

Abstract
Staphylococcus aureus adhesion to the host's skin and mucosae enables asymptomatic colonization and the establishment of infection. This process is facilitated by cell wall-anchored adhesins that bind to host ligands. Therapeutics targeting this process could provide significant clinical benefits; however, the development of anti-adhesives requires an in-depth knowledge of adhesion-associated factors and an assay amenable to high-throughput applications. Here, we describe the development of a sensitive and robust whole cell assay to enable the large-scale profiling of S. aureus adhesion to host ligands. To validate the assay, and to gain insight into cellular factors contributing to adhesion, we profiled a sequence-defined S. aureus transposon mutant library, identifying mutants with attenuated adhesion to human-derived fibronectin, keratin, and fibrinogen. Our screening approach was validated by the identification of known adhesion-related proteins, such as the housekeeping sortase responsible for covalently linking adhesins to the cell wall. In addition, we also identified genetic loci that could represent undescribed anti-adhesive targets. To compare and contrast the genetic requirements of adhesion to each host ligand, we generated a S. aureus Genetic Adhesion Network, which identified a core gene set involved in adhesion to all three host ligands, and unique genetic signatures. In summary, this assay will enable high-throughput chemical screens to identify anti-adhesives and our findings provide insight into the target space of such an approach.
Keywords: high-throughput screening, anti-adhesives, antivirulence, Staphylococcus aureus, MRSA, antibiotic resistance, cell wall-anchored proteins, methicillin-resistant Staphylococcus aureus (MRSA), bacterial adhesion, virulence factor, drug discovery
An important component of bacterial virulence is adhesion to the host's skin and mucosae (1, 2, 3, 4); as such, the development of anti-adhesive therapeutics could provide significant clinical benefits (1, 3, 5). However, such an approach requires an in-depth knowledge of adhesion-associated factors in clinically relevant strains (6). In addition, sensitive and robust assays are required to enable high-throughput chemical screening campaigns.
This study is focused on the opportunistic pathogen Staphylococcus aureus. The organism's arsenal of virulence factors enables a variety of diseases ranging from skin and soft tissue infections to severe systemic infections (7). Strains of S. aureus possess numerous cell wall-anchored (CWA) surface proteins (8) covalently attached to the peptidoglycan through the action of sortases, mainly the housekeeping transpeptidase sortase A (SrtA) (9, 10). CWA proteins perform many functions, including adhesion, immune evasion, iron acquisition, and biofilm formation (8). As such, the pathogenicity of SrtA-deficient S. aureus strains is significantly impaired in several animal models of infection (11, 12, 13). In regard to adhesion, a subset of CWA proteins recognize different host ligands, which enables S. aureus strains to colonize and cause infections in distinct sites within the body (8). Therefore, specific anti-adhesives will be required in different clinical contexts.
One bottleneck in the study of S. aureus adhesion is the lack of a high-throughput and whole cell assay. Rather than focusing on a single target (e.g. SrtA), a whole cell assay would permit an unbiased phenotypic approach for identifying anti-adhesives, enabling the sampling of a larger target space. Furthermore, whole cell assays provide inhibitors with guaranteed biological activity, which is often a limiting factor of target-based approaches (14, 15). Existing adhesion detection methods have largely relied on staining of adhered bacteria with crystal violet, or the manual enumeration of colony-forming units (16, 17, 18, 19, 20). Although these assays are capable of detecting strains with reduced adhesion, they lack the feasibility, consistency, sensitivity, and/or speed required for high-throughput applications. To overcome these limitations, we describe the development of an ELISA to detect S. aureus adhesion to host ligands in a high-throughput manner.
To validate our high-throughput screening assay, and to gain insight into the genetic requirements of S. aureus host cell adhesion, we performed a large-scale genetic screen, profiling an arrayed transposon mutant library of methicillin-resistant S. aureus (MRSA) USA300 (21). Specifically, we investigated the genetic requirements of S. aureus adhesion to three different host cell ligands: fibronectin, keratin, and fibrinogen. These ligands are implicated in distinct and clinically relevant scenarios, as detailed below.
Fibronectin
S. aureus can invade, survive, and replicate within professional and non-professional phagocytic cells (22, 23). The interaction of S. aureus with fibronectin underlies the organism's ability to invade non-professional phagocytic host cells, which is mediated by the fibronectin-binding proteins (FnBPs) A and B (24, 25, 26, 27, 28, 29). Host cell invasion facilitates the establishment of chronic and difficult to treat infections (24, 30, 31, 32); the intracellular lifestyle of S. aureus protects the bacterium from antibiotics (33) and the host's immune system (34). Furthermore, endothelial host cell invasion can cause inflammation, endocarditis, and crossover into blood vessels, resulting in systemic infections (35). Interfering with S. aureus fibronectin adhesion could provide significant clinical impact; in this instance, anti-adhesives would likely act as adjuvants (36) potentiating antibiotics in infected individuals and could reduce the occurrence of chronic infections.
Keratin
The ability of S. aureus to colonize the skin and nasal cavity is facilitated by the CWA adhesin clumping factor B (ClfB), which recognizes analogous binding motifs within keratin and cornified envelope proteins such as loricrin, located within the epidermis (1, 37, 38, 39, 40, 41). S. aureus persistently colonizes the nasal cavity of ∼20% of the adult population (42, 43) and the majority (∼80%) of aggressive hospital infections are caused by these colonizing isolates (1, 44, 45, 46). Interfering with ClfB-mediated adhesion to the skin could enable the development of S. aureus decolonizing agents (1).
Fibrinogen
Finally, the interaction of S. aureus with fibrinogen/fibrin is important during the early stages of infection (47). S. aureus adhesion to fibrinogen is associated with the colonization of prosthetic joints and/or indwelling devices (48), cell clumping, and the formation of a protective fibrin/fibrinogen shield; the latter two of which are important for virulence and immune evasion (49, 50). Although S. aureus produces an array of factors interacting with fibrinogen, clumping factor A (ClfA) is the adhesin enabling the abovementioned pathogenic traits (51). Consequently, ClfA has been the focus of S. aureus vaccine efforts (52).
Although the molecular basis and importance of the abovementioned S. aureus host-ligand interactions are well-described, it is likely that numerous cellular factors contribute to adhesion, which are all candidate anti-adhesive targets. Indeed, virulence gene expression is controlled by intricate networks of regulators (53) and the surface stability of adhesins is governed by the controlled production of extracellular proteases, which degrade adhesins and/or host ligands (54, 55, 56, 57, 58). As such, our high-throughput genetic screen identified 20 adhesion defective mutants and we used this information to construct a S. aureus Genetic Adhesion Network. Overall, we identified a core gene set involved in adhesion to all three host ligands and we delineated unique genetic signatures. Although the genetic screen validated our high-throughput whole cell assay, identifying a significant proportion of know adhesion-associated factors, we also identified previously undescribed genetic loci that warrant further investigation. These new loci may represent undescribed drug targets and this assay is well suited to identify new small molecule inhibitors of all the targets identified in this study.
In summary, this whole cell assay will enable strategies to inhibit S. aureus host cell adhesion, providing a means to profile large chemical libraries, and also sheds light on the target space of such an approach. The identification of anti-adhesive therapeutics could provide an important complement to traditional antibacterial chemotherapy, for use as single or combinatorial therapies, potentially reducing the need for antibiotics and/or enhancing antibiotic action (36) during infection.
Results
The development of a whole cell high-throughput assay to detect surface adhered S. aureus
To enable the large-scale profiling of S. aureus adhesion to host ligands, here we describe the development of a whole cell high-throughput assay (Fig. 1A). We selected the predominant community-associated MRSA strain in North America: MRSA USA300 (59, 60), which is designated CMRSA-10 in Canada (59, 61, 62, 63, 64). Due to the clinical importance of this lineage, a genetic resource (the Nebraska Transposon Mutant Library: the NTML) has been developed to enable the study of gene function and regulation using the MRSA USA300 JE2 strain as a background (21). Transposon mutants from the NTML will herein be referred to by their respective gene name followed by ::Tn.
Figure 1.

An ELISA-based approach for the large-scale profiling of Staphylococcus aureus adhesion to host ligands.A, schematic diagram of the high-throughput S. aureus adhesion assay. B, recognition of surface-adhered MRSA USA300 by the primary antibody is not dependent on the presence of cell wall-anchored proteins. A Nunc™ MaxiSorp™ microtiter plate was coated with MRSA USA300 JE2 and isogenic JE2 srtA::Tn (devoid of CWAs) propagated to an OD600nm of 0.6. The cells were harvested, washed, and standardized to the various optical densities shown. Using the described ELISA, detection of surface adhered JE2 srtA::Tn was equivalent to that of the parental strain, indicating that recognition by the primary antibody is independent of CWA surface proteins. The values shown are the average mean ± S.D. of three duplicate biological replicates.
To detect surface-adhered MRSA USA300, we developed an ELISA-based approach, selecting a mouse monoclonal primary antibody with affinity for intact and UV-treated S. aureus, and a horseradish peroxidase-conjugated secondary antibody. To optimize the assay, we first assessed detection of MRSA USA300 JE2 adhered to the surface of MaxiSorp microtiter plates (i.e. nonspecific and high-affinity binding to an abiotic surface) (Fig. 1B). The ELISA successfully detected the surface-adhered bacteria, and we then compared detection of an isogenic srtA::Tn strain (21) adhered to this abiotic surface, which was equally well-recognized (Fig. 1B). This strain is devoid of the majority of CWA proteins (including the immunoglobulin binding protein A), revealing that the primary antibody recognizes factors other than CWA surface proteins and is, therefore, suitable to study CWA adhesin-mediated adhesion.
We next investigated S. aureus adhesion to host ligands, using microtiter plates coated with either human-derived fibronectin, keratin, or fibrinogen. It is well described that the affinity of S. aureus for host ligands changes throughout the cell cycle (37, 65, 66, 67, 68, 69), which needs to be taken into consideration when profiling adhesion to different ligands. Thus, we harvested S. aureus at different stages in the cell cycle, with standardized and equivalent colony-forming units, and assessed adhesion to each host ligand (Fig. 2, A–C). To further validate the assay, we also profiled isogenic mutants devoid of the keratin adhesin ClfB (clfB::Tn (21)), fibronectin adhesins FnBPA/B (ΔfnbA and fnbB (70)), fibrinogen adhesin ClfA (clfA::Tn (21)), as well as srtA::Tn (21), which lacks the majority of CWA proteins (Fig. 2, A–C). As anticipated, S. aureus lacking CWA proteins (srtA::Tn) was severely impaired in adhesion to all three ligands. In line with previous studies, fibronectin adhesion was maximal during the earlier stages of growth (OD600nm 0.2-0.5) (67, 68) and significantly decreased thereafter (Fig. 2A). CWA protein-mediated keratin adhesion (Fig. 2B) was maximal at an OD600nm > 0.6 (37, 65, 66). Although ClfB is also capable of adhering to fibrinogen (71), we confirmed loss of the gene encoding ClfA significantly impairs fibrinogen binding later in the growth cycle (OD600nm >1.0) (65, 69) (Fig. 2C). In addition, ClfA-mediated fibrinogen adhesion was isolated by the inclusion of CaCl2. The presence of Ca2+ is known to interfere with the interaction of both ClfA (72) and ClfB (65, 72) with fibrinogen. However, ClfB is more sensitive (65, 72) (IC50= 0.8 mm, versus 2-3 mm for ClfA) and a ClfA subinhibitory level of Ca2+ (1.7 mm) was selected when assessing fibrinogen adhesion. Finally, to provide a robust and sensitive screening window, we titrated each host ligand and selected a single concentration for subsequent studies (Fig. 2, D and E).
Figure 2.

Optimization of the whole cell adhesion assay. The affinity of S. aureus for host ligands changes throughout the cell cycle; the strains were sampled at the different OD600nm shown and standardized to an OD600nm of 1.0. Adhesion to fibronectin (A), keratin (B), and fibrinogen (C), was assessed using the described ELISA (as shown in Fig. 1A), by measurement of the A450nm. For each host ligand, subsequent adhesion studies were performed with strains grown to the OD600nm range highlighted by the gray box. This range was selected based on the OD600nm where MRSA USA300 JE2 adhesion was maximal and presented an optimum ratio of separation between MRSA USA300 JE2 and the isogenic mutants of each respective ligand. To optimize the screening window, dose-dependent ELISAs were performed with MRSA USA300 JE2 and isogenic JE2 srtA::Tn adhering to human-derived fibronectin (D), keratin (E), and fibrinogen (F). The strains were grown to the specified OD600nm highlighted by the gray box and standardized to an OD600nm of 1.0. The dashed lines indicate the chosen concentration of each ligand for the subsequent transposon library adhesion screens. The values shown are the mean of three technical replicates.
Validation of the high-throughput whole cell S. aureus adhesion assay
To validate the use of our assay for the large-scale profiling of S. aureus adhesion, we took a functional genomics approach to identify genetic loci contributing to adhesion to fibronectin, keratin, or fibrinogen. We used an MRSA USA300 transposon library comprised of 1,920 annotated mutants generated by the insertion of the mariner-based bursa aurealis transposon into genes that are not considered to be essential for growth under standard laboratory conditions (21). In total, this library provides coverage of ≥75% of the predicted MRSA USA300 genes (73). For profiling of the individual mutants, the library was arrayed into a 96-well–microtiter plate glycerol stock. A cryo-replicator was used to inoculate the library into tryptic soy broth (TSB) in 96-well–round bottom microtiter plates. After 18 h at 37 °C, these cultures were used to inoculate fresh TSB, and the strains were grown at 37 °C until the OD600nm range shown in Fig. 2A was reached, which depends on the host ligand being profiled. The growth of each mutant was measured, revealing reproducible growth (Fig. 3), and enabled the identification of strains with growth defects. The bacterial cells were then applied to microtiter plates coated with the individual host ligands and the ELISA was performed, which reproducibly detected adhesion (Fig. 3). To circumvent the limitations of plate-to-plate variation, the growth and adhesion values were normalized by the interquartile mean (IQM) (74). The IQM represents the mean of the inner two quartiles of ranked data, thereby preventing any influence from outliers (74). We then determined the ratio between adhesion and growth for each mutant (Fig. 4). A hit was defined as a mutant that provided a ratio less than 4 standard deviations for keratin and fibronectin, and less than 6 standard deviations for fibrinogen, from the IQM of the data set. Because the mutants were sampled at a higher OD600nm in the fibrinogen screen, the variance was lower and a higher cut-off value was used.
Figure 3.
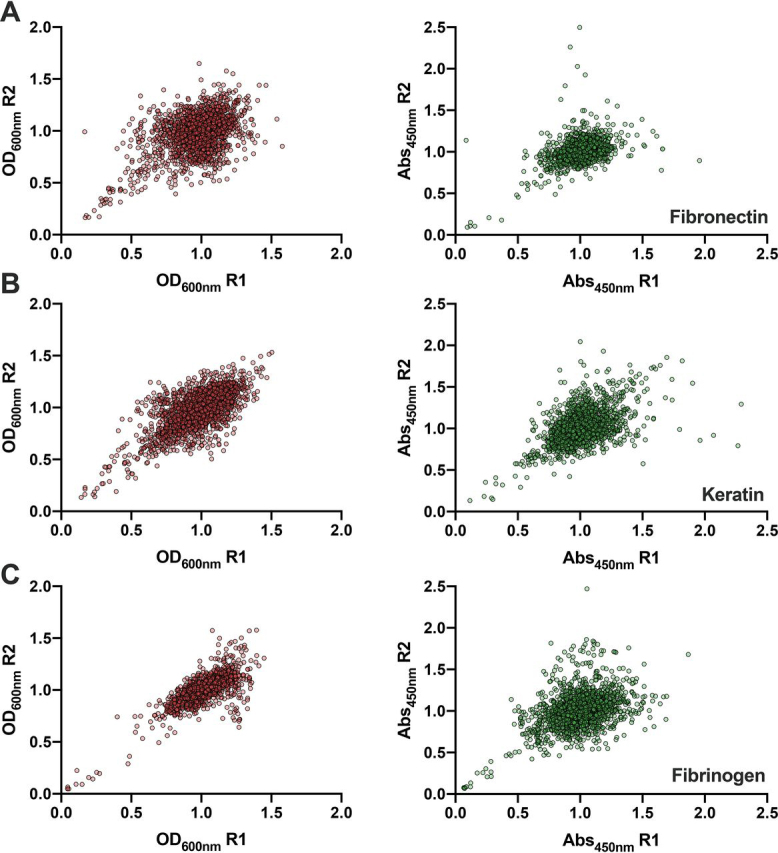
Replica plots of normalized growth and adhesion values from the large-scale profiling of an MRSA USA300 transposon library (21). Growth (OD600nm) is shown on the left (red) and adhesion (A450nm) values are shown on the right (green). A, fibronectin; B, keratin; C, fibrinogen. R1 = replicate 1 and R2 = replicate 2.
Figure 4.
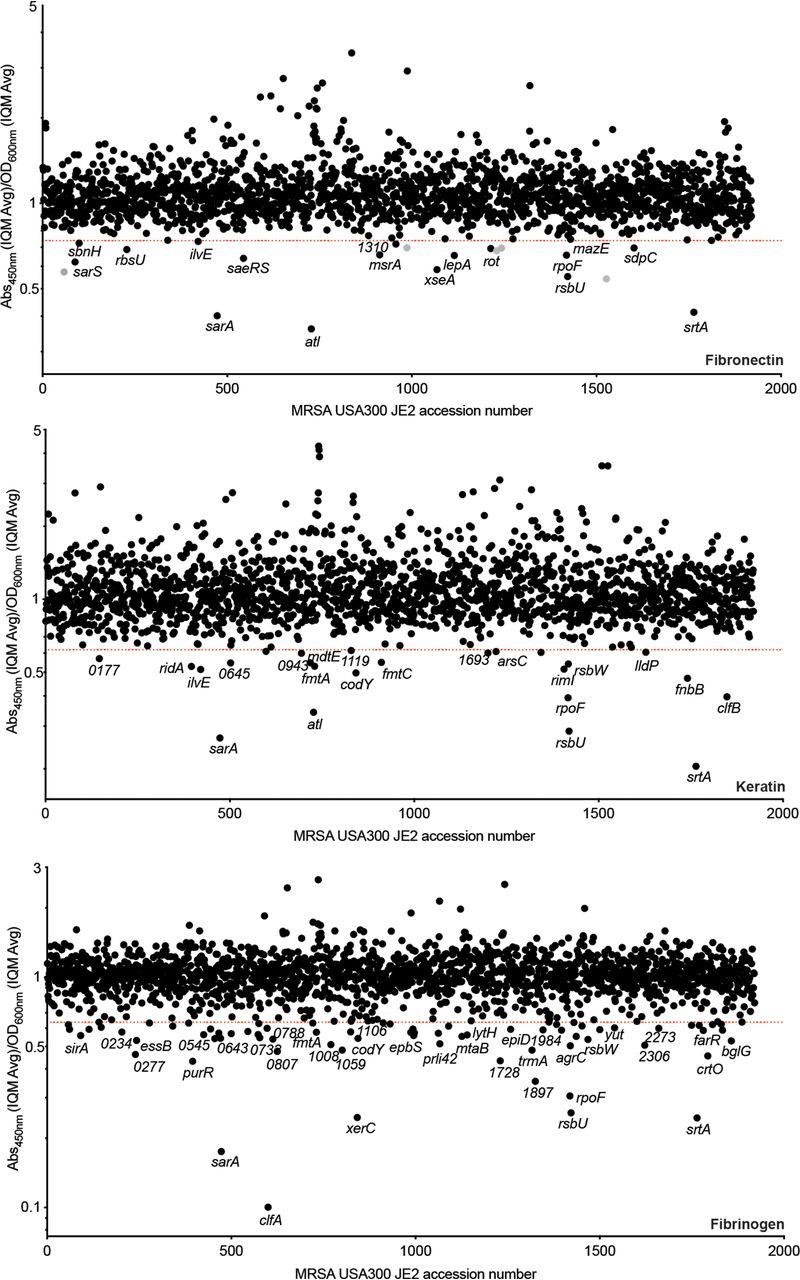
Identifying genetic loci associated with S. aureus adhesion to human-derived fibronectin, keratin, and fibrinogen, respectively. The values shown in the index plots are the ratio between the average of duplicate adhesion values (A450nm) and duplicate growth values (OD600nm) (see Fig. 3). The strains are ordered based on their associated SAUSA300 accession number (21). The screens were performed with ligand concentrations and strains sampled at the given time points depicted in Fig. 2. Strains falling below the red lines exhibited a ratio less than 4 standard deviations (fibronectin and keratin) and 6 standard deviations (fibrinogen) from the IQM (75) of the data set.
As anticipated, the assay was validated by the identification of known adhesion-related determinants, such as the gene encoding SrtA, which was identified in all three screens. We also identified disruptions within genes encoding key host ligand adhesins: clfB in the keratin screen and clfA in the fibrinogen screen (Fig. 4). Because there are two fibronectin binding proteins (FnBPA/B), disruption of either gene alone is not sufficient to impair fibronectin adhesion (Fig. 4).
Construction of a S. aureus genetic adhesion network highlights the anti-adhesive target space
All adhesion attenuated mutants identified in the primary adhesion screens (Fig. 4) were compiled into a sublibrary and profiled for adhesion to all three host ligands, as described above. For mutants that showed confirmed adhesion-related phenotypes, the transposons were transduced using phage 80α into a fresh JE2 background, to eliminate the possibility of secondary-site mutations. Transposon insertions were confirmed by PCR and the newly generated mutants were then profiled to assess dose-dependent adhesion to each host ligand (Fig. S1). A single concentration of each ligand was selected to compare the relative adhesion of each mutant to the parental strain (Table 1). This information was used to create a visual representation of the contribution of each gene to adhesion (Fig. 5). By comparing adhesion of each strain to each of the three polymers we observed similarities and differences between the three screens. In total, we identified 20 genetic loci associated with adhesion to at least one of the three host ligands; 12 for fibronectin, 9 for keratin, and 13 for fibrinogen. We also identified a core gene set required for adhesion to all three host ligands and we identified undescribed genetic loci. Importantly, these genes represent potential targets that could be identified in future screens of chemical libraries. To further strengthen our findings, we profiled poorly characterized adhesion-defective mutants (sarS, rpiR, fmtC, fmtA, recD2, hypo, and ydiL) using crystal violet as an alternative detection method (Fig. S2). By employing this method, with the exception of sarS in the fibronectin screen, we confirmed that all of the mutants displayed reduced adhesion, which is not a reflection of reduced affinity of the detecting antibodies.
Table 1.
The S. aureus Genetic Adhesion Network Genes associated with S. aureus adhesion to a minimum of one of three host ligands (keratin, fibronectin, and fibrinogen)
Adhesion was assessed by titrating each host ligand (Fig. S1). For creation of the Genetic Adhesion Network, a final concentration was selected for each ligand, as indicated. The value listed for each mutant strain is the average percentage of adhesion relative to wild-type JE2 (three biological replicates), with lower values indicating a greater reduction in adhesion to a specific ligand. Genes mazE (SAUSA300_2026), recD2 (SAUSA300_1576), rpiR (SAUSA300_2264), and ydiL (SAUSA300_1984) are provisional names provided on the basis of their predicted structure and function. One gene, labeled hypo, is unannotated (SAUSA300_0602). p values were calculated using the two-tailed unpaired Student's t test; statistically significant (*, p ≤ 0.05) decreases in adhesion, compared with wild-type JE2, were used to create the Genetic Adhesion Network (Fig. 5).
| Gene | Locus | Keratin (0.125 μg/well) % adhesion | Keratin p value | Fibronectin (0.125 μg/well) % adhesion | Fibronectin p value | Fibrinogen (0.25 μg/well) % adhesion | Fibrinogen p value |
|---|---|---|---|---|---|---|---|
| agrC | SAUSA300_1991 | 106.8 | 0.57 | 102.4 | 0.75 | 77.0 | 0.040* |
| arlR | SAUSA300_1308 | 107.4 | 0.33 | 111.5 | 0.25 | 57.9 | 0.00096* |
| atl | SAUSA300_0955 | 51.6 | 0.026* | 23.7 | 0.0015* | 50.7 | 0.0044* |
| clfA | SAUSA300_0772 | 104.0 | 0.72 | 130.1 | 0.20 | 44.4 | 0.00050* |
| clfB | SAUSA300_2565 | 42.2 | 0.0055* | 81.4 | 0.45 | 101.9 | 0.26 |
| codY | SAUSA300_1148 | 68.3 | 0.038* | 49.4 | 0.012* | 97.2 | 0.80 |
| fmtA | SAUSA300_0959 | 60.5 | 0.028* | 73.2 | 0.12 | 76.7 | 0.049* |
| fmtC | SAUSA300_1255 | 96.1 | 0.44 | 82.7 | 0.13 | 54.5 | 0.00093* |
| hypo | SAUSA300_0602 | 100.4 | 0.95 | 100.4 | 0.97 | 65.1 | 0.026* |
| mazE | SAUSA300_2026 | 98.9 | 0.92 | 68.9 | 0.0058* | 96.3 | 0.75 |
| recD2 | SAUSA300_1576 | 41.8 | 0.00029* | 38.4 | 0.0019* | 20.6 | 0.000082* |
| rpiR | SAUSA300_2264 | 116.6 | 0.050 | 67.1 | 0.0056* | 136.9 | 0.020 |
| rpoF | SAUSA300_2022 | 18.6 | 0.0013* | 25.7 | 0.0012* | 35.2 | 0.0018* |
| rsbU | SAUSA300_2025 | 16.5 | 0.0012* | 17.0 | 0.00082* | 35.3 | 0.0016* |
| saeR | SAUSA300_0691 | 108.1 | 0.43 | 79.6 | 0.030* | 121.7 | 0.056 |
| saeS | SAUSA300_0690 | 104.9 | 0.44 | 86.0 | 0.037* | 117.5 | 0.016 |
| sarA | SAUSA300_0605 | 26.0 | 0.00016* | 29.6 | 0.000051* | 17.7 | 0.000085* |
| sarS | SAUSA300_0114 | 104.3 | 0.55 | 70.4 | 0.014* | 116.1 | 0.14 |
| srtA | SAUSA300_2467 | 20.9 | 0.0015* | 21.4 | 0.00083* | 34.4 | 0.0016* |
| ydiL | SAUSA300_1984 | 102.8 | 0.86 | 115.7 | 0.068 | 69.0 | 0.015* |
Figure 5.
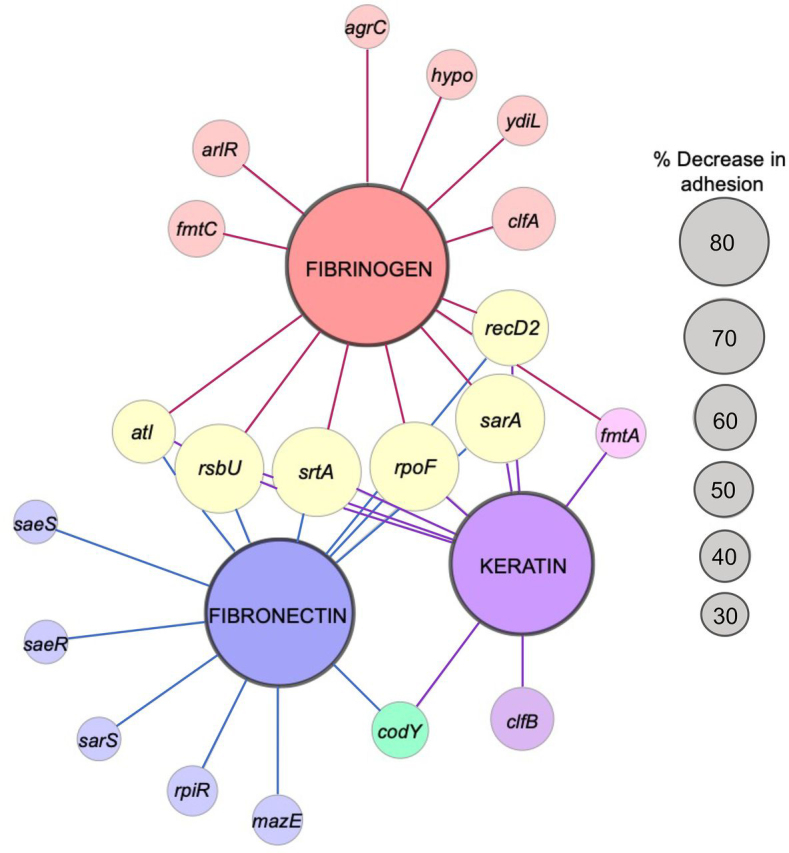
The S. aureus Genetic Adhesion Network and anti-adhesive target space. The larger central nodes identify the host ligand in each screen (fibrinogen, fibronectin, and keratin). The smaller nodes radiating from these points indicate transposons inserted into genes that caused a significant reduction (Student's t test, p value ≤ 0.05; see Table 1 and Fig. S1) in adhesion to the host ligand they are linked. The node size for each gene reflects the percent adhesion compared with the parental strain (i.e. the larger the node the more pronounced the attenuated adhesion). The figure was generated using Cytoscape. Genes mazE (SAUSA300_2026), recD2 (SAUSA300_1576), rpiR (SAUSA300_2264), and ydiL (SAUSA300_1984) are names provided on the basis of their predicted structure and function. One gene, labeled hypo (hypothetical protein), is unannotated (SAUSA300_0602).
Assessing the proteolytic activity of adhesion attenuated mutants
We identified a number of genes associated with virulence gene regulation, and three were identified in all three screens: rpoF (the alternative sigma factor B; SigB; σB) (75), rsbU (75) (increases SigB activity; rsbW (75) was also identified, which is a polar mutation), and the gene encoding the global staphylococcal accessory regulator (sarA) (76). These genes are known to impact the expression of a wide variety of virulence-related genes, including the genes encoding adhesins (77, 78). In addition to positively regulating adhesin gene expression (76, 79), a number of the identified regulators (e.g. SarA and CodY) also repress extracellular protease production (54). Therefore, disruptions within these repressors would enhance the mutant's proteolytic capabilities. Some of these proteases are known to impact the stability of surface proteins (55, 56) and/or host ligands, enabling the bacterium to detach and disseminate (54, 57, 66). The NTML has previously been profiled to identify mutants with protease activity, which highlighted 12 mutants (brnQ, ilvE, sarA, xerC, hslU, codY, rpoF, rsbW, rsbV, rsbU, NE1833) with increased production, many of which had previously been associated with this phenomenon (21). As shown in Fig. 5, a number of these mutants also exhibit reduced adhesion. We assessed the level of proteolytic activity of the newly generated mutants on casein plates (Fig. 6), by measuring the zone of clearance, which enabled us to compare the extent of protease production for each mutant. Overall, we identified varying degrees of proteolysis, with five mutants exhibiting enhanced protease production and six with decreased levels (Fig. 6).
Figure 6.

Assessing the level of proteolytic activity in the adhesion attenuated mutants. Protease activity was assessed using TSA containing 1.5% Difco Skim Milk. The zones of clearance were measured using ImageJ. Each strain was measured with at least three biological replicates. Welch's t test (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001) was used to compare the mean length for each mutant to the mean of the parental strain (red dashed line).
Discussion
The antibiotic resistance crisis signifies an urgent need for orthogonal and complementary therapeutics to control and treat bacterial infections. Antivirulence approaches (80), including the inhibition of bacterial host cell adhesion (1), could reduce the severity of this global health crisis. However, the development of such strategies requires sensitive and robust assays amenable to high-throughput applications, and an in-depth understanding of the underlying processes. Here, we present an ELISA-based approach (Fig. 1) for the high-throughput profiling of S. aureus adhesion to host ligands. We performed a genetic screen to confirm the assay was able to detect genetic loci previously associated with adhesion, and to ascertain the MRSA USA300 anti-adhesive target space. Overall, the NTML enabled us to assess the contribution of ≥75% of predicted MRSA USA300 genes (73). During creation of the NTML, the authors acknowledged 579 open reading frames were not interrupted, a significant fraction of which are likely indispensable for growth (21). Therefore, profiling the NTML enabled assessment of a large proportion of the dispensable genes in MRSA USA300.
In this work we identified 20 genetic loci (1% of the NTML) associated with adhesion to at least one of the three clinically important host ligands. As anticipated, many of the identified genes have previously been implicated in adhesion. For example, in addition to identifying the srtA mutant, which is devoid of CWAs, the clfB mutant was identified in the keratin screen, and clfA in the fibrinogen screen. In addition, we also identified the gene encoding the major peptidoglycan hydrolase, autolysin (Atl), which reduced adhesion to all three host ligands (Fig. 5 and Fig. S1). Atl, and the functionally interchangeable enzyme in Staphylococcus epidermidis (AtlE) (81), have been described as staphylococcal adhesins (82). These proteins have been associated with adhesion to polystyrene (82) and host proteins including fibronectin (83, 84), fibrinogen (84), vitronectin (84, 85), heparin (83), gelatin (83), heat shock cognate 71-kDa protein (Hsc70) (86), and thrombospondin 1 (86). Here, we show that keratin is a host cell ligand also affected by loss of Atl.
Generation of a Genetic Adhesion Network identified a core subset of genes (n = 6) required for adhesion to all three ligands (Fig. 5 and Table 1), which included expected genes such as srtA (11), sarA (87, 88, 89), rpoF and rpoF-related genes (rsbU) (77, 78), and atl (83, 84). Inhibitors of these elements could represent anti-adhesives capable of simultaneously interfering with the interaction of S. aureus with fibronectin, keratin, and fibrinogen.
To the best of our knowledge, the adhesion-associated roles of sarS, rpiR, fmtC, fmtA, recD2, hypo, and ydiL (Fig. 5, Fig. S2, Table 1) remain unknown. Of note, we were unable to confirm the adhesion-associated defect of sarS using an alternative detection method (Fig. S2). In addition, genomic assessment and operon predictions (90) showed that some of these genes are located within an operon (e.g. recD2), and thus we cannot rule out polar effects due to transposon insertion. Complementation studies are required to further confirm the adhesion phenotypes.
Based on our current knowledge, we summarize the findings of this study by broadly categorizing each identified genetic locus (Fig. 5) into four likely and/or known groups: 1) adhesin gene expression (saeRS, codY, rsbU, rpoF, sarA); 2) adhesin surface presentation (srtA, arlRS); 3) extracellular protease production (rpoF, rsbU, codY, saeRS, sarA); and 4) unknown/unconfirmed roles (sarS, rpiR, mazE (rpoF), fmtC, fmtA, recD2, hypo, ydiL). In terms of adhesin expression (Group 1), we identified a number of genes associated with virulence gene regulation: rpoF (75, 77, 78), rsbU (75, 77, 78), and sarA (76, 87, 88, 89). These genes are known to impact the expression of genes encoding adhesins (77, 78). Furthermore, deletion of codY reduced the affinity of S. aureus for keratin and fibronectin (Fig. 5) and CodY has been shown to positively influence the expression of the fibronectin adhesin fnbA (79). In regard to adhesin surface presentation (Group 2), SrtA covalently attaches adhesins to the peptidoglycan (9, 10), and disruption of the ArlRS two-component system reduces fibrinogen adhesion due to increased production of the Giant Staphylococcal Surface Protein (encoded by the ebh gene), preventing fibrinogen recognition by ClfA (91). As described, in addition to positively regulating adhesin gene expression (76, 79), a number of the identified regulators (e.g. SarA and CodY) also repress extracellular protease production (Group 3) (54, 55, 56, 57, 66), which can degrade surface proteins and/or host ligands. Indeed, increased protease production (e.g. due to loss of SarA repression (55)) attenuates virulence, and complete loss of protease activity causes a hypervirulent phenotype, which could be due to the increased stability of surface-associate virulence factors (57, 58). Because we identified a number of genes that have previously been associated with protease production, we assessed the proteolytic activity of the newly created mutants (Fig. 6) (54). Overall, we identified a significant enrichment of mutants with varying degrees of protease production. Importantly, in addition to degrading adhesins, extracellular proteases can also degrade host ligands. This study sampled both possibilities simultaneously, because we applied the bacterial cells and the spent growth medium, containing secreted proteases, to immobilized ligands. Proteolytic assessment could be used to provide insight into the targets of candidate anti-adhesives identified using the described whole cell assay; for example, if the inhibitors enhance proteolysis, they could be targeting the genetic loci identified in Fig. 6. Finally, whereas the role of MazE (Group 4) in S. aureus adhesion is currently unknown, the mazE gene resides in the mazEF/pemIK toxin-antitoxin loci, which directly precedes the σB-operon. Disruption of this gene could impact the expression of rpoF.
In summary, here we describe the development and validation of a sensitive and robust whole cell assay to identify anti-adhesive therapeutics. We also provide the first comprehensive functional genomic approach to ascertain the factors directly contributing to S. aureus host cell adhesion. Delineating the genetic requirements of S. aureus adhesion has provided a more in-depth understanding of the interaction of this organism with the host, and sheds light on the MRSA USA300 anti-adhesive target space. The development of anti-adhesive therapeutics would complement our antibiotic arsenal, offering the potential to reduce the severity of the antibiotic resistance crisis using a more targeted approach. As shown in Table 1 and Fig. 5, this assay is capable of detecting mutants with varying degrees of adhesion, which suggests it will be well suited for chemical library screening, enabling the identification of anti-adhesives with differing degrees of potencies.
Materials and methods
Bacterial strains, plasmids, and growth conditions
The NTML (NR-48501) and MRSA USA300 JE2 (21) were obtained from the Network on Antimicrobial Resistance in S. aureus (NARSA) repository through BEI resources, NIAID, National Institutes of Health (RRID:SCR_013698). The NTML was constructed in the MRSA USA300 JE2 strain, which was used as a background for all purposes of this study. Regeneration of the adhesion-attenuated mutants of interest was accomplished by transducing the transposons into the parental JE2 strain using phage 80α transduction. Successful transductants were selected on erythromycin (5-10 μg/ml) and confirmed by PCR using gene- and transposon-specific primers (Table 2). Unless otherwise stated, S. aureus strains were propagated in BD BactoTM Tryptic Soy Broth. For profiling of the NTML, the strain library was arrayed in a 96-well–microtiter plate glycerol (25% v/v) stock format at −80 °C. A cryo-replicator and replicator press (EnzyScreen) were used to inoculate 100 μl of TSB in 96-well–round bottom microtiter plates. All strains were grown at 37 °C with aeration at 220 rpm (25 mm throw), with the exception of microtiter plates that were aerated with 900 rpm using an incubator with a 3-mm throw (INFORS HT Multitron).
Table 2.
Primers used in this study
| Primers | Sequence (5′-3′) | Transposon primer used | Source |
|---|---|---|---|
| Confirmation of bursa aurealis transposon insertion site | |||
| Upstream | CTCGATTCTATTAACAAGGG | Bae et al. (2008) | |
| Buster | GCTTTTTCTAAATGTTTTTTAAGTAAATCAAGTAC | Bae et al. (2008) | |
| New_Upstream | CTTCAAACTTGACTTCAGC | This study | |
| New_Buster | CCAGTCTGGATCCAGTTG | This study | |
| accC_Tn_check | TTCGTTGTTTAATTGCG | Buster | This study |
| arlR_Tn_check | TTGATTACGGTGCAGA | Upstream | This study |
| atl_Tn_check | GTTGCATTAACGCTTGTAG | Upstream | This study |
| clfA_Tn_check | AACACGCAATTCGGAA | Buster | This study |
| clfB_Tn_check | TTGAAAAAAAGAATTGATTATTTGTC | Buster | This study |
| codY_Tn_check | ACGAGAGAGTTAAACACG | Upstream | This study |
| fmtA_Tn_check | GGTTGCGCCGTCTAAAC | fmtA_r versus_check | This study |
| fmtA_rversus_check | CACCCTTCGTATTGTAAGG | fmtA_Tn_check | This study |
| fmtC_Tn_check | GGCATCGCTTGTTATTC | Upstream | This study |
| fnbA_Tn_check | CTTAGGTACGGCATTAG | Buster | This study |
| fnbB_Tn_check | GCAATCTTAGATACGGC | Buster | This study |
| GNAT4_Tn_check | GAGACTTGTTTCGACAG | Upstream | This study |
| graR_Tn_check | TGGGTGATATGGATGC | Upstream | This study |
| hypo_rversus_check | GCAAGTGGCAACTCTATTG | New_Buster | This study |
| ilvE_new_check | GCAATCACCATGTCACAAG | ilvE_rversus_check | This study |
| ilvE_rversus_check | GTACAACGACTCTCCAAC | ilvE_new_check | This study |
| mazE_Tn_check | TGATTAGACGAGGAGATG | Upstream | This study |
| PutLipo_Tn_check | GACACTGGGATGTTTAC | Upstream | This study |
| RecD2_Tn_check | TGTCAGACCCTACACT | Upstream | This study |
| rot_Tn_check | GCATAAGTTAGCACATACAA | Buster | This study |
| rpiR_Tn_check | CGCGTTAAACAACAATAGC | New_Upstream | This study |
| rpoF_Tn_check | GCGAAAGAGTCGAAATC | Buster | This study |
| rsbU_Tn_check | GCCTGAAGACATTGTCG | Upstream | This study |
| rsbW_rversus_check | TTATCGAAATGCGCG | Buster | This study |
| sarA_Tn_check | GAGTTGTTATCAATGGTCAC | New_Upstream | This study |
| sarS_Tn_check | AGTGCATATACAAGGAGA | NaT_fwd_check | This study |
| sdcS_NaT_Tn_check | GTGCGGGACAACTTATTG | NaT_fwd_check | This study |
| NaT_fwd_check | GCACTGATTAAGTTTACCC | SdcS_NaT_Tn_check | This study |
| spa_rversus_check | CTAGGTGTAGGTATTGCATC | New_Buster | This study |
| srtA_Tn_check | TTATTTGACTTCTGTAGCTACAAAGATTTTACG | Upstream | This study |
| srtB_Tn_check | TTAACTTACCTTAATTATTTTTGCGAC | Upstream | This study |
| vraG_Tn_check | TGGCGTTAATTATGACC | Upstream | This study |
| xerC_Tn_check | GAATCATATTCAAGATGCGT | Upstream | This study |
| yabJ_rversus_check | CACAACAAGATTACCGG | Upstream | This study |
| YdiL_Tn_check | GGGCATCATTGCTAACTG | New_Buster | This study |
| YfeH_Tn_check | ATGTTGCGACATTAGG | Upstream | This study |
Antibodies and reagents
Surface adhered S. aureus was detected using a mouse anti-S. aureus monoclonal primary antibody raised against UV-inactivated S. aureus (Invitrogen) and a secondary anti-mouse goat IgM, HRP-conjugated antibody (Invitrogen). HRP colorimetric substrate (1-stepTM Ultra 3,3‘,5,5‘-tetramethylbenzidine) and NuncTM Immunosorbent (MaxisorpTM) 96-well–microtiter plates were purchased from Thermo Scientific. MaxisorpTM plates were coated with keratin derived from human epidermis and fibronectin and fibrinogen from human plasma (Millipore Sigma). BSA (BSA, HyCloneTM) was solubilized in PBS.
The ELISA-based S. aureus adhesion assay
For coating MaxisorpTM plates with host ligands, 100 μl of keratin (diluted in carbonate buffer: 15 mm Na2HCO3, 35 mm NaHCO3 (pH 9.6)), fibrinogen, or fibronectin (diluted in PBS (pH 7.4)), were applied and incubated at 4 °C for 18 h. The plates were blocked with 300 μl of 2% BSA (w/v) for 1.5 h at room temperature. The microtiter plate lids were removed, and the bacterial cultures were irradiated under UV light for 20 min and stored at 4 °C. 100 μl of each culture was applied to the host ligand-coated microtiter plate wells, the plates were sealed, agitated for 5 min (900 rpm), and incubated for 1 h with gentle shaking (100 rpm). 100 μl of the primary antibody (2 μg/ml in 1% BSA) was applied to each well and the plates were agitated for 5 min (600 rpm), followed by 1 h incubation with gentle shaking (100 rpm). 100 μl of the HRP-conjugated secondary antibody (125 ng/ml in 1% BSA) was applied as described for the primary antibody. Adhesion was assessed using 100 μl of room temperature equilibrated colorimetric substrate (3,3‘,5,5‘-tetramethylbenzidine), which was agitated for 1 min (600 rpm), followed by gentle shaking (100 rpm) for 20 min. The reaction was quenched by oxidation with 100 μl of 2 m H2SO4. Absorbance was measured at 450 nm using a BioTek Synergy H1 microplate reader. Between each described step, the plates were incubated at room temperature and washed four times with 300 μl of PBS (pH 7.4), with the exception of host ligand aspiration, where the plates were only washed once with 300 μl of PBS. When assessing fibrinogen adhesion, the PBS was supplemented with 1.7 mm CaCl2.
To optimize the described assay, the strains were sampled at different stages of the growth cycle. Overnight saturated cultures were diluted 1:100 in TSB and the bacterial cells were harvested via centrifugation (4000 × g, 4 °C) at intervals of 0.2 from an OD600nm of 0.2 to 1.2. The cells were washed with PBS and standardized to an OD600nm of 1.0. Simultaneously, the polymer and antibodies concentrations were varied until an optimal signal to noise ratio was achieved, by assessment of the Z-factor (92). The parental MRSA USA300 JE2 strain and an isogenic srtA::Tn mutant strain were used as positive and negative controls, respectively.
MRSA USA300 primary antibody recognition was determined by coating a Nunc™ MaxiSorp™ microtiter plate with MRSA USA300 JE2 and isogenic JE2 srtA:Tn. Overnight saturated cultures were diluted 1:100 in TSB and the strains were propagated to an OD600nm of 0.6. The cells were harvested via centrifugation (4000 × g, 4 °C), washed with PBS, and standardized in PBS to an OD600nm of 0.5, 0.25, 0.125, 0.0625, and 0.0313. The standardized cultures were applied to an uncoated Nunc™ MaxiSorp™ microtiter plate and incubated for 1 h with gentle agitation (100 rpm). Following incubation, the plates were blocked, and the ELISA was performed as described.
Crystal violet adhesion assay
Nunc™ MaxiSorp™ microtiter plates were coated with ligands, blocked, and washed as described. Overnight cultures were diluted 1:100 in 10 ml of TSB in 50-ml tubes. To assess fibrinogen and keratin adhesion, 100 μl of each culture, propagated to the respective OD600nm, was applied to ligand-coated microtiter plate wells, the plates were sealed, agitated for 5 min (900 rpm), and incubated for 1 h with gentle shaking (100 rpm). The bacterial cultures were irradiated under UV light for 20 min. For fibronectin adhesion, due to the lower OD600nm, 200 μl of each culture was applied to fibronectin-coated microtiter plate wells and the plates were treated as described, this step was repeated twice. Adhesion was assessed by staining adherent cells with 100 μl of filtered 0.5% (w/v) crystal violet for 2 min, washing three times in 300 μl of PBS (pH 7.4), and solubilizing in 0.7% (v/v) acetic acid. Absorbance was measured at 595 nm using a BioTek Synergy H1 microplate reader.
Profiling the NTML to identify adhesion attenuated mutants
The NTML was profiled using a cryo-replicator (EnzyScreen) to inoculate 100 μl of TSB (5 μg/ml of erythromycin), followed by incubation at 37 °C for 18 h with aeration (900 rpm). These saturated cultures were then used to inoculate 100 μl of TSB and the strains were grown to the appropriate OD600nm for each host ligand (Fig. 2). The adhesion assay was performed as described above. Data were ranked and normalization was performed by using the IQM method (74). Ranked data were divided into four quartiles where the mean of the inner two quartiles represents the IQM (74). Normalization per plate was achieved by comparing each strain to the IQM (74).
To confirm the adhesion attenuated phenotypes, the newly generated transposon mutants were propagated in microtiter plates as described. MaxiSorp™ plates were coated with increasing concentrations of fibronectin, keratin, or fibrinogen, starting at a high dose of 1 μg/well (10 μg/ml). The ELISA was performed as described above, with a minor modification. Following UV irradiation, 50 μl of each culture and 50 μl of PBS were applied to the host ligand-coated microtiter plate wells. MRSA USA300 JE2 was included in each plate as a control. Each strain was assessed using a minimum of three biological replicates. The resulting data were used to generate the genetic adhesion network, which was created using Cytoscape (93). GraphPad Prism 8 was used for statistical analysis.
Assessing mutant proteolytic activity
Proteolytic activity was assessed on TSA containing 1.5% Difco Skim Milk. Cultures were inoculated into 100 μl of TSB and propagated overnight at 37 °C with aeration (900 rpm). Each strain was point inoculated onto the skim milk agar, from the overnight culture, using a cryo-replicator. Plates were incubated at 37 °C for 21 h before being imaged. Zones of clearing were assessed using ImageJ (94), by measuring from the edge of the spotted colony to the edge of the zone of clearance. Each strain was measured with at least three biological replicates. GraphPad Prism 8 was used for statistical analysis. Welch's t test was used to compare the mean length for each mutant to the mean of the parental strain (JE2).
Data availability
The authors confirm that the data supporting the findings of this study are available within this article (and its supporting material). The raw data from the primary genome-wide adhesion screens are available upon request. Please contact the corresponding author (gcox@uoguelph.ca).
Acknowledgments
We thank Maritza Vatta for technical assistance with aspects of the adhesion assay. We also thank Dr. David Heinrichs and Dr. Mariya Goncheva for assistance and advice with the transduction.
Gouvernement du Canada (NFRFE-2018-01058) to Georgina Cox
J. P. Bickell Foundation (Medical Research Grant) to Georgina Cox
Canada Foundation for Innovation (CFI) (JELF 37730) to Georgina Cox
Edited by Joseph M. Jez
Footnotes
This article contains supporting information.
Author contributions—L. E. P., A. C. L., and G. C. data curation; L. E. P., A. C. L., and G. C. formal analysis; L. E. P., A. C. L., and J. M. validation; L. E. P., A. C. L., and J. M. investigation; L. E. P. and A. C. L. visualization; L. E. P. and G. C. methodology; A. C. L. and G. C. writing-review and editing; G. C. conceptualization; G. C. resources; G. C. supervision; G. C. funding acquisition; G. C. writing-original draft; G. C. project administration.
Funding and additional information—This work was supported by New Frontiers in Research Fund-Exploration Grant NFRFE-2018-01058, a Medical Research Grant from the J. P. Bickell Foundation, and the Canada Foundation for Innovation Grant JELF 37730 (to G. C.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- CWA
- cell wall-anchored
- SrtA
- sortase A
- MRSA
- methicillin-resistant S. aureus
- FnBP
- fibronectin-binding protein
- ClfB
- clumping factor B
- ClfA
- clumping factor A
- TSB
- tryptic soy broth
- IQM
- interquartile mean
- Atl
- autolysin
- HRP
- horseradish peroxidase.
Supplementary Material
References
- 1.Leonard A.C., Petrie L.E., Cox G. Bacterial anti-adhesives: inhibition of Staphylococcus aureus nasal colonization. ACS Infect. Dis. 2019;5:1668–1681. doi: 10.1021/acsinfecdis.9b00193. 31374164. [DOI] [PubMed] [Google Scholar]
- 2.Johnny W.P. Bacterial pathogenesis. In: Barron S., editor. Medical Microbiology. 4 Ed. 1996. Galveston, TX. [Google Scholar]
- 3.Ofek I., Hasty D.L., Sharon N. Anti-adhesion therapy of bacterial diseases: prospects and problems. FEMS Immunol. Med. Microbiol. 2003;38:181–191. doi: 10.1016/S0928-8244(03)00228-1. 14522453. [DOI] [PubMed] [Google Scholar]
- 4.Pizarro-Cerdá J., Cossart P. Bacterial adhesion and entry into host cells. Cell. 2006;124:715–727. doi: 10.1016/j.cell.2006.02.012. 16497583. [DOI] [PubMed] [Google Scholar]
- 5.Sattin S., Bernardi A. Glycoconjugates and glycomimetics as microbial anti-adhesives. Trends Biotechnol. 2016;34:483–495. doi: 10.1016/j.tibtech.2016.01.004. 26875976. [DOI] [PubMed] [Google Scholar]
- 6.Spaulding C.N., Klein R.D., Schreiber H.L.T., Janetka J.W., Hultgren S.J. Precision antimicrobial therapeutics: the path of least resistance? NPJ Biofilms Microbiomes. 2018;4:4. doi: 10.1038/s41522-018-0048-3. 29507749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crossley K.B. Wiley-Blackwell; Chichester, UK: 2010. Staphylococci in human disease. [Google Scholar]
- 8.Foster T.J., Geoghegan J.A., Ganesh V.K., Höök M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014;12:49–62. doi: 10.1038/nrmicro3161. 24336184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazmanian S.K., Ton-That H., Schneewind O. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 2001;40:1049–1057. doi: 10.1046/j.1365-2958.2001.02411.x. 11401711. [DOI] [PubMed] [Google Scholar]
- 10.Ilangovan U., Ton-That H., Iwahara J., Schneewind O., Clubb R.T. Structure of sortase, the transpeptidase that anchors proteins to the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 2001;98:6056–6061. doi: 10.1073/pnas.101064198. 11371637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazmanian S.K., Liu G., Jensen E.R., Lenoy E., Schneewind O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5510–5515. doi: 10.1073/pnas.080520697. 10805806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jonsson I.M., Mazmanian S.K., Schneewind O., Verdrengh M., Bremell T., Tarkowski A. On the role of Staphylococcus aureus sortase and sortase-catalyzed surface protein anchoring in murine septic arthritis. J. Infect. Dis. 2002;185:1417–1424. doi: 10.1086/340503. 11992276. [DOI] [PubMed] [Google Scholar]
- 13.Weiss W.J., Lenoy E., Murphy T., Tardio L., Burgio P., Projan S.J., Schneewind O., Alksne L. Effect of srtA srtB gene expression on the virulence of Staphylococcus aureus in animal models of infection. J. Antimicrob. Chemother. 2004;53:480–486. doi: 10.1093/jac/dkh078. 14762051. [DOI] [PubMed] [Google Scholar]
- 14.Matano L.M., Morris H.G., Wood B.M., Meredith T.C., Walker S. Accelerating the discovery of antibacterial compounds using pathway-directed whole cell screening. Bioorg. Med. Chem. 2016;24:6307–6314. doi: 10.1016/j.bmc.2016.08.003. 27594549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Payne D.J., Gwynn M.N., Holmes D.J., Pompliano D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug. Discov. 2007;6:29–40. doi: 10.1038/nrd2201. 17159923. [DOI] [PubMed] [Google Scholar]
- 16.Herman-Bausier P., El-Kirat-Chatel S., Foster T.J., Geoghegan J.A., Dufrêne Y.F. Staphylococcus aureus fibronectin-binding protein A mediates cell-cell adhesion through low-affinity homophilic bonds. MBio. 2015;6:e00413–e00415. doi: 10.1128/mBio.00413-15. 26015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oh K.B., Oh M.N., Kim J.G., Shin D.S., Shin J. Inhibition of sortase-mediated Staphylococcus aureus adhesion to fibronectin via fibronectin-binding protein by sortase inhibitors. Appl. Microbiol. Biotechnol. 2006;70:102–106. doi: 10.1007/s00253-005-0040-8. 16010573. [DOI] [PubMed] [Google Scholar]
- 18.Weidenmaier C., Kokai-Kun J.F., Kristian S.A., Chanturiya T., Kalbacher H., Gross M., Nicholson G., Neumeister B., Mond J.J., Peschel A. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat. Med. 2004;10:243–245. doi: 10.1038/nm991. 14758355. [DOI] [PubMed] [Google Scholar]
- 19.Pickering A.C., Vitry P., Prystopiuk V., Garcia B., Höök M., Schoenebeck J., Geoghegan J.A., Dufrenê Y.F., Fitzgerald J.R. Host-specialized fibrinogen-binding by a bacterial surface protein promotes biofilm formation and innate immune evasion. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007816. 31216354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roche F.M., Meehan M., Foster T.J. The Staphylococcus aureus surface protein SasG and its homologues promote bacterial adherence to human desquamated nasal epithelial cells. Microbiology. 2003;149:2759–2767. doi: 10.1099/mic.0.26412-0. 14523109. [DOI] [PubMed] [Google Scholar]
- 21.Fey P.D., Endres J.L., Yajjala V.K., Widhelm T.J., Boissy R.J., Bose J.L., Bayles K.W. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio. 2013;4:e00537–12. doi: 10.1128/mBio.00537-12. 23404398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flannagan R.S., Heit B., Heinrichs D.E. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol. 2016;18:514–535. doi: 10.1111/cmi.12527. 26408990. [DOI] [PubMed] [Google Scholar]
- 23.Grosz M., Kolter J., Paprotka K., Winkler A.C., Schäfer D., Chatterjee S.S., Geiger T., Wolz C., Ohlsen K., Otto M., Rudel T., Sinha B., Fraunholz M. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell Microbiol. 2014;16:451–465. doi: 10.1111/cmi.12233. 24164701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Josse J., Laurent F., Diot A. Staphylococcal adhesion and host cell invasion: fibronectin-binding and other mechanisms. Front. Microbiol. 2017;8 doi: 10.3389/fmicb.2017.02433. 29259603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran Van Nhieu G., Isberg R.R. Bacterial internalization mediated by beta 1 chain integrins is determined by ligand affinity and receptor density. EMBO J. 1993;12:1887–1895. doi: 10.1002/j.1460-2075.1993.tb05837.x. 8491181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha B., Francois P.P., Nusse O., Foti M., Hartford O.M., Vaudaux P., Foster T.J., Lew D.P., Herrmann M., Krause K.H. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell Microbiol. 1999;1:101–117. doi: 10.1046/j.1462-5822.1999.00011.x. 11207545. [DOI] [PubMed] [Google Scholar]
- 27.Fowler T., Wann E.R., Joh D., Johansson S., Foster T.J., Höök M. Cellular invasion by Staphylococcus aureus involves a fibronectin bridge between the bacterial fibronectin-binding MSCRAMMs and host cell beta1 integrins. Eur J. Cell Biol. 2000;79:672–679. doi: 10.1078/0171-9335-00104. 11089915. [DOI] [PubMed] [Google Scholar]
- 28.Grundmeier M., Hussain M., Becker P., Heilmann C., Peters G., Sinha B. Truncation of fibronectin-binding proteins in Staphylococcus aureus strain Newman leads to deficient adherence and host cell invasion due to loss of the cell wall anchor function. Infect. Immun. 2004;72:7155–7163. doi: 10.1128/IAI.72.12.7155-7163.2004. 15557640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinha B., Francois P., Que Y.A., Hussain M., Heilmann C., Moreillon P., Lew D., Krause K.H., Peters G., Herrmann M. Heterologously expressed Staphylococcus aureus fibronectin-binding proteins are sufficient for invasion of host cells. Infect. Immun. 2000;68:6871–6878. doi: 10.1128/iai.68.12.6871-6878.2000. 11083807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Löffler B., Tuchscherr L., Niemann S., Peters G. Staphylococcus aureus persistence in non-professional phagocytes. Int. J. Med. Microbiol. 2014;304:170–176. doi: 10.1016/j.ijmm.2013.11.011. 24365645. [DOI] [PubMed] [Google Scholar]
- 31.Sinha B., Fraunholz M. Staphylococcus aureus host cell invasion and post-invasion events. Int. J. Med. Microbiol. 2010;300:170–175. doi: 10.1016/j.ijmm.2009.08.019. 19781990. [DOI] [PubMed] [Google Scholar]
- 32.Strobel M., Pförtner H., Tuchscherr L., Volker U., Schmidt F., Kramko N., Schnittler H.J., Fraunholz M.J., Löffler B., Peters G., Niemann S. Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin. Microbiol. Infect. 2016;22:799–809. doi: 10.1016/j.cmi.2016.06.020. 27393124. [DOI] [PubMed] [Google Scholar]
- 33.Darouiche R.O., Hamill R.J. Antibiotic penetration of and bactericidal activity within endothelial cells. Antimicrob. Agents Chemother. 1994;38:1059–1064. doi: 10.1128/aac.38.5.1059. 8067738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foster T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005;3:948–958. doi: 10.1038/nrmicro1289. 16322743. [DOI] [PubMed] [Google Scholar]
- 35.Sinha B., Herrmann M. Mechanism and consequences of invasion of endothelial cells by Staphylococcus aureus. Thromb. Haemost. 2005;94:266–277. doi: 10.1160/TH05-04-0235. 16113815. [DOI] [PubMed] [Google Scholar]
- 36.Wright G.D. Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol. 2016;24:862–871. doi: 10.1016/j.tim.2016.06.009. 27430191. [DOI] [PubMed] [Google Scholar]
- 37.O'Brien L.M., Walsh E.J., Massey R.C., Peacock S.J., Foster T.J. Staphylococcus aureus clumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: implications for nasal colonization. Cell Microbiol. 2002;4:759–770. doi: 10.1046/j.1462-5822.2002.00231.x. 12427098. [DOI] [PubMed] [Google Scholar]
- 38.Walsh E.J., O'Brien L.M., Liang X., Hook M., Foster T.J. Clumping factor B, a fibrinogen-binding MSCRAMM (microbial surface components recognizing adhesive matrix molecules) adhesin of Staphylococcus aureus, also binds to the tail region of type I cytokeratin 10. J. Biol. Chem. 2004;279:50691–50699. doi: 10.1074/jbc.M408713200. 15385531. [DOI] [PubMed] [Google Scholar]
- 39.Wertheim H.F., Walsh E., Choudhurry R., Melles D.C., Boelens H.A., Miajlovic H., Verbrugh H.A., Foster T., van Belkum A. Key role for clumping factor B in Staphylococcus aureus nasal colonization of humans. PLoS Med. 2008;5:e17. doi: 10.1371/journal.pmed.0050017. 18198942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mulcahy M.E., Geoghegan J.A., Monk I.R., O'Keeffe K.M., Walsh E.J., Foster T.J., McLoughlin R.M. Nasal colonisation by Staphylococcus aureus depends upon clumping factor B binding to the squamous epithelial cell envelope protein loricrin. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1003092. 23300445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakr A., Bregeon F., Mege J.L., Rolain J.M., Blin O. Staphylococcus aureus nasal colonization: an update on mechanisms, epidemiology, risk factors, and subsequent infections. Front. Microbiol. 2018;9 doi: 10.3389/fmicb.2018.02419. 30349525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wertheim H.F., Melles D.C., Vos M.C., van Leeuwen W., van Belkum A., Verbrugh H.A., Nouwen J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005;5:751–762. doi: 10.1016/S1473-3099(05)70295-4. 16310147. [DOI] [PubMed] [Google Scholar]
- 43.Bornstein E. Eradication of Staphylococcus aureus and MRSA in the nares: A historical perspective of the ecological niche, with suggestions for future therapy considerations. Adv. Microbiol. 2017;7:420–449. doi: 10.4236/aim.2017.76034. [DOI] [Google Scholar]
- 44.Wertheim H.F., Vos M.C., Ott A., van Belkum A., Voss A., Kluytmans J.A., van Keulen P.H., Vandenbroucke-Grauls C.M., Meester M.H., Verbrugh H.A. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet. 2004;364:703–705. doi: 10.1016/S0140-6736(04)16897-9. 15325835. [DOI] [PubMed] [Google Scholar]
- 45.Kluytmans J.A., Wertheim H.F. Nasal carriage of Staphylococcus aureus and prevention of nosocomial infections. Infection. 2005;33:3–8. doi: 10.1007/s15010-005-4012-9. 15750752. [DOI] [PubMed] [Google Scholar]
- 46.von Eiff C., Becker K., Machka K., Stammer H., Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia: study group. N. Engl. J. Med. 2001;344:11–16. doi: 10.1056/NEJM200101043440102. 11136954. [DOI] [PubMed] [Google Scholar]
- 47.Cheng A.G., Kim H.K., Burts M.L., Krausz T., Schneewind O., Missiakas D.M. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 2009;23:3393–3404. doi: 10.1096/fj.09-135467. 19525403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moreillon P., Entenza J.M., Francioli P., McDevitt D., Foster T.J., François P., Vaudaux P. Role of Staphylococcus aureus coagulase and clumping factor in pathogenesis of experimental endocarditis. Infect. Immun. 1995;63:4738–4743. doi: 10.1128/IAI.63.12.4738-4743.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas S., Liu W., Arora S., Ganesh V., Ko Y.P., Hook M. The complex fibrinogen interactions of the Staphylococcus aureus coagulases. Front. Cell Infect. Microbiol. 2019;9:106. doi: 10.3389/fcimb.2019.00106. 31041195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Higgins J., Loughman A., van Kessel K.P., van Strijp J.A., Foster T.J. Clumping factor A of Staphylococcus aureus inhibits phagocytosis by human polymorphonuclear leucocytes. FEMS Microbiol. Lett. 2006;258:290–296. doi: 10.1111/j.1574-6968.2006.00229.x. 16640587. [DOI] [PubMed] [Google Scholar]
- 51.Crosby H.A., Kwiecinski J., Horswill A.R. Staphylococcus aureus aggregation and coagulation mechanisms, and their function in host-pathogen interactions. Adv. Appl. Microbiol. 2016;96:1–41. doi: 10.1016/bs.aambs.2016.07.018. 27565579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Redi D., Raffaelli C.S., Rossetti B., De Luca A., Montagnani F. Staphylococcus aureus vaccine preclinical and clinical development: current state of the art. New Microbiol. 2018;41:208–213. 29874390. [PubMed] [Google Scholar]
- 53.Gordon C.P., Williams P., Chan W.C. Attenuating Staphylococcus aureus virulence gene regulation: a medicinal chemistry perspective. J. Med. Chem. 2013;56:1389–1404. doi: 10.1021/jm3014635. 23294220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gimza B.D., Larias M.I., Budny B.G., Shaw L.N. Mapping the global network of extracellular protease regulation in Staphylococcus aureus. mSphere. 2019;4 doi: 10.1128/mSphere.00676-19. 00676–19. 31645429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zielinska A.K., Beenken K.E., Mrak L.N., Spencer H.J., Post G.R., Skinner R.A., Tackett A.J., Horswill A.R., Smeltzer M.S. sarA-mediated repression of protease production plays a key role in the pathogenesis of Staphylococcus aureus USA300 isolates. Mol. Microbiol. 2012;86:1183–1196. doi: 10.1111/mmi.12048. 23075270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mrak L.N., Zielinska A.K., Beenken K.E., Mrak I.N., Atwood D.N., Griffin L.M., Lee C.Y., Smeltzer M.S. saeRS and sarA act synergistically to repress protease production and promote biofilm formation in Staphylococcus aureus. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0038453. 22685571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kolar S.L., Ibarra J.A., Rivera F.E., Mootz J.M., Davenport J.E., Stevens S.M., Horswill A.R., Shaw L.N. Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. Microbiologyopen. 2013;2:18–34. doi: 10.1002/mbo3.55. 23233325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paharik A.E., Salgado-Pabon W., Meyerholz D.K., White M.J., Schlievert P.M., Horswill A.R. The Spl serine proteases modulate Staphylococcus aureus protein production and virulence in a rabbit model of pneumonia. mSphere. 2016;1 doi: 10.1128/mSphere.00208-16. 00208–16. 27747296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tenover F.C., Goering R.V. Methicillin-resistant Staphylococcus aureus strain USA300: origin and epidemiology. J. Antimicrob. Chemother. 2009;64:441–446. doi: 10.1093/jac/dkp241. 19608582. [DOI] [PubMed] [Google Scholar]
- 60.Diekema D.J., Richter S.S., Heilmann K.P., Dohrn C.L., Riahi F., Tendolkar S., McDanel J.S., Doern G.V. Continued emergence of USA300 methicillin-resistant Staphylococcus aureus in the United States: results from a nationwide surveillance study. Infect. Control Hosp. Epidemiol. 2014;35:285–292. doi: 10.1086/675283. 24521595. [DOI] [PubMed] [Google Scholar]
- 61.Nichol K.A., Adam H.J., Roscoe D.L., Golding G.R., Lagace-Wiens P.R.S., Hoban D.J., Zhanel G.G., Zhanel G.G., Hoban D.J., Adam H.J., Karlowsky J.A., Baxter M.R., Nichol K.A., Lagace-Wiens P.R.S., Walkty A., on behalf of the Canadian Antimicrobial Resistance Alliance (CARA) Changing epidemiology of methicillin-resistant Staphylococcus aureus in Canada. J. Antimicrob. Chemother. 2013;68:i47–i55. doi: 10.1093/jac/dkt026. [DOI] [PubMed] [Google Scholar]
- 62.Alam M.T., Read T.D., Petit R.A., 3rd, Boyle-Vavra S., Miller L.G., Eells S.J., Daum R.S., David M.Z. Transmission and microevolution of USA300 MRSA in U.S. households: evidence from whole-genome sequencing. MBio. 2015;6 doi: 10.1128/mBio.00054-15. 25759497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Al-Rawahi G.N., Reynolds S., Porter S.D., Forrester L., Kishi L., Chong T., Bowie W.R., Doyle P.W. Community-associated CMRSA-10 (U.S.A.-300) is the predominant strain among methicillin-resistant Staphylococcus aureus strains causing skin and soft tissue infections in patients presenting to the emergency department of a Canadian tertiary care hospital. J. Emerg. Med. 2010;38:6–11. doi: 10.1016/j.jemermed.2007.09.030. 18325716. [DOI] [PubMed] [Google Scholar]
- 64.Nimmo G.R. USA300 abroad: global spread of a virulent strain of community-associated methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 2012;18:725–734. doi: 10.1111/j.1469-0691.2012.03822.x. 22448902. [DOI] [PubMed] [Google Scholar]
- 65.Ni Eidhin D., Perkins S., Francois P., Vaudaux P., Höök M., Foster T.J. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol. Microbiol. 1998;30:245–257. doi: 10.1046/j.1365-2958.1998.01050.x. 9791170. [DOI] [PubMed] [Google Scholar]
- 66.McAleese F.M., Walsh E.J., Sieprawska M., Potempa J., Foster T.J. Loss of clumping factor B fibrinogen binding activity by Staphylococcus aureus involves cessation of transcription, shedding and cleavage by metalloprotease. J. Biol. Chem. 2001;276:29969–29978. doi: 10.1074/jbc.M102389200. 11399757. [DOI] [PubMed] [Google Scholar]
- 67.Shinji H., Yosizawa Y., Tajima A., Iwase T., Sugimoto S., Seki K., Mizunoe Y. Role of fibronectin-binding proteins A and B in in vitro cellular infections and in vivo septic infections by Staphylococcus aureus. Infect. Immun. 2011;79:2215–2223. doi: 10.1128/IAI.00133-11. 21422173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greene C., McDevitt D., Francois P., Vaudaux P.E., Lew D.P., Foster T.J. Adhesion properties of mutants of Staphylococcus aureus defective in fibronectin-binding proteins and studies on the expression of fnb genes. Mol. Microbiol. 1995;17:1143–1152. doi: 10.1111/j.1365-2958.1995.mmi_17061143.x. 8594333. [DOI] [PubMed] [Google Scholar]
- 69.Wolz C., Goerke C., Landmann R., Zimmerli W., Fluckiger U. Transcription of clumping factor A in attached and unattached Staphylococcus aureus in vitro and during device-related infection. Infect. Immun. 2002;70:2758–2762. doi: 10.1128/iai.70.6.2758-2762.2002. 12010960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goncheva M.I., Flannagan R.S., Sterling B.E., Laakso H.A., Friedrich N.C., Kaiser J.C., Watson D.W., Wilson C.H., Sheldon J.R., McGavin M.J., Kiser P.K., Heinrichs D.E. Stress-induced inactivation of the Staphylococcus aureus purine biosynthesis repressor leads to hypervirulence. Nat. Commun. 2019;10:775. doi: 10.1038/s41467-019-08724-x. 30770821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walsh E.J., Miajlovic H., Gorkun O.V., Foster T.J. Identification of the Staphylococcus aureus MSCRAMM clumping factor B (ClfB) binding site in the alphaC-domain of human fibrinogen. Microbiology. 2008;154:550–558. doi: 10.1099/mic.0.2007/010868-0. 18227259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.O'Connell D.P., Nanavaty T., McDevitt D., Gurusiddappa S., Hook M., Foster T.J. The fibrinogen-binding MSCRAMM (clumping factor) of Staphylococcus aureus has a Ca2+-dependent inhibitory site. J. Biol. Chem. 1998;273:6821–6829. doi: 10.1074/jbc.273.12.6821. 9506984. [DOI] [PubMed] [Google Scholar]
- 73.Diep B.A., Gill S.R., Chang R.F., Phan T.H., Chen J.H., Davidson M.G., Lin F., Lin J., Carleton H.A., Mongodin E.F., Sensabaugh G.F., Perdreau-Remington F. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367:731–739. doi: 10.1016/S0140-6736(06)68231-7. 16517273. [DOI] [PubMed] [Google Scholar]
- 74.Mangat C.S., Bharat A., Gehrke S.S., Brown E.D. Rank ordering plate data facilitates data visualization and normalization in high-throughput screening. J. Biomol. Screen. 2014;19:1314–1320. doi: 10.1177/1087057114534298. 24828052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Senn M.M., Giachino P., Homerova D., Steinhuber A., Strassner J., Kormanec J., Fluckiger U., Berger-Bachi B., Bischoff M. Molecular analysis and organization of the sigmaB operon in Staphylococcus aureus. J. Bacteriol. 2005;187:8006–8019. doi: 10.1128/JB.187.23.8006-8019.2005. 16291674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chan P.F., Foster S.J. Role of SarA in virulence determinant production and environmental signal transduction in Staphylococcus aureus. J. Bacteriol. 1998;180:6232–6241. doi: 10.1128/.180.23.6232-6241.1998. 9829932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Entenza J.M., Moreillon P., Senn M.M., Kormanec J., Dunman P.M., Berger-Bachi B., Projan S., Bischoff M. Role of sigmaB in the expression of Staphylococcus aureus cell wall adhesins ClfA and FnbA and contribution to infectivity in a rat model of experimental endocarditis. Infect. Immun. 2005;73:990–998. doi: 10.1128/IAI.73.2.990-998.2005. 15664942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bischoff M., Dunman P., Kormanec J., Macapagal D., Murphy E., Mounts W., Berger-Bächi B., Projan S. Microarray-based analysis of the Staphylococcus aureus sigmaB regulon. J. Bacteriol. 2004;186:4085–4099. doi: 10.1128/JB.186.13.4085-4099.2004. 15205410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pohl K., Francois P., Stenz L., Schlink F., Geiger T., Herbert S., Goerke C., Schrenzel J., Wolz C. CodY in Staphylococcus aureus: a regulatory link between metabolism and virulence gene expression. J. Bacteriol. 2009;191:2953–2963. doi: 10.1128/JB.01492-08. 19251851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dickey S.W., Cheung G.Y.C., Otto M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017;16:457–471. doi: 10.1038/nrd.2017.23. 28337021. [DOI] [PubMed] [Google Scholar]
- 81.Biswas R., Voggu L., Simon U.K., Hentschel P., Thumm G., Götz F. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol. Lett. 2006;259:260–268. doi: 10.1111/j.1574-6968.2006.00281.x. 16734789. [DOI] [PubMed] [Google Scholar]
- 82.Heilmann C., Hussain M., Peters G., Götz F. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol. Microbiol. 1997;24:1013–1024. doi: 10.1046/j.1365-2958.1997.4101774.x. 9220008. [DOI] [PubMed] [Google Scholar]
- 83.Porayath C., Suresh M.K., Biswas R., Nair B.G., Mishra N., Pal S. Autolysin mediated adherence of Staphylococcus aureus with fibronectin, gelatin and heparin. Int. J. Biol. Macromol. 2018;110:179–184. doi: 10.1016/j.ijbiomac.2018.01.047. 29398086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heilmann C., Hartleib J., Hussain M.S., Peters G. The multifunctional Staphylococcus aureus autolysin aaa mediates adherence to immobilized fibrinogen and fibronectin. Infect. Immun. 2005;73:4793–4802. doi: 10.1128/IAI.73.8.4793-4802.2005. 16040992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kohler T.P., Gisch N., Binsker U., Schlag M., Darm K., Völker U., Zahringer U., Hammerschmidt S. Repeating structures of the major staphylococcal autolysin are essential for the interaction with human thrombospondin 1 and vitronectin. J. Biol. Chem. 2014;289:4070–4082. doi: 10.1074/jbc.M113.521229. 24371140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hirschhausen N., Schlesier T., Schmidt M.A., Götz F., Peters G., Heilmann C. A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell Microbiol. 2010;12:1746–1764. doi: 10.1111/j.1462-5822.2010.01506.x. 20642807. [DOI] [PubMed] [Google Scholar]
- 87.Xiong Y.Q., Bayer A.S., Yeaman M.R., Van Wamel W., Manna A.C., Cheung A.L. Impacts of sarA and agr in Staphylococcus aureus strain Newman on fibronectin-binding protein A gene expression and fibronectin adherence capacity in vitro and in experimental infective endocarditis. Infect. Immun. 2004;72:1832–1836. doi: 10.1128/iai.72.3.1832-1836.2004. 14977998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chien Y., Manna A.C., Projan S.J., Cheung A.L. SarA, a global regulator of virulence determinants in Staphylococcus aureus, binds to a conserved motif essential for sar-dependent gene regulation. J. Biol. Chem. 1999;274:37169–37176. doi: 10.1074/jbc.274.52.37169. 10601279. [DOI] [PubMed] [Google Scholar]
- 89.Cheung A.L., Koomey J.M., Butler C.A., Projan S.J., Fischetti V.A. Regulation of exoprotein expression in Staphylococcus aureus by a locus (sar) distinct from agr. Proc. Natl. Acad. Sci. U.S.A. 1992;89:6462–6466. doi: 10.1073/pnas.89.14.6462. 1321441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Price M.N., Huang K.H., Alm E.J., Arkin A.P. A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 2005;33:880–892. doi: 10.1093/nar/gki232. 15701760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walker J.N., Crosby H.A., Spaulding A.R., Salgado-Pabón W., Malone C.L., Rosenthal C.B., Schlievert P.M., Boyd J.M., Horswill A.R. The Staphylococcus aureus ArlRS two-component system is a novel regulator of agglutination and pathogenesis. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003819. 24367264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang J.H., Chung T.D., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. 10838414. [DOI] [PubMed] [Google Scholar]
- 93.Franz M., Lopes C.T., Huck G., Dong Y., Sumer O., Bader G.D. Cytoscape.js: a graph theory library for visualisation and analysis. Bioinformatics. 2016;32:309–311. doi: 10.1093/bioinformatics/btv557. 26415722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. 22930834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Uncited reference
- 95.Bae T., Glass E.M., Schneewind O., Missiakas D. Generating a collection of insertion mutations in the Staphylococcus aureus genome using bursa aurealis. Methods Mol. Biol. 2008;416:103–116. doi: 10.1007/978-1-59745-321-9_7. 18392963. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within this article (and its supporting material). The raw data from the primary genome-wide adhesion screens are available upon request. Please contact the corresponding author (gcox@uoguelph.ca).