

Abstract
Purpose:
The purpose of this study was to evaluate the rational combination of TORC1/2 inhibitor TAK-228 and Aurora A kinase inhibitor alisertib in preclinical models of triple-negative breast cancer (TNBC) and to conduct a phase I dose escalation trial in patients with advanced solid tumors.
Experimental Design:
TNBC cell lines and patient-derived xenograft (PDX) models were treated with alisertib, TAK-228, or the combination and evaluated for changes in proliferation, cell cycle, mTOR pathway modulation, and terminal cellular fate, including apoptosis and senescence. A phase I clinical trial was conducted in patients with advanced solid tumors treated with escalating doses of alisertib and TAK-228 using a 3+3 design to determine the maximum tolerated dose (MTD).
Results:
The combination of TAK-228 and alisertib resulted in decreased proliferation and cell-cycle arrest in TNBC cell lines. Treatment of TNBC PDX models resulted in significant tumor growth inhibition and increased apoptosis with the combination. In the phase I dose escalation study, 18 patients with refractory solid tumors were enrolled. The MTD was alisertib 30 mg b.i.d. days 1 to 7 of a 21-day cycle and TAK-228 2 mg daily, continuous dosing. The most common treatment-related adverse events were neutropenia, fatigue, nausea, rash, mucositis, and alopecia.
Conclusions:
The addition of TAK-228 to alisertib potentiates the antitumor activity of alisertib in vivo, resulting in increased cell death and apoptosis. The combination is tolerable in patients with advanced solid tumors and should be evaluated further in expansion cohorts with additional pharmacodynamic assessment.
Introduction
The Aurora kinases are nuclear proteins that play an important role in spindle pole organization and mitotic progression. The 3 Aurora kinases (Aur A, B, and C) are distinguished by their locations and specific mitotic functions (1). Overexpression of Aur A and B correlates with cancer progression in many tumor types (2), and multiple Aurora kinase inhibitors are in various stages of clinical development (3).
Alisertib is a small-molecule, selective inhibitor of Aur A, which induces G2–M cell-cycle arrest, induction of apoptosis, and tumor growth inhibition in preclinical models (4–9). In triple-negative breast cancer (TNBC) models, we previously demonstrated that the induction of apoptosis in response to alisertib is dependent on p53 and p73 activity, and knockdown of these proteins leads to a switch in the phenotypic response from apoptosis to cellular senescence (10). Senescence is a physiologic response to cellular stress, which results in permanent growth arrest (11, 12). Despite growth arrest observed in senescent cells, senescence is also associated with secretion of growth factors into the tumor microenvironment that can stimulate progression of adjacent cancer cells resulting in chemotherapy resistance (13). In TNBC patient-derived xenograft (PDX) models treated with alisertib to resistance or demonstrating de novo resistance, an increase in senescent cells is observed that is accompanied by a decrease in apoptosis (10, 14).
Clinically, alisertib monotherapy has minimal activity in TNBC, with a median progression-free survival of 1.5 months in 14 TNBC patients treated in a phase II trial (15). Mechanisms of resistance to Aurora A inhibition may be similar to other situations where we observe treatment-related senescence, such as with adriamycin (16, 17), a drug with known activity in breast cancer and other malignancies. Studies of alisertib in combination with other agents to overcome this resistance have shown promise in TNBC, such as the combination of alisertib and fulvestrant in women with endocrine-resistant ER+ metastatic breast cancer (18), for which a phase II clinical trial is ongoing (NCT02860000).
In preclinical TNBC models, enrichment of the mTOR signaling pathway has been linked to intrinsic and acquired resistance to alisertib (14). Similar findings have been documented in other tumor types where alisertib treatment leads to upregulation of the PI3K/Akt/mTOR pathway (4–8, 19, 20). TAK-228 (MLN0128) is a potent and selective mTOR kinase inhibitor targeting TORC1 and TORC2, with preclinical activity in a variety of cancers (21–23). TAK-228 has been investigated in a number of clinical trials including in combination with exemestane or fulvestrant in patients with metastatic ER+/HER2− breast cancer where the combination was found to be tolerable with promising antitumor activity (24, 25). We selected TAK-228 for combination with alisertib based on broad mTOR pathway blockade related to its unique TORC1/2 inhibition profile.
The purpose of this study was to investigate the antitumor activity of the combination of TAK-228 and alisertib in preclinical models of TNBC and to assess the safety, tolerability, and preliminary efficacy in human patients with advanced solid tumors in a phase I clinical trial.
Materials and Methods
Cell culture and reagents
Human TNBC cell lines MDA-MB-468, MDA-MB-231, HCC1937, BT-20, and CAL51 were obtained from ATCC and cultured as previously described (26). Cells were passaged for 6 months or less. Cell lines were authenticated at the Barbara Davis Center Molecular Biology Service Center (Aurora, CO) and screened for Mycoplasma every 3 months.
TAK-228 was dissolved in 100% DMSO for in vitro use and prepared in 5% NMP+ 15% PVP+ 80% HPLC H2O for use in vivo. Alisertib was dissolved in 100% DMSO for in vitro use and prepared in a 1:1 mixture of 2% NaHCO2 and 10% hydroxypropyl β–cyclo-dextran (HPBCD) for use in vivo. TAK-228 and alisertib were kindly provided by the NCI Cancer Therapy Evaluation Program for preclinical work and by Takeda for the phase I clinical trial.
Cell viability assays
Two cell lines sensitive to alisertib (MDA-MB-468 and MDA-MB-231) and 2 cell lines resistant to alisertib (HCC1937 and BT-20) were selected for evaluation. Cells were plated in 96-well white-walled plates and incubated overnight. Cells were then exposed to increasing concentrations of alisertib (25 nmol/L-125 nmol/L) alone and in combination with TAK-228 at 50 nmol/L (sensitive cell lines) or 125 nmol/L (resistant cell lines). Proliferation was assessed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) at 96 hours according to the manufacturer’s instructions. Proliferation was normalized to the no-drug control and graphed using Prism software (GraphPad).
Immunoblotting analysis
Cells from the alisertib-sensitive MDA-MB-468 TNBC cell line and the alisertib-resistant HCC1937 cell line were seeded in 6-well plates. Following 24-hour incubation, cells were exposed to alisertib 50–100 nmol/L, TAK-228 50–100 nmol/L, or the combination for 24–72 hours. Cells were harvested with trypsin/EDTA and lysed in cell lysis buffer (Cell Signaling Technology) as described previously (27). Fifty micrograms of protein were electrophoresed on 4% to 12% Bis-Tris precast gels (Life Technologies) and transferred to a nitrocellulose membrane using Pierce G2 Fast Blotter (Pierce). Membranes were blocked and then incubated overnight as previously described (27), with the following primary antibodies at 1:1,000 dilutions: S6RP, pS6RP, 4EBP1, p4EBP1, survivin, and a-tubulin (Cell Signaling Technology). Blots were washed and incubated with the appropriate secondary goat anti-rabbit and goat anti-mouse immunoglobulin G (H + L) DyLight-conjugated antibodies (Cell Signaling Technology) followed by additional washes as described previously (27). Blots were then developed using the Odyssey Infrared Imaging System (LI-COR).
Cell-cycle analysis
Cells were transduced with the FUCCI (fluorescent ubiquitination-based cell-cycle indicator) system as previously described (28). The FUCCI system consists of 2 fluorescent polypeptides that are degraded in a cell-cycle–specific manner to distinguish cells in the G1 phase (labeled with red fluorescent protein) and S/G2–M phase (labeled with a green fluorescent protein; ref. 29). TP53 wild-type CAL51 cells were selected for use in the cell-cycle assays as p53 dysfunction has been shown to affect the cell-cycle transition in FUCCI models (30). CAL51 FUCCI cells were plated and treated with TAK-228 100 nmol/L, alisertib 100 nmol/L, or the combination. Cell-cycle status according to the FUCCI biosensor system was assessed at 0 to 96 hours using live-cell microscopy with an environmentally controlled Olympus IX81 fluorescence microscope. Relative growth of CAL51 cells was compared across treatment arms and graphed using Prism software (GraphPad).
Treatment with TAK-228 and alisertib in TNBC PDX models
PDX models CU_TNBC_004 (TP53 E294* mutant) and CU_TNBC_007 (TP53 N196K mutant) were generated as previously described (10, 26, 31). The CU_TNBC_004 model was previously identified as intrinsically resistant to alisertib (14). Twelve-gauge trocars were used to inject tumor sections subcutaneously into the bilateral flanks of each mouse. Tumor measurements and weights were collected twice weekly using digital calipers and weighing scale and recorded (Studylog Systems). Tumor volumes were calculated using the following formula: volume – (length × width2) × 0.52. Once tumors reached an average of 150 to 300 mm3, mice were randomized into one of 4 treatment groups: vehicle control, TAK-228 0.5 mg/kg daily, alisertib 30 mg/kg daily, and TAK-228/alisertib combination. All treatments were administered by oral gavage (100 μL). Tumor growth curves were plotted and tumor growth inhibition (TGI = [1 – (average of final treated tumor volume/average of final vehicle tumor volume)] × 100) was calculated. At the end of the treatment, mice were euthanized with CO2 followed by cervical dislocation and tumor samples were collected. Animal studies were performed in a facility accredited by the American Association for Accreditation of Laboratory Animal Care in accordance with NIH guidelines and approved by the University of Colorado Institutional Animal Care and Use Committee.
Senescence-associated β-galactosidase (SA-β-gal) staining
PDX tumor samples were processed using Tissue-Tek optimum cutting temperature (OCT) compound (Sakura Finetek) and frozen in liquid nitrogen prior to being cut into slides by the University of Colorado Cancer Center (UCCC) Pathology Core Facility. Slides were fixed and stained for SA-β-gal activity at pH of 6.0 using the Senescence β-gal Staining Kit (Cell Signaling Technology), as described previously (14). Hematoxylin and eosin (H&E) staining was performed on matched slides by the UCCC Pathology Core Facility and reviewed by a pathologist, confirming the presence of tumor cells. Representative images were acquired using a Zeiss microscope at 40 × magnification.
IHC analysis
PDX tumor tissue was formalin-fixed and paraffin-embedded (FFPE) by the UCCC Histology Core Facility. IHC assays were performed on 5-μm FFPE tumor sections using the Ventana XT autostainer (Ventana Medical Systems). Antigen retrieval consisted of incubation with Ventana’s CC1 antigen retrieval solution for 20 minutes. Primary antibodies Ki-67 (Thermo Fisher) and cleaved caspase-3 (Cell Signaling Technology) were incubated with tissue for 1 hour at 37°C, then labeled with biotinylated goat anti-rabbit IgG secondary antibody (Vector Laboratories) for 32 minutes at room temperature, and detected with horseradish peroxidase (HRP) DAB Map system (Ventana) followed by hematoxylin and bluing reagent counterstain (Ventana). Images were captured at 10 × magnification using the ScanScope XT whole-slide imaging system (Aperio Technologies).
Immunofluorescence
Slides created from OCT blocks as described above were fixed and locked with 1% BSA and 1 × PBS for 1 hour at 37°C and subsequently incubated with BAX (Thermo Fisher) and DR5 (Santa Cruz) primary antibodies. Following PBS wash, slides were incubated with secondary antibodies AlexaFluor 488 and AlexaFluor 555 (Life Technologies) for 1 hour at 37°C and washed in PBS. Counterstaining with DAPI in 1 × PBS was performed for 5 minutes, followed by PBS wash. Slides were mounted with Fluoromount-G mounting media (SouthernBiotech) and imaged using the Olympus FV-1000 confocal microscope at 60 × magnification, as described previously (14).
Phase I clinical trial patient selection
Patients greater than 18 years of age with an incurable solid tumor malignancy, refractory to standard therapy, were eligible. Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1; adequate hematologic, hepatic, and renal function; HgbA1c ≤7%; fasting serum glucose ≤130 mg/dL and fasting triglycerides ≤300 mg/dL; and normal cardiac function were required. Patients with an underlying condition associated with excessive daytime sleepiness (including uncontrolled sleep apnea), other severe cardiac or pulmonary disease, or risk of malabsorption of oral medications were excluded.
The protocol was approved by the local institutional review board and conducted in accordance with the Belmont Report. All patients signed a written consent prior to enrollment according to federal and institutional guidelines.
Study design
The primary objective of the phase I clinical trial was to determine the maximum tolerated dose (MTD) and dose-limiting toxicity (DLT) of TAK-228 in combination with alisertib.
Eligible patients were treated with the combination of TAK-228 by mouth daily on a continuous schedule and alisertib by mouth twice daily (b.i.d.) on days 1 to 7 of a 21-day cycle at escalating doses. During cycle 1 only, a single dose of TAK-228 was administered between days −7 and −3, and a single dose of alisertib was administered day 1 to allow for single-agent PK analysis for each drug. Combination treatment was initiated cycle 1 day 2.
The starting dose for TAK-228 was 1 mg, which was 75% lower than the MTD of TAK-228 in combination with exemestane or fulvestrant (24). The starting dose of alisertib was 30 mg, which was 40% lower than the MTD of alisertib alone according to this dosing schedule (32). These doses were selected to accommodate potential overlapping toxicities including mucositis, while remaining within a range of potential therapeutic activity for each single agent. Dose escalation was conducted according to a standard 3+3 design.
The MTD was defined as the highest dose at which less than 1/3 of at least 6 evaluable patients experienced a treatment-related DLT within 28 days of the first dose of TAK-228. DLT was defined as any of the following considered at least possibly related to treatment with TAK-228 or alisertib: (1) grade 4 neutropenia lasting >7 days; (2) grade 3 neutropenia accompanied by fever and/or infection; (3) thrombocytopenia with significant bleeding; (4) any other grade ≥4 hematologic toxicity; (5) inability to administer at least 75% of planned doses of either drug within cycle 1 due to drug-related toxicity; and (6) grade 3 or higher nonhematologic toxicity, despite adequate treatment, with the exception of short-duration (<72 hours) grade 3 hyperglycemia, grade 3 rash, or grade 3 nausea, vomiting, or diarrhea. Toxicity was graded according to the NCI Common Terminology Criteria for Adverse Effects (NCI CTCAE) version 4.03.
Blood samples were taken predose and 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours after a single dose of TAK-228 in cycle 1 between day −7 and day −3, and predose and 1, 2, 4, 6, 8, and 24 hours after a single dose of alisertib on cycle 1 day 1 for the assessment of single-agent PK parameters. Assessment of combination PK was performed predose and at 0.25, 0.5, 1, 2, 4, 6, 8, and 24 hours post-dose on cycle 1 day 7 and day 1 of all subsequent cycles. Alisertib and TAK-228 were measured in human plasma using a validated LC/MS/MS assay carried out in the UCCC Pharmacology Shared Resource.
Serial tumor biopsies were performed prior to treatment, after a 7-day lead-in of alisertib, and following alisertib and TAK-228 treatment on cycle 2 day 7 in two patients treated at the MTD. Samples were evaluated by IHC for Ki-67 and cleaved caspase-3, as described above.
Tumor imaging was performed every 3 cycles or as clinically indicated. Patients with measurable disease who underwent restaging imaging were evaluable for response assessment using RECIST v1.1. Responses were evaluated as best response to treatment. Patients continued on study until disease progression or unacceptable toxicity, with duration of time on study and reason for discontinuation summarized.
Statistical analysis
Preclinical analyses were performed using Prism version 5.0. Comparisons were preformed using a t test with P = 0.05 as the cutoff for statistical significance. In the phase I clinical trial, statistics were descriptive in nature, with no formal statistical hypothesis tested.
Results
Effect of TAK-228 and alisertib exposure in vitro on cellular proliferation and mTOR pathway effectors
The addition of TAK-228 to alisertib treatment resulted in a statistically significant decrease in proliferation as compared with single-agent alisertib in cell lines sensitive and resistant to alisertib (Fig. 1A). This effect was most pronounced in the alisertib-sensitive MDA-MB-468 cell line, but was also observed in resistant models.
Figure 1.

Effect of alisertib and TAK-228 on proliferation, downstream effectors, and cell-cycle arrest in TNBC cell lines. A, Relative proliferation as compared with vehicle control cells according to CellTiter-Glo at 96 hours with increasing concentrations of alisertib alone and in combination with TAK-228 at 50 nmol/L (alisertib-sensitive cell lines) and 125 nmol/L (alisertib-resistant cell lines). Degree of statistical significance as compared with single-agent alisertib: *, P ≤ 0.05; **, P ≤ 0.01. B, Immunoblots showing effects of alisertib and TAK-228 on apoptosis and downstream effectors of mTOR at doses of 50 nmol/L at 24 hours and 100 nmol/L at 72 hours. C, CAL51 cells transduced with the FUCCI system treated with alisertib and TAK-228 alone and in combination at 100 nmol/L and observed over 96 hours using an environmentally controlled Olympus IX81 fluorescence microscope. Cells appear red in G1 phase, indicating arrest, and green in S/G2–M phase. D, Growth curves based on time-lapse images over 96 hours for CAL51 FUCCI cells. Degree of statistical significance of combination therapy as compared with single-agent treatment arms at 72 and 96 hours: *, P ≤ 0.05; **, P ≤ 0.01.
Treatment with alisertib alone resulted in an increase in the mTOR pathway effector pS6RP, confirming upregulation of the mTOR pathway with Aur A inhibition in both alisertib-sensitive and -resistant TNBC cell lines (Fig. 1B). Treatment with TAK-228 alone or TAK-228 added to alisertib resulted in a decrease in both pS6RP and the related protein p4EBP1. We observed an increase in the antiapoptotic protein survivin in cells treated with alisertib and a decrease in survivin in cells treated with TAK-228 alone or TAK-228 added to alisertib (Fig. 1B).
Effect of TAK-228 and alisertib treatment on cell cycle in CAL51 FUCCI cells
Figure 1C depicts CAL51 cells transduced with the FUCCI system, with assessment of fluorescence over time following treatment. We observed cells in G1 (red) and S/G2–M (green) phase across the 96-hour time points in the groups treated with no drug, alisertib, or TAK-228, which indicates continued progression through the cell cycle despite treatment. In contrast, treatment with TAK-228 and alisertib resulted in the absence of cells in S/G2–M phase (green), consistent with complete G1 cycle arrest that is maintained across all time points (Fig. 1C). Treatment with the combination of TAK-228 and alisertib also resulted in a significant decrease in relative growth as assessed using the FUCCI system as compared with no drug or single-agent treatment at 72 hours (combo vs. no drug P < 0.01, combo vs. TAK-228 P = 0.007, combo vs. alisertib P = 0.0007). This was not preserved in the TAK-228–treated cells at 96 hours (combo vs. no drug P < 0.01, combo vs. TAK-228 P = 0.106, combo vs. alisertib P = 0.014; Fig. 1D).
TAK-228 and alisertib treatment results in TGI in TNBC PDX models
In the alisertib-resistant CU_TNBC_004 PDX model, a combination effect was observed following treatment with TAK-228 0.5 mg/kg daily and alisertib 30 mg/kg daily (Fig. 2A). In this model, TGI of the combination of TAK-228 and alisertib daily was 94.27%, as compared with 54.47% with TAK-228 and 35.09% with alisertib, with a statistically significant decrease in tumor volume in the combination arm as compared with the alisertib (P < 0.0001) and TAK-228 (P < 0.0001) arms. In the second PDX model, CU_TNBC_007, there was a trend toward decreased tumor volume with the combination, though this was not statistically significant compared with TAK-228 (P = 0.0874) or alisertib (P = 0.4366) alone. Greater TGI was observed with the combination (TGI 77.33%), as compared with TAK-228 (30.73%) and alisertib (51.74%; Fig. 2B).
Figure 2.
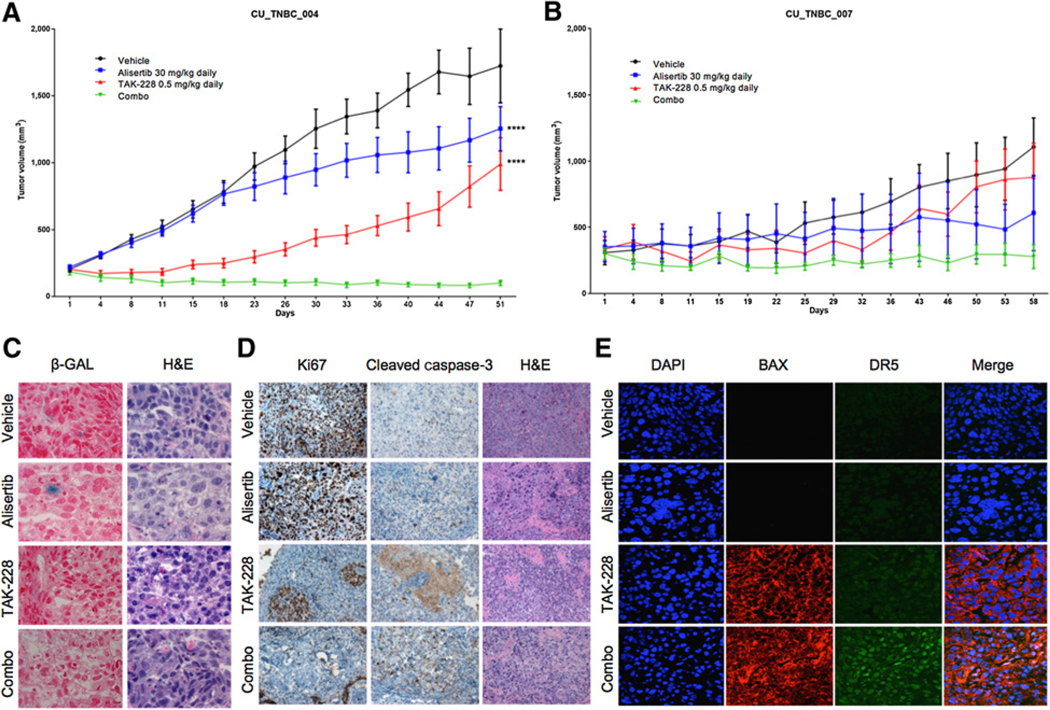
In vivo effects of alisertib and TAK-228 in TNBC PDX models. A, Effect of alisertib 30 mg/kg p.o. daily, TAK-228 0.5 mg/kg p.o. daily, and the combination in the CU_TNBC_004 alisertib-resistant PDX model. TGI compared with vehicle was 35.1% for alisertib, 54.5% for TAK-228, and 94.3% for the combination. Degree of statistical significance of combination treatment as compared with single-agent treatment arms: ****, P < 0.0001. B, Effect of alisertib 30 mg/kg daily, TAK-228 0.5 mg/kg daily, and the combination in the alisertib-resistant CU_TNBC_007 PDX model. TGI compared with vehicle was 51.7% for alisertib, 30.7% for TAK-228, and 77.3% for the combination. C, Senescence assessed by SA-β-gal staining on tumors from CU_TNBC_004 treated with alisertib, TAK-228, or the combination. Representative images at 40× magnification; senescent cells appear blue. D, IHC analysis of Ki67, cleavedcaspase-3, and H&E on tumor tissues at day38 to assess cell turnover and apoptosis. Images at 10×. E, IF analysis of tumors for DAPI, BAX, and DR5 for assessment of apoptosis. Images at 60× using confocal microscopy.
In vivo effects on senescence and apoptosis
Assessment of tissues from the CU_TNBC_004 PDX model for senescence by SA-β-gal staining demonstrated scattered senescent cells following treatment with alisertib alone, consistent with prior published results. Senescent cells were not observed with TAK-228 alone or in combination with alisertib. Extensive necrosis was observed on H&E in tumors treated with TAK-228 in combination with alisertib (Fig. 2C).
Treatment with TAK-228 and alisertib resulted in a decrease in Ki67 as a marker of cellular proliferation and an increase in cleaved caspase-3 as a marker of apoptosis in the CU_TNBC_004 model (Fig. 2D). Immunofluorescence staining revealed an increase in cytoplasmic BAX, also a marker of apoptosis, in tumors treated with TAK-228 alone or in combination with alisertib. Expression of the apoptosis mediator DR5 was observed in tumors treated with the combination of TAK-228 and alisertib (Fig. 2E).
Phase I clinical trial: Patient characteristics and MTD
A total of 18 patients were treated in the phase I trial at the UCCC; relevant characteristics are listed in Table 1. The majority of patients were heavily pretreated, with 44% of patients having previously received 5 or more lines of therapy.
Table 1.
Patient demographics and baseline characteristics.
| Characteristic | Number of patients (%) N = 18 |
|---|---|
| Age | |
| Median (range) | 65 (48–76) |
| Gender | |
| Male | 3 (17%) |
| Female | 15 (83%) |
| Race/ethnicity | |
| Caucasian | 14 (78%) |
| African American | 1 (6%) |
| Asian | 1 (6%) |
| Hispanic | 2 (11%) |
| Tumor type | |
| Ovarian | 4 (22%) |
| Breast (all ER+ HER2−) | 5 (28%) |
| Prostate | 2 (11%) |
| Other (fallopian tube, endometrial, peritoneal, pancreatic, rectal, gastric, gallbladder) | 7 (39%) (1 of each tumor type) |
| Baseline ECOG performance status | |
| 0 | 4 (22%) |
| 1 | 14 (78%) |
| Prior lines of therapy for metastatic disease | |
| 2 | 1 (6%) |
| 3 | 4 (22%) |
| 4 | 5 (28%) |
| 5 or more | 8 (44%) |
Three patients were treated at dose level 1 (TAK-228 1 mg daily and alisertib 30 mg b.i.d. days 1–7), with no DLT observed. Four patients were treated at dose level 2 (TAK-228 2 mg daily and alisertib 30 mg b.i.d. days 1–7), including one patient who elected to discontinue study treatment prior to completion of the first cycle. No DLT was observed. At dose level 3 (TAK-228 2 mg daily and alisertib 40 mg b.i.d. days 1–7), one patient was replaced due to rapid clinical progression prior to completion of the first cycle. Two of 6 evaluable patients had a documented DLT. One patient experienced intolerable grade 2 gastroesophageal reflux disease (GERD) and grade 2 nausea leading to an inability to administer at least 75% of doses in cycle 1. A second patient experienced grade 3 fatigue. As such, an alternate dose level termed dose level 2.5 (TAK-228 3 mg daily and alisertib 30 mg b.i.d. days 1–7) was evaluated. The first 2 patients treated experienced DLT with grade 3 fatigue. Thus, TAK-228 2 mg daily and alisertib 30 mg b.i.d. days 1 to 7, as evaluated in dose level 2, was determined to be the MTD and recommended phase II dose (RP2D) of the combination (Table 2). This dose level was then expanded to include 2 additional patients, with a total of 6 patients receiving treatment at the MTD.
Table 2.
Treatment-related adverse events attributed to one or both drugs occurring in at least 10% of patients across cycles.
| Number (%) | Dose level 1 | Dose level 2a | Dose level 3 | Dose level 2.5 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TAK-228 1 mg | TAK-228 2 mg | TAK-228 2 mg | TAK-228 3 mg | |||||||
| Alisertib 30 mg | Alisertib 30 mg | Alisertib 40 mg | Alisertib 30 mg | Overall | ||||||
| (N = 3) |
(N = 6) |
(N = 7) |
(N = 2) |
(N = 18) |
||||||
| Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | |
| Neutropenia | 0 | 0 | 2 (33) | 1 (17) | 2 (29) | 3 (43) | 0 | 1 (50) | 4 (22) | 5 (28) |
| Fatigue | 1 (33) | 0 | 1 (17) | 0 | 1 (14) | 1 (14) | 2 (100) | 2 (100) | 5 (28) | 3 (17) |
| Nausea | 1 (33) | 0 | 0 | 0 | 4 (57) | 1 (14) | 0 | 0 | 5 (28) | 1 (6) |
| Mucositis | 0 | 0 | 1 (17) | 1 (17) | 2 (29) | 0 | 0 | 0 | 3 (17) | 1 (6) |
| Rash | 0 | 0 | 1 (17) | 0 | 1 (14) | 1 (14) | 1 (50) | 0 | 3 (17) | 1 (6) |
| Alopecia | 1 (33) | 0 | 2 (33) | 0 | 1 (14) | 0 | 0 | 0 | 4 (22) | 0 |
| Somnolence | 1 (33) | 0 | 0 | 0 | 0 | 0 | 1 (50) | 0 | 2 (11) | 0 |
| Hyperglycemia | 1 (33) | 0 | 0 | 0 | 0 | 0 | 1 (50) | 0 | 2 (11) | 0 |
| Diarrhea | 0 | 0 | 2 (33) | 0 | 0 | 0 | 0 | 0 | 2 (11) | 0 |
| Vomiting | 0 | 0 | 0 | 0 | 2 (29) | 0 | 0 | 0 | 2 (11) | 0 |
| Dose-limiting toxicity (DLT) | 0/3b | 0/5b | 2/6b | 2/2b | 4/16b | |||||
| DLT events | Grade 2 nauseac | Grade 3 fatigue | ||||||||
| Grade 3 fatigue | Grade 3 fatigue | |||||||||
Determined maximum tolerated dose.
Number of patients evaluable for DLT assessment.
Unable to administer at least 75% of doses in cycle 1 due to this toxicity.
Toxicity profile of TAK-228 and alisertib in patients with advanced solid tumors
Sixteen of the 18 patients (89%) receiving at least one dose of study treatment experienced an adverse effect attributed to study therapy. Of these patients, 13 (72%) experienced adverse effects at least possibly related to TAK-228, and 15 (83%) experienced adverse effects at least possibly related to alisertib. Rash was the most common toxicity attributed to TAK-228 alone; neutropenia and fatigue were the most common toxicities attributed to alisertib alone. The most common toxicities attributed to both drugs were fatigue, nausea, and mucositis (Table 2). Most toxicities were grade 1 or 2 and manageable with supportive care. Neutropenia was the only grade 4 toxicity documented; no grade 5 toxicities were reported. Dose reduction of alisertib was required in 2 patients; 1 required 2 reductions for cytopenias (dose level 3), and the second required 2 reductions and ultimate discontinuation for fatigue (dose level 2.5). Dose reduction of TAK-228 was required in 3 patients, including 1 patient treated at the MTD (dose level 2) for mucositis, 1 at dose level 2.5 for fatigue, and 1 at dose level 3 for rash.
The mTOR pathway is important in glucose homeostasis, and a known adverse effect of mTOR inhibitor therapy is hyperglycemia. Hyperglycemia was documented in one patient at dose level 1 and one at dose level 2.5 (13%, both grade 2). Due to structural similarity to benzodiazepines, Aurora A kinase inhibitors can have GABA receptor activity and benzodiazepine-like central nervous system (CNS) effects. Somnolence related to alisertib was documented in one patient at dose level 1 and one at dose level 2.5 (13%, both grade 1). An additional patient experienced grade 1 cognitive changes (dose level 2). None of these events required dose modification. However, in one patient with recurrent grade 3 fatigue requiring multiple dose reductions of alisertib (dose level 2.5), CNS drug effects were thought to possibly contribute.
Pharmacokinetic analysis
No significant drug interaction between the agents was identified in PK analysis (Table 3). At the MTD of TAK-228 of 2 mg, the Cmax of the single-agent lead-in dose was 24.2 (±8.8) ng/mL, which was similar to the Cmax following combination dosing with alisertib of 24.7 (±13.6) ng/mL. The AUC0–24h of TAK-228 2 mg alone and in combination with alisertib was also comparable: 172.9 (±106.9) ng/mL × hours as compared with 128.2 (±72.7) ng/mL hours. The half-life of single-agent TAK-228 at the 2 mg dose was 6.8 (±4.2) hours, similar to 6.0 (±2.1) hours in combination with alisertib.
Table 3.
Pharmacokinetic parameters of TAK-228 and alisertib alone and in combination across phase I dose levels.
| TAK-228 pharmacokinetic parameters | ||||
|---|---|---|---|---|
| Tmax (h) | Cmax (ng/mL) | AUC (ng/mL × h) | Half-life (h) | |
| Run-in dose | ||||
| 1 mg cohort | 1 ± 0 | 7.7 ± 2.6 | 46.7 ± 13.1 | 9.8 ± 3.7 |
| 2 mg cohort | 1.1 ± 0.5 | 24.2 ± 8.8 | 172.9 ± 106.9 | 6.8 ± 4.2 |
| 3 mg cohort | 1.3 ± 1.1 | 25.7 ± 9.4 | 154.5 ± 109.8 | 6.6 ± 7.8 |
| Combo dose | ||||
| 1 mg cohort | 1.2 ± 0.8 | 8.6 ± 4.2 | 47.4 ± 13.8 | 9.1 ± 1.7 |
| 2 mg cohort | 1.5 ± 1.2 | 24.7 ± 13.6 | 128.2 ± 72.7 | 6.0 ± 2.1 |
| Alisertib pharmacokinetic parameters | ||||
| Tmax (h) | Cmax (ng/mL) | AUC0–8h (ng/mL × h) | AUCss 0–8h (ng/mL × h)a | |
| Run-in dose | ||||
| 30 mg cohort | 4.9 ± 3.0 | 579 ± 202 | 2,633 ± 607 | 7,505 ± 1,731 |
| 40 mg cohort | 10.6 ± 9.5 | 706 ± 268 | 2,623 ± 1,566 | 7,477 ± 4,464 |
| Combo dose | ||||
| 30 mg cohort | 1.3 ± 1.6 | 1,049 ± 363 | 6,119 ± 2,331b | |
| 40 mg cohort | 1.5 ± 1.0 | 1,306 ± 287 | 7,672 ± 1508b | |
The AUC at steady state (AUCss) was estimated using an accumulation factor based on a 12-hour dosing interval and a 20-hour half-life for alisertib as reported in earlier clinical trials.
Since alisertib was dosed twice per day during combination dosing, the AUC0–8 h should be compared with the AUCss 0–8 h for more accurate comparison.
Accumulation of alisertib was expected with b.i.d. dosing for 7 days given its half-life of approximately 20 hours. As such, an accumulation factor was utilized to estimate steady-state AUC (AUCss) when comparing data from the single lead-in dose to b.i.d. dosing with the combination. At the MTD of 30 mg b.i.d., utilizing the accumulation factor, the alisertib AUCss 0–8h was 7,505 (±1,731) ng/mL × hours, as compared with an AUC0–8h of 6,119 (±2,331) ng/mL × hours when combined with TAK-228. Differences in Tmax and Cmax between the single-dose, single-agent alisertib lead-in and b.i.d. combination dosing with TAK-228 were attributed to these differences in accumulation.
Antitumor activity
The median duration of study protocol treatment was 2.3 months (range, 0.4–13.4 months; Fig. 3A). A patient with ER+/HER2− breast cancer who had received 5 prior lines of therapy remained on study for 15 cycles (over 10 months), and a patient with castrate-resistant prostate cancer who had received 9 prior lines of therapy remained on study for 18 cycles (over 13 months).
Figure 3.

Antitumor activity of TAK-228 and alisertib in patients with refractory solid tumors. A, Duration of time on study in months, each bar represents a patient. Dose levels indicated by color of bar. *, Patient discontinued study related to toxicity. B, Best response on imaging in patients with measurable disease per RECIST. Dose levels indicated by color of bar. *, Progression per RECIST related to new lesion.
Ten patients with measurable disease were evaluable for RECIST response assessment. No patients had partial response to therapy. Best response of stable disease per RECIST was observed in 7 of 10 evaluable patients, with a 29% decrease in RECIST-measurable lesions documented in a patient with fallopian tube cancer. Progressive disease was documented in 3 patients related to the development of new lesions (Fig. 3B). The median duration of stable disease among those meeting RECIST criteria for such was approximately 4 months.
The most common reason for study discontinuation was radiographic progression for 10 patients, treatment-related toxicity for 4 patients, clinical progression for 3 patients, and patient preference for 1 patient. Six of these patients discontinued study treatment prior to first restaging imaging due to toxicity (3), clinical progression (2), or patient preference (1).
Pharmacodynamics
Serial tumor biopsies were performed in 2 patients with metastatic ER+/HER2− breast cancer treated at the MTD. (Supplementary Fig. S1). Patient 1 had disease progression on imaging at week 6 and patient 2 had stable disease on imaging and remained on study for 5 cycles prior to progression. Consistent with our preclinical observations, we observed a significant decrease in Ki67 expression following treatment with the combination of alisertib and TAK-228 in patient 2, which was associated with cleaved caspase expression, indicative of treatment-induced apoptosis. These changes were not observed in patient 1 who had early disease progression consistent with de novo resistance to treatment.
Discussion
The addition of TAK-228 to alisertib resulted in increased anticancer activity in preclinical models of TNBC, including cell lines and PDX models. In the phase I dose escalation study, the combination was found to be tolerable with a manageable side effect profile in patients with advanced solid tumors. The MTD was defined as TAK-228 2 mg oral once daily with continuous dosing in combination with alisertib 30 mg oral b.i.d. days 1 to 7 of a 21-day cycle.
The association of treatment-induced senescence with the development of de novo and acquired resistance to alisertib has been previously reported (10), as has mTOR pathway activation in models with acquired resistance to Aur A inhibition (14). Our preclinical findings add to a growing body of literature further supporting a role for mTOR pathway activation in resistance to Aur A inhibition. In clinical pharmacodynamic analysis, the combined inhibition of mTOR and Aur A led to decreased cell proliferation and increased apoptosis in tumor tissues from a patient treated at the MTD with stabilization of disease on study treatment. These findings need to be confirmed in a larger set of serial tumor biopsies, which will be collected in expansion cohorts.
Indications of translation of this mechanistic effect into clinical benefit were not directly assessed in the accompanying phase I study focused on safety and tolerability, though further evaluation of the efficacy is ongoing in expansion cohorts. However, of note in preliminary efficacy evaluation is the finding of 2 heavily pretreated patients (1 with ER+/HER2− breast cancer and 1 with castrate-resistant prostate cancer) remaining on treatment with prolonged stable disease for approximately 1 year. The clinical benefit noted in ER+/HER2− breast cancer is supported by the known activity of mTOR inhibitor everolimus (33) and potential activity of alisertib, as will be further elucidated in an ongoing phase II clinical trial evaluating alisertib with or without fulvestrant (NCT02860000). And though mTOR inhibitors are not FDA approved for treatment of prostate cancer, Akt inhibition in combination with abiraterone has shown promise in a phase II trial (34), with a phase III study ongoing (NCT03072238).
The adverse-effect profile of the combination was as anticipated according to known single-agent toxicities. Grade 3 fatigue was the most common DLT event, but was observed only at doses above the MTD. Though a total of 7 dose reductions were required on study, only one (for mucositis) occurred at the MTD. The pharmacodynamics of alisertib have been extensively studied in skin and tumor samples across single-agent clinical trials, with exposure-pharmacodynamic analyses supporting pharmacodynamically active exposures over a dose range of 30 to 50 mg b.i.d. for 7 days of a 21-day cycle (35). Furthermore, mechanism-based toxicities of neutropenia (alisertib) and hyperglycemia and rash (TAK-228) were observed at and below the MTD in this trial. And pharmacokinetic parameters of combination therapy at the MTD are similar to PK of each drug at the single-agent MTD (32, 36). Overall, these data indicate that alisertib and TAK-228 can be safely combined at pharmacokinetically and pharmacodynamically active doses at the MTD.
Additional combinations of mTOR inhibition and cell-cycle inhibition are being explored both preclinically and clinically, with combinations with cyclin-dependent kinase (CDK) inhibitors the most developed. Two early-phase trials have specifically evaluated such combinations in advanced ER+/HER2− breast cancer with hormonal therapy, and similar to this trial, neutropenia, stomatitis, and fatigue were the most common reported adverse events in both. The severity of these events was more significant in the study of letrozole or fulvestrant plus CDK 4/6 inhibitor palbociclib and dual PI3K/mTOR inhibitor gedatolisib, with the rate of those grade ≥3 in severity over 50% (37). Interestingly, a clinical benefit rate of 41.1% at 24 weeks was observed in the phase II portion of the study evaluating exemestane plus CDK 4/6 inhibitor ribociclib and mTOR inhibitor everolimus, which exceeded the primary endpoint threshold of the trial (>10%; ref. 38). Additional studies are evaluating mTOR/CDK inhibitor combinations in other tumor types (NCT03740334 and NCT03065062; refs. 39, 40).
An intermittent, pulse-dosing schedule of alisertib has recently been evaluated in combination with fulvestrant in patients with ER+/HER2− metastatic breast cancer. In this trial, alisertib was administered on days 1 to 3, 8 to 10, and 15 to 17 on a 28-day schedule with alisertib 50 mg b.i.d. identified as the RP2D. Interestingly, patients participating in this study had no grade ≥3 neutropenia or stomatitis, as compared with single-agent alisertib according to the standard dosing schedule, where grade 3 to 4 stomatitis was documented in 15% and grade 3 to 4 neutropenia in 57% (18). We chose to administer alisertib b.i.d. days 1 to 7 of a 21-day cycle in combination with continuously dosed TAK-228 in this trial based on available data at the time of trial design and with the hypothesis that higher alisertib doses followed by a break would result in more robust anticancer activity with a more favorable toxicity profile. However, consideration could be given to evaluating this combination with an alternate schedule of alisertib in the future. The toxicity profile will be closely evaluated in the expansion cohorts to help guide these future plans.
In conclusion, these data suggest TAK-228 and alisertib are a rational combination for the treatment of solid tumor malignancies that is safe and tolerable, with further efficacy assessments warranted. Expansion cohorts of patients with refractory solid tumors of all primary sites, as well as a cohort of patients with refractory pancreatic adenocarcinoma, will provide further information on this efficacy. In addition, pharmacodynamic assessment in these patients will help better delineate the mechanism of the combination. Functional imaging with diffusion-weighted magnetic resonance imaging will give a sense of single-agent versus combination effect on cellular metabolism, and biopsy samples at baseline, on single-agent therapy, and on combination therapy will be compared for evaluation of on-target activity as well as apoptosis and senescence. Further evaluation of additional dosing schedules is also planned. Together, these results will continue to provide more information on the clinical setting most likely to benefit from the combination of TAK-228 and alisertib for the treatment of cancer.
Supplementary Material
Translational Relevance.
Senescence and upregulation of genes in the PI3K/AKT/mTOR pathway have been observed in triple-negative breast cancer (TNBC) patient-derived xenograft (PDX) models treated with Aurora A kinase inhibitor alisertib to resistance. This study evaluates the addition of TORC1/2 inhibitor TAK-228 to alisertib in preclinical TNBC models, and translation of these findings to a first-in-human phase I dose escalation trial. The preclinical work suggests a combination effect of TAK-228 and alisertib, adding to a growing body of literature further supporting a role for mTOR pathway activation in resistance to Aurora A inhibition. In the clinical phase of this study, the combination was found to be safe and tolerable at the maximum tolerated dose (MTD), with 2 patients with heavily pretreated cancers remaining on study treatment for approximately 1 year. Pharmacodynamic assessment on tumor samples mirror preclinical findings in TNBC PDX models. Additional pharmacodynamic evaluation is ongoing in expansion cohorts.
Acknowledgments
The authors thank Drs. Peter Kabos and Carol Sartorius for original passage of the TNBC PDX models at the UCCC. Preclinical work was supported by CTEP MTA 15–2-00025, PI Diamond and Tentler. Both preclinical and clinical work were supported by Takeda Pharmaceutical Company. Dr. S. Gail Eckhardt received funding from the CPRIT Scholar Award RR160093.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Disclosure of Potential Conflicts of Interest
C.L. O’Bryant is an employee/paid consultant for AstraZeneca and Pfizer and reports receiving commercial research grants from Tesaro and Pfizer. W.A. Messersmith reports receiving commercial research grants from Takeda. No potential conflicts of interest were disclosed by the other authors.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Invest New Drugs 2012;30:2411–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem 2004;279:52175–82. [DOI] [PubMed] [Google Scholar]
- 3.Falchook GS, Bastida CC, Kurzrock R. Aurora kinase inhibitors in oncology clinical trials: current state of the progress. Semin Oncol 2015;42:832–48. [DOI] [PubMed] [Google Scholar]
- 4.Yuan CX, Zhou ZW, Yang YX, He ZX, Zhang X, Wang D, et al. Inhibition of mitotic Aurora kinase A by alisertib induces apoptosis and autophagy of human gastric cancer AGS and NCI-N78 cells. Drug Des Dev Ther 2015;9:487–508. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Ding YH, Zhou ZW, Ha CF, Zhang XY, Pan ST, He ZX, et al. Alisertib, an Aurora kinase A inhibitor, induces apoptosis and autophagy but inhibits epithelial to mesenchymal transition in human epithelial ovarian cancer cells. Drug Des Dev Ther 2015;9:425–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang F, Li H, Yan XG, Zhou ZW, Yi ZG, He ZX, et al. Alisertib induces cell cycle arrest and autophagy and suppresses epithelial-to-mesenchymal transition involving PI3K/Akt/mTOR and sirtuin 1-mediated signaling pathways in human pancreatic cancer cells. Drug Des Dev Ther 2015;9:575–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niu NK, Wang ZL, Pan ST, Ding HQ, Au GH, He ZX, et al. Pro-apoptotic and pro-autophagic effects of the Aurora kinase A inhibitor alisertib (MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the activation of mitochondria-mediated pathway and inhibition of p38 MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Dev Ther 2015;9:1555–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li JP, Yang YX, Liu QL, Pan ST, He ZX, Zhang X, et al. The investigational Aurora kinase A inhibitor alisertib (MLN8237) induces cell cycle G2–M arrest, apoptosis, and autophagy via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer cells. Drug Des Dev Ther 2015;9:1627–52. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Zhou N, Singh K, Mir MC, Parker Y, Lindner D, Dreicer R, et al. The investigational Aurora kinase A inhibitor MLN8237 induces defects in cell viability and cell-cycle progression in malignant bladder cancer cells in vitro and in vivo. Clin Cancer Res 2013;19:1717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tentler JJ, Ionkina AA, Tan AC, Newton TP, Pitts TM, Glogowska MJ, et al. p53 family members regulate phenotypic response to Aurora kinase A inhibition in triple-negative breast cancer. Mol Cancer Ther 2015;14:1117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev 2014;28:99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007;8:729–40. [DOI] [PubMed] [Google Scholar]
- 13.Jackson JG, Pant V, Li Q, Chang LL, Quintas-Cardama A, Garza D, et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012;21:793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ionkina AA, Tentler JJ, Kim J, Capasso A, Pitts TM, Ryall KA, et al. Efficacy and molecular mechanisms of differentiated response to the Aurora and angiogenic kinase inhibitor ENMD-2076 in preclinical models of p53-mutated triple-negative breast cancer. Front Oncol 2017;7:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melichar B, Adenis A, Lockhart AC, Bennouna J, Dees EC, Kayaleh O, et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: a five-arm phase 2 study. Lancet Oncol 2015;16:395–405. [DOI] [PubMed] [Google Scholar]
- 16.Di X, Shiu RP, Newsham IF, Gewirtz DA. Apoptosis, autophagy, accelerated senescence and reactive oxygen in the response of human breast tumor cells to adriamycin. Biochem Pharmacol 2009;77:1139–50. [DOI] [PubMed] [Google Scholar]
- 17.Elmore LW, Di X, Dumur C, Holt SE, Gewirtz DA. Evasion of a single-step, chemotherapy-induced senescence in breast cancer cells: implications for treatment response. Clin Cancer Res 2005;11:2637–43. [DOI] [PubMed] [Google Scholar]
- 18.Haddad TC, D’Assoro A, Suman V, Opyrchal M, Peethambaram P, Liu MC, et al. Phase I trial to evaluate the addition of alisertib to fulvestrant in women with endocrine-resistant, ER+ metastatic breast cancer. Breast Cancer Res Treat 2018;168:639–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z, Wang F, Zhou ZW, Xia HC, Wang XY, Yang YX, et al. Alisertib induces G2–M arrest, apoptosis, and autophagy via PI3K/Akt/mTOR- and p38 MAPK-mediated pathways in human glioblastoma cells. Am J Transl Res 2017;9:845–73. [PMC free article] [PubMed] [Google Scholar]
- 20.Ren BJ, Zhou ZW, Zhu DJ, Ju YL, Wu JH, Ouyang MZ, et al. Alisertib induces cell cycle arrest, apoptosis, autophagy and suppresses EMT in HT29 and Caco-2 cells. Int J Mol Sci 2015;17:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ingels A, Zhao H, Thong AE, Saar M, Valta MP, Nolley R, et al. Preclinical trial of a new dual mTOR inhibitor, MLN0128, using renal cell carcinoma tumorgrafts. Int J Cancer 2014;134:2322–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gokmen-Polar Y, Liu Y, Toroni RA, Sanders KL, Mehta R, Badve S, et al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res Treat 2012;136:673–82. [DOI] [PubMed] [Google Scholar]
- 23.Lou HZ, Weng XC, Pan HM, Pan Q, Sun P, Liu LL, et al. The novel mTORC1/2 dual inhibitor INK-128 suppresses survival and proliferation of primary and transformed human pancreatic cancer cells. Biochem Biophys Res Commun 2014;450:973–8. [DOI] [PubMed] [Google Scholar]
- 24.Diamond J, Borges VF, Kabos P, Krill-Jackson E, Graham R, Hoffman A, et al. Phase 1b/2 safety and efficacy of TAK-228, plus exemestane or fulvestrant in postmenopausal women with ER+/HER2-metastatic breast cancer. Ann Oncol 2016;27(suppl_6):277P. [Google Scholar]
- 25.Diamond JR Potter D, Salkeni M, Silverman P, Haddad T, Forget F, et al. Phase 2 safety and efficacy results of TAK-228 in combination with exemestane or fulvestrant in postmenopausal women with ER-positive/HER2-negative metastaticbreast cancer previously treated with everolimus. 2018; San Antonio, TX. Cancer Res 2019;79 Suppl 4:PD1–09. [Google Scholar]
- 26.Diamond JR, Eckhardt SG, Tan AC, Newton TP, Selby HM, Brunkow KL, et al. Predictive biomarkers of sensitivity to the aurora and angiogenic kinase inhibitor ENMD-2076 in preclinical breast cancer models. Clin Cancer Res 2013;19:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klauck PJ, Bagby SM, Capasso A, Bradshaw-Pierce EL, Selby HM, Spreafico A, et al. Antitumor activity of the polo-like kinase inhibitor, TAK-960, against preclinical models of colorectal cancer. BMC Cancer 2018;18:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chittajallu DR, Florian S, Kohler RH, Iwamoto Y, Orth JD, Weissleder R, et al. In vivo cell-cycle profiling in xenograft tumors by quantitative intravital microscopy. Nat Methods 2015;12:577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008;132:487–98. [DOI] [PubMed] [Google Scholar]
- 30.Marcus JM, Burke RT, DeSisto JA, Landesman Y, Orth JD. Longitudinal tracking of single live cancer cells to understand cell cycle effects of the nuclear export inhibitor, selinexor. Sci Rep 2015;5:14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bagby S, Messersmith WA, Pitts TM, Capasso A, Varella-Garcia M, Klauck PJ, et al. Development and maintenance of a preclinical patient derived tumor xenograft model for the investigation of novel anti-cancer therapies. J Vis Exp 2016;115:54393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cervantes A, Elez E, Roda D, Ecsedy J, Macarulla T, Venkatakrishnan K, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective aurora a kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2012;18:4764–74. [DOI] [PubMed] [Google Scholar]
- 33.Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Bono JS, De Giorgi U, Rodrigues DN, Massard C, Bracarda S, Font A, et al. Randomized phase II study evaluating Akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN loss. Clin Cancer Res 2019;25:928–36. [DOI] [PubMed] [Google Scholar]
- 35.Venkatakrishnan K, Zhou X, Ecsedy J, Mould DR, Liu H, Danaee H, et al. Dose selection for the investigational anticancer agent alisertib (MLN8237): pharmacokinetics, pharmacodynamics, and exposure-safety relationships. J Clin Pharmacol 2015;55:336–47. [DOI] [PubMed] [Google Scholar]
- 36.Ghobrial IM, Siegel DS, Vij R, Berdeja JG, Richardson PG, Neuwirth R, et al. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: a phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenstrom’s macroglobulinemia. Am J Hematol 2016;91:400–5. [DOI] [PubMed] [Google Scholar]
- 37.Forero-Torres A, Han H, Dees EC, Wesolowski R, Bardia A. Phase Ib study of gedatolisib in combination with palbociclib and endocrine therapy in women with estrogen receptor positive metastatic breast cancer. J Clin Oncol 2018;36(15_suppl):1040. [Google Scholar]
- 38.Bardia A, Hurvitz SA, DeMichele A, Clark AS, Zelnak AB. Triplet therapy (continuous ribociclib, everolimus, exemestane) in HER+/HER2-advanced breast cancer postprogression on a CDK4/6 inhibitor: efficacy, safety and biomarker results. J Clin Oncol. 2019;37(15_suppl):1016. [Google Scholar]
- 39.Weinberg BA, Wang H, Witkiewicz AK, Marshall J, Knudsen ES. A phase I/II study of ribociclib plus everolimus in patients with metastatic pancreatic adenocarcinoma refractory to chemotherapy. J Clin Oncol. 2018;36(15_suppl):TPS4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tawfik B, Gerber DE, Burtness B, Hughes RS, Myers LL. Phase II trial of ribociclib and everolimus in p16 low anaplastic thyroid cancer. J Clin Oncol 2017;35(15_suppl):TPS6098. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.