

We carried out a genome-wide association study in Latin Americans and identified novel face morphology loci..
Abstract
To characterize the genetic basis of facial features in Latin Americans, we performed a genome-wide association study (GWAS) of more than 6000 individuals using 59 landmark-based measurements from two-dimensional profile photographs and ~9,000,000 genotyped or imputed single-nucleotide polymorphisms. We detected significant association of 32 traits with at least 1 (and up to 6) of 32 different genomic regions, more than doubling the number of robustly associated face morphology loci reported until now (from 11 to 23). These GWAS hits are strongly enriched in regulatory sequences active specifically during craniofacial development. The associated region in 1p12 includes a tract of archaic adaptive introgression, with a Denisovan haplotype common in Native Americans affecting particularly lip thickness. Among the nine previously unidentified face morphology loci we identified is the VPS13B gene region, and we show that variants in this region also affect midfacial morphology in mice.
INTRODUCTION
Because of its evolutionary, biomedical, and forensic significance, there is an increasing interest in the characterization of the genetic basis of human facial variation. Thus far, nine genome-wide association (GWA) studies (GWASs) have reported ~50 genetic loci associated with facial features in humans (1–12), although only about a dozen of these loci show significant association signals across independent studies, representing the most robust findings so far. Other than stochastic variation, the limited overlap of findings from independent GWASs could relate to various methodological and biological factors, including a different genetic architecture of facial variation across human populations.
We recently initiated a research program focused on the genetic basis of physical appearance in a sample of ~7000 individuals recruited in Latin America the Consortium for the Analysis of the Diversity and Evolution of Latin America (CANDELA cohort) (13). Our study sample is mostly of mixed European-Native American ancestry and shows extensive phenotypic variation and high genetic diversity (13, 14). This sample has high power for the genetic analysis of physical appearance in a considerably understudied population (4, 15–17). In a previous analysis of 14 facial features (characterized mainly using a discrete, categorical scoring approach), we reported genome-wide significant associations with single-nucleotide polymorphisms (SNPs) at six genomic loci (4). These loci harbor candidate genes known to be involved in mammalian craniofacial development/evolution, and four have now been replicated in other study samples, using a range of phenotyping approaches (10, 18).
From an evolutionary perspective, facial projection shows extensive variation across extinct and extant mammals, with aspects of brow-ridge, midface, and mandible projection being defining features of primate and hominid taxa (19). Such changes in facial projection have often been proposed to result from the action of selection, including various environmental adaptations, and possibly (particularly in hominids) social factors (20). In modern humans, the facial profile varies greatly between individuals and populations and has been shown to be highly informative in various perception domains, including gender and age attribution, individual recognition, and attractiveness (21).
Here, we present results stemming from GWA analyses of the facial profile in the CANDELA cohort. We evaluated 59 quantitative traits (i.e., landmark-based measurements) and found significant associations at 32 genomic regions (9 previously unidentified, 4 of which replicate in a European meta-analysis). We provide robust evidence of association for 23 loci that have been implicated by previous GWASs, more than doubling the number of well-validated face morphology loci. We find that associated SNPs are strongly enriched in enhancers active during the late organogenesis phase of human development, including many that are specifically active during craniofacial development. In the region of association in 1p12 (overlapping WARS2/TBX15), we find a Denisovan haplotype affecting face morphology, particularly lip thickness. Among the four previously unidentified face morphology loci that replicate in Europeans is VPS13B (associated with nose shape), and we demonstrate that variants of the homologous region in the mouse (Vps13b) also influence nasal morphology.
RESULTS
Study sample and phenotyping
We examined individuals from the CANDELA study cohort, collected in five Latin American countries (13). Two operators placed 19 landmarks and 22 semi-landmarks (fig. S1), primarily along the midfacial contour, on right profile two-dimensional (2D) photographs of 6169 subjects (3408 women and 2761 men). After Procrustes superposition, we defined 59 measurements (distances, ratios, and angles) based on the landmarks/semi-landmarks. These measurements quantify aspects of the morphology of the upper, middle, and lower face, with similar measurements having been examined in a range of morphological studies of the human face, including genetic, medical, anthropological, and forensic applications (Fig. 1 and table S1). We found these quantitative traits to be reliably assessed (see Methods and table S2), approximately normally distributed and with extensive variation in the CANDELA sample. The individuals studied were genotyped on Illumina’s OmniExpress BeadChip. After quality control filters, we retained 671,038 SNPs for further analyses.
Fig. 1. Face profile features showing genome-wide significant association.
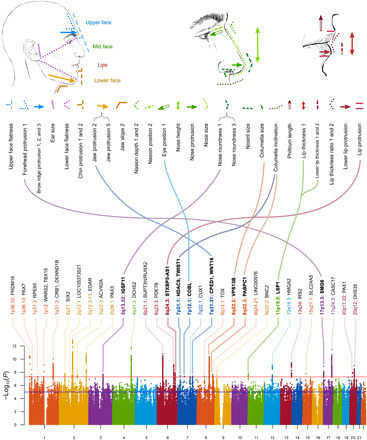
Top: Drawings indicate the features for which the 32 traits listed below were measured in the CANDELA individuals (as defined in table S1). Bottom: Aggregate of the GWAS signals detected (across all traits). The chromosomal region (and nearest candidate gene) showing strongest association to a trait is indicated above each GWAS peak (bold type marks the previously unidentified face morphology regions identified here). Curved lines in the middle of the figure connect the previously unidentified face morphology region to their associated traits.
Other than the high correlation (r > 0.8) seen for similar traits from the same anatomical structure (e.g., nose roundedness and lip thickness), most traits were found to have a low to moderate correlation (table S3). On the basis of genome-wide SNP data, average admixture proportions in the CANDELA individuals examined were estimated as 51% European, 45% Native American, and 4% sub-Saharan African. Most traits showed a low to moderate (but generally significant) correlation with age, body mass index (BMI), sex, and continental genetic ancestry (table S4). The strongest effects for age, BMI, and sex were seen, respectively, for measures of lip thickness (r ~ 0.33), traits affected by neck thickness (r ~ 0.49), and brow-ridge protrusion (r ~ 0.51). The strongest effect of Native American/European ancestry estimates [themselves strongly negatively correlated (13)] was for a measure of nasion position (r ~ 0.38). In addition, several measures of nose/chin protrusion and lip thickness had correlations ~0.20 with Native American/European ancestry. Subcontinental genetic ancestry (North Europe v. Iberia; Central Andes v. Southern South America) also correlated significantly with certain traits, particularly of the midface (table S4), consistent with previous observations (14).
On the basis of a kinship matrix derived from the SNP data, we estimated narrow-sense trait heritability (h2) using LDAK5 (22). We found moderate (but mostly highly significant) heritability values for all traits, ranging from 0.20 to 0.61 (table S3), consistent with independent estimates (2, 18). The highest heritability (>0.60) was observed for measures of brow-ridge, nose, and lip protrusion. Genetic correlations among traits were approximately proportional to phenotypic correlations (except for traits with low h2), suggesting a similar role for genetic and environmental effects across traits (table S3).
GWA analyses
We performed imputation using 1000 Genomes Phase 3 data (23), which allowed us to include up to 8,703,729 autosomal SNPs in the GWA analyses. For association testing, we used multiple linear regression, as implemented in PLINK (24), with an additive genetic model adjusting for age, sex, BMI, landmarking operator, and the first six principal components (PCs) computed from the SNP data. A total of 32 genomic regions (including 2684 SNPs) showed genome-wide significant association (P value < 5 × 10−8) with at least one trait (and up to six) (Figs. 1 and 2, Table 1, and table S5). These regions often also showed association with additional traits at a genome-wide suggestive significance threshold (P value < 10−5; Fig. 2). Conversely, of the 59 traits examined, 32 showed genome-wide significant association with at least one genomic region (and up to five regions), as well as genome-wide suggestive association with regions significantly associated with other traits (Fig. 2). Index SNPs (i.e., the one with the smallest P value in a region) at loci showing strongest association with a trait explain 0.4 to 0.9% of trait variance (median = 0.52%), in line with previous estimates of genetic effects in GWASs of facial variation (1, 15, 18).
Fig. 2. Thirty-two chromosomal regions associated with face profile traits.
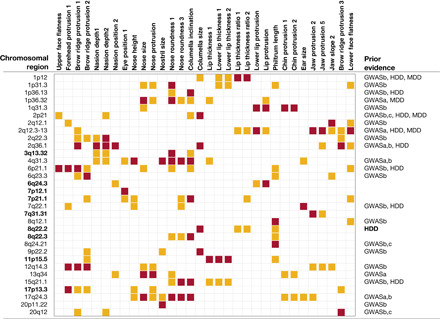
The 32 traits showing genome-wide significant association are listed at the top (Fig. 1 and table S1). For ease of visualization, association P values are represented by colored boxes as follows: red, genome-wide significant (<5 × 10−8); orange, genome-wide suggestive (<10−5) (P values are provided in table S5). Chromosomal regions in bold were previously unidentified (Fig. 1 and Table 1). Prior evidence: GWASa, associated with a similar face trait; GWASb, associated with a different facial trait; GWASc, associated with a facial development disorder [e.g., nonsyndromic cleft lip/palate (NSCL/P)]; HDD, human developmental defect reported (e.g., Mendelian disorder); MDD, model developmental defect (related phenotype involving a homologous region reported in animal models).
Table 1. Features of the nine chromosomal regions newly associated with face profile traits in the CANDELA sample.
Candidate genes where the index SNP is intragenic are underlined. Association P values in bold are genome-wide significant (P ≤ 5 × 10−8). Rep., European meta-analysis; AFR, sub-Saharan African; EUR, European (from 1000 genomes); NAM, Native American (14); CAN, CANDELA. “# SNPs” refers to the total number of genome-wide significant SNPs in that chromosomal region (P value ≤ 5 × 10−8).
|
Chromosomal region |
Nearest candidate gene |
Index SNP | Association P value |
Rep. P value |
Minor allele frequency |
# SNPs |
||||||||||||
|
Forehead protrusion 1 |
Eye position 1 |
Nose roundness 1 |
Columella inclination |
Columella size |
Lip thickness 1 |
Lower lip thickness 1 |
Lower lip thickness 2 |
Lip protrusion |
Jaw protrusion 2 |
AFR | EUR | NAM | CAN | |||||
| 3q13.32 | IGSF11 | rs7633584 | 7.3 × 10−3 | 5.2 × 10−1 | 1.8 × 10−8 | 4.4 × 10−6 | 4.2 × 10−1 | 1.6 × 10−1 | 3.8 × 10−1 | 2.0 × 10−1 | 8.2 × 10−2 | 2.6 × 10−1 | 2.4 × 10−4 | 0.29 | 0.05 | 0.39 | 0.22 | 11 |
| 6q24.3 | STXBP5-AS1 | rs6570789 | 9.8 × 10−1 | 8.0 × 10−1 | 2.0 × 10−1 | 2.2 × 10−1 | 4.1 × 10−2 | 7.9 × 10−2 | 1.2 × 10−2 | 4.2 × 10−2 | 1.2 × 10−8 | 3.3 × 10−3 | 9.2 × 10−3 | 0.40 | 0.55 | 0.92 | 0.68 | 11 |
| 7p12.1 | INTERGENIC (COBL) | rs17134499 | 6.2 × 10−1 | 4.1 × 10−8 | 9.9 × 10−1 | 6.4 × 10−1 | 1.3 × 10−1 | 5.6 × 10−1 | 6.5 × 10−1 | 6.6 × 10−1 | 6.3 × 10−1 | 9.8 × 10−1 | 6.6 × 10−2 | 0.32 | 0.02 | 0.03 | 0.03 | 3 |
| 7p21.1 | HDAC9 | rs1178103 | 9.4 × 10−1 | 1.1 × 10−4 | 2.4 × 10−4 | 3.3 × 10−8 | 1.2 × 10−4 | 1.9 × 10−2 | 4.1 × 10−3 | 7.4 × 10−3 | 3.0 × 10−3 | 2.6 × 10−1 | 4.9 × 10−4 | 0.13 | 0.18 | 0.38 | 0.26 | 1 |
| 7q31.31 | CPED1, WNT16 | rs6950680 | 4.9 × 10−4 | 7.2 × 10−1 | 9.8 × 10−1 | 5.6 × 10−1 | 2.1 × 10−1 | 6.9 × 10−1 | 4.7 × 10−1 | 3.4 × 10−1 | 8.2 × 10−1 | 3.6 × 10−8 | 2.0 × 10−4 | 0.21 | 0.36 | 0.04 | 0.21 | 2 |
| 8q22.2 | VPS13B | rs75976055 | 5.7 × 10−2 | 8.3 × 10−1 | 8.8 × 10−2 | 3.0 × 10−2 | 4.2 × 10−11 | 8.3 × 10−1 | 2.8 × 10−2 | 9.3 × 10−2 | 3.7 × 10−2 | 3.0 × 10−1 | 5.0 × 10−5 | 0.08 | 0.13 | 0.31 | 0.38 | 598 |
| 8q22.3 | PABPC1 | 8:101737776:AT:ATT | 1.0 × 10−2 | 8.5 × 10−1 | 4.9 × 10−7 | 1.1 × 10−8 | 3.6 × 10−3 | 1.7 × 10−1 | 6.9 × 10−1 | 9.6 × 10−1 | 3.9 × 10−1 | 1.3 × 10−1 | – | 0.23 | 0.00 | 0.00 | 0.02 | 17 |
| 11p15.5 | LSP1 | rs907613 | 4.9 × 10−1 | 3.7 × 10−1 | 7.7 × 10−1 | 5.4 × 10−1 | 7.1 × 10−1 | 4.2 × 10−8 | 4.0 × 10−8 | 1.4 × 10−8 | 4.9 × 10−1 | 9.8 × 10−1 | 4.0 × 10−2 | 0.09 | 0.33 | 0.41 | 0.39 | 8 |
| 17p13.3 | SMG6 | rs8069947 | 1.2 × 10−9 | 1.8 × 10−1 | 2.2 × 10−1 | 3.8 × 10−1 | 6.8 × 10−1 | 2.7 × 10−2 | 2.6 × 10−2 | 5.0 × 10−2 | 4.2 × 10−1 | 7.1 × 10−1 | 2.3 × 10−2 | 0.26 | 0.49 | 0.59 | 0.54 | 41 |
To account for the number of traits and SNPs tested, we also calculated adjusted genome-wide significance thresholds following a global false discovery rate (FDR) procedure (25), at two overall significance levels (α = 1% and α = 5%). The commonly used threshold of 5 × 10−8 was slightly more stringent than the calculated FDR thresholds (see Methods); therefore, the associations identified as significant above remain significant after FDR adjustment for multiple testing.
Integration of face profile GWAS hits with the literature
For 24 of the 32 regions showing significant trait associations, there is substantial literature supporting an involvement in craniofacial morphology (summarized in Fig. 2 and table S5). This evidence is of five broad types, with certain loci being implicated by more than one line of evidence: (i) Some hits represent direct replications of previous GWAS findings. That is, the same (or a similar) facial trait has been associated with the same genomic region in previous studies. The most prominent example is nasion position (measured in various ways), which has been repeatedly associated with SNPs in the PAX3 region on 2q35 (1, 2, 10, 15, 18). Here, we replicate this finding in that we observe an association of this region with measures sensitive to nasion position (table S1). (ii) Some of the genome regions showing association have been detected in previous face morphology GWAS but for traits other than those associated here, suggesting an effect of these regions on various aspects of facial morphology. A prime example is the WARS2/TBX15 gene region in 1p12. We initially reported association of SNPs in this region with rolling of the helix (in the outer ear) (15); this region was subsequently associated with midface morphology and frontal bossing (10, 18), and in the current study, we find it associated with two measures of lip thickness (traits 35 and 36; Fig. 2 and table S1). (iii) Certain genomic regions identified here have been implicated by previous GWASs of craniofacial anomalies, particularly nonsyndromic cleft lip/palate (NSCL/P). For instance, SNPs in 8q24.21, downstream of MYC, have been strongly associated with NSCL/P (26), and here we find this region associated with philtrum length, consistent with genes in the region being involved in the development of this part of the midface. (iv) For some of the associated chromosomal regions highlighted here, there is evidence in the literature of their involvement in complex craniofacial abnormalities, including Mendelian disorders. For instance, the 8q22.2 region associated here with columella size (trait 27) has been involved in Cohen syndrome (OMIM #216550). Last, (v) some of the regions associated here have been previously implicated in craniofacial development in animal models. For instance, the Tabby mouse mutant (15) presents a range of craniofacial abnormalities caused by a duplication involving the Tbx15 gene in the 1p12 homologous region, and here we found this locus to be associated with lower lip thickness and lip thickness ratio. In Supplementary Notes, we provide comments on the 23 genomic regions detected here for which GWAS evidence of involvement in with nonpathological craniofacial variation has been reported previously.
Regulatory annotation enrichment analysis
Index SNPs at the GWAS hits identified here occur primarily in noncoding regions (table S5). We therefore evaluated the potential enrichment of SNPs in these regions within enhancers identified by chromatin 25-state annotations in 150 human tissues and cell types across embryonic, fetal, and adult time points (including multiple stages of human craniofacial development) (27). The index SNPs we identified are not necessarily causal and are often in strong linkage disequilibirium (LD) with other SNPs within a region (e.g., Figs. 3 and 4). We therefore identified proxy variants in LD with the index variant (r2 > 0.8) up to 1 Mb away. We then used the program GREGOR (28) to evaluate the enrichment of these SNPs (relative to an equal number of randomly selected SNPs) in regulatory annotations for each of the tissues and cell types mentioned above. We found a ~2-fold and ~4-fold enrichment of SNPs at face GWAS hits in all craniofacial enhancers and in craniofacial-specific enhancers, respectively (Bonferroni-corrected P values < 10−50) (fig. S2). Craniofacial-specific enhancers have not been detected in any other human tissues or stages of development and represent a small fraction of all genome annotations (i.e., ~8% of all craniofacial enhancers) (27). The observed enrichment of SNPs in the craniofacial-specific enhancers suggests that these regions play a regulatory role specific to facial development rather than being broadly involved in skeletal biology or embryonic development. Contrasting samples from early and late embryonic stages, we observe that samples from the later stages have the highest (~2-fold; Bonferroni-corrected P value < 10−20) enrichment (fig. S2). This supports previous analyses suggesting that gene expression regulation in late embryonic and early fetal development has the greatest impact on nonpathological adult face morphology (18, 27). Below, we comment on specific regulatory elements present in selected genome regions associated with face morphology.
Fig. 3. Evidence of association, Denisovan introgression, and selection in the WARS2-TBX15 region.
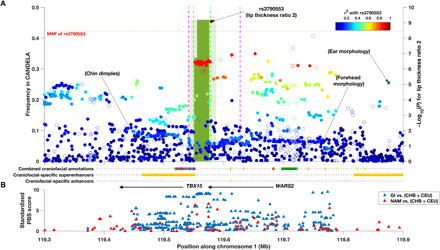
(A) Association P values (right y-axis) with lip thickness ratio 2 and the location and frequency (left y-axis) of introgressed haplotypes inferred in the CANDELA data. Filled dots indicate the SNPs retained for introgression analysis. Dots are plotted accordingly to the significance of their association and have been colored to reflect r2 with the index SNP for association with that trait (rs3790553). The location of the index SNPs associated previously with ear morphology (15), chin dimples (7), and forehead shape (10) is indicated. SNPs associated with facial features in the European meta-analysis (18) are outside the region shown here. The green bins show the introgressed region, and their height displays the allelic frequency and their shade refers to the confidence level at which the tract was called (pale, 20%; dark, 90%). The dashed lines denote the bounds of the Denisovan introgression region previously estimated with the same approach used here (green) (31) or using a Conditional Random Field approach (purple) (32). (B) Standardized PBS scores obtained for genotyped SNPs when contrasting Native Americans (NAM) to CHB + CEU (red triangles) or Greenland Inuits (GI) to CHB + CEU (blue triangles). The location of the TBX15 and WARS2 genes is indicated by black arrows along with various annotations inferred from human embryonic tissue (27) (the color key for combined craniofacial annotations can be found in table S6).
Fig. 4. Features of four genomic regions newly associated with face features in the CANDELA sample and replicating in Europeans.
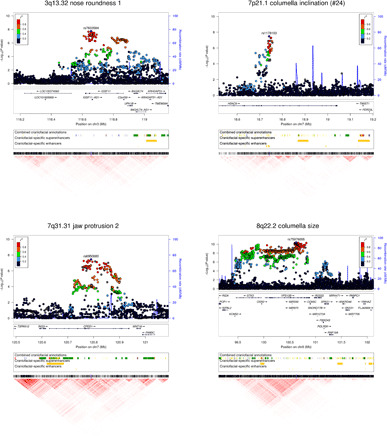
Regional association P values (index SNP is labeled) are shown at the top. Regulatory annotations from human embryonic tissue (27) (combined chromatin states at various Carnegie stages and craniofacial-specific enhancers and superenhancers; the color key for combined craniofacial annotations is in table S6) are provided below each association plot. A heatmap of pairwise LD (r2) in the region is shown at the bottom of each panel. Similar plots for the other previously unidentified regions, detected in the CANDELA sample but not replicating in the European meta-analysis, are shown in Supplementary Notes.
Association in 1p12 involves SNPs in a region with evidence of adaptive introgression from archaic humans
SNPs in 1p12 overlapping the WARS2/TBX15 gene region were associated here with two measures of lip thickness ratio [Figs. 1 and 2; lip thickness ratio referring to the thickness of the upper lip relative to total lip thickness: upper lip thickness/(upper + lower lip thickness); see table S1]. This genomic region has been previously reported to be associated with features of the outer ear (15) and face (7, 10, 18), but this is the first time it is associated with lip thickness. SNPs in 1p12 have also been implicated in GWASs of anthropometric traits and fat distribution (29). The WARS2/TBX15 gene region has been reported to show a strong signal of selection in Greenland Inuit, as evidenced by extreme Population Branch Statistics (PBS) values (30). This observation has been interpreted as resulting from adaptation to a low environmental temperature (30). Furthermore, the selection signal in this region was shown to overlap with a tract of introgression from archaic humans, most likely Denisovans (31), pointing to the possibility of adaptive introgression.
We evaluated evidence for Denisovan introgression in the WARS2/TBX15 gene region in the CANDELA data using a Hidden Markov Model approach (31). We inferred two overlapping introgressed tracts called at confidence levels 20 to 99%, with decreasing size (Fig. 3 and fig. S3). At 99% confidence, the largest and most frequent (34%) introgression tract inferred in the CANDELA data almost exactly matches the 28-kb tract previously inferred in Greenlandic Inuits and various Asian and Native American populations (31). This tract is included within a longer tract of introgression reported previously using an alternative (Conditional Random Field) approach (Fig. 3) (32). We also estimated PBS values in Native American populations related to the ancestral populations contributing to admixture in Latin Americans (14). As previously observed in Greenlandic Inuit (30), we found extreme PBS values in that region, peaking in the tract of Denisovan introgression (Fig. 3). Notably, the SNPs showing significant association with lip thickness in the CANDELA data overlap with the signal of selection and the tract of Denisovan introgression (Fig. 3). The index SNP (rs3790553) is included in the introgressed haplotype inferred at 50% confidence. By contrast, SNPs previously associated with ear morphology (15) and facial features (10, 18) are located over 90 kb away from the tract of Denisovan introgression and have an association r2 with this tract < 0.23 (Fig. 3). Furthermore, the derived allele at rs3790553 is carried by 91% of the introgressed haplotypes, while the ancestral allele of that SNP is carried by 99.7% of the chromosomes in which the introgression tract was not inferred. This level of association was not seen for any of the index SNPs reported in previous GWAS implicating the WARS2/TBX15 region. We examined the effect of the Denisovan haplotype on the profile traits evaluated in the CANDELA sample and found five significant associations (P values < 10−3; fig. S4). Among these, a positive effect is seen on lip thickness ratio and upper lip protrusion, while a negative effect is seen on lower lip thickness and protrusion. There is also a positive effect (albeit not significant) on upper lip thickness. These opposite effects of the Denisovan haplotype on the upper v. lower lips effectively add up in the lip thickness ratio measures, consistent with them showing strongest associations.
Features of loci not significantly associated in previous face morphology GWASs
As shown in Fig. 2, among the 32 genomic regions showing association, 9 have not been genome-wide significant in previous GWASs of nonpathological facial features. We sought replication of the index SNPs from these nine regions in a recent facial morphology GWA meta-analysis that included data for over 10,000 Europeans (18). Traits examined in the meta-analysis consist of pairwise distances between 13 landmarks placed on 3D images. Although we placed in the CANDELA 2D photos the same nine right-profile landmarks used in the European meta-analysis, only 3 of the 59 measurements we obtained correspond to distances included in the European meta-analysis. Considering this limited overlap, we examined association P values in the full set of 36 right-profile distances measured in the European meta-analysis (Table 1). One of our nine index SNPs is not polymorphic in Europeans; therefore, we could not evaluate replication for this SNP. Of the eight index SNPs that are polymorphic in Europeans, four showed replication P values exceeding a Bonferroni-corrected threshold for significance for at least one facial distance (Table 1). Although most of the distances with significant association in the European meta-analysis do not correspond to those showing association in the CANDELA data, they often share landmarks. For example, SNP rs75976055 is associated here with a measure of columella size incorporating the Subnasale landmark (table S1), and the strongest replication P value (5 × 10−5) was observed in the European meta-analysis for the distance between the Subnasale and Labial inferius landmarks.
Figure 4 shows regional association plots for the four previously unidentified facial morphology loci with evidence of replication in the European meta-analysis. In 3q13.32, SNPs in the IGSF11 (immunoglobulin superfamily member 11) region show significant association with nose roundness (and suggestive association with columella inclination). The index SNP in this region (rs7633584) is located within an intron of IGSF11. IGSF11 is a cell adhesion molecule playing a role in osteoclast differentiation and bone resorption (33), but no association with morphological features has been reported previously.
In 7p21.1, an SNP significantly associated with columella inclination is intronic to HDAC9 (Histone deacetylase 9). HDAC9 is an enzyme involved in gene expression regulation through histone acetylation. SNPs in this gene region have been suggestively associated with the vertical position of the sublabial sulcus, relative to the central midface (5). The region of association identified here overlaps with two craniofacial-specific enhancers (Fig. 4). Although HDAC9 does not appear to be expressed in developing mouse craniofacial tissues, many enhancers located within HDAC9 have been shown to form long-range interactions with the neighboring TWIST1 gene during the development of limbs and pharyngeal arches in mice (34). TWIST1 is a transcription factor important in limb and craniofacial development (34), and mutations of TWIST1 have been shown to result in Saethre-Chotzen syndrome, defined by its facial and cranial gestalt (OMIM #101400). These observations suggest that the effect of SNPs showing association with profile traits in the CANDELA individuals could be mediated through regulatory elements within HDAC9 controlling TWIST1 expression during craniofacial development.
SNPs in 7q31.31 associated with jaw protrusion overlap CPED1 (Cadherin-like and PC-esterase domain containing 1), a gene of unknown function. The index SNP in this region (rs6950680) is intronic to CPED1, with a number of regulatory elements active in embryonic craniofacial tissue being present within CEPD1 (Fig. 4). Variants in this region have been associated with height (35) and bone mineral density (36). Of potential relevance, the WINT16 gene is located less than 30 kb downstream of CEPD1. WINT signaling, in concert with other morphogenetic signaling pathways, plays critical roles in embryonic development, including midfacial morphogenesis, and has been implicated in orofacial clefting (37).
Last, in 8q22.2, we find that 597 SNPs overlapping VPS13B (vacuolar protein sorting 13 homolog B) are significantly associated with columella size. Mutations in VPS13B have been reported to cause Cohen syndrome (OMIM #216550), a recessive multisystem disorder with characteristic facial dysmorphism that can include a high nasal bridge, prominent nose, short philtrum, and malar hypoplasia. Below, we describe mouse follow-up analyses we carried out for this gene region.
The five loci with no evidence of replication in the European meta-analysis include genes and regulatory features of potential relevance for craniofacial development. These are discussed in Supplementary Notes.
Mouse follow-up of the VPS13B hit
Since nose development and structure in humans and mice share features and as soft tissues constituting the nose are tightly integrated with the nasal aperture, we conducted geometric morphometric analyses of the facial region of mice skulls to evaluate the potential impact of Vps13b variants. We first carried out geometric morphometric analyses in a recently described mouse Vps13b-KO model of Cohen syndrome (38), compared to matched wild-type (WT) controls (Supplementary Notes). We found that Vps13b genotype has a significant phenotypic effect and explains 11% of the total Procrustes variance in facial and palatal shape (Supplementary Notes and table S3-2). The magnitude of this effect is two times stronger in males (Procrustes distance = 0.030) than in females (Procrustes distance = 0.017). The changes mainly involve the nasal region, resulting in an elongation and widening of the nasal tip and the dorsal aspect of the maxillae (Fig. 5A and movie S1). Concomitant with these changes, there is a narrowed and elevated palate.
Fig. 5. Vps13b and mouse craniofacial shape.
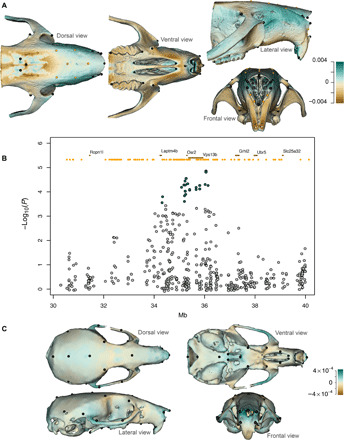
(A) Shape changes in the Vps13b KO mutant male mice relative to WT. Effect is predicted according to the expected marginal means at the populational average centroid size. Darker colors represent strong positive (blue) or negative (brown) deviation from the WT, given in Procrustes distance. Positive distance represents expansion from the WT, whereas negative distance represents contraction from the WT. Mesh warping is amplified by a factor of 2 to facilitate the visualization (see also movie S1). (B) Regional association plot for the VPS13 homologous region in chromosome 15 among outbred mice. Significantly associated SNPs are shown as green dots (FDR < 5%). Ensembl annotations for genes (brown) and enhancers active in embryonic facial prominence (orange) are shown at the top. (C) Phenotypic effect associated with allele dosage at the Vps13b index SNP among outbred mice. Skull zones with an expansion/contraction relative to the mean shape are shown in brown/blue (see also movie S2). To facilitate visualization, shape variation has been magnified by ×20.
In addition to characterizing craniofacial variation in the Vps13b-KO mice, we reexamined data from a GWAS on cranioskeletal variation in outbred mice (39). The published analysis did not report a significant association of univariate craniofacial phenotypes with SNPs in the Vps13b region (39). However, reanalyzing these data using a multivariate mixed model, we found that 22 SNPs in a 1.82-Mb region of chromosome 15 overlapping Vps13b (Fig. 5B) are significantly associated with craniofacial variation (FDR < 5%). SNPs in this region affect the protrusion/shrinkage of the nasal region, in a similar manner as seen for the Vps13b-KO mice (Fig. 5C and movie S2).
DISCUSSION
Craniofacial form is largely determined during embryonic development through the concerted actions of cranial neural crest cells, mesodermally derived mesenchyme, and ectoderm. The cranial neural crest cell population is the major driver of facial outgrowth and differentiates into the cartilage and bony elements of the face (among other tissues) that principally determine facial structure (40). There has been considerable interest in understanding the role that cranial neural crest genes play in species-specific differences in facial structure, much of which appears to be determined by genetic variation in enhancers that regulate key genes in this cell type (41). Furthermore, neural crest cell–dependent facial form is dictated early in development by signals received from the developing forebrain as well as the overlying ectoderm (42), implying that any a priori assumptions about tissue-specific functions of genes controlling variation in facial form should be avoided. GWASs provide a hypothesis-free approach to pinpointing important genetic variation underlying population-level craniofacial shape differences.
Our previous study on the CANDELA cohort focused on 14 categorical traits scored onto a few discrete categories. Here, we consider a much larger set of phenotypes (a total of 59), and we assessed these quantitatively, based on landmarks and semi-landmarks placed on face profile pictures. Although examined on the same study cohort, the different phenotyping approach used here adds substantial power to the GWA analyses. Statistically, quantitative traits are more informative than categorical ones. Focusing the landmarking on the profile also has several advantages. The landmarks are relatively prominent and easy to assess, and the process is less susceptible to external factors, such as head rotation. In addition to the main landmarks, we also use a set of semi-landmarks to capture in fuller detail variation along the facial contour.
The number of previously reported face GWAS hits replicated here, despite major differences across studies in phenotyping approaches and phenotype definitions, is noticeable: a total of 23, including 7 of the regions that have been repeatedly associated with facial features in previous studies. The fact that we detect significant genetic effects at genome regions previously associated with facial features different from those examined here is consistent with the view that many loci involved in craniofacial development have various effects on facial morphology and that certain facial traits can be affected by overlapping sets of genes (10). Although stochastic effects and differences in facial phenotype characterization across studies could explain the lack of replication of some of the loci associated previously, the genetic architecture of facial morphology in human populations probably also plays a role. Evidence for the impact of genetic structure on facial variation across human populations was observed here in the form of an effect of continental and subcontinental genetic ancestry on profile features, pointing to the existence of trait alleles with differentiated frequencies between continents, as well as between different areas within continents. A prominent example of such continental differentiation in trait allele frequency is the EDAR region associated here (and in the literature) with measures of facial flatness and jaw protrusion (4). The associated EDAR variants are essentially absent in Europe, and therefore, this effect is not detectable in Europeans (4). Similarly, SNPs associated at one of the previously unidentified genome regions identified here are not polymorphic in Europeans, and therefore, we could not test for replication in the European meta-analysis. This variable genetic architecture of facial features across human populations emphasizes the need for further studies in non-Europeans for a fuller characterization of the genetic basis of facial variation in modern humans.
Natural selection appears to have played a major role in the evolution of craniofacial morphology in many mammalian species, particularly in relation to various environmental adaptations, including diet (19). Natural selection has also long been proposed to have played a role on the evolution of the human face (20). However, thus far, there is little evidence for genomic signatures of selection at human face morphology loci (18). From this perspective, it is interesting that we find an overlap of the signal of association of lip thickness ratio at the WARS2/TBX15 region with a signal of selection, also coinciding with a tract of Denisovan introgression (30, 31). There is increasing evidence for adaptive archaic introgression in modern humans, particularly for pigmentation and immune response genes, and for the phenotypic effects of these introgressed tracts being mediated through their regulatory impact on the genome (43). It has thus been suggested that the strong signal of selection seen in the WARS2/TBX15 region could have been driven by adaptation to a cold environment (30). Archaic humans are thought to have been adapted to cold conditions by the time they encountered modern humans migrating from less extreme latitudes, and this could have favored adaptive introgression around WARS2/TBX15 (31). Consistent with the possibility of adaptive pressure to a cold environment, association studies have implicated the WARS2/TBX15 region in body fat distribution, height, and waist-hip ratio (29). However, the SNPs associated with these body features do not overlap with the tract of Denisovan introgression (31). Similarly, SNPs in 1p12 previously associated with ear and face features are located some distance away from the tract of introgression (10, 15, 18) and are not in LD with it (Fig. 3). The Denisovan haplotype has a low frequency in Europeans and a high frequency in East Asians and is nearly fixed in Native Americans (31), and we observe it at a frequency of ~50% in the CANDELA data (in line with the European/Native American admixture of these samples), thus facilitating the detection of any associated phenotypic effects. It is possible that the facial trait association that we see in the CANDELA data for SNPs in 1p12 could relate to an impact of variants in this region on facial fat distribution (perhaps as part of a broader effect on body fat distribution) in East Asians and Native Americans and that this could have been relevant in adaptation to a cold environment. The WARS2/TBX15 region includes a bivalent transcription factor as well as a superenhancer (Fig. 3), hallmarks of critical patterning transcription factors responsible for specifying tissue/cell-type identity (44), in this case active during early human craniofacial development. It will therefore be important to directly examine the role of variants in the WARS2/TBX15 regions (including the Denisovan haplotype) on gene expression regulation and to evaluate the impact of any such changes in gene expression on body fat distribution.
Of the previously unidentified face morphology loci we detected, evidence is most compelling for VPS13B, mutations of which result in Cohen syndrome. Consistent with the effect on columella size that we detect in the CANDELA individuals, patients with Cohen syndrome typically exhibit a beak-shaped, prominent nose (OMIM #216550). In agreement with these phenotypic effects in humans, here we show that mice deficient in Vps13b present a cranioskeletal dysmorphology that includes anteriorly lengthened nasal bones. Other cranioskeletal changes with similarities across humans and mice have been reported for rare mutations at a number of loci, for instance, in Williams syndrome (45). These homologous changes probably reflect genetic effects on related cranial neural crest–derived bony elements of the skull (45). Furthermore, our confirmation that common variants of Vps13b affect midfacial morphology mice establishes a specific role for this gene in craniofacial development, beyond the various phenotypic effects associated with VPS13B mutations and Cohen syndrome. VPS13B is a widely expressed transmembrane protein, and on the basis of its homology to the yeast VPS13 protein, it is thought to function in vesicle-mediated transport and sorting of proteins within the cell. By forming a complex with the small guanosine triphosphatase RAB6 at the Golgi, VPS13B strongly colocalizes with the cis-Golgi matrix protein GM130 and is required to maintain Golgi integrity (46). VPS13B also plays a role in Golgi protein glycosylation (47) and has been identified as a tethering factor functioning in transport from early endosomes to recycling endosomes (47). Facial dysmorphism is a common feature of disorders of glycosylation (48) and likely reflects the importance of glycosylation as a modulator of many extracellular signaling pathways that are highly active during embryogenesis.
The observation of an effect of Vps13b on morphology in outbred mice suggests that variation in this gene could play a role in natural craniofacial variation in Mus as well as Homo. In this context, it seems relevant that the VPS13B region has been highlighted in genome scans for selective sweeps both in dog breeds (49) and in comparisons of Neanderthal and modern human sequences (50). This suggests that certain gene regions affecting facial variation within species could also have played a role in craniofacial differentiation across species. This appears to be the case for the SUPT3H/RUNX2 region. We initially detected an association of SNPs in this gene region with nose bridge breadth (15). Subsequently, an effect for SNPs in SUPT3H/RUNX2 on nose morphology (10) and chin dimples (7) has been reported, and here we find an association of this region with forehead protrusion. The ratio of glutamines to alanines in a functional domain of RUNX2 was shown to correlate with facial length in Carnivora (51), and this glutamine/alanine ratio has recently been shown to also correlate with facial length in anthropoid primates (52). It will therefore be of interest to more broadly examine the extent to which the loci being implicated in human facial variation could play a role in natural variation in other species, as well as in the differentiation of facial features between taxa. Of particular interest for our species is the role that these genes could have played in the differentiation of anatomically modern humans from more archaic forms, which might also provide insights into the evolutionary forces involved.
METHODS
Study subjects
We used CANDELA consortium (13) data from 6282 volunteers recruited in five Latin American countries (Brazil, 630; Chile, 1794; Colombia, 1419; Mexico, 1265; Peru, 1174). All volunteers provided written informed consent. Ethics approvals were obtained from Universidad Nacional Autónoma de México (México), Universidad de Antioquia (Colombia), Universidad Perúana Cayetano Heredia (Perú), Universidad de Tarapacá (Chile), Universidade Federal do Rio Grande do Sul (Brazil), and University College London (UK).
Phenotyping
On each of the 6282 right side face profile 2D digital photographs (13), we manually placed 19 landmarks [commonly used in face morphology studies (53)] and 22 semi-landmarks (fig. S1) using TpsDig (54). Then, we slid the semi-landmarks using TpsUtil and TpsRelw (54) and performed a full Procrustes superimposition using MorphoJ (55). From those 41 Procrustes-adjusted landmarks and semi-landmarks, we obtained 59 facial measurements using MATLAB 9.6.0 (table S1). The measurements include distances, angles, and ratios, on the overall face and on specific regions of the upper, middle, or lower face, including features of the nose, lips, and eyes. The measurement protocol used was devised building on our previous analyses (4) and seeks to include features reported to be variable in human populations (56). While GWAS of facial landmarks commonly analyze all pairwise distances (1–3, 6, 18), or linear combinations of landmark coordinates such as PCs (10), the measurements obtained here are akin to those commonly used in certain anthropological, medical, or forensic applications (57). Broadly, 35 features were measured according to one or more definitions (up to five for jaw protrusion) and covering either the whole profile (face flatness) or a specific region of the face: forehead (4 measurements), ear (inclination and size), eye (4 measurements), nose (18 measurements), lips and philtrum (15 measurements), jaw (8 measurements), and chin (3 measurements). We assessed the reliability of our phenotyping protocol by calculating concordance correlation coefficient (CCC) for all landmarks, semi-landmarks, and measurements (58). For four pairs of series, the CCC was computed as , where S12 is the covariance between the two series of that pair and and Yi are, respectively, the variance and the average of the ith series of that pair. The pairs of series were INTER (inter-observer: two operators landmarked the same 200 photographs), INTRA 1 (intra-observer 1: operator P.J. landmarked twice the same 50 photographs 2 weeks apart), INTRA 2 (intra-observer 2: operator B.B. landmarked twice the same 100 photographs 2 weeks apart), and ALT (alternative picture: operator B.B. landmarked two different photographs of the same 100 individuals 2 weeks apart). All CCC results are given in table S2.
DNA genotyping and quality control
DNA samples from participants were genotyped on the Illumina HumanOmniExpress chip including 730,525 SNPs. PLINK v1.9 (24) was used to exclude SNPs and individuals with >5% missing data, markers with minor-allele frequency < 1%, monozygous twins or sample duplicates [PLINK Identity-By-Descent (IBD) estimate > 0.95], and those who failed the X or Y chromosome sex concordance check (sex estimated from X chromosome heterozygosity or Y chromosome call rate not matching recorded sex information). After applying these filters, 636,195 autosomal SNPs and 7010 individuals (1853 from Colombia, 693 from Brazil, 1891 from Chile, 1288 from Mexico, and 1285 from Peru) were retained for further analysis. Because of the admixed nature of the study sample, there is an inflation in Hardy-Weinberg P values (4). We therefore did not exclude markers based on Hardy-Weinberg deviation but performed stringent quality controls at software and biological levels (4). For example, we checked for batch effects by genotyping a control sample on each plate and comparing its genotype calls across batches; the consistency rate (i.e., matched proportion of genotypes) was ≥0.999 in all cases after SNP-level QC. We also assessed the accuracy of our genotypes independently by comparing against genotype calls obtained on a subset of samples by high-coverage sequencing, and consistency was again ≥0.999.
SNP genotype imputation
The chip genotype data of the CANDELA sample were phased using SHAPEIT2 (59). IMPUTE2 (60) was then used to impute genotypes at untyped SNPs using variant positions from the 1000 Genomes Phase 3 data, which include haplotype information for 2504 individuals across the world at 77,818,332 autosomal variant positions. Positions that are monomorphic in 1000 Genomes Latin American samples [CLM (Colombian in Medellin), MXL (Mexican in Los Angeles), PEL (Peruvian in Lima), and PUR (Puerto Rican from Puerto Rico)] were excluded, leading to 11,218,392 autosomal biallelic variants being imputed in our dataset. Of these, we removed 64,681 SNPs based on the following criteria that may indicate poor imputation and/or genotyping: (i) imputation quality scores < 0.4, (ii) low concordance value (<0.7) with chip genotypes, or (iii) a large gap between info [a cross-validation metric implemented in IMPUTE2 (60), indicating imputation certainty] and concordance values (info_type0-concord_type0 > 0.1). In the latter two cases, these SNPs were also excluded from the chip dataset. The IMPUTE2 (60) genotype probabilities at each locus were converted into most probable genotypes using PLINK (24) (at the default setting of >0.9 certainty). SNPs with a proportion of samples with uncalled genotypes > 5% and minor-allele frequency < 1% were excluded. The final imputed dataset contained genotypes for 8,780,546 autosomal biallelic SNPs (638,266 genotyped SNPs and 8,142,280 imputed SNPs).
We have estimated the imputation accuracy of the final dataset using the median info score. The overall median info score was 99.2%. It is slightly higher (99.5%) for “European” variants (defined as variants with MAF (Minor Allele Frequency) > 20% in highly European individuals and MAF < 5% in highly Native American individuals) as well as for “Native American” variants (99.3%). For the sake of comparison, the rare variants (MAF < 5% in CANDELA; ~30% of the imputed dataset) have a median info score of 97.4%. We have therefore concluded that imputation was as reliable for Native American variants as for European variants and was high overall for the entire imputed dataset including rare variants as observed from the median values. (As SNPs with MAF < 1% in the AMR reference population or in the CANDELA samples are excluded, we consider a threshold of MAF < 5% for “rare” SNPs.)
In addition, we sought to verify the quality of imputed genotypes by comparing with an independently sequenced dataset. A set of Native American samples collected for a previous study (14) was sequenced at high coverage and variants were filtered. We calculated the concordance for these samples as the proportion of imputed genotypes that match the sequence data exactly. The overall median concordance was high (99.4%) and remained high in subcategories: 98.9% for SNPs common in Native American samples (MAF > 5% in that population) and 99.8% for rare SNPs in Native American samples (MAF < 5%).
Sample filtering
For all traits together, we initially performed the following data quality control on volunteers with available genotype data regardless of the studied trait. We discarded from the dataset any volunteer (i) with missing age information, (ii) with discordant information between recorded age and age computed based on birth date, (iii) older than 45 years, or (iv) younger than 18 years. Also, as the African ancestry is underrepresented in our population, we discarded from the dataset 23 individuals whose African continental ancestry share (computed using Admixture) (61) was higher than either their Native American or European shares.
The filtering of individuals was then performed for each trait independently to maximize the number of samples per trait (a global filtering, e.g., on phenotype outliers, would discard 942 individuals from the dataset). To that end, we first removed from the dataset any individual presenting an outlier phenotype (trait value lower or greater than the trait value average for that sex plus or minus three times the SD).
Then, among the remaining individuals for that trait, we identified highly related pairs and removed one member of each pair. The threshold for highly related pairs was set to an IBD probability estimate equal to 0.1, to exclude third-degree relatives and higher. The threshold, however, needed adjustment because of inflation in IBD estimates due to the admixed nature of the samples. In our previous study (4), we observed that highly Native samples have higher IBD values due to drift and inbreeding in the founder populations: Among unrelated CANDELA samples with >95% European ancestry, median IBD is <0.01, but among samples with >95% Native ancestry, median IBD value is ~0.2. Therefore, the actual IBD threshold was adjusted according to the ethnic composition of the participants, so that it is always 0.1 above the expected IBD level between unrelated samples of that ethnic composition. For example, that threshold was relaxed to 0.30 (0.1 + 0.2) for the specific case of a large (~240 individuals) genetic group mainly composed of Peruvian and Chilean samples and of very high Native American ancestry (average Native American ancestry is equal to 94%).
Once highly related pairs are identified, we recursively removed individuals involved in multiple pairs, starting with those involved in many pairs, and update the count until each individual is included in at most one pair. For these remaining pairs, we removed the sample with greater age deviation from the average sampling age.
Genetic PCs were computed on the resulting set of individuals for each trait. Among these individuals, genetic outliers were identified by computing, for each sample, the distance to the nearest neighbor based on the 10 leading PCs [obtained with PLINK (24)], with the ith PC weighted by . Samples whose distance to nearest-neighbor distance was greater than the average distance plus three times the SD were discarded as genetic outliers.
Such sample filtering was applied independently for each of the 59 phenotypes, yielding similar numbers of samples qualified for downstream analyses (from 5686 to 5731; median, 5719). Some of these analyses use the set of all samples that were qualified at least for one trait (5764 samples).
Statistical genetic analyses
Primary GWA was implemented using PLINK v1.9 (24). For each phenotypic trait independently, we performed a multiple linear regression analysis with sex, age, landmarking operator, BMI, and the six leading genetic PCs (computed on the samples qualified for this trait) as covariates. The association analyses were tested on the imputed data. Q-Q plots for all traits showed no sign of inflation, the genomic control factor λ being ~1 in all cases, indicating that our study performs appropriate subject QC, such as exclusion of relatives and outliers, and appropriately controls for population stratification.
To account for multiple testing across all the traits, we estimated the FDR threshold with the Benjamini-Hochberg procedure (25). Applying the FDR procedure on all 59 traits and all tested SNPs (i.e., a total of 507,440,483 tests), the adjusted genome-wide significance threshold was 6.72 × 10−7 at α = 5% and 7.08 × 10−8 at α = 1%. Both P value thresholds were larger than the commonly used GWAS genome-wide significance threshold of 5 × 10−8, indicating that this threshold of 5 × 10−8 is more stringent than the FDR significance thresholds adjusted for all traits, and therefore, its use controls for testing multiple traits. Therefore, we used the commonly threshold of 5 × 10−8 across all traits.
To estimate narrow-sense heritability (h2) and genetic correlations between traits, we have computed a genomic relationship matrix gathering the 5764 samples qualified for at least one trait, on 634,753 genotyped SNPs (MAF > 1%, autosomes only) using LDAK5 (22) with default parameters (option --power set to −0.25). For each of the 59 traits, we further used the submatrix gathering all samples qualified for this specific trait. We have obtained h2 by partitioning the phenotypic variance into additive genetic () and residual () variances using REML in LDAK5 (22) on a linear mixed model y ~ sex + age + BMI + rater + g + e, where sex, age, BMI, and rater are fixed effects and g and e are random (respectively additive genetic and the residual) effects. Testing h2 = 0 was done by fitting the same model with GCTA (Genome-wide Complex Trait Analysis software) (62). We have used the same tools on a bivariate model (with the same terms) to estimate the genetic correlation () for each pair of traits.
Cranioskeletal analysis of Vps13btm1.Ics KO mice
Mice were bred and housed at the animal facility of the University of Burgundy, and animal procedures were approved by the local ethics committee under the reference number 821231010. A total of 51 mice, balanced between genders and genotypes, were euthanized at 18.7 ± 1.2 weeks old. Skulls were cleaned with dermestid beetles and then scanned at 29 μm using a Skyscan 1174 microCT. Brain removal before scanning damaged some specimens, and they were not included further in the study. Prior analyses reveal three additional gross outliers that were removed from the data. A final sample size of 14 females (9 WT and 5 KO) and 23 males (12 WT and 11 KO) was retained for the study.
Mesh surfaces of each skull were obtained in Avizo 2019 using a Marching cube algorithm. A set of 11 paired landmarks and 4 unpaired landmarks on the face and the palate was digitized on these meshes using the R package digit3DLand version 0.1.5. Seven additional curve paired semi-landmarks and five unpaired curve semi-landmarks were also digitized (Supplementary Notes and fig. S3-1). Sliding of these curve semi-landmarks on the 3D surface of the skulls was performed using the R package Morpho 2.6. Before sliding, meshes were decimated to 500,000 faces and smoothed using a surface preserving Laplace algorithm in the R package Rvcg version 0.98. The final set of 44 landmarks and semi-landmarks was superimposed using a full generalized Procrustes analysis with bilateral object symmetry (63) in the Morpho package.
A linear model accounting for centroid size as well as genotype by sex effects was fitted on the symmetric shapes. Mutant mice were generally smaller and lighter than their WT counterparts (Supplementary Notes). Therefore, skull centroid size was included as a covariate in the modeling of shape variation to control for size-related effects. Since sample size was small compared to the number of variables, phenotypic effects were evaluated with Procrustes analysis of variance (ANOVA) (64) computed with the R package geomorph version 3.1.3 and processed with 1000 residual permutations. Shape changes were visualized as shape differences between expected marginal genotype means predicted at the population average centroid size for WT and KO and visualized with a twofold amplification.
Analysis of genetic association in outbred mice
We reanalyzed the data of Pallares et al. (39), which include genotypes for 70,000 SNPs obtained from 692 mice and raw 3D coordinate data for 44 landmarks (17 pairs of symmetric landmarks and 10 landmarks on the median plane). Craniofacial variation was modeled only on the basis of the 67 non-null PCs obtained from the 44 × 3 symmetric tangent coordinates (which include 51 dimensions redundant due to symmetry) (63).
Association between SNP genotype and the set of 67 PCs representing skull shape was evaluated with a multivariate mixed model, incorporating centroid size as covariate. Population structure was modeled from the genomic relatedness matrix (65). The variance components estimated from the model were used to correct both genotypes and phenotypes (66). Association was tested on the basis of Pillai trace statistics obtained from the multivariate regression between the corrected genotypes and PC scores. FDR was computed on the basis of 100 permutations of corrected PC scores following the approach of Nicod et al. (66) and used to identify SNPs exceeding an FDR threshold of 5%.
Regulatory annotation enrichment analysis
We extracted chromatin enhancer annotations (ChromHMM states 13 to 18) identified in 127 non-craniofacial tissues analyzed by the Roadmap Epigenomics Consortium (67), as well as those obtained in 24 primary human embryonic craniofacial tissues, 4.5 to 8 post-conception weeks, and two samples derived from cultured HEPM (human embryonic palatal mesenchyme) cells (27). We also extracted the superenhancer annotations reported for craniofacial tissue using the ROSE (Ranking Of Super Enhancer) approach (27, 44). The SNPs included in the GWAS were filtered for association P values < 5 × 10−8, and their enrichment in enhancer annotations was evaluated using the approach implemented in GREGOR (28). Testing of each associated SNP included a minimum of 500 neighboring SNPs with r2 ≥ 0.8, in windows up to 1 Mb [based on 1000 Genome Project European data (23)].
Selection signal near WARS2-TBX15
To test for signals of selection at the WARS2-TBX15 region in Native Americans, we computed PBS scores (68) using CANDELA individuals with more than 95% Native American ancestry (127 samples). As PBS in an FST-based statistic, we inferred allele frequencies by accounting for non-Native American ancestry. To do so, we used the RFMix software (69) to assign local ancestry at each SNP. Three continental populations consisting of phased haplotypes for 175 Native Americans, 107 IBS (Iberian Population in Spain; 1000 Genomes Project Phase 3), and 101 YRI (Yoruba in Ibadan, Nigeria; 1000 Genomes Project Phase 3) were used as reference samples. We ran RFMix (69) with the phase correction feature enabled and performed two rounds of the EM (expectation maximization) algorithm. We used the default window size of 0.2 cM, the default number of trees (100), and the default model of admixture occurring eight generations ago. Genetic distances were obtained from the HapMap Phase II genetic map. For each SNP, we then computed Native American allele frequency fNat as
where fNatCANDELA is the allele frequency for CANDELA individuals with >95% Native American ancestry, fEur is the allele frequency for European individuals, and fAfr is the allele frequency of sub-Saharan African individuals. The ancestry proportion for each reference populations was estimated as
where i is a haplotype at each site and P(Anc) is the posterior probability of assignment of a given ancestry. Using these estimated allele frequencies, we computed PBS scores for Native Americans using CEU (Northern Europeans from Utah, USA; 1000 Genomes Project Phase 3) and CHB (Han Chinese in Beijing, China; 1000 Genomes Project Phase 3) as reference populations. SNPs not polymorphic in at least two populations were excluded.
Detection of Denisovan introgression near WARS2-TBX15 and association with face profile traits
The imputed genotypes of the 7026 CANDELA samples were first phased in a 3-Mb window around the WARS2-TBX15 region (chromosome 1, from 118 to 121 Mb) using SHAPEIT2 (59) with default parameters and no reference panel. We have retained 2383 biallelic SNPs among the 9582 variants in the central megabase of that window, complying with the following rules: (i) those with read depth greater than or equal to 20 in Homo denisovensis (downloaded from http://cdna.eva.mpg.de/), (ii) those with filter PASS in the VCFs of Denisova and 1000 Genomes sequences, (iii) those for which ancestral allele information was consistent throughout VCFs (Variant Call Formats), (iv) those for which ancestral allele is not different from the two reported alleles in modern or archaic samples, (v) those that have the same polymorphism reported in modern and both archaic samples, and (vi) those that were located within the 119 to 120 Mb. This selection contained the leading SNP for our GWAS hit in that region (rs3790553) as well as three other close SNPs that were found to be significantly associated with facial traits in former studies: rs1766786 [associated with chin dimples in Pickrell et al. (7)], rs72691108 [associated with frontal bossing phenotype in Claes et al. (10)], and rs17023457 [associated with two ear phenotypes—folding of antihelix and antitragus size—in Adhikari et al. (15)]. We ran the same hidden Markov model as in a previous study of Greenlandic Inuit (admixtureHMM) (31) on the 14,052 CANDELA chromosomes after coding them according to the human-chimp ancestral allele (0/1 for ancestral/derived allele, following the authors’ procedure). The purpose of admixtureHMM is to detect segments of the test chromosomes that are more likely to originate from archaic than modern humans. We used the allele frequencies of the Denisovan sample and, as proxy for modern humans, those of the 108 YRI samples from the 1000 Genomes Phase 3. We ran the admixtureHMM with default parameters, except for confidence level for which we have tested four values (20, 50, 90, and 99%). The putative introgression tracts that were called at each of these thresholds and with a minor allele frequency ≥ 1% were further considered for association analyses. We have then tested for association between the genotypes (0, 1, or 2—the number of copies of an introgression tract) and the 59 traits using the same linear model as for GWA tests (see the “Statistical genetic analyses” section), hence accounting for sex, age, BMI, landmarking operator, and six genetic PCs.
URLs
For HaploReg, see https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php. For NCBI, see www.ncbi.nlm.nih.gov. For UCSC (University of California Santa Cruz Genome Browser), see https://genome.ucsc.edu. For Ensembl, see www.ensembl.org. For GTEx, see https://gtexportal.org/. For GWAS catalog, see www.ebi.ac.uk/gwas/. For Denisovan sample, see http://cdna.eva.mpg.de/.
For the R package FastMan used to draw the Manhattan plot in Fig. 1, see https://github.com/kaustubhad/fastman. For the R package digit3DLand version 0.1.5, see https://github.com/morphOptics/digit3DLand. For the R package Morpho 2.6, see https://cran.r-project.org/web/packages/Morpho/index.html. For the R package Rvcg version 0.98, see https://cran.r-project.org/web/packages/Rvcg/index.html. For the R package geomorph version 3.1.3, see https://cran.r-project.org/package=geomorph.
Acknowledgments
We thank the volunteers for their enthusiastic support for this research. We also thank A. Alvarado, M. Ballesteros Romero, R. Cebrecos, M. Á. Contreras Sieck, F. de Ávila Becerril, J. De la Piedra, M. Teresa Del Solar, W. Flores, M. Granados Riveros, R. Paim, R. Gunski, J. Felisberto Menezes Cavalheiro, E. Correa de Souza Jr., W. Hart, I. Jafet Moreno, P. León-Mimila, F. Quispealaya, D. Rogel Diaz, R. Rojas, and V. Sarabia for assistance with volunteer recruitment, sample processing, and data entry. We are very grateful to the institutions that allowed the use of their facilities for the assessment of volunteers, including Escuela Nacional de Antropología e Historia and Universidad Nacional Autónoma de México (México), Universidade Federal do Rio Grande do Sul (Brazil), 13° Companhia de Comunicações Mecanizada do Exército Brasileiro (Brazil), Pontificia Universidad Católica del Perú, and Universidad de Lima and Universidad Nacional Mayor de San Marcos (Perú). We also acknowledge the Centre de Calcul Intensif d’Aix-Marseille for granting access to high-performance computing resources and the Gismo analytical platform of the University of Burgundy for scanning the mouse skulls. We thank M. Fumagalli and Q. Li for comments on the manuscript and D. Speed for advice on heritability calculations. We acknowledge M. A. Ikram, T. E. C. Nijsten, M. A. de Jong, S. Boehringer, M. K. Lee, E. Feingold, M. L. Marazita, L. Paternoster, H. Thompson, G. C. Sharp, S. Lewis, S. Richmond, A. Zhurov, and L. Pallares for facilitating access to published datasets. We are grateful to E. Bellini for the face drawing incorporated in Fig. 1. We thank L. Pallares and A. Palmer for sharing the GWA mouse data. Funding: Work leading to this publication was funded by grants from the National Natural Science Foundation of China (#31771393), the Scientific and Technology Committee of Shanghai Municipality (18490750300), the Ministry of Science and Technology of China (2020YFE0201600), the Shanghai Municipal Science and Technology Major Project (2017SHZDZX01) and the 111 Project (B13016), the Leverhulme Trust (F/07 134/DF), BBSRC (BB/I021213/1), the Excellence Initiative of Aix-Marseille University–A*MIDEX (a French “Investissements d’Avenir” program), Universidad de Antioquia (CODI sostenibilidad de grupos 2013-2014 and MASO 2013-2014), Conselho Nacional de Desenvolvimento Científico e Tecnológico, Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (Apoio a Núcleos de Excelência Program), Fundação de Aperfeiçoamento de Pessoal de Nível Superior, the National Institute of Dental and Craniofacial Research (R01-DE027023, U01-DE020078, R01-DE016148, and X01-HG007821), and a Santander Research and Scholarship Award. B.B. is supported by a doctoral scholarship from Ecole Doctorale 251 Aix-Marseille Université. Author contributions: B.B., C.C.S.d.C., C.G., C.J., C.M., E.W., F.R., G.B., G.P., H.V.-R., J.C., J.G.-V., L.P., L.S.-F., M.-C.B., M.D., M.F.-G., M.H., P.E.-M., P.J., R.B., R.G.-J., S.C.-Q., T.H., V.A.-A., V.G., V.V., V.R., and W.A. contributed to volunteer recruitment or data collection. B.B., C.V.-G., J.M.-R., K.A., N.N., P.F., S.P., and Z.X. performed analyses. B.Y., D.B., K.A., L.D., M.K., N.N., R.G.-J., and T.C. provided guidance on aspects of study design. A.R.-L., C.C., C.T.-R., D.B., L.F., F.L., S.M.W., J. R.S., E.S., L.J.H., P.G.H., T.D.S., and M.K. obtained funding or granted access to resources. A.R.-L. and D.B. designed the project. A.R.-L., B.B., K.A., J.C., N.N. and P.F. wrote the paper with input from coauthors. A.R.-L. coordinated the study. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Raw genotype or phenotype data cannot be made available due to restrictions imposed by the ethics approval. Summary statistics obtained here have been deposited at GWAS central and can be accessed with the link: www.gwascentral.org/study/HGVST4323 (available from the Spring 2021 release).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabc6160/DC1
REFERENCES AND NOTES
- 1.Paternoster L., Zhurov A. I., Toma A. M., Kemp J. P., Pourcain B. S., Timpson N. J., McMahon G., McArdle W., Ring S. M., Smith G. D., Richmond S., Evans D. M., Genome-wide association study of three-dimensional facial morphology identifies a variant in PAX3 associated with nasion position. Am. J. Hum. Genet. 90, 478–485 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu F., van der Lijn F., Schurmann C., Zhu G., Chakravarty M. M., Hysi P. G., Wollstein A., Lao O., de Bruijne M., Ikram M. A., van der Lugt A., Rivadeneira F., Uitterlinden A. G., Hofman A., Niessen W. J., Homuth G., de Zubicaray G., McMahon K. L., Thompson P. M., Daboul A., Puls R., Hegenscheid K., Bevan L., Pausova Z., Medland S. E., Montgomery G. W., Wright M. J., Wicking C., Boehringer S., Spector T. D., Paus T., Martin N. G., Biffar R., Kayser M., A genome-wide association study identifies five loci influencing facial morphology in Europeans. PLOS Genet. 8, e1002932 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cole J. B., Manyama M., Kimwaga E., Mathayo J., Larson J. R., Liberton D. K., Lukowiak K., Ferrara T. M., Riccardi S. L., Li M., Mio W., Prochazkova M., Williams T., Li H., Jones K. L., Klein O. D., Santorico S. A., Hallgrimsson B., Spritz R. A., Genomewide association study of African children identifies association of SCHIP1 and PDE8A with facial size and shape. PLOS Genet. 12, e1006174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adhikari K., Fuentes-Guajardo M., Quinto-Sánchez M., Mendoza-Revilla J., Chacón-Duque J. C., Acuña-Alonzo V., Jaramillo C., Arias W., Lozano R. B., Pérez G. M., Gómez-Valdés J., Villamil-Ramírez H., Hunemeier T., Ramallo V., de Cerqueira C. C. S., Hurtado M., Villegas V., Granja V., Gallo C., Poletti G., Schuler-Faccini L., Salzano F. M., Bortolini M.-C., Canizales-Quinteros S., Cheeseman M., Rosique J., Bedoya G., Rothhammer F., Headon D., González-José R., Balding D., Ruiz-Linares A., A genome-wide association scan implicates DCHS2, RUNX2, GLI3, PAX1 and EDAR in human facial variation. Nat. Commun. 7, 11616 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee M. K., Shaffer J. R., Leslie E. J., Orlova E., Carlson J. C., Feingold E., Marazita M. L., Weinberg S. M., Genome-wide association study of facial morphology reveals novel associations with FREM1 and PARK2. PLOS ONE 12, e0176566 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaffer J. R., Orlova E., Lee M. K., Leslie E. J., Raffensperger Z. D., Heike C. L., Cunningham M. L., Hecht J. T., Kau C. H., Nidey N. L., Moreno L. M., Wehby G. L., Murray J. C., Laurie C. A., Laurie C. C., Cole J., Ferrara T., Santorico S., Klein O., Mio W., Feingold E., Hallgrimsson B., Spritz R. A., Marazita M. L., Weinberg S. M., Genome-wide association study reveals multiple loci influencing normal human facial morphology. PLOS Genet. 12, e1006149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pickrell J. K., Berisa T., Liu J. Z., Segurel L., Tung J. Y., Hinds D. A., Detection and interpretation of shared genetic influences on 42 human traits. Nat. Genet. 48, 709–717 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crouch D. J. M., Winney B., Koppen W. P., Christmas W. J., Hutnik K., Day T., Meena D., Boumertit A., Hysi P., Nessa A., Spector T. D., Kittler J., Bodmer W. F., Genetics of the human face: Identification of large-effect single gene variants. Proc. Natl. Acad. Sci. U.S.A. 115, E676–E685 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cha S., Lim J. E., Park A. Y., Do J.-H., Lee S. W., Shin C., Cho N. H., Kang J.-O., Nam J. M., Kim J.-S., Woo K.-M., Lee S.-H., Kim J. Y., Oh B., Identification of five novel genetic loci related to facial morphology by genome-wide association studies. BMC Genomics 19, 481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Claes P., Roosenboom J., White J. D., Swigut T., Sero D., Li J., Lee M. K., Zaidi A., Mattern B. C., Liebowitz C., Pearson L., Gonzalez T., Leslie E. J., Carlson J. C., Orlova E., Suetens P., Vandermeulen D., Feingold E., Marazita M. L., Shaffer J. R., Wysocka J., Shriver M. D., Weinberg S. M., Genome-wide mapping of global-to-local genetic effects on human facial shape. Nat. Genet. 50, 414–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Claes P., Liberton D. K., Daniels K., Rosana K. M., Quillen E. E., Pearson L. N., McEvoy B., Bauchet M., Zaidi A. A., Yao W., Tang H., Barsh G. S., Absher D. M., Puts D. A., Rocha J., Beleza S., Pereira R. W., Baynam G., Suetens P., Vandermeulen D., Wagner J. K., Boster J. S., Shriver M. D., Modeling 3D facial shape from DNA. PLOS Genet. 10, e1004224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Indencleef K., Roosenboom J., Hoskens H., White J. D., Shriver M. D., Richmond S., Peeters H., Feingold E., Marazita M. L., Shaffer J. R., Weinberg S. M., Hens G., Claes P., Six NSCL/P loci show associations with normal-range craniofacial variation. Front. Genet. 9, 502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruiz-Linares A., Adhikari K., Acuña-Alonzo V., Quinto-Sanchez M., Jaramillo C., Arias W., Fuentes M., Pizarro M., Everardo P., de Avila F., Gómez-Valdes J., León-Mimila P., Hunemeier T., Ramallo V., de Cerqueira C. C. S., Burley M.-W., Konca E., de Oliveira M. Z., Veronez M. R., Rubio-Codina M., Attanasio O., Gibbon S., Ray N., Gallo C., Poletti G., Rosique J., Schuler-Faccini L., Salzano F. M., Bortolini M.-C., Canizales-Quinteros S., Rothhammer F., Bedoya G., Balding D., Gonzalez-José R., Admixture in Latin America: Geographic structure, phenotypic diversity and self-perception of ancestry based on 7,342 individuals. PLOS Genet. 10, e1004572 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chacón-Duque J.-C., Adhikari K., Fuentes-Guajardo M., Mendoza-Revilla J., Acuña-Alonzo V., Barquera R., Quinto-Sánchez M., Gómez-Valdés J., Martínez P. E., Villamil-Ramírez H., Hünemeier T., Ramallo V., de Cerqueira C. C. S., Hurtado M., Villegas V., Granja V., Villena M., Vásquez R., Llop E., Sandoval J. R., Salazar-Granara A. A., Parolin M.-L., Sandoval K., Peñaloza-Espinosa R. I., Rangel-Villalobos H., Winkler C. A., Klitz W., Bravi C., Molina J., Corach D., Barrantes R., Gomes V., Resende C., Gusmão L., Amorim A., Xue Y., Dugoujon J.-M., Moral P., González-José R., Schuler-Faccini L., Salzano F. M., Bortolini M.-C., Canizales-Quinteros S., Poletti G., Gallo C., Bedoya G., Rothhammer F., Balding D., Hellenthal G., Ruiz-Linares A., Latin Americans show wide-spread Converso ancestry and imprint of local Native ancestry on physical appearance. Nat. Commun. 9, 5388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adhikari K., Reales G., Smith A. J. P., Konka E., Palmen J., Quinto-Sanchez M., Acuña-Alonzo V., Jaramillo C., Arias W., Fuentes M., Pizarro M., Lozano R. B., Pérez G. M., Gómez-Valdés J., Villamil-Ramírez H., Hunemeier T., Ramallo V., de Cerqueira C. C. S., Hurtado M., Villegas V., Granja V., Gallo C., Poletti G., Schuler-Faccini L., Salzano F. M., Bortolini M.-C., Canizales-Quinteros S., Rothhammer F., Bedoya G., Calderón R., Rosique J., Cheeseman M., Bhutta M. F., Humphries S. E., Gonzalez-José R., Headon D., Balding D., Ruiz-Linares A., A genome-wide association study identifies multiple loci for variation in human ear morphology. Nat. Commun. 6, 7500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adhikari K., Fontanil T., Cal S., Mendoza-Revilla J., Fuentes-Guajardo M., Chacón-Duque J.-C., Al-Saadi F., Johansson J. A., Quinto-Sanchez M., Acuña-Alonzo V., Jaramillo C., Arias W., Lozano R. B., Perez G. M., Gómez-Valdes J., Villamil-Ramirez H., Hunemeier T., Ramallo V., de Cerqueira C. C. S., Hurtado M., Villegas V., Granja V., Gallo C., Poletti G., Schuler-Faccini L., Salzano F. M., Bortolini M.-C., Canizales-Quinteros S., Rothhammer F., Bedoya G., Gonzalez-José R., Headon D., López-Otín C., Tobin D. J., Balding D., Ruiz-Linares A., A genome-wide association scan in admixed Latin Americans identifies loci influencing facial and scalp hair features. Nat. Commun. 7, 10815 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adhikari K., Mendoza-Revilla J., Sohail A., Fuentes-Guajardo M., Lampert J., Chacón-Duque J. C., Hurtado M., Villegas V., Granja V., Acuña-Alonzo V., Jaramillo C., Arias W., Lozano R. B., Everardo P., Gómez-Valdés J., Villamil-Ramírez H., de Cerqueira C. C. S., Hunemeier T., Ramallo V., Schuler-Faccini L., Salzano F. M., Gonzalez-José R., Bortolini M.-C., Canizales-Quinteros S., Gallo C., Poletti G., Bedoya G., Rothhammer F., Tobin D. J., Fumagalli M., Balding D., Ruiz-Linares A., A GWAS in Latin Americans highlights the convergent evolution of lighter skin pigmentation in Eurasia. Nat. Commun. 10, 358 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiong Z., Dankova G., Howe L. J., Lee M. K., Hysi P. G., de Jong M. A., Zhu G., Adhikari K., Li D., Li Y., Pan B., Feingold E., Marazita M. L., Shaffer J. R., Aloney K. M., Xu S.-H., Jin L., Wang S., de Vrij F. M. S., Lendemeijer B., Richmond S., Zhurov A., Lewis S., Sharp G. C., Paternoster L., Thompson H., Gonzalez-Jose R., Bortolini M. C., Canizales-Quinteros S., Gallo C., Poletti G., Bedoya G., Rothhammer F., Uitterlinden A. G., Ikram M. A., Wolvius E., Kushner S. A., Nijsten T. E. C., Palstra R.-J. T. S., Boehringer S., Medland S. E., Tang K., Ruiz-Linares A., Martin N. G., Spector T. D., Stergiakouli E., Weinberg S. M., Liu F., Kayser M.; On behalf of the International Visible Trait Genetics (VisiGen) , Novel genetic loci affecting facial shape variation in humans. eLife 8, e49898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Usui K., Tokita M., Creating diversity in mammalian facial morphology: A review of potential developmental mechanisms. Evodevo 9, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lacruz R. S., Stringer C. B., Kimbel W. H., Wood B., Harvati K., O’Higgins P., Bromage T. G., Arsuaga J.-L., The evolutionary history of the human face. Nat. Ecol. Evol. 3, 726–736 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Davidenko N., Silhouetted face profiles: A new methodology for face perception research. J. Vis. 7, 6 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Speed D., Cai N.; UCLEB Consortium, Johnson M. R., Nejentsev S., Balding D. J., Reevaluation of SNP heritability in complex human traits. Nat. Genet. 49, 986–992 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.1000 Genomes Project Consortium, Auton A., Brooks L. D., Durbin R. M., Garrison E. P., Kang H. M., Korbel J. O., Marchini J. L., McCarthy S., McVean G. A., Abecasis G. R., A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang C. C., Chow C. C., Tellier L. C., Vattikuti S., Purcell S. M., Lee J. J., Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Roy. Statist. Soc. Ser. B 57, 289–300 (1995). [Google Scholar]
- 26.Birnbaum S., Ludwig K. U., Reutter H., Herms S., Steffens M., Rubini M., Baluardo C., Ferrian M., de Assis N. A., Alblas M. A., Barth S., Freudenberg J., Lauster C., Schmidt G., Scheer M., Braumann B., Bergé S. J., Reich R. H., Schiefke F., Hemprich A., Pötzsch S., Steegers-Theunissen R. P., Pötzsch B., Moebus S., Horsthemke B., Kramer F.-J., Wienker T. F., Mossey P. A., Propping P., Cichon S., Hoffmann P., Knapp M., Nöthen M. M., Mangold E., Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat. Genet. 41, 473–477 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Wilderman A., VanOudenhove J., Kron J., Noonan J. P., Cotney J., High-resolution epigenomic atlas of human embryonic craniofacial development. Cell Rep. 23, 1581–1597 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt E. M., Zhang J., Zhou W., Chen J., Mohlke K. L., Chen Y. E., Willer C. J., GREGOR: Evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 31, 2601–2606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heid I. M., Jackson A. U., Randall J. C., Winkler T. W., Qi L., Steinthorsdottir V., Thorleifsson G., Zillikens M. C., Speliotes E. K., Mägi R., Workalemahu T., White C. C., Bouatia-Naji N., Harris T. B., Berndt S. I., Ingelsson E., Willer C. J., Weedon M. N., Luan J., Vedantam S., Esko T., Kilpeläinen T. O., Kutalik Z., Li S., Monda K. L., Dixon A. L., Holmes C. C., Kaplan L. M., Liang L., Min J. L., Moffatt M. F., Molony C., Nicholson G., Schadt E. E., Zondervan K. T., Feitosa M. F., Ferreira T., Allen H. L., Weyant R. J., Wheeler E., Wood A. R.; MAGIC, Estrada K., Goddard M. E., Lettre G., Mangino M., Nyholt D. R., Purcell S., Smith A. V., Visscher P. M., Yang J., Mc Carroll S. A., Nemesh J., Voight B. F., Absher D., Amin N., Aspelund T., Coin L., Glazer N. L., Hayward C., Heard-Costa N. L., Hottenga J.-J., Johansson A., Johnson T., Kaakinen M., Kapur K., Ketkar S., Knowles J. W., Kraft P., Kraja A. T., Lamina C., Leitzmann M. F., Mc Knight B., Morris A. P., Ong K. K., Perry J. R. B., Peters M. J., Polasek O., Prokopenko I., Rayner N. W., Ripatti S., Rivadeneira F., Robertson N. R., Sanna S., Sovio U., Surakka I., Teumer A., van Wingerden S., Vitart V., Zhao J. H., Cavalcanti-Proença C., Chines P. S., Fisher E., Kulzer J. R., Lecoeur C., Narisu N., Sandholt C., Scott L. J., Silander K., Stark K., Tammesoo M.-L., Teslovich T. M., Timpson N. J., Watanabe R. M., Welch R., Chasman D. I., Cooper M. N., Jansson J.-O., Kettunen J., Lawrence R. W., Pellikka N., Perola M., Vandenput L., Alavere H., Almgren P., Atwood L. D., Bennett A. J., Biffar R., Bonnycastle L. L., Bornstein S. R., Buchanan T. A., Campbell H., Day I. N. M., Dei M., Dörr M., Elliott P., Erdos M. R., Eriksson J. G., Freimer N. B., Fu M., Gaget S., Geus E. J. C., Gjesing A. P., Grallert H., Grässler J., Groves C. J., Guiducci C., Hartikainen A.-L., Hassanali N., Havulinna A. S., Herzig K.-H., Hicks A. A., Hui J., Igl W., Jousilahti P., Jula A., Kajantie E., Kinnunen L., Kolcic I., Koskinen S., Kovacs P., Kroemer H. K., Krzelj V., Kuusisto J., Kvaloy K., Laitinen J., Lantieri O., Lathrop G. M., Lokki M.-L., Luben R. N., Ludwig B., Mc Ardle W. L., Mc Carthy A., Morken M. A., Nelis M., Neville M. J., Paré G., Parker A. N., Peden J. F., Pichler I., Pietiläinen K. H., Platou C. G. P., Pouta A., Ridderstråle M., Samani N. J., Saramies J., Sinisalo J., Smit J. H., Strawbridge R. J., Stringham H. M., Swift A. J., Teder-Laving M., Thomson B., Usala G., van Meurs J. B. J., van Ommen G.-J., Vatin V., Volpato C. B., Wallaschofski H., Walters G. B., Widen E., Wild S. H., Willemsen G., Witte D. R., Zgaga L., Zitting P., Beilby J. P., James A. L., Kähönen M., Lehtimäki T., Nieminen M. S., Ohlsson C., Palmer L. J., Raitakari O., Ridker P. M., Stumvoll M., Tönjes A., Viikari J., Balkau B., Ben-Shlomo Y., Bergman R. N., Boeing H., Smith G. D., Ebrahim S., Froguel P., Hansen T., Hengstenberg C., Hveem K., Isomaa B., Jørgensen T., Karpe F., Khaw K.-T., Laakso M., Lawlor D. A., Marre M., Meitinger T., Metspalu A., Midthjell K., Pedersen O., Salomaa V., Schwarz P. E. H., Tuomi T., Tuomilehto J., Valle T. T., Wareham N. J., Arnold A. M., Beckmann J. S., Bergmann S., Boerwinkle E., Boomsma D. I., Caulfield M. J., Collins F. S., Eiriksdottir G., Gudnason V., Gyllensten U., Hamsten A., Hattersley A. T., Hofman A., Hu F. B., Illig T., Iribarren C., Jarvelin M.-R., Kao W. H. L., Kaprio J., Launer L. J., Munroe P. B., Oostra B., Penninx B. W., Pramstaller P. P., Psaty B. M., Quertermous T., Rissanen A., Rudan I., Shuldiner A. R., Soranzo N., Spector T. D., Syvanen A.-C., Uda M., Uitterlinden A., Völzke H., Vollenweider P., Wilson J. F., Witteman J. C., Wright A. F., Abecasis G. R., Boehnke M., Borecki I. B., Deloukas P., Frayling T. M., Groop L. C., Haritunians T., Hunter D. J., Kaplan R. C., North K. E., O’Connell J. R., Peltonen L., Schlessinger D., Strachan D. P., Hirschhorn J. N., Assimes T. L., Wichmann H.-E., Thorsteinsdottir U., van Duijn C. M., Stefansson K., Cupples L. A., Loos R. J. F., Barroso I., Mc Carthy M. I., Fox C. S., Mohlke K. L., Lindgren C. M., Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat. Genet. 42, 949–960 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fumagalli M., Moltke I., Grarup N., Racimo F., Bjerregaard P., Jorgensen M. E., Korneliussen T. S., Gerbault P., Skotte L., Linneberg A., Christensen C., Brandslund I., Jorgensen T., Huerta-Sanchez E., Schmidt E. B., Pedersen O., Hansen T., Albrechtsen A., Nielsen R., Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 349, 1343–1347 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Racimo F., Gokhman D., Fumagalli M., Ko A., Hansen T., Moltke I., Albrechtsen A., Carmel L., Huerta-Sanchez E., Nielsen R., Archaic adaptive introgression in TBX15/WARS2. Mol. Biol. Evol. 34, 509–524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sankararaman S., Mallick S., Patterson N., Reich D., The combined landscape of denisovan and Neanderthal ancestry in present-day humans. Curr. Biol. 26, 1241–1247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim H., Takegahara N., Walsh M. C., Middleton S. A., Yu J., Shirakawa J., Ueda J., Fujihara Y., Ikawa M., Ishii M., Kim J., Choi Y., IgSF11 regulates osteoclast differentiation through association with the scaffold protein PSD-95. Bone Res. 8, 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirsch N., Eshel R., Yaacov R. B., Shahar T., Shmulevich F., Dahan I., Levaot N., Kaplan T., Lupianez D. G., Birnbaum R. Y., Unraveling the transcriptional regulation of TWIST1 in limb development. PLOS Genet. 14, e1007738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kichaev G., Bhatia G., Loh P. R., Gazal S., Burch K., Freund M. K., Schoech A., Pasaniuc B., Price A. L., Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 104, 65–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medina-Gomez C., Kemp J. P., Trajanoska K., Luan J., Chesi A., Ahluwalia T. S., Mook-Kanamori D. O., Ham A., Hartwig F. P., Evans D. S., Joro R., Nedeljkovic I., Zheng H.-F., Zhu K., Atalay M., Liu C.-T., Nethander M., Broer L., Porleifsson G., Mullin B. H., Handelman S. K., Nalls M. A., Jessen L. E., Heppe D. H. M., Richards J. B., Wang C., Chawes B., Schraut K. E., Amin N., Wareham N., Karasik D., Van der Velde N., Ikram M. A., Zemel B. S., Zhou Y., Carlsson C. J., Liu Y., McGuigan F. E., Boer C. G., Bønnelykke K., Ralston S. H., Robbins J. A., Walsh J. P., Zillikens M. C., Langenberg C., Li-Gao R., Williams F. M. K., Harris T. B., Akesson K., Jackson R. D., Sigurdsson G., den Heijer M., van der Eerden B. C. J., van de Peppel J., Spector T. D., Pennell C., Horta B. L., Felix J. F., Zhao J. H., Wilson S. G., de Mutsert R., Bisgaard H., Styrkársdóttir U., Jaddoe V. W., Orwoll E., Lakka T. A., Scott R., Grant S. F. A., Lorentzon M., van Duijn C. M., Wilson J. F., Stefansson K., Psaty B. M., Kiel D. P., Ohlsson C., Ntzani E., van Wijnen A. J., Forgetta V., Ghanbari M., Logan J. G., Williams G. R., Bassett J. H. D., Croucher P. I., Evangelou E., Uitterlinden A. G., Ackert-Bicknell C. L., Tobias J. H., Evans D. M., Rivadeneira F., Life-course genome-wide association study meta-analysis of total body BMD and assessment of age-specific effects. Am. J. Hum. Genet. 102, 88–102 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reynolds K., Kumari P., Rincon L. S., Gu R., Ji Y., Kumar S., Zhou C. J., Wnt signaling in orofacial clefts: Crosstalk, pathogenesis and models. Dis. Model. Mech. 12, dmm037051 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Da Costa R., Bordessoules M., Guilleman M., Carmignac V., Lhussiez V., Courot H., Bataille A., Chlémaire A., Bruno C., Fauque P., Thauvin C., Faivre L., Duplomb L., Vps13b is required for acrosome biogenesis through functions in Golgi dynamic and membrane trafficking. Cell. Mol. Life Sci. 77, 511–529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pallares L. F., Carbonetto P., Gopalakrishnan S., Parker C. C., Ackert-Bicknell C. L., Palmer A. A., Tautz D., Mapping of craniofacial traits in outbred mice identifies major developmental genes involved in shape determination. PLOS Genet. 11, e1005607 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soldatov R., Kaucka M., Kastriti M. E., Petersen J., Chontorotzea T., Englmaier L., Akkuratova N., Yang Y., Häring M., Dyachuk V., Bock C., Farlik M., Piacentino M. L., Boismoreau F., Hilscher M. M., Yokota C., Qian X. Y., Nilsson M., Bronner M. E., Croci L., Hsiao W.-Y., Guertin D. A., Brunet J.-F., Consalez G. G., Ernfors P., Fried K., Kharchenko P. V., Adameyko I., Spatiotemporal structure of cell fate decisions in murine neural crest. Science 364, eaas9536 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Prescott S. L., Srinivasan R., Marchetto M. C., Grishina I., Narvaiza I., Selleri L., Gage F. H., Swigut T., Wysocka J., Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell 163, 68–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu J., Verzi M. P., Robinson A. S., Tang P. L.-F., Hua L. L., Xu S.-M., Kwok P.-Y., Black B. L., Endothelin signaling activates Mef2c expression in the neural crest through a MEF2C-dependent positive-feedback transcriptional pathway. Development 142, 2775–2780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dannemann M., Racimo F., Something old, something borrowed: Admixture and adaptation in human evolution. Curr. Opin. Genet. Dev. 53, 1–8 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Whyte W. A., Orlando D. A., Hnisz D., Abraham B. J., Lin C. Y., Kagey M. H., Rahl P. B., Lee T. I., Young R. A., Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Segura-Puimedon M., Sahun I., Velot E., Dubus P., Borralleras C., Rodrigues A. J., Valero M. C., Valverde O., Sousa N., Herault Y., Dierssen M., Perez-Jurado L. A., Campuzano V., Heterozygous deletion of the Williams-Beuren syndrome critical interval in mice recapitulates most features of the human disorder. Hum. Mol. Genet. 23, 6481–6494 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Seifert W., Kuhnisch J., Maritzen T., Lommatzsch S., Hennies H. C., Bachmann S., Horn D., Haucke V., Cohen syndrome-associated protein COH1 physically and functionally interacts with the small GTPase RAB6 at the Golgi complex and directs neurite outgrowth. J. Biol. Chem. 290, 3349–3358 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duplomb L., Duvet S., Picot D., Jego G., El Chehadeh-Djebbar S., Marle N., Gigot N., Aral B., Carmignac V., Thevenon J., Lopez E., Riviere J. B., Klein A., Philippe C., Droin N., Blair E., Girodon F., Donadieu J., Bellanne-Chantelot C., Delva L., Michalski J. C., Solary E., Faivre L., Foulquier F., Thauvin-Robinet C., Cohen syndrome is associated with major glycosylation defects. Hum. Mol. Genet. 23, 2391–2399 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Peanne R., de Lonlay P., Foulquier F., Kornak U., Lefeber D. J., Morava E., Perez B., Seta N., Thiel C., Van Schaftingen E., Matthijs G., Jaeken J., Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet. 61, 643–663 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Vaysse A., Ratnakumar A., Derrien T., Axelsson E., Pielberg G. R., Sigurdsson S., Fall T., Seppälä E. H., Hansen M. S. T., Lawley C. T., Karlsson E. K.; LUPA Consortium, Bannasch D., Vilà C., Lohi H., Galibert F., Fredholm M., Häggström J., Hedhammar A., André C., Lindblad-Toh K., Hitte C., Webster M. T., Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLOS Genet. 7, e1002316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Green R. E., Krause J., Briggs A. W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M. H.-Y., Hansen N. F., Durand E. Y., Malaspinas A.-S., Jensen J. D., Marques-Bonet T., Alkan C., Prüfer K., Meyer M., Burbano H. A., Good J. M., Schultz R., Aximu-Petri A., Butthof A., Höber B., Höffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E. S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Ž., Gušic I., Doronichev V. B., Golovanova L. V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R. W., Johnson P. L. F., Eichler E. E., Falush D., Birney E., Mullikin J. C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Pääbo S., A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sears K. E., Goswami A., Flynn J. J., Niswander L. A., The correlated evolution of Runx2 tandem repeats, transcriptional activity, and facial length in carnivora. Evol. Dev. 9, 555–565 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Ritzman T. B., Banovich N., Buss K. P., Guida J., Rubel M. A., Pinney J., Khang B., Ravosa M. J., Stone A. C., Facing the facts: The Runx2 gene is associated with variation in facial morphology in primates. J. Hum. Evol. 111, 139–151 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Caple J., Stephan C. N., A standardized nomenclature for craniofacial and facial anthropometry. Int. J. Leg. Med. 130, 863–879 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Rohlf F. J., The tps series of software. Hystrix It. J. Mamm. 26, 9–12 (2015). [Google Scholar]
- 55.Klingenberg C. P., MorphoJ: An integrated software package for geometric morphometrics. Mol. Ecol. Resour. 11, 353–357 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Ritz-Timme S., Gabriel P., Tutkuviene J., Poppa P., Obertová Z., Gibelli D., De Angelis D., Ratnayake M., Rizgeliene R., Barkus A., Cattaneo C., Metric and morphological assessment of facial features: A study on three European populations. Forensic Sci. Int. 207, 239.e1–239.e239238 (2011). [DOI] [PubMed] [Google Scholar]
- 57.Farkas L. G., Katic M. J., Forrest C. R., Comparison of craniofacial measurements of young adult African-American and North American white males and females. Ann. Plast. Surg. 59, 692–698 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Lin L. I., A concordance correlation coefficient to evaluate reproducibility. Biometrics 45, 255–268 (1989). [PubMed] [Google Scholar]
- 59.O’Connell J., Gurdasani D., Delaneau O., Pirastu N., Ulivi S., Cocca M., Traglia M., Huang J., Huffman J. E., Rudan I., McQuillan R., Fraser R. M., Campbell H., Polasek O., Asiki G., Ekoru K., Hayward C., Wright A. F., Vitart V., Navarro P., Zagury J. F., Wilson J. F., Toniolo D., Gasparini P., Soranzo N., Sandhu M. S., Marchini J., A general approach for haplotype phasing across the full spectrum of relatedness. PLOS Genet. 10, e1004234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Howie B., Fuchsberger C., Stephens M., Marchini J., Abecasis G. R., Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alexander D. H., Novembre J., Lange K., Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang J., Lee S. H., Goddard M. E., Visscher P. M., GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klingenberg C. P., Barluenga M., Meyer A., Shape analysis of symmetric structures: Quantifying variation among individuals and asymmetry. Evolution 56, 1909–1920 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Goodall C. R., Procrustes methods in the statistical analysis of shape (with discussion and rejoinder). J. Roy. Stat. Soc. Ser. B 53, 285–339 (1991). [Google Scholar]
- 65.Kang H. M., Sul J. H., Service S. K., Zaitlen N. A., Kong S.-Y., Freimer N. B., Sabatti C., Eskin E., Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nicod J., Davies R. W., Cai N., Hassett C., Goodstadt L., Cosgrove C., Yee B. K., Lionikaite V., MC Intyre R. E., Remme C. A., Lodder E. M., Gregory J. S., Hough T., Joynson R., Phelps H., Nell B., Rowe C., Wood J., Walling A., Bopp N., Bhomra A., Hernandez-Pliego P., Callebert J., Aspden R. M., Talbot N. P., Robbins P. A., Harrison M., Fray M., Launay J.-M., Pinto Y. M., Blizard D. A., Bezzina C. R., Adams D. J., Franken P., Weaver T., Wells S., Brown S. D. M., Potter P. K., Klenerman P., Lionikas A., Mott R., Flint J., Genome-wide association of multiple complex traits in outbred mice by ultra-low-coverage sequencing. Nat. Genet. 48, 912–918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y.-C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G. N., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J. M., Li W., Marra M. A., Mc Manus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L.-H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yi X., Liang Y., Huerta-Sanchez E., Jin X., Cuo Z. X. P., Pool J. E., Xu X., Jiang H., Vinckenbosch N., Korneliussen T. S., Zheng H., Liu T., He W., Li K., Luo R., Nie X., Wu H., Zhao M., Cao H., Zou J., Shan Y., Li S., Yang Q., Asan, Ni P., Tian G., Xu J., Liu X., Jiang T., Wu R., Zhou G., Tang M., Qin J., Wang T., Feng S., Li G., Huasang, Luosang J., Wang W., Chen F., Wang Y., Zheng X., Li Z., Bianba Z., Yang G., Wang X., Tang S., Gao G., Chen Y., Luo Z., Gusang L., Cao Z., Zhang Q., Ouyang W., Ren X., Liang H., Zheng H., Huang Y., Li J., Bolund L., Kristiansen K., Li Y., Zhang Y., Zhang X., Li R., Li S., Yang H., Nielsen R., Wang J., Wang J., Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maples B. K., Gravel S., Kenny E. E., Bustamante C. D., RFMix: A discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet. 93, 278–288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boehringer S., van der Lijn F., Liu F., Gunther M., Sinigerova S., Nowak S., Ludwig K. U., Herberz R., Klein S., Hofman A., Uitterlinden A. G., Niessen W. J., Breteler M. M. B., van der Lugt A., Wurtz R. P., Nothen M. M., Horsthemke B., Wieczorek D., Mangold E., Kayser M., Genetic determination of human facial morphology: Links between cleft-lips and normal variation. Eur. J. Hum. Genet. 19, 1192–1197 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ludwig K. U., Bohmer A. C., Bowes J., Nikolic M., Ishorst N., Wyatt N., Hammond N. L., Golz L., Thieme F., Barth S., Schuenke H., Klamt J., Spielmann M., Aldhorae K., Rojas-Martinez A., Nothen M. M., Rada-Iglesias A., Dixon M. J., Knapp M., Mangold E., Imputation of orofacial clefting data identifies novel risk loci and sheds light on the genetic background of cleft lip ± cleft palate and cleft palate only. Hum. Mol. Genet. 26, 829–842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qiao L., Yang Y., Fu P., Hu S., Zhou H., Peng S., Tan J., Lu Y., Lou H., Lu D., Wu S., Guo J., Jin L., Guan Y., Wang S., Xu S., Tang K., Genome-wide variants of Eurasian facial shape differentiation and a prospective model of DNA based face prediction. J. Genet. Genomics 45, 419–432 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Beaty T. H., Murray J. C., Marazita M. L., Munger R. G., Ruczinski I., Hetmanski J. B., Liang K. Y., Wu T., Murray T., Fallin M. D., Redett R. A., Raymond G., Schwender H., Jin S.-C., Cooper M. E., Dunnwald M., Mansilla M. A., Leslie E., Bullard S., Lidral A. C., Moreno L. M., Menezes R., Vieira A. R., Petrin A., Wilcox A. J., Lie R. T., Jabs E. W., Wu-Chou Y. H., Chen P. K., Wang H., Ye X., Huang S., Yeow V., Chong S. S., Jee S. H., Shi B., Christensen K., Melbye M., Doheny K. F., Pugh E. W., Ling H., Castilla E. E., Czeizel A. E., Ma L., Field L. L., Brody L., Pangilinan F., Mills J. L., Molloy A. M., Kirke P. N., Scott J. M., Arcos-Burgos M., Scott A. F., A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 42, 525–529 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fatemifar G., Hoggart C. J., Paternoster L., Kemp J. P., Prokopenko I., Horikoshi M., Wright V. J., Tobias J. H., Richmond S., Zhurov A. I., Toma A. M., Pouta A., Taanila A., Sipila K., Lähdesmäki R., Pillas D., Geller F., Feenstra B., Melbye M., Nohr E. A., Ring S. M., Pourcain B. S., Timpson N. J., Smith G. D., Jarvelin M.-R., Evans D. M., Genome-wide association study of primary tooth eruption identifies pleiotropic loci associated with height and craniofacial distances. Hum. Mol. Genet. 22, 3807–3817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schuler S., Hauptmann J., Perner B., Kessels M. M., Englert C., Qualmann B., Ciliated sensory hair cell formation and function require the F-BAR protein syndapin I and the WH2 domain-based actin nucleator Cobl. J. Cell Sci. 126, 196–208 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Goetz S. C., Anderson K. V., The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331–344 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu X., Wang F., Zhao Y., Yang P., Chen J., Sun H., Liu L., Li W., Pan L., Guo Y., Kou Z., Zhang Y., Zhou C., He J., Zhang X., Li J., Han W., Li J., Liu G., Gao S., Yang Z., A gain-of-function mutation in Tnni2 impeded bone development through increasing Hif3a expression in DA2B mice. PLOS Genet. 10, e1004589 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei B., Jin J.-P., TNNT1, TNNT2, and TNNT3: Isoform genes, regulation, and structure-function relationships. Gene 582, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yakar S., Werner H., Rosen C. J., Insulin-like growth factors: Actions on the skeleton. J. Mol. Endocrinol. 61, T115–T137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wittkopp N., Huntzinger E., Weiler C., Sauliere J., Schmidt S., Sonawane M., Izaurralde E., Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survival. Mol. Cell. Biol. 29, 3517–3528 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabc6160/DC1