

Abstract
Increasing evidence of immune system activation during the progression of hypertension and renal injury has led to a need for new methods to study individual cell types. Transfer of immune cells serves as a powerful tool to isolate effects of specific subsets. Transfer studies in Rag1−/− mice have demonstrated an important role of T-cell activation in hypertension, but this approach has yielded limited success in rat models. Using the T-cell-deficient Dahl salt-sensitive (SS) rat, SSCD247−/−, we hypothesized that splenocyte transfer from SS wild-type animals into SSCD247−/− animals would populate the T-cell compartment. The Dahl SS background provides a model for studying salt-sensitive hypertension; therefore, we also tested whether the dietary salt content of the donor would confer differential salt sensitivity in the recipient. To test this, donors were maintained on either a low-salt or a high-salt diet, and at postnatal day 5 the recipients received splenocyte transfer from one of these groups before a high-salt diet challenge. We showed that splenocyte transfer elevated blood pressures while rats were fed low salt and exacerbated the salt-sensitive increase in pressure when they were fed fed high salt. Furthermore, transfer of splenocytes conferred exacerbated renal damage. Lastly, we confirmed the presence of T cells in the circulation and in the spleen, and that infiltration of immune cells, including T cells, macrophages and B cells, into the kidney was elevated in those receiving the transfer. Interestingly, the source of the splenocytes, from donors fed either a low-salt or a high-salt diet, did not significantly affect these salt-sensitive phenotypes.
Keywords: salt-sensitive hypertension, T cell, transfer
1 |. INTRODUCTION
Since the 1960s, transfer experiments have demonstrated that circulating factors from hypertensive donor animals can induce an elevation in blood pressure in normotensive recipients (Masson, Kashii, & Panisset, 1964; Okuda & Grollman, 1967; Olsen, 1980). Seminal work Guzik et al. (2007) reinvigorated the field by demonstrating that the attenuation of angiotensin II-induced hypertension seen with genetic deletion of recombination activating 1 (Rag1) in mice, which deletes mature T and B cells, was lost when animals received a T-cell transfer. Various investigators have used this technique to examine different aspects of T-cell function in hypertensive disease progression. Bone marrow transfer techniques have also been used to isolate genetic influences of haematopoietic cells experimentally; however, these chimeric animals are also exposed to lethal doses of irradiation to eliminate the host immune system (Batchu et al., 2016; Chen et al., 2018a; Meissner et al., 2017; Rudemiller et al., 2015). Therefore, the Rag1−/− animals have provided opportunities for simpler study design, because they possess the immunological space for transferred T or B cells to populate the recipient without competition from host cells. Given that either T or B cells could populate the recipient, T-cell isolation before transfer is necessary to attribute phenotypic changes to T cells specifically.
Researchers studying hypertensive disease progression have used different approaches to transfer these cells, including negative selection of T cells using magnetic bead sorting (Barhoumi et al., 2011; Chen et al., 2018b; Guzik et al., 2007; Hoch et al., 2009; Iulita et al., 2019; Ji et al., 2014; Kinsey et al., 2009; Kvakan et al., 2009; Marvar et al., 2010; Mian, Barhoumi, Briet, Paradis, & Schiffrin, 2016; Pons et al., 2013; Senchenkova, Russell, Kurmaeva, Ostanin, & Granger, 2011; Trott et al., 2014; Wu et al., 2014, 2016), positive selection for T-regulatory cells (Barhoumi et al., 2011; Cornelius et al., 2015; Kasal et al., 2012; Mian et al., 2016; Turner et al., 2010) or flow-assisted cell (FAC) sorting (Chen et al., 2018b; Kassan, Galan, Partyka, Trebak, & Matrougui, 2011; Kvakan et al., 2009; Yang et al., 2008). In addition, cell culture techniques have been used to enrich specific T-cell subsets before transfer (Chen et al., 2018b; Hayakawa et al., 2000; McRae, Russell, Chia, & Dwyer, 2013; Meyers, Tomaszewski, Glass, & Chen, 1998; Wallace et al., 2012; Wang et al., 2006). Important work has demonstrated that the sex of the donor and of the recipient can influence the hypertensive phenotype of the recipient, which has revealed an integral role of sex hormones in the development of hypertension (Ji et al., 2014; Pollow et al., 2014).
Previous work from our laboratory has used SSRag1−/− animals to demonstrate that the adaptive immune system potentiates Dahl salt-sensitive (SS) hypertension (Mattson et al., 2013). Given that the Dahl SS rat is a genetic animal model of salt-sensitive hypertension with phenotypes that parallel human salt-sensitive disease and end-organ damage, experimental results from this model might provide additional insight into human disease. Further work that used genetic deletion of CD247, which encodes the CD3 zeta chain and therefore deletes CD3+ T cells, demonstrated that elimination of T cells specifically attenuates the salt-sensitive increase in blood pressure of the Dahl SS rat (Rudemiller et al., 2014). To our knowledge, there is only one study using a T-cell transfer technique to enhance salt sensitivity. Liu et al. (2017) transferred CD8+ T cells from deoxycorticosterone acetate (DOCA)–salt-treated mice to mice which then only received the high-salt (HS) diet. They showed that transfer of these cells induced a hydrochlorothiazide-sensitive, salt-sensitive increase in pressure not observed in non-transfer control animals. Alternatively, a study by Pons et al. (2013) was able to show that Wistar rats that were tolerized ta peptide fragment of HSP70 during the washout period of an l-NAME plus high-salt model of salt-sensitive hypertension did not develop salt-dependent increases in blood pressure. Furthermore, transfer of T cells from these animals was able to confer the same protection in the recipients. These studies strengthen the evidence for the role of T cell in salt-sensitive hypertension; however, evidence has yet to demonstrate that transfer of T cells can potentiate salt-sensitive renal damage or genetic salt sensitivity.
In the present study, we test the hypothesis that freshly isolated splenocytes from wild-type Dahl SS animals, a genetic model of salt-sensitive hypertension and renal injury, transferred into SSCD247−/−, T-cell-deficient recipients will populate the T-cell compartment to potentiate the salt-sensitive phenotypes. Researchers in our laboratory initially attempted adoptive immune cell transfer studies using isolated splenocytes, thymocytes, circulating T cells and renal T cells by I.V. and I.P. injection in the adult rat. We were not able to produce reliable or reproducible reconstitution of the adopted cells in the recipients, and the number of cells that engrafted was near negligible. Given that the host immune system in the rat starts to reach maturity by postnatal day 7 in the thymus, day 14 in the bone marrow, day 21 in the lymph nodes and day 21 in the spleen (Parker, Picut, Swanson, & Toot, 2015), it may follow that adoptive transfer after these time points might not provide efficient engraftment owing to lack of immunological space. Therefore, we decided that a transfer at an earlier time point, in this case postnatal day 5, would be able to produce sufficient engraftment. In this study, we undertook splenocyte transfer at this time point.
Recent work from our laboratory has demonstrated that the transcriptomic profile of T cells both from the circulation and in the kidney is different between low-salt (LS)-fed and HS-fed Dahl rats (Abais-Battad et al., 2019). The renal T cells from HS-fed Dahl rats show an upregulation in inflammatory pathways, which might contribute to the progressive nature of renal damage that accumulates during HS challenge. Given that the clonality of T cells may influence the progression of hypertensive disease (Trott et al., 2014), the exposure of a T-cell donor to different levels of salt during life might influence the salt sensitivity of the recipient in this method of an adoptive transfer experiment. Therefore, in this study we test whether the dietary salt content of the donor confers differential salt sensitivity of the recipient.
2 |. METHODS
2.1 |. Ethical approval
All studies were approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee (assurance no. A3102–01).
2.2 |. Splenocyte transfer into T-cell-deficient Dahl SS rats
Colonies of Dahl SS and SSCD247−/− (SS/JrHsdMcwi and SSCD247em1Mcwi) rats were maintained at the Medical College of Wisconsin and fed a diet containing 0.4% NaCl (LS; AIN-76A purified rodent diet #113755; Dyets Inc., Easton, Bethlehem, PA, USA) ad libitum. The SSCD247−/− males and females were mated, and confirmation of pregnancy was determined by vaginal swab. The experimental strategy is depicted in Figure 1a. On gestational day 5, a separate group of littermate SS male animals, which served as splenocyte donors, were either maintained on the LS diet or switched to a diet containing 4.0% NaCl (HS; AIN-76A purified rodent diet #113756; Dyets Inc.) after a baseline overnight urine collection. Dams were allowed to deliver undisturbed. On postnatal day 5, the donor SS animals, maintained on either an LS or an HS diet for 21 days, were euthanized and splenocytes isolated (described in subsection 2.3). A mixed population of 107 freshly isolated, naive, splenocytes from either the LS-fed or the HS-fed animals was injected I.P. into littermate SSCD247−/− pups. A third group of pups did not receive a transfer of splenocytes and served as naive controls.
FIGURE 1.
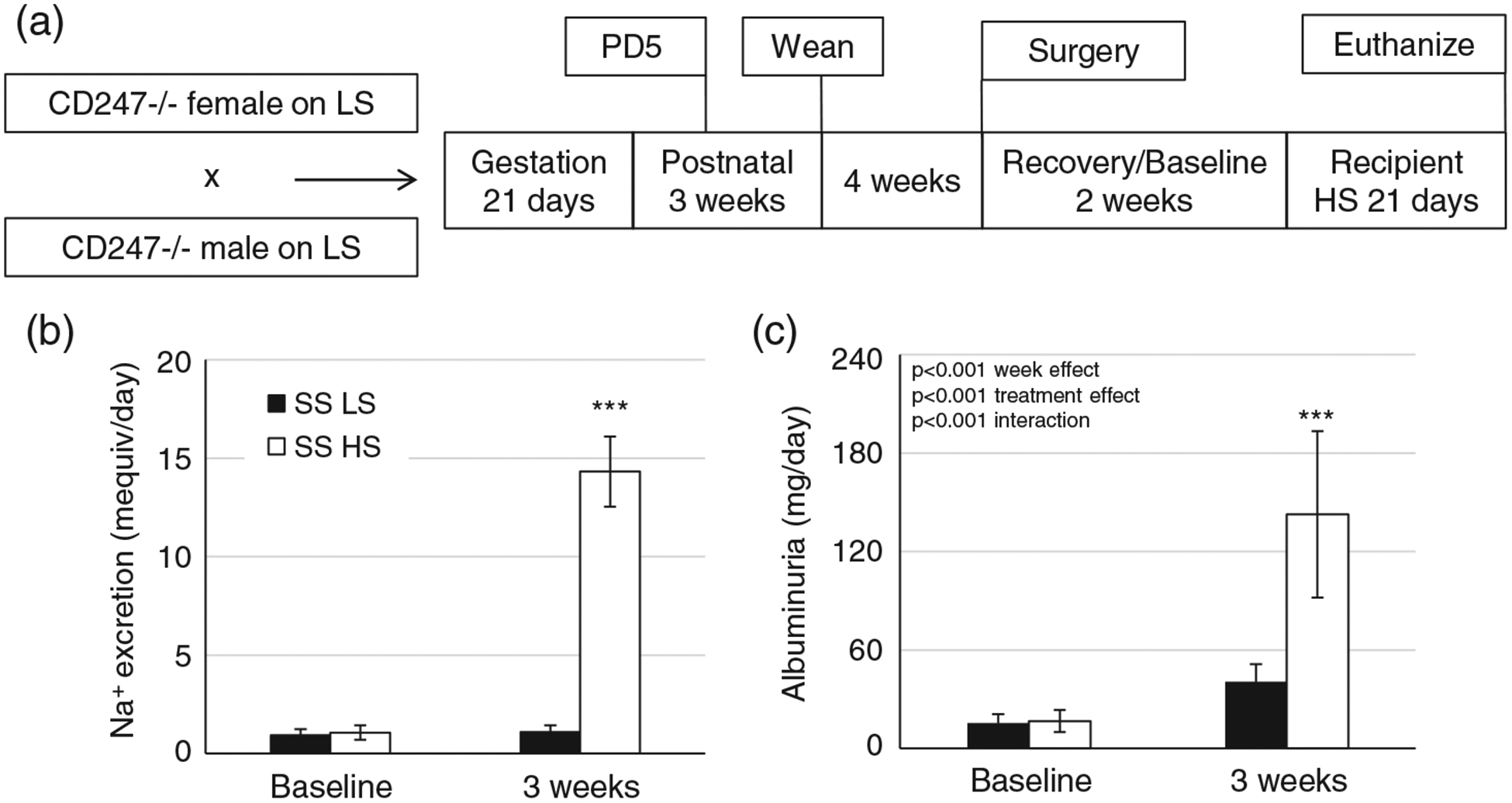
(a) Schematic diagram for the study design of the recipient animals. The SSCD247−/− animals maintained on a low-salt (LS) diet were mated. On postnatal day 5, some recipient pups received a splenocyte transfer from either LS- or high-salt (HS)-fed animals, whereas other pups did not receive a transfer. All the pups were weaned to, and maintained on, an LS diet. After telemetry implantation surgery at 7 weeks of age and subsequent recovery and baseline recordings, all animals were switched to an HS diet for 21 days. (b,c) Sodium excretion (b) and albuminuria (c) of the salt-sensitive (SS) donors (n = 11/11) at baseline and after 21 days of the LS (SS LS) or HS (SS HS) diet. ***P < 0.001 versus SS LS. Two-way repeated-measures ANOVA with the Holm–Sidak post hoc test was used, and the P-values for the main effects are reported in each graph
At 7 weeks of age, male recipients [SSCD247−/− + LS splenocytes (LSS), SSCD247−/− + HS splenocytes (HSS) or SSCD247−/− naive (Control)] were implanted with radiotelemeter catheters in the carotid artery as previously described (Abais-Battad et al., 2019; Fehrenbach et al., 2019) for continuous blood pressure monitoring. Antibiotics (cefazolin 25 mg kg−1); (Henry Schein Inc., Melville, NY, USA) and analgesics (buprenorphine SR 0.3 mg kg−1); (ZooPharm, Fort Collins, CO, USA) were administered subcutaneously after surgery. After recovery, baseline blood pressure recordings and a baseline overnight urine collection, animals were switched to an HS diet for 3 weeks. Weekly urine collections were used to monitor renal damage via albumin excretion. At the end of the HS period, animals were deeply anaesthetized by inhalation of isoflurane balanced O2, aortic blood collected, kidneys flushed with heparinized saline, and the kidneys and spleen extracted. The deeply anaesthetized animals were then euthanized by pneumothorax.
2.3 |. Splenocyte isolation for donation
Donor animals were deeply anaesthetized by inhalation of isoflurane balanced O2. A mid-line incision was made, the spleen excised, and the anaesthetized animals were euthanized by pneumothorax. Spleens were then chopped with small surgical scissors into small pieces in a 70 μm strainer placed in a 5 cm Petri dish containing 5 ml of wash buffer (2% Heat-Inactivated Fetal Bovine Serum (HI-FBS) and 2 mM ethylenediaminetetraacetic acid dulbecco’s phosphate buffered saline (EDTA DPBS)). The end of the plunger from a 1 ml syringe was used to push the tissue through the strainer, and additional wash buffer was used to ensure that freely isolated cells passed through the strainer. The resulting cell suspension was pelleted, and red blood cells were lysed with red cell lysis buffer (eBioscience, ThermoFisher Scientific, Waltham, Massachusetts, USA). At this point, samples from two or three separate LS or HS donors were pooled and passed through a 40 μm strainer to ensure single-cell suspension. The cell concentration was determined by counting on a haemocytometer, and the cells were pelleted and resuspended at a concentration of 50 million cells ml−1. Ten million cells (200 μl) were aliquoted into an insulin syringe for injection.
2.4 |. Urinalysis
Overnight urine collections were performed on days −1 and 21 of the HS challenge period for donor animals and on days −1, 7, 14 and 21 of the HS challenge for the recipients. Urine electrolytes were measured by flame photometry (model 410; Corning Inc, Corning, New York, USA). Urine creatinine values were measured by an autoanalyser with an assay based on the Jaffé reaction (ACE; Alfa Wasserman, Fairfield, NJ, USA). Urine albumin was quantified with a fluorescent assay that used Albumin Blue 580 dye (Sigma-Aldrich, St Louis, MO, USA) and a fluorescent plate reader (FL-600; BioTek, Winooski, VT, USA). Urine protein excretion was quantified using Weichselbaum’s biuret reagent and an autoanalyser (ACE; Alfa Wasserman). Urinary kidney injury marker 1 (KIM1; R&D Systems, Minneapolis, MN, USA; #RKM100) and nephrin (Ethos Biosciences Inc., Philadelphia, PA, USA; #1036) measurements were performed according to the manufacturer’s instructions.
2.5 |. Circulating and renal immune cell isolation, followed by flow cytometry analysis
Immune isolation was performed as previously described in detail (Fehrenbach, Abais-Battad, Dasinger, Lund, & Mattson, 2019). Circulating immune cells were isolated using Histopaque-1083 (Sigma-Aldrich). For renal immune cell isolation, whole kidneys were minced and digested in RPMI 1640 containing L-glutamine, Hepes, collagenase type IV and DNase, and passed through 100, 70 and 40 μm filters. Mononuclear cells were isolated by Percoll (Sigma-Aldrich) density gradient centrifugation. Isolated cells were first incubated with CD32 Fc receptor blocking antibody, followed by an incubation with a cocktail of extracellular antibodies, including anti-CD45 (Biolegend, San Diego, CA, USA; #202214, clone OX-1) for leukocytes, anti-CD3 (eBioscience; #46-0030-82, clone G4.18) for T cells, anti-CD4 (BioLegend; #201518, clone W3/25) for helper T cells, anti-CD8 (BioLegend; #201703, clone OX-8) for cytotoxic T cells, anti-CD45R (BD Bioscience; #554881, clone HIS24) for B cells, and anti-CD11b/c (eBioscience; #50-0110-82, clone OX-42) for mononuclear myeloid-derived cells.
All cells were then analysed by flow cytometry (LSRII; Becton Dickenson, Franklin Lakes, NJ, USA) with FACSDIVA software (Becton Dickinson) and FlowJo Software (TreeStar Inc., Ashland, OR, USA). To quantify the numbers of the various immune cells, unstained single cells were counted using a haemocytometer. Afterwards, the number of live cells was determined as the 4′,6-diamidino-2-phenylindole (DAPI)-negative percentage, and the percentage of cells staining CD45+ was used to calculate the number of leukocytes. A downstream gating strategy (Figure 2) was used to determine the profile of these CD45+ leukocytes.
FIGURE 2.

Flow cytometry gating strategy. After gating for the general cell population, those that are singlets and those negative for DAPI staining, CD45+ cells were used to gate for total leucocytes. From this population, gates were placed for CD11b/c+ monocytes/macrophages, CD45R+ B cells, and CD3+ T cells along with their CD4+ helper and CD8+ cytotoxic subsets
2.6 |. Histological analysis
After extraction, the right kidney and the spleen were fixed in 10% neutral buffered formalin, paraffin embedded, cut into 4-μm-thick sections and mounted. Kidney slices were stained with Periodic Acid-Schiff (PAS), whereas anti-CD3+ antibody binding was used to visualize the presence of T cells in the spleen.
2.6.1 |. CD3 staining
All slides were deparaffinized and stained on the Leica BondRx automated staining platform. Antigen was retrieved using Retrieval Citrate Buffer Solution (Leica Microsystems, Buffalo Grove, IL, USA; AR9661), blocked with peroxidase (DAKO, Santa Clara, CA, USA; S200389), and avidin and biotin blocking kit applied (Vector Laboratories Inc., Burlingame, CA, USA; SP-2001). After protein block (DAKO; X090930–2), primary anti-CD3 (1:200; DAKO; A0452) and secondary donkey anti-rabbit-biotin (1:500) were applied. Streptavidin-horseradish peroxidase and 3,3′-diaminobenzidine were added to each section and slides counterstained with 15% Aniline Blue. Slides were dehydrated, cleared and mounted with synthetic mounting media. Stained kidney and spleen images were then obtained using Nikon Coolscan. The percentage of outer medullary casts in the kidney and presence of T cells in the spleen were determined by colour inclusion via ImageJ (Image J, US National Institutes of Health, Bethesda, MD, USA) software in a blinded manner. Glomerular injury was determined by assigning >40 glomeruli per kidney categorical scores on a scale of one (best) to four (worst) based on the degree of glomerulosclerosis and mesangial expansion. All histological analysis was performed in a blinded manner.
2.7 |. Statistical analysis
One-way ANOVA with the Holm–Sidak post hoc test or two-way repeated-measures ANOVA with the Holm–Sidak post hoc test were used where appropriate. Data are expressed as means ± SD. A value of P < 0.05 was considered statistically significant. Sigmaplot (Systat Software Inc., San Jose, CA, USA) was used for all statistical analysis.
3 |. RESULTS
3.1 |. Donor Dahl SS animals fed high salt develop renal damage
Sodium excretion was dramatically elevated in HS-fed donor rats compared with donors maintained on an LS diet (P < 0.001; Figure 1b), as a consequence of the increased sodium intake. Albuminuria was used to monitor the renal damage phenotype of donor animals, which, as previously reported (De Miguel, Das, Lund, & Mattson, 2010; Fehrenbach et al., 2019), demonstrated that Dahl SS animals develop renal damage when on an HS diet (Figure 1c).
3.2 |. Rate of constitution of the T-cell compartment is not determined by salt intake of the donors
Constitution of the T-cell compartment was assessed via flow cytometry of peripheral blood mononuclear cells. CD3+ T cells were almost undetectable in Control animals (Figure 3a). Splenocyte transfer resulted in constitution of T cells to ~50–60,000 CD3+ cells ml−1 (P < 0.001 versus Control), although rats receiving the transfer from HS-fed fed donors had similar levels of these CD3+ cells to those receiving a transfer from LS-fed donors (P > 0.05; Figure 3a). Interestingly, this technique resulted in constitution of primarily CD4+ T cells (80% of CD3+ T cells), with far fewer CD8+ cells (17% of CD3+ T cells; Figure 3b). The circulating CD11b/c+ monocyte count was not different between any of the groups (P > 0.05; Figure 3c), although the number of CD45R+ B cells was lower in animals receiving the transfer (P < 0.001 versus Control; Figure 3d). CD3+ staining of splenic tissue confirmed the engraftment of T cells into the spleen, which was not present in Control animals (P < 0.001 versus Control; Figure 3e).
FIGURE 3.

(a–d) Quantification of circulating peripheral blood mononuclear cells, including CD3+ T cells (a), CD4+ helper T cells and CD8+ cytotoxic T cells (b), CD11b/c+ myeloid cells (c) and CD45R+ B cells (d). (e,f) Splenic T cells stained with anti-CD3 (e) quantified in panel (f). Recipient groups of SSCD247−/− rats included naive control rats (Control; n = 7), rats receiving a splenocyte transfer from donor rats maintained on a low-salt diet (LSS; n = 11) and rats receiving splenocyte transfer from donor rats maintained on a high-salt diet (HSS; n = 13). **P < 0.01 and ***P < 0.001 versus Control. One-way ANOVA with the Holm–Sidak post hoc test was used in panels (c,d), and the P-value for the effect of treatment is reported in each graph
3.3 |. Splenocyte transfer exacerbates SS hypertension independent of donor dietary salt content
All animals had stable mean arterial pressures (MAPs) while on the LS diet; however, those receiving a transfer of either LS or HS splenocytes demonstrated elevated LS pressures (P < 0.05 versus Control). There was no difference between LS or HS splenocyte transfer in the development of salt-sensitive hypertension (P > 0.05 all days, LSS versus HSS); however, both sets of recipients developed significantly higher pressures by day 8 of HS compared with those not receiving a transfer (day 8 P < 0.05 interaction between donor group and days of HS diet versus Control; Figure 4a). Control animals did not develop significantly elevated pressures from baseline until day 13 (P < 0.05), whereas LSS and HSS animals experienced a significant elevation by day 3 (P < 0.05) of the HS challenge.
FIGURE 4.
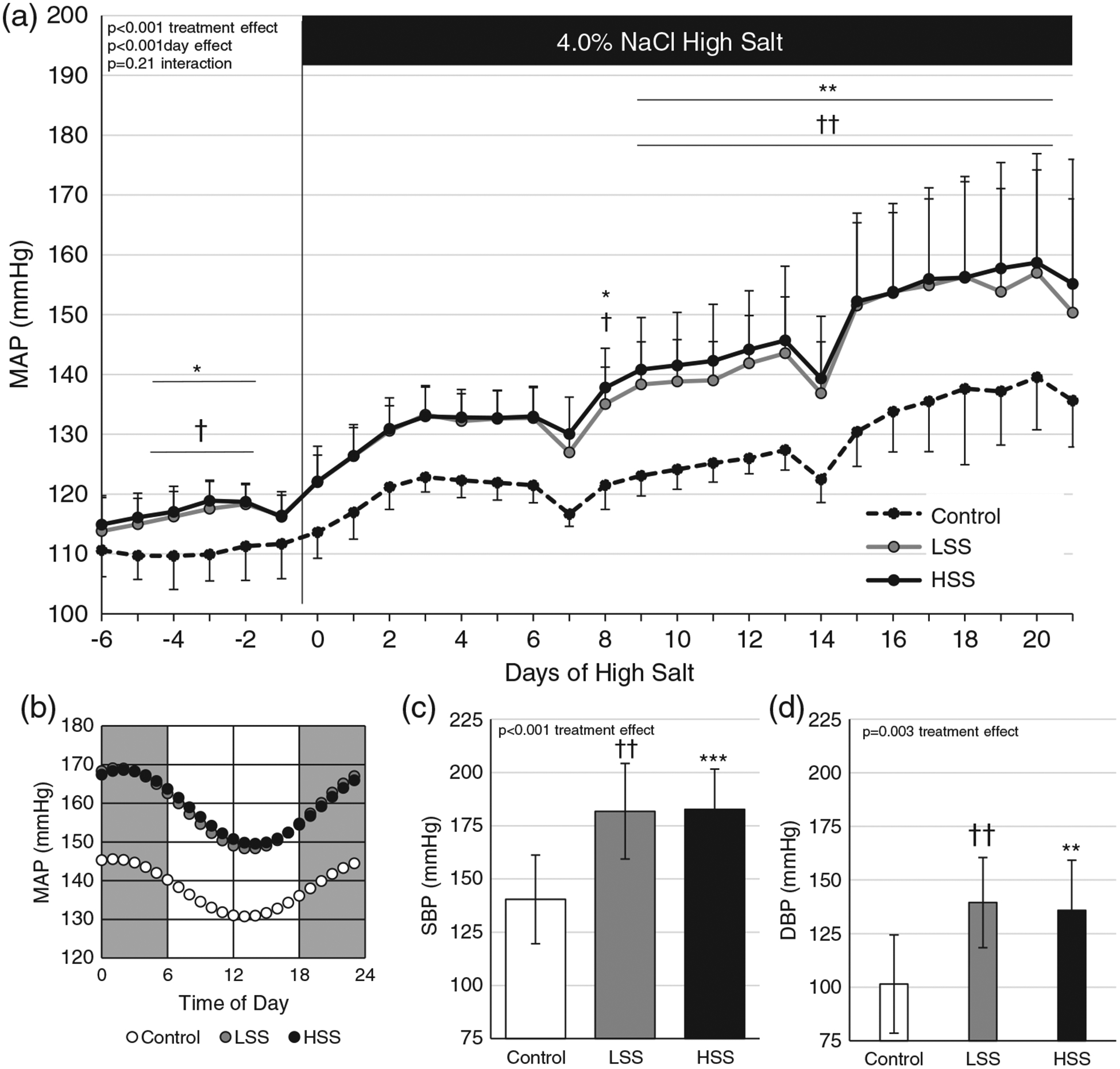
(a) Mean arterial pressure (MAP) during 6 days of baseline recording on a low-salt (LS) diet and 21 days of a high-salt (HS) diet. (b) Diurnal MAP imputed from the last 80 h of blood pressure recordings. (c,d) Systolic (SBP) and diastolic (DBP) blood pressures on day 20 of the HS diet. Recipient groups of SSCD247−/− rats included naive control rats (Control; n = 7), rats receiving a splenocyte transfer from donor rats maintained on an LS diet (LSS; n = 10) and rats receiving splenocyte transfer from donor rats maintained on an HS diet (HSS; n = 12). *P < 0.05, **P < 0.01 and ***P < 0.001, HSS versus Control; †P < 0.05 and ††P < 0.01, LSS versus Control. Two-way repeated-measures ANOVA with the Holm–Sidak post hoc test was used for panel (a), whereas a one-way ANOVA with the Holm–Sidak post hoc test was used in panels (c,d), and the P-values for the main effects are reported in each graph
The last 80 h of blood pressure for each animal, binned hourly, was fitted to a cosine curve using Microsoft Excel Solver tool constrained to a 24 h period. The fitted function was used to calculate imputed values over the 24 h period, and the values were averaged for each group and presented in Figure 4b, demonstrating that transfer of splenocytes did not affect the phase shift (P > 0.05) or amplitude (P > 0.05) of the curves. Blood pressures were disaggregated into systolic (Figure 4d) and diastolic (Figure 4e) pressures, demonstrating the same pattern as seen for the MAP.
3.4 |. Splenocyte transfer exacerbates renal damage
Splenocyte transfer increased renal mass/body mass by the end of the HS period compared with Control animals (P < 0.05 LSS versus Control, P < 0.01 HSS versus Control), but there was no significant difference between LSS and HSS groups (P > 0.05; Figure 5a). There was no significant difference in spleen mass/body mass across any of the groups (P > 0.05; Figure 5a). Creatinine, sodium and potassium excretion rates were also similar across all groups (P > 0.05; Figure 5b–d).
FIGURE 5.
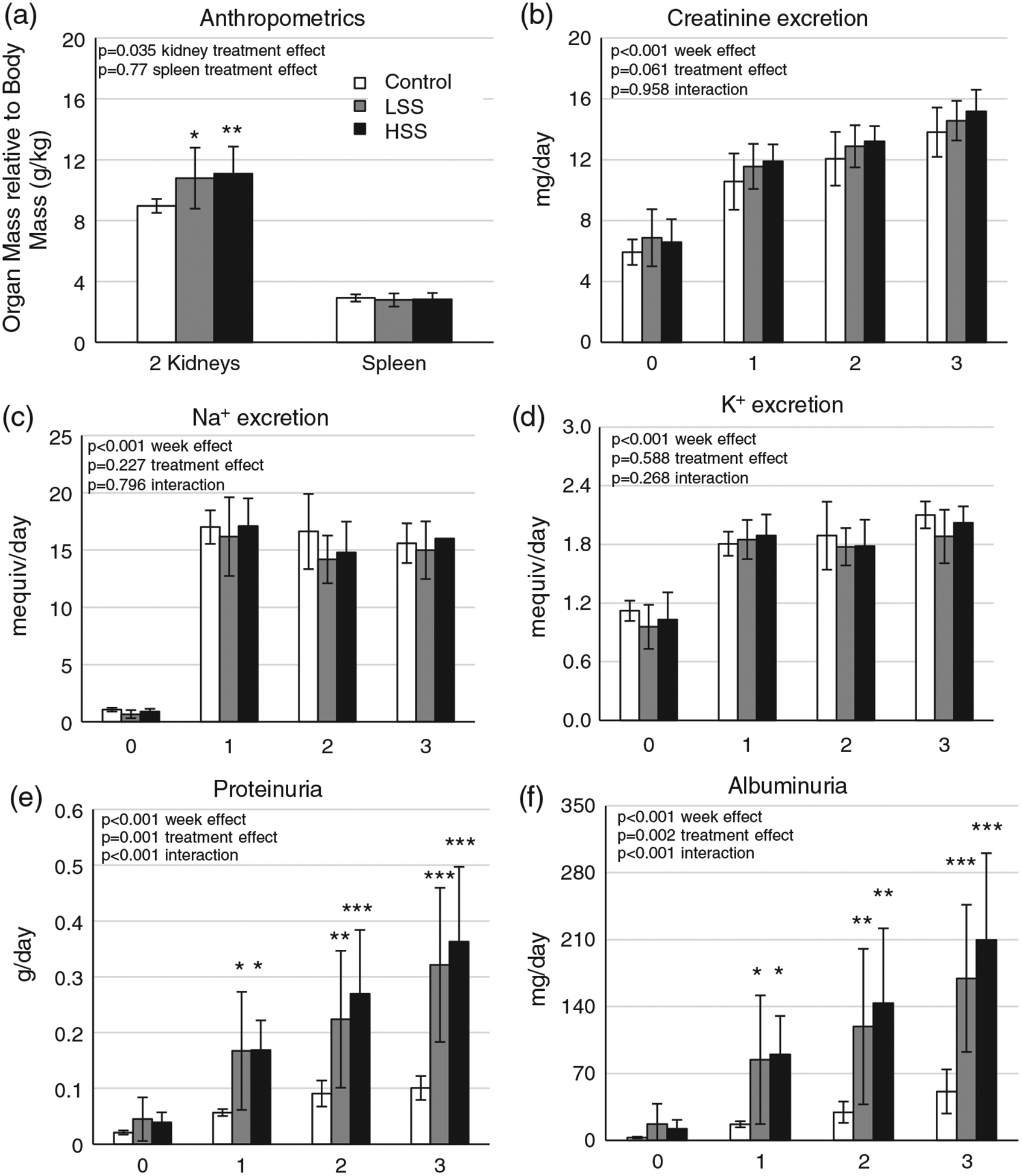
(a) Spleen and total kidney weight. (b–f) Urinary excretion of creatinine (b), sodium (c), potassium (d), protein (e) and albumin (f). Recipient groups of SSCD247−/− rats included naive control rats (Control; n = 7), rats receiving a splenocyte transfer from donor rats maintained on a low-salt diet (LSS; n = 11) and rats receiving splenocyte transfer from donor rats maintained on a high-salt diet (HSS; n = 13). *P < 0.05, **P < 0.01 and ***P < 0.001 versus Control. One-way ANOVA with the Holm–Sidak post hoc test was used in panel (a), whereas two-way repeated-measures ANOVA with the Holm–Sidak post hoc test was used for panels (c,d), and the P-values for the main effects are reported in each graph
The level of proteinuria between the recipients receiving splenocytes from LS and HS donors was not different (P > 0.05); however, transfer of splenocytes, irrespective of donor diet, exacerbated the salt-sensitive increase in protein excretion (Figure 5e; P < 0.001 interaction between donor group and weeks of HS diet) and in albuminuria (Figure 5f; P < 0.001 interaction between donor group and weeks of HS diet) compared with Control. The magnitude of albuminuria seen in LSS and HSS recipients (~175–200 mg day−1) was similar to that of the original donors that were fed HS (~150 mg day−1).
Histological analysis of medullary protein casts demonstrated a similar exacerbation of damage with both transfer groups compared with Control (P < 0.01 HSS versus Control, P < 0.05 LSS versus Control) but no difference between recipients of LSS or HSS (P > 0.05; Figure 6a,b). Likewise, LSS and HSS rats demonstrated an increased frequency of damaged glomeruli (score of three or four, P < 0.001; Figure 6c) compared with Controls. Lastly, urinary KIM1, an additional marker of renal damage, was increased in LSS and HSS rats on day 21 of HS diet (Figure 6d; P < 0.01). Nephrin excretion at this time point (Figure 6e) was also increased in those receiving the transfer, albeit significantly increased only in HSS rats.
FIGURE 6.
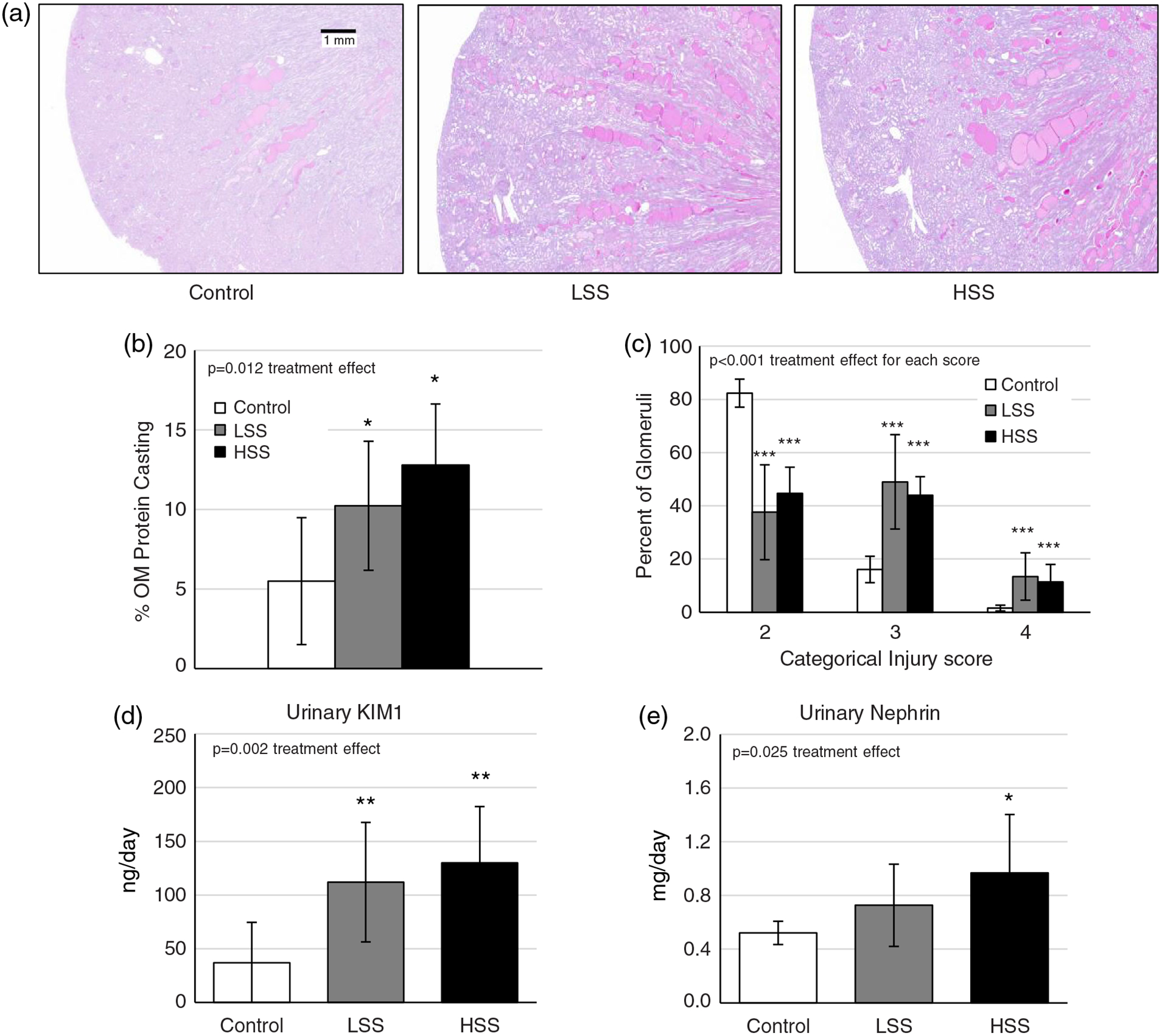
(a–c) Histological images of kidney damage using Periodic Acid–Schiff (PAS) staining (a), with outer medulla (OM) protein casting quantified in panel (b) and glomerular injury score quantified in panel (c). (d,e) Urinary kidney injury marker 1 (KIM1; d) and nephrin (e) excretion were measured on day 21 of high-salt (HS) diet. Recipient groups of SSCD247−/− rats included naive control rats (Control; n = 7), rats receiving a splenocyte transfer from donor rats maintained on a low-salt diet (LSS; n = 9) and rats receiving splenocyte transfer from donor rats maintained on an HS diet (HSS; n = 10). To quantify glomerular injury scores, n = 7 per group were used. *P < 0.05, **P < 0.01 and ***P < 0.001 versus Control. One-way ANOVA with the Holm–Sidak post hoc test was used, and the P-value for the effect of treatment is reported in each graph
3.5 |. Increased renal T-cell infiltration is accompanied by increased macrophage and B-cell infiltration
Similar to what was seen in circulating populations, CD3+ T cells were almost completely undetectable in Control kidneys (Figure 7a). Infiltration of T cells was present in transfer recipients (P < 0.001 versus Control), but no difference was observed between animals receiving splenocytes from LS or HS fed donors (P > 0.05). Of these infiltrating CD3+ T cells, 93% were CD4+, whereas only 6% were CD8+. Interestingly, both groups of transfer recipients showed increased CD45R+ B-cell (P < 0.001 versus Control) and CD11b/c+ macrophage (P < 0.05 versus Control) infiltration compared with those receiving no transfer, but did not differ between the two recipient groups (P > 0.05; Figure 7).
FIGURE 7.
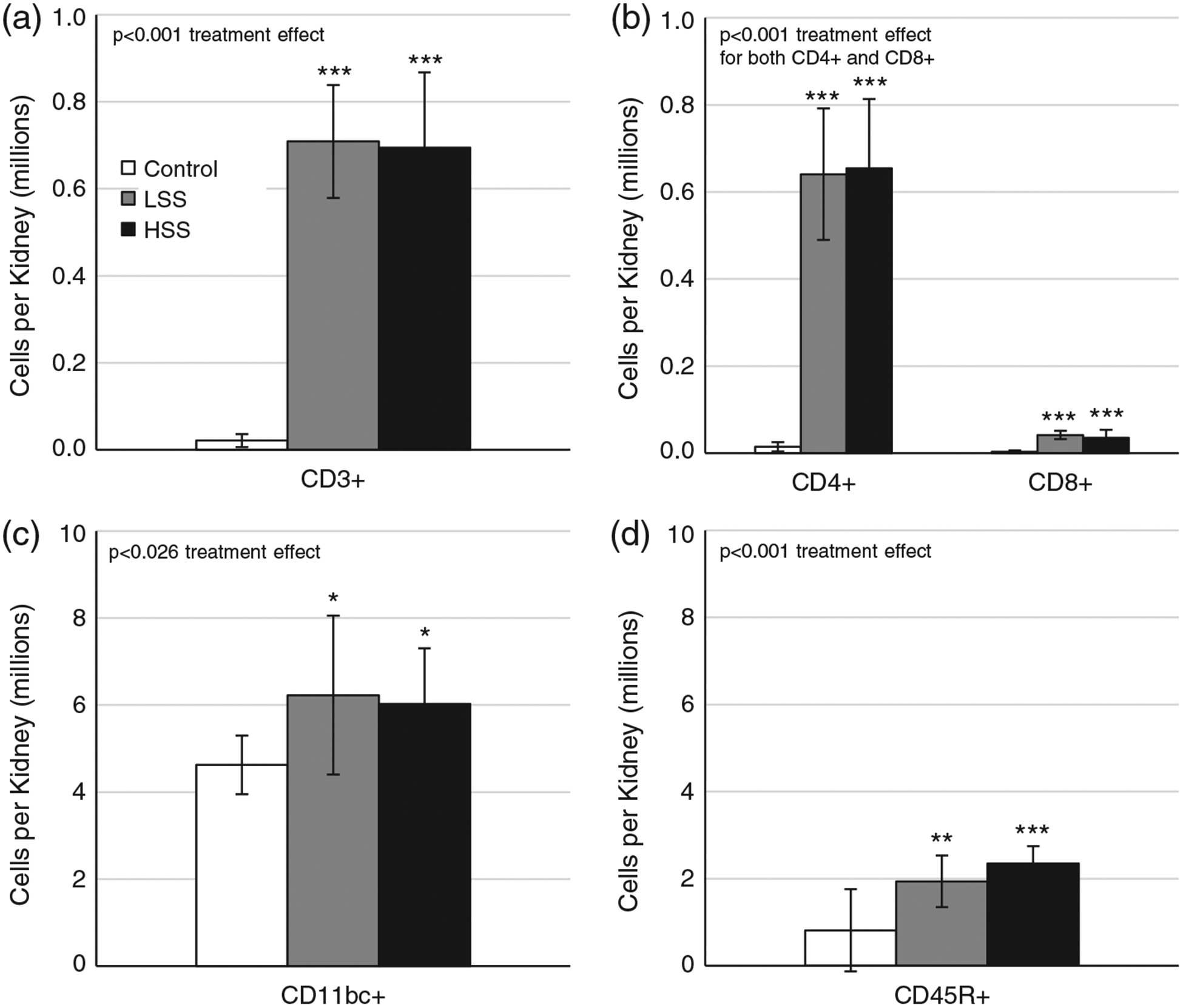
(a–d) Quantification of infiltrating immune cells, including CD3+ T cells (a), CD4+ helper T cells and CD8+ cytotoxic T cells (b), CD11b/c+ myeloid cells (c) and CD45R+ B cells (d). Recipient groups of SSCD247−/− rats included naive control rats (Control; n = 7), rats receiving a splenocyte transfer from donor rats maintained on a low-salt diet (LSS; n = 11) and rats receiving splenocyte transfer from donor rats maintained on a high-salt diet (HSS; n = 13). *P < 0.05, **P < 0.01 and ***P < 0.001 versus Control. One-way ANOVA with the Holm–Sidak post hoc test was used, and the P-value for the effect of treatment is reported in each graph
4 |. DISCUSSION
Over the past 50 years, a model has developed to include adaptive immune system activation and inflammation as contributory factors to the progression of hypertensive disease (Harrison, 2013). In humans, treatment with the immunosuppressive drug mycophenolate mofetil lowered blood pressure in patients with the inflammatory diseases psoriasis or rheumatoid arthritis (Herrera, Ferrebuz, MacGregor, & Rodriguez-Iturbe, 2006). The effect of mycophenolate mofetil treatment on blood pressure was later confirmed in animal models of hypertension and cemented the adaptive system as a central player that needed further investigation (Bravo, Quiroz, Ferrebuz, Vaziri, & Rodríguez-Iturbe, 2007; Muller et al., 2002). These pharmacological studies were followed by experiments in lymphocyte-deficient animals (Crowley et al., 2010), which also showed that reducing adaptive activation attenuated angiotensin II-dependent increases in blood pressure. As the field narrowed down on a more specific immune cell type, it had already been noted that thymectomy in the Lyon hypertensive rat prevented spontaneous elevation in blood pressure, providing specific evidence for the contribution of the T cell (Bataillard, Freiche, Vincent, Sassard, & Touraine, 1986). This, paired with the results from Harrison’s group (Guzik et al., 2007), pointed to an integral role of the T cell, which we confirmed in the T-cell-deficient CD247−/− animal (Rudemiller et al., 2014). Others have shown that immunological priming of the T cell may influence the reactivity of these cells, because transfer of kidney-derived, HSP70-reactive T cells induced renal damage in recipients (Weiss, Madaio, Tomaszewski, & Kelly, 1994).
The role of the T cell continues to be an attractive field of study, providing a therapeutic target for the prevention of progressive hypertensive disease. As previously reviewed (Mattson, 2019), the renal damage accumulating during salt-sensitive hypertension contributes to the progressive nature of the disease pathology, primarily through continued immune activation. Experimental data from animal studies continues to show that reducing immune activation attenuates the disease progression and even lowers blood pressure. Immune cell transfer studies provide a tool to identify therapeutic targets in immune cell signalling more specifically. The adoptive transfer technique has previously been used to validate the role of the T cell in DOCA–salt-induced salt-sensitive hypertension (Batchu et al., 2016; Liu et al., 2017). In the present study, our primary findings demonstrate, in a genetic rat model of hypertension, that T-cell transfer can indeed be used to exacerbate salt-sensitive hypertension. Moreover, we have shown that populating the T-cell compartment is sufficient to enhance the salt-induced renal damage irrespective of the original dietary salt content of the donor. It is becoming clear to us that immune system activation functions less during the initial induction of a hypertensive state but more in amplifying the disease through progressively accumulating renal damage.
Contrary to our hypothesis, the diet of the splenocyte donor did not affect the development of disease in the recipient. Transfer of splenocytes from either HS- or LS-fed donors exacerbated salt-sensitive hypertension and renal damage to an approximately equal extent. This is important, because there is considerable evidence that an increased sodium concentration can drive a Th17-type response in both human and murine naive T cells (Kleinewietfeld et al., 2013; Wu et al., 2013). Furthermore, an HS diet, which alters the microbiome of the gut, can also drive expansion of the Th17 cell type (Wilck et al., 2017). However, the present results demonstrate that this enhanced inflammatory activity attributable to an HS diet is not conserved by adoptive transfer. Therefore, the inflammatory consequence of a high-sodium environment appears to be transient and is not converted to long-term immunological memory in the spleen. To strengthen this claim, we have recently reported that the transcriptomic profile of T cells is remarkably similar between SS animals fed either an LS or an HS diet (Abais-Battad et al., 2019). Therefore, additional cues, beyond strictly an increase in sodium, are necessary for the T-cell-induced exacerbation of SS hypertension. This is consistent with recent work by Evans et al. (2017), who demonstrated that servo control of blood pressure to the left kidney of SS rats fed an HS diet conferred protection from salt-induced renal immune cell infiltration and renal damage.
Based on this evidence, either an increase in renal perfusion pressure and/or renal damage is necessary for T cells to potentiate the salt-sensitive phenotypes, with little to no effect of an HS diet without these cues. For these reasons, T cells are recruited to the kidney in response to increased pressure and/or damage but are not preprogrammed to elicit a pro-inflammatory response attributable to an HS diet. Thus, the magnitude of T-cell recruitment to the kidney and its response to the kidney environment might be the defining relationship between T-cell infiltration and the potentiation of SS hypertension, rather than its exposure to previous environments. Hence, the absence of a differential response between LSS and HSS recipients is less surprising. We have recently reported that not only is production of reactive oxygen species crucial for the full development of the salt-sensitive phenotypes in the Dahl rat, but p67phox-dependent NOX2-derived reactive oxygen species from haematopoietic cells are responsible for a significant amount of salt-induced renal damage and elevation in blood pressure (Abais-Battad et al., 2020). Although not specifically interrogated, we believe that T cells are a major source of reactive oxygen species in the kidney, because the p67 subunit of NOX2 is upregulated in T cells compared with whole renal tissue homogenate and further upregulated when these rats are on an HS diet (De Miguel, Guo, Lund, Feng, & Mattson, 2011).
Interestingly, the constitution of the T-cell compartment observed does not appear to reach the same levels in immune-competent, wild-type, Dahl SS rats as previously reported (Abais-Battad, Lund, Fehrenbach, Dasinger, & Mattson, 2018; Fehrenbach et al., 2019). This does show, however, that limited constitution is sufficient to influence disease and provides the first evidence that immune cell transfer in the Dahl SS rat is sufficient to affect the progression of hypertensive disease. We believe that studies building on these results will be able to examine more specific aspects of T-cell signalling and activation in the Dahl SS rat.
New Findings.
-
What is the central question of this study?
Recruitment of immune cells to the kidney potentiates hypertensive pathology, but more refined methods are needed to assess these cells functionally. Adoptive transfer studies of immune cells have been limited in rat models and especially in the study of salt-sensitive hypertension. We tested the hypothesis that splenocyte transfer into T-cell-deficient rats is sufficient to exacerbate salt-sensitive hypertension.
-
What is the main finding and its importance?
We demonstrate that transfer of splenocytes into T-cell-deficient animals exacerbates salt-sensitive hypertension, and an enrichment in the CD4+ compartment specifically induces this phenomenon.
ACKNOWLEDGEMENTS
We would like to thank Mary Cherian-Shaw for her technical assistance. This work was supported by the National Heart, Lung, and Blood Institute (grant nos F31HL144084, HL137748 and HL116264), the American Heart Association (grant no. 18POST33990140) and the Georgia Research Alliance.
Funding information
National Heart, Lung, and Blood Institute, Grant/Award Numbers: F31HL144084, HL137748, HL116264
Footnotes
COMPETING INTERESTS
None declared.
REFERENCES
- Abais-Battad JM, Alsheikh AJ, Pan X, Fehrenbach DJ, Dasinger JH, Lund H, … Mattson DL (2019). Dietary effects on Dahl salt-sensitive hypertension, renal damage, and the T lymphocyte transcriptome. Hypertension, 74, 854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abais-Battad JM, Lund H, Dasinger JH, Fehrenbach DJ, Cowley AW Jr., & Mattson DL (2020). NOX2-derived reactive oxygen species in immune cells exacerbates salt-sensitive hypertension. Free Radical Biology and Medicine, 146, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abais-Battad JM, Lund H, Fehrenbach DJ, Dasinger JH, & Mattson DL (2018). Rag1-null Dahl SS rats reveal that adaptive immune mechanisms exacerbate high protein-induced hypertension and renal injury. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 315, R28–R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, … Schiffrin EL (2011). T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension, 57, 469–476. [DOI] [PubMed] [Google Scholar]
- Bataillard A, Freiche JC, Vincent M, Sassard J, & Touraine JL (1986). Antihypertensive effect of neonatal thymectomy in the genetically hypertensive LH rat. Thymus, 8, 321–330. [PubMed] [Google Scholar]
- Batchu N, Hughson A, Wadosky KM, Morrell CN, Fowell DJ, & Korshunov VA (2016). Role of Axl in T-lymphocyte survival in salt-dependent hypertension. Arteriosclerosis, Thrombosis, and Vascular Biology, 36, 1638–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo Y, Quiroz Y, Ferrebuz A, Vaziri ND, & Rodríguez-Iturbe B (2007). Mycophenolate mofetil administration reduces renal inflammation, oxidative stress, and arterial pressure in rats with lead-induced hypertension. American Journal of Physiology-Renal Physiology, 293, F616–F623. [DOI] [PubMed] [Google Scholar]
- Chen G, Zuo S, Tang J, Zuo C, Jia D, Liu Q, … Yu Y (2018a). Inhibition of CRTH2-mediated Th2 activation attenuates pulmonary hypertension in mice. Journal of Experimental Medicine, 215, 2175–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH, Ruan CC, Ge Q, Ma Y, Xu JZ, Zhang ZB, … Gao PJ (2018b). Deficiency of complement C3a and C5a receptors prevents angiotensin II-induced hypertension via regulatory T cells. Circulation Research, 122, 970–983. [DOI] [PubMed] [Google Scholar]
- Cornelius DC, Castillo J, Porter J, Amaral LM, Campbell N, Paige A, … LaMarca B (2015). Blockade of CD40 ligand for intercellular communication reduces hypertension, placental oxidative stress, and AT1-AA in response to adoptive transfer of CD4+ T lymphocytes from RUPP rats. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 309, R1243–R1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, & Ruiz P (2010). Lymphocyte responses exacerbate angiotensin II-dependent hypertension. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 298, R1089–R1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miguel C, Das S, Lund H, & Mattson DL (2010). T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 298, R1136–R1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miguel C, Guo C, Lund H, Feng D, & Mattson DL (2011). Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. American Journal of Physiology-Renal Physiology, 300, F734–F742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans LC, Petrova G, Kurth T, Yang C, Bukowy JD, Mattson DL, & Cowley AW Jr. (2017). Increased perfusion pressure drives renal T-cell infiltration in the Dahl salt-sensitive rat. Hypertension, 70, 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrenbach DJ, Abais-Battad JM, Dasinger JH, Lund H, & Mattson DL (2019). Salt-sensitive increase in macrophages in the kidneys of Dahl SS rats. American Journal of Physiology-Renal Physiology, 317, F361–F374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, … Harrison DG (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. Journal of Experimental Medicine, 204, 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DG (2013). The mosaic theory revisited: Common molecular mechanisms coordinating diverse organ and cellular events in hypertension. Journal of the American Society of Hypertension, 7, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa S, Fujikawa T, Fukuoka H, Chisima F, Karasaki-Suzuki M, Ohkoshi E, … Sakurai I (2000). Murine fetal resorption and experimental pre-eclampsia are induced by both excessive Th1 and Th2 activation. Journal of Reproductive Immunology, 47, 121–138. [DOI] [PubMed] [Google Scholar]
- Herrera J, Ferrebuz A, MacGregor EG, & Rodriguez-Iturbe B (2006). Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. Journal of the American Society of Nephrology, 17, S218–S225. [DOI] [PubMed] [Google Scholar]
- Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, … Harrison DG (2009). Regulation of T-cell function by endogenously produced angiotensin II. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 296, R208–R216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iulita MF, Duchemin S, Vallerand D, Barhoumi T, Alvarez F, Istomine R, … Girouard H (2019). CD4+ regulatory T lymphocytes prevent impaired cerebral blood flow in angiotensin II-induced hypertension. Journal of the American Heart Association, 8, e009372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Zheng W, Li X, Liu J, Wu X, Zhang MA, … Sandberg K (2014). Sex-specific T-cell regulation of angiotensin II-dependent hypertension. Hypertension, 64, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, … Schiffrin EL (2012). T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension, 59, 324–330. [DOI] [PubMed] [Google Scholar]
- Kassan M, Galan M, Partyka M, Trebak M, & Matrougui K (2011). Interleukin-10 released by CD4+CD25+ natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 31, 2534–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, … Okusa MD (2009). Regulatory T cells suppress innate immunity in kidney ischemiareperfusion injury. Journal of the American Society of Nephrology, 20, 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, … Hafler DA (2013). Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature, 496, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, … Muller DN (2009). Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation, 119, 2904–2912. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B, … Mu S (2017). CD8+ T cells stimulate Na-Cl co-transporter NCC in distal convoluted tubules leading to salt-sensitive hypertension. Nature Communications, 8, 14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, … Harrison DG (2010). Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circulation Research, 107, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson GM, Kashii C, & Panisset JC (1964). Transfer of experimental renal hypertension and vascular disease. Canadian Medical Association Journal, 90, 231–235. [PMC free article] [PubMed] [Google Scholar]
- Mattson DL (2019). Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nature Reviews Nephrology, 15, 290–300. [DOI] [PubMed] [Google Scholar]
- Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, & Jacob H (2013). Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 304, R407–R414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae JL, Russell PA, Chia JS, & Dwyer KM (2013). Overexpression of CD39 protects in a mouse model of preeclampsia. Nephrology, 18, 351–355. [DOI] [PubMed] [Google Scholar]
- Meissner A, Miro F, Jiménez-Altayó F, Jurado A, Vila E, & Planas AM (2017). Sphingosine-1-phosphate signalling—a key player in the pathogenesis of Angiotensin II-induced hypertension. Cardiovascular Research, 113, 123–133. [DOI] [PubMed] [Google Scholar]
- Meyers CM, Tomaszewski JE, Glass JD, & Chen CW (1998). The nephritogenic T cell response in murine chronic graft-versus-host disease. Journal of Immunology, 161, 5321–5330. [PubMed] [Google Scholar]
- Mian MO, Barhoumi T, Briet M, Paradis P, & Schiffrin EL (2016). Deficiency of T-regulatory cells exaggerates angiotensin II-induced microvascular injury by enhancing immune responses. Journal of Hypertension, 34, 97–108. [DOI] [PubMed] [Google Scholar]
- Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, … Luft FC (2002). Immunosuppressive treatment protects against angiotensin II-induced renal damage. American Journal of Pathology, 161, 1679–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, & Grollman A (1967). Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Texas Reports on Biology and Medicine, 25, 257–264. [PubMed] [Google Scholar]
- Olsen F (1980). Transfer of arterial hypertension by splenic cells from DOCA–salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathologica et Microbiologica Scandinavica. Section C, Immunology, 88, 1–5. [DOI] [PubMed] [Google Scholar]
- Parker GA, Picut CA, Swanson C, & Toot JD (2015). Histologic features of postnatal development of immune system organs in the Sprague-Dawley rat. Toxicologic Pathology, 43, 794–815. [DOI] [PubMed] [Google Scholar]
- Pollow DP, Uhrlaub J, Romero-Aleshire M, Sandberg K, Nikolich-Zugich J, Brooks HL, & Hay M (2014). Sex differences in T-lymphocyte tissue infiltration and development of angiotensin II hypertension. Hypertension, 64, 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons H, Ferrebuz A, Quiroz Y, Romero-Vasquez F, Parra G, Johnson RJ, & Rodriguez-Iturbe B (2013). Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt-sensitive hypertension. American Journal of Physiology-Renal Physiology, 304, F289–F299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL, & PhysGen Knockout Program. (2014). CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension, 63, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudemiller NP, Lund H, Priestley JR, Endres BT, Prokop JW, Jacob HJ, … Mattson DL (2015). Mutation of SH2B3 (LNK), a genome-wide association study candidate for hypertension, attenuates Dahl salt-sensitive hypertension via inflammatory modulation. Hypertension, 65, 1111–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senchenkova EY, Russell J, Kurmaeva E, Ostanin D, & Granger DN (2011). Role of T lymphocytes in angiotensin II-mediated microvascular thrombosis. Hypertension, 58, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, … Harrison DG (2014). Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension, 64, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JE, Paust HJ, Steinmetz OM, Peters A, Riedel JH, Erhardt A, … Panzer U (2010). CCR6 recruits regulatory T cells and Th17 cells to the kidney in glomerulonephritis. Journal of the American Society of Nephrology, 21, 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace K, Novotny S, Heath J, Moseley J, Martin JN Jr., Owens MY, & LaMarca B (2012). Hypertension in response to CD4+ T cells from reduced uterine perfusion pregnant rats is associated with activation of the endothelin-1 system. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 303, R144–R149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YM, Zhang GY, Wang Y, Hu M, Wu H, Watson D, … Alexander SI (2006). Foxp3-transduced polyclonal regulatory T cells protect against chronic renal injury from adriamycin. Journal of the American Society of Nephrology, 17, 697–706. [DOI] [PubMed] [Google Scholar]
- Weiss RA, Madaio MP, Tomaszewski JE, & Kelly CJ (1994). T cells reactive to an inducible heat shock protein induce disease in toxin-induced interstitial nephritis. Journal of Experimental Medicine, 180, 2239–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, … Müller DN (2017). Salt-responsive gut commensal modulates TH17 axis and disease. Nature, 551, 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, … Kuchroo VK (2013). Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature, 496, 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L, … Harrison DG (2016). Immune activation caused by vascular oxidation promotes fibrosis and hypertension. Journal of Clinical Investigation, 126, 50–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, … Harrison DG (2014). Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circulation Research, 114, 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Kim SJ, Kim N, Oh JE, Lee JG, Chung NH, … Kim YS (2008). NKT cells inhibit the development of experimental crescentic glomerulonephritis. Journal of the American Society of Nephrology, 19, 1663–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]