

Abstract
Growing evidence suggests overlap between Alzheimer’s disease (AD) and Parkinson’s disease (PD) pathophysiology in a subset of patients. Indeed, 50–80% of autopsy cases with a primary clinicopathological diagnosis of Lewy body disease (LBD) – most commonly manifesting during life as PD – have concomitant amyloid-beta and tau pathology, the defining pathologies of AD. Here we evaluated common genetic variants in genome-wide association with AD as predictors of concomitant AD pathology in the brains of people with a primary clinicopathological diagnosis of PD or Dementia with Lewy Bodies (DLB), diseases both characterized by neuronal Lewy bodies. In the first stage of our study, 127 consecutive autopsy-confirmed cases of PD or DLB from a single center were assessed for AD neuropathological change (ADNC), and these same cases were genotyped at 20 single nucleotide polymorphisms (SNPs) found by genome-wide association study to associate with risk for AD. In these 127 Training set individuals, we developed a logistic regression model predicting the presence of ADNC, using backward stepwise regression for model selection and 10-fold cross-validation to estimate performance. The best-fit model generated a risk score for ADNC (ADNC-RS) based on age at disease onset and genotype at three SNPs (APOE, BIN1, and SORL1 loci), with an area under the receiver operating curve (AUC) of 0.751 in our Training set. In the replication stage of our study, we assessed model performance in a separate Test set of the next 81 individuals genotyped in our center. In the Test set, the AUC was 0.781, and individuals with ADNC-RS in the top quintile had four-fold increased likelihood of having AD pathology at autopsy compared with those in each of the lowest two quintiles. Finally, in the validation stage of our study, we applied our ADNC-RS model to 70 LBD individuals from 20 Alzheimer’s Disease Research Centers (ADRC) whose autopsy and genetic data were available in the National Alzheimer’s Coordinating Center (NACC) database. In this Validation set, the AUC was 0.754. Thus, in patients with autopsy-confirmed PD or DLB, a simple model incorporating three AD-risk SNPs and age at disease onset substantially enriches for concomitant AD pathology at autopsy, with implications for identifying LBD patients in which targeting amyloid-beta or tau is a therapeutic strategy.
Keywords: Parkinson’s disease, Alzheimer’s disease, Genetics, Dementia
INTRODUCTION
Alzheimer’s (AD) and Parkinson’s diseases (PD) are the two most common neurodegenerative disorders, together affecting >6 million individuals worldwide [10, 27]. AD is defined neuropathologically by the presence of amyloid-beta (Aβ) plaques and tau neurofibrillary tangles (NFT), while PD is defined by the presence of Lewy bodies composed of alpha-synuclein (aSyn). The average age of a patient receiving an AD clinical diagnosis is ~80 years old [2], while the average age of a patient receiving a PD clinical diagnosis is ~60 years old [36].
PD is not the only disease defined by aSyn Lewy bodies. Rather, PD belongs to a group of “synucleinopathies” collectively called the Lewy body diseases (LBD). The LBD comprise PD, with or without dementia, dementia with Lewy bodies (DLB), and multiple system atrophy (MSA) [19], with the first two entities (PD and DLB) demonstrating neuronal aSyn Lewy bodies, while MSA shows aSyn inclusions in glia. Importantly, the distinction between DLB and PD with dementia (PDD) is clinical, based on the timing of development of dementia [31]. On neuropathological examination, DLB and PDD patients are nearly indistinguishable at the individual level. Furthermore, DLB and PDD share preclinical features, and shared genetic variants confer an increased risk in both disorders [4, 14, 28].
Despite traditional separation between AD and the LBD, growing evidence suggests a dynamic interaction between their pathophysiologies. Fifty to 80% of patients with a primary clinicopathological diagnosis of LBD have concomitant Aβ and tau pathology [38]. At autopsy, up to 40% of PD patients exhibit enough Aβ and NFT to qualify for a secondary diagnosis of AD [14]. Mechanistically, in vitro and in vivo studies suggest that aSyn, tau, and Aβ may interact synergistically in events leading to disease development [5, 44]. From a practical viewpoint, these findings suggest that LBD patients may be at-risk for developing AD.
Genetic risk factors for developing AD have been identified through family studies and genome-wide association studies (GWAS). In a recent AD GWAS comparing >50,000 cases with >100,000 controls, 25 distinct loci were associated with risk for AD [24]. However, the genetic heritability (h2) reported for this study was only 0.071, and various genetic risk scores composed of AD GWAS-nominated variants have poor predictive value for AD in the general population [11].
We reasoned that the high prevalence of AD within the LBD population might enhance the ability of AD genetic risk variants to predict the development of AD pathology. Accordingly, we genotyped all common genetic variants reported in two or more AD GWAS to associate with AD risk in a single-center cohort of 208 consecutive cases with a primary clinicopathological diagnosis of either PD or DLB. We tested these AD risk variants for their ability to predict concomitant AD pathology in these cases, validating our best model in an additional 70 LBD cases from the multi-center National Alzheimer’s Coordinating Center (NACC) database.
MATERIALS AND METHODS
Participants:
Clinical and neuropathological data from all autopsy cases enrolled between February 1985 and July 2019 at the University of Pennsylvania (Penn) Center for Neurodegenerative Disease Research brain bank were assessed [45]. A clinicopathological diagnosis was assigned to each case primarily determined by neuropathology and secondarily accounting for clinical history. We note that although some cases were banked decades ago, all cases have been reassessed using modern criteria and techniques. Those with (1) a primary clinicopathological diagnosis of PD, PDD, or DLB and (2) DNA available for genetic studies were included in the analysis. All cases included in this study had a clinicopathological diagnosis of DLB or PD with or without dementia; we excluded MSA to focus on primary neuronal synucleinopathies [17]. Of 1922 accessioned cases, 208 cases met the above criteria, with 127 cases having complete genotype data at the outset of our study (Training set), and 81 more cases genotyped during the course of our study (Test set).
The National Alzheimer’s Coordinating Center (NACC) database is a multi-center collection of clinical and neuropathological data from over 42,000 de-identified individuals across 39 past and present Alzheimer’s Disease Research Centers (ADRCs), as of March 2020 [3]. Through the Alzheimer’s Disease Genetics Consortium (ADGC), genetic data are also available for some of these individuals. Individuals with (1) evidence of Lewy body neuropathology, (2) a presumptive etiological diagnosis of Lewy body disease, (3) from a non-Penn ADRC, and (4) SNP genotypes for AD GWAS loci available through the ADGC, were included in the validation stage of our analysis [22, 23]. Cases with genetic mutations for familial AD were excluded. Seventy cases from twenty ADRCs met criteria and were included in the analysis. Informed consent was obtained from all participants prior to death at each ADRC.
Prior to conducting these studies, approval was obtained from the Penn Institutional Review Board, and informed consent was obtained from all participants prior to death. All procedures in these studies adhere to the tenets of the Declaration of Helsinki.
Immunohistochemistry and Neuropathological Staging
For the Penn cases, neuropathological characterization of defined brain regions (frontal neocortex, temporal neocortex, parietal neocortex, occipital neocortex, anterior cingulate gyrus, hippocampus including entorhinal cortex, amygdala, basal ganglia, thalamus, midbrain, pons, medulla, cerebellum) was conducted on all cases as previously described [1, 45]. Briefly, each brain region was assessed by hematoxylin and eosin stain in addition to immunohistochemical stains (NAB228 for Aβ generated by Dr. Trojanowski, PHF1 for phosphorylated tau gifted by Peter Davies, SYN303 for aSyn generated by Dr. Trojanowski, and 1D3 for phosphorylated TDP-43 gifted by Manuela Neumann and Elisabeth Kremmer) in order to assign a semi-quantitative score (none, rare, mild, moderate, or severe) for tau, Aβ, aSyn, and TDP-43 pathologies. For the NACC cases, neuropathological characterization of brain regions was conducted at individual ADRCs in accordance with established guidelines [21]. For all cases, an AD Neuropathologic Change (ADNC) score was also assigned in accordance with the National Institute on Aging’s guidelines for the neuropathologic assessment of AD. Absence of AD co-pathology was defined by an ADNC of None or Low, while presence of AD co-pathology was defined by an ADNC score of Intermediate or High [32].
Genotyping of AD Risk Variants
Single nucleotide polymorphisms (SNPs) in genome-wide association with AD risk were nominated from the literature. Three AD genome-wide association studies (GWAS) together examining >70,000 AD subjects and >380,000 controls were used to identify candidate SNPs [16, 24, 25]. SNPs reaching genome-wide significance (p < 5 × 10−8) in at least two of these three major GWAS were included in our study. Twenty independent loci reached criteria for inclusion.
For the Penn cases, SNP genotype was determined by Illumina Global Screening Arrays (GSA), or TaqMan SNP Genotyping Assays, as previously described [7]. In some cases, proxy SNPs (D’>0.8 in the EUR reference population from 1000 Genomes Project Phase 3 [41]) were substituted, as indicated in the text. For the NACC cases, SNP genotyping was completed by the Alzheimer’s Disease Genetic Consortium (ADGC) via Illumina or Affymetrix high-density microarrays, as previously described [33].
Association of Individuals Risk Variants with ADNC
Logistic regression models were used to test for association between genotype at each SNP and the presence or absence of AD co-pathology in the Penn cohort (N=208). Because the APOE locus has three alleles reported to have differential effects in AD [13, 39], we considered the number of APOE E2 and APOE E4 alleles separately. Additional analyses were performed with sex and age at disease onset as covariates in the logistic regression.
Logistic Regression Model Predicting ADNC
Penn autopsy cases were split into Training (N=127, 61%) and Test (N=81, 39%) sets, which is within the range of optimal allocation proportions for large data sets with high data accuracy [8]. The Training set comprised the first batch of 127 cases genotyped, for which data was available at the outset of the study, while the Test set comprised the next 81 cases genotyped, for which genetic data was obtained during the subsequent replication step of our study. There was no overlap between Training and Test sets. Backwards stepwise regression was used to develop a binary classifier to predict the presence or absence of AD co-pathology in the Training set, with age at disease onset and sex as covariates in the model. Comparison of Akaike information criterion (AIC) at each step was used to determine model fitness, and we estimated predictive performance by ten-fold cross-validation (100 iterations). The Hosmer and Lemeshow goodness-of-fit test [12] was used to evaluate the final logistic regression model developed in the Training set.
Model performance at predicting AD co-pathology was assessed in both the Penn Training and Test sets using receiver operating characteristic (ROC) curves, generating an area under the curve (AUC) for both the Training and Test sets.
The best-fit model was also applied to LBD subjects from the NACC database, with ROC curve analyses.
Development of the ADNC-RS
An ADNC Risk Score (ADNC-RS) was calculated for each case based on the best model developed in the Penn-based Training set by multiplying the age at disease onset or the risk allele dose by the respective regression coefficient. The risk score can be used to calculate the probability of AD co-pathology using the formula: p = e^(Risk Score) / (1 + e^(Risk Score)). Specifically, because our model is a logistic regression model, the output (Risk Score) is in log odds. Log odds may be converted to odds by taking the antilog (eRisk Score). The odds may then be converted to probability using the standard probability (p) formula (p = odds/(1 + odds)). Scores and probabilities were generated using the “predict” function in the “caret” package in R [20] from the logistic regression model.
Additional Details Regarding Statistical Analysis
Analyses were conducted in R (http://www.r-project.org) and Prism 8 (http://www.graphpad.com/scientific-software/prism); R-scripts as well as Penn-based datasets are available in the Supplementary Methods as an Online Resource. The “caret” package was used for cross-validation and model generation [30]. The “ROCR” and “pROC” packages were used for creating and analyzing receiver operating characteristic (ROC) curves [37, 40]. T-test, Wilcoxon rank-sum, or Fisher’s exact tests were used to assess differences between clinical variables, as indicated by the distributions of data. For all statistical tests, power was set at 0.8, alpha was set to 0.05, and all tests were two-sided.
RESULTS
Penn LBD Cohort Characteristics:
Two hundred and eight participants from Penn with a primary clinicopathological diagnosis of PD or DLB were included in this analysis. The mean age at clinical disease onset was 64.51 years (SEM 0.70) and at death was 77.67 years (SEM 0.55). The majority of these subjects (n=163/208 (78.4%)) received clinicopathological diagnoses of PD; 108 of these PD individuals had dementia at the time of death, and 55 did not. Additional diagnoses for this cohort, as well as clinical and demographic details, are shown in Tables 1 and 2.
Table 1.
Demographic and Clinical Characteristics of Cohort. Data represent mean (SEM) unless otherwise noted.
| Characteristic | Whole Cohort | None/Low ADNC | Intermediate/High ADNC | p-value b |
|---|---|---|---|---|
| N | 208 | 129 | 79 | |
| Age at Onset, y | 64.51 (0.70) | 62.52 (0.93) | 67.76 (0.93) | 0.000 |
| Age at Death, y | 77.67 (0.55) | 76.95 (0.75) | 78.84 (0.79) | 0.149 |
| Disease Duration, y | 13.80 (0.61) | 15.27 (0.85) | 11.41 (0.75) | 0.000 |
| Sex, (F/M), N (%) | 45(22)/163(78) | 30(23)/99(77) | 15(19)/64(81) | 0.494 |
| Race, N (%) White | 199 (97.6) | 122 (97.6) | 77 (97.5) | 0.999 |
| Non-White | 5 (2.4) | 3 (2.4) | 2 (2.5) | |
| Last MMSEa | 20.07 (0.74) | 21.29 (0.92) | 18.08 (1.18) | 0.021 |
| Last UPDRS-IIIa | 45.47 (1.91) | 43.96 (2.17) | 49.10 (3.82) | 0.222 |
| Dementia Diagnosis During | 55(26.4)/ | 47(36.4)/ | 8(10)/ | 0.000 |
| Life, (No/Yes) N (%) | 153(73.6) | 82(63.6) | 71(89.9) | |
| Lewy Body Distribution, N (%) | ||||
| Diffuse or Neocortical | 120 (58.0) | 56 (43.4) | 64 (82.1) | |
| Transitional or Limbic | 58 (28.0) | 47 (36.4) | 11 (14.1) | 0.000 |
| Brainstem Predominant | 29 (14.0) | 26 (20.2) | 3 (3.8) | |
Available Data (N=100 MMSE, N=68 UPDRS-III).
Comparison between None/Low ADNC and Intermediate/High ADNC groups.
p<0.05.
Table 2.
Clinicopathological Diagnosis of Cohort.
| Clinicopathological Diagnosis | Whole Cohorta | None/Low ADNCb | Intermediate/High ADNCb |
|---|---|---|---|
| Parkinson’s disease | 55 (26.44) | 47 (85.45) | 8 (14.55) |
| Parkinson’s disease dementia | 108 (51.92) | 62 (57.41) | 46 (42.59) |
| Dementia with Lewy bodies | 45 (21.63) | 20 (44.44) | 25 (55.55) |
Data represent N (%) of whole cohort.
Data represent N (%) of ADNC category.
Only 43/208 (20.67%) of this LBD cohort had no ADNC at autopsy, while more than one-third had intermediate or high levels of ADNC (Fig. 1a, Supplementary Table 1, Online Resource). Among the group with no ADNC, 16.8% are identified as Primary Age Related Tauopathy (PART). In the Penn LBD cohort, virtually all individuals with amyloid plaques also demonstrated NFT; indeed, only 4 individuals had amyloid plaques without evidence of NFTs. Representative immunohistochemical sections of anterior cingulate and middle frontal cortex demonstrating Lewy body pathology with co-occurring Aβ and tau NFT are shown in Fig. 1b.
Fig 1.
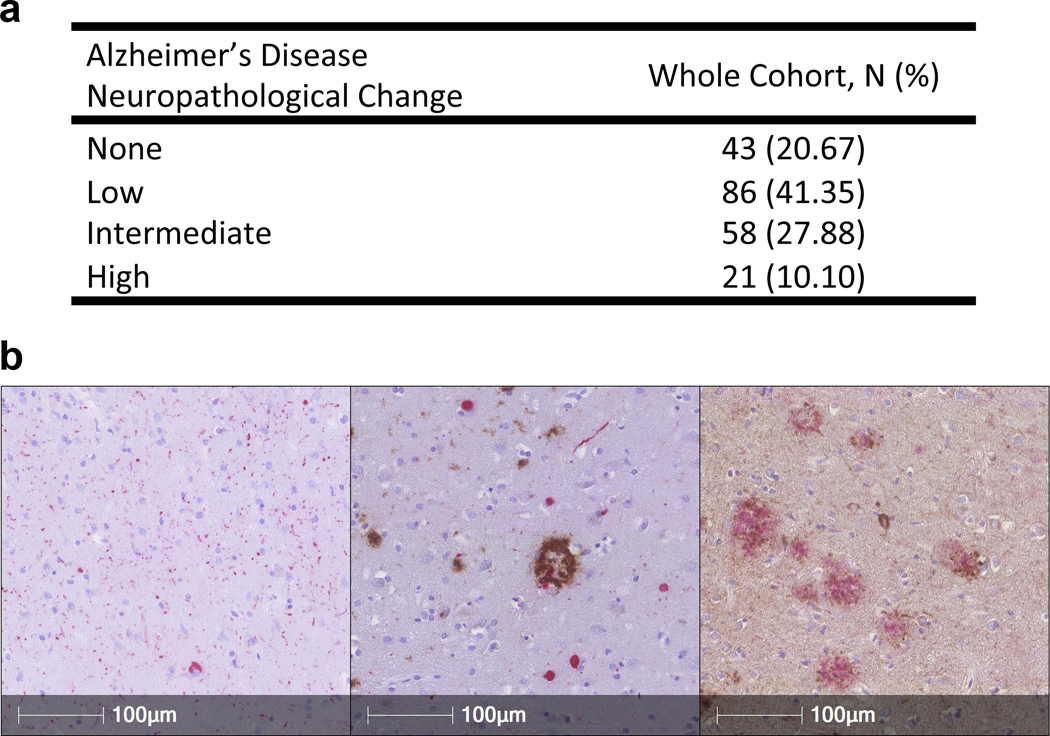
Alzheimer’s Disease Neuropathological Change (ADNC) scores in N = 208 cases from Penn with a primary clinicopathological diagnosis of PD or DLB. a Number of subjects and % of whole cohort at each level of ADNC. b Representative immunohistochemical sections (160X) demonstrating Lewy body aSyn pathology alone in anterior cingulate (left panel, in red), concomitant Aβ (brown) and aSyn (red) pathology in anterior cingulate (middle panel), and Aβ (red) and tau NFT (brown) pathology in middle frontal cortex (right panel). aSyn pathology was detected with the MJFR13 antibody against phosphorylated aSyn. Aβ pathology was detected with the NAB228 antibody, and tau NFT’s were detected with the 17028 rabbit polyclonal anti-tau antibody.
Clinical Differences in PD/DLB patients with vs. without ADNC
Compared to PD/DLB subjects with absent or low levels of ADNC, subjects with intermediate-to-high levels of ADNC were older at disease onset (67.76 vs. 62.52 years, p<0.001) and had shorter disease duration (11.41 vs. 15.27 years, p<0.001). They were also more cognitively impaired, with lower MMSE scores (18.08 vs. 21.29, p=0.02), and greater rates of clinical dementia (89.9% vs. 63.6%, p<0.001, Table 1), prior to death. The mean time between the last MMSE and death was 2.66 years (SD 2.37).
Association of Individual AD Risk SNPs with ADNC in PD/DLB patients
Twenty genetic loci have been robustly associated with risk for developing AD by multiple GWAS [16, 24, 25] (Table 3). As shown in Table 4, the number of APOE E4 alleles associated with increased risk for ADNC in PD/DLB (nominal p<0.001). One other locus near SORL1, represented by rs11218343, approached but did not meet the significance threshold for association with ADNC (nominal p=0.06). Adjusting for age at onset and sex minimally affected these results (Supplementary Table 2, Online Resource).
Table 3.
Genetic loci nominated from Alzheimer’s disease GWAS literature.
| Variant | Chr. | Positiona | Nearest Geneb | Maj/ Min | MAFc | Reported OR (95%CI) for ADd |
|---|---|---|---|---|---|---|
| rs3818361e | 1 | 207611623 | CR1 | G/A | 0.278 | 1.20 (1.13–1.27) |
| rs6733839 | 2 | 127135234 | BIN1 | C/T | 0.399 | 1.20 (1.17–1.23) |
| rs7570320f | 2 | 233167045 | INPP5D | C/A | 0.369 | 0.92 (0.87–0.97) |
| rs9271100g | 6 | 32608701 | HLA-DRB5/DRB1 | C/T | 0.273 | 1.11 (1.06–1.17) |
| rs75932628 | 6 | 41161514 | TREM2 | C/T | 0.010 | 2.08 (1.73–2.49) |
| rs10948363h | 6 | 47520026 | CD2AP | A/G | 0.278 | 1.09 (1.06–1.12) |
| rs1476679i | 7 | 100406823 | NYAP1j | C/T | 0.323 | 0.92 (0.90–0.95) |
| rs11762262k | 7 | 143410783 | EPHA1 | C/T | 0.207 | 0.91 (0.86–0.96) |
| rs17057043l | 8 | 27362793 | PTK2B | G/A | 0.343 | 1.11 (1.06–1.16) |
| rs11136000m | 8 | 27607002 | CLU | C/T | 0.369 | 0.88 (0.85–0.90) |
| rs11257240n | 10 | 11677075 | ECHDC3 | T/G | 0.350 | 1.07 (1.02–1.12) |
| rs920573o | 11 | 60157486 | MS4A6A | G/A | 0.475 | 0.90 (0.86–0.95) |
| rs3851179 | 11 | 86157598 | PICALM | C/T | 0.429 | 0.88 (0.86–0.90) |
| rs11218343 | 11 | 121564878 | SORL1 | T/C | 0.040 | 0.80 (0.75–0.85) |
| rs17125944p | 14 | 52933911 | FERMT2 | T/C | 0.081 | 1.14 (1.09–1.18) |
| rs10498633q | 14 | 92460608 | SLC24A4 | G/T | 0.182 | 0.92 (0.89–0.94) |
| rs11854073r | 15 | 58680796 | ADAM10 | G/A | 0.308 | 0.93 (0.91–0.95) |
| rs3752246 | 19 | 1056493 | ABCA7 | C/G | 0.187 | 1.15 (1.11–1.18) |
| rs7274581s | 20 | 56443204 | CASS4 | T/C | 0.091 | 0.88 (0.85–0.92) |
GRCh38.
Based on position of top SNP in reference to RefSeq assembly.
1000 Genomes, CEU population.
Reported by Kunkle et al (2019).
proxy rs4844610 (D’=1.0),
proxy rs10933431 (D’=0.84),
proxy rs9271058 (D’=1.0),
proxy rs9473117 (D’=1.0),
proxy rs12539172 (D’=0.98),
Previously the ZCWPW1 locus,
proxy rs10808026 (D’=1.0),
proxy for rs73223431 (D’=1.0),
proxy rs9331896 (D’=1.0),
proxy rs7920721 (D’=0.95),
proxy rs7933202 (D’=0.94),
proxy rs17125924,
proxy rs12881735 (D’=1.0),
proxy rs593742 (D’=0.88),
proxy rs6024870 (D’=0.94).
Table 4.
Associations between individual genetic loci and presence of concomitant AD pathology. Logistic regression coefficients (β), standard error (SE), and nominal p-values are shown. No covariates are included. TREM2 is omitted from analysis due to lack of genetic variation at this SNP in the Training set.
| Locus | β | SE | Nominal p-value |
|---|---|---|---|
| APOE E4 | 1.049 | 0.269 | 0.000 |
| APOE E2 | −0.667 | 0.538 | 0.215 |
| CR1 | 0.308 | 0.242 | 0.203 |
| BIN1 | 0.191 | 0.206 | 0.354 |
| INPP5D | −0.180 | 0.187 | 0.335 |
| HLA-DRB5/DRB1 | 0.068 | 0.229 | 0.768 |
| CD2AP | 0.007 | 0.230 | 0.976 |
| NYAP1a | −0.149 | 0.281 | 0.596 |
| EPHA1 | −0.255 | 0.264 | 0.333 |
| PTK2B | −0.109 | 0.215 | 0.610 |
| CLU | −0.057 | 0.225 | 0.799 |
| ECHDC3 | −0.147 | 0.217 | 0.499 |
| MS4A6A | 0.313 | 0.211 | 0.138 |
| PICALM | 0.009 | 0.212 | 0.967 |
| SORL1 | 1.204 | 0.650 | 0.064 |
| FERMT2 | 0.195 | 0.394 | 0.620 |
| SLC24A4 | −0.022 | 0.302 | 0.942 |
| ADAM10 | 0.321 | 0.253 | 0.204 |
| ABCA7 | 0.040 | 0.252 | 0.875 |
| CASS4 | −0.028 | 0.281 | 0.921 |
Previously the ZCWPW1 locus.
p<0.05.
Development of a Model Predicting Concomitant AD Pathology in PD/DLB Individuals
In a Training set consisting of the first 127 PD/DLB individuals genotyped at Penn, we developed a logistic regression model to predict concomitant AD pathology (defined as intermediate-to-high levels of ADNC). We began by including genotypes at all 20 AD risk SNPs [16, 24, 25] (Table 3), age at disease onset, and sex in the model. We then used backward stepwise regression, with model selection based on the Akaike Information Criterion (AIC). For each model, we also estimated predictive performance by ten-fold cross-validation (100 iterations) within the Training set (Fig. 2a).
Fig 2.

Backward stepwise logistic regression model selection for predicting concomitant Alzheimer’s Disease (AD) pathology in N = 127 cases (Training set) with a clinicopathologic diagnosis of PD or DLB from Penn. Concomitant AD pathology is defined as an AD Neuropathological Change (ADNC) score of Intermediate or High. a Akaike information criterion (AIC, left axis) at each step during model selection and the corresponding area under the receiver operating characteristics curve (AUC, right axis), estimated by ten-fold cross-validation, within the Training set are shown. Initial model included all AD risk SNPs, sex, and age at disease onset as predictors; sequential elimination of predictors and effect on AIC and AUC are shown from left to right. As the Training set cases showed no genetic variability at the TREM2 locus, this locus was not included in the model. b Coefficients (β), standard error (SE), and p-values for the four predictors included in the best model (lowest AIC) for predicting concomitant AD in LBD cases.
Our best model (by AIC) incorporated only four predictors: age at disease onset, number of APOE E4 alleles, and genotype at the BIN1 and SORL1 loci (Fig. 2b). The Hosmer-Lemeshow goodness-of-fit test for this model produced a χ2(8, N = 127) = 7.578, p = 0.4758, indicating fit. The area under the receiver operator curve (AUC) for this model in our Training set data (ten-fold cross-validation) was 0.751 (Fig. 3a), whereas the AUC for a shuffled version of our dataset in which ADNC positive vs. negative status was permuted (null model) was 0.479 (Supplementary Fig. 1, Online resource).
Fig 3.

Performance characteristics of the best model for predicting concomitant Alzheimer’s disease (AD) pathology among Penn cases with a clinicopathological diagnosis of PD or DLB. Receiver operating characteristics (ROC) curves and areas under the curve (AUC) of the final model (with age at onset, number of APOE4 alleles, BIN1 genotype, and SORL1 genotype as predictors) in the Training (a) and Test (b) cohorts are shown. c The Alzheimer’s Disease Neuropathological Change Risk Score (ADNC-RS) calculated from the best logistic regression model is shown for both the Training set and Test set cohorts. Individuals positive for ADNC showed higher average ADNC-RS. d The probability of concomitant AD pathology was calculated from the ADNC Risk Score for each case. Values above 0.5 have a high probability of concomitant AD pathology, while values below 0.5 have a low probability of concomitant AD pathology. The prevalence of concomitant AD pathology at each quintile of ADNC Risk Score in the Training (e) and Test (f) cohorts demonstrates fourfold enrichment for the presence of ADNC for individuals in the top quintile vs. individuals in the first two quintiles of risk. *p<0.05.
Model Performance in Test Set
We applied the best model developed in our Penn-based Training set to a Penn-based Test set of 81 PD/DLB individuals whose data were never used to develop the predictor. Despite differences in the proportion of cases with concomitant AD pathology in the Training set (46%) vs. the Test set (26%), our model performed equally well in the Test set, with an AUC of 0.781 (Fig. 3b).
We additionally performed a subgroup analysis, applying our predictor only in the subset of our 208 Penn cases with a clinicopathological diagnosis of PD or PDD (N=163), which minimally affected the results (AUC = 0.728, Supplementary Fig. 2, Online Resource).
Development of an ADNC Risk Score
In order to develop a clinically-useful tool, we used our logistic regression model to generate a continuous risk score (vs. binary outcome predictor) for concomitant AD pathology (ADNC risk score, or ADNC-RS). An ADNC-RS was calculated for each case using the following formula:
ADNC − RS = −7.97717 + 0.0636(Age at onset) + 1.04327(APOE E4 alleles) + 0.45498(BIN1 risk alleles) + 1.48933(SORL1 risk alleles).
The distribution of ADNC-RS across both the Penn-based Training and Test sets is shown in Fig. 3c. The ADNC-RS was significantly higher for PD/DLB individuals with concomitant AD pathology in both the Training (M 0.241 (SEM 0.129) vs. M −0.622 (SEM 0.111), p<0.001) and Test sets (M 0.378 (SEM 0.139) vs. M −0.470 (SEM 0.113), p<0.001), compared to those without concomitant AD pathology. For each case the ADNC-RS was used to determine the probability of AD co-pathology; the distribution of predicted probability of AD co-pathology is shown in Fig. 3d. We found, importantly, that individuals with ADNC-RS in the highest quintile were four times more likely to have AD pathology than individuals with ADNC-RS in the lowest two quintiles. This enrichment was observed in both the Training set (Fig. 3e) and the Test set (Fig. 3f) individuals.
Validation of the ADNC Risk Score in LBD cases from the National Alzheimer’s Coordinating Center (NACC) Database
Having demonstrated that our logistic regression predictor and its associated ADNC-RS performed well in Penn-based individuals from both our Training and Test sets, we sought to validate this predictor in a national multi-site setting.
The National Alzheimer’s Coordinating Center (NACC) is a national database of clinical and neuropathological data from over 42,000 de-identified individuals across 39 past and present Alzheimer’s Disease Research Centers (ADRCs), as of March 2020. Genetic information for some patients is also available through the Alzheimer’s Disease Genetics Consortium (ADGC). Seventy individuals from 20 non-Penn ADRCs with autopsy-confirmed LBD neuropathology and presumed clinical etiology of LBD were included in this analysis. The mean age at disease onset was 70.49 years (SEM 1.03), and mean age at death was 80.41 years (SEM 0.98). Since many NACC patients are recruited from memory disorder clinics, this group was highly enriched for patients with dementia during life (n=58/70 (82.9%)) and intermediate/high ADNC at autopsy (n=62/70 (88.6%)), compared to the Penn-based PD and DLB cohort. Additional clinical and demographic details are shown in Table 5.
Table 5.
Demographic and Clinical Characteristics of NACC Validation Set. Data represent mean (SEM) unless otherwise noted.
| Characteristic | Whole Dataset | None/Low ADNC | Intermediate/High ADNC | p-value b |
|---|---|---|---|---|
| N | 70 | 8 | 62 | |
| Age at Onset, y | 70.49 (1.03) | 69.50 (3.55) | 70.61 (1.08) | 0.634 |
| Age at Death, y | 80.41 (0.98) | 80.25 (4.03) | 80.44 (0.99) | 0.939 |
| Disease Duration, y | 9.93 (0.54) | 10.75 (1.89) | 9.82 (0.56) | 0.474 |
| Sex, (F/M), N (%) | 23 (32.9)/ 47 (67.1) | 1 (12.5)/ 7 (87.5) | 22 (37.5)/ 40 (62.5) | 0.2572 |
| Race, N (%) White | 70 (100.0) | 8 (100.0) | 62 (100.0) | >0.999 |
| Non-White | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Last MMSEa | 19.34 (1.377) | 27.2 (0.58) | 18.03 (1.47) | 0.013 |
| Dementia Diagnosis During | 12 (17.1)/ | 5 (62.5)/ | 7 (11.3)/ | 0.003 |
| Life, (No/Yes) N (%) | 58 (82.9) | 3 (37.5) | 55 (88.7) | |
| Lewy Body Distribution, N (%) | ||||
| Diffuse or Neocortical | 44 (62.9) | 5 (62.5) | 39 (62.9) | |
| Transitional or Limbic | 10 (14.3) | 2 (25.0) | 8 (12.9) | 0.558 |
| Brainstem Predominant | 4 (5.7) | 1 (12.5) | 3 (4.8) | |
| Amygdala | 11 (15.7) | 0 (0.0) | 11 (17.7) | |
| Olfactory bulb | 1 (1.4) | 0 (0.0) | 1 (1.6) | |
Available data (N = 35).
Comparison between None/Low ADNC and Intermediate/High ADNC groups.
p<0.05.
Despite these differences in prevalence of ADNC, applying our best Penn-derived model to the NACC Validation set resulted in a ROC AUC of 0.754 (Fig. 4a), indicating comparable performance to that seen in our Penn-based Training (0.751) and Test sets (0.781). LBD individuals from the NACC database with AD co-pathology exhibited higher average ADNC-RS than those without AD co-pathology (M 0.552 (SEM 0.109) vs. M −0.179 (SEM 0.244), p=0.018)) (Fig. 4b). Despite the NACC database’s enrichment for patients with ADNC, higher ADNC-RS continued to correlate with a higher prevalence of AD co-pathology (Fig. 4d).
Fig 4.

Performance characteristics of the best model for predicting concomitant Alzheimer’s disease (AD) pathology among non-Penn, NACC cases with neuropathological evidence of Lewy bodies and presumed clinical diagnosis of LBD. a Receiver operating characteristic (ROC) curve and area under the curve (AUC) of the final model (developed in the Penn-based Training set, with age at disease onset, number of APOE4 alleles, BIN1 genotype, and SORL1 genotype as predictors) are shown. b The Alzheimer’s Disease Neuropathological Change Risk Score (ADNC-RS) calculated from the final model is shown for LBD cases from the NACC. Individuals positive for ADNC showed higher average ADNC-RS. c Despite the NACC database’s enrichment for ADNC-positive individuals compared to the Penn-based cases (Training and Test sets combined), the ADNC-RS correlated with prevalence of ADNC in both groups.
DISCUSSION
In this study, we performed an in-depth analysis of 208 PD/DLB cases from Penn in order to determine whether common genetic variants associated with risk for AD by GWAS might predict which individuals would develop concomitant AD pathology. We first demonstrated that concomitant AD pathology is highly prevalent in PD/DLB patients, with over one-third of the Penn cohort exhibiting intermediate-to-high levels of ADNC. We next evaluated a set of 20 common genetic variants found by multiple AD GWAS to associate with risk for AD, examining their association with ADNC in PD/DLB and developing a best-fit logistic regression model predicting the presence of intermediate-to-high ADNC in these primary neuronal synucleinopathies. A best-fit predictor incorporating only age at disease onset and genotype at 3 SNPs achieved moderately high performance (AUC 0.75 – 0.78) in both the Training set in which it was developed and a held-out Test set. From our logistic regression model, we developed a continuous metric, the ADNC-RS, and demonstrated that this simple tool could identify a population of LBD individuals at very high risk for development of concomitant AD pathology. Finally, we applied our logistic regression model and associated ADNC-RS calculator to LBD cases from the national, multi-site NACC database, validating its performance (AUC = 0.754) in a set of 70 cases recruited outside of Penn.
Our findings have clinical implications. Both “proteinopathies” defining ADNC – plaques composed of Aβ and NFT composed of tau – are targetable with drugs in clinical trials now, and, in clinical AD, immunological approaches targeting Aβ have shown enough promise to proceed to Phase III trials [6, 35]. However, within the clinical AD spectrum, the need to identify individuals ever-earlier in the course of pathophysiology [43] in order to see benefit with these therapies has created considerable problems with feasibility, not to mention potential burden to the healthcare system should any of these therapeutics attain FDA approval. These practical issues have been compounded by the fact that genetic risk scores based on AD GWAS-nominated variants achieve only very modest predictive value in the general population, where the absolute prevalence of AD is relatively low [11]. The performance of such genetics-based risk scores may be vastly improved in a population enriched for the presence of AD pathology, however [9].
Patients with primary clinical diagnoses of PD during life (and LBD at autopsy) represent exactly such an AD pathology-enriched population. Indeed, the prevalence of concomitant AD pathology in this group has been reported to range from 38% to 70%, depending on the definition of AD pathology used, and on whether clinical diagnosis of PD or primary pathological diagnoses of LBD is used [38, 42]. Our study corroborates these findings, with ~38% of PD/DLB individuals from Penn demonstrating an intermediate to high degree of ADNC, and only ~20% showing no ADNC. As a consequence, in this enriched population, the logistic regression model developed here achieves an AUC of ~0.781.
More important from a practical perspective, we use the predictors (and associated weights) identified in our model to develop a risk score for ADNC (the ADNC-RS) that can identify those PD/DLB individuals most likely to exhibit ADNC at autopsy. Indeed, in both our Penn-based Training and Test sets, those individuals with ADNC-RS in the top 20% are four times more likely to develop ADNC than LBD individuals with ADNC-RS in the bottom 40%, while in the NACC Validation set, higher ADNC-RS still correlated with higher likelihoods of individuals having ADNC, despite the NACC database’s bias towards individuals with ADNC. Because the ADNC-RS requires knowledge of only the age at disease onset and genotype at 3 AD risk SNPS, it can be easily calculated in most settings using results from a simple blood sample. Thus, the ADNC-RS developed here might serve as a screening step enriching for those PD/DLB individuals who warrant assessment for development of ADNC using more expensive modalities such as Aβ or tau imaging. Moreover, as plasma biomarkers for AD are emerging now [15, 18, 29, 34] future studies incorporating plasma biomarkers with the clinico-genetic predictor described here may further improve accuracy.
How certain can we be of our model and associated risk score? While the definitive answer to this question will lie in future studies investigating other cohorts, several aspects of our current study increase confidence. First, we nominate candidate genetic variants for inclusion in model development in an unbiased manner, starting with all loci reported to associate with risk for AD across two or more major GWAS studies. Second, we use strict criteria that are widely accepted in the field for defining ADNC. Third, in the first two stages of our study, we employ a Training set/Test set design in our analyses, with each group defined by consecutive genotyping of autopsy cases diagnosed with PD or DLB. Such a design guards against over-fitting, and our results confirm that we are not over-fitting the Training set data, since performance in the Test set is as high as in the Training set. Indeed, because completion of our Test set cases followed completion of our Training set cases, these two subgroups had different levels of concomitant AD pathology (46% of cases with concomitant AD pathology in the Training set vs. 26% in the Test set), but the ADNC-RS performed equally well in enriching for individuals with AD co-pathology in both subgroups. Finally, we validated our findings in a multi-site group of LBD individuals recruited outside of Penn, finding that the ADNC-RS performed equally well in a dataset highly enriched with concomitant AD pathology (88.6% of cases).
Limitations of the current study should be considered alongside the previously-mentioned strengths. In particular, although our sample sizes of 208 neuropathologically-characterized PD/DLB cases from Penn and 70 LBD cases from NACC are not small, a larger sample, across multiple centers, would be a valuable addition to the work presented here. In addition, further investigations of the cognitive consequences of ADNC in PD or DLB patients would add clinical depth to our findings. Third, because the focus of this study was neuropathological, we defined our cohort neuropathologically, rather than using a clinicopathological diagnosis of PD. That said, a subset analysis of the 163 individuals in our Penn-based LBD cohort with a clinical diagnosis of PD yielded near-identical results. In the future, however, a clinically-defined study in a PD population, verifying the presence or absence of ADNC by imaging, could extend the current work. Finally, we recognize that the LBD cases in our NACC Validation set may differ clinically from the PD/DLB cases characterized at Penn, because most NACC participants are recruited at memory disorders clinics. That said, we selected for only the NACC LBD cases whose clinical diagnosis was presumed to be LBD (n=70 out of 559 NACC cases with autopsy and genetic data). Moreover, in thinking about the potential clinical uses of our predictor, we are encouraged by its high performance in this Validation set, since heterogeneity is the norm rather than the exception in most clinical contexts.
In addition to the clinical implications discussed above, the biological implications of our study are also worth considering. Specifically, the genetic loci identified in our final model predicting ADNC in LBD individuals were APOE, BIN1, and SORL1. Many functions for APOE have been reported, but a consistent finding over many years is that the APOE E4 allele (included in our predictive model) encodes a form of this protein that binds Aβ less efficiently [46]. BIN1 encodes a protein that functions in beta-secretase 1 trafficking, which in turn can impact the production of Aβ. SORL1 encodes the sortilin-related receptor 1, which also functions in intracellular trafficking, including the sorting of APP to the retromer pathway for degradation or to the endosome-lysosome system, where APP is cleaved to generate Aβ. Collectively, the fact that our best predictive model incorporates these three genetic loci underscores the importance of Aβ production and processing in the development of ADNC among LBD individuals. In addition, direct interaction between BIN1 and tau regulates tau phosphorylation, which may affect the development of AD pathology via a different route [26]. Interestingly, among these three genetic loci, the SORL1 locus exerted the strongest effect in our model, with a coefficient of ~1.5 compared to ~1 for the APOE locus. As the SNP at the SORL1 locus is relatively rare (minor allele frequency of 0.04), the contribution to AUC may be seen in only a small fraction of individuals, however. In contrast, in the general population, among AD common genetic risk, APOE has by far the largest effect size.
In summary, we present our findings from a study of 208 PD/DLB cases at Penn, validated in 70 additional LBD cases from the multi-site NACC database, demonstrating that age at disease onset and genotype at 3 SNPs is sufficient to identify a subset of LBD individuals at very high risk for development of concomitant AD pathology. The development of molecular tools such as the ADNC-RS reported here may in turn be permissive for strategies to target Aβ and tau accumulation in PD and other LBD.
Supplementary Material
ACKNOWLEDGMENTS:
We would like to acknowledge Nick Cullen and Marijan Posavi for their assistance with formulating analyses in this paper. We thank Travis Unger for technical assistance. We additionally thank our patients and their families for their generosity in contributing to this research.
This research was supported by the NIH (RO1 NS115139, P30 AG010124, U19 AG062418) and a Biomarkers Across Neurodegenerative Diseases (BAND) grant from the Michael J. Fox Foundation/Alzheimer’s Association/Weston Institute. Alice Chen-Plotkin is additionally supported by the Parker Family Chair.
The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P30 AG062428–01 (PI James Leverenz, MD) P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P30 AG062421–01 (PI Bradley Hyman, MD, PhD), P30 AG062422–01 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Thomas Wisniewski, MD), P30 AG013854 (PI Robert Vassar, PhD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P30 AG062429–01(PI James Brewer, MD, PhD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG053760 (PI Henry Paulson, MD, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P30 AG049638 (PI Suzanne Craft, PhD), P50 AG005136 (PI Thomas Grabowski, MD), P30 AG062715–01 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), P50 AG047270 (PI Stephen Strittmatter, MD, PhD).
The Alzheimer’s Disease Genetic Consortium (ADGC) supported collection and genotyping of samples used in this study through National Institute on Aging (NIA) grants U01AG032984 and RC2AG036528.
Samples from the National Centralized Repository for Alzheimer’s Disease and Related Dementias (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Arnold SE, Toledo JB, Appleby DH, Xie SX, Wang L-S, Baek Y, et al. (2013) Comparative survey of the topographical distribution of signature molecular lesions in major neurodegenerative diseases. J Comp Neurol 521:4339–55. doi: 10.1002/cne.23430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes J, Dickerson B, Frost C, Dipstat MA, Jiskoot LC, Wolk D, et al. (2015) Alzheimer’s disease first symptoms are age dependent: evidence from the NACC dataset. Alzheimers Dement 11:1349–1357. doi: 10.1016/j.jalz.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beekly DL, Ramos EM, Van Belle G, Deitrich W, Clark AD, Jacka ME, et al. (2004) The National Alzheimer’ s Coordinating Center (NACC) Database: An Alzheimer Disease Database. Alzheimer Dis Assoc Disord 18:270–277 [PubMed] [Google Scholar]
- 4.Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, Escott-Price V, et al. (2014) Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet. doi: 10.1093/hmg/ddu334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, Laferla FM (2010) Neurobiology of Disease Synergistic Interactions between A, Tau, and-Synuclein: Acceleration of Neuropathology and Cognitive Decline. doi: 10.1523/JNEUROSCI.0490-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Congdon EE, Sigurdsson EM (2018) Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Deerlin VM, Sleiman PMA, Martinez-Lage M, Chen-Plotkin A, Wang L-S, Graff-Radford NR, et al. (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42:234–239. doi: 10.1038/ng.536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobbin KK, Simon RM (2011) Optimally splitting cases for training and testing high dimensional classifiers. BMC Med Genomics. doi: 10.1186/1755-8794-4-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Escott-Price V, Sims R, Bannister C, Harold D, Vronskaya M, Majounie E, et al. (2015) Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain. doi: 10.1093/brain/awv268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guttmacher AE, Collins FS, Nussbaum RL, Ellis CE (2003) Alzheimer’s Disease and Parkinson’s Disease. N Engl J Med 348:1356–1364. doi: 10.1056/NEJM2003ra020003 [DOI] [PubMed] [Google Scholar]
- 11.Harris SE, Davies G, Luciano M, Payton A, Fox HC, Haggarty P, et al. (2014) Polygenic risk for alzheimer’s disease is not associated with cognitive ability or cognitive aging in non-demented older people. J Alzheimer’s Dis. doi: 10.3233/JAD-131058 [DOI] [PubMed] [Google Scholar]
- 12.Hosmer DW, Lemeshow S (1980) Goodness of fit tests for the multiple logistic regression model. Commun Stat - Theory Methods. doi: 10.1080/03610928008827941 [DOI] [PubMed] [Google Scholar]
- 13.Huang YWA, Zhou B, Wernig M, Südhof TC (2017) ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell. doi: 10.1016/j.cell.2016.12.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irwin DJ, Hurtig HI (2018) The Contribution of Tau, Amyloid-Beta and Alpha-Synuclein Pathology to Dementia in Lewy Body Disorders. J Alzheimers Dis Park 8. doi: 10.4172/2161-0460.1000444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janelidze S, Mattsson N, Palmqvist S, Smith R, Beach TG, Serrano GE, et al. (2020) Plasma P-tau181 in Alzheimer’s disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat Med. doi: 10.1038/s41591-020-0755-1 [DOI] [PubMed] [Google Scholar]
- 16.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. (2019) Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51:404–413. doi: 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jellinger KA (2014) Neuropathology of multiple system atrophy: New thoughts about pathogenesis. Mov Disord. doi: 10.1002/mds.26052 [DOI] [PubMed] [Google Scholar]
- 18.Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, et al. (2020) Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: a diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. doi: 10.1016/S1474-4422(20)30071-5 [DOI] [PubMed] [Google Scholar]
- 19.Kövari E, Horvath J, Bouras C (2009) Neuropathology of Lewy body disorders. Brain Res. Bull. 80:203–210 [DOI] [PubMed] [Google Scholar]
- 20.Kuhn M (2008) Building predictive models in R using the caret package. J Stat Softw 28. doi: 10.18637/jss.v028.i05 [DOI] [Google Scholar]
- 21.Kukull W, Bigio E, Cairns N, Kofler J, Montine T, Nelson P SJ (2015) Neuropathology Data Set Coding Guidebook. In: Natl. Alzheimer’s Coord. Cent https://www.alz.washington.edu/NONMEMBER/NP/npguide10.pdf [Google Scholar]
- 22.Kukull WA (2015) NACC Uniform Dataset Researchers Data Dictionary. In: Natl. Alzheimer’s Coord. Cent https://www.alz.washington.edu/WEB/rdds_uds.pdf [Google Scholar]
- 23.Kukull WA (2015) Researchers Data Dictionary Genetic Data. In: Natl. Alzheimer’s Coord. Cent. https://www.alz.washington.edu/WEB/rdd_gen.pdf [Google Scholar]
- 24.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51:414–430. doi: 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45:1452–1458. doi: 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lasorsa A, Malki I, Cantrelle FX, Merzougui H, Boll E, Lambert JC, et al. (2018) Structural basis of tau interaction with BIN1 and regulation by tau phosphorylation. Front Mol Neurosci. doi: 10.3389/fnmol.2018.00421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marras C, Beck JC, Bower JH, Roberts E, Ritz B, Ross GW, et al. (2018) Prevalence of Parkinson’s disease across North America. npj Park Dis. doi: 10.1038/s41531-018-0058-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mata IF, Samii A, Schneer SH, Roberts JW, Griffith A, Leis BC, et al. (2008) Glucocerebrosidase gene mutations: A risk factor for Lewy body disorders. Arch Neurol. doi: 10.1001/archneurol.2007.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattsson N, Andreasson U, Zetterberg H, Blennow K, Weiner MW, Aisen P, et al. (2017) Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. doi: 10.1001/jamaneurol.2016.6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Max Kuhn Contributions from Jed Wing A, Weston S, Williams A, Keefer C, Engelhardt A, Cooper T, et al. (2019) Package “caret”: Classification and Regression Training. https://cran.r-project.org/web/packages/caret/caret.pdf [Google Scholar]
- 31.McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, et al. (2017) Diagnosis and management of dementia with Lewy bodies. Neurology 89:88–100. doi: 10.1212/WNL.0000000000004058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach NIH Public Access. Acta Neuropathol 123:1–11. doi: 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Naj AC, Jun G, Beecham GW, Wang L, Vardarajan BN, Buros J, et al. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33, and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43:436–441. doi: 10.1038/ng.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Doré V, et al. (2018) High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature. doi: 10.1038/nature25456 [DOI] [PubMed] [Google Scholar]
- 35.Panza F, Lozupone M, Logroscino G, Imbimbo BP (2019) A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 15:73–78. doi: 10.1038/s41582-018-0116-6 [DOI] [PubMed] [Google Scholar]
- 36.Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. (2015) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30:1591–1601. doi: 10.1002/mds.26424 [DOI] [PubMed] [Google Scholar]
- 37.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. (2011) pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12. doi: 10.1186/1471-2105-12-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. (2018) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141:2181–2193. doi: 10.1093/brain/awy146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. doi: 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sing T, Sander O, Beerenwinkel N, Lengauer T (2015) Package “ROCR”: Visualizing the Performance of Scoring Classifiers [DOI] [PubMed] [Google Scholar]
- 41.Slowikowski K (2019) Proxysnps: Get proxy SNPs for a SNP in the 1000 Genomes Project. In: Github. https://github.com/slowkow/proxysnps [Google Scholar]
- 42.Smith C, Malek N, Grosset K, Cullen B, Gentleman S, Grosset DG (2019) Neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. J Neurol Neurosurg Psychiatry. doi: 10.1136/jnnp-2019-321111 [DOI] [PubMed] [Google Scholar]
- 43.Sperling R, Mormino E, Johnson K (2014) The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron. doi: 10.1016/j.neuron.2014.10.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spires-Jones TL, Attems J, Dietmar ·, Thal R (2017) Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol 134:187–205. doi: 10.1007/s00401-017-1709-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Toledo JB, Van Deerlin VM, Lee EB, Suh E, Baek Y, Robinson JL, et al. (2014) A platform for discovery: The University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimer’s Dement 10:477–484.e1. doi: 10.1016/j.jalz.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G (2019) Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol 15:501–518. doi: 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.