I | Analysis of mitochondrial fusion in vitro | pg xx |
| Generating stable cell lines expressing fluorescent mitochondrial matrix targeted proteins | ||
| Mitochondrial isolation | ||
| Fusion assay | ||
| II | Analysis of mitochondrial fission in vitro | pg xx |
| Purification of Drp1 | ||
| Liposome preparation | ||
| Visualization of Drp1 tubulation by EM | ||
| GTP time-course to assess Drp1 constriction by EM | ||
| III | Analysis of mitochondrial dynamics in vivo | pg xx |
| Targeting fluorescent proteins to mitochondria | ||
| Analysis of mitochondrial network continuity using fluorescence loss in photobleaching (FLIP) | ||
| Analysis of mitochondrial network morphology using particle counting within a resolvable region of the network | ||
| Resolving Drp1-marked mitochondrial fission events | ||
| Resolving fusion events using FRAP |
Rationale
Over the past two decades significant advances in our understanding of the molecular machines that drive mitochondrial fission and fusion have been made. The first decade of the 21st century yielded seminal discoveries identifying the core components of mitochondrial fission and fusion machineries as dynamin related proteins (DRPs), which are large mechano-chemical, membrane-remodeling GTPases. Dnm1/Drp1 (yeast/mammals) was identified as the sole mitochondrial fission DRP, whereas two fusion DRPs, Fzo1/Mfn1 (and Mfn2) and Mgm1/OPA1 (yeast/mammals) were discovered to drive fusion of the outer and inner mitochondrial membranes (OMM and IMM), respectively (Chan, 2012; Hoppins et al., 2007). In the last decade, new technologies and approaches have yielded a more complex model of fission and fusion. In vivo and in vitro studies suggest that mitochondrial fission is carried out through multiple constriction steps, which are marked by the endoplasmic reticulum (ER) (Friedman et al., 2011) and potentially two additional fission DRPs, dynamin-2 (Dyn2) and the cleaved, soluble form of Mgm1/OPA1 (Anand et al., 2014; Faelber et al., 2019; Lee et al., 2016), working in concert with Drp1. In situ and in vitro studies also suggest that there are distinct stages of OMM docking leading mitochondrial fusion (Brandt et al., 2016; Qi et al., 2016).
Invaluable insights into the mechanisms and regulation of mitochondrial fission and fusion have been gained through the use of highly complementary in vitro and in vivo approaches. Recent technological advances in both electron and light microscopy have greatly enhanced our ability to study the mitochondrial fission and fusion machines and their regulators. Advances in electron microscopy (EM) techniques, including cryo-EM tomography, state of the art detectors, and improvements to imaging and analysis pipelines, have provided high resolution snapshots of fission and fusion sites in cells (Brandt et al., 2016; Friedman et al., 2011), as well as specific stages of these processes reconstituted in vitro using isolated mitochondria or pure protein and phospholiposomes (Engelhart & Hoppins, 2019; Faelber et al., 2019; Francy et al., 2015; Hoppins et al., 2011; Ingerman et al., 2005; Kalia et al., 2018; Mears et al., 2011; Meeusen et al., 2004). Advances in fluorescence microscopy and fluorophores have facilitated the widespread use of multi-channel imaging due to expanded coverage of the light spectrum, increased the duration of imaging due to increased fluorophore photostability and decreased phototoxicity of specific imaging modalities, and allowed for the development of mitochondrial content mixing assays using photoswitchable or photoactivatable fluorescent probes (Karbowski et al., 2014; Owens & Edelman, 2015; Twig et al., 2006). Together these approaches have led to major advances in our understanding of mitochondrial fission and fusion: for example, the findings that inner and outer membrane dynamics can be uncoupled during fission and fusion, mitochondrial fission and fusion events are spatially coupled to the ER, and adaptor proteins and specific lipids alter Dnm1/Drp1 structure and activity (Bustillo-Zabalbeitia et al., 2014; Cho et al., 2017; Clinton et al., 2016; Friedman et al., 2011; Guo et al., 2018; Hoppins et al., 2011; Kalia et al., 2018; Meeusen et al., 2004).
The combined use of both in vitro and in vivo methods will continue to be a powerful and necessary approach to dissect the complex membrane remodeling processes of mitochondrial fission and fusion and the contributions of the ever-growing number of protein and membrane factors that directly impact these processes. In this chapter, we will supplement the in vitro methods published in the previous edition (Ingerman et al., 2007). Here we will focus on more recently developed methods to examine the mechanisms and regulation of mitochondrial dynamics in vitro and in vivo.
I. Analysis of mitochondrial fusion in vitro
Definition
The ability to examine mitochondrial fusion in vitro allows for deeper investigations into the mechanism driving the process and provides a reductionist approach to examine how individual factors impact the process. An in vitro fusion assay using mitochondria isolated from yeast has been detailed in the previous edition (Ingerman et al., 2007). Here we will describe the method for an in vitro fusion assay optimized for mammalian mitochondria (Engelhart & Hoppins, 2019; Hoppins et al., 2011; Samanas & Hoppins, in press)
Materials
Cell Lines:
Mouse embryonic fibroblasts (MEFs) (such as ATCC CRL-2991/2992/2993); Retroviral packaging cell line Platinum-E (Plat-E) cells (Cell Biolabs, Inc. #RV-101).
Plasmids:
Plasmids encoding two different mitochondrial matrix targeted fluorescent proteins, such as Addgene #58425 (pclbw-mitoTagRFP) and #58426 (pclbw-mitoCFP) (Mishra et al., 2014).
Cell culture media and reagents:
DMEM + high glucose and GlutaMAX- (Gibco/Fisher Scientific) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin; Opti-MEM- (Gibco #31985–062); FuGENE- HD transfection reagent (Promega #E2311); Puromycin and blasticidin antibiotics; Polybrene infection reagent (hexadimethrine bromide); Freezing medium (DMEM + glucose supplemented with 20% FBS and 10% DMSO).
Other solutions and reagents:
Sterile leur lock syringes; 0.45 μm PES membrane syringe filters; No. 1.5 coverslips or glass bottom dishes (such as MatTek #P35G-1.5–20-C); 0.1 M Tris-MOPS pH 7.4 (dissolve 1.21 g Tris powder in about 50 mL water, adjust the pH of the solution to 7.4 with MOPS powder and bring volume to 100 mL, filter sterilize and store at 4°C); 1 M sucrose (dissolve 85.58 g sucrose in water to a total volume of 250 mL, filter sterilize and store at 4°C); 0.1 M EGTA-Tris (dissolve 7.62 g EGTA in about 50 mL water, adjust the pH of the solution to 7.4 with Tris powder and bring the final volume to 100 mL, filter sterilize and store at RT); 3 M sorbitol (dissolve 136.5 g sorbitol in water to a final volume of 250 mL, filter sterilize and store at 4°C); 0.5 M PIPES pH 6.8 (dissolve 15.12 g PIPES in about 50 mL water, adjust the pH of the solution to 6.8 with KOH and bring the final volume to 100 mL, filter sterilize and store at RT); 1 M magnesium acetate (dissolve 10.72 g magnesium acetate in water to a final volume of 50 mL, filter sterilize and store at RT); 5 M potassium acetate (dissolve 24.54 g potassium acetate in water to a final volume of 50 mL, filter sterilize and store at RT); Mitochondrial Isolation Buffer (MIB): 0.01 M Tris-MOPS pH 7.4, 0.2 M sucrose, 1 mM EGTA-Tris (make fresh each day and keep on ice or at 4°C); Cytosol Buffer: 20 mM PIPES pH 6.8, 150 mM potassium acetate, 5 mM magnesium acetate, 0.2 M sorbitol (make fresh each day and store on ice); 30 mg/mL creatine kinase (resuspend creatine kinase powder in cytosol buffer at working concentration of 30 mg/mL); 1 M creatine phosphate (dissolve 211.11 mg creatine phosphate in 1 mL cytosol buffer, and store 10 μL aliquots at −80°C); 250 mM GTP (dissolve 25 mg GTP in 191 μL cytosol buffer, adjust the pH of the solution to 7.0 with 2 M NaOH, and store 5 μL aliquots at −80°C); 250 mM ATP (dissolve 25 mg ATP in 197 μL cytosol buffer, adjust the pH of the solution to 7.0 with 2 M NaOH, and store 5 μL aliquots at −80°C); Fusion Buffer: 1.2 mg/ml creatine kinase, 40 mM creatine phosphate, 1.5 mM ATP, 1.5 mM GTP in cytosol buffer (dilute 250 mM ATP/GTP stock to 25 mM in cytosol buffer); 3% low melt agarose: dissolve 30 mg low melt agarose in 1 mL cytosol buffer by warming the solution to 90°C, keep at 65°C.
All solutions should be prepared using ultrapure water and analytical grade reagents. Filter sterilization should be performed with 0.2 μm filters. Dispose of all reagents according to local guidelines.
Equipment
Kontes Potter-Elvehjem tissue grinder, 4 mL (such as Fisher Scientific #K885510-0020); Motor driven homogenizer (such as Caframo #BDC3030); Single well glass slides and covers (such as Fisher Scientific #S175201); Fluorescence widefield or confocal microscope.
Protocols
Generating stable cell lines expressing fluorescent mitochondrial matrix targeted proteins
NOTE: For all experiments, cells are maintained at 37°C and 5% CO2.
TIMING: 6 days
Split Plat-E cells, which have been maintained in complete media supplemented with 1 mg/mL puromycin and 10 mg/mL blasticidin, and plate at approximately 80% confluency (approximately 3.5 × 105 cells in each well of a 6 well dish) in 2.5 mL complete media (no antibiotics).
The next day, transfect Plat-E cells with high purity, low endotoxin plasmid DNA, such as mitochondria-targeted RFP (mtRFP) or CFP (mtCFP). Combine 175 μL Opti-MEM with 3 μg of DNA and mix. Add 10 μL of FuGENE- HD directly to the liquid and mix vigorously. Incubate at RT for 15 min before dropping the mixture onto the Plat-E cells. Change the media on the Plat-E cells 8–24 hr later. Supernatant containing the virus will be collected ~48, 56, 72 and 80 hr post-transfection.
Plate MEF cells 12–24 hr before the 48 hr viral infection at about 70% confluency (approximately 1 × 105 cells per well of 6 well dish). This is usually done at the same time the media on the transfected Plat-E cells is changed.
For the 48 hr viral transfection, aspirate media from MEF cells and discard. Collect media from Plat-E cells with a sterile syringe. Pass this virus-containing media through a 0.45 μm PES membrane syringe filter onto the MEF cells. Add polybrene reagent to virus-containing media on the MEF cells at a final concentration of 1 mg/mL. Replace media on Plat-E cells. Incubate for 6–10 hr. For the 56 hr transfection, repeat procedure and incubate overnight. The 48 hr and 56 hr transfections are usually done at the beginning and end of the workday, respectively.
For the 72 and 80 hr infections, repeat step 4.
The day after the 80 hr infection, trypsinize and move all MEF cells from the 6-well dish to a 10 cm plate.
-
Assess expression of mtCFP and mtRFP by fluorescence microscopy.
CRITICAL STEP: Approximately 95% of the cells should be expressing the fluorescent protein to ensure that most isolated mitochondria are visible following the fusion reaction.
Freeze several tubes at 2 × 106 cells per mL in freezing media. These can be thawed onto a 15 cm dish and split two days later.
NOTE: To generate a large number of cells expressing either mtRFP or mtCFP and thus reduce the number of cell passages, perform the experiment in triplicate so that there are 3 wells of a 6 well dish for each cell line.
Mitochondrial isolation
TIMING: 2–3 hr
-
Seed five 15 cm plates of each mtRFP and mtCFP cells so that they are ~95% confluent on the day of the assay. At minimum, a wild type population of both mtRFP and mtCFP is required as a control to be performed in parallel with additional experiments.
CRITICAL STEP: To obtain high quality and high quantities of mitochondria, a total cell pellet > 150 μL for both mtRFP and mtCFP is required.
Make 25 mL MIB and 1 mL cytosol buffer fresh the day of the assay and store at 4°C or on ice.
After removing most media from the plates, use a cell scraper to detach cells into ~ 5 mL of media. Combine cells from all five plates of the mtRFP cells into one 50 mL conical tube and the cells from all five plates of the mtCFP cells into another tube.
Pellet cells by centrifugation at 300 × g for 10 min at 4°C.
Return tubes to ice and aspirate media. Finger flick the tube to loosen the cell pellet and then resuspend cells in 5 mL cold MIB.
Pellet cells by centrifugation at 300 × g for 5 min at 4°C. Aspirate all buffer.
Resuspend cells in one pellet volume MIB and transfer to ice cold homogenizers, one for each cell population.
Homogenize cells on ice with a Teflon pestle operating at 400 rpm. Slowly move the pestle up and down 12–15 times, avoiding the formation of bubbles in the sample by keeping the pestle below the buffer at all times.
Transfer homogenate to a cold 1.5 mL microcentrifuge tube. To remove nuclei and unbroken cells, spin the homogenate at 300 × g for 5 min at 4°C and then transfer the supernatant to a new pre-chilled 1.5 mL microcentrifuge tube.
Repeat the homogenization of the cell pellet recovered from the 300 × g spin. Following the 300 × g spin of the second homogenization, combine supernatants. Discard the pellet.
With the combined supernatants, centrifuge 300 × g for 5 min at 4°C to remove any remaining nuclei or unbroken cells. Transfer the post-nuclear supernatant to a clean, cold 1.5 mL microcentrifuge tube and discard the small pellet.
To isolate mitochondria, spin the supernatant at 7400 × g for 10 min at 4°C. The post-mitochondrial supernatant can be transferred to a cold 1.5 mL microcentrifuge tube and saved as the crude cytosol fraction.
Wash crude mitochondria in 500 μL MIB and re-pellet by centrifugation at 7400 × g for 10 min at 4°C to wash the mitochondria. Aspirate all supernatant and resuspend the mitochondrial pellet in one pellet volume MIB.
Determine the mitochondrial protein concentration by Bradford assay.
Fusion assay
TIMING: 2–3 hr
For each reaction condition, mix 10–15 μg of mitochondrial protein for each color of mitochondria together in a 1.5 mL microcentrifuge tube. If the same mitochondrial pair is being tested under multiple conditions, a master mix can be made. Pellet the mitochondrial mixture by centrifugation at 7400 × g for 10 min at 4°C. Move the tubes to ice and incubate for 10 min.
-
Remove the supernatant from all tubes. Add 10 μL of appropriate fusion reaction buffer to each tube on ice to cover the mitochondrial pellet. Cytosol buffer can be used as a negative control. Crude cytosol or pure protein can be added to the standard fusion buffer to test the effect of these factors. Resuspend the mitochondrial pellet in fusion buffer by pipetting up and down, avoiding the formation of bubbles. Leave on ice until all tubes have been resuspended.
CRITICAL STEP: All buffers and reagents must be in good condition for fusion to work. Creatine kinase deteriorates over time and nucleotides will deteriorate more quickly if not stored under the correct conditions.
Incubate at 37°C for 60 min.
Slides can be prepared in one of two ways: (A) Create an agarose bed in single well depression slides by pipetting 30–40 μL of hot low melt agarose into the depression and immediately placing a plain glass slide on top to create a flat surface. These can be prepared during the incubation step above. Just before use, move the top slide off of the agarose bed by pushing to the side (do not lift up). Clean off the excess agarose from around the depression with a razor blade and a delicate task wipe. At the end of the 60 min incubation, move tubes to ice. Pipet 4 μL reaction mixture onto the agarose bed and drop a coverslip to cover the agarose bed. Seal with nail polish. (B) Following the fusion incubation step above, pipet 3 μL of the fusion reaction onto the middle of a plain slide. Mix with 3 μL of warm low melt agarose by pipetting up and down 2–3 times and drop a coverslip on top of this mixture while still warm. Seal with nail polish. Collect images of each reaction with a confocal or widefield fluorescence microscope with the appropriate excitation and emission settings to detect the matrix-targeted fluorophores. Collect Z stacks to capture the entire mitochondrial volume in both colors; we recommend 10–12 steps of 0.2 μm each. Collect multiple fields of view from different positions on the agarose bed so that at least 400 total mitochondria are imaged (Fig. 1). CRITICAL STEP: The isolated mitochondria often vary in brightness. Ensure that exposure settings are sufficient to capture the dimmest mitochondria in the sample.
Figure 1. Assessing mitochondrial fusion in vitro.
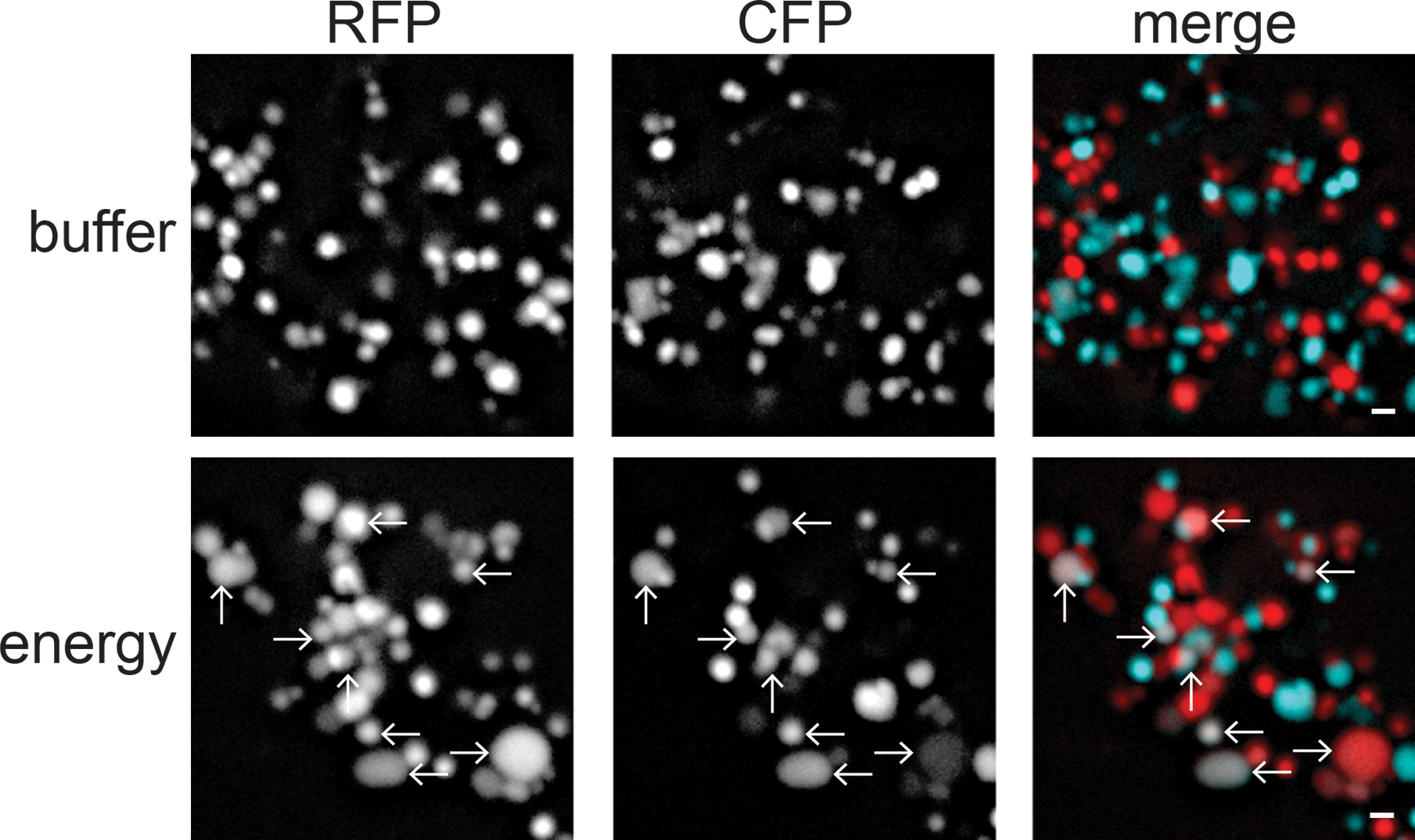
Images of mitochondria from negative control and fusion reactions. Following incubation with buffer, mitochondria with matrix RFP or matrix CFP remain separate (top panel). Following incubation with fusion buffer (energy), some organelles contain both matrix RFP and CFP, and are a result of mitochondrial fusion in vitro (arrows, lower panel). Scale bar = 1 μm.
Analysis and statistics
Fusion efficiency is quantified by dividing the number of fused mitochondria (those with both fluorophores in three dimensions) by the total number of mitochondria in a given field of view (Fig. 1). Count at least 4 fields and a minimum of 400 total mitochondria. Data are represented as relative to wild type controls performed in parallel. Standard deviation is calculated from at least three independent biological replicates (different passage number and different mitochondrial prep). Statistical significance is determined by paired t-test analysis.
Anticipated Results
Mitochondria isolated from wild type control cells typically have 15–20% of total mitochondria that are fused.
II. Analysis of mitochondrial fission in vitro
Rationale
Our understanding of mitochondrial fission has been greatly impacted by studies examining the activity and structure of Dnm1/Drp1 using reductionist systems in vitro. It is well established that the GTPase cycle of Dnm1/Drp1 is tightly connected to the conformation and dynamics of Dnm1/Drp1 oligomers. Thus, being able to examine the biochemical activities as well as structural properties of Dnm1/Drp1 are critical to unravel the mechanism of mitochondrial fission. Assays to measure the GTPase cycle kinetics and biochemical activities of Dnm1/Drp1 have been detailed in the previous edition as well as in a recent methods chapter (Clinton et al., in press; Ingerman et al., 2007). Here, we will focus on Drp1-mediated liposome tubulation and constriction assays visualized by EM.
Materials
Solutions, Reagents & Consumables:
BL21(DE3)Star chemically competent E. coli; pCal-N-Ek-Drp1 vector (Francy et al., 2015); Luria Broth (LB); Ampicillin; Isopropyl-β-D-thiogalactopyranoside (IPTG); Lysis/Wash Buffer: 0.5 M L-Arginine pH 7.4, 0.3 M NaCl, 5 mM MgCl2, 2 mM CaCl2, 1 mM Imidazole, 10 mM β-Mercaptoethanol; Elution Buffer: 0.5 M L-Arginine pH 7.4, 0.3 M NaCl, 2.5 mM EGTA, 10 mM β-Mercaptoethanol; Size Exclusion Chromatography (SEC) Buffer: 25 mM HEPES (KOH) pH 7.5, 0.15 M KCl, 5 mM MgCl2, 10 mM β-Mercaptoethanol; Calmodulin Affinity Resin; Prescission Protease; Amicon Ultra-15 30 MWCO Centrifugal Filter; Pefabloc-SC; Coomassie; Assembly Buffer: 25 mM HEPES (KOH) pH 7.5, 150 mM KCl, 10 mM β-Mercaptoethanol; 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC); 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE); Heart Cardiolipin; Guanosine Triphosphate (GTP); Methyleneguanosine 5′-triphosphate (GMPPCP); MgCl2; Carbon-coated EM grids; Parafilm; Filter Paper; Uranyl Acetate
Equipment:
Swinging Basket Centrifuge; Sonicator; Ultracentrifuge; AKTA Pure Chromatography System; HiLoad 16/600 Superdex 200 Prep Grade; Lipid Extruder with filter supports and 1.0 μm polycarbonate filters; Electron Microscope - FEI Technai Spirit or F20; High-precision tweezers; imaging software (ImageJ or alternative)
Protocols
Purification of Drp1
NOTE: All chromatography steps should be performed at 4°C.
TIMING: 3 days
Expression of Drp1 in E. coli
-
1
Transform BL21(DE3)Star chemically competent E. coli with the pCal-N-EK-Drp1 vector. Using a single colony of the BL21(DE3)Star cells containing the pCal-N-Ek-Drp1 plasmid, inoculate LB containing 100 μg/mL ampicillin and grow overnight at 37°C with shaking at 200 rpm.
-
2
The following day, inoculate 2 L fresh LB containing 100 μg/mL ampicillin with the starter culture (1:50 dilution). Incubate at 37°C with shaking at 200 rpm.
-
3
Monitor cell density and when an OD600 of 0.6–0.8 is achieved, induce cells with 1 mM IPTG. Incubate overnight (~24 hr) at 18 °C while shaking at 200 rpm.
-
4
The next morning, harvest the culture by centrifugation at 4,300 × g for 20 min at 4°C, and discard the media.
-
5
Resuspend the cell pellet in Lysis/Wash Buffer (5 mL per gram cell paste) with 1 mM Pefabloc SC and 200 μg/mL lysozyme.
-
6
Lyse cells with a probe sonicator on ice. (Note: Other cell disruption methods including French Press and microfluidizers also work.)
CRITICAL STEP: The resuspended cells will appear cloudy and after cell disruption the solution should be noticeably more transparent. If the solution remains extremely cloudy, sonication should be repeated to ensure efficient lysis.
-
7
Pellet the cell debris by ultracentrifugation at 150,000 × g for 1 hr at 4°C.
-
8
Collect the clarified cell lysate and store on ice or at 4°C to load onto affinity resin.
Affinity Chromatography
-
9
Prepare a Calmodulin Affinity Resin column with 4 mL of slurry (50% EtOH), which yields a final column volume of 2 mL. Wash the resin with at least 5 column volumes Lysis/Wash Buffer.
-
10
Apply the clarified lysate from the bacterial expression to the column. This can be done by gravity or inline using an FPLC system. Collect the flow through.
-
11
Following sample loading, wash the column with 50 mL Wash Buffer to remove impurities.
-
12
Elute bound protein with Elution buffer in a series of 1 mL fractions.
CRITICAL STEP: Additional binding and elution steps can be performed with the collected flow through to maximize protein yield.
-
13
Identify the fractions containing the bulk of your CBP-tagged Drp1 using Coomassie dye.
-
14
Pool all protein-containing fractions. Add 2 μL Prescission Protease per milliliter of pooled protein to remove the affinity tag, and incubate overnight at 4°C.
-
15
The next morning, concentrate the pooled protein down to 5 mL or less using an Amicon Ultra-15 30 MWCO centrifugal filter.
-
16
Centrifuge the concentrated protein at 10,000 × g for 20 min at 4°C to clear protein solution before loading on the sizing column.
Size Exclusion Chromatography
-
17
Equilibrate the Superdex 200 column with one column volume of SEC Buffer.
-
18
Inject the protein onto the column and run the sample at 0.5–1.0 mL/min.
-
19
Collect fractions and identify the fractions containing the purified Drp1 protein.
-
20
Add glycerol as a cryoprotectant to a final concentration of 5%, and concentrate the protein using a 30 MWCO concentrator.
-
21
Aliquot the purified Drp1 and flash freeze in liquid nitrogen. Store at −80°C.
Liposome preparation
NOTE: Drp1 has been shown to preferentially interact with negatively charged lipid templates (Bustillo-Zabalbeitia et al., 2014; Francy et al., 2015; Macdonald et al., 2014; Stepanyants et al., 2015), and cardiolipin (CL) will be used for this example. Additional factors can be implemented in additional experiments, including changing lipid composition to alter charge, flexibility, and curvature of membrane templates.
TIMING: 2–3 hr
Combine a mixture of 40 mol% DOPC, 35 mol% DOPE, and 25 mol% CL dissolved in chloroform in a clean glass test tube.
Evaporate the solvent with dry nitrogen gas while rotating the tube to form a thin lipid film. Remove residual solvent with a centrifugal evaporator for 1 h at 37°C.
Add Assembly Buffer preheated to 37°C such that the final lipid concentration is 1–2 mM. Incubate 30 min at 37°C with gentle agitation to fully resuspend the lipid mixture.
Transfer lipids to a plastic test tube and place the tube in liquid nitrogen until completely frozen (roughly 30 s). Then place in a 37°C water bath until fully thawed (roughly 1–2 min). Repeat for a total of 4 freeze-thaw cycles.
Prepare a lipid extruder by soaking 4 filter supports and a 1.0 μm polycarbonate filter in Assembly Buffer and assembling the extruder according to manufacturer instructions.
Extrude the lipid solution through the filter 21 times using gentle, constant pressure.
Lipids can be stored at 4 °C and should be used within 2–3 days.
Visualization of Drp1 tubulation by EM
TIMING: 1–2 days
Incubate CL containing liposomes with Drp1 (2 μM final) and incubate for 1 h at RT.
Prepare carbon-coated mesh grids, filter paper and tweezers. The grids may be glow discharged or plasma cleaned before the sample is applied to increase their hydrophilicity. NOTE: If you want to make a control grid with liposome alone, you may need to pre-coat the grid with bovine serum albumin or equivalent protein to promote lipid association.
On a piece of parafilm, add 5–10 μL of sample along with two drops of 2% uranyl acetate (Fig. 2A).
Place the grid, carbon-side down, onto the drop of sample. Allow the grid to remain on the drop for 30–60 s.
Blot the grid on a piece of filter paper to remove excess buffer.
Wash the grid on the first drop of 2% uranyl acetate by touching the grid to the drop and then immediately blotting it on a piece of filter paper. Repeat wash.
Place the grid on the second drop of 2% uranyl acetate for 30–60 s.
Remove excess stain by blotting the grid on a piece of filter paper.
Store in a grid box under vacuum until an electron microscope is prepared to image the grid. CRITICAL STEP: Drp1 helical oligomers on liposomes will be discernable at lower magnification (~4,000–6,000 X), and higher magnification (~30,000 – 50,000 X) can be used to identify specific features of polymers (i.e. size, abundance, and helical patterns). In the absence of Drp1 self-assembly, no discernible features are obvious.
Save images to a format that can be used to assess Drp1-lipid tube features qualitatively and quantitatively.
Figure 2. Visualizing Drp1 tubulation by EM.
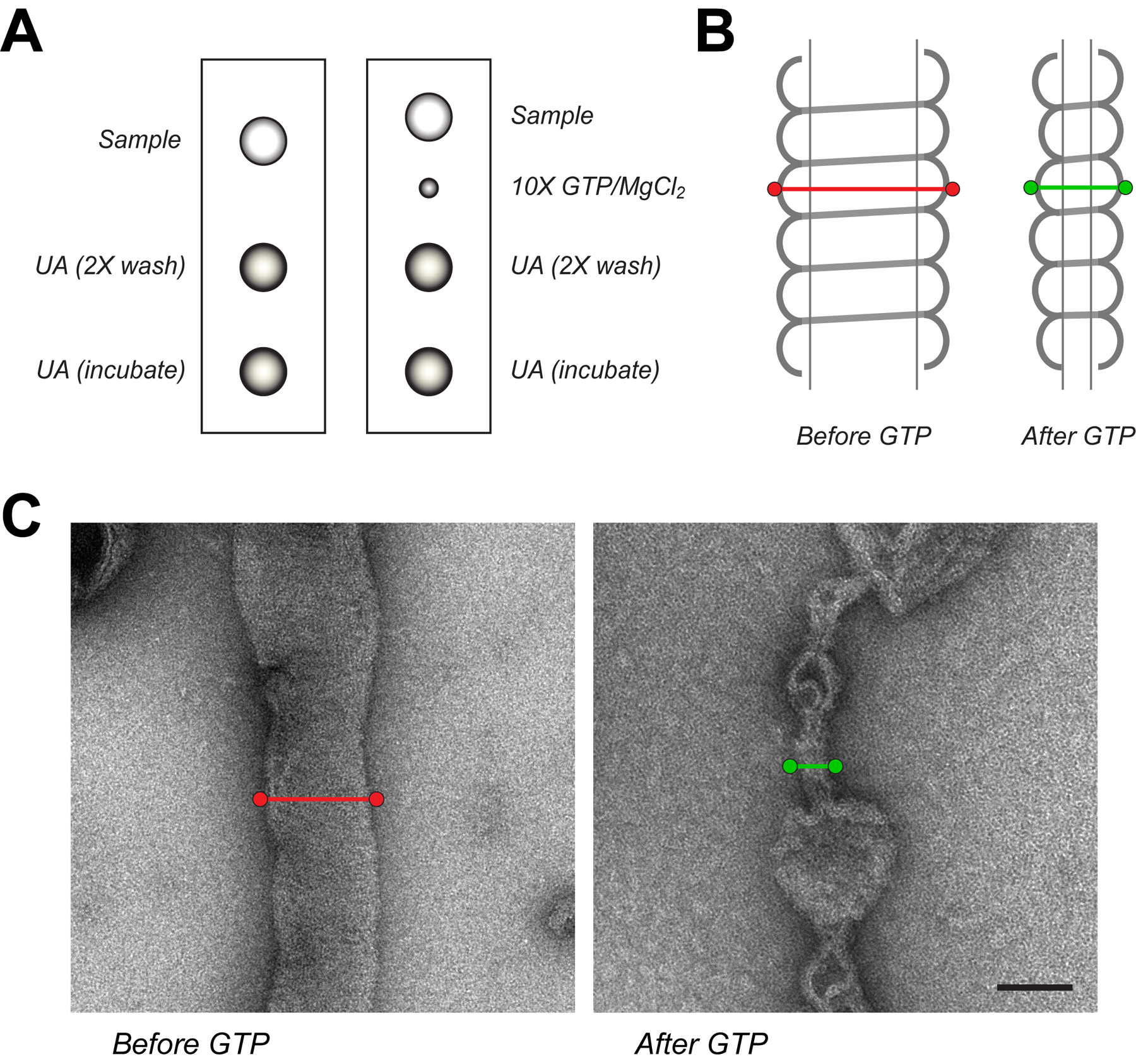
(A) To prepare negative stain grids, a drop of sample containing Drp1-lipid tubes alone or with nucleotide is placed on parafilm with two separate drops for washes and a final incubation with 2% uranyl acetate. For assessment of nucleotide interactions at shorter time points after addition, a separate drop of nucleotide and MgCl2 (10X) is placed adjacent to the sample drop. The grid is initially placed on the sample drop and the nucleotide can be added at the desired time before stopping the reaction with stain washes and incubation. (B) Measurements of protein-lipid tube diameters are made by tracing a line orthogonal to the helical axis at the edge of detectable protein before (red line) and after (green line) nucleotide addition. (C) Representative micrographs highlight the morphological change that occurs when GTP is added to pre-decorated Drp1-lipid tubes. Scale bar, 100 nm.
GTP time-course to assess Drp1 constriction by EM
NOTE: The effect of nucleotide binding and hydrolysis on membrane deformation can be investigated by adding GMP-PCP and GTP, respectively, in the presence of MgCl2.
TIMING: 1–2 days
Pre-assemble Drp1 lipid tubes in Assembly Buffer by incubating CL containing liposomes with Drp1 for 1 hr at RT as above.
Prepare carbon-coated mesh grids, filter paper and tweezers.
To examine the effect of GTP binding, add GMPPCP (1 mM final) and MgCl2 (2 mM final) to Drp1 lipid tubes and incubate for desired length of time.
To examine the effect of GTP hydrolysis, add GTP (1 mM final) and MgCl2 (2 mM final) to Drp1 lipid tubes and incubate for desired length of time.
For GTP incubations lasting longer than 2 min, add 5–10 μL of sample containing a mixture of Drp1, lipid and nucleotide along with two drops of 2% uranyl acetate on a piece of parafilm (Fig. 2A).
Roughly 1 min prior to the desired end of the incubation with nucleotide, place the grid, carbon-side down, onto the sample drop, and allow the incubation to proceed on the grid for the remaining 30–60 s.
To stop the incubation, transfer the grid to the first drop of 2% uranyl acetate by touching the grid to the drop and then immediately blotting it on a piece of filter paper. Repeat wash and proceed to final incubation with stain (step 13).
For GTP incubations lasting less than 2 min, prepare a 10X stock solution of GTP and MgCl2.
Prepare a 9 μL drop of preassembled Drp1-lipid sample and a 1 μL drop of the GTP/MgCl2 solution along with two drops of 2% uranyl acetate on a piece of parafilm (Fig. 2A).
1 min prior to the desired end of the incubation with nucleotide, place the grid, carbon-side down, onto the Drp1-lipid sample drop, and allow the incubation to proceed.
At the desired time preceding the end of the incubation, drag the Drp1-lipid sample drop onto the GTP/MgCl2 drop to begin the hydrolysis reaction on the grid. (EXAMPLE: For a 30 s GTP incubation, the grid would rest on the Drp1-lipid drop for 30 s before dragging the grid to the GTP/MgCl2 drop to incubate for an additional 30 s).
To stop the incubation, transfer the grid to the first drop of 2% uranyl acetate by touching the grid to the drop and then immediately blotting it on a piece of filter paper. Repeat wash.
For all samples, place the washed grid on the second drop of 2% uranyl acetate for an additional 30–60 s.
Remove excess stain by blotting the grid on a piece of filter paper.
Store in a grid box under vacuum until an electron microscope is prepared to image the grid.
-
As above, ultrastructural changes can be assessed from EM images.
NOTE: A more detailed analysis without the use of heavy metal stains can be performed using cryo-electron microscopy (cryo-EM). Detailed structural features, including changes in the underlying lipid morphology, can be examined and quantified.
Related Techniques
Drp1 (and Dnm1) interactions with various lipids, nucleotides and adaptor proteins regulate its structure and activity. Therefore, the above protocol can be adapted to examine the impact of each of these factors independently or in combination. For liposomes, the composition of the lipid species can be altered to test specific interactions. As highlighted above, the different effects of GTP binding and hydrolysis can be dissected using nucleotide analogs. Other nucleotide states, including GDP-bound, can be assessed. To reconstitute Drp1 recruitment to membranes by adaptor proteins, these integral proteins must be incorporated with lipid templates. This can be done using the full-length proteins with the transmembrane domain (TMD) intact (Macdonald et al., 2016), but this can be difficult without the use of detergents. As an alternative, these adaptors can be tethered to the surface of lipid templates through use of an affinity tag (Clinton et al., 2016; Osellame et al., 2016). For example, His-tagged Mitochondrial fission factor (His-Mff) will associate onto liposomes containing NTA(Ni2+) lipids (Clinton & Mears, 2017). In this way, lipid-proximal protein interactions can be investigated for a range of lipid and protein compositions.
Analysis and Statistics
Ultrastructural changes can be quantified using image analysis software, such as ImageJ (http://imagej.nih.gov/ij/; Schneider et al., 2012).
Protein decoration can be measured when compared with naked lipid templates.
Diameters of tubular segments can be measured from the outermost portion of the assemblies (Fig. 2B).
CRITICAL STEP: It is ideal to have a blinded colleague measure these morphological changes independently to ensure that the trends are consistent and unbiased. Standard deviation is calculated from the diameters of at least 100 tubular segments for each condition.
Anticipated results
Drp1 readily tubulates CL-containing liposomes (Fig. 2C)(Francy et al., 2015; Stepanyants et al., 2015). The diameters of the helical assemblies vary widely, which reflects the inherent flexibility of the Drp1 polymers. This is similar to previous results with Dnm1 (Mears et al., 2011). Additionally, the average size of Drp1 tubes is dependent on the splice variant used in the studies (Macdonald et al., 2016). Regardless of the starting geometry, addition of GTP, and associated hydrolysis, will lead to a narrowing of Drp1/Dnm1 helical polymers within seconds (Fig. 2B–C) (Francy et al., 2015; Mears et al., 2011). Following constriction, the polymers dissociate from the lipids and will reassemble transiently. Therefore, the relative abundance of Drp1-lipid tubes will be diminished when GTP is added. Conversely, the use of a non-hydrolysable GTP analog, GMPPCP, stabilizes Drp1/Dnm1 tubules and no constriction is observed with GTP binding. Collectively, measurements of these structures by EM can be used to assess morphological changes in Drp1 in response to specific lipid and nucleotide interactions.
III. Analysis of mitochondrial dynamics in vivo
Rationale
Mitochondrial network morphology is an efficient readout for the steady-state balance of mitochondrial fission-fusion rates within the cell. A fragmented mitochondrial network indicates that fission rates dominate fusion rates. Conversely, an elongated, hyperconnected network signifies that fusion rates are dominant. Here, we describe two complementary approaches to quantify mitochondrial continuity (by photobleaching analysis) and fragmentation (by particle analysis).
The steady-state balance of fission/fusion rates can be affected by changes in metabolism, signaling pathways, cell state, etc. Therefore, a more direct test of whether factors play a role in fission or fusion is to catalog their spatiotemporal localization to fission/fusion events. We also describe techniques to resolve fission (using Drp1 as a marker) and fusion (by photobleaching) in live cells by analyzing changes in fluorescence intensity across line-scans.
Materials
Cell Lines:
HeLa cells (or an appropriate cell line)
Plasmids:
Plasmids encoding TOM20-EGFP (Cho et al., 2017), TOM20-mCh (XhoI/SalI subcloned from TOM20-EGFP into pmCherry-N1), mito-EGFP (Song et al., 2009), mCh-Drp1 (Addgene 49152) and mito-BFP (Addgene 49151) (Friedman et al., 2011).
Cell culture media and reagents:
DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Gibco/ThermoFisher), Opti-MEM (Gibco/ThermoFisher), Lipofectamine 2000 (Invitrogen), FluoroBrite DMEM + 10% FBS + 1x GlutaMAX (Gibco/ThermoFisher)
Other reagents and solutions:
No. 1.5 glass bottom microscope dishes (such as MatTek #P35G-1.5–20-C), No. 1.5 coverslips, glass slides, fibronectin, PBS, paraformaldehyde, Triton X-100, normal donkey serum, antiTom20 antibody (c-11415, Santa Cruz Biotech), Prolong Antifade Mountant (Invitrogen), NucBlue Fixed Cell ReadyProbes Reagent (Invitrogen)
Equipment:
Laser-scanning confocal microscope outfitted with resonant scanning mirrors, epifluorescence or confocal fluorescence microscope and the ability to Z-section, and spinning disc confocal microscope. The microscope appropriate for each application is specified in the protocol. All microscopes should be outfitted with 63x and/or 100x objectives. Additionally, all confocal microscopes used for live cell imaging should be outfitted with an incubation chamber, 405/488/561/640 nm lasers, and appropriate filters.
Protocols
Exposure to light during imaging can lead to phototoxicity. Therefore, care should be taken to minimize the exposure times and laser or light power required for individual experiments to minimize the phototoxic stress to cells, which can alter mitochondrial structure and function.
Targeting fluorescent proteins to mitochondria
A widely used technique to visualize mitochondria in live cells is the targeting of a fluorescent protein to the mitochondrial matrix. In yeast, amino acids 1–69 of Su9 from Neurospora crassa are a commonly used mitochondrial matrix targeting sequence. This sequence is fused to the N-terminus of a fluorescent protein. Matrix-targeted GFP, dsRED, BFP, and TagBFP have been used successfully (Friedman et al., 2011; Meeusen et al., 2004; Ping et al., 2016; Westermann & Neupert, 2000). For a visual marker of the OMM, fluorescent protein fusions to the OMM protein OM45 are commonly used (Cerveny et al., 2001; Meeusen et al., 2004).
In mammalian cells, the mitochondrial targeting sequences from human Cox8 (amino acids 1–29) and S. cerevisiae Cox4 (amino acids 1–21) are commonly used to target fluorescent proteins to the matrix (Chen et al., 2003; Friedman et al., 2011; Mishra et al., 2014). To visualize the OMM, plasmids expressing fluorescent proteins fused to the last 37 amino acids of OMP25 or Tom20-fluourescent protein fusions have been used successfully (Cho et al., 2017; Nemoto & De Camilli, 1999; Song et al., 2009).
Analysis of mitochondrial network continuity using fluorescence loss in photobleaching (FLIP)
There are two major caveats associated with assessing mitochondrial network morphology by light microscopy: 1) Discrete compartments may appear to be continuous (Twig et al., 2006) and 2) The microtubule organizing center (MTOC)/perinuclear region of the cytoplasm is too crowded to resolve. The second caveat has been circumvented by assessing mitochondrial morphology within a region of interest (ROI) outside of the MTOC (4.3.3).
FLIP has been utilized to demonstrate the continuity of the ER (Dayel et al., 1999). Here, we describe a comprehensive method to simultaneously measure continuity of two major compartments of the mitochondrial network using FLIP. In this procedure, the outer mitochondrial membrane (OMM) is labeled with TOM20-GFP and the matrix is labeled with mito-mCherry (mCh) to monitor OMM and matrix compartment dynamics. Briefly, reference images of both GFP and mCh channels are captured prior to photobleaching. Next, GFP and mCh are simultaneously photobleached repeatedly in a small region of the mitochondrial network within a single cell. As diffusion brings fresh fluorophores into the targeted area and photobleached fluorophores out of the targeted area, fluorescence is lost from all regions that are continuous. Finally, post-photobleached images are captured for comparison to reference images. The level of OMM and matrix continuity are measured as the percentage of mitochondrial network area in post-photobleached images compared to reference images. Smaller percentages will indicate a more interconnected network, whereas larger percentages will indicate a more fragmented network. Importantly, this approach allows for simultaneous monitoring of OMM and matrix continuity, which provides a direct assay for resolving conditions that induce an uncoupling of OMM and matrix dynamics.
FLIP overcomes both caveats described above and provides a direct assessment of mitochondrial network continuity in living cells. FLIP also overcomes the undersampling of elongated morphology phenotypes, as well as subjectivity, that is associated with ROI particle analysis (4.3.3). However, one limitation of the FLIP approach is that it might undersample fragmented mitochondrial networks leading to muted differences between fragmented and normal network phenotypes (use 4.3.3).
Transient transfection of HeLa cells with plasmids encoding for fluorescently-tagged OMM or matrix markers
NOTE: The desired confluency is 70–90% at the time of imaging. For all experiments, live cells are maintained at 37°C and 5% CO2.
TIMING: 3 days
Cover the glass coverslip region of microscope dishes with 10 μg/mL of fibronectin/PBS. Incubate at 37°C for 5 hr.
Remove fibronectin/PBS solution. Rinse plate with 1 mL PBS to remove excess soluble fibronectin. Seed 0.5 × 105 HeLa cells/mL in DMEM + 10% FBS + antibiotics (2 mL total volume). Incubate overnight.
Initiate transient transfection ~24 hr before imaging. Transfect combinations of 100 ng TOM20-EGFP + 100 ng of mito-mCh per microscope dish to monitor OMM or matrix continuity, respectively. Add plasmids into 250 μL Opti-MEM. Separately, add 5 μL of Lipofectamine 2000 into 250 μL Opti-MEM. Wait 5 min.
Gently mix solutions with plasmids and Lipofectamine 2000. Incubate at RT for 18 min.
Remove media from cells and rinse with 1 mL of Opti-MEM. Add 1.5 mL of Opti-MEM to cells followed by gently adding transfection mix to cells dropwise.
After 5 hr, remove Opti-MEM and rinse cells once with 1 mL of full media. Add 2 mL of full media. Incubate overnight.
Live-cell imaging: Capture pre- and post-bleach images of green and red fluorescence using a laser-scanning confocal microscope
TIMING: 1 hr per microscope dish
Prepare imaging media (FluoroBrite DMEM + 10% FBS + 1x GlutaMAX). Warm to 37°C.
Remove growth media and rinse plate with 1 mL of imaging media. Add 2 mL of imaging media to the microscope dish and load into the incubation chamber of the microscope.
Identify cells expressing both TOM20-EGFP and mito-mCh.
Capture whole cell Z-stack images of green and red fluorescence (Pre-bleach).
-
Position a 100 μm2 ROI within the dense perinuclear region of the mitochondrial network that resides over the microtubule organizing center (MTOC). Simultaneously excite the cell using 50 iterations of 25% 561 nm laser power and 25% 473 nm with a 4 ms pixel dwell time to photobleach TOM20-EGFP and mito-mCh.
NOTE: Photobleaching parameters will vary with different microscopes.
Capture whole cell Z-stack images of green and red fluorescence (Post-bleach) immediately after photobleaching to assess diffusion within continuous compartments and to minimize the risk of capturing content-mixing due to fusion events occurring after photobleaching.
Data Analysis: Colocalization using ImageJ
Open pre-bleach and post-bleach green and red fluorescence Z-stack images as a stack in ImageJ (https://imagej.nih.gov/ij/; Schneider et al., 2012).
Process the four Z-stacks into max intensity projection images: Image > Stacks > ZProjection… > Projection type: Max Intensity
Convert to 8-bit for thresholding: Image > Type > 8-bit.
Plugins > Filters > Otsu Thresholding.
Process > Make Binary.
Obtain total mitochondrial area for each max intensity projection: Analyze > Analyze Particles…
-
Calculate the percentages of photobleached OMM and matrix:
- OMM - (Post-bleach TOM20-EGFP Area/Pre-bleach TOM20-EGFP Area)*100
- Matrix - (Post-bleach mito-mCh Area/Pre-bleach mito-mCh Area)*100
CRITICAL: Although OMM and IMM dynamics have been assumed to be highly coupled, simultaneous imaging of both the OMM and IMM with high spatiotemporal resolution in recent studies has shown that constriction of these two membranes are not synchronized suggesting that these are regulated by non-overlapping mechanisms (Cho et al., 2017). Therefore, it is important to assess the continuity of the OMM and IMM in order to identify future factors that differentially regulate one membrane over the other.
Statistical Analysis:
Use student’s T-test when comparing two conditions or one-way ANOVA with multiple comparisons when comparing 3 or more conditions (similar to assessing %FRAP recovery – see Kamerkar et al. 2018).
Analysis of mitochondrial network morphology using particle counting within a resolvable region of the network.
Particle analysis within an ROI that lies outside of the MTOC has been the conventional approach to assess mitochondrial morphology over the last decade and is useful for assessing fragmented mitochondrial morphology phenotypes (Losón et al., 2013). However, this approach cannot resolve dense mitochondrial networks and under-samples elongated mitochondrial morphology phenotypes (see Kamerkar et al., 2018; Lee et al., 2016; Yu et al., 2019 and use 4.2.2 for these limitations).
TOM20 immunofluorescence in HeLa cells
TIMING: 3 days including the curing process
Seed 0.5 × 105 HeLa cells/mL on fibronectin-coated coverslips in a 6-well plate (2 mL total volume) and aim to fix cells at 70–90% confluency.
Gently add and quickly remove 2 mL of PBS (37°C) to remove cell debris.
Immediately add 2 mL of 4% paraformaldehyde/PBS (37°C). Incubate for 10 min.
Remove fixative.
Wash cells with 2 mL of PBS (RT). Incubate for 2 min. Repeat two more times.
Add 2 mL of 0.1% Triton X-100/PBS (RT). Incubate for 5 min.
Wash cells with 2 mL of PBS (RT). Incubate for 2 min.
Carefully remove coverslips and place (cell side facing up) on a sheet of parafilm that is sitting flat on top of a wet paper towel.
Carefully add 100 μL of blocking buffer (5% normal donkey serum/PBS – RT) dropwise onto the coverslip and then cover to attenuate evaporation. Incubate for 30 min.
Carefully remove blocking buffer.
Add 100 μL of blocking buffer + 1:1000 TOM20 antibody (RT) dropwise onto the coverslip and then cover to attenuate evaporation. Incubate for 30 min. NOTE: This step can take place overnight (4°C). Optimize volume and incubation chamber to attenuate evaporation that might be associated with the longer incubation time.
Transfer coverslips back to 6-well plate for washing. Repeat step 5.
Repeat step 8. Add 100 μL of blocking buffer + 1:400 donkey anti-mouse Alexa Fluor-488 antibody (RT) dropwise onto the coverslip and then cover to attenuate evaporation. Incubate for 60 min.
Transfer coverslips back to 6-well plate for washing. Repeat step 5.
Mount coverslips onto microscope slides using Prolong Antifade Mountant with NucBlue Stain. Allow mountant to cure overnight at RT away from light. This mountant is ideal because it has a similar refractive index to glass coverslips after 20+ hr of curing.
Immunofluorescence imaging: Capture high-resolution Z-stack serial images with a confocal microscope using 63x or 100x objectives
Capture Z-stack serial images of TOM20 immunofluorescence and DAPI fluorescence through the volume of the cell.
Data Analysis: Particle analysis using ImageJ
Open Z-stack images as a stack in ImageJ.
Process the Z-stack into one max intensity projection image: Image > Stacks > ZProjection… > Projection type: Max Intensity
Create a 230 μm2 region of interest (ROI) to sample the mitochondrial network: Edit > Selection > Specify…
Place ROI over a resolvable region of the mitochondrial network and crop the image: Image > crop.
Convert to 8-bit for thresholding: Image > Type > 8-bit.
Plugins > Filters > Otsu Thresholding.
Process > Make Binary.
Obtain mitochondria/ROI and total mitochondrial area/ROI: Analyze > Analyze Particles… NOTE: Area/Mitochondrion = (mitochondria/ROI)/(total mitochondrial area/ROI)
Statistical Analysis:
Use student’s T-test when comparing two conditions or one-way ANOVA with multiple comparisons when comparing 3 or more conditions (Lee et al. 2016; Kamerkar et al. 2018).
Visualizing fission and fusion events
Our ability to resolve mitochondrial fission and fusion by light microscopy is complicated by the inability to resolve membranes directly to determine the difference between a continuous mitochondrion or two overlapping mitochondria. Therefore, we must rely on coincidence detectors to resolve mitochondrial fission and fusion. For mitochondrial fission, we use the well-established fission factor Drp1 for two reasons, 1) Drp1 punctum localize to the overwhelming majority of fission sites and 2) the Drp1 punctum splits into two Drp1 puncta, which is coincident with a mitochondrion dividing into two daughter mitochondria (Fig. 3) (Lee et al., 2016). For mitochondrial fusion, we can use FRAP to generate a heterogeneous population of mitochondria in which all mitochondria are labeled with matrix targeted BFP and MOM (or MIM) some mitochondria are either photobleached or unbleached. Fusion is then detected by content-mixing. Post hoc analyses of fluorescent intensities using line-scans can be used to verify the coincidence of 1) Drp1 splitting and mitochondrial fission (Fig. 3), or 2) fluorescence content-mixing and mitochondrial fusion.
Figure 3. Assessing Drp1 localization to mitochondrial fission events using line-scans.
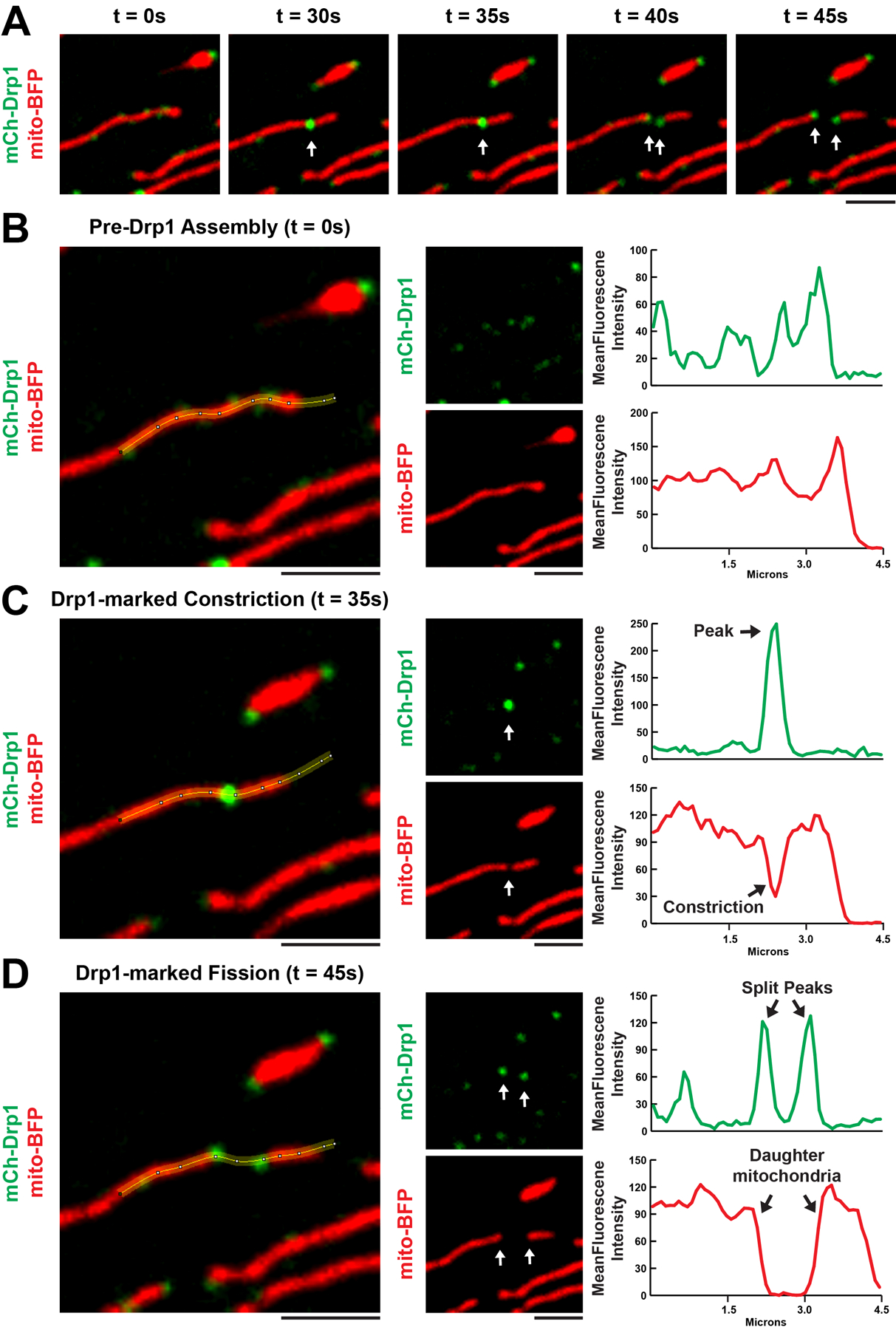
(A) Time-lapse images of a mitochondrial fission event in HeLa cells expressing exogenous mCh-Drp1 (green) and mito-BFP (red) captured with a 100x objective attached to a spinning disc confocal microscope. The arrows highlight the simultaneous splitting of a Drp1 punctum with mitochondrial fission. (B, C, D) Segmented lines overlaid on the t = 0s, 35s, 45s merges from A (left panel) together with individual channels (middle panel) and the corresponding graphs of mean fluorescence intensities (MFI) across the segmented line (right panel). (B) Low MFI Drp1 puncta localized across the highlighted mitochondrion. (C) The low MFI Drp1 puncta (from B) coalesce into a high MFI Drp1 punctum that colocalizes with a mitochondrial constriction. (D) The high MFI Drp1 punctum (from C) splits into two puncta which remain associated with the ends of two newly created daughter mitochondria. Scale bars = 2 μm.
Resolving Drp1-marked mitochondrial fission events
Transient transfection of HeLa cells with plasmids encoding for mCh-Drp1 together with mito-BFP (IMM marker). Note: This plasmid combination allows for the potential assessment of a third and fourth marker in green and far red fluorescence.
TIMING: 3 days
Repeat steps 1–6 of the transient transfection of HeLa cells described above using 75 ng mCh-Drp1 and 75 ng mito-BFP.
Live-cell imaging: Capture time-lapse movies using a 100x objective on a spinning disc confocal microscope
TIMING: 1 hr per microscope dish
Prepare imaging media (FluoroBrite DMEM + 10% FBS + 1x GlutaMAX). Warm to 37°C.
Remove growth media and rinse plate with 1 mL of imaging media. Add 2 mL of imaging media to the microscope dish and load into the incubation chamber of the microscope.
Identify cells expressing all of the fluorescently-tagged markers.
Capture images every 5 s for the duration of two min.
CRITICAL: The frequency of image capture and movie duration are both optimized to yield data with an endpoint that occurs before significant photobleaching. These conditions will differ for different microscopes. Movies with a higher frequency image capture and a longer duration are now achievable through the improvement of spinning disc confocal microscopes, the incorporation of resonant scanning mirrors into laser-scanning confocal microscopes, and the development of super-resolution microscopy systems specialized for multi-channel live-cell imaging, such as the Zeiss Airyscan confocal microscope.
Data analysis: Localization of factors to fission events with line-scan analysis of fluorescent intensities using ImageJ
Open a time-lapse movie as a merged stack in ImageJ and identify fission events.
Convert to 8-bit for thresholding: Image > Type > 8-bit.
Draw a segmented line through the longitudinal axis of the mitochondrion that undergoes fission. Set the thickness of the line to be the width of the mito-BFP fluorescence signal.
Obtain the mean fluorescence intensities across the segmented line: Analyze > Plot Profile. Click List on the Plot Profile to obtain the raw data points and load into a spreadsheet such as Excel.
Repeat step 4 for each fluorescence channel.
Repeat steps 4 and 5 for timepoints leading up to and after fission.
Interpretation of line-scans: Mitochondrial constrictions that eventually undergo fission will be identified by dips in mito-BFP fluorescence intensities that overlap with mCh-Drp1 fluorescence intensity peaks. Fission will be verified when the mitochondrial constriction fluorescence intensity drop to 0 coincides with the resolution of two new and distinct Drp1 fluorescence peaks (Fig. 3).
Resolving fusion events using FRAP
Transient transfection of HeLa cells with plasmids encoding for TOM20-mCh (OMM) and mito-BFP (IMM) to assess OMM fusion, or mito-mCh (IMM) and Tom20-BFP to assess IMM fusion. mCh-tagged constructs will be photobleached to generate a heterogenous population of bleached and unbleached mitochondria, whereas BFP-tagged constructs will be used as a reference marker for total mitochondria.
TIMING: 3 days
-
Repeat steps 1–6 of the transient transfection protocol for HeLa cells described above using 100 ng TOM20-mCh and 75 ng mito-BFP, or 50 ng mito-mCh and 100 ng TOM20-BFP.
NOTE: A major advantage of using FRAP (over photoactivatable/photoswitchable fluorophores) to generate mitochondrial heterogeneity is the potential to evaluate a third and fourth marker tagged to fluorophores excited by 473 and 640 nm lasers.
Live-cell imaging: Photobleach multiple discrete mitochondria in the cell periphery then capture a post-bleach time-lapse movie to track photobleached mitochondria and observe whether they fuse and recover fluorescence.
TIMING: 1 hr per microscope dish
Prepare imaging media (FluoroBrite DMEM + 10% FBS + 1x GlutaMAX). Warm to 37°C.
Remove growth media and rinse plate with 1 mL of imaging media. Add 2 mL of imaging media to the microscope dish and load into the incubation chamber of the microscope.
-
Identify a cell that is expressing all of the fluorescently-tagged markers. Create ROIs for five discrete mitochondria in the cell periphery.
NOTE: Quickly accomplish steps 4–6 once the ROIs have been established to minimize the chance that mitochondria will traffic outside of the ROI.
Capture whole-cell images of all channels (Pre-bleach).
-
Photobleach mCh-tagged markers within the ROIs using 15 iterations of 25% 561 nm laser power with a 4 ms pixel dwell time.
NOTE: Photobleaching parameters will vary between microscope.
Capture time-lapse movies of all channels (Post-bleach).
Data Analysis:
Localization of factors to fusion events with line-scan analysis of fluorescent intensities using ImageJ
Open Post-bleach time-lapse movie as a merged stack in ImageJ and identify fusion events.
Convert to 8-bit for thresholding: Image > Type > 8-bit.
Draw a continuous segmented line through the longitudinal axis of the two mitochondria that eventually fuse. Set the thickness of the line to be the width of the BFP fluorescence signal.
Obtain the mean fluorescence intensities across the segmented line: Analyze > Plot Profile. Click List on the Plot Profile to obtain the raw data points and load into a spreadsheet such as Excel.
Repeat step 4 for each fluorescence channel.
Repeat steps 4 and 5 for timepoints leading up to and after fusion.
Interpretation of line-scans: The photobleached mitochondrion will only display BFP fluorescence, whereas unbleached mitochondrion will display both mCh and BFP fluorescence. Fusion will be verified by line-scan analysis to show mCh fluorescence recovery in the photobleached mitochondrion.
Alternatives approaches
The methods outlined above can be performed using alternative cell lines (for example, COS-7, U2OS, MEFs, etc.), which will require optimization of transfection parameters. Additionally, the methods can be applied to the analysis of mitochondrial continuity and dynamics in yeast. As described above, fluorescent proteins can be targeted to the mitochondrial matrix and OMM in yeast and used to visualize mitochondrial dynamics and membrane continuity. In addition, Dnm1-GFP expressed from its endogenous locus as a yEGFP fusion (for example, using pKT128, Addgene #8729 to tag Dnm1) (Sheff & Thorn, 2004) or from plasmid pHS20 (Sesaki & Jensen, 1999) can be used in combination with matrix-targeted dsRED to mark fission events in yeast.
Alternative approaches available to assess mitochondrial network morphology and connectivity: 1) A population of cells are binned based upon a qualitative assessment of network morphology (fragmented, short, long, net-like, collapsed) (Kamerkar et al., 2018; Losón et al., 2013; Otera et al., 2016; Yu et al., 2019). 2) FRAP has been used to assess network continuity with the readout being the extent of recovery within the photobleached ROI (Kamerkar et al., 2018; Losón et al., 2013; Osellame et al., 2016). 3) Green-to-red photoswitchable fluorophores, such as Dendra2 and Maple, have been used to assess network continuity with the readout being the spread of red fluorescence beyond the photoswitched ROI (Losón et al., 2013; Twig et al., 2006).
Alternative approaches available to generate fluorescence heterogeneity in the mitochondrial population with the goal of resolving mitochondrial fusion in living cells. Photoactivatable dark-to-green fluorophores, such as PAGFP, and green-to-red photoswitchable fluorophores, such as Dendra2 and Maple, have been used to capture content-mixing after two heterogeneous mitochondria fuse (Karbowski et al., 2014; Liu et al., 2009; Twig et al., 2006). However, fewer markers can be simultaneously imaged in combination with photoactivatable/photoswitchable fluorophores compared to using FRAP to generate mitochondrial heterogeneity to assess mitochondrial fusion because photoactivatable/photoswitchable fluorophores require that the 405 nm laser is dedicated to photoactivation or photoswitching.
Acknowledgements
SH is supported by the NIH (R01 GM 118509); LLL is supported by the NIH (R01 GM120303); JEL is supported by the HHMI Faculty Award of G.K. Voeltz; JAM is supported by the NIH (R01 GM125844 and R01 CA208516).
References
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E et al. (2014) The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. Journal of Cell Biology, 204, 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt T, Cavellini L, Kühlbrandt W, & Cohen MM (2016) A mitofusin-dependent docking ring complex triggers mitochondrial fusion in vitro. Elife, 5, e14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustillo-Zabalbeitia I, Montessuit S, Raemy E, Basañez G, Terrones O, & Martinou JC (2014) Specific interaction with cardiolipin triggers functional activation of Dynamin-Related Protein 1. PLoS One, 9, e102738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveny KL, McCaffery JM, & Jensen RE (2001) Division of mitochondria requires a novel DNM1-interacting protein, Net2p. Molecular Biology of the Cell, 12, 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC (2012) Fusion and fission: interlinked processes critical for mitochondrial health. Annual Review of Genetics, 46, 265–287. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, & Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. Journal of Cell Biology, 160, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B, Cho HM, Jo Y, Kim HD, Song M, Moon C et al. (2017) Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nature Communications, 8, 15754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton RW, Bauer B, & Mears JA Purification of dynamin-related protein 1 for structural and functional studies. Methods in Molecular Biology, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton RW, Francy CA, Ramachandran R, Qi X, & Mears JA (2016) Dynamin-related Protein 1 Oligomerization in Solution Impairs Functional Interactions with Membrane-anchored Mitochondrial Fission Factor. Journal of Biological Chemistry, 291, 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton RW, & Mears JA (2017) Using Scaffold Liposomes to Reconstitute Lipid-proximal Protein-protein Interactions In Vitro. Journal of Visualized Experiments, e54971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayel MJ, Hom EF, & Verkman AS (1999) Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophysical Journal, 76, 2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhart EA, & Hoppins S (2019) A catalytic domain variant of mitofusin requiring a wildtype paralog for function uncouples mitochondrial outer-membrane tethering and fusion. Journal of Biological Chemistry, 294, 8001–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faelber K, Dietrich L, Noel JK, Wollweber F, Pfitzner AK, Mühleip A et al. (2019) Structure and assembly of the mitochondrial membrane remodelling GTPase Mgm1. Nature, 571, 429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francy CA, Alvarez FJ, Zhou L, Ramachandran R, & Mears JA (2015) The mechanoenzymatic core of dynamin-related protein 1 comprises the minimal machinery required for membrane constriction. Journal of Biological Chemistry, 290, 11692–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, & Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science, 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Li D, Zhang S, Yang Y, Liu JJ, Wang X et al. (2018) Visualizing Intracellular Organelle and Cytoskeletal Interactions at Nanoscale Resolution on Millisond Timescales. Cell, 175, 1430–1442. e17. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ et al. (2011) The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Molecular Cell, 41, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, & Nunnari J (2007) The machines that divide and fuse mitochondria. Annual Review of Biochemistry, 76, 751–780. [DOI] [PubMed] [Google Scholar]
- Ingerman E, Meeusen S, Devay R, & Nunnari J (2007) In vitro assays for mitochondrial fusion and division. Methods in Cell Biology, 80, 707–720. [DOI] [PubMed] [Google Scholar]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE et al. (2005) Dnm1 forms spirals that are structurally tailored to fit mitochondria. Journal of Cell Biology, 170, 1021–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM et al. (2018) Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature, 558, 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerkar SC, Kraus F, Sharpe AJ, Pucadyil TJ, & Ryan MT (2018) Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nature Communications, 9, 5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Cleland MM, & Roelofs BA (2014) Photoactivatable green fluorescent protein-based visualization and quantification of mitochondrial fusion and mitochondrial network complexity in living cells. Methods in Enzymology, 547, 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Westrate LM, Wu H, Page C, & Voeltz GK (2016) Multiple dynamin family members collaborate to drive mitochondrial division. Nature, 540, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weaver D, Shirihai O, & Hajnóczky G (2009) Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO Journal, 28, 3074–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losón OC, Song Z, Chen H, & Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular Biology of the Cell, 24, 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald PJ, Francy CA, Stepanyants N, Lehman L, Baglio A, Mears JA et al. (2016) Distinct Splice Variants of Dynamin-related Protein 1 Differentially Utilize Mitochondrial Fission Factor as an Effector of Cooperative GTPase Activity. Journal of Biological Chemistry, 291, 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald PJ, Stepanyants N, Mehrotra N, Mears JA, Qi X, Sesaki H et al. (2014) A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Molecular Biology of the Cell, 25, 1905–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, & Hinshaw JE (2011) Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nature Structural & Molecular Biology, 18, 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen S, McCaffery JM, & Nunnari J (2004) Mitochondrial fusion intermediates revealed in vitro. Science, 305, 1747–1752. [DOI] [PubMed] [Google Scholar]
- Mishra P, Carelli V, Manfredi G, & Chan DC (2014) Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metabolism, 19, 630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto Y, & De Camilli P (1999) Recruitment of an alternatively spliced form of synaptojanin 2 to mitochondria by the interaction with the PDZ domain of a mitochondrial outer membrane protein. EMBO Journal, 18, 2991–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osellame LD, Singh AP, Stroud DA, Palmer CS, Stojanovski D, Ramachandran R et al. (2016) Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. Journal of Cell Science, 129, 2170–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Miyata N, Kuge O, & Mihara K (2016) Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. Journal of Cell Biology, 212, 531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GC, & Edelman DB (2015) Photoconvertible fluorescent protein-based live imaging of mitochondrial fusion. Methods in Molecular Biology, 1313, 237–246. [DOI] [PubMed] [Google Scholar]
- Ping HA, Kraft LM, Chen W, Nilles AE, & Lackner LL (2016) Num1 anchors mitochondria to the plasma membrane via two domains with different lipid binding specificities. Journal of Cell Biology, 213, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Yan L, Yu C, Guo X, Zhou X, Hu X et al. (2016) Structures of human mitofusin 1 provide insight into mitochondrial tethering. Journal of Cell Biology, 215, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanas NB, & Hoppins S Cell-free Analysis of Mitochondrial Fusion by Fluorescence Microscopy. Methods in Molecular Biology, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, & Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nature Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, & Jensen RE (1999) Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. Journal of Cell Biology, 147, 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff MA, & Thorn KS (2004) Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast, 21, 661–670. [DOI] [PubMed] [Google Scholar]
- Song Z, Ghochani M, McCaffery JM, Frey TG, & Chan DC (2009) Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Molecular Biology of the Cell, 20, 3525–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanyants N, Macdonald PJ, Francy CA, Mears JA, Qi X, & Ramachandran R (2015) Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Molecular Biology of the Cell, 26, 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Graf SA, Wikstrom JD, Mohamed H, Haigh SE, Elorza A et al. (2006) Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. American Journal of Physiology-Cell Physiology, 291, C176–84. [DOI] [PubMed] [Google Scholar]
- Westermann B, & Neupert W (2000) Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast, 16, 1421–1427. [DOI] [PubMed] [Google Scholar]
- Yu R, Jin SB, Lendahl U, Nistér M, & Zhao J (2019) Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO Journal, 38, e99748. [DOI] [PMC free article] [PubMed] [Google Scholar]