

Abstract
Cross-electrophile coupling reactions of two Csp3–X bonds remain challenging. Herein we report an intramolecular nickel-catalyzed cross-electrophile coupling reaction of 1,3-diol derivatives. Notably, this transformation is utilized to synthesize a range of mono- and 1,2-disubstituted alkylcyclopropanes, including those derived from terpenes, steroids, and aldol products. Additionally, enantioenriched cyclopropanes are synthesized from the products of proline-catalyzed and Evans aldol reactions. A procedure for direct transformation of 1,3-diols to cyclopropanes is also described. Calculations and experimental data are consistent with a nickel-catalyzed mechanism that begins with stereoablative oxidative addition at the secondary center.
Graphical Abstract
Cross-electrophile coupling (XEC) reactions have the potential to construct carbon-carbon bonds in an efficient manner. To favor cross-reactivity, reactions often pair two substrates of different reactivity, in part to differentiate oxidative addition events.1 For example, development of aryl-alkyl XEC reactions have been fruitful.1d,e,2 Reactions that combine two substrates with similar reactivity can be challenging and result in homocoupled products.3 As such, many known examples of nickel-catalyzed XEC reactions that forge Csp3–Csp3 bonds employ one activated substrate as a coupling partner, e.g., allylic or benzylic electrophiles.1d,e,4,5,6,7 There are few examples of nickel-catalyzed XEC reactions that engage two unactivated alkyl electrophiles.8,9 Development of an XEC reaction that employs readily-available, unactivated alkyl electrophiles as both reactive partners would significantly expand the scope of these transformations.
A 1,3-diol is a compelling functional group motif for use in XEC reactions. Largely due to their prevalence in polyketides, 1,3-diols have robust, well-established synthetic routes for their preparation.10 For example, substituted 1,3-diols are easily accessed through the reduction of aldol products. We hypothesized that sulfonates derived from 1,3-diols would be engaged by a nickel catalyst and undergo an intramolecular XEC reaction to form cyclopropanes. Furthermore, since aldol reactions can provide outstanding levels of enantioselectivity,11 this strategy could provide straightforward and predictable access to enantioenriched cyclopropanes, leveraging a well-established and powerful field. Additionally, these reactions would complement traditional asymmetric cyclopropanation routes that typically engage alkene starting materials.12
Alkyl sulfonates have a history of use in nickel-catalyzed cross-coupling (XC) and XEC reactions.13,14,15 Notably, sulfonates utilized in these reactions are frequently generated in situ from the corresponding alcohols. In the context of cross-coupling reactions, nickel-catalyzed Negishi couplings of benzylic mesylates and Kumada reactions of cyclic sulfates have been reported (Scheme 1a).16,17 The use of alkyl sulfonates as electrophiles in nickel-catalyzed XEC reactions developed contemporaneously, beginning with homocoupling reactions.18 Chemoselective XEC reactions have utilized alkyl sulfonates with aryl and vinyl halides and pseudohalides (Scheme 1b).19,20,21 Cross-selective pairing of two alkyl sulfonates was demonstrated using methyl tosylate (Scheme 1c).7a,22 However, to the best of our knowledge, no cross-selective XEC reaction of two primary or secondary alkyl sulfonates has been reported. Based on the accessibility of 1,3-diols and reactivity of sulfonates, we sought to develop a nickel-catalyzed XEC reaction of two alkyl mesylates for cyclopropane synthesis (Scheme 1c).12
Scheme 1.
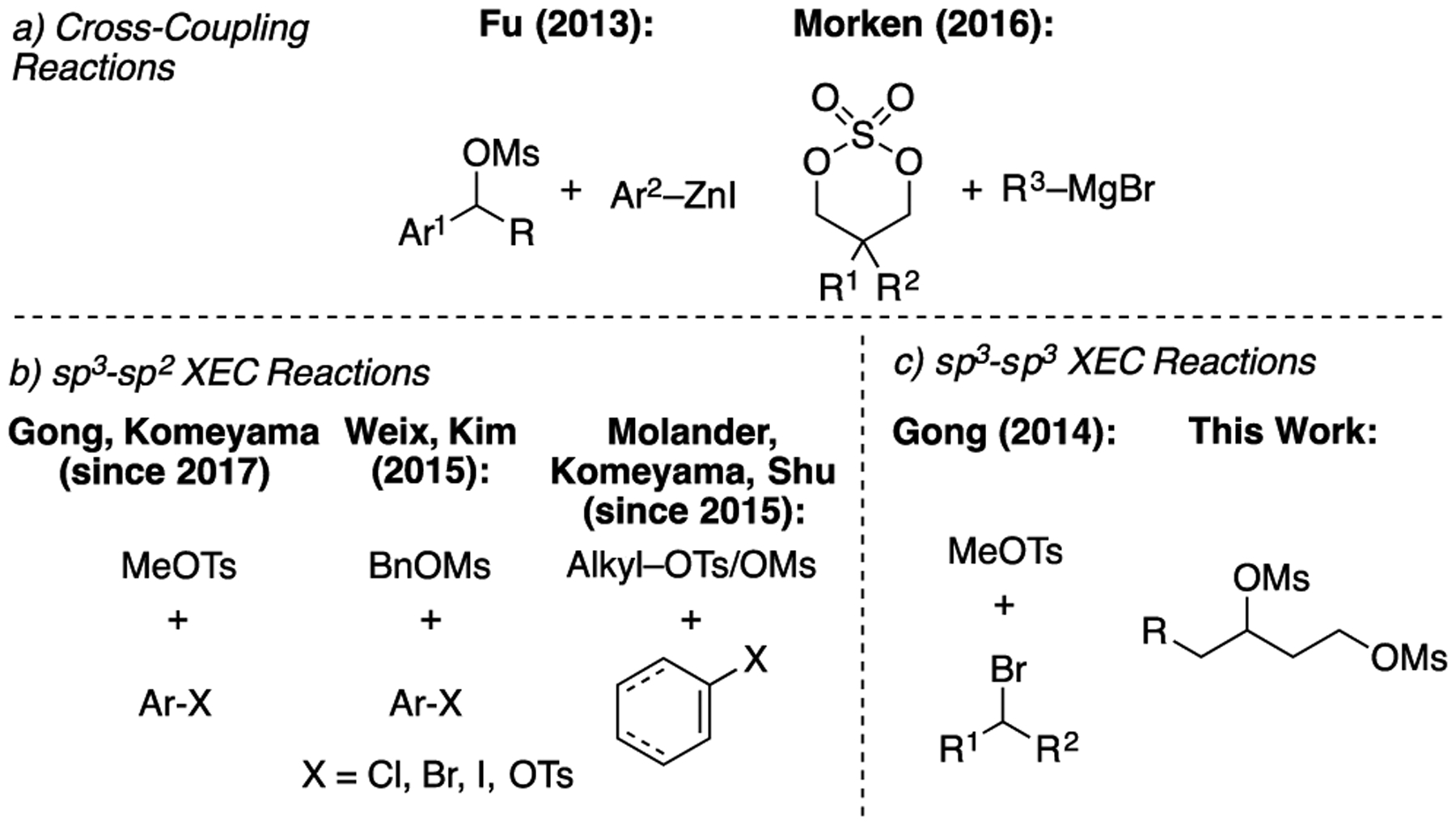
Nickel-Catalyzed XC and XEC Reactions of Alkyl Sulfates and Sulfonates
We began our investigation by employing 1,3-dimesylate 1. This substrate was designed to provide an alkylcyclopropane with low volatility to facilitate isolation and analysis. We evaluated a series of ligands in the presence of Ni(cod)2 and methylmagnesium iodide (MeMgI) in DCM/PhMe (Table 1). The diphosphine ligands rac-BINAP and dppm, in addition to the pyridyl ligand Bphen, produced the highest yields of cyclopropane 2 (entries 1–3). Additionally, a 70% yield of the desired cyclopropane was achieved utilizing bench-stable ((R)-BINAP)NiCl2 as the catalyst (entry 9). In general, across a range of substrates, rac-BINAP and dppm provided robust reaction yields, and so we selected these ligands for further experiments.23
Table 1.
Ligand Evaluation of XEC Reaction of Unactivated 1,3-Dimesylates
 | ||
|---|---|---|
| Entry | Deviation from reaction conditions | Yielda (%) |
| 1 | none | 75 |
| 2 | BPhen | 78 |
| 3 | dppm | 71 |
| 4 | PCy3 | 56 |
| 5 | DPEPhos | 44 |
| 6 | SiMes*HBF4 | 33 |
| 7 | Xantphos | 16 |
| 8 | no ligand | 13 |
| 9 | ((R)-BINAP)NiCI2 | 70 |
| 10 | PhMgBr | 5 |
| 11 | PhMgBr + Mgl2 | 17 |
| 12 | no Ni, no ligand | 5 |
| 13 | no MeMgl | 0 |
Yield determined by 1H NMR based on comparison to PhTMS as internal standard.
Following the ligand evaluation, we next investigated the importance of the Grignard reagent and nickel catalyst. Modifying the Grignard reagent to phenylmagnesium bromide almost completely shut down the reaction, with 5% of the desired cyclopropane observed (entry 10). Adding MgI2 to PhMgBr reaction conditions provided a similar result (entry 11). A control reaction without nickel and ligand (MeMgI only) produced a 5% yield of the desired cyclopropane (entry 12), while a control reaction in the absence of MeMgI provided no conversion to the desired cyclopropane and only recovered starting material (entry 13).
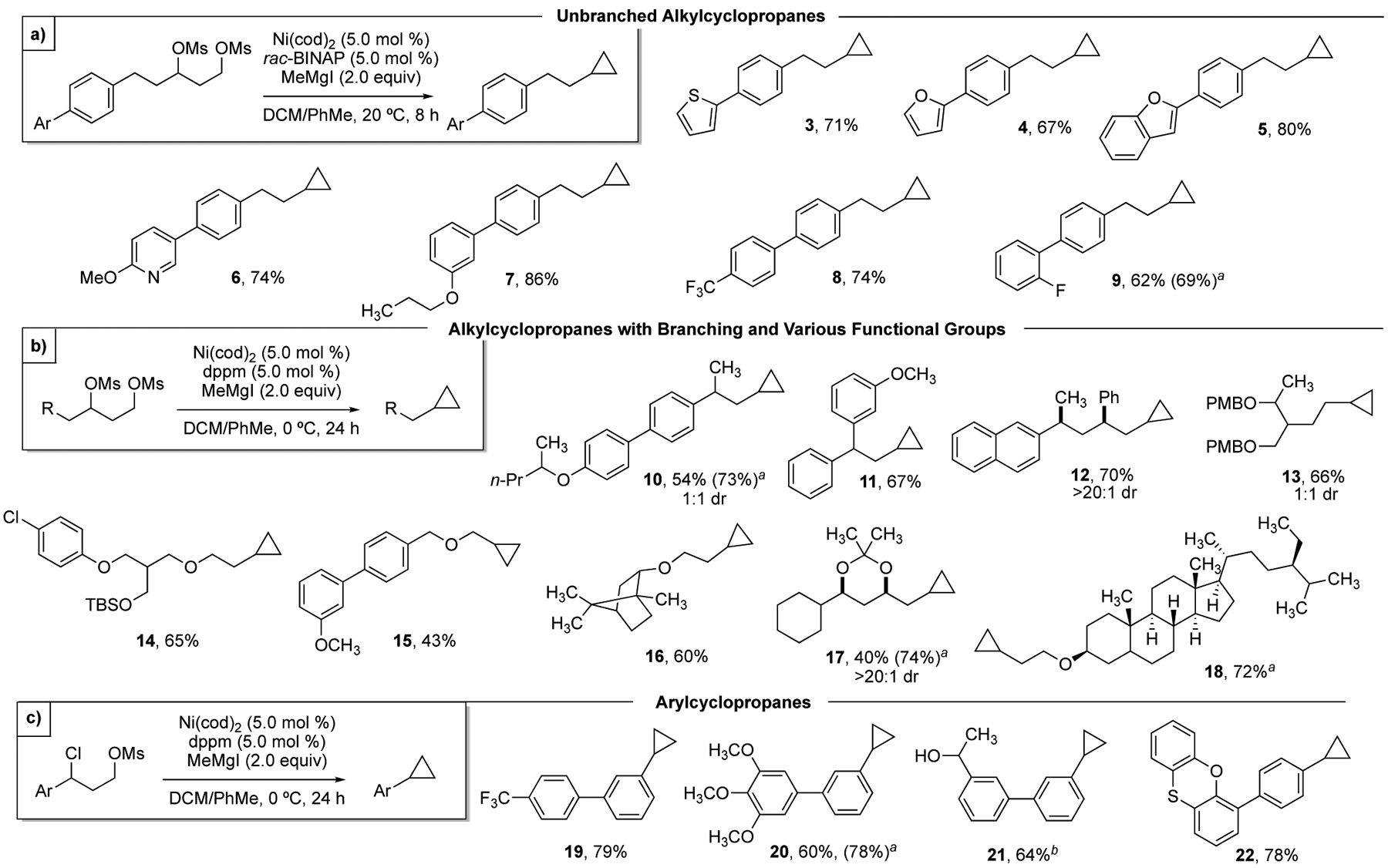
With optimized conditions in hand, we investigated the tolerance of various substituted aromatic and heterocyclic groups (Scheme 2a). Isolated yields are reported, however for certain substrates volatility or polarity complicated isolation. Therefore, yield determined by 1H NMR by comparison to an internal standard is also reported. Heterocycles such as thiophene, furan, and benzofuran were well tolerated under the reaction conditions (3–5), as was the substituted heterocycle 2-methoxypyridine (6). Electron-donating groups were well tolerated in the synthesis of 2 and 7, as well as electron-withdrawing groups such as aryl CF3 and aryl fluoride (8,9).
Scheme 2.
Isolated yields of reaction products. aYield determined by 1H NMR based on comparison to PhTMS as internal standard. b3.0 equiv of MeMgI
Our next efforts focused on testing the impact of steric bulk near the forming cyclopropane (Scheme 2b). For compounds such as 11, standard reaction conditions employing rac-BINAP as the ligand provided modest yields.23 Fortunately, we found that for these more hindered substrates, using dppm as the ligand and performing the reaction at 0 °C provided good yields. A series of alkyl and aryl groups were well tolerated in the β-position relative to the cyclopropane (10–12). For example, the diol precursor for cyclopropane 12 was prepared by a stereospecific Kumada ring-opening reaction.24 We were also interested in the functional group compatibility of the reaction. Compounds containing benzylic ethers (PMB), aryl chlorides, alkyl ethers, and silyl protecting groups (TBS) underwent smooth cyclopropane formation (13–15).
We next sought out to confirm our hypothesis that aromatic groups were not required for desired reactivity. To confirm this, we successfully synthesized cyclopropanes containing only sp3 centers such as borneol derivative 16 and β-sitosterol derivative 18. Acetonide 17 was formed smoothly from the corresponding tetraol derivative, demonstrating tolerance to a typical protecting group employed in polyketide synthesis. These results confirm that – in contrast to our laboratory’s previously published XC and XEC reactions – this XEC reaction does not require benzylic or allylic electrophiles to engage the nickel catalyst.5a,5b,25
Based on these results, we concluded that arene coordination would no longer be critical for reactivity, but the reaction could nonetheless provide a strategy for synthesis of arylcyclopropanes. In our prior work, synthesis was restricted to cyclopropanes bearing extended aromatic rings, due to requirements to activate substrates for oxidative addition.5a,5b,25 To thoroughly investigate the generality of this XEC reaction, we evaluated simple benzylic substrates. Notably, under standard conditions for preparation of the 1,3-dimesylates, benzylic mesylates undergo substitution to afford the benzylic chlorides.26 Subjection of 1,3-chloromesylates to our standard reaction conditions provided cyclopropanes bearing a range of substituted aromatic substituents (Scheme 2c; 19–22).
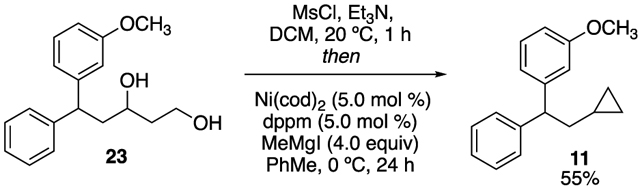
The potential impact of this transformation would be expanded if 1,3-diols could be employed as starting materials for the reaction. We were encouraged that other XC and XEC reactions that employ sulfonates generated in situ have been reported.16,20a A procedure was developed such that diol 23 was treated with MsCl and base, followed by addition to catalyst and Grignard reagent. Cyclopropane 11 was formed in good yield, similar to that observed when employing the corresponding 1,3-dimesylate. Therefore, this method allows direct conversion of a 1,3-diol to the corresponding cyclopropane (eq 1).
 |
(1) |
In order to elucidate a potential reaction mechanism, our first question was whether the mechanism involves oxidative addition (OA) of an alkyl mesylate, or if alkyl iodides are formed as intermediates in the reaction.27 A control experiment demonstrated that under the reaction conditions, in the absence of nickel catalyst, MeMgI converts 1,3-dimesylate 24 to 1,3-diiodide 25 (Table 2, entry 1). Monitoring the XEC reaction at early conversion produced 1,3-diiodide 25 in 56% yield and cyclopropane 7 in 18% yield (entry 2). Therefore, we hypothesized that 1,3-diiodide 25 is formed in situ and is a reactive intermediate in the nickel-catalyzed XEC reaction. Indeed, subjecting 25 to XEC reaction conditions provided clean conversion to cyclopropane 7 (eq 2).
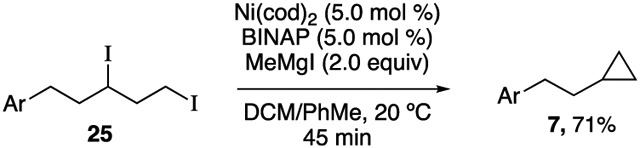 |
(2) |
Table 2.
Formation of 1,3-diiodide 25 in situ
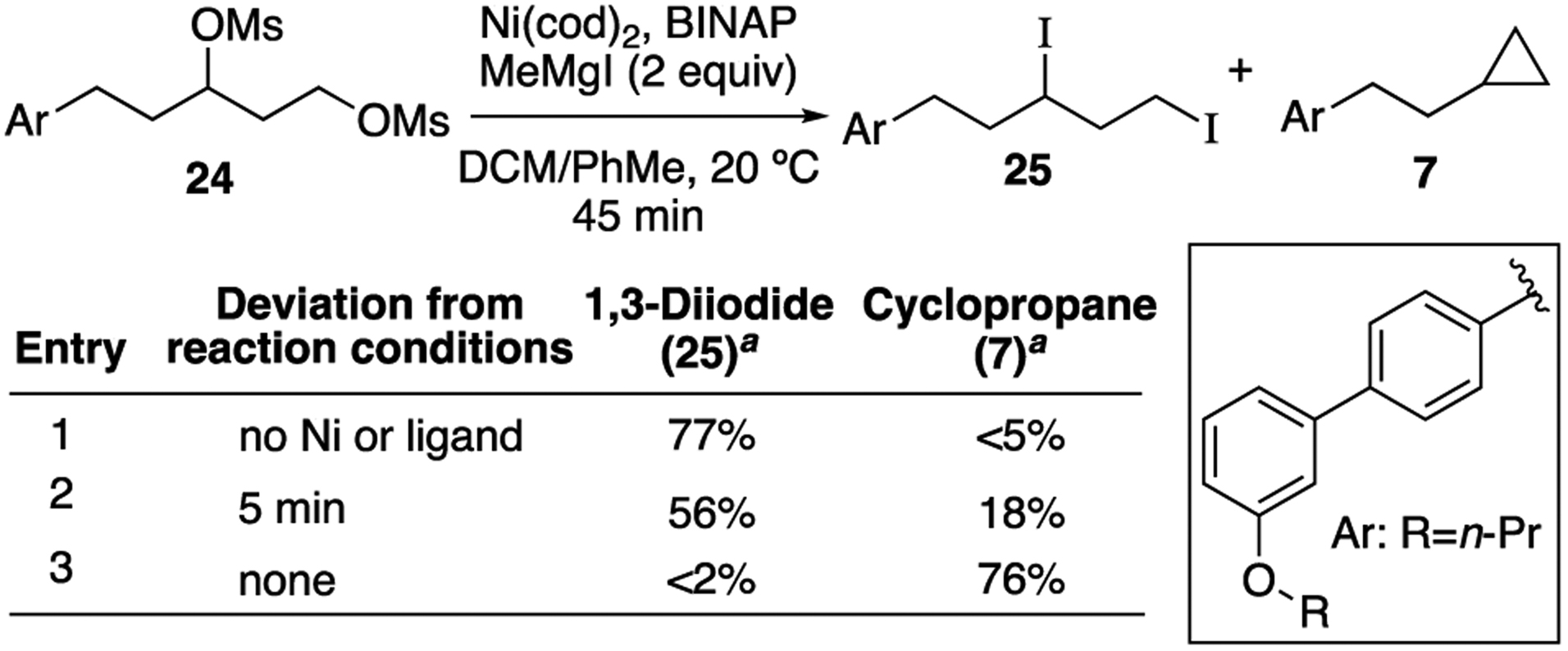
|
Yield determined by 1H NMR based on comparison to PhTMS as internal standard. See SI for details.
Our second question was whether oxidative addition initiates at the primary or secondary center. To begin to address this question, we calculated the relative barrier heights for oxidative addition of 1° and 2° alkyl iodides, via both radical and SN2 mechanisms (Figure 1).28 For both the secondary and primary center, the barrier heights for reaction via the radical pathway are significantly lower. The calculated lowest energy transition state (TS3) engages the secondary center by halogen atom abstraction to generate a 2° alkyl radical intermediate (INT7).29,30,31 This barrier height is lower for reaction at the secondary center, because the more substituted center is better able to stabilize the incipient alkyl radical.29,32,33,34 Therefore, we propose that the XEC reaction begins with iodine atom abstraction at the 2° alkyl iodide moiety, and proceeds through a 2° alkyl radical (INT7). This alkyl radical recombines with the nickel catalyst to form organonickel species INT8.
Figure 1.
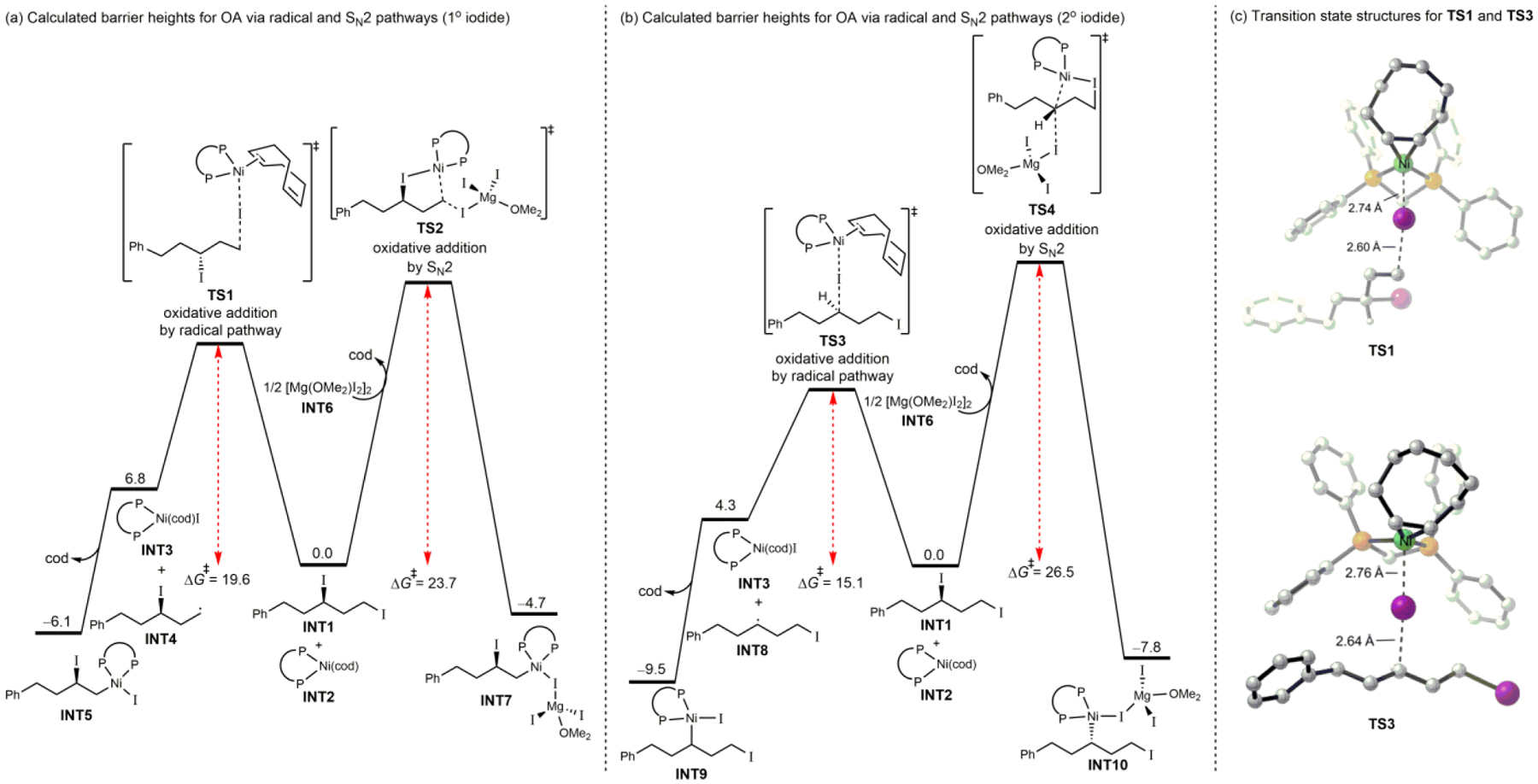
Comparison of DFT-computed barriers for Ni(0)-catalyzed C–I oxidative addition, employing dppm as the ligand. Free energies in toluene are in kcal/mol and were calculated under the M06/def2-TZVPP-SMD(toluene)//B3LYP-D3(BJ)/def2-SVP-SMD(toluene) level of theory.
To challenge these computational findings, we designed a series of competition experiments that probe the relative rates of the nickel-catalyzed XEC reaction at 1° and 2° centers.34 In a competition experiment, equimolar amounts of 1,3-dimesylate 24 and 1° mesylate 26 were subjected to XEC reaction conditions.35 Cyclopropane 7 was favored with a 7:1 ratio, relative to total products generated from reaction of 1° mesylate 26 (Scheme 3a). Similarly, in a competition experiment between 1,3-dimesylate 24 and 2° mesylate 27, cyclopropane 7 was favored, although to a lesser extent, with a 3:1 ratio (Scheme 3b). Therefore, in both cases, the reaction of the 1,3-dimesylate is favored over reaction of the monomesylate. Furthermore, comparison of the product distributions demonstrates that the secondary center engages faster than the primary center under the XEC conditions. These results are consistent with the calculated transition state barriers. They are also consistent with prior studies comparing the rates of reactions of substituted alkyl halides with nickel catalysts.29,34 Nickel-catalyzed halogen atom abstraction provides the basis for the observed preference for reaction of the 2° over 1° alkyl iodide.
Scheme 3.
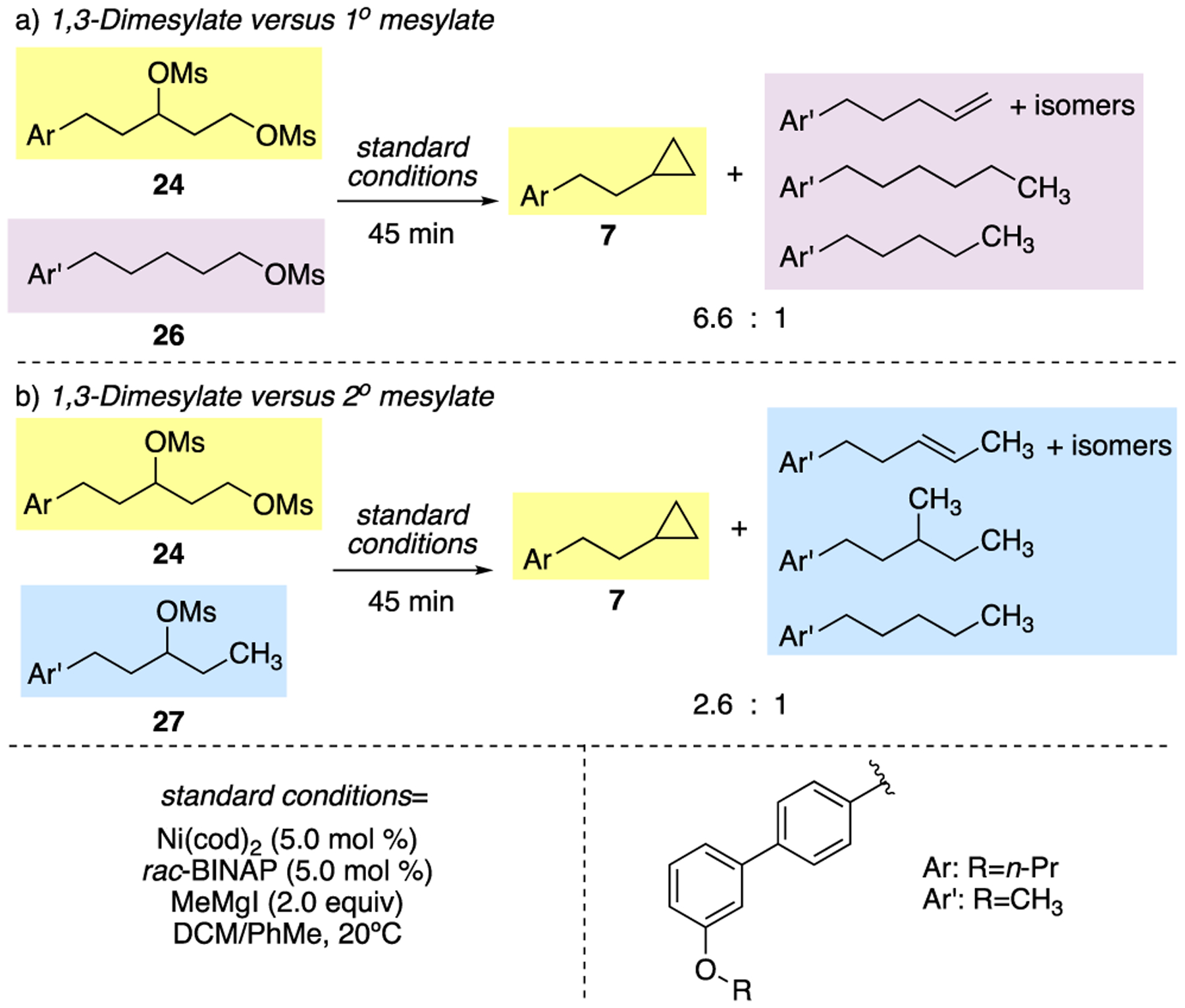
Competition experiments.
To further corroborate our proposed mechanism, we sought to determine the stereochemical outcome of the XEC reaction. Since oxidative addition of the 2° alkyl iodide is predicted to proceed via the alkyl radical, reactions are expected to be stereoablative.4 Consistent with this hypothesis, we observe a stereoconvergent XEC reaction to form 1,2-disubstituted cyclopropane 29 (Scheme 4). Either diastereomer of 1,3-dimesylate 28 provides trans-cyclopropane 29, consistent with epimerization via an alkyl radical intermediate.
Scheme 4.
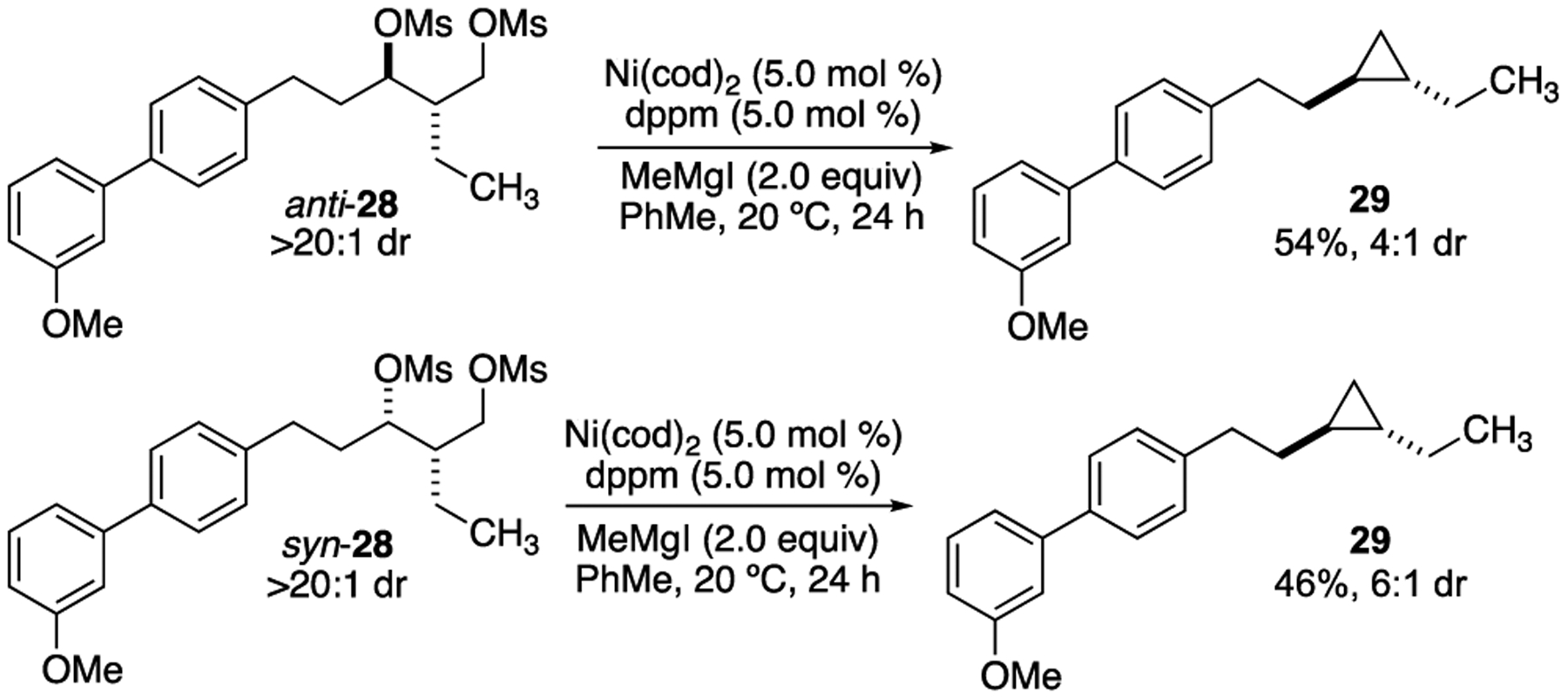
Stereoconvergent XEC reactions
Given the yield and diastereoselectivity observed in Scheme 4, we set out to apply our transformation to a range of 1,2-disubstituted alkylcyclopropanes (Scheme 5). The majority of corresponding diols were prepared by aldol or Claisen transformations.36 Compounds 30–36 were formed in moderate to good yield with preference for the trans diastereomer. Functional groups including dibenzofuran (31), methoxy phenyls (32), aryl ethers (33 and 35), and acetals (34 and 36) were tolerated in this alkyl series. Paralleling our strategy in Scheme 2, we also demonstrated arylcyclopropanes can be successfully synthesized from the corresponding benzylic chlorides (37–38).
Scheme 5.
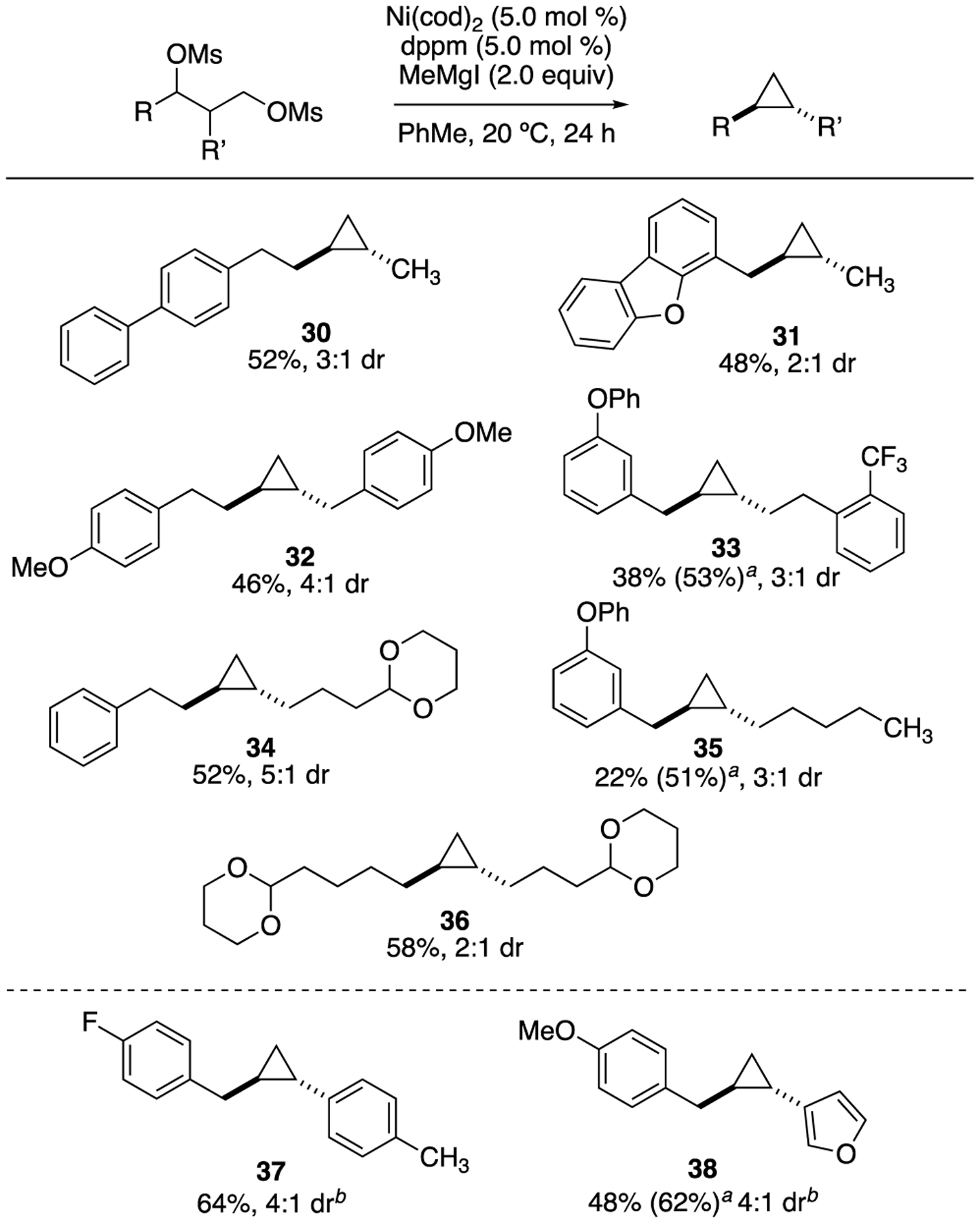
1,2-Disubstituted cyclopropanes
aYield determined by 1H NMR based on comparison to PhTMS as internal standard. bReaction stirred at 0 °C
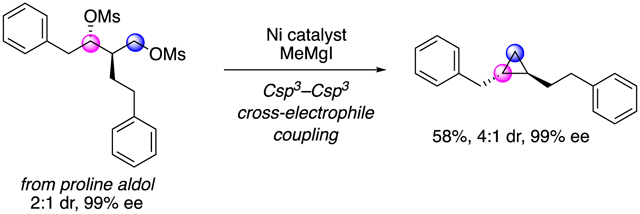
Based on our understanding of the reaction mechanism and observed levels of diastereoselectivity, we set out to synthesize enantioenriched 1,2-disubstituted cyclopropanes (Scheme 6). Proline-catalyzed and Evans aldol reactions were employed to prepare the corresponding substituted 1,3-diols with high enantioselectivity.37,38 Utilizing our transformation, alkylcyclopropane 40 and arylcyclopropane 42 were both formed with high enantiomeric excess. Configuration at C2 is conserved through the XEC reaction. The reaction favors formation of the trans-cyclopropane, setting the configuration of C1. Notably, compounds 40 and 42 do not bear the signature directing groups or acyl substitution required for direct synthesis by other asymmetric cyclopropanation methods.12
Scheme 6.
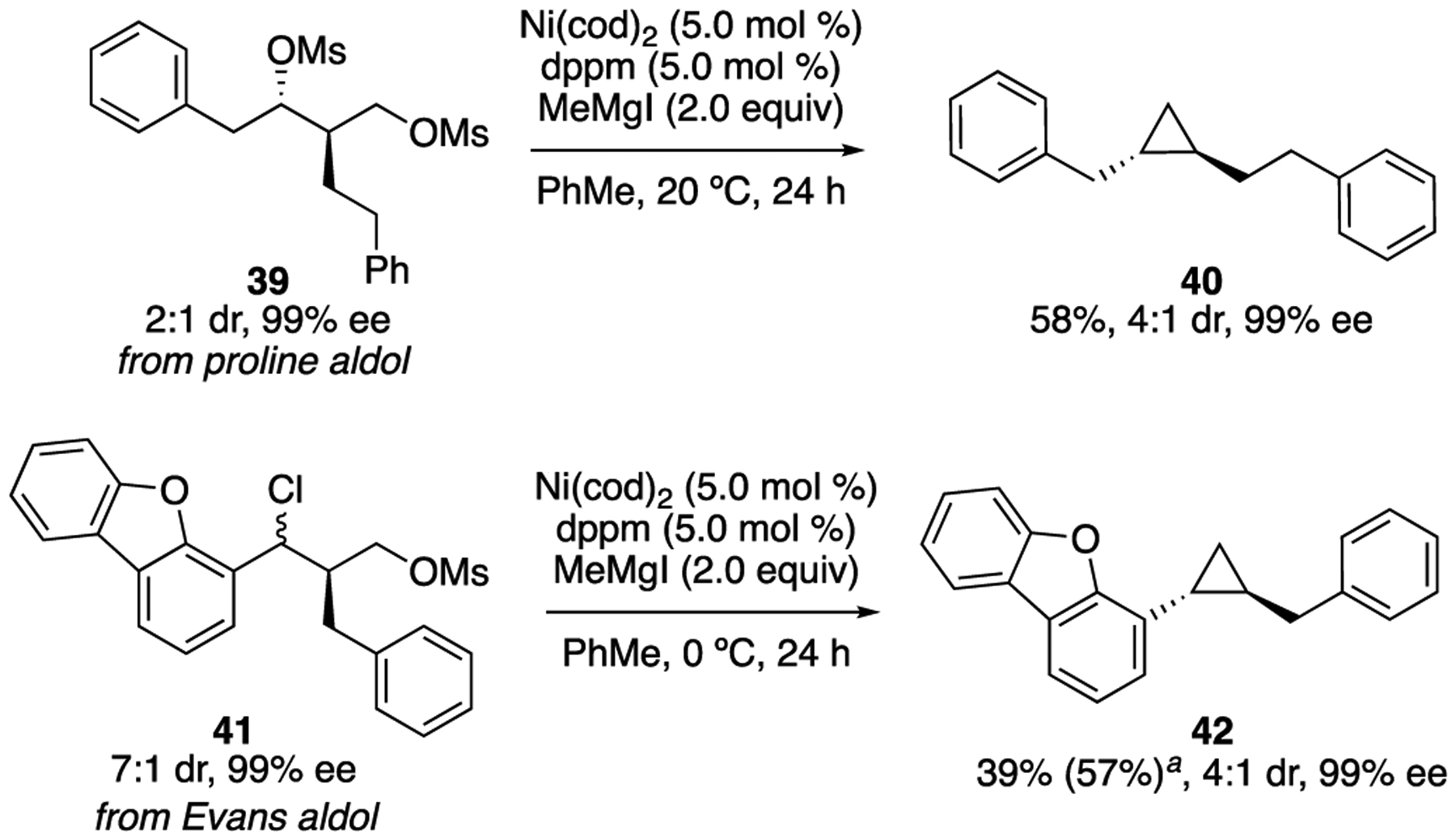
Enantioenriched 1,2-disubstituted cyclopropanes
aYield determined by 1H NMR based on comparison to PhTMS as internal standard
In summary, we report a nickel-catalyzed cross-electrophile coupling reaction of 1,3-dimesylates for the synthesis of alkylcyclopropanes. This transformation does not require activation of either electrophilic partner, and engages two alkyl mesylates. This method is tolerant of various functional groups and also allows access to arylcyclopropanes without extended aromatics. Furthermore, direct transformation of a 1,3-diol to the corresponding cyclopropane was established. Initial experimental and computational investigations are consistent with formation of 1,3-alkyl iodides that react with the nickel catalyst by iodine atom abstraction to provide 2° alkyl radicals. Synthesis of 1,2-disubstituted cyclopropanes is a stereoconvergent process consistent with the proposed mechanism of oxidative addition, and favors the trans diastereomer. The products of enantioselective aldol reactions were transformed to the corresponding enantioenriched cyclopropanes, therefore capitalizing on outstanding strategies of 1,3-diol synthesis.
Supplementary Material
ACKNOWLEDGMENT
Financial support from NIH NIGMS (R01GM100212, E.R.J.), National Natural Science Foundation of China (21702182, 21873081, X.H.), the Fundamental Research Funds for the Central Universities (2019QNA3009, X.H.) and Zhejiang University (X.H.) is gratefully acknowledged. Calculations were performed on the high-performance computing system at Department of Chemistry, Zhejiang University. We thank the Pronin laboratory (UC Irvine) for use of GC instrument, and the Rychnovsky laboratory (UC Irvine) for use of HPLC instrument. We also thank the Vanderwal laboratory (UC Irvine) for diisopropylamine and notably Bonnie Pak for guidance on synthesis of dibutylboron triflate. Abraham Paz is acknowledged for synthesis of starting material SI-59.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website.
Full experimental procedures and characterization data for all new compounds, as well as copies of 1H and 13C NMR spectra for each (PDF). Full details of computational experiments.
REFERENCES
- 1.(a) Biswas S; Weix DJ Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Weix DJ Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res 2015, 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Knappke CEI; Grupe S; Gärtner D; Corpet M; Gosmini C; Jacobi von Wangelin A Reductive Cross-Coupling Reactions between Two Electrophiles. Chem. Eur. J 2014, 20, 6828–6842. [DOI] [PubMed] [Google Scholar]; (e) Wang X; Dai Y; Gong H Nickel-Catalyzed Reductive Couplings. Top. Curr. Chem 2016, 374, 43. [DOI] [PubMed] [Google Scholar]
- 2.For an example, see: Everson DA; Jones BA; Weix DJ Replacing Conventional Carbon Nucleophiles with Electrophiles: Nickel-Catalyzed Reductive Alkylation of Aryl Bromides and Chlorides. J. Am. Chem. Soc 2012, 134, 6146–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For a recent discussion and solution in the context of vinyl electrophiles, see: Olivares AM; Weix DJ Multimetallic Ni- and Pd-Catalyzed Cross-Electrophile Coupling to form Highly Substituted 1,3-Dienes. J. Am. Chem. Soc 2018, 140, 2446–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucas EL; Jarvo ER Stereospecific and Stereoconvergent Cross-Couplings Between Alkyl Electrophiles. Nat. Rev. Chem 2017, 1, 0065. [Google Scholar]
- 5.(a) Tollefson EJ; Erickson LW Jarvo ER Stereospecific Intramolecular Reductive Cross-Electrophile Coupling Reactions for Cyclopropane Synthesis. J. Am. Chem. Soc 2015, 137, 9760. [DOI] [PubMed] [Google Scholar]; (b) Erickson LW Lucas EL; Tollefson EJ; Jarvo ER Nickel-Catalyzed Cross-Electrophile Coupling of Alkyl Fluorides: Stereospecific Synthesis of Vinylcyclopropanes. J. Am. Chem. Soc 2016, 138, 14006–14011. [DOI] [PubMed] [Google Scholar]; (c) Konev MO; Hanna LE; Jarvo ER Intra- and Intermolecular Nickel-Catalyzed Reductive Cross-Electrophile Coupling Reactions of Benzylic Esters with Aryl Halides. Angew. Chem. Int. Ed 2006, 55, 6730–6733. [DOI] [PubMed] [Google Scholar]
- 6.For allylic XEC with alkyl halides, see: [Google Scholar]; (a) Dai Y; Wu F; Zang Z; You H; Gong H Ni-Catalyzed Reductive Allylation of Unactivated Alkyl Halides with Allylic Carbonates. Chem. Eur. J 2012, 18, 808–812. [DOI] [PubMed] [Google Scholar]; (b) Yu Y; Chen H; Qian Q; Yao K; Gong H An Extension of Nickel-Catalyzed Reductive Coupling Between Tertiary Alkyl Halides with Allylic Carbonates. Tetrahedron 2018, 74, 5651–5658. [Google Scholar]
- 7.Examples of other activated Csp3 electrophiles that have been paired with alkyl halides in Ni-catalyzed XEC reactions: [Google Scholar]; (a) methyl p-toluenesulfonate: Liang Z; Xue W; Lin K; Gong H Nickel-Catalyzed Reductive Methylation of Alkyl Halides and Acid Chlorides with Methyl p-Tosylate. Org. Lett 2014, 16, 5620–5623. [DOI] [PubMed] [Google Scholar]; (b) Primary alkyl Katritzky salts: Ni S; Li C-X; Mao L; Wang Y; Han J; Pan Y Ni-Catalyzed Deaminative Cross-Electrophile Coupling of Katritzky Salts with Halides via C–N Bond Activation. Sci. Adv 2019, 5: eaaw9516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Togni’s reagent: Chen Y; Ma G; Gong H Copper-Catalyzed Reductive Trifluoromethylation of Alkyl Iodides with Togni’s Reagent. Org. Lett 2018, 20, 4677–4680. [DOI] [PubMed] [Google Scholar]
- 8.(a) Gu J; Wang X; Xue W; Gong H Nickel-Catalyzed Reductive Coupling of Alkyl Halides with Other Electrophiles: Concept and Mechanistic Considerations. Org. Chem. Front 2015, 2, 1411–1421. [Google Scholar]; (b) Yu X; Yang T; Wang S; Xu H; Gong H Nickel-Catalyzed Reductive Cross-Coupling of Unactivated Alkyl Halides. Org. Lett 2011, 13, 2138–2141. [DOI] [PubMed] [Google Scholar]; (c) Xu H; Zhao C; Qian Q; Deng W; Gong H Nickel-Catalyzed Cross-Coupling of Unactivated Alkyl Halides Using Bis(pinacolato)diboron as Reductant. Chem. Sci 2013, 4, 4022–4029. [Google Scholar]; (d) Xue W; Xu H; Liang Z; Qian Q; Gong H Nickel-Catalyzed Reductive Cyclization of Alkyl Dihalides. Org. Lett 2014, 16, 4984–4987. [DOI] [PubMed] [Google Scholar]
- 9.A successful strategy is to employ only one partner that will generate an alkyl radical under photocatalytic conditions. For a lead reference, see: Smith RT; Zhang X; Rincón JA; Agejas J; Mateos C; Barberis M; Garcia-Cerrada S; de Frutos O; MacMillan DWC Metallaphotoredox-Catalyzed Cross-Electrophile Csp3-Csp3 Coupling of Aliphatic Bromides. J. Am. Chem. Soc 2018, 140, 17433–17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For selected reviews, see: [Google Scholar]; a) Rychnovsky SD Oxo Polyene Macrolide Antibiotics. Chem. Rev 1995, 95, 2021–2040. [Google Scholar]; b) Bode SE; Wolberg M; Muüller M Stereoselective Synthesis of 1,3-Diols. Synthesis 2006, 4, 557–588. [Google Scholar]; c) Gupta P; Mahajan N; Taneja SC Recent Advances in the Stereoselective Synthesis of 1,3-Diols using Biocatalysts. Catal. Sci. Technol 2013, 3, 2462–2480. [Google Scholar]
- 11.For selected reviews, see: [Google Scholar]; (a) Yamashita Y; Yasukawa T; Yoo WJ; Kitanosono, Kobayashi, S. Catalytic Enantioselective Aldol Reactions Chem Soc. Rev 2018, 47, 4388–4480. [DOI] [PubMed] [Google Scholar]; (b) Cowden CJ; Paterson I Asymmetric Aldol Reactions Using Boron Enolates Organic Reactions; Paquette LA, et al. , Ed.; Wiley: New York, 1997; Vol. 51; pp 1–200. [Google Scholar]
- 12.Due to their interesting structural and biological properties, several strategies for cyclopropane synthesis have been reported and frequently employ carbenes and carbenoids. For reviews, see: [Google Scholar]; (a) Ebner C; Carreira E Cyclopropanation Strategies in Recent Total Syntheses. Chem. Rev 2017, 117, 11651–11679. [DOI] [PubMed] [Google Scholar]; (b) Lebel H; Marcoux J–F; Molinaro C; Charette AB Stereoselective Cyclopropanation Reactions. Chem. Rev 2003, 103, 977–1050. [DOI] [PubMed] [Google Scholar]; (c) Bartoli G; Bencivenni G; Dalpozzo R Asymmetric Cyclopropanation Reactions. Synthesis 2014, 46, 979–1029. [Google Scholar]; (d) Wu W; Lin Z; Jiang H Recent Advances in the Synthesis of Cyclopropanes. Org. Biomol. Chem 2018, 16, 7315–7329. [DOI] [PubMed] [Google Scholar]
- 13.For review of phenol derivatives, aryl triflates and aryl sulfonates, in traditional cross-coupling reactions, see: [Google Scholar]; Rosen BM; Quasdorf KW; Wilson DA; Zhang N; Resmerita A-M; Garg NK; Percec V Nickel-Catalyzed Cross-Couplings Involving Carbon–Oxygen Bonds. Chem. Rev 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) for a lead reference, see: Hofmayer MS; Lutter FH; Grokenberger L; Hammann JM; Knochel P Practical Ni-Catalyzed Cross-Coupling of Unsaturated Zinc Pivalates with Unsaturated Nonaflates and Triflates. Org. Lett 2019, 21, 36–39. [DOI] [PubMed] [Google Scholar]
- 14.For reviews and a lead reference for aryl triflates in XEC, see: [Google Scholar]; reference 1.; (b) Huang L; Ackerman LKG; Kang K; Parson AM; Weix DJ LiCl-Accelerated Multimetallic Cross-Coupling of Aryl Chlorides with Aryl Triflates. J. Am. Chem. Soc 2019, 141, 10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For representative lead references for XC and XEC reactions of aryl and alkyl sulfonates with alternative metal catalysts, see: [Google Scholar]; reference 1d. Cu:; (b) Burns DH; Miller JD; Chan H–K; Delaney MO Scope and Utility of a New Soluble Copper Catalyst [CuBr–LiSPh–LiBr–THF]: A Comparison with Other Copper Catalysts in Their Ability to Couple One Equivalent of a Grignard Reagent with an Alkyl Sulfonate. J. Am. Chem. Soc 1997, 119, 2125–2133. [Google Scholar]; (c) Terao J; Kambe N Cross-Coupling Reaction of Alkyl Halides with Grignard Reagents Catalyzed by Ni, Pd, or Cu Complexes with π-Carbon Ligands. Acc. Chem. Res 2008, 41, 1545–1554. [DOI] [PubMed] [Google Scholar]; (d) Liu J–H Yang C–T; Lu X–Y; Zhang Z–Q; Xu L; Cui M; Lu X; Xiao B; Fu Y; Liu L Copper-Catalyzed Reductive Cross-Coupling of Nonactivated Alkyl Tosylates and Mesylates with Alkyl and Aryl Bromides. Chem. Eur. J 2014, 20, 15334–15338. Fe: [DOI] [PubMed] [Google Scholar]; (e) Furstner A; Leitner A; Mendez M;M; Krause H Iron-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc 2002, 124, 13856–13863. [DOI] [PubMed] [Google Scholar]; (f) Atack TC; Lecker RM Cook SP Iron-Catalyzed Borylation of Alkyl Electrophiles. J. Am. Chem. Soc 2014, 136, 9521–9523. [DOI] [PubMed] [Google Scholar]
- 16.Do HQ; Chandrashekar ERR; Fu GC Nickel/Bis(oxazoline)-Catalyzed Asymmetric Neigishi Arylations of Racemic Secondary Benzylic Electrophiles to Generate Enantioenriched 1,1-Diarylalkanes. J. Am. Chem. Soc 2013, 135, 16288–16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eno MS; Lu A; Morken JP Nickel-Catalyzed Asymmetric Kumada Cross-Coupling of Symmetric Cyclic Sulfates. J. Am. Chem. Soc 2016, 138, 7824–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Prinsell MR; Everson DA; Weix DJ Nickel-Catalyzed, Sodium Iodide-Promoted Reductive Dimerization of Alkyl Halides, Alkyl Pseudohalides, and Allylic Acetates. Chem. Commun 2010, 46, 5743–5745. [DOI] [PubMed] [Google Scholar]; (b) Komeyama K; Tsunemitsu R; Michiyuki T; Yoshida H; Osaka I Ni/Co-Catalyzed Homo-Coupling of Alkyl Tosylates. Molecules 2019, 24, 1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Methyl sulfonates: [Google Scholar]; (a) Wang J; Zhao J; Gong H Nickel-Catalyzed Methylation of Aryl Halides/Tosylates with Methyl Tosylate. Chem. Commun 2017, 53, 10180–10183. [DOI] [PubMed] [Google Scholar]; (b) Komeyama K; Yamahata Y; Osaka I Nickel and Nucleophilic Cobalt-Catalyzed Trideuteriomethylation of Aryl Halides Using Trideuteriomethyl p-Toluenesulfonate. Org. Lett 2018, 20, 4375–4378. [DOI] [PubMed] [Google Scholar]
- 20.(a) Benzylic sulfonatesAckerman LKG; Anka-Lufford LL; Naodovic M; Weix DJ Cobalt Co-Catalysis for Cross-Electrophile Coupling: Diarylmethanes from Benzyl Mesylates and Aryl Halides. Chem. Sci 2015, 6, 1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jung H-S; Kim S-H Alternative Approach Toward the Generation of Benzylic Zinc Reagent: Direct Oxidative Addition of Active Zinc into the Carbon–Oxygen Bond of Benzyl Mesylates. Synlett 2015, 26, 666–670. [Google Scholar]
- 21.Primary and secondary alkyl sulfonates: [Google Scholar]; (a) Molander GA; Traister KM; O’Neill BT Engaging Nonaromatic, Heterocyclic Tosylates in Reductive Cross-Coupling with Aryl and Heteroaryl Bromides. J. Org. Chem 2015, 80, 2907–2911. [DOI] [PubMed] [Google Scholar]; (b) Komeyama K; Ohata R; Kiguchi S; Osaka I Highly Nucleophilic Vitamin B12-Assisted Nickel-Catalyzed Reductive Coupling of Aryl Halides and Non-Activated Alkyl Tosylates. Chem. Commun 2017, 53, 6401–6404. [DOI] [PubMed] [Google Scholar]; (c) Duan J; Du Y-F; Pang X; Shu X-Z Ni-Catalyzed Cross-Electrophile Coupling Between Vinyl/Aryl and Alkyl Sulfonates: Synthesis of Cycloalkenes and Modification of Peptides. Chem. Sci 2019, 10, 8706–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.XEC of primary alkyl tosylates with secondary alkyl bromides was reported during preparation of this manuscript: Komeyama K; Michiyuki T; Osaka I Nickel/Cobalt-Catalyzed C(sp3)–C(sp3) Cross-Coupling of Alkyl Halides with Alkyl Tosylates. ACS Catal. 2019, 9, 9285–9291. [Google Scholar]
- 23.See Supporting Information for details.
- 24.Tollefson EJ; Dawson DD; Osborne CA; Jarvo ER Stereospecific Cross-Coupling Reactions of Aryl-Substituted Tetrahydrofurans, Tetrahydropyrans, and Lactones. J. Am. Chem. Soc 2014, 136, 14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen P-P; Lucas EL; Greene MA; Zhang S; Tollefson EJ; Erickson LE; Taylor BL; Jarvo ER; Hong X A Unified Explanation for Chemoselecivity and Stereospecificity of Ni-Catalyzed Kumada and Cross-Electrophile Coupling Reactions of Benzylic Ethers: A Combined Computational and Experimental Study. J. Am. Chem. Soc 2019, 141, 5835–5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding R He Y; Wang X; Xu J; Chen Y; Feng M; Qi C Treatment of Alcohols with Tosyl Chloride Does Not Always Lead to the Formation of Tosylates. Molecules 2011, 16, 5665–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Related nickel-catalyzed reactions of alkyl sulfonates likely proceed through the corresponding alkyl halides, generated in situ. For example, [Google Scholar]; See reference 16.; See reference 7a.; See reference 8c.; See reference 21c.; (e) A related Fe-catalyzed reaction: Ito S; Fujiwara Y.-i.; Nakamua E; Nakamura M Iron-Catalyzed Cross-Coupling of Alkyl Sulfonates with Arylzinc Reagents. Org. Lett 2009, 11, 4306–4309. [DOI] [PubMed] [Google Scholar]
- 28.The computational details and level of theory are included in the Supporting Information.
- 29.Kehoe R; Mahadevan M; Manzoor A; McMurray G; Wiendefeld P; Baird MC Reactions of the Ni(0) Compound Ni(PPh3)4 with Unactivated Alkyl Halides: Oxidative Addition Reactions Involving Radical Processes and Nickel(I) Intermediates. Organometallics 2018, 37, 2450–2467. [Google Scholar]
- 30.For related mechanisms for oxidative addition of aryl halides, see [Google Scholar]; (a) Tsou TT; Kochi JK Mechanism of Oxidative Addition. Reactions of Nickel(0) Complexes with Aromatic Halides J. Am. Chem. Soc 1979, 101, 6319–6332. [Google Scholar]; (b) Funes-Ardoiz I; Nelson DJ; Maseras F Halide Abstraction Competes with Oxidative Addition in the Reactions of Aryl Halides with [Ni(PMenPh(3-n))4] Chem – Eur J. 2017, 23, 16728–16733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.For a general discussion, see Komiya S; Hirano M Activation of Substrates with Polar Single Bonds Current Methods in Inorganic Chemistry, Vol. 3; Kurosawa H, Yamamoto A, Eds.; Elsevier Science B.V.: Amsterdam, 2003; pp 115–186. [Google Scholar]
- 32.Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Books: Sausalito, 2006. [Google Scholar]
- 33.Hammond GS J. Am. Chem. Soc 1955, 77, 334–338. [Google Scholar]
- 34.Dudnik AS; Fu GC Nickel-Catalyzed Coupling Reactions of Alkyl Electrophiles, Including Unactivated Tertiary Halides, to Generate Carbon-Boron Bonds. J. Am. Chem. Soc 2012, 134, 10693–10697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Control experiments establish that alkyl iodides are rapidly formed from mesylates 26 and 27 under the reaction conditions. See Supporting Information for details.
- 36.(a) von Richter V Chem. Berichte 1869, 2, 552–553. [Google Scholar]; (b) Claisen L; Claparede A Condensationen von Ketonen mit Aldehyden. Chem. Berichte 1881, 14, 2460–2468. [Google Scholar]; (c) Heathcock CH; Buse CT; Kleschick WA; Pirrung MC; Sohn JE; Lampe J Acyclic Stereocontrol. 7. Stereoselective Synthesis of 2-Alkyl-3-hydroxy Carbonyl Compounds by Aldol Condensation. J. Org. Chem 1980, 45, 1066–1081. [Google Scholar]
- 37.Northrup AB; MacMillan DWC The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes. J. Am. Chem. Soc 2002, 124, 6798–6799. [DOI] [PubMed] [Google Scholar]
- 38.Evans DA; Bartoli J; Shih TL Enantioselective Aldol Condensations. 2. Erythro-Selective Chiral Aldol Condensations via Boron Enolates. J. Am. Chem. Soc 1981, 103, 2127–2129. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.