

Abstract
Background:
Type 2 diabetes (T2D) is characterized by impaired tissue sensitivity to insulin action (ie, insulin resistance) and impaired β-cell insulin secretion. Because obesity contributes importantly to the development of insulin resistance, we sought to determine whether insulin secretory defects would predominate in non-obese compared to obese T2D.
Methods:
We measured β-cell function and secretory capacity using the glucose-potentiated arginine test in T2D subjects early in the disease course classified as non-obese (BMI <30; n = 12) or obese (BMI ≥30 kg/m2; n = 28) and additionally compared responses from non-obese T2D with a non-diabetic control group (n = 12).
Results:
The acute insulin response to glucose potentiation of arginine-induced insulin release was less in non-obese T2D than in controls and associated with impaired β-cell sensitivity to glucose (PG50). Proinsulin secretory ratios were increased in nonobese T2D when compared to obese T2D. Obese T2D subjects had reduced insulin sensitivity (M/I) while non-obese T2D subjects had insulin sensitivity that was comparable to controls.
Conclusions:
In non-obese T2D, insulin secretory defects predominate with impaired β-cell sensitivity to glucose and proinsulin processing in the absence of insulin resistance. Future studies should consider whether different β-cell secretory phenotypes and tissue sensitivity to insulin explain the varying responsiveness to T2D interventions.
Keywords: β-cell function, insulin secretion, insulin sensitivity, type 2 diabetes
1 |. INTRODUCTION
Type 2 diabetes (T2D) develops in the setting of progressive impairment of glucose tolerance due to failure of compensatory mechanisms for regulating glucose homeostasis. Normally, to maintain glucose tolerance, changes in insulin sensitivity must be accompanied by changes in insulin secretion. Classically, T2D is associated with an obese phenotype characterized by impaired tissue sensitivity to insulin action (ie, insulin resistance). To compensate for the resistance to insulin, the pancreatic β-cells increase production and release of insulin to maintain normal blood glucose. With time and in susceptible individuals, β-cells fail to maintain increased insulin secretion resulting in glucose intolerance that progresses to clinical T2D. Autopsy studies have demonstrated that obese individuals without diabetes have a 50% increase in relative β-cell volume as compared to lean individuals without diabetes, while all those with diabetes (obese and lean) exhibited a >40% decrease in β-cell volume compared to body weight matched non-diabetic control subjects attributed to increased β-cell apoptosis with resulting decline in β-cell mass.1 However, the β-cell functional defects that develop early in the course of T2D and contribute to, or may be a consequence of, these pathologic observations,2 may not be the same in obese and non-obese patients,3 yet the therapeutic algorithm and assessment of novel therapies remains the same for all individuals with T2D.
Functional β-cell mass is best assessed in vivo from the β-cell secretory capacity, determined by glucose potentiation of arginine-induced insulin release.4 Previous studies have demonstrated decreased β-cell secretory capacity in obese T2D5 and in non-obese T2D,6 and increased proinsulin secretion relative to insulin in obese and non-obese T2D.7 While these studies clearly demonstrate insulin secretion defects present in advanced T2D with significant hyperglycaemia [fasting glucose ≥180 mg/dL (10 mmol/L)], we sought to examine mechanisms of β-cell dysfunction early in the disease course in non-obese compared with obese subjects. To do this, we evaluated β-cell function and secretory capacity using the glucose-potentiated arginine (GPA) test in a cohort of patients with early T2D who enrolled in a previously reported clinical trial.8
2 |. SUBJECTS AND METHODS
2.1 |. Subjects
As part of a study designed to investigate the role of incretin-based vs sulfonylurea therapy in early T2D, subjects were recruited with fasting glucose between 110 and 159 mg/dL (6.1–8.8 mmol/L) off all anti-diabetic agents and underwent a standard 75 g oral glucose tolerance test to confirm the presence of diabetes.8 For the present analysis, subjects with T2D were classified as obese (BMI ≥30; n = 28) or non-obese (BMI <30 kg/m2; n = 12) with additional comparison made of the non-obese T2D subjects to a non-diabetic control group (n = 12). The non-diabetic control group was gender and body weight matched to the non-obese T2D group (Table 1), and selected from subjects studied simultaneously in our laboratory using the same GPA test methodology9,10 as the study in early T2D. All study protocols were approved by the University of Pennsylvania Institutional Review Board, and all subjects provided written informed consent to participate.
TABLE 1.
Subject characteristics
| Obese T2D n = 28 | Non-obese T2D n = 12 | Non-diabetic controls n = 12c | |
|---|---|---|---|
| Sex, male/female | 14/14* | 11/1 | 10/2 |
| Ancestry, Caucasian, # | 11 | 5 | 9 |
| African American, # | 17 | 4 | 3 |
| Asian, # | 0 | 3 | 0 |
| Age, years | 54.7 ± 1.8 | 58.8 ± 2.5 | 32.2 ± 2.3† |
| Diabetes duration, years | 3.9 ± 0.8 | 4.8 ± 1.5 | NA |
| BMI, kg/m2 | 35.5 ± 0.8* | 25.0 ± 0.7 | 25.1 ± 0.8 |
| Fasting glucose, mg/dLa | 130 ± 3 | 126 ± 3 | 91 ± 3† |
| Fasting insulin, μU/mLb | 21.5 ± 2.7* | 13.1 ± 1.9 | 7.5 ± 0.9† |
| Fasting proinsulin, pmol/L | 29.2 ± 2.9 £ | 20.6 ± 2.0 | 8.3 ± 1.6† |
| One-hour glucose, mg/dLa | 240 ± 8 | 255 ± 14 | 125 ± 13‡ |
| Two-hour glucose, mg/dLa | 237 ± 10 | 228 ± 13 | 93 ± 7‡ |
| HbA1c, % | 6.5 ± 0.1 | 6.5 ± 0.1 | ND |
Note: Data are means ± SE. T2D, type 2 diabetes; NA, not applicable; ND, not done. One-hour and two-glucose levels were derived from a standard 75 g oral glucose tolerance test (OGTT).
To convert units to mmol/L, multiply by 0.05551.
To convert units to pmol/L, multiply by 7.175.
n = 8 for the OGTT.
P ≤ .05 when comparing obese T2D to non-obese T2D subjects;
P ≤ .05 when comparing non-obese T2D to non-diabetic controls;
P < .1 when comparing obese T2D to non-obese T2D subjects;
P < .001 when comparing non-obese T2D to non-diabetic controls.
2.2 |. Glucose-potentiated arginine Test
All subjects fasted overnight after 20:00 for 12 hours. By 07:00, one catheter was placed in an antecubital vein for infusions, and one catheter was placed in a contralateral hand or forearm vein for blood sampling, with the hand or arm warmed by a heating pad to promote arterialization of venous blood. Patency of the catheters was maintained with slow infusions of 0.9% saline. At least 20 minutes of acclimatization to catheter placement transpired prior to testing. After baseline blood sampling at −5 and −1 minute, 5 g arginine hydrochloride (10% solution) was injected over a 1-minute period. Blood samples were collected at 2, 3, 4, and 5 minutes post-injection. After this baseline arginine stimulation test, a hyperglycaemic clamp technique11 utilizing a variable rate of a 20% glucose solution was performed to achieve a plasma glucose level of ~230 mg/dL (12.8 mmol/L). Blood samples were taken every 5 minutes and measured at the bedside using an automated glucose analyser (YSI 2300; Yellow Springs Instruments, Yellow Springs, Ohio) to adjust the infusion rate and achieve the desired level. After 45-minutes of the glucose infusion, the arginine stimulation test was performed again. It has been demonstrated that the first administration of arginine has no effect on the subsequent response to arginine using this protocol.12 Then, after a 2-hour period without glucose infusion, a hyperglycaemic clamp was performed to achieve a plasma glucose level of ~340 mg/dL (18.9 mmol/L). Forty-five minutes after initiation of the glucose infusion, another arginine stimulation test was performed. Samples were collected on ice in tubes containing EDTA and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, Missouri), centrifuged at 4°C, separated, and frozen at −80°C for subsequent analysis. Plasma insulin and proinsulin were measured in duplicate by double-antibody radioimmunoassays (Millipore, Billerica, Massachusetts).
2.3 |. Calculations
The GPA test enables characterization of glucose-dependent insulin secretion from the glucose dose-response curve for the acute insulin response to arginine preformed at fasting, ~230 mg/dL, and ~340 mg/dL glucose levels. Under fasting conditions, the arginine stimulation test measures first-phase insulin release to a maximally stimulating dose of the non-glucose secretagogue, arginine, as the acute insulin response to arginine (AIRarg) determined as the mean of the 2-, 3-, 4- and 5-minutes insulin levels minus the mean of the baseline values.5,11 The AIRarg during the ~230 mg/dL (12.8 mmol/L) glucose clamp enables determination of glucose potentiation of arginine-induced insulin release (AIRpot).11 The AIRarg performed at ~340 mg/dL (18.9 mmol/L) glucose clamp allows for determination of the β-cell secretory capacity (AIRmax), as the AIRarg is maximal at plasma glucose concentrations >315 mg/dL (17.5 mmol/L).13 Between 60 and 250 mg/dL (3.3–13.9 mmol/L) the magnitude of AIRarg is a linear function of the plasma glucose level, whereby the glucose potentiation slope (GPS) can be calculated, and using the y-intercept (b), the plasma glucose level at which half-maximal insulin secretion (PG50) can be derived by solving the equation ½(AIRmax) = (GPS·PG50) + b and provides a measure of β-cell sensitivity to glucose.5 Insulin sensitivity (M/I) was determined by dividing the mean glucose infusion rate required during the ~230 mg/dL (12.8 mmol/L) glucose clamp (M) by the mean pre-stimulus insulin level (I) between 40 and 45 minutes of the glucose infusion.14 The disposition index (DI) relating insulin secretion to the prevailing insulin sensitivity was determined by multiplying each of the acute insulin responses and the M/I.14 The fasting proinsulin-to-insulin (PI/I) ratio was calculated as the molar concentration of proinsulin divided by the molar concentration of insulin times 100. We examined the proinsulin secretory ratio (PISR) as an estimate of β-cell granule content in response to each injection of arginine by dividing each acute proinsulin responses by the respective acute insulin response.15
2.4 |. Statistical analysis
All data are expressed as means ± SE. Comparison of the non-obese T2D subjects were made with the obese T2D subjects and with the non-diabetic controls using two-tailed independent Student’s t tests or the Mann-Whitney U test for nonparametric data with the Statistica software (StatSoft, Inc., Tulsa, Oklahoma). Significance was considered at P ≤ .05 (two-tailed).
3 |. RESULTS
There were more females in the obese than the non-obese T2D group that was similar in gender distribution to the non-diabetic control group, and the subjects with diabetes were older than the controls (Table 1). For comparable fasting glucose, fasting insulin was higher in obese compared to non-obese T2D subjects (Table 1). Fasting glucose, insulin and proinsulin were higher in non-obese T2D than in controls (Table 1). There was no difference in oral glucose tolerance or glycaemic control as measured by HbA1c between the obese and non-obese T2D subjects (Table 1). One- and two-hour glucose during oral glucose tolerance testing was higher in non-obese T2D than in controls (Table 1). There was a trend towards higher AIRarg and AIRpot in obese than non-obese T2D, while AIRpot was lower and AIRmax trended lower in non-obese T2D compared to non-diabetic controls (Table 2; Figure 1). This resulted in β-cell sensitivity to glucose (PG50) being impaired in the non-obese T2D subjects compared to non-diabetic controls (Table 2; Figure 1). Acute proinsulin responses to arginine were comparable in the obese and non-obese T2D subjects while APRpot trended lower and APRmax was reduced in non-obese T2D compared to the non-diabetic controls (Table 2). Fasting PI/I trended higher in the non-obese T2D compared to control group, and the PISRs were elevated in non-obese compared to obese T2D under fasting and hyperglycaemic clamp conditions, and trended higher in non-obese T2D compared to non-diabetic controls during maximal glucose potentiation (Table 2). Obese T2D subjects exhibited decreased insulin sensitivity (M/I) while non-obese T2D subjects had insulin sensitivity that was comparable to that in the non-diabetic control group (Table 2; Figure 1). Accounting for the relationship between β-cell function and insulin sensitivity, the DI was similar in the obese and non-obese T2D subjects, but was significantly impaired under glucose-potentiated conditions in the non-obese T2D subjects when compared to the non-diabetic control group (Table 2).
TABLE 2.
Metabolic parameters during the glucose-potentiated arginine test
| Obese T2D n = 28 | Non-obese T2D n = 12 | Non-diabetic controls n = 12 | |
|---|---|---|---|
| AIRarg, μU/mLa | 51.2 ± 7.0£ | 32.3 ± 5.3 | 33.5 ± 4.5 |
| AIRpot, μU/mLa | 123 ± 16£ | 81 ± 17 | 137 ± 18† |
| AIRmax, μU/mLa | 189 ± 31 | 127 ± 22 | 191 ± 26¥ |
| PG50, mg/dLb | 193 ± 11 | 208 ± 21 | 160 ± 8† |
| APRarg, pmol/L | 8.5 ± 0.8 | 7.7 ± 2.3 | 6.9 ± 1.5 |
| APRpot, pmol/L | 14.9 ± 0.9 | 17.9 ± 2.9 | 25.0 ± 2.7¥ |
| APRmax, pmol/L | 16.3 ± 1.3 | 18.5 ± 2.0 | 26.2 ± 2.4† |
| Fasting PI/I | 20.4 ± 2.1 | 27.7 ± 5.2 | 16.3 ± 2.9¥ |
| PISR1 | 2.8 ± 0.4* | 5.0 ± 1.4 | 3.2 ± 0.6 |
| PISR2 | 2.1 ± 0.2* | 4.9 ± 1.5 | 2.9 ± 0.3 |
| PISR3 | 1.7 ± 0.2* | 2.6 ± 0.3 | 1.8 ± 0.1¥ |
| M/I, mg kg−1·min−1/μU/mL | 0.21 ± 0.02* | 0.44 ± 0.06 | 0.45 ± 0.06 |
| DI1, mg kg−1 min−1 | 8.8 ± 1.2 | 12.6 ± 2.4 | 14.2 ± 2.6 |
| DI2, mg kg−1 min−1 | 20.5 ± 2.2 | 28.6 ± 4.5 | 54.5 ± 8.2† |
| DI3, mg kg−1 min−1 | 33.5 ± 4.8 | 45.6 ± 7.3 | 79.2 ± 14.3† |
Note: Data are means ± SE. AIRarg, acute insulin response to arginine; AIRpot, acute insulin response to glucose-potentiated [~230 mg/dL (12.8 mmol/L) glucose clamp] arginine; AIRmax, acute insulin response to maximally glucose-potentiated [~340 mg/dL (18.9 mmol/L) glucose clamp] arginine; APRarg, acute proinsulin response to arginine; APRpot, acute proinsulin response to glucose-potentiated arginine; APRmax, acute proinsulin response to maximally glucose-potentiated arginine; PI/I, molar ratio of proinsulin/insulin; PISR, molar ratio of the APR/AIR under fasting (PISR1), glucose-potentiated (PISR2), and maximally glucose-potentiated (PISR3) conditions; PG50, plasma glucose at which half-maximal insulin secretion is achieved, a measure of β-cell sensitivity to glucose; M/I, mean glucose infusion rate divided by second-phase insulin levels during the ~230 mg/dL glucose clamp, a measure of insulin sensitivity (Reference 14); DI, disposition index under fasting (DI1), glucose-potentiated (DI2), and maximally glucose-potentiated (DI3) conditions (Reference 14).
To convert units to pmol/L, multiply by 7.175.
To convert units to mmol/L, multiply by 0.05551.
P ≤ .05 when comparing obese T2D to non-obese T2D subjects;
P < .1 when comparing obese T2D to non-obese T2D subjects;
P ≤ .05 when comparing non-obese T2D subjects to non-diabetic controls;
P < .1 when comparing non-obese T2D subjects to non-diabetic controls.
FIGURE 1.
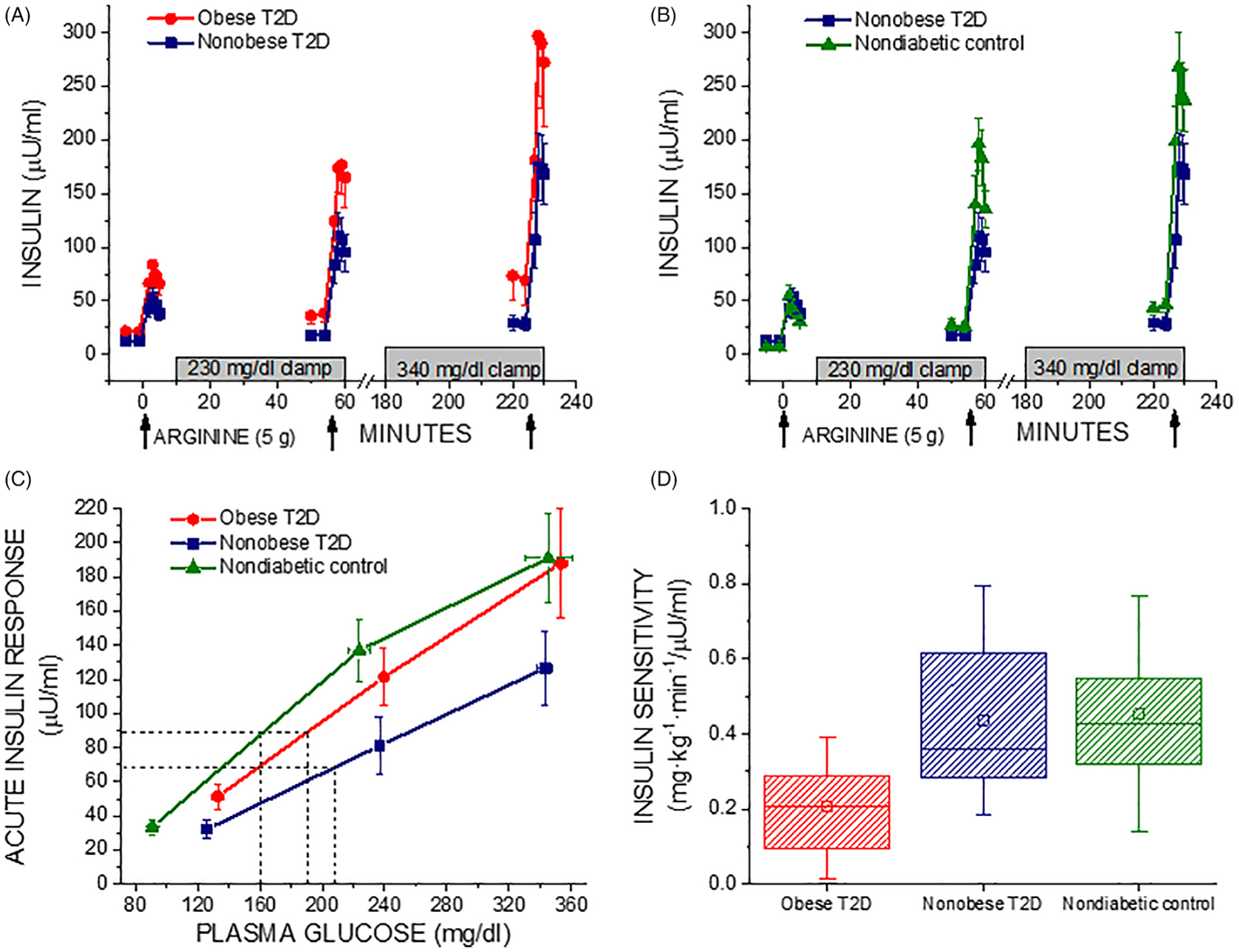
A and B, Plasma insulin in response to bolus injections of arginine (arrows) administered under fasting, ~230 mg/dL (12.8 mmol/L), and ~340 mg/dL (18.9 mmol/L) hyperglycemic clamp conditions in obese T2D compared to non-obese T2D subjects (A) and non-obese T2D compared to non-diabetic control subjects (B). C, Acute insulin responses (AIRs) as a function of the pre-stimulus plasma glucose concentration. The glucose potentiation slope (GPS), calculated as the difference in the AIR at fasted and ~230 mg/dL (12.8 mmol/L) glucose levels divided by the difference in plasma glucose, is down shifted in obese T2D and in particular non-obese T2D (0.66 ± 0.11 and 0.43 ± 0.11) relative to control (0.79 ± 0.12; P < .05 vs nonobese T2D). β-Cell sensitivity to glucose is determined as the PG50, the plasma glucose concentration at which half maximal insulin secretion was achieved, using the y-intercept (b) of the GPS to solve the equation AIRmax/2 = GPS·PG50 + b as represented by the dashed lines, and is right shifted in T2D, especially for the non-obese T2D group (P < .05 vs control), indicating impaired β-cell sensitivity to glucose. D, Insulin sensitivity (M/I) was reduced in obese T2D compared to non-obese T2D (P < .05) that was not different from control. In A, B, and C, data are given as mean ± SE, and in D as median and IQR (box) and mean (open squares) and range (error bars)
4 |. DISCUSSION
These data demonstrate that in non-obese T2D, early in the disease course insulin secretion defects predominate with impaired β-cell sensitivity to glucose and less efficient processing of proinsulin, together contributing to compromised β-cell function. We did not find a significant reduction in insulin sensitivity (M/I) in the non-obese T2D subjects, supporting a primary β-cell defect as the aetiology of their impaired glucose regulation. In contrast, at a similar stage in progression of abnormal fasting glucose, the obese T2D subjects maintained more robust insulin secretory responses and efficient proinsulin processing than the non-obese T2D, yet their insulin sensitivity was markedly reduced. While we did not study a group of obese non-diabetic controls that would be expected to exhibit even greater insulin secretory responses for a comparable degree of insulin resistance, our obese T2D subjects studied early in the disease course maintained relatively preserved β-cell secretory capacity that nonetheless could not adequately compensate for the presence of marked insulin resistance.
Previous studies that have involved patients with more advanced T2D and significant hyperglycaemia [fasting glucose ≥180 mg/dL (10 mmol/L)] demonstrated decreased β-cell secretory capacity in obese T2D5 and in non-obese T2D,6 with resulting estimates of β-cell sensitivity to glucose not different than in controls. The impaired β-cell sensitivity to glucose seen in the non-obese T2D subjects reported here likely represents an earlier stage defect whereby earlier in the disease course and in the absence of significant hyperglycaemia [fasting glucose <160 mg/dL (8.9 mmol/L)] the β-cell secretory capacity is relatively preserved,16 allowing for the dose-response curve for glucose-dependent insulin secretion to right-shift (Figure 1). Similar to the greater impairment of in vivo insulin secretion observed here for non-obese compared to obese T2D, previous ex vivo assessment of islets isolated from donors with T2D also demonstrated more pronounced impairment of glucose stimulated insulin secretion in islets from non-obese compared with obese donors.17 The requirement for higher levels of glucose to potentiate insulin secretion may possibly represent a protective mechanism against the burden of increase β-cell secretory demand imparted by chronically elevated glucose levels.
Increased β-cell secretory demand has also been associated with increased proinsulin secretion relative to insulin in both obese and non-obese T2D with more advanced disease,7 whereas we observed disproportionately increased proinsulin secretion only in non-obese when compared to obese T2D early in the disease course. In a previous report involving patients with established T2D and in the presence of hyperglycaemia (HbA1c ~8.6%–8.9%), disproportionately increased proinsulin secretion was evidenced in both lean and obese subjects with T2D when compared to BMI-matched controls.18 In the present report, patients had early T2D with better glycaemic control (HbA1c ~6.5%) and elevated PISRs were present in the non-obese compared to obese T2D subjects, suggesting that greater β-cell dysfunction is present at the onset of T2D in non-obese individuals. This finding together with the decreased β-cell sensitivity to glucose present in the non-obese T2D group supports a primary defect in β-cell function that may in fact represent a protective mechanism lowering insulin secretion by the β-cell in response to glucose when proinsulin processing and insulin production are impaired.19 In a recent longitudinal cohort study of individuals with T2D treated with oral anti-hyperglycaemic agents, increasing fasting PI/I ratio was associated with increasing HbA1c without change in homeostatic model assessment of insulin resistance.20 Thus, worsening proinsulin processing appears related to progressive β-cell dysfunction during deterioration of glucose tolerance in T2D.
Our results are consistent with other efforts to better define the heterogeneity of T2D by creating subgroup classifications or phenotypes based on patient characteristics and supported by genomic associations with the potential to inform a more personalized approach to disease management. Ahlqvist and colleagues performed a cluster analysis using clinical variables from a Swedish cohort of adult-onset diabetes, which after separating autoimmune type 1 diabetes (6.4%), divided T2D into clusters of insulin-deficient (17.5%), insulin-resistant (15.3%), obesity-related (21.6%), and age-related (39.1%) diabetes that carried varying genetic associations and risk for diabetes complications.21 Taking a genomic clustering approach, Udler and colleagues identified five clusters of T2D loci and traits, with two characterized by β-cell secretory defects and three characterized by insulin resistance phenotypes with either obesity, a ‘lipodystrophy-like’ fat distribution, or disrupted liver lipid metabolism.22 While these studies relied on fasting measures of glucose, C-peptide, insulin and proinsulin to characterize β-cell function and insulin sensitivity, using more sophisticated physiologic measures in the present study our non-obese T2D subjects likely correspond to the insulin-deficient and β-cell secretory defect clusters, respectively, while our obese T2D subjects may represent the insulin resistant obesity related clusters. While no clinically useful test of genetic differences in T2D has been developed yet, future work to determine whether subgroups of T2D with varying β-cell secretory phenotypes and tissue sensitivity to insulin respond differently to interventions should consider incorporating more specific mechanistic measures such as those derived from glucose-potentiation of arginine-induced insulin secretion reported here.
Our study is limited not only by the absence of an obese non-diabetic control group, but also by more women in the obese T2D since female sex may be protective against β-cell dysfunction in African American ancestry,23 and the younger age of our non-obese control group that was derived from a convenience sample.24 In addition, a higher maximum glucose (eg, >450 mg/dL) for potentiation of arginine-induced insulin secretion might further increase AIRmax in T2D,5 but that would result in a higher PG50 and only strengthen the reported impairment in β-cell sensitivity to glucose observed in the non-obese T2D group reported here.
In conclusion, the present evidence supports the characteristic association of obese T2D with insulin resistance, and that decreased β-cell sensitivity to glucose and impaired proinsulin processing represent the predominant early pathophysiologic defects in non-obese T2D. These findings help to differentiate a phenotype for the early presentation of T2D wherein future therapeutic interventions may target β-cell dysfunction as the primary defect in non-obese individuals and insulin resistance as the dominate problem in obese individuals, and so personalize the approach to T2D care based on the underlying primary pathophysiology.
ACKOWLEDGEMENTS
We thank Dr. Heather Collins of the University of Pennsylvania Diabetes Research Center for performance of the radioimmunoassays, and Huong-Lan Nguyen of the University of Pennsylvania Institute for Diabetes, Obesity & Metabolism for laboratory assistance.
This work was supported by the Pennsylvania Department of Health, Bureau of Health Statistics and Research Grant 4100043362 (Penn Center for Excellence in Regenerative Medicine), Public Health Services Research Grants UL1 TR000003 (University of Pennsylvania Clinical and Translational Research Center) and P30 DK19525 (University of Pennsylvania Diabetes Research Center), the Joanne and Raymond Welsh Research Fellowship (to L.G.), and the Human Metabolism Resource of the University of Pennsylvania Institute for Diabetes, Obesity & Metabolism.
Funding information National Center for Advancing Translational Sciences, Grant/Award Number: UL1 TR000003; National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Number: P30 DK19525; Pennsylvania Department of Health, Grant/Award Number: 4100043362
Footnotes
CONFLICT OF INTEREST
The authors have no relevant conflict of interest to disclose.
REFERENCES
- 1.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. [DOI] [PubMed] [Google Scholar]
- 2.Leahy JL, Hirsch IB, Peterson KA, Schneider D. Targeting beta-cell function early in the course of therapy for type 2 diabetes mellitus. J Clin Endocrinol Metab. 2010;95:4206–4216. [DOI] [PubMed] [Google Scholar]
- 3.Vaag A, Lund SS. Non-obese patients with type 2 diabetes and prediabetic subjects: distinct phenotypes requiring special diabetes treatment and (or) prevention? Appl Physiol Nutr Metab. 2007;32:912–920. [DOI] [PubMed] [Google Scholar]
- 4.Kahn SE, Carr DB, Faulenbach MV, Utzschneider KM. An examination of beta-cell function measures and their potential use for estimating beta-cell mass. Diabetes Obes Metab. 2008;10(suppl 4):63–76. [DOI] [PubMed] [Google Scholar]
- 5.Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D Jr. Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74:1318–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Haeften TW, Van Maarschalkerweerd WW, Gerich JE, Van der Veen EA. Decreased insulin secretory capacity and normal pancreatic B-cell glucose sensitivity in non-obese patients with NIDDM. Eur J Clin Invest. 1991;21:168–174. [DOI] [PubMed] [Google Scholar]
- 7.Roder ME, Porte D, Schwartz RS, Kahn SE. Disproportionately elevated proinsulin levels reflect the degree of impaired B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1998;83:604–608. [DOI] [PubMed] [Google Scholar]
- 8.Gudipaty L, Rosenfeld NK, Fuller CS, Gallop R, Schutta MH, Rickels MR. Effect of exenatide, sitagliptin, or glimepiride on beta-cell secretory capacity in early type 2 diabetes. Diabetes Care. 2014;37: 2451–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rickels MR, Goeser ES, Fuller C, et al. Loss-of-function mutations in ABCA1 and enhanced beta-cell secretory capacity in young adults. Diabetes. 2015;64:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rickels MR, Mueller R, Teff KL, Naji A. Beta-cell secretory capacity and demand in recipients of islet, pancreas, and kidney transplants. J Clin Endocrinol Metab. 2010;95:1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward WK, Halter JB, Beard JC, Porte D Jr. Adaptation of B and a cell function during prolonged glucose infusion in human subjects. Am J Physiol Endocrinol Metab. 1984;246:E405–E411. [DOI] [PubMed] [Google Scholar]
- 12.Larsson H, Ahren B. Glucose-dependent arginine stimulation test for characterization of islet function: studies on reproducibility and priming effect of arginine. Diabetologia. 1998;41:772–777. [DOI] [PubMed] [Google Scholar]
- 13.Seaquist ER, Robertson RP. Effects of hemipancreatectomy on pancreatic alpha and beta cell function in healthy human donors. J Clin Invest. 1992;89:1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guldstrand M, Ahren B, Adamson U. Improved beta-cell function after standardized weight reduction in severely obese subjects. Am J Physiol Endocrinol Metab. 2003;284:E557–E565. [DOI] [PubMed] [Google Scholar]
- 15.Kahn SE, Halban PA. Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes. 1997;46:1725–1732. [DOI] [PubMed] [Google Scholar]
- 16.Kahn SE. Clinical review 135 - the importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86:4047–4058. [DOI] [PubMed] [Google Scholar]
- 17.Rosengren AH, Braun M, Mahdi T, et al. Reduced insulin exocytosis in human pancreatic beta-cells with gene variants linked to type 2 diabetes. Diabetes. 2012;61:1726–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roder ME, Dinesen B, Hartling SG, et al. Intact proinsulin and beta-cell function in lean and obese subjects with and without type 2 diabetes. Diabetes Care. 1999;22:609–614. [DOI] [PubMed] [Google Scholar]
- 19.Steiner DF, Park SY, Stoy J, Philipson LH, Bell GI. A brief perspective on insulin production. Diabetes Obes Metab. 2009;11(suppl 4): 189–196. [DOI] [PubMed] [Google Scholar]
- 20.Russo GT, Giorda CB, Cercone S, et al. Beta cell stress in a 4-year follow-up of patients with type 2 diabetes: a longitudinal analysis of the BetaDecline study. Diabetes Metab Res Rev. 2018;34:e3016. [DOI] [PubMed] [Google Scholar]
- 21.Ahlqvist E, Storm P, Karajamaki A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6: 361–369. [DOI] [PubMed] [Google Scholar]
- 22.Udler MS, Kim J, von Grotthuss M, et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLoS Med. 2018;15:e1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mauvais-Jarvis F, Sobngwi E, Porcher R, et al. Ketosis-prone type 2 diabetes in patients of sub-Saharan African origin: clinical pathophysiology and natural history of beta-cell dysfunction and insulin resistance. Diabetes. 2004;53:645–653. [DOI] [PubMed] [Google Scholar]
- 24.Fritsche A, Madaus A, Stefan N, et al. Relationships among age, proinsulin conversion, and beta-cell function in nondiabetic humans. Diabetes. 2002;51(suppl 1):S234–S239. [DOI] [PubMed] [Google Scholar]