

Abstract
Heterotrimeric G-proteins are signaling switches broadly divided into four families based on the sequence and functional similarity of their Gα subunits: Gs, Gi/o, Gq/11, and G12/13. Artificial mutations that activate Gα subunits of each of these families have long been known to induce oncogenic transformation in experimental systems. With the advent of next-generation sequencing, activating hotspot mutations in Gs, Gi/o, or Gq/11 proteins have also been identified in patient tumor samples. In contrast, patient tumor-associated G12/13 mutations characterized to date lead to inactivation rather than activation. By using bioinformatic pathway analysis and signaling assays, here we identified cancer-associated hotspot mutations in Arg-200 of Gα13 (encoded by GNA13) as potent activators of oncogenic signaling. First, we found that components of a G12/13-dependent signaling cascade that culminates in activation of the Hippo pathway effectors YAP and TAZ is frequently altered in bladder cancer. Up-regulation of this signaling cascade correlates with increased YAP/TAZ activation transcriptional signatures in this cancer type. Among the G12/13 pathway alterations were mutations in Arg-200 of Gα13, which we validated to promote YAP/TAZ-dependent (TEAD) and MRTF-A/B-dependent (SRE.L) transcriptional activity. We further showed that this mechanism relies on the same RhoGEF-RhoGTPase cascade components that are up-regulated in bladder cancers. Moreover, Gα13 Arg-200 mutants induced oncogenic transformation in vitro as determined by focus formation assays. In summary, our findings on Gα13 mutants establish that naturally occurring hotspot mutations in Gα subunits of any of the four families of heterotrimeric G-proteins are putative cancer drivers.
Keywords: GTPase, G-protein, G-protein-coupled receptor (GPCR), cancer biology, oncogene, Bladder Cancer
Heterotrimeric G-proteins are critical transducers of signaling triggered by a large family of G-protein–coupled receptors (GPCRs). Essentially, GPCRs promote GTP loading on the α-subunits of G-proteins (1, 2), which triggers signaling downstream. Heterotrimeric G-proteins are composed of a nucleotide-binding Gα subunit and an obligatory Gβγ dimer, and they are classified into four families based on the nature of the Gα subunits. These four families are Gs, Gi/o, Gq/11, and G12/13, and Gα subunits of each one of them have distinct actions on specific effectors. For example, Gs members stimulate adenylyl cyclase activity, whereas Gi/o family members tend to inhibit it; Gq/11 members stimulate phospholipase C enzymes and a subgroup of RhoGEFs; and G12/13 members stimulate a different subgroup of RhoGEFs (3, 4). Signaling is terminated upon GTP hydrolysis mediated by the intrinsic GTPase of Gα subunits.
The role of heterotrimeric G-proteins in cancer-related signaling has been documented for decades. Early studies identified cancer-associated mutations in Gαs that disrupted its GTPase activity, rendering the G-protein constitutively active (5). This seminal finding spurred a wave of studies exploring whether analogous mutations introduced artificially in other Gα subunits would also promote their ability to induce oncogenic transformation. It was found that GTPase-deficient mutants of most, if not all, Gα subunits tested led to oncogenic transformation in vitro, regardless of the G-protein family they belonged to. For example, Gαi2, Gαo, and Gαz (Gi/o family); Gαq (Gq/11 family); and Gα12 and Gα13 (G12/13 family), in addition to Gαs (Gs family), all promoted oncogenic transformation as assessed by in vitro assays using fibroblasts (6, 7, 8, 9, 10, 11, 12, 13). In most cases, transformation in vitro correlated well with tumor growth in vivo using mouse xenografts. Thus, one theme emerging from these studies was that enhancement of GPCR/G-protein signaling tends to favor oncogenicity.
Despite these initial observations and the identification of some mutations in G-proteins in tumors (5, 14), only with the recent advent of deep-sequencing techniques has it become obvious that dysregulation of the GPCR/G-protein signaling axis in cancer is highly prevalent (15, 16, 17). The mutational landscape of GPCR/G-protein signaling components in cancer supports the theme that G-protein hyperactivation in cancer tends to be pro-oncogenic. There are many examples of GPCRs that are either overexpressed or contain activating mutations (15, 17, 18, 19, 20), and negative regulators of G-protein activity have also been shown to bear loss-of-function mutations (21). As for G-proteins themselves, it is now known that hyperactive G-protein mutants can be very frequent in certain types of cancers. The most striking example is uveal melanoma, in which ∼90% of tumors contain activating mutations in Gαq (encoded by GNAQ) or Gα11 (GNA11) (22, 23). Similarly, activating mutations in Gαs (GNAS) can be as frequent as 70% in certain subtypes of pancreatic ductal carcinomas (24, 25), and activating mutations in Gαi2 can be as frequent as 24% in epitheliotropic intestinal T-cell lymphomas (26). Thus, for representative members of three of four families of G-proteins (Gq/11, Gs, and Gi/o) the oncogenic activity in vitro caused by artificially introduced mutations has found a counterpart in prevalent mutations in cancer. Interestingly, findings so far suggest that the remaining family of G-proteins (G12/13) might be an exception to this trend. For example, mutations in Gα13 in some types of lymphoma are frequent, but they lead to inactivation rather than activation (27, 28). This suggests that, at least in these lymphomas, Gα13 activity is tumor-suppressive. This is the opposite of the oncogene function previously suggested from experiments in vitro with a constitutively active artificial Gα13 mutation (7, 9).
Activation of Gα12 or Gα13 proteins leads to activation of RhoA-dependent transcriptional programs, including those mediated by the activation of the Hippo pathway effectors YAP and TAZ. The cascade of events involves the direct activation of a subgroup of RhoGEFs, composed of p115-RhoGEF, PDZ-RhoGEF, and LARG, by active, GTP-loaded Gα subunits of G12/13 proteins, which in turn activates RhoA, RhoB, and RhoC GTPases (29). Through mechanisms that involve the remodeling of the actin cytoskeleton, Rho GTPases induce transcriptional responses that include those regulated by YAP/TAZ, which serve as co-factors for the TEA domain–containing transcription factor family (TEADs), and via myocardin-related transcription factors A and B (MRTF-A/B), which serve as co-activators for the transcriptional factor SRF (30, 31, 32, 33, 34, 35, 36). Although the G12/13-YAP/TAZ signaling axis has been shown to be oncogenic in some contexts (35, 36, 37), no cancer-associated mutation that activates Gα12 or Gα13 has been characterized to date. Prompted by the finding that the G12/13-YAP/TAZ signaling axis is up-regulated in bladder cancer, here we characterized the effect of hotspot mutations in Gα13 identified in this cancer type. We found that mutations in the Arg-200 of Gα13, a residue required to hydrolyze GTP, lead to activation of YAP/TAZ-dependent and MRTF-A/B-dependent transcription through a RhoGEF–Rho GTPase cascade and that they promote oncogenic transformation in vitro. This implies that naturally occurring hotspot mutations in Gα subunits of any of the four families of heterotrimeric G-proteins are putative cancer drivers.
Results and discussion
G12/13 pathway up-regulation correlates with increased Yap/TAZ transcriptional activity in bladder cancer
We mined data from the Cancer Genome Atlas (TCGA) through cBioportal to explore genomic alterations in components of a G12/13-YAP/TAZ pathway (Fig. 1A). More specifically, we queried the G-proteins Gα12 (GNA12) and Gα13 (GNA13); the RhoGEFs p115-RhoGEF (ARHGEF1), PDZ-RhoGEF (ARHGEF11), and LARG (ARHGEF12); the Rho GTPases RhoA (RHOA), RhoB (RHOB), and RhoC (RHOC); and the Hippo pathway effectors YAP (YAP1) and TAZ (WWTR1). We found that these genes were altered in a large portion (∼40%) of the TCGA bladder cancers (TCGA-BLCA) (Fig. 1B). The alterations appeared to be largely mutually exclusive and trending toward up-regulation. For example, both heterotrimeric G-proteins, two of the three RhoGEFs, and both Hippo effectors displayed amplifications as the dominant feature. For RhoA (RHOA) and RhoB (RHOB), the main feature was that they were mutated, and several of these mutations are classified as putative drivers in cBioportal (38). Although not all RhoA/RhoB mutations have been characterized, some of them have been previously proposed to lead to signaling activation, like Ala-161 mutations in RhoA (39) or the E172K mutation in RhoB (40). Thus, although LARG (ARHGEF12) and RhoC (RHOC) are exceptions to the overall trend, these observations suggest that the G12/13-YAP/TAZ pathway might be up-regulated in bladder cancer.
Figure 1.

G12/13-YAP/TAZ signaling axis is up-regulated in many bladder cancers.A, diagram of a G12/13-YAP/TAZ signaling cascade. B, many bladder cancers show genetic alterations in the G12/13-YAP/TAZ signaling cascade that collectively suggest up-regulation. Data mined from the TCGA-BLCA data set is displayed as an oncoprint representation. C, up-regulated expression of the G12/13 pathway correlates with YAP/TAZ activation in bladder cancers. The correlation between the expression of G12/13 pathway components and a 24-gene transcriptional signature regulated by YAP/TAZ was assessed by ssGSEA. The solid line represents the linear regression fit of the data, with 95% confidence intervals indicated in gray. D, comparison of the correlation coefficient observed in C (vertical dotted line) with a null distribution generated from bootstrapping correlation coefficients for the G12/13 pathway and 10,000 randomly selected 24-gene signatures. The statistical significance p value was determined as described under “Experimental procedures.”
Motivated by these observations, we carried out a bioinformatic analysis of gene expression data to establish a potential correlation between up-regulation of the G12/13 pathway and YAP/TAZ activation. For this, we turned to a previously characterized 24-gene signature that depends on YAP/TAZ (41) and analyzed its relationship to the expression levels of the rest of the upstream components of the proposed G12/13 pathway. We used single-sample gene set enrichment analysis (ssGSEA) to quantify relative enrichment of each pathway across over 400 primary tumors in the TCGA-BLCA RNA-Seq data set. We found a strong correlation between the activation scores of the G12/13 pathway and the activation scores for YAP/TAZ (Fig. 1C). We then tested the observed correlation coefficient against a null distribution of correlations between ssGSEA-quantified activity of the G12/13 pathway and 10,000 random 24-gene signatures, resulting in a significant p value of 1e−4 (Fig. 1D). Taken together, these observations indicate that up-regulation of the G12/13 pathway in bladder cancer correlates with increased transcriptional output of the downstream effectors YAP/TAZ.
Gα13 Arg-200 mutants induce YAP/TAZ activity via a RhoGEF–Rho GTPase axis
Although overexpression of WT G12/13 family Gα proteins has been found before to be sufficient to promote transformation (7, 8), a recent study also found that the mutation frequency of GNA13 in the TCGA-BLCA data set is statistically higher than background mutation frequency (q = 0.007) (42). Moreover, the distribution of mutations across the sequence of Gα13 suggested a hotspot at Arg-200 (Fig. 2A). The presence of an arginine in this position is absolutely conserved across Gα subunits (Fig. 2A), and its mutation in several other Gα subunits leads to increased activity and favors oncogenic transformation (5, 11, 13, 14). From studies in other Gα proteins, it has been found that this arginine is crucial for GTPase activity and that it cannot be replaced by other amino acids, even if they preserve the positive charge of the side chain like in the case of lysine (5, 43, 44, 45). Thus, we hypothesized that Gα13 Arg-200 mutations identified in bladder cancer would similarly induce the formation of an active G-protein that increases downstream signaling, including YAP/TAZ (Fig. 2B). Because mutation of this arginine to any other residue is expected to have similar consequences (5, 43), we focused our efforts on characterizing Gα13 R200K and Gα13 R200G because these are the two mutants most frequently found in bladder cancer. Before assessing the impact of these mutants in cell signaling assays, we validated that they adopted an active conformation by using a well-validated assay that relies on protection from trypsin hydrolysis (Fig. S1) (44, 46). Next, we expressed Gα13 R200K and Gα13 R200G in HEK293T cells and assessed activation of YAP/TAZ using a TEAD reporter assay (Fig. 2C). We compared the effect of expressing these two mutants with that of Gα13 WT as well as with that of Gα13 Q226L, an artificial mutant previously shown to enhance downstream signaling including YAP/TAZ-dependent TEAD transcriptional activity (34, 37). Whereas expression of Gα13 WT led to a modest increase of TEAD activity, expression of Gα13 R200K and Gα13 R200G led to a significantly larger increase comparable with that observed in cells expressing the control mutant Gα13 Q226L (Fig. 2C). To determine whether the observed increase in TEAD activity by Gα13 mutants was mediated by YAP/TAZ, we knocked down both proteins simultaneously using a previously validated siRNA sequence (47, 48). As expected, depletion of YAP and TAZ led to a large suppression of TEAD activation by Gα13 R200K, R200G, or Q226L (Fig. 2D). To further map the cascade of events leading to YAP/TAZ activation by Gα13 mutants, we blocked the pathway that putatively operates in bladder cancer at different levels. First, inhibition of the Rho GTPases RhoA, RhoB, and RhoC by expression of Clostridium botulinum C3 toxin efficiently suppressed TEAD activation by Gα13 R200K, R200G, or Q226L (Fig. 2E). Then we tested the effect of a fragment of p115-RhoGEF that works as a dominant-negative by preventing the binding of active Gα13 to its target RhoGEFs that operate upstream of Rho GTPases in the pathway (49). Expression of this dominant-negative construct, consisting of p115-RhoGEF's RGS homology (RH) domain (p115RH), but not a control construct, inhibited TEAD activation by Gα13 R200K, R200G, or Q226L (Fig. 2F). To further validate the specificity of these manipulations, we tested their impact on Gα13-mediated activation of another transcriptional output not controlled by YAP/TAZ but still dependent on Rho GTPase activation (i.e. the transcriptional activation of SRF via MRTF-A/B) (Fig. 2B). As expected, bladder cancer–associated mutants Gα13 R200K and R200G led to robust activation of the SRF reporter, comparable with that of the positive control Gα13 Q226L, which was suppressed by inhibition of the activation of RhoGEFs or Rho GTPases but not upon YAP/TAZ depletion (Fig. 2, G–I). Taken together, these results demonstrate that Gα13 hotspot mutations in Arg-200 found in bladder cancer are bona fide activating mutations that lead to induction of YAP/TAZ-dependent transcription via a RhoGEF–Rho GTPase cascade.
Figure 2.
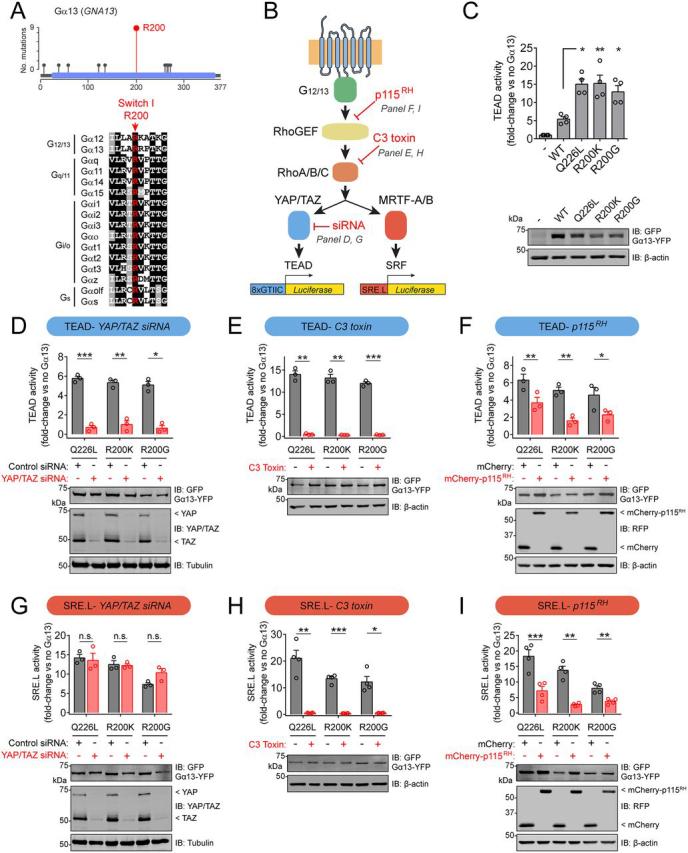
Hotspot mutations in Gα13 Arg-200 cause constitutive G-protein activation and lead to enhanced YAP/TAZ-dependent and MRTF-A/B-dependent transcription.A, top, lollipop plot of Gα13 residues mutated in bladder cancer. Bottom, alignment of Gα switch I region showing in red the fully conserved arginine that corresponds to Gα13 Arg-200. B, diagram of a G12/13 signaling cascade culminating in the activation of transcriptional regulators and specific luciferase-based reporters used to measure their activity. Manipulations implemented in other panels of this figure to inhibit specific steps of the pathway are indicated in red. C, Gα13 Arg-200 mutants activate YAP/TAZ-dependent transcription. HEK293T cells were transfected with plasmids for the expression of the indicated Gα13 constructs and TEAD reporter (8xGTIIC) activity determined as described under “Experimental procedures.” Results are mean ± S. E. (error bars), n = 4. *, p < 0.05; **, p < 0.01, analysis of variance with Tukey post hoc test. D and G, YAP/TAZ depletion abolishes TEAD reporter (D) but not SRE.L reporter (G) activation caused by Gα13 Arg-200 mutants. HEK293T cells were transfected with plasmids for the expression of the indicated Gα13 constructs and with the indicated siRNAs, and TEAD reporter or SRE.L reporter activity was determined as described under “Experimental procedures.” Results are mean ± S. E., n = 3. *, p < 0.05; **, p < 0.01; ***, p < 0.001, n.s., not significant, Student's t test. E and H, Rho GTPase blockade abolishes TEAD reporter (E) and SRE.L reporter (H) activation caused by Gα13 Arg-200 mutants. HEK293T cells were transfected with plasmids for the expression of the indicated Gα13 constructs with or without a plasmid for the expression of C3 toxin. TEAD reporter or SRE.L reporter activity was determined as described under “Experimental procedures.” Results are mean ± S. E., n = 3–4. *, p < 0.05; **, p < 0.01; ***, p < 0.001, Student's t test. F and I, blocking Gα13-mediated activation of RhoGEFs with a dominant-negative construct (p115RH) inhibits TEAD reporter (F) and SRE.L reporter (I) activation caused by Gα13 Arg-200 mutants. HEK293T cells were transfected with plasmids for the expression of the indicated Gα13 constructs and a plasmid for the expression of mCherry-p115RH or mCherry as negative control. TEAD reporter or SRE.L reporter activity was determined as described under “Experimental procedures.” Results are mean ± S. E., n = 3–4. *, p < 0.05; **, p < 0.01; ***, p < 0.001, Student's t test. For all panels showing reporter activation results, an immunoblot of lysates of cells used in one of the experiments is shown below the graph.
Gα13 Arg-200 mutants induce oncogenic transformation in vitro
Finally, we sought to determine whether the Gα13 hotspot mutations in Arg-200 described above would be sufficient to promote oncogenic transformation in vitro. For this, we used focus formation assays with NIH3T3 cells. This widely used system is particularly well-suited to analyze the putative oncogenic activity of Gα13 Arg-200 mutants because it has been used for the vast majority of Gα oncogenic mutants reported to date as a good proxy for tumor growth in mice, including for the oncogenic activity of artificial activating mutations introduced in Gα13 (7). First, we assessed whether Gα13 R200K and Gα13 R200G mutants also lead to increased signaling activity in NIH3T3 cells. Surprisingly, we found that whereas Gα13 R200K and Gα13 R200G led to robust increases in the MRTF-A/B-dependent SRE.L reporter, they had no significant effect on the activity of the YAP-TAZ–dependent TEAD reporter (Fig. S2). These results confirm that Gα13 R200K and Gα13 R200G behave as active G-proteins but that the downstream signaling consequences are cell type–specific. Next, we generated NIH3T3 cell lines stably expressing Gα13 WT, Gα13 R200K, and Gα13 R200G at comparable levels by lentiviral transduction and selection with the appropriate agents (Fig. 3A). Both Gα13 R200K and Gα13 R200G induced the formation of numerous foci, whereas Gα13 WT only had a modest effect (Fig. 3, B and C).
Figure 3.

Gα13 Arg-200 mutants induce NIH3T3 cell transformation in vitro.A, generation of NIH3T3 cell lines stably expressing the indicated Gα13 proteins. Lentiviral particles for the expression of Gα13 were generated in HEK293T cells and used to transduce NIH3T3 cells, followed by antibiotic selection. Lysates of each one of the cell lines were immunoblotted as indicated. Images were generated by splicing lanes from the same membrane, and the vertical dotted line indicates the position of the boundary between the two segments that were merged. B and C, Gα13 Arg-200 mutants promote focus formation in NIH3T3 cells more efficiently than Gα13 WT. Cells were seeded on plates and stained with crystal violet 10 days later. Images of a representative experiment are shown in B, whereas C shows the quantification of foci. Results are mean ± S. E. (error bars), n = 6. **, p < 0.01; ***, p < 0.001; n.s., not significant, analysis of variance with Tukey post hoc test.
Conclusions
Recent reports have suggested that mutations in Gα13 are putative oncogene drivers in bladder cancer based on bioinformatics predictions (17, 42, 50), but no other experimental evidence to support the predictions has been provided. The results presented here provide the missing experimental evidence that supports the idea of Gα13 hotspot mutations as putative drivers in bladder cancer and suggest that pharmacological blockade of the pathway activated downstream might be a viable therapeutic avenue. Moreover, our findings on Gα13 mutants establish that naturally occurring hotspot mutations in Gα subunits of any of the four families of heterotrimeric G-proteins (i.e. in Gs, Gi/o, Gq/11, and, now, G12/13) are putative cancer drivers, thereby providing definitive confirmation of a long-held tenet.
Experimental procedures
Data processing
Data for the oncoprint in Fig. 1B were obtained through cBioportal (38) by querying the term GNA13 on March 23rd, 2020 in the data set “Bladder Cancer (TCGA, Cell 2017).” For the lollipop plot in Fig. 2A, data were obtained from all of the data sets classified as Bladder Urothelial Carcinoma in cBioportal. TCGA-BLCA RNA-Seq count matrix (generated with STAR 2-Pass and HTSeq-Counts) and available metadata were downloaded through the Genomic Data Commons gdc-client (51, 52). We performed a variance-stabilizing transformation of the data using the R package DESeq2 (version 1.23.10) followed by a log transformation (53).
Pathway-level correlation analysis
Pathway activity in TCGA-BLCA was measured for G12/13 and YAP/TAZ signatures—represented by key selected genes (for G12/13: GNA12, GNA13, ARHGEF1, ARHGEF11, ARHGEF12, RHOA, RHOB, and RHOC; for YAP/TAZ: YAP1, WWTR1, MYOF, AMOTL2, LATS2, CTGF, CYR61, ANKRD1, ASAP1, AXL, F3, IGFBP3, CRIM1, FJX1, FOXF2, GADD45A, CCDC80, NT5E, DOCK5, PTPN14, ARHGEF17, NUAK2, TGFB2, and RBMS3)—through gene set variation analysis using the ssGSEA method and a Gaussian kernel from the R package GSVA (version 1.34.0) (54). We then measured the Pearson correlation between the activities of each pathway across all primary tumors. We further tested the significance of the observed correlation coefficient by comparing it with a null distribution generated through bootstrapping 10,000 random 24-gene signatures and measuring the correlation of their ssGSEA-quantified activity with the G12/13 pathway.
Plasmid constructs and siRNAs
pGL3b-8xGTIIC-luciferase (TEAD reporter) (55) was from Addgene (catalog no. 34615). pGL3-SRE.L (56) was a gift from Richard Neubig (Michigan State University). pCMV-Beta (Clontech, 631719) was a gift from Matthew Layne (Boston University). Plasmid pCS2-Nanoluc encoding nanoluciferase driven by the cytomegalovirus promoter was a gift from Daniel Cifuentes (Boston University). pcDNA3.1-Gα13(EE) was from the cDNA Resource Center (GNA130EI00). pcDNA3.1-Gα13(EE)-YFP was generated as described previously (57). Lentiviral pLVX-puro-Gα13(EE)-YFP plasmids were generated by inserting Gα13(EE)-YFP into the EcoRI site of a modified pLVX-puro plasmid (pLVX-puro-MCS+). Plasmid pEF-C3 was a gift from Silvio Gutkind (University of California, San Diego). mCherry-p115RH (also known as Lck-mCherry-RGS (49)) and mCherry (also known as Lck-mCherry) plasmids were a gift from Joachim Goedhart (University of Amsterdam). All point mutations were generated using QuikChange II (Agilent, 200523). siRNA used for knockdown of YAP and TAZ was UGUGGAUGAGAUGGAUACA (47, 48), and the control siRNA was from Qiagen (catalog no. 1027310).
Trypsin protection assays
This assay was carried out as described previously (29), except that HEK293T lysates were treated with trypsin for 5 min.
Reporter assays in HEK293T and NIH3T3 cells
TEAD and SRE.L reporter assays in HEK293T cells (ATCC, CRL-3216) were performed as described previously (58) using the Dual-Glo Luciferase Assay System (Promega, E2920) to determine luciferase activity and fluorescein di-β-d-galactopyranoside (Marker Gene Technologies) for β-gal activity. Cells were transfected with the following plasmids using calcium phosphate: pGL3-SRE.L (0.5 μg) or pGL3b-8xGTIIC-luciferase (0.5 μg) and pCMV-Beta (0.5 μg), along with plasmid pcDNA3.1-Gα13(EE)-YFP (0.6 μg of WT or 0.15–0.2 μg of mutant plasmid per well). In some experiments, 0.1 μg of pEF-C3, mCherry-p115RH, or mCherry was also co-transfected. Six hours after transfection, medium was replaced by Dulbecco's modified Eagle's medium with 0.5% fetal bovine serum. 16-24 h later, cells were washed with PBS and harvested by gentle scraping. When using siRNA, cells were first reverse-transfected with 20 pmol of YAP/TAZ or control siRNA using Lipofectamine RNAiMAX (Invitrogen, 13778075) the day before calcium phosphate transfection. For NIH3T3 cells (ATCC, CRL-1658), the same procedures were followed except that the plasmids pGL3-SRE.L (0.25 μg) or pGL3b-8xGTIIC-luciferase (0.25 μg), pCS2-Nanoluc (0.05 μg), and 0.5 μg of WT or 0.3–0.6 μg of mutant plasmid per well were transfected using Turbofect (ThermoScientific, R0531) and that firefly luciferase and nanoluciferase activities were determined using the Nano-Glo Dual-Luciferase reporter assay system (Promega, N1610).
NIH3T3 focus formation
Lentivirus packaging, transduction, and selection (1 μg/ml puromycin) were carried out as described previously (57). All surviving clones were pooled and maintained in the presence of 0.5 μg/ml puromycin. For focus formation assays, NIH3T3 cell lines were seeded (200,000 cells/plate) in 10-cm plates coated with 0.1% gelatin. Medium was replaced at days 5, 7, and 9 after seeding. At day 10, cells were washed with cold PBS, fixed with cold 100% methanol, and stained with crystal violet (0.05% (w/v) in 20% (v/v) methanol). After washing with 20% (v/v) methanol, plates were imaged using a flatbed scanner, and distinct visible colonies (∼1.5 mm2 or larger) with dark blue staining were manually counted in the whole plate.
Immunoblotting
Cell pellets were lysed and immunoblotted as described previously (58) using the following antibodies: GFP (1:1,000; Clontech JL-8), red fluorescent protein (1:1,000; Rockland Immunochemicals 600-401-379), YAP/TAZ (1:1,000; Cell Signaling Technology, D24E4), α-tubulin (1:2,500; Sigma T6074), β-actin (1:2,000; LI-COR 926-42212), and Glu-Glu (1:1,000; Biolegend 901801). The secondary antibodies were goat anti-rabbit Alexa Fluor 680 (1:10,000; Life Technologies A21077), goat anti-mouse Alexa Fluor 680 (1:10,000; Life Technologies A28183), goat anti-mouse IRDye 800 (1:10,000; LI-COR 926-32210), and goat anti-rabbit IRDye 800 (1:10,000; LI-COR 926-32211).
Data availability
All data are contained in the article except the raw data used for the genomics analysis, which corresponds to the Cancer Genome Atlas data set named TCGA-BLCA and was accessed and/or downloaded directly from cBioPortal or Genomic Data Commons as indicated under “Experimental procedures.”
HHS | NIH | National Institute of General Medical Sciences (NIGMS) (R01GM130120) to Mikel Garcia-Marcos
American Cancer Society (ACS) (PF-19-084-01-CDD) to Marcin Maziarz
American Cancer Society (ACS) (RSG-17-138-01-CSM) to Xaralabos Varelas
HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI) (R01HL12439) to Xaralabos Varelas
Edited by Henrik G. Dohlman
Footnotes
This article contains supporting information.
Author contributions—M. M., A. F., and M. G.-M. formal analysis; M. M., A. F., J. Z., L. D., and Z. Z. investigation; M. M. and M. G.-M. writing-original draft; M. M. and M. G.-M. writing-review and editing; A. F. data curation; A. F. and M. G.-M. visualization; S. M., X. V., and M. G.-M. supervision; X. V. and M. G.-M. funding acquisition; M. G.-M. conceptualization; M. G.-M. project administration.
Funding and additional information—This work was primarily supported by National Institutes of Health Grant R01GM130120 (to M. G.-M.). M. M. was supported by American Cancer Society Postdoctoral Fellowship PF-19-084-01-CDD. X. V. was supported by National Institutes of Health Grant R01HL12439 and American Cancer Society Research Scholar Grant RSG-17-138-01-CSM. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G-protein–coupled receptor
- GEF
- guanine nucleotide exchange factor
- TEAD
- TEA domain–containing transcription factor
- MRTF
- myocardin-related transcription factors
- SRF
- serum response factor
- SRE
- serum response element
- RH
- RGS homology
- YAP
- Yes-associated protein
- TAZ
- transcriptional co-activator with PDZ-binding motif
- TCGA
- the Cancer Genome Atlas
- BLCA
- bladder cancer
- ssGSEA
- single-sample gene set enrichment analysis.
Supplementary Material
References
- 1.Oldham W.M., Hamm H.E. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. 2008;9:60–71. doi: 10.1038/nrm2299. 18043707. [DOI] [PubMed] [Google Scholar]
- 2.Gilman A.G. G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. 3113327. [DOI] [PubMed] [Google Scholar]
- 3.Neves S.R., Ram P.T., Iyengar R. G protein pathways. Science. 2002;296:1636–1639. doi: 10.1126/science.1071550. 12040175. [DOI] [PubMed] [Google Scholar]
- 4.Dorsam R.T., Gutkind J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. 17251915. [DOI] [PubMed] [Google Scholar]
- 5.Landis C.A., Masters S.B., Spada A., Pace A.M., Bourne H.R., Vallar L. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–696. doi: 10.1038/340692a0. 2549426. [DOI] [PubMed] [Google Scholar]
- 6.Kalinec G., Nazarali A.J., Hermouet S., Xu N., Gutkind J.S. Mutated α subunit of the Gq protein induces malignant transformation in NIH 3T3 cells. Mol. Cell. Biol. 1992;12:4687–4693. doi: 10.1128/mcb.12.10.4687. 1328859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu N., Voyno-Yasenetskaya T., Gutkind J.S. Potent transforming activity of the G13 α subunit defines a novel family of oncogenes. Biochem. Biophys. Res. Commun. 1994;201:603–609. doi: 10.1006/bbrc.1994.1744. 8002992. [DOI] [PubMed] [Google Scholar]
- 8.Xu N., Bradley L., Ambdukar I., Gutkind J.S. A mutant α subunit of G12 potentiates the eicosanoid pathway and is highly oncogenic in NIH 3T3 cells. Proc. Natl. Acad. Sci. U. S. A. 1993;90:6741–6745. doi: 10.1073/pnas.90.14.6741. 8393576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voyno-Yasenetskaya T.A., Pace A.M., Bourne H.R. Mutant α subunits of G12 and G13 proteins induce neoplastic transformation of Rat-1 fibroblasts. Oncogene. 1994;9:2559–2565. 8058319. [PubMed] [Google Scholar]
- 10.Wong Y.H., Chan J.S., Yung L.Y., Bourne H.R. Mutant α subunit of Gz transforms Swiss 3T3 cells. Oncogene. 1995;10:1927–1933. 7761094. [PubMed] [Google Scholar]
- 11.Pace A.M., Wong Y.H., Bourne H.R. A mutant α subunit of Gi2 induces neoplastic transformation of Rat-1 cells. Proc. Natl. Acad. Sci. U. S. A. 1991;88:7031–7035. doi: 10.1073/pnas.88.16.7031. 1651490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ram P.T., Horvath C.M., Iyengar R. Stat3-mediated transformation of NIH-3T3 cells by the constitutively active Q205L Gαo protein. Science. 2000;287:142–144. doi: 10.1126/science.287.5450.142. 10615050. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S.K., Gallego C., Lowndes J.M., Pleiman C.M., Sable C., Eisfelder B.J., Johnson G.L. Analysis of the fibroblast transformation potential of GTPase-deficient gip2 oncogenes. Mol. Cell. Biol. 1992;12:190–197. doi: 10.1128/mcb.12.1.190. 1729598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyons J., Landis C.A., Harsh G., Vallar L., Grünewald K., Feichtinger H., Duh Q.Y., Clark O.H., Kawasaki E., Bourne H.R. Two G protein oncogenes in human endocrine tumors. Science. 1990;249:655–659. doi: 10.1126/science.2116665. 2116665. [DOI] [PubMed] [Google Scholar]
- 15.O'Hayre M., Vázquez-Prado J., Kufareva I., Stawiski E.W., Handel T.M., Seshagiri S., Gutkind J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer. 2013;13:412–424. doi: 10.1038/nrc3521. 23640210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kan Z., Jaiswal B.S., Stinson J., Janakiraman V., Bhatt D., Stern H.M., Yue P., Haverty P.M., Bourgon R., Zheng J., Moorhead M., Chaudhuri S., Tomsho L.P., Peters B.A., Pujara K. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. 20668451. [DOI] [PubMed] [Google Scholar]
- 17.Wu V., Yeerna H., Nohata N., Chiou J., Harismendy O., Raimondi F., Inoue A., Russell R.B., Tamayo P., Gutkind J.S. Illuminating the Onco-GPCRome: novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 2019;294:11062–11086. doi: 10.1074/jbc.REV119.005601. 31171722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright S.C., Kozielewicz P., Kowalski-Jahn M., Petersen J., Bowin C.F., Slodkowicz G., Marti-Solano M., Rodríguez D., Hot B., Okashah N., Strakova K., Valnohova J., Babu M.M., Lambert N.A., Carlsson J. A conserved molecular switch in Class F receptors regulates receptor activation and pathway selection. Nat. Commun. 2019;10:667. doi: 10.1038/s41467-019-08630-2. 30737406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore A.R., Ceraudo E., Sher J.J., Guan Y., Shoushtari A.N., Chang M.T., Zhang J.Q., Walczak E.G., Kazmi M.A., Taylor B.S., Huber T., Chi P., Sakmar T.P., Chen Y. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016;48:675–680. doi: 10.1038/ng.3549. 27089179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chua V., Lapadula D., Randolph C., Benovic J.L., Wedegaertner P.B., Aplin A.E. Dysregulated GPCR signaling and therapeutic options in uveal melanoma. Mol. Cancer Res. 2017;15:501–506. doi: 10.1158/1541-7786.MCR-17-0007. 28223438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiGiacomo V., Maziarz M., Luebbers A., Norris J.M., Laksono P., Garcia-Marcos M. Probing the mutational landscape of regulators of G protein signaling proteins in cancer. Sci. Signal. 2020;13 doi: 10.1126/scisignal.aax8620. 32019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Raamsdonk C.D., Bezrookove V., Green G., Bauer J., Gaugler L., O'Brien J.M., Simpson E.M., Barsh G.S., Bastian B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. 19078957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Raamsdonk C.D., Griewank K.G., Crosby M.B., Garrido M.C., Vemula S., Wiesner T., Obenauf A.C., Wackernagel W., Green G., Bouvier N., Sozen M.M., Baimukanova G., Roy R., Heguy A., Dolgalev I. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010;363:2191–2199. doi: 10.1056/NEJMoa1000584. 21083380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ideno N., Yamaguchi H., Ghosh B., Gupta S., Okumura T., Steffen D.J., Fisher C.G., Wood L.D., Singhi A.D., Nakamura M., Gutkind J.S., Maitra A. GNAS(R201C) induces pancreatic cystic neoplasms in mice that express activated KRAS by inhibiting YAP1 signaling. Gastroenterology. 2018;155:1593–1607.e12. doi: 10.1053/j.gastro.2018.08.006. 30142336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu J., Matthaei H., Maitra A., Dal Molin M., Wood L.D., Eshleman J.R., Goggins M., Canto M.I., Schulick R.D., Edil B.H., Wolfgang C.L., Klein A.P., Diaz L.A., Jr., Allen P.J., Schmidt C.M. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3002543. 21775669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nairismägi M.L., Tan J., Lim J.Q., Nagarajan S., Ng C.C., Rajasegaran V., Huang D., Lim W.K., Laurensia Y., Wijaya G.C., Li Z.M., Cutcutache I., Pang W.L., Thangaraju S., Ha J. JAK-STAT and G-protein-coupled receptor signaling pathways are frequently altered in epitheliotropic intestinal T-cell lymphoma. Leukemia. 2016;30:1311–1319. doi: 10.1038/leu.2016.13. 26854024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Hayre M., Inoue A., Kufareva I., Wang Z., Mikelis C.M., Drummond R.A., Avino S., Finkel K., Kalim K.W., DiPasquale G., Guo F., Aoki J., Zheng Y., Lionakis M.S., Molinolo A.A. Inactivating mutations in GNA13 and RHOA in Burkitt's lymphoma and diffuse large B-cell lymphoma: a tumor suppressor function for the Gα13/RhoA axis in B cells. Oncogene. 2016;35:3771–3780. doi: 10.1038/onc.2015.442. 26616858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muppidi J.R., Schmitz R., Green J.A., Xiao W., Larsen A.B., Braun S.E., An J., Xu Y., Rosenwald A., Ott G., Gascoyne R.D., Rimsza L.M., Campo E., Jaffe E.S., Delabie J. Loss of signalling via Gα13 in germinal centre B-cell-derived lymphoma. Nature. 2014;516:254–258. doi: 10.1038/nature13765. 25274307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aittaleb M., Boguth C.A., Tesmer J.J. Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol. Pharmacol. 2010;77:111–125. doi: 10.1124/mol.109.061234. 19880753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miralles F., Posern G., Zaromytidou A.I., Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. 12732141. [DOI] [PubMed] [Google Scholar]
- 31.Cen B., Selvaraj A., Prywes R. Myocardin/MKL family of SRF coactivators: key regulators of immediate early and muscle specific gene expression. J. Cell. Biochem. 2004;93:74–82. doi: 10.1002/jcb.20199. 15352164. [DOI] [PubMed] [Google Scholar]
- 32.Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J.D., Wang C.-Y., Chinnaiyan A.M., Lai Z.-C., Guan K.-L. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. 18579750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu O.M., Miyamoto S., Brown J.H. Myocardin-related transcription factor A and Yes-associated protein exert dual control in G protein-coupled receptor- and RhoA-mediated transcriptional regulation and cell proliferation. Mol. Cell. Biol. 2016;36:39–49. doi: 10.1128/mcb.00772-15. 26459764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu F.X., Zhao B., Panupinthu N., Jewell J.L., Lian I., Wang L.H., Zhao J., Yuan H., Tumaneng K., Li H., Fu X.D., Mills G.B., Guan K.L. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–791. doi: 10.1016/j.cell.2012.06.037. 22863277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu O.M., Benitez J.A., Plouffe S.W., Ryback D., Klein A., Smith J., Greenbaum J., Delatte B., Rao A., Guan K.L., Furnari F.B., Chaim O.M., Miyamoto S., Brown J.H. YAP and MRTF-A, transcriptional co-activators of RhoA-mediated gene expression, are critical for glioblastoma tumorigenicity. Oncogene. 2018;37:5492–5507. doi: 10.1038/s41388-018-0301-5. 29887596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yagi H., Asanoma K., Ohgami T., Ichinoe A., Sonoda K., Kato K. GEP oncogene promotes cell proliferation through YAP activation in ovarian cancer. Oncogene. 2016;35:4471–4480. doi: 10.1038/onc.2015.505. 26804165. [DOI] [PubMed] [Google Scholar]
- 37.Park H.W., Kim Y.C., Yu B., Moroishi T., Mo J.S., Plouffe S.W., Meng Z., Lin K.C., Yu F.X., Alexander C.M., Wang C.Y., Guan K.L. Alternative Wnt signaling activates YAP/TAZ. Cell. 2015;162:780–794. doi: 10.1016/j.cell.2015.07.013. 26276632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., Antipin Y., Reva B., Goldberg A.P., Sander C., Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. 22588877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagata Y., Kontani K., Enami T., Kataoka K., Ishii R., Totoki Y., Kataoka T.R., Hirata M., Aoki K., Nakano K., Kitanaka A., Sakata-Yanagimoto M., Egami S., Shiraishi Y., Chiba K. Variegated RHOA mutations in adult T-cell leukemia/lymphoma. Blood. 2016;127:596–604. doi: 10.1182/blood-2015-06-644948. 26574607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hess J.M., Bernards A., Kim J., Miller M., Taylor-Weiner A., Haradhvala N.J., Lawrence M.S., Getz G. Passenger hotspot mutations in cancer. Cancer Cell. 2019;36:288–301.e14. doi: 10.1016/j.ccell.2019.08.002. 31526759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y., Xu X., Maglic D., Dill M.T., Mojumdar K., Ng P.K., Jeong K.J., Tsang Y.H., Moreno D., Bhavana V.H., Peng X., Ge Z., Chen H., Li J., Chen Z. Comprehensive molecular characterization of the Hippo signaling pathway in cancer. Cell Rep. 2018;25:1304–1317.e5. doi: 10.1016/j.celrep.2018.10.001. 30380420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R., Castro M.A.A., Gibb E.A., Kanchi R.S., Gordenin D.A., Shukla S.A., TCGA Research Network Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171:540–556.e25. doi: 10.1016/j.cell.2017.09.007. 28988769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freissmuth M., Gilman A.G. Mutations of GS α designed to alter the reactivity of the protein with bacterial toxins: substitutions at ARG187 result in loss of GTPase activity. J. Biol. Chem. 1989;264:21907–21914. 2557345. [PubMed] [Google Scholar]
- 44.Kleuss C., Raw A.S., Lee E., Sprang S.R., Gilman A.G. Mechanism of GTP hydrolysis by G-protein α subunits. Proc. Natl. Acad. Sci. U. S. A. 1994;91:9828–9831. doi: 10.1073/pnas.91.21.9828. 7937899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berman D.M., Wilkie T.M., Gilman A.G. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein α subunits. Cell. 1996;86:445–452. doi: 10.1016/s0092-8674(00)80117-8. 8756726. [DOI] [PubMed] [Google Scholar]
- 46.Leyme A., Marivin A., Casler J., Nguyen L.T., Garcia-Marcos M. Different biochemical properties explain why two equivalent Gα subunit mutants cause unrelated diseases. J. Biol. Chem. 2014;289:21818–21827. doi: 10.1074/jbc.M114.549790. 24982418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaulk S.G., Lattanzi V.J., Hiemer S.E., Fahlman R.P., Varelas X. The Hippo pathway effectors TAZ/YAP regulate dicer expression and microRNA biogenesis through Let-7. J. Biol. Chem. 2014;289:1886–1891. doi: 10.1074/jbc.C113.529362. 24324261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang C.S., Stampouloglou E., Kingston N.M., Zhang L., Monti S., Varelas X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. 2018;19 doi: 10.15252/embr.201643577. 29661856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reinhard N.R., Mastop M., Yin T., Wu Y., Bosma E.K., Gadella T.W.J., Jr., Goedhart J., Hordijk P.L. The balance between Gαi-Cdc42/Rac and Gα12/13-RhoA pathways determines endothelial barrier regulation by sphingosine-1-phosphate. Mol. Biol. Cell. 2017;28:3371–3382. doi: 10.1091/mbc.E17-03-0136. 28954861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bailey M.H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M.C., Kim J., Reardon B., Kwok-Shing Ng P., Jeong K.J., Cao S., Wang Z., Gao J. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;174:1034–1035. doi: 10.1016/j.cell.2018.07.034. 30096302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jensen M.A., Ferretti V., Grossman R.L., Staudt L.M. The NCI Genomic Data Commons as an engine for precision medicine. Blood. 2017;130:453–459. doi: 10.1182/blood-2017-03-735654. 28600341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson S., Fitzsimons M., Ferguson M., Heath A., Jensen M., Miller J., Murphy M.W., Porter J., Sahni H., Staudt L., Tang Y., Wang Z., Yu C., Zhang J., Ferretti V. Developing cancer informatics applications and tools using the NCI genomic data commons API. Cancer Res. 2017;77:e15–e18. doi: 10.1158/0008-5472.CAN-17-0598. 29092930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. 25516281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. 23323831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., Elvassore N., Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. 21654799. [DOI] [PubMed] [Google Scholar]
- 56.Wells C.D., Gutowski S., Bollag G., Sternweis P.C. Identification of potential mechanisms for regulation of p115 RhoGEF through analysis of endogenous and mutant forms of the exchange factor. J. Biol. Chem. 2001;276:28897–28905. doi: 10.1074/jbc.M102913200. 11384980. [DOI] [PubMed] [Google Scholar]
- 57.Maziarz M., Park J.C., Leyme A., Marivin A., Garcia-Lopez A., Patel P.P., Garcia-Marcos M. Revealing the activity of trimeric G-proteins in live cells with a versatile biosensor design. Cell. 2020;182:770–785.e16. doi: 10.1016/j.cell.2020.06.020. 32634377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maziarz M., Leyme A., Marivin A., Luebbers A., Patel P.P., Chen Z., Sprang S.R., Garcia-Marcos M. Atypical activation of the G protein Gαq by the oncogenic mutation Q209P. J. Biol. Chem. 2018;293:19586–19599. doi: 10.1074/jbc.RA118.005291. 30352874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained in the article except the raw data used for the genomics analysis, which corresponds to the Cancer Genome Atlas data set named TCGA-BLCA and was accessed and/or downloaded directly from cBioPortal or Genomic Data Commons as indicated under “Experimental procedures.”
HHS | NIH | National Institute of General Medical Sciences (NIGMS) (R01GM130120) to Mikel Garcia-Marcos
American Cancer Society (ACS) (PF-19-084-01-CDD) to Marcin Maziarz
American Cancer Society (ACS) (RSG-17-138-01-CSM) to Xaralabos Varelas
HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI) (R01HL12439) to Xaralabos Varelas