

Abstract
The effect of chronic of hyperinsulinemia in the fetal liver is poorly understood. Here, we produced hyperinsulinemia with euglycemia for ∼8 days in fetal sheep [hyperinsulinemic (INS)] at 0.9 gestation. INS fetuses had increased insulin and decreased oxygen and amino acid (AA) concentrations compared with saline-infused fetuses [control (CON)]. Glucose (whole body) utilization rates were increased, as expected, in INS fetuses. In the liver, however, there were few differences in genes and metabolites related to glucose and lipid metabolism and no activation of insulin signaling proteins (Akt and mTOR). There was increased p-AMPK activation and decreased mitochondrial mass (PGC1A expression, mitochondrial DNA content) in INS livers. Using an unbiased multivariate analysis with 162 metabolites, we identified effects on AA and one-carbon metabolism in the INS liver. Expression of the transaminase BCAT2 and glutaminase genes GLS1 and GLS2 was decreased, supporting decreased AA utilization. We further evaluated the roles of hyperinsulinemia and hypoxemia, both present in INS fetuses, on outcomes in the liver. Expression of PGC1A correlated only with hyperinsulinemia, p-AMPK correlated only with hypoxemia, and other genes and metabolites correlated with both hyperinsulinemia and hypoxemia. In fetal hepatocytes, acute treatment with insulin activated p-Akt and decreased PGC1A, whereas hypoxia activated p-AMPK. Overall, chronic hyperinsulinemia produced greater effects on amino acid metabolism compared with glucose and lipid metabolism and a novel effect on one-carbon metabolism in the fetal liver. These hepatic metabolic responses may result from the downregulation of insulin signaling and antagonistic effects of hypoxemia-induced AMPK activation that develop with chronic hyperinsulinemia.
Keywords: fetus, insulin signaling, liver, metabolism
INTRODUCTION
Relatively little is known about the effects of chronic hyperinsulinemia in the late gestation fetal liver. This is important because late gestation is a critical developmental window for the maturation of metabolic pathways, including the activation of gluconeogenesis, lipid oxidation, and antioxidant defenses in the fetal liver (21, 25, 41). These and other pathways support the function of the liver postnatally as the major site of carbohydrate metabolism and triglyceride synthesis. Insulin regulates these pathways via canonical signaling involving Akt and mTOR with its downstream targets S6K and S6 (26, 64, 65). As a result, insulin signaling and action in the adult liver increases glucose uptake, glycogen storage, glycolysis, amino acid uptake, protein synthesis, and lipid synthesis and suppresses glucose production (52, 60, 79). In the fetal liver, however, the response to insulin may be unique due to the immaturity of these pathways and because the fetus receives a constant supply of glucose and amino acids from the placenta in the umbilical circulation rather than ingestion of meals with increased lipid content after birth (28, 80). Furthermore, in contrast to the adult liver whereby insulin suppresses glucose production and increases glucose uptake (57, 79), insulin’s effects in the fetal liver may be limited to promoting glucose utilization due to the developmental absence of hepatic glucose production (24).
Fetuses from diabetic pregnancies are exposed to increased glucose concentrations, which result in increased fetal insulin secretion and fetal hyperinsulinemia (66). Consequently, these fetuses have an increased risk for fetal overgrowth, being born large for gestational age, and some are characterized by fetal hypoxemia and a higher risk of an intrauterine fetal demise (9, 23, 73, 74). The effects in the liver that result from hyperinsulinemia in these fetuses are largely unknown. Exposure to chronic hyperinsulinemia may disrupt the normal development of hepatic metabolism in the fetus. Given the key role of the liver in coordinating substrate metabolism, it is important to determine the specific metabolic effects of hyperinsulinemia in the fetal liver.
Several studies in sheep and monkey models support a role for experimental hyperinsulinemia on fetal whole body glucose metabolism and growth (8, 18, 20, 30, 31, 53, 54, 71). However, only a few studies have measured the liver specific effects (70, 78), and none have done so under chronic conditions with euglycemia. We have shown that fetuses exposed to reduced glucose supply and hypoglycemia for 8 wk have an early activation of hepatic glucose production and that a physiological twofold increase in insulin concentrations for 1 wk decreases gluconeogenic gene expression in the liver (78). Although we did not measure the liver response to insulin in normal fetuses in that study, we have shown that an acute supraphysiological increase in insulin concentrations during a 4-h hyperinsulinemic-euglycemic clamp produced robust responses, including the activation of Akt signaling and gene expression in the liver of late-gestation fetal sheep (37, 76). Another study in the rhesus macaque fetus has shown that chronic hyperinsulinemia for 3 wk in late gestation decreased gluconeogenic enzyme activity and increased fatty acid synthase activity in the liver (70). However, fetal glycemia was not maintained, and fetal concentrations of glucose decreased during hyperinsulinemia (70). The confounding effect of hypoglycemia is important because other studies in fetal sheep have shown that insulin’s effects on promoting whole (fetal) body rates of oxygen consumption, glucose utilization, and glucose oxidation are maximal when glucose supply and glucose concentrations are maintained or increased in the fetus (18, 19, 29, 31). Overall, these studies provide limited information about the signaling and metabolic responses in the fetal liver during chronic hyperinsulinemia in the absence of decreased glucose concentrations.
The goal of this study was to determine the effect of chronic experimental hyperinsulinemia under euglycemic conditions on signaling and metabolic pathways in the developing fetal liver. We used a model in late-gestation fetal sheep (0.9 gestation) that produced a twofold increase in fetal plasma insulin concentrations whereas fetal glucose concentrations were maintained at baseline values with an exogenous glucose infusion (1, 7). These hyperinsulinemic (INS) fetuses had an ∼50% increase in total glucose delivery (1, 7), representing the sum of the net fetal uptake rate from the placenta and the exogenous glucose infusion rate necessary to maintain glycemia. This design prevents hypoglycemia and allows for the study of insulin-specific responses independent of fetal glucose concentrations. It also permits maximal insulin-stimulated glucose utilization that is not limited by glucose supply. Additionally, INS fetuses had 30% lower arterial oxygen concentrations, raising the possibility that hypoxemia may have additional effects in the fetal liver during hyperinsulinemia (1, 7). Our hypothesis was that chronic fetal hyperinsulinemia would activate insulin signaling and anabolic pathways in the fetal liver and that some of these responses would be unique relative to those expected in the adult liver. To test this, we used a series of targeted pathway analyses to evaluate insulin signaling, glucose metabolism, lipid synthesis, and mitochondrial function. Next, to complement these targeted studies, we used a global multivariate metabolomics analysis to identify novel targets of insulin action in the fetal liver. Finally, we used isolated fetal hepatocytes to isolate the acute effects of insulin and hypoxemia, both of which were present in vivo in INS fetuses. Our results provide novel information about the chronic effect of hyperinsulinemia in the fetal liver.
METHODS
Experimental model of chronic fetal hyperinsulinemia.
The liver tissue samples used in this study were obtained from fetuses described in our previous publications where other in vivo metabolic data and responses in the skeletal muscle and pancreas were reported (1, 7). Experiments were designed and reported with reference to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (43). Briefly, pregnant Columbia-Rambouillet ewes with singleton pregnancies were studied under regulatory compliance at the University of Colorado, which is accredited by the American Association for the Accreditation of Laboratory Animal Care International (AAALAC). To produce experimental hyperinsulinemia with euglycemia, late-gestation fetuses were surgically instrumented with indwelling catheters placed in the umbilical vein, descending aorta, and femoral veins at ∼120 dGA. Animals were allowed to recover from surgery for a minimum of 5 days before infusions were started. INS fetuses (n = 7) received a continuous intravenous (iv) infusion of Humulin R insulin that was increased over the first 3 days to produce a twofold increase in fetal concentrations. A concurrent infusion of dextrose with variable rate was used prevent a fall in glucose concentrations based on daily measurement of fetal glucose concentrations. Fetuses in the control (CON) group (n = 8) received a saline infusion adjusted to match the infusion rate in the INS fetuses. One day before the end of the infusion period, metabolic studies were performed. To measure glucose metabolism, [6,6-2H2]glucose was infused with a 3-mL bolus, followed by a continuous infusion at 3 mL/h (30 mg/mL) (4, 39). Umbilical blood flow was measured by steady-state diffusion, using ethanol as a tracer, as previously reported (7). After isotopic steady state was reached (∼120 min), four consecutive steady-state fetal arterial blood samples were collected for measurement of nutrients, hormones, and tracer enrichments. The data in Table 1 represent the average of these data. On the final day of study, pregnant ewes and their fetuses were euthanized, and fetal liver tissue samples were collected and snap-frozen in liquid nitrogen.
Table 1.
Fetal characteristics
| Variable (n = 8 CON and 7 INS fetuses) | CON | INS |
|---|---|---|
| Fetal gestational age, days | 134.4 ± 0.5 | 133.6 ± 0.8 |
| Infusion period, days | 8.1 ± 0.3 | 7.6 ± 0.3 |
| Fetal weight, g | 3,264 ± 216 | 3,152 ± 228 |
| Fetal liver weight, g | 2.30 ± 0.23 | 2.18 ± 0.16 |
| Fetal artery | ||
| Insulin, plasma, ng/mL | 0.39 ± 0.07 | 0.97 ± 0.12* |
| Glucose, plasma, mM | 1.18 ± 0.09 | 1.18 ± 0.09 |
| Lactate, plasma, mM | 2.30 ± 0.23 | 2.18 ± 0.16 |
| Amino acids, plasma, mM | 4.51 ± 0.12 | 3.35 ± 0.307* |
| Oxygen content, blood, mM | 3.16 ± 0.09 | 1.98 ± 0.2* |
| Norephinephrine, plasma, pg/mL | 607 ± 98 | 1,218 ± 166* |
| Cortisol, plasma, ng/mL | 7.97 ± 1.03 | 9.38 ± 1.04 |
| Glucagon, plasma, pg/mL | 31.5 ± 4.2 | 42.8 ± 6.2 |
| IGF-I, plasma, ng/mL | 123.3 ± 8.2 | 156.5 ± 13.4* |
Glucose tracer enrichments and calculations.
Glucose tracer enrichments [molar percent excess (MPE)] were measured in the fetal artery and umbilical vein plasma samples (4, 39). Briefly, glucose was converted to the aldonitrile peracetate derivative for gas chromatography-mass spectrometry (GC-MS) analysis. Glucose [6,6-2H2] enrichment was monitored at m/z of 330/328 ratio. Glucose MPE was calculated as the difference in peak area ratios between unenriched (baseline) and enriched samples. Fetal glucose utilization rate was calculated as previously described (32, 33, 76). Fetal glucose production rate was calculated as the difference between fetal glucose utilization rate and net fetal glucose uptake rate (33). One fetus in the CON group had an umbilical venous catheter that did not draw, which prevented the measuring of umbilical blood flow and uptake rates, leaving seven CON fetuses for the glucose tracer-based measurements.
Hepatic gene expression.
RNA was extracted from the fetal liver and used in real time PCR as described (5, 76). Briefly, cDNA was diluted 1:10 and used in a 10-μL reaction with primers and 1× SYBR green master mix (Roche) for qPCR using the LightCycler 480 (Roche) and analyzed using the absolute quantification/2nd derivative maximum analysis method with relative standard curves produced from serial fourfold dilutions of a pooled liver cDNA sample (76, 77). A complete listing of the genes measured is provided in Table 2, including the gene name and HGNC (HUGO Gene Nomenclature Committee)-approved gene symbol. All primers are designed to span introns and avoid alternative spliced variants using ovine gene sequences in NCBI. The selected amplicon was blasted against the sheep genome to confirm 100% identity. Primer efficiency was tested, and the presence of a single peak in the amplicon melt curve analysis was confirmed. Results were normalized to RPS15 mRNA expression which, was similar among all samples (5, 76–78).
Table 2.
PCR primer sequences
| Officiala | Commonb | Gene Name | Forward Primer | Reverse Primer |
|---|---|---|---|---|
| Insulin and nutrient signaling | ||||
| INSR-A* | Insulin receptor, transcript a | CCCGAAGACCGACTCTCA | AGGCCTGGG ATGAAAAC | |
| INSR-B* | Insulin receptor, transcript b | CCGAAGACCGACTCTCAGAT | CAACAGGGCCTGAAGATGAT | |
| TAT | Tyrosine aminotransferase | CCTGCCGACAGATCCTGAAG | TCCTCCCGACTGGATAAGCA | |
| CDKN1A | P21 | Cyclin-dependent kinase inhibitor 1a | CCGAGACTTTCTGAACCGCT | GCAATCAGTGGAGTGAGGCT |
| Glucose metabolism | ||||
| PFKL* | Phosphofructokinase, liver isoform | TGGTGGCTCCATGCTGGGGA | GCAGGGCGTGGATGCTGTGA | |
| PFKFB1 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 1 | GGCCCTGAATGAGATTGATGCGG | TCCCTTAGGATAGCGGTAGCGG | |
| PKLR* | Pyruvate kinase, liver, red cell isoform | TGGCGGGAAAGCCCGTTGTC | CCAGAACGGCGTTGGCCACA | |
| PKM* | Pyruvate kinase, muscle isoform | ACCACGCAGAGACCATCAAG | GGTCCTTTAGTGTCCAGGGC | |
| PDK1 | Pyruvate dehydrogenase kinase 1 | TGGAGCATCACGCTGACAAA | CTCAGAGGAACACCACCTCC | |
| PDK2 | Pyruvate dehydrogenase kinase 2 | TACATGGCCTCTCCTGACCT | AAGCATGTGGTAGAGGTGGG | |
| PDK4* | Pyruvate dehydrogenase kinase 4 | CCCAGAGGACCAAAAGGCAT | GGGTCAGCTGTACAGGCATC | |
| LDHA* | Lactate dehydogenase A | CATGGCCTGTGCCATCAGTA | GGAAAAGGCTGCCATGTTGG | |
| LDHB* | Lactate dehydogenase B | GAGGGAGCGATCCCAAACAA | CAGAATGCTGATGGCACACG | |
| PCK1* | Phosphoenolpyruvate carboxykinase 1, cytosolic | TGTCCGAGGAGGATTTTGAG | ATGCCAATCTTGGACAGAGG | |
| PCK2* | Phosphoenolpyruvate carboxykinase 2, mitochondrial | GCCTGTGCTTCAGGCCCTGG | TGCATGGCCACTGGCACACC | |
| G6PC* | Glucose-6-phosphatase catalytic-subunit | GGATTCTGGATCGTGCAACT | ATCCAATGGCGAAACTGAAC | |
| PC* | Pyruvate carboxylase | GCACAGCATGGGGCTTGGCT | AACTGGGCCAGGTCCCCCAC | |
| Lipid metabolism | ||||
| SREBF1* | SREBP1C | Sterol regulatory element binding transcription factor 1, transcript c | ATGGATTGCACGTTCGAAG | CGGGAATTCGCTGTCTTG |
| ACACA* | ACC1 | Acetyl-CoA carboxylase alpha | CGATGTCAACCTCCCTGCCGC | ATCGCCCCAGGGAGAGACCC |
| FASN* | Fatty acid synthase | GACACATCCTTTGAGCAGCA | TTTGCCATTTCCAGGAATC | |
| MLXIPL | CHREBP | Carbohydrate-response element-binding protein | CAAGTGGCGCATCTACTACAAG | AAGAGCTGCTCGCACCAT |
| Amino acid metabolism | ||||
| BCAT1 | Branched-chain amino acid transaminase 1 | CATCCTGGACTTGGCACACA | CAGGCGGTACCTGAACCAAA | |
| BCAT2* | Branched-chain amino acid transaminase 2 | TGTCCTCCGTTTCCACAAGG | AGCTTTACACCGGGAGCATC | |
| BCKDK* | Branched-chain keto acid dehydrogenase kinase | AAAGTGGGTGGACTTTGCCA | GCATCGGGATGAAGGGGAAA | |
| GLUD1* | Glutamate dehydrogenase 1 | AGCGCTCTGCCAGGCAAATCAT | GCGGCCGTTCTCAGGTCCAG | |
| ASNS* | Asparagine synthetase | AAGCCATGACGGAAGATGGG | ATGTCCTGGCGAAAAAGGCT | |
| GLS | Glutaminase 1 | CCCAGAAGGCACAGACATGGTTGG | GGGCAGAAGCCACCATTAGCCA | |
| GLS2 | Glutaminase 2 | CTGGTGCCATTGTTGTGAGC | ATGTGGCATTGCTGAAACCC | |
| Mitochondrial function | ||||
| PPARGC1A* | PGC1A | PPARG coactivator-1α | GTGACTCTGGGGTCAGAGGA | CACCAAACCCACAGAGAACC |
| ESRRA* | Estrogen-related receptor-α | GAGCGCGAGGAGTATGTTCT | AGGGCCTCGTGTAGAGCTTC | |
| YY1* | YY1 transcription factor | ACCTGGCATTGACCTCTCAG | GGGCAAGCTATTGTTCTTGG | |
| CYCS* | Cytochrome c, somatic | AACCTCCATGGTCTGTTTGG | CTCCATCAGCGTCTCCTCTC | |
| IDH1 | isocitrate dehydrogenase 1 | GACATGGTGGCCCAAGCTAT | CATCATGCCGAGAGAGCCAT | |
| IDH2 | Isocitrate dehydrogenase 2 | CTGGCCACCCAGAAGTACAG | CCATTGGGGCTCTTCCACAT | |
| NRF1 | Nuclear respiratory factor 1 | ACGGAAACGTCCTCATGTGT | ATAGCTTGCTGTCCCACTCG | |
| NFE2L2 | NRF2 | Nuclear factor, erythroid 2 like 2 | GCTTTTGGCAGAGACATTCC | GCATTGAAGACTGGGCTCTC |
| Reference gene | ||||
| RPS15* | Ribosomal protein S15 | ATCATTCTGCCCGAGATGGTG | CGGGCCGGCCATGCTTTACG | |
| Mitochondrial/nuclear DNA | ||||
| HBB | Hemoglobin subunit beta | CCCATGGCAAGAAGGTGCTA | CGTGCAGCTTATCACAGTGC | |
| MT-CYT | Cytochrome b | TATACACGCAAACGGGGCAT | GCTGTGGCTATTGTCGCAAA | |
Hepatic protein expression.
Whole cell protein lysates were prepared from liver tissue, and Western immunoblotting was performed using previously described methods (4, 5, 76–78). Antibodies against phosphorylated and/or total forms of IRβ, Akt (S473), mTOR (S2448), S6K (S421, T424), S6 (S235/S236), AMP-activated protein kinase (AMPK; T172), ERK (T202/Y204), and pyruvate dehydrogenase (PDH; S293), and actin proteins were used as previously described (4, 5, 77, 78). Specificity was confirmed by the presence of a single immunoreactive band at the expected molecular weight. Results were quantified on each blot and expressed as a ratio of phosphorylated to total protein or relative to actin for proteins without a measured phosphorylated protein.
Hepatic metabolite measurements.
Fetal liver tissue samples were used in metabolomic profiling at the University of Colorado School of Medicine Biological Mass Spectrometry Core Facility (15, 59, 81). Briefly, liver tissue (25 mg) samples were extracted in ice-cold lysis-extraction buffer (methanol-acetonitrile-water, 5:3:2). Analyses were performed using a Vanquish UHPLC system coupled online to a Q Exactive mass spectrometer (Thermo). Samples were resolved over a Kinetex C18 column (2.1 × 150 mm, 1.7 µm; Phenomenex, Torrance, CA) at 25°C using a 3-min isocratic condition of 5% acetonitrile, 95% water, and 0.1% formic acid flowing at 250 µL/min or using a 9 min gradient at 400 µL/min from 5 to 95% B (A: water-0.1% formic acid; B: acetonitrile-0.1% formic acid) (58). Mass spectrometry analysis and data elaboration were performed as described (14). Metabolite assignments were performed using MAVEN (11). Metabolites were excluded if more than half the samples contained a zero value, and metabolites where less than half the samples contained a zero value were replaced with the half-minimum value for the metabolite. Peak intensity values for liver metabolites were analyzed with MetaboAnalyst 4.0 (10). Sample normalization was performed using normalization by mean with data autoscaling. Normalized data were used in univariate and multivariate analyses. Metabolites related to specific pathways were analyzed with univariate t tests, and heatmaps were generated for data visualization in GraphPad Prism.
Multivariate analysis was used, with all 162 metabolites identified to determine the global effect of hyperinsulinemia using MetaboAnalyst. Principal component analysis was performed using partial least squares discriminant analysis (PLS-DA). The 20 metabolites with the highest variable importance in projection (VIP) scores were identified and used to generate a heatmap with hierarchical clustering of metabolites. For pathway analysis, the top 20 VIP ranked metabolites were used in the Pathway Analysis module in MetaboAnalyst with the Human KEGG pathway term library. Pathways with adjusted Holm P < 0.05 were declared significant and corresponded to a false discovery rate (FDR) adjusted to P < 0.01. Impact scores are calculated from the pathway topology analysis.
Hepatic glycogen and triglyceride concentrations.
Fetal liver glycogen content was measured as described and expressed as milligrams per gram of wet tissue weight (46). Fetal liver triglyceride content was measured (no. TR22421; ThermoFisher Infinity Triglyceride Reagent) following lipid extraction and normalized to wet tissue weight (50, 75).
Assays of hepatic mitochondrial mass.
DNA was isolated from the fetal liver using phenol-chloroform extraction. DNA was diluted to 10 ng/μL and used in 10-μL reactions with primers and 1× SYBR green master mix (Roche) for quantitative PCR (qPCR) using the LightCycler 480 (Roche). Data were analyzed with LightCycler 480 software using the absolute quantification/second derivative maximum analysis method with relative standard curves produced from serial fourfold dilutions of a pooled liver DNA sample (76, 77). Primers were developed for real-time PCR assays for the HBB (nuclear gene) and MT-CYT (mitochondrial gene) (Table 2). All primers were designed within an exon and avoided alternative spliced variants using ovine gene sequences. The mitochondrial DNA ratio was calculated as MT-CYT expression expressed relative to HBB. Fetal liver citrate synthase activity was measured in liver tissue homogenates (69). Samples were homogenized in buffer containing 250 mM sucrose, 10 mM Tris Base, and 1 mM EGTA. Citrate synthase activity was measured colorimetrically in 30-s intervals for 3 min, and rates were calculated using the established reaction equation (no. 701040; Cayman). Each reaction contained 15 μg of protein, and the assay was performed in duplicate. Fetal liver thiobarbituric acid-reactive substances (TBARS) content was measured colorimetrically (no. 700870; Cayman) in protein lysate samples prepared as described above and expressed relative to protein content.
Primary fetal hepatocytes.
Primary hepatocytes were isolated from livers from late-gestation (0.9 gestation) fetal sheep that were studied under basal conditions as part of other ongoing studies in our laboratory. Briefly, a portion of the right lobe of the fetal liver was perfused and digested with collagenase, and hepatocytes were separated by centrifugation (75, 76). Hepatocytes were plated in DMEM with 1.1 mM glucose supplemented with 2 mM glutamine, 2.2 mM lactate, 1 mM pyruvate, 1× nonessential amino acids, 100 U/mL penicillin-streptomycin, 1 nM insulin, 100 nM dexamethasone, and 10% FBS on collagen coated Primaria (BD Falcon) plates. After a 4-h attachment period, cells were washed and media replaced with serum-free DMEM plus 0.2% BSA (SF media). The next day, for signaling experiments, hepatocytes were washed and incubated in fresh SF media (basal) at ambient oxygen conditions with 5% CO2 for 4 h. Insulin was added at 1 or 10 nM doses for the final 30 min and compared with cells with only SF media. Another treatment group included exposing hepatocytes to low-oxygen conditions for 4 h in an incubator set to 3% oxygen (12, 51). Cells were lysed for Western blotting for measurement of phosphorylated and total forms of Akt (S473) and AMPK (T172). For gene expression studies, hepatocytes were washed and incubated with SF media (basal) or in SF with 100 nM insulin at ambient oxygen conditions and 5% CO2 and collected for RNA and subsequent real time PCR. A third treatment group included exposing hepatocytes 3% oxygen conditions for 24 h. Gene expression was normalized to RPS15 expression and expressed as a Log2 fold change relative to a basal treatment in ambient oxygen conditions (represented by 0 in graphs). Studies were performed in two preparations of isolated primary fetal hepatocytes with duplicate wells per treatment.
Statistical analyses.
Data were analyzed by two-sided t test using GraphPad Prism to compare CON versus INS groups. Data are presented as means ± SE, and significance was considered when P ≤ 0.05. For hepatocyte signaling data, a two-sided t test was used. For hepatocyte gene expression, a one-sided t test relative to the basal treatment was used since these experiments were designed to test whether the gene expression change observed in vivo in the liver was changed in the same direction in vitro.
RESULTS
Effect of hyperinsulinemia on whole (fetal) body glucose metabolism.
The characteristics of CON and INS fetuses are summarized in Table 1 (1, 7). Fetal body weight, age, and length of infusion were similar between groups. Plasma insulin concentrations were twofold higher, by design, in INS compared with CON fetuses. Plasma glucose and lactate concentrations were similar between CON and INS groups. Plasma total amino acid concentrations, representing the sum of all individual amino acids, were 25% lower in INS fetuses. Blood oxygen content was 37% lower in INS compared with CON fetuses. Plasma norepinephrine concentrations were increased twofold in INS fetuses (Table 1). There were no differences in plasma cortisol or glucagon concentrations. Plasma IGF-I concentrations were increased in INS fetuses.
To determine the effect of hyperinsulinemia on whole body fetal glucose metabolism, we infused [6,6-2H2]glucose into the fetus to measure fetal glucose utilization. Fetal glucose utilization rates were increased in INS compared with CON fetuses (Fig. 1). These rates were similar to the total glucose entry rates in fetuses in both CON and INS groups (21), demonstrating the absence of significant endogenous fetal glucose production in either group (Fig. 1). This is consistent with the expected absence of endogenous fetal hepatic glucose production at this developmental time point when euglycemia is maintained (21, 24, 41). In the following studies, we evaluated the effect of hyperinsulinemia on signaling and metabolic pathways in the fetal liver.
Fig. 1.

Chronic hyperinsulinemia increases glucose utilization in the fetus. Rates of fetal (whole body) glucose utilization and endogenous glucose production measured in control (CON) and hyperinsulinemic (INS) fetuses. Means ± SE are shown (n = 8 CON and 7 INS). *P < 0.05 in CON vs. INS.
Fetal hepatic glucose metabolism.
We first sought to determine whether the increase in whole body fetal glucose utilization rates in INS fetuses (shown in Fig. 1) was associated with pathways known to increase glucose utilization in the liver. There was no change in the glycolytic genes PFKL, PKLR, or PKM, yet there was a twofold increase in the bifunctional gene PFKFB1 (Fig. 2A). Expression of genes encoding the different pyruvate dehydrogenase kinase protein isoforms PDK1, PDK2, and PDK4 each were decreased by 20–50% in the INS fetal liver. However, despite the decrease in mRNA expression of these upstream kinase inhibitors of PDH, there was no decrease in the phosphorylation of the PDH protein (data not shown). Expression of the lactate dehydrogenase gene LDHB, but not LDHA, was decreased by 20% in the INS fetal liver. At the metabolite level, there were no differences in glucose, intermediates in glycolysis, lactate, and pyruvate, or products of the pentose phosphate pathways (P > 0.18 for metabolites shown; Fig. 2B). There also were no differences in hepatic glycogen concentrations in CON and INS fetal livers (Fig. 2C). In line with the absence of glucose production (Fig. 1), there was no difference in expression of the gluconeogenic genes PCK1, PCK2, PC, or G6PC (Fig. 2A).
Fig. 2.
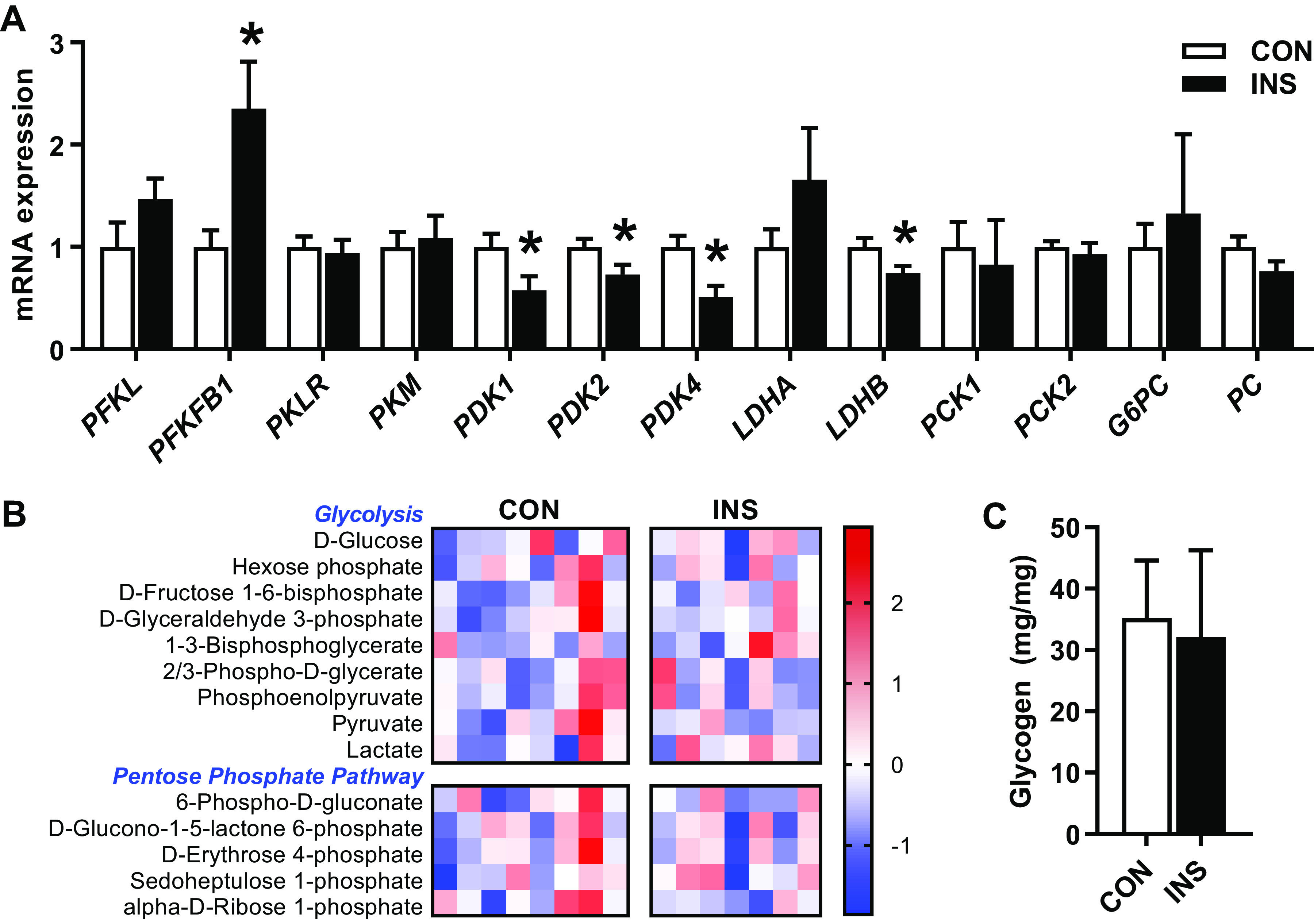
Effect of chronic hyperinsulinemia on glucose metabolism pathways in the fetal liver. A: expression of genes involved in glycolysis, glucose oxidation, lactate metabolism, and gluconeogenesis measured in liver of control (CON) and hyperinsulinemic (INS) fetuses. B: heatmap showing normalized relative intensity of metabolites related to glycolysis and pentose phosphate pathway. Quantitative changes are color coded from blue (low) to red (high). C: fetal hepatic glycogen content. Means ± SE are shown (n = 8 CON and 7 INS). *P < 0.05 in CON vs. INS.
Insulin signaling pathway in the fetal liver.
We next determined the effect of hyperinsulinemia on expression and activation (phosphorylation) of components in the insulin-signaling pathway (26, 64). Protein expression of the insulin receptor β-subunit IRβ and Akt was not different (Fig. 3, A and B). There was no detectable phosphorylation of Akt in CON or INS livers (data not shown) and no increase in the phosphorylation of mTOR, S6K, S6, or ERK proteins in the INS fetal liver (Fig. 3, A and C). Expression of both insulin receptor splice variants, INSR-A and INSR-B, was decreased by 50% in INS fetal liver (Fig. 3D). To further assess hepatic insulin action, we measured the expression of two insulin target genes (60). The expression of tyrosine aminotransferase (TAT) is normally suppressed by insulin and was decreased by 60%, and yet expression of p21 cell cycle regulator (CDKN1A), normally increased with insulin, was not different in the INS fetal liver (Fig. 3D).
Fig. 3.

Effect of chronic hyperinsulinemia on insulin- and nutrient-signaling pathways in the fetal liver. A: protein expression was measured by Western blotting in liver samples of control (CON) and hyperinsulinemic (INS) fetuses for IRb and Akt and for phosphorylated and total mammalian target of rapamycin (mTOR; S2448), S6K (S421, T424), S6 (S235/S236), and ERK (T202/Y204). B: results were quantified for IRb and Akt expression and normalized to actin. C: results were quantified, and the ratio of phosphorylated to total protein was calculated for mTOR, p70, S6, and ERK. D: expression of genes involved in insulin signaling in the fetal liver. Means ± SE are shown (n = 8 CON and 7 INS). *P < 0.05 in CON vs. INS. INSR-A and INSR-B, insulin receptor genes A and B, respectively; TAT, tyrosine aminotransferase.
Fetal hepatic lipid metabolism.
Insulin promotes fat storage by converting excess glucose into lipids in the adult liver (64, 79). Because INS fetuses have higher insulin concentrations and increased glucose utilization, we tested the effect on the lipogenic pathway. We found no increase in the expression of the lipogenic genes ACC1 or FASN or transcription factor sterol regulatory element-binding protein-1c (SREBP1C) in the INS compared with CON fetal liver (Fig. 4A). Interestingly, expression of carbohydrate-responsive element-binding protein (CHREBP), a glucose-responsive transcription factor and regulator of lipogenic gene expression, was decreased by 50% in the INS fetal liver (Fig. 4A) Hepatic triglyceride content and glycerol-3-phosphate levels were similar between groups (Fig. 4B), and yet phosphoethanolamine, a metabolite in glycerophospholipid synthesis, was increased by 20% (P < 0.05), and choline levels tended to be increased (P = 0.06) (Fig. 4C). We found no differences in carnitine or in the profile acyl-carnitines representing products of lipid oxidation (Fig. 4C).
Fig. 4.
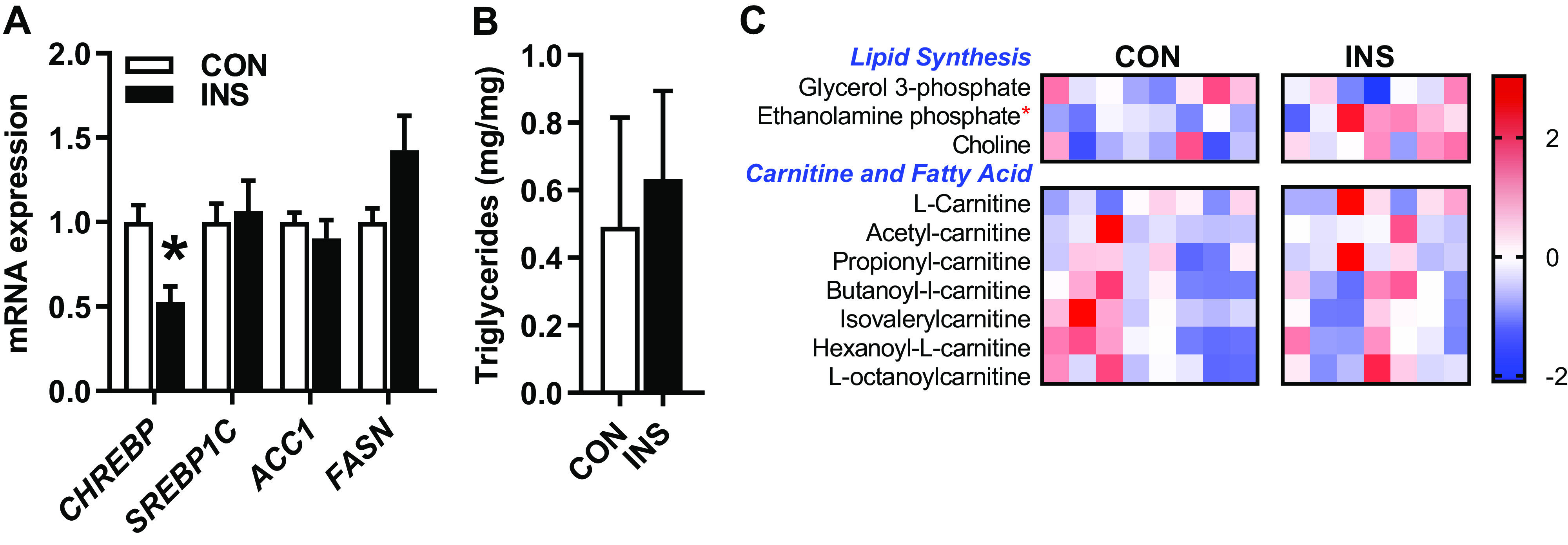
Effect of chronic hyperinsulinemia on fetal hepatic lipid metabolism pathways. A: expression of genes involved in lipid synthesis in liver of control (CON) and hyperinsulinemic (INS) fetuses. B: triglyceride content expressed per mg of tissue. C: heatmap showing normalized relative intensity of metabolites related to lipid synthesis and oxidation. Quantitative changes are color coded from blue (low) to red (high). Means ± SE are shown (n = 8 CON, 7 INS). *P < 0.05 in CON vs. INS. ACC1, acetyl-CoA carboxylase 1; CHREBP, carbohydrate-responsive element-binding protein; FASN, fatty acid synthase; SREBP1C, sterol regulatory element-binding protein-1c.
Fetal hepatic oxidative metabolism and mitochondrial mass.
We next sought to determine whether insulin promoted oxidative metabolism and energy production based on the profile of intermediates in the TCA cycle and energy status markers. We found no differences in levels of TCA cycle intermediates (P > 0.10 for metabolites shown; Fig. 5A). We also found no differences in the levels of nucleotides or energy metabolites measured in CON versus INS fetal livers (see Supplemental Table S1; Supplemental Material for this article can be found online at https://doi.org/10.6084/m9.figshare.12485444.v1). However, the ratio of AMP to ATP was increased (P = 0.08; Fig. 5B). Consistent with this, we found that the phosphorylation of AMPK protein, which is regulated by increased AMP/ATP ratio (27), was increased by more than twofold in INS fetal livers (Fig. 5, C and D).
Fig. 5.
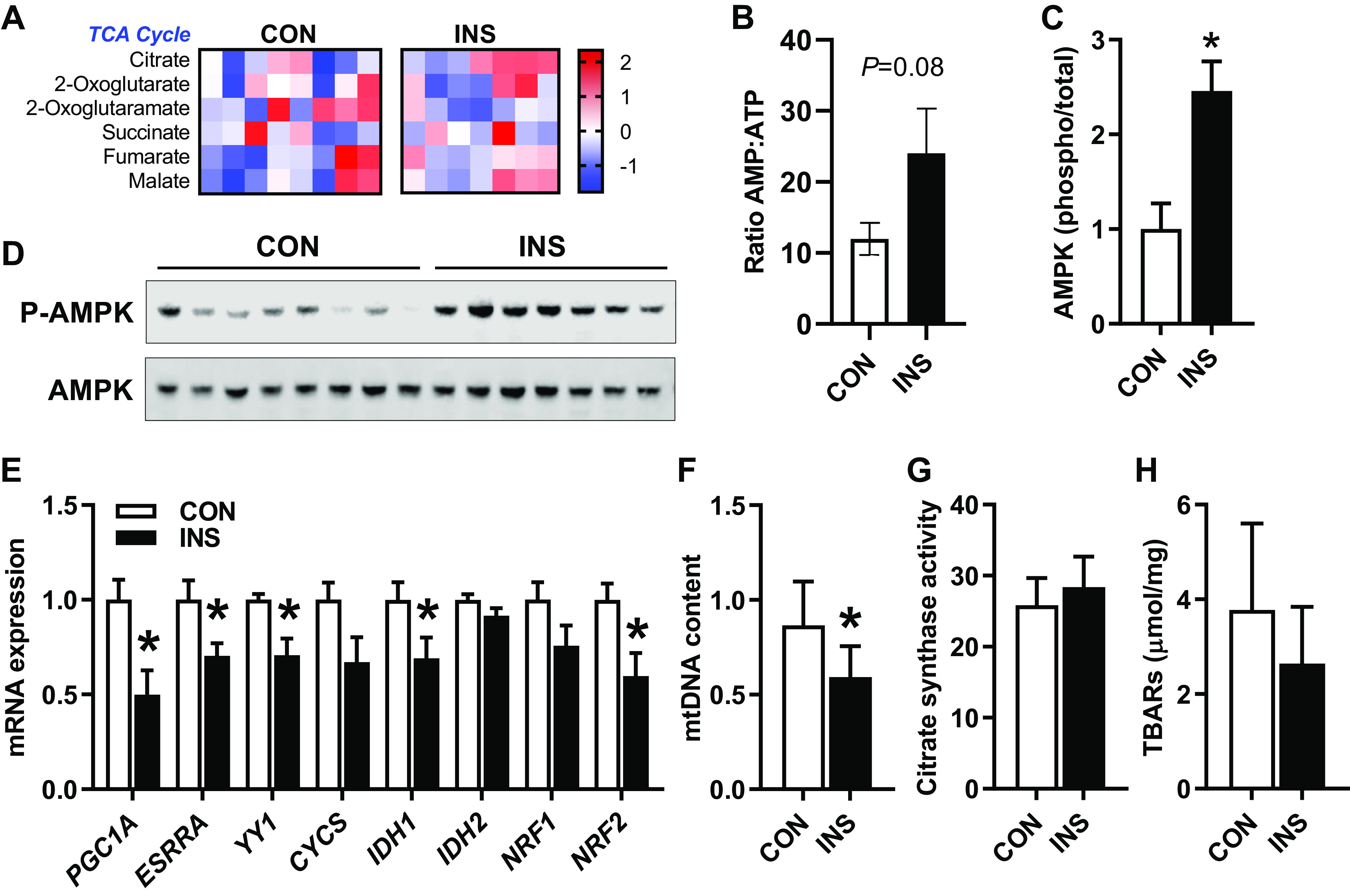
Effect of chronic hyperinsulinemia on markers of fetal hepatic mitochondrial substrate metabolism and function. A: heatmap showing normalized relative intensity of metabolites in the TCA cycle in liver of control (CON) and hyperinsulinemic (INS) fetuses. Quantitative changes are color coded from blue (low) to red (high). B: ratio of the relative intensity of AMP/ATP. C: protein expression measured by Western blotting for phosphorylated and total AMP-activated protein kinase (AMPK; T172). Results were quantified, and the ratio of phosphorylated to total protein was AMPK. D: representative blots for phosphorylated and total AMPK. E: expression of genes involved in mitochondrial biogenesis and function. F: ratio of MT-CYB to HBB DNA as markers of mitochondrial DNA (mtDNA) and nuclear DNA, respectively, measured in DNA samples from CON and INS fetal livers. G: citrate synthase activity expressed relative to protein content in CON and INS fetal livers (nmol·min−1·mg−1 protein). H: thiobarbituric acid-reactive substances (TBARS) relative to protein in CON and INS fetal livers (μmol/mg protein). Means ± SE are shown (n = 8 CON and 7 INS). *P < 0.05 in CON vs. INS. CYCS, cytochrome c, somatic; IDH1 and -2, isocitrate dehydrogenase 1 and 2; NRF1 and -2, nuclear respiratory factor 1 and 2, respectively; PGC1A, peroxisome proliferator-activated receptor-γ coactivator-1α.
We next evaluated markers for and pathways regulating mitochondrial mass. Hepatic expression of the transcriptional coactivator PGC1A and two of its target genes, ESRRA and YY1 (35), was decreased in the INS fetal liver (Fig. 5E). Expression of IDH1 and NRF2 also was decreased, whereas expression of CYCS, IDH2, or NRF1 was not changed. To determine whether these changes in gene expression produced changes in mitochondrial mass and number, we measured mitochondrial DNA content and citrate synthase activity (44, 68). The ratio of mitochondrial DNA to nuclear DNA (MT-CYT to HBB) was decreased by 30% in INS fetal livers (Fig. 5F). However, there was no difference in citrate synthase activity between groups (Fig. 5G). Furthermore, there were no differences in hepatic TBAR concentrations, a marker of oxidative stress via lipid peroxidation between CON and INS fetuses (Fig. 5H).
Multivariate analysis of metabolites identified effects on amino acid and one-carbon metabolism.
To identify the global effect of fetal hyperinsulinemia in the liver, we extended our study of metabolite regulation and subjected the full set of 162 metabolites from our metabolomics data set to an unbiased multivariate analysis. PLS-DA analysis showed distinct separation between groups (Fig. 6A). The top 20 metabolites with the highest variable in projection (VIP) scores are displayed on a heatmap (Fig. 6B). Using this list of metabolites, we identified an enrichment in pathways associated with the metabolism of amino acids (Fig. 6C). We also found decreased levels of S-adenosyl-l-homocysteine, l-homocysteine, and cystathione (Fig. 6B), supporting effects of fetal hyperinsulinemia on one-carbon metabolism.
Fig. 6.
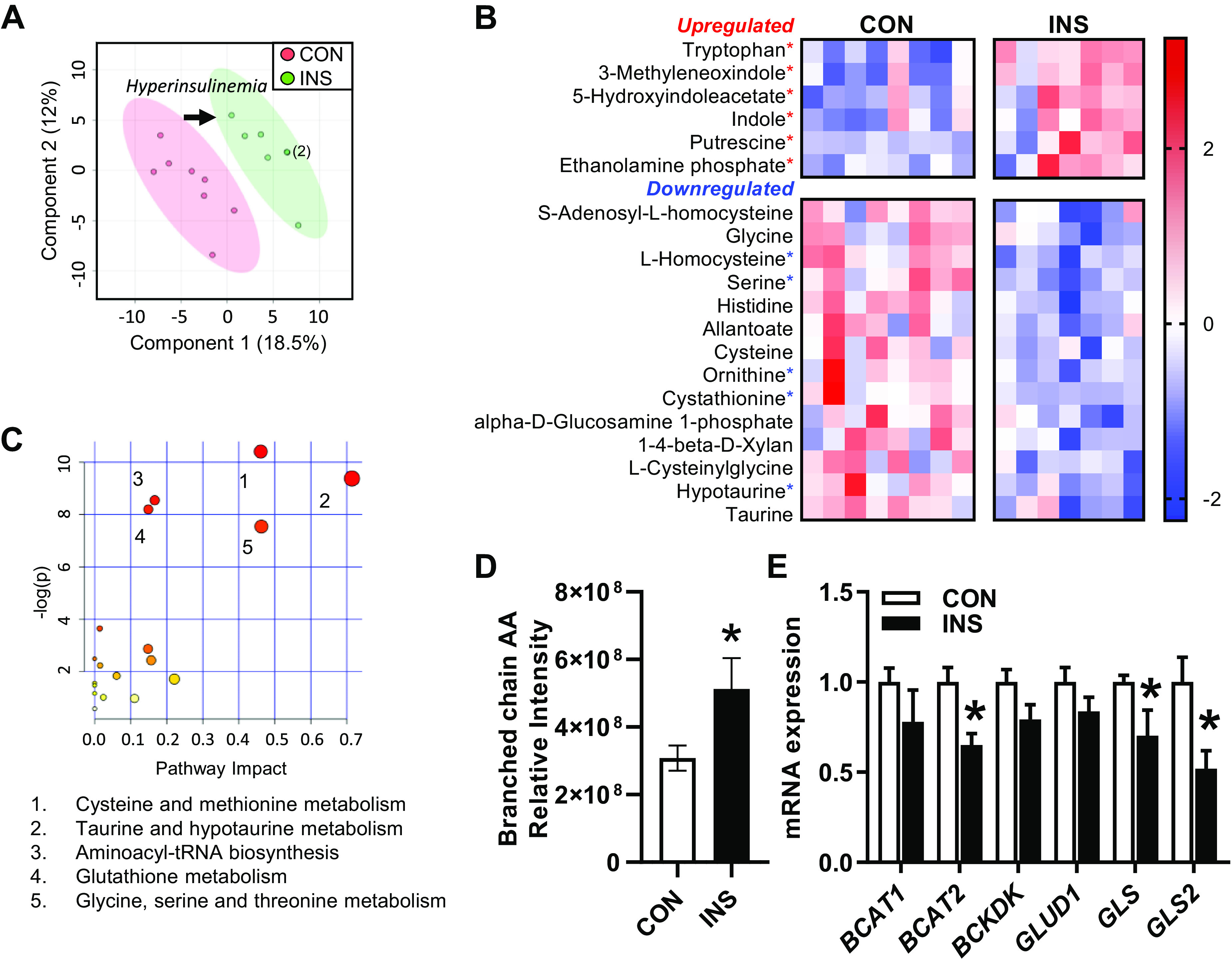
Global effect of chronic hyperinsulinemia on fetal hepatic metabolomic profile. Liver samples from control (CON) and hyperinsulinemic (INS) fetuses were subject to global metabolomics profiling. A: partial least squares discriminant analysis (PLS-DA) was used to identify metabolites with changes in abundance that defined separation of samples between the groups. Two-dimensional plot is shown. Arrow indicates shift in metabolome with hyperinsulinemia. Two samples in the INS group are overlapping as indicated by the (2). B: heatmap showing hierarchical clustering of the top 20 metabolites with the highest variable in projection scores. Metabolites that are significant by univariate analysis are indicated (*P < 0.05). Row values are normalized for each metabolite, and quantitative changes are color coded from blue (low) to red (high). C: pathway analysis enrichment results are plotted with the impact score on the x-axis and significance [−log (P value)] on y-axis. The identity of the pathways with Holm adjusted P < 0.05 are listed in rank order of significance. D: relative intensity of the branched-chain amino acids (AA; sum of Leu, Ile, and Val). E: expression of genes involved in amino acid metabolism in the fetal liver. Means ± SE are shown (n = 8 CON and 7 INS). *P < 0.05 in CON versus INS.
Amino acids are the largest substrate for oxidative metabolism in the fetal liver (36, 49, 72). Given the decrease in several amino acids (Fig. 6B), we further examined the effects of hyperinsulinemia on amino acid metabolism. Interestingly, branched-chain amino acid (BCAA) concentrations were increased in the INS fetal liver (Fig. 6D). We measured the expression of genes regulating BCAA metabolism. Expression of the mitochondrial form of the branched-chain aminotransferase BCAT2 was 30% lower in INS fetal livers, whereas there was no change in the cytosolic form BCAT1 or BCKDK, the kinase that inhibits branched-chain ketoacid dehydrogenase activity (Fig. 6E). The decrease in BCAT2 and increased concentrations of BCAA support decreased BCAA catabolism during hyperinsulinemia. We also evaluated glutamine-glutamate metabolism. There was no difference in expression of glutamate dehydrogenase (GLUD1), and yet expression of the glutaminase genes GLS and GLS2 were both decreased in the INS fetal liver (Fig. 6E). Decreased expression of the glutaminase genes may function to decrease conversion of glutamine to glutamate and thus decrease anaplerotic flux into the TCA cycle (67). However, there was no difference in glutamine or glutamate levels (see Supplemental Fig. S1).
Role of insulin versus oxygen.
In the INS fetus, insulin concentrations were increased and oxygen concentrations decreased. This combination raises the possibility that hypoxemia may antagonize the anabolic effects of insulin. To investigate this, we evaluated the relationships between hyperinsulinemia and hypoxemia, with the key factors identified to be up- or downregulated in the INS fetal liver (Fig. 7A). We focused on l-homocysteine as a marker for one-carbon metabolism, serine and tryptophan for AA metabolism, expression of PFKFB1 and CHREBP for glucose metabolism, PGC1A for mitochondrial function, and phosphorylation of AMPK for hypoxemia effects. l-Homocysteine and serine concentrations were negatively correlated and tryptophan concentrations positively correlated with both hyperinsulinemia and hypoxemia. The mRNA expression of PFKBP1 was positively correlated, whereas CHREBP expression was negatively correlated with both hyperinsulinemia and hypoxemia. PGC1A expression was negatively correlated with hyperinsulinemia but not hypoxemia. There was no association with p-AMPK expression and hyperinsulinemia (P = 0.25), and yet p-AMPK expression was associated with the degree of hypoxemia. The remaining variables were tightly correlated with both hyperinsulinemia and hypoxemia.
Fig. 7.
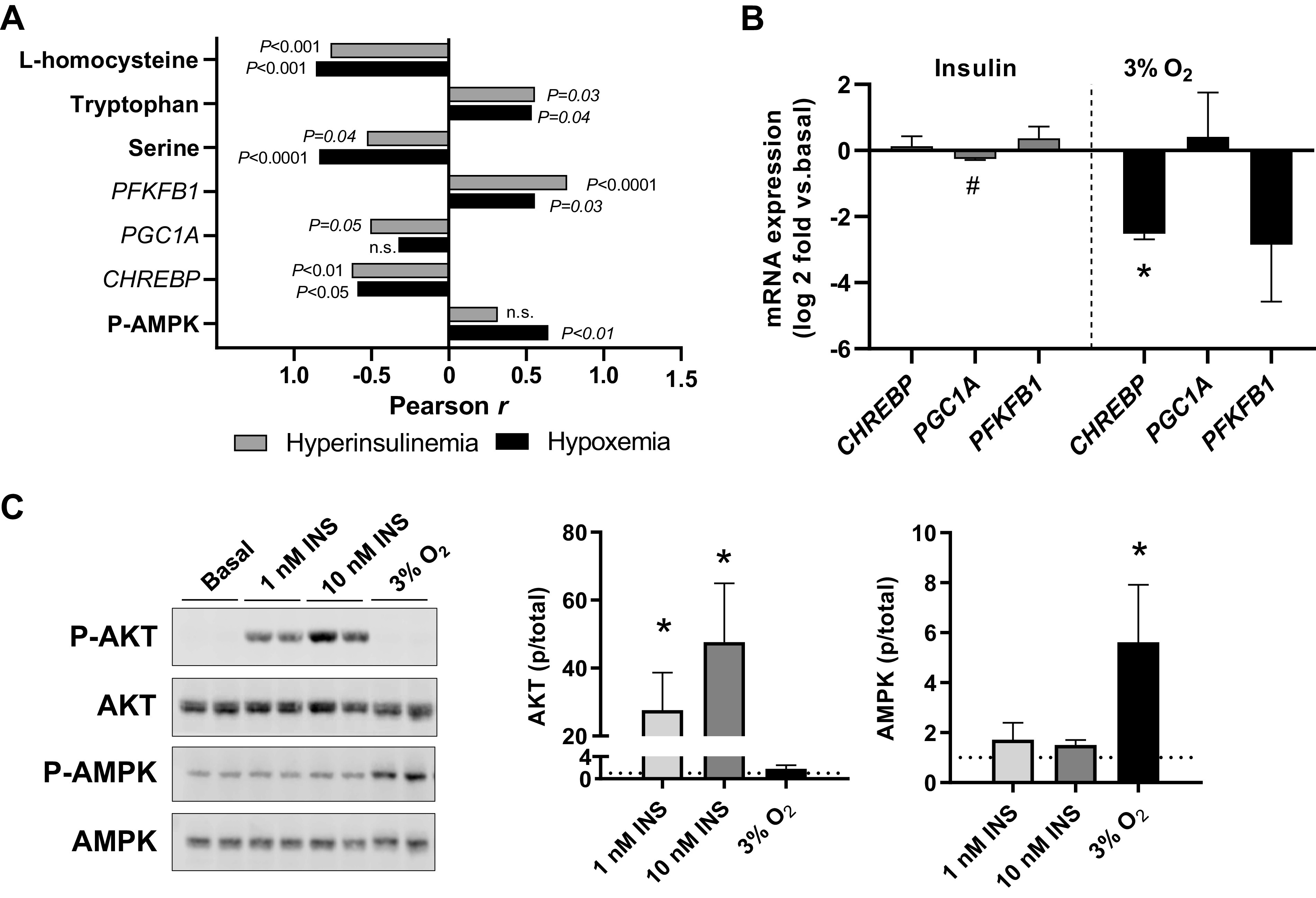
Role of hyperinsulinemia and hypoxemia on metabolic and signaling effects in the hyperinsulinemic (INS) fetal liver. A: the association between key metabolites, genes, and phosphorylation of AMP-activated protein kinase (p-AMPK) with hyperinsulinemia (gray bars) or degree of hypoxemia (black bars) represented by multiplying fetal oxygen concentrations by −1. Correlation coefficients are shown. B: isolated primary fetal hepatocytes were treated for 24 h with 100 nM insulin (at ambient air conditions) or 3% oxygen (3% O2). RNA was isolated and gene expression measured. Results are expressed as Log2 fold change relative to a basal treatment (represented by 0 in graphs) in ambient air. Means and SE shown (n = 2 primary cell preparations). #P < 0.10 and *P < 0.05 using 1-sided t test relative to basal treatment. C: isolated primary fetal hepatocytes were studied under basal conditions in ambient air (Basal) and compared with hepatocytes treated with insulin at 1 and 10 nM doses for 30 min or hypoxia with 3% O2 for 4 h. Samples were analyzed by Western blotting for phosphorylated (p-Akt) and total Akt (S473) and AMPK (T172) protein expression. Means and SE shown (n = 2 primary cell preparations). *P < 0.05 relative to basal treatment, indicated with dotted line equal to 1.0. CHREBP, carbohydrate-responsive element-binding protein; PGC1A, peroxisome proliferator-activated receptor-γ coactivator-1α; PFKFB1, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 1.
Regulation of signaling and gene expression in isolated fetal hepatocytes.
Finally, we used isolated primary fetal hepatocytes to experimentally test in vitro the contribution of hyperinsulinemia and hypoxemia that are present in vivo in INS fetuses. The expression of PGC1A tended to decrease with insulin treatment (Fig. 7B). Hypoxia treatment decreased expression of CHREBP. Next, we measured acute signaling responses to insulin on Akt activation and to hypoxia on AMPK activation. Hepatocytes treated with 1 and 10 nM doses of insulin for 30 min increased phosphorylation of Akt dose dependently (Fig. 7C). Hepatocytes exposed to 3% oxygen for 4 h had a fivefold increase in phosphorylation of AMPK compared with basal hepatocytes exposed only to ambient air (∼21% oxygen; Fig. 7C).
DISCUSSION
Our results demonstrate that chronic hyperinsulinemia produced greater effects on amino acid metabolism compared with glucose and lipid metabolism, a novel effect on one-carbon metabolism, and downregulation of insulin receptor downstream signaling in the fetal liver. Using our global metabolomics analysis, we identified a robust effect of hyperinsulinemia on decreased abundance of several metabolites associated with amino acid and one-carbon metabolism. However, we found few transcriptional changes for genes regulating glucose metabolism and little effect on relative levels of metabolites in glucose utilization pathways, including glycolysis, glycogen synthesis, or pentose phosphate pathway in the INS fetal liver. We also found no effect on lipogenesis, triglyceride synthesis, or lipid oxidation and an absence of activation of insulin receptor downstream signaling proteins in the INS fetal liver. These results support a potential downregulation of hepatic insulin signaling in the INS fetal liver that may mediate the observed lack of response on targets in glucose and lipid metabolism. We also found that phosphorylation of AMPK was increased. Thus, given the presence of hypoxemia with chronic hyperinsulinemia in the INS fetus, the overall metabolic response in the fetal liver may occur in part through the downregulation of insulin receptor signaling and a novel mechanism involving hypoxemia-induced AMPK activation that antagonizes insulin’s effects on substrate utilization.
We expected increased glucose utilization and increased lipid synthesis in response to hyperinsulinemia in the fetal liver, given that insulin signaling activates these pathways in the adult (52, 57, 79). However, we found inconsistent effects on gene expression and steady-state metabolite concentrations associated with pathways for glucose utilization or lipid synthesis in the INS fetal liver. First, there were no robust increases in major regulatory genes for glycolysis (PFKL, PKM, and PKL) or lipogenesis (SREBP1C, FASN, and ACC1). Second, we found no increase in hepatic glycogen or triglyceride concentrations, both products of biosynthetic pathways stimulated by insulin and glucose in humans and postnatal animal models (64, 79). We did find an effect of INS on increasing glycerophospholipid synthesis, a necessary pathway to maintain cell membrane integrity. There was no effect on the profile of acyl-carnitines, which is congruent with the notion that lipids are a minor fuel for oxidative metabolism in the fetus and that hepatic lipid oxidation pathways are not active until birth (25). Third, there were no differences in metabolites associated with glycolysis or the pentose phosphate pathway, a pathway that also would be predicted to be increased in the presence of insulin and glucose. Fourth, expression of three PDK isoforms (PDK1, PDK2, and PDK4) and LDHB was decreased, and yet there was no increase in PDH protein phosphorylation, no differences in hepatic lactate or pyruvate concentrations, and no increase in lactate concentrations in INS fetuses (7). Expression of PFKFBP1, the major fetal and hepatic isoform in the family of bifunctional PFKFBP genes, was increased; however, its role in regulating F2,6-P levels or glycolytic flux is not clear given that this enzyme is exquisitely regulated by posttranslational modifications (61, 63). Finally, under normal conditions, the fetal liver consumes only ∼15% of the total glucose taken up by the fetus from the placenta (36), whereas the remaining 85% is used by other fetal organs and tissues. Accordingly, other organs, in addition to the liver, may be responsible for the robust increase in whole body glucose utilization in INS fetuses.
Despite the presence of a nearly threefold increase in fetal arterial insulin concentrations, we did not observe increased phosphorylation and expression of classic insulin-signaling target proteins, including Akt (26, 64), mTOR, and the mTOR target proteins S6K and S6 (65). Using isolated hepatocytes, we confirmed that acute higher doses of insulin phosphorylate Akt. This is consistent with our previous studies demonstrating that acute (3–4 h) high-dose insulin infusions in vivo robustly activate Akt and mTOR signaling pathways in the liver and muscle of fetal sheep (6, 37, 76). The lack of activation of Akt and mTOR in response to chronic insulin in the INS fetal liver could be explained three ways: first, a reflection of the relative lower dose of insulin compared with doses used in acute infusion studies (76); second, lower amino acid concentrations (7), which may reduce activation of these signaling cascades (65); and third, feedback to suppress signaling due to prolonged insulin infusion period over 10 days. In support of the latter, expression of the insulin receptor genes (INSR-A and INSR-B) was decreased in the INS fetal liver. Thus, we speculate that the chronicity of the hyperinsulinemia led to a marked downregulation of hepatic insulin signaling.
There was evidence for decreased mitochondrial mass in the INS fetal liver. The ratio of mitochondrial to nuclear DNA was lower in INS fetal livers, suggesting fewer mitochondria. In support, expression of the transcriptional coactivator PGC1A and its target genes YYI and ESRRA, along with other functional mitochondrial regulators IDH1 and NRF2, was decreased (13). The decrease in PGC1A expression is likely a direct effect of insulin, as it was associated with hyperinsulinemia and suppressed with insulin in our studies using isolated fetal sheep hepatocytes. In addition, insulin has been shown to suppress PGC1A in mouse liver (45) and liver of fetal sheep with hypoglycemia (78) and decrease mitochondrial function in mouse hepatocytes (47). The lack of decrease in citrate synthase activity raises the possibility that alternate compensatory mechanisms are in place to normalize TCA cycle activity in the INS fetal liver. Alternatively, decreased PGC1A and mtDNA reflect feedback mechanisms to limit mitochondrial mass in the presence of insulin that accelerates oxidative metabolism.
We identified a novel and robust effect in the INS fetal liver on amino acid metabolism. Concentrations of amino acids in the INS fetal liver were decreased and may represent decreased utilization or depletion that results from increased utilization. Our gene expression data support decreased amino acid utilization, as INS fetal livers had decreased expression of BCAT2 and both glutaminase genes, GLS and GLS2, suggesting decreased BCAA catabolism and decreased flux of glutamine into the TCA cycle for oxidative metabolism (67). However, the impact of this potential change in hepatic substrate utilization is unclear because under normal conditions amino acids are the major carbon source for fetal hepatic oxidative metabolism, followed by lactate and glucose (36, 49, 72). Thus, if the preference for amino acids as the substrate for hepatic mitochondrial oxidation decreases, then we would expect compensatory increases in glucose utilization to maintain normal oxidative metabolism. We recognize that an increase in glucose or amino acid flux may not be detectable based on our end-point tissue analysis and steady-state metabolite concentrations. Additionally, several of the intermediary metabolic enzymes we measured are regulated posttranslationally or allosterically, independent of their mRNA expression. Thus, hepatic glucose utilization may be increased in the INS fetus but is not detectable based on the methods we used. To determine the utilization of specific substrates in the INS fetal liver, future studies are needed to directly measure hepatic amino acid flux and glucose utilization in vivo. At the fetal whole body level, our in vivo data demonstrate decreased concentrations of nearly all amino acids yet no difference in whole body amino acid uptake rates in the INS fetus (7). Additional studies are needed to test the responses to hyperinsulinemia in the liver and skeletal muscle on amino acid and glucose utilization that allow the fetus to maintain normal oxygen consumption rates despite a 40% increase in total carbon (sum of glucose and amino acids) substrate uptake (3, 7).
The significance of decreased intermediates in one-carbon metabolism is novel and warrants further investigation. One-carbon metabolism involves the folate and methionine cycles and integrates input signals from nutrients like glucose amino acids (serine, glycine) with output in the form of intermediates for nucleotide, lipid, and protein synthesis and redox balance (16, 48). Thus, decreased flux in one-carbon metabolism may limit flux in other pathways and contribute to the lack of effects on increased substrate metabolism in the INS fetal liver. In addition, intermediates in one-carbon metabolism are important substrates for methylation reactions and may underlie changes in DNA methylation, epigenetics, and developmental programming in offspring exposed to hyperinsulinemia and diabetic pregnancies (2, 34, 42, 56).
In addition to being hyperinsulinemic, INS fetuses also are hypoxemic, consistent with other models of fetal hyperinsulinemia (8, 53, 55). Fetal hypoxemia may underlie increased catecholamine signaling, as increased fetal norepinephrine concentrations were associated with decreased fetal oxygenation (1). Furthermore, our prior data support a suppressive effect of catecholamines on pancreatic insulin secretion and insulin-stimulated myogenesis in INS fetuses (1, 7). Thus, the confounding effects of fetal hypoxemia and hypercatecholamenemia likely antagonize insulin’s effects on anabolic and biosynthetic pathways. Our data support that hypoxemia-induced p-AMPK activation may mediate these effects. We found that p-AMPK was correlated with hypoxemia but not hyperinsulinemia, and p-AMPK was increased with low oxygen in isolated fetal hepatocytes. AMPK is activated under low-nutrient conditions, including hypoglycemia and hypoxemia, and functions to promote the breakdown of glucose and lipids and inhibit their synthesis and storage (22). Thus, hypoxemia-induced AMPK activation may prevent insulin-stimulated lipid synthesis, as AMPK phosphorylates and inactivates the ACC enzymes, thus suppressing fatty acid synthesis (22). Furthermore, AMPK inhibits protein synthesis via inhibition of the mTORC1 complex (22), which may contribute to the lack of signaling in the mTOR targets we measured. In terms of mitochondrial function, AMPK may increase PGC1A expression and mitochondrial biogenesis (22) or promote mitophagy (17). We speculate that AMPK activation in the INS fetal livers may function in the later role given the reduction in hepatic mtDNA content and decrease in PGC1A and targets related to mitochondrial biogenesis.
Both fetal hyperinsulinemia and hypoxemia were associated with decreased CHREBP and increased PFKFBP1 expression in the INS fetal liver. Decreased CHREBP expression may result from increased PFKFBP1 and potentially increased F-2,6-BP levels (61, 63, 82). However, as supported by our data in isolated hepatocytes, hypoxemia may drive the decrease in CHREBP expression. This decrease is paradoxical, as CHREBP would have been predicted to be increased in the INS fetal liver, as it is normally glucose responsive (40, 62) and activates regulatory enzymes in glycolysis and lipogenesis (62). We speculate that hypoxemia-induced inhibition of CHREBP may prevent the expected insulin-stimulated increase in expression of genes (PKL, ACC1, FASN) that are transcriptional targets of both insulin and CHREBP. Alternatively, the absence of increased expression of these genes may reflect the downregulation of insulin signaling that results from chronic hyperinsulinemia.
Perspectives and significance.
Our results demonstrate that chronic hyperinsulinemia produced greater effects on amino acid metabolism compared with glucose and lipid metabolism and a novel effect on one-carbon metabolism. These effects may be the result of the downregulation of hepatic insulin signaling in combination with an antagonistic effect of hypoxemia that is present in fetuses with hyperinsulinemia. Our data support that fetuses exposed to hyperinsulinemia have increased glucose utilization, which might support increased fetal growth, although this potential for growth likely may be limited by a downregulation of hepatic insulin signaling and antagonized by decreased oxygen supply. Further studies are needed to discern the specific effects of hypoxemia and decreased insulin signaling in the fetal liver. This is important for understanding metabolic effects in fetuses from diabetic pregnancies, as these fetuses have increased fetal concentrations of insulin (66) and often develop hypoxemia (9, 23, 73, 74).
GRANTS
This work was supported by NIH Grants R01-DK-108910 (S.R.W.), F32-DK-120070 (A.K.J.), R01-DK-088139 (P.J.R), R01-HD-093701 (P.J.R), and R01-HD079404 (L.D.B.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.J.R., A.D., L.D.B., and S.R.W. conceived and designed research; P.J.R., A.K.J., S.L.B., and S.R.W. performed experiments; A.K.J., S.L.B., and S.R.W. analyzed data; P.J.R., A.K.J., A.D., W.W.H., L.D.B., and S.R.W. interpreted results of experiments; S.R.W. prepared figures; S.R.W. drafted manuscript; P.J.R., A.K.J., L.D.B., and S.R.W. edited and revised manuscript; P.J.R., A.K.J., S.L.B., A.D., W.W.H., L.D.B., and S.R.W. approved final version of manuscript.
ENDNOTE
At the request of the authors, readers are herein alerted to the fact that additional materials related to this article may be found at https://doi.org/10.6084/m9.figshare.12485444.v1. These materials are not a part of this article and have not undergone peer review by the American Physiological Society (APS). APS and the journal editors take no responsibility for these materials, for the website address, or for any links to or from it.
REFERENCES
- 1.Andrews SE, Brown LD, Thorn SR, Limesand SW, Davis M, Hay WW Jr, Rozance PJ. Increased adrenergic signaling is responsible for decreased glucose-stimulated insulin secretion in the chronically hyperinsulinemic ovine fetus. Endocrinology 156: 367–376, 2015. doi: 10.1210/en.2014-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bansal A, Simmons RA. Epigenetics and developmental origins of diabetes: correlation or causation? Am J Physiol Endocrinol Metab 315: E15–E28, 2018. doi: 10.1152/ajpendo.00424.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown LD, Hay WW Jr. Effect of hyperinsulinemia on amino acid utilization and oxidation independent of glucose metabolism in the ovine fetus. Am J Physiol Endocrinol Metab 291: E1333–E1340, 2006. doi: 10.1152/ajpendo.00028.2006. [DOI] [PubMed] [Google Scholar]
- 4.Brown LD, Kohn JR, Rozance PJ, Hay WW Jr, Wesolowski SR. Exogenous amino acids suppress glucose oxidation and potentiate hepatic glucose production in late gestation fetal sheep. Am J Physiol Regul Integr Comp Physiol 312: R654–R663, 2017. doi: 10.1152/ajpregu.00502.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown LD, Rozance PJ, Bruce JL, Friedman JE, Hay WW Jr, Wesolowski SR. Limited capacity for glucose oxidation in fetal sheep with intrauterine growth restriction. Am J Physiol Regul Integr Comp Physiol 309: R920–R928, 2015. doi: 10.1152/ajpregu.00197.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown LD, Rozance PJ, Thorn SR, Friedman JE, Hay WW Jr. Acute supplementation of amino acids increases net protein accretion in IUGR fetal sheep. Am J Physiol Endocrinol Metab 303: E352–E364, 2012. doi: 10.1152/ajpendo.00059.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown LD, Wesolowski SR, Kailey J, Bourque S, Wilson A, Andrews SE, Hay WW Jr, Rozance PJ. Chronic hyperinsulinemia increases myoblast proliferation in fetal sheep skeletal muscle. Endocrinology 157: 2447–2460, 2016. doi: 10.1210/en.2015-1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carson BS, Philipps AF, Simmons MA, Battaglia FC, Meschia G. Effects of a sustained insulin infusion upon glucose uptake and oxygenation of the ovine fetus. Pediatr Res 14: 147–152, 1980. doi: 10.1203/00006450-198002000-00016. [DOI] [PubMed] [Google Scholar]
- 9.Catalano PM, Shankar K. Obesity and pregnancy: mechanisms of short term and long term adverse consequences for mother and child. BMJ 356: j1, 2017. doi: 10.1136/bmj.j1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong J, Soufan O, Li C, Caraus I, Li S, Bourque G, Wishart DS, Xia J. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46: W486–W494, 2018. doi: 10.1093/nar/gky310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clasquin MF, Melamud E, Rabinowitz JD. LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr Protoc Bioinformatics Chapter 14: 11, 2012. doi: 10.1002/0471250953.bi1411s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Culpepper C, Wesolowski SR, Benjamin J, Bruce JL, Brown LD, Jonker SS, Wilkening RB, Hay WW Jr, Rozance PJ. Chronic anemic hypoxemia increases plasma glucagon and hepatic PCK1 mRNA in late-gestation fetal sheep. Am J Physiol Regul Integr Comp Physiol 311: R200–R208, 2016. doi: 10.1152/ajpregu.00037.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 450: 736–740, 2007. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 14.D’Alessandro A, Nemkov T, Yoshida T, Bordbar A, Palsson BO, Hansen KC. Citrate metabolism in red blood cells stored in additive solution-3. Transfusion 57: 325–336, 2017. doi: 10.1111/trf.13892. [DOI] [PubMed] [Google Scholar]
- 15.D’Alessandro A, Slaughter AL, Peltz ED, Moore EE, Silliman CC, Wither M, Nemkov T, Bacon AW, Fragoso M, Banerjee A, Hansen KC. Trauma/hemorrhagic shock instigates aberrant metabolic flux through glycolytic pathways, as revealed by preliminary (13)C-glucose labeling metabolomics. J Transl Med 13: 253, 2015. doi: 10.1186/s12967-015-0612-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ducker GS, Rabinowitz JD. One-Carbon Metabolism in Health and Disease. Cell Metab 25: 27–42, 2017. doi: 10.1016/j.cmet.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461, 2011. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fowden AL, Forhead AJ. Insulin deficiency alters the metabolic and endocrine responses to undernutrition in fetal sheep near term. Endocrinology 153: 4008–4018, 2012. doi: 10.1210/en.2012-1063. [DOI] [PubMed] [Google Scholar]
- 19.Fowden AL, Hay WW Jr. The effects of pancreatectomy on the rates of glucose utilization, oxidation and production in the sheep fetus. Q J Exp Physiol 73: 973–984, 1988. doi: 10.1113/expphysiol.1988.sp003231. [DOI] [PubMed] [Google Scholar]
- 20.Fowden AL, Hughes P, Comline RS. The effects of insulin on the growth rate of the sheep fetus during late gestation. Q J Exp Physiol 74: 703–714, 1989. doi: 10.1113/expphysiol.1989.sp003322. [DOI] [PubMed] [Google Scholar]
- 21.Fowden AL, Mundy L, Silver M. Developmental regulation of glucogenesis in the sheep fetus during late gestation. J Physiol 508: 937–947, 1998. doi: 10.1111/j.1469-7793.1998.937bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell 66: 789–800, 2017. doi: 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gauster M, Desoye G, Tötsch M, Hiden U. The placenta and gestational diabetes mellitus. Curr Diab Rep 12: 16–23, 2012. doi: 10.1007/s11892-011-0244-5. [DOI] [PubMed] [Google Scholar]
- 24.Girard J Gluconeogenesis in late fetal and early neonatal life. Biol Neonate 50: 237–258, 1986. doi: 10.1159/000242605. [DOI] [PubMed] [Google Scholar]
- 25.Girard J Metabolic adaptations to change of nutrition at birth. Biol Neonate 58, Suppl 1: 3–15, 1990. doi: 10.1159/000243294. [DOI] [PubMed] [Google Scholar]
- 26.Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol 19: 31–44, 2018. doi: 10.1038/nrm.2017.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardie DG AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 25: 1895–1908, 2011. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hay WW Jr Energy and substrate requirements of the placenta and fetus. Proc Nutr Soc 50: 321–336, 1991. doi: 10.1079/PNS19910042. [DOI] [PubMed] [Google Scholar]
- 29.Hay WW Jr, DiGiacomo JE, Meznarich HK, Hirst K, Zerbe G. Effects of glucose and insulin on fetal glucose oxidation and oxygen consumption. Am J Physiol Endocrinol Metab 256: E704–E713, 1989. doi: 10.1152/ajpendo.1989.256.6.E704. [DOI] [PubMed] [Google Scholar]
- 30.Hay WW Jr, Meznarich HK. The effect of hyperinsulinaemia on glucose utilization and oxidation and on oxygen consumption in the fetal lamb. Q J Exp Physiol 71: 689–698, 1986. doi: 10.1113/expphysiol.1986.sp003027. [DOI] [PubMed] [Google Scholar]
- 31.Hay WW Jr, Meznarich HK, DiGiacomo JE, Hirst K, Zerbe G. Effects of insulin and glucose concentrations on glucose utilization in fetal sheep. Pediatr Res 23: 381–387, 1988. doi: 10.1203/00006450-198804000-00008. [DOI] [PubMed] [Google Scholar]
- 32.Hay WW Jr, Sparks JW, Battaglia FC, Meschia G. Maternal-fetal glucose exchange: necessity of a three-pool model. Am J Physiol 246: E528–E534, 1984. doi: 10.1152/ajpendo.1984.246.6.E528. [DOI] [PubMed] [Google Scholar]
- 33.Hay WW Jr, Sparks JW, Quissell BJ, Battaglia FC, Meschia G. Simultaneous measurements of umbilical uptake, fetal utilization rate, and fetal turnover rate of glucose. Am J Physiol Endocrinol Metab 240: E662–E668, 1981. doi: 10.1152/ajpendo.1981.240.6.E662. [DOI] [PubMed] [Google Scholar]
- 34.Heerwagen MJ, Miller MR, Barbour LA, Friedman JE. Maternal obesity and fetal metabolic programming: a fertile epigenetic soil. Am J Physiol Regul Integr Comp Physiol 299: R711–R722, 2010. doi: 10.1152/ajpregu.00310.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herzog B, Cardenas J, Hall RK, Villena JA, Budge PJ, Giguère V, Granner DK, Kralli A. Estrogen-related receptor alpha is a repressor of phosphoenolpyruvate carboxykinase gene transcription. J Biol Chem 281: 99–106, 2006. doi: 10.1074/jbc.M509276200. [DOI] [PubMed] [Google Scholar]
- 36.Houin SS, Rozance PJ, Brown LD, Hay WW Jr, Wilkening RB, Thorn SR. Coordinated changes in hepatic amino acid metabolism and endocrine signals support hepatic glucose production during fetal hypoglycemia. Am J Physiol Endocrinol Metab 308: E306–E314, 2015. doi: 10.1152/ajpendo.00396.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones AK, Brown LD, Rozance PJ, Serkova NJ, Hay WW Jr, Friedman JE, Wesolowski SR. Differential effects of intrauterine growth restriction and a hypersinsulinemic-isoglycemic clamp on metabolic pathways and insulin action in the fetal liver. Am J Physiol Regul Integr Comp Physiol 316: R427–R440, 2019. doi: 10.1152/ajpregu.00359.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones AK, Rozance PJ, Brown LD, Goldstrohm DA, Hay WW Jr, Limesand SW, Wesolowski SR. Sustained hypoxemia in late gestation potentiates hepatic gluconeogenic gene expression but does not activate glucose production in the ovine fetus. Am J Physiol Endocrinol Metab 317: E1–E10, 2019. doi: 10.1152/ajpendo.00069.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabashima T, Kawaguchi T, Wadzinski BE, Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci USA 100: 5107–5112, 2003. doi: 10.1073/pnas.0730817100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalhan S, Parimi P. Gluconeogenesis in the fetus and neonate. Semin Perinatol 24: 94–106, 2000. doi: 10.1053/sp.2000.6360. [DOI] [PubMed] [Google Scholar]
- 42.Kalhan SC One carbon metabolism in pregnancy: Impact on maternal, fetal and neonatal health. Mol Cell Endocrinol 435: 48–60, 2016. doi: 10.1016/j.mce.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG; NC3Rs Reporting Guidelines Working Group . Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579, 2010. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F, Hey-Mogensen M. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 590: 3349–3360, 2012. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 447: 1012–1016, 2007. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 46.Limesand SW, Rozance PJ, Smith D, Hay WW Jr. Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab 293: E1716–E1725, 2007. doi: 10.1152/ajpendo.00459.2007. [DOI] [PubMed] [Google Scholar]
- 47.Liu H-Y, Yehuda-Shnaidman E, Hong T, Han J, Pi J, Liu Z, Cao W. Prolonged exposure to insulin suppresses mitochondrial production in primary hepatocytes. J Biol Chem 284: 14087–14095, 2009. doi: 10.1074/jbc.M807992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Locasale JW Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer 13: 572–583, 2013. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marconi AM, Battaglia FC, Meschia G, Sparks JW. A comparison of amino acid arteriovenous differences across the liver and placenta of the fetal lamb. Am J Physiol 257: E909–E915, 1989. doi: 10.1152/ajpendo.1989.257.6.E909. [DOI] [PubMed] [Google Scholar]
- 50.McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, Grove KL. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 119: 323–335, 2009. doi: 10.1172/JCI32661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mesarwi OA, Shin MK, Bevans-Fonti S, Schlesinger C, Shaw J, Polotsky VY. Hepatocyte hypoxia inducible factor-1 mediates the development of liver fibrosis in a mouse model of nonalcoholic fatty liver disease. PLoS One 11: e0168572, 2016. doi: 10.1371/journal.pone.0168572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 6: 87–97, 2000. doi: 10.1016/S1097-2765(05)00015-8. [DOI] [PubMed] [Google Scholar]
- 53.Milley JR The effect of chronic hyperinsulinemia on ovine fetal growth. Growth 50: 390–401, 1986. [PubMed] [Google Scholar]
- 54.Milley JR, Papacostas JS, Tabata BK. Effect of insulin on uptake of metabolic substrates by the sheep fetus. Am J Physiol Endocrinol Metab 251: E349–E356, 1986. doi: 10.1152/ajpendo.1986.251.3.E349. [DOI] [PubMed] [Google Scholar]
- 55.Milley JR, Rosenberg AA, Philipps AF, Molteni RA, Jones MD Jr, Simmons MA. The effect of insulin on ovine fetal oxygen extraction. Am J Obstet Gynecol 149: 673–678, 1984. doi: 10.1016/0002-9378(84)90257-6. [DOI] [PubMed] [Google Scholar]
- 56.Monteiro LJ, Norman JE, Rice GE, Illanes SE. Fetal programming and gestational diabetes mellitus. Placenta 48, Suppl 1: S54–S60, 2016. doi: 10.1016/j.placenta.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 57.Moore MC, Coate KC, Winnick JJ, An Z, Cherrington AD. Regulation of hepatic glucose uptake and storage in vivo. Adv Nutr 3: 286–294, 2012. doi: 10.3945/an.112.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nemkov T, Hansen KC, D’Alessandro A. A three-minute method for high-throughput quantitative metabolomics and quantitative tracing experiments of central carbon and nitrogen pathways. Rapid Commun Mass Spectrom 31: 663–673, 2017. doi: 10.1002/rcm.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nemkov T, Reisz JA, Gehrke S, Hansen KC, D’Alessandro A. High-throughput metabolomics: isocratic and gradient mass spectrometry-based methods. Methods Mol Biol 1978: 13–26, 2019. doi: 10.1007/978-1-4939-9236-2_2. [DOI] [PubMed] [Google Scholar]
- 60.O’Brien RM, Granner DK. Regulation of gene expression by insulin. Physiol Rev 76: 1109–1161, 1996. doi: 10.1152/physrev.1996.76.4.1109. [DOI] [PubMed] [Google Scholar]
- 61.Okar DA, Manzano A, Navarro-Sabatè A, Riera L, Bartrons R, Lange AJ. PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem Sci 26: 30–35, 2001. doi: 10.1016/S0968-0004(00)01699-6. [DOI] [PubMed] [Google Scholar]
- 62.Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr 27: 179–192, 2007. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- 63.Ros S, Schulze A. Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab 1: 8, 2013. doi: 10.1186/2049-3002-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rui L Energy metabolism in the liver. Compr Physiol 4: 177–197, 2014. doi: 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell 168: 960–976, 2017. [Cell 169: 361–371, 2017]. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwartz R Hyperinsulinemia and macrosomia. N Engl J Med 323: 340–342, 1990. doi: 10.1056/NEJM199008023230512. [DOI] [PubMed] [Google Scholar]
- 67.Simon J, Nuñez-García M, Fernández-Tussy P, Barbier-Torres L, Fernández-Ramos D, Gómez-Santos B, Buqué X, Lopitz-Otsoa F, Goikoetxea-Usandizaga N, Serrano-Macia M, Rodriguez-Agudo R, Bizkarguenaga M, Zubiete-Franco I, Gutiérrez-de Juan V, Cabrera D, Alonso C, Iruzubieta P, Romero-Gomez M, van Liempd S, Castro A, Nogueiras R, Varela-Rey M, Falcón-Pérez JM, Villa E, Crespo J, Lu SC, Mato JM, Aspichueta P, Delgado TC, Martínez-Chantar ML. Targeting hepatic glutaminase 1 ameliorates non-alcoholic steatohepatitis by restoring very-low-density lipoprotein triglyceride assembly. Cell Metab 31: 605–622.e10, 2020. doi: 10.1016/j.cmet.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spinazzi M, Casarin A, Pertegato V, Ermani M, Salviati L, Angelini C. Optimization of respiratory chain enzymatic assays in muscle for the diagnosis of mitochondrial disorders. Mitochondrion 11: 893–904, 2011. doi: 10.1016/j.mito.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 69.Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc 7: 1235–1246, 2012. doi: 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- 70.Susa JB, Gruppuso PA, Widness JA, Domenech M, Clemons GK, Sehgal P, Schwartz R. Chronic hyperinsulinemia in the fetal rhesus monkey: effects of physiologic hyperinsulinemia on fetal substrates, hormones, and hepatic enzymes. Am J Obstet Gynecol 150: 415–420, 1984. doi: 10.1016/S0002-9378(84)80150-7. [DOI] [PubMed] [Google Scholar]
- 71.Susa JB, Neave C, Sehgal P, Singer DB, Zeller WP, Schwartz R. Chronic hyperinsulinemia in the fetal rhesus monkey. Effects of physiologic hyperinsulinemia on fetal growth and composition. Diabetes 33: 656–660, 1984. doi: 10.2337/diab.33.7.656. [DOI] [PubMed] [Google Scholar]
- 72.Teng C, Battaglia FC, Meschia G, Narkewicz MR, Wilkening RB. Fetal hepatic and umbilical uptakes of glucogenic substrates during a glucagon-somatostatin infusion. Am J Physiol Endocrinol Metab 282: E542–E550, 2002. doi: 10.1152/ajpendo.00248.2001. [DOI] [PubMed] [Google Scholar]
- 73.Teramo KA Obstetric problems in diabetic pregnancy – The role of fetal hypoxia. Best Pract Res Clin Endocrinol Metab 24: 663–671, 2010. doi: 10.1016/j.beem.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 74.Teramo KA, Widness JA. Increased fetal plasma and amniotic fluid erythropoietin concentrations: markers of intrauterine hypoxia. Neonatology 95: 105–116, 2009. doi: 10.1159/000153094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thorn SR, Baquero KC, Newsom SA, El Kasmi KC, Bergman BC, Shulman GI, Grove KL, Friedman JE. Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates. Diabetes 63: 2702–2713, 2014. doi: 10.2337/db14-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thorn SR, Brown LD, Rozance PJ, Hay WW Jr, Friedman JE. Increased hepatic glucose production in fetal sheep with intrauterine growth restriction is not suppressed by insulin. Diabetes 62: 65–73, 2013. doi: 10.2337/db11-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thorn SR, Regnault TR, Brown LD, Rozance PJ, Keng J, Roper M, Wilkening RB, Hay WW Jr, Friedman JE. Intrauterine growth restriction increases fetal hepatic gluconeogenic capacity and reduces messenger ribonucleic acid translation initiation and nutrient sensing in fetal liver and skeletal muscle. Endocrinology 150: 3021–3030, 2009. doi: 10.1210/en.2008-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thorn SR, Sekar SM, Lavezzi JR, O’Meara MC, Brown LD, Hay WW Jr, Rozance PJ. A physiological increase in insulin suppresses gluconeogenic gene activation in fetal sheep with sustained hypoglycemia. Am J Physiol Regul Integr Comp Physiol 303: R861–R869, 2012. doi: 10.1152/ajpregu.00331.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Titchenell PM, Lazar MA, Birnbaum MJ. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol Metab 28: 497–505, 2017. doi: 10.1016/j.tem.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ward Platt M, Deshpande S. Metabolic adaptation at birth. Semin Fetal Neonatal Med 10: 341–350, 2005. doi: 10.1016/j.siny.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 81.Wesolowski SR, Mulligan CM, Janssen RC, Baker PR II, Bergman BC, D’Alessandro A, Nemkov T, Maclean KN, Jiang H, Dean TA, Takahashi DL, Kievit P, McCurdy CE, Aagaard KM, Friedman JE. Switching obese mothers to a healthy diet improves fetal hypoxemia, hepatic metabolites, and lipotoxicity in non-human primates. Mol Metab 18: 25–41, 2018. doi: 10.1016/j.molmet.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu C, Khan SA, Peng LJ, Lange AJ. Roles for fructose-2,6-bisphosphate in the control of fuel metabolism: beyond its allosteric effects on glycolytic and gluconeogenic enzymes. Adv Enzyme Regul 46: 72–88, 2006. doi: 10.1016/j.advenzreg.2006.01.010. [DOI] [PubMed] [Google Scholar]