

The protective role of nitric oxide in calcific aortic valve disease is mediated by S-nitrosylation of proteins.
Abstract
Calcific aortic valve disease (CAVD) is an increasingly prevalent condition, and endothelial dysfunction is implicated in its etiology. We previously identified nitric oxide (NO) as a calcification inhibitor by its activation of NOTCH1, which is genetically linked to human CAVD. Here, we show NO rescues calcification by an S-nitrosylation–mediated mechanism in porcine aortic valve interstitial cells and single-cell RNA-seq demonstrated NO regulates the NOTCH pathway. An unbiased proteomic approach to identify S-nitrosylated proteins in valve cells found enrichment of the ubiquitin-proteasome pathway and implicated S-nitrosylation of USP9X (ubiquitin specific peptidase 9, X-linked) in NOTCH regulation during calcification. Furthermore, S-nitrosylated USP9X was shown to deubiquitinate and stabilize MIB1 for NOTCH1 activation. Consistent with this, genetic deletion of Usp9x in mice demonstrated CAVD and human calcified aortic valves displayed reduced S-nitrosylation of USP9X. These results demonstrate a previously unidentified mechanism by which S-nitrosylation–dependent regulation of a ubiquitin-associated pathway prevents CAVD.
INTRODUCTION
The estimated global prevalence of calcific aortic valve disease (CAVD) is 12.6 million cases per year and affects ~2% of individuals over 70 years of age (1). Inadequate cardiac output due to left ventricular outflow obstruction caused by the stenotic aortic valve (AoV) results in decreased exercise capacity, progressive left ventricular hypertrophy, and ultimately heart failure and death (2). CAVD is an active and progressive condition and is the main contributor to the development of AoV stenosis in adults (2). Because of the limited molecular understanding of disease mechanisms, no pharmacological treatment is available to effectively prevent the onset or progression of CAVD, and AoV replacement remains the only treatment option (3, 4).
A healthy AoV is able to fully open during systole and close during diastole to create unidirectional blood flow. In CAVD, the valve leaflets cannot open to their full extent because of increased stiffness. The diseased AoV is characterized by thick, fibrotic leaflets, often covered with calcific nodules on the aortic surface (2). Each valve leaflet is composed of three stratified layers of specialized extracellular matrix (ECM), which provide biomechanical strength to withstand a constant oscillating hemodynamic environment. The ECM is interspersed with valve interstitial cells (VICs) and is encapsulated by a monolayer of valve endothelial cells (VECs). There is bidirectional communication between VECs and VICs, which regulates the behavior of each cell type (5, 6). Turnover of the valve ECM is regulated by VICs, which are largely quiescent and fibroblast-like in healthy valves (7, 8). CAVD is proposed to be driven by a phenotypic switch of VICs from fibroblast-like to myofibroblast-like and osteoblast-like cells that deposit bone-like matrix restricting the movement of valve cusps. Valvular heart disease is often triggered by injury and/or impaired VEC-VIC signaling (6, 9).
We previously demonstrated that VEC-derived nitric oxide (NO), a small lipophilic second messenger molecule, plays an important role in AoV development and disease including VIC calcification (10). In vivo and in vitro studies demonstrated that NO can activate the NOTCH1 signaling pathway to inhibit calcification, and mutations in NOTCH1 are associated with bicuspid aortic valve and CAVD in humans (10, 11). Our laboratory established a genetic interaction between endothelial NO synthase and Notch1 in mice that was important for CAVD pathogenesis (10). However, the mechanistic basis of NO-mediated NOTCH1 activation and protection from CAVD was not defined. NO exerts most of its cellular influence either by soluble guanylate cyclase (sGC)/cyclic guanosine 3′,5′-monophosphate (cGMP) pathway activation or by S-nitrosylation, a reversible posttranslational modification of Cys-thiol group (12). S-nitrosylation regulates a wide range of critical cellular functions including metabolism, membrane trafficking, enzyme activity, and stability through both allosteric and active-site modification (13). In addition, S-nitrosylation also regulates other posttranslational modifications such as phosphorylation and ubiquitination, which, in turn, influence protein function and degradation that are important for cardiovascular and various other diseases (13, 14). The ubiquitin-proteasome pathway has been implicated in almost every aspect of cellular function and signaling in eukaryotes (15). As expected, the ubiquitin-proteasome pathway also regulates aspects of the NOTCH signaling pathway (16, 17), which is important for AoV development and disease (11). However, the relationship between these two pathways in the development of valve disease is not clear.
Here, we demonstrated how S-nitrosylation protects the valve leaflet from calcification by inhibiting myofibroblast activation of aortic VICs (AVICs). Mass spectrometry (MS) analysis of S-nitrosylated proteins in AVICs revealed involvement of the ubiquitin-proteasome pathway in the calcific process. We identified S-nitrosylation–mediated activation of a deubiquitinase, USP9X (ubiquitin specific peptidase 9, X-linked), a NOTCH1 signaling regulator in AVICs. We further showed that S-nitrosylation of USP9X stabilizes MIB1, which activates NOTCH1 signaling in neighboring cells to prevent calcification. Our data also revealed the involvement of USP9X in CAVD using a murine model harboring a conditional deletion of Usp9x in endothelial and endothelial-derived cells. In humans, we also observed reduced S-nitrosylation of USP9X in calcified AoVs with an associated reduction in MIB1 and activated NOTCH1 expression, consistent with the proposed mechanism. Overall, we demonstrated a previously unrecognized mechanism that prevents CAVD mediated by S-nitrosylation and the ubiquitin-proteasome pathway.
RESULTS
NO inhibits calcification in AVICs via S-nitrosylation
We and others previously reported that NO donor inhibits spontaneous calcification of cultured porcine AVICs (pAVICs) in vitro (10, 18). In addition, our laboratory found that the presence of NO donor was associated with the activation of Notch1 signaling, but the underlying mechanism was not known (10). NO functions either by activating the sGC/cGMP pathway or by modulating redox-dependent, posttranslational modification, such as S-nitrosylation. To define the NO-mediated mechanism involved in the inhibition of calcification, we specifically activated either the sGC/cGMP pathway, by adding cGMP analog 8-Br-PET-cGMP, or S-nitrosylation, with the addition of S-nitrosoglutathione (GSNO) (Fig. 1). While inhibition of spontaneous calcification of pAVICs after 5 days of culture on a stiff surface (plastic tissue culture plates) was noted in the presence of NO donor, calcific nodules were continued to be observed after activation of the sGC/cGMP pathway, suggesting that NO-mediated inhibition of calcification is independent of this pathway under these in vitro conditions (Fig. 1, A to C and E). Similar to the NO donor treatment, calcification was blocked in the presence of an S-nitrosylating agent (GSNO), consistent with the involvement of S-nitrosylation in the inhibition of calcification (Fig. 1, D and E). RUNX2, a marker of osteoblast differentiation, was down-regulated in the presence of NO donor and GSNO but unaltered after activation of the sGC/cGMP pathway with 8-Br-PET-cGMP (Fig. 1, F to J, and fig. S1, A and B). We confirmed the activation of the sGC/cGMP pathway by measuring phosphorylated vasodilator-stimulated phosphoprotein (pVASP), a known target of the sGC/cGMP pathway (19). pVASP was increased in the presence of 8-Br-PET-cGMP, but its expression was unchanged in the presence of NO donor and GSNO, indicating no activation of the sGC/cGMP pathway (fig. S1, A and B).
Fig. 1. Calcification of pAVICs is attenuated with the addition of DETA NONOate (NO donor) or GSNO (S-nitrosylating agent).
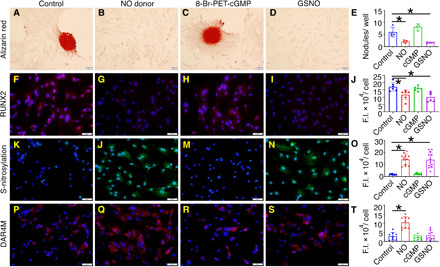
(A to D) Alizarin red staining and nodule formation of pAVICs cultured for 5 days under control conditions or with the addition of NO donor, 8-Br-PET-cGMP (cGMP analog), or GSNO. (E) Quantification of nodule number. (F to I) RUNX2 (red) expression with nuclear 4′,6-diamidino-2-phenylindole (DAPI) staining (blue) is shown in four conditions and quantified in (J). (K to N) Total cellular S-nitrosylated proteins (green) counterstained with DAPI and quantified in (O). (P to S) DAR4M staining (red), which measures NO, is shown with DAPI (blue) counterstaining and is quantified in (T). Scale bars, 50 μm. *P ≤ 0.05 (two-tailed). For all experiments, Mann-Whitney test was performed. F.I., fluorescence intensity.
Furthermore, we found evidence of increased S-nitrosylation of proteins in pAVICs treated with NO donor and GSNO compared to untreated cells or those treated with 8-Br-PET-cGMP, which suggests that S-nitrosylation is important for NO-mediated suppression of calcification in pAVICs (Fig. 1, K to O). We also assessed the amount of NO in pAVICs by diaminorhodamine-4M (DAR4M) staining in the unlikely event that GSNO was degraded into glutathione and NO to activate the sGC/cGMP pathway. NO was significantly increased after addition of NO donor and was not significantly altered after treatment with either GSNO or 8-Br-PET-cGMP (Fig. 1, P to T).
In addition, NO-mediated S-nitrosylation can also inhibit cell cycle progression (20), which would reduce the confluency of pAVICs in culture after NO and GSNO treatment. To test whether reduced confluency of pAVICs contributes to the rescue of calcification, we cultured NO- and GSNO-treated pAVICs for 10 days until similar confluency was achieved compared to control. Calcific nodules were not observed in confluent cultured pAVICs after NO and GSNO treatment (fig. S1C). In addition, expression of both markers for osteogenic activation, RUNX2, and for myofibroblast activation, smooth muscle actin (SMA), was reduced after NO and GSNO treatment (fig. S1D). In this study, we focused on RUNX2 as a marker of osteogenic activity.
NO inhibits pAVIC activation and regulates NOTCH1 target genes
We next evaluated the transcriptional changes that occurred in cultured pAVICs with the addition of NO donor using single-cell RNA sequencing (scRNA-seq). The gene expression of pAVICs cultured in the presence of NO donor for 5 days was compared to that of untreated pAVICs. Unbiased clustering of the gene expression from both of these cell populations using Uniform Manifold Approximation and Projection (UMAP) demonstrated two distinct populations and suggested that the effect of NO on pAVICs is uniform and not specific to a subpopulation of cells (Fig. 2A).
Fig. 2. Gene expression analysis of pAVICs exposed to NO donor or GSNO by scRNA-seq.
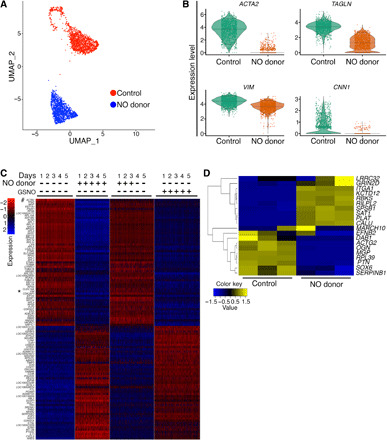
(A) UMAP plot of scRNA-seq expression data shows two distinct populations of pAVICs cultured in the presence (blue) or absence (red) of NO donor. (B) Violin plots show down-regulation of ACTA2, TAGLN, VIM, and CNN1 with NO donor. Each dot in the violin plots represents gene expression in individual cells. Horizontal lines represent 0.05, 0.5, and 0.95 quantiles of gene expression. (C) Heatmap represents Z-scored expression of top 100 differentially expressed genes in individual cells from the four culture conditions on day 5. # and * indicate ACTA2 and VIM, respectively. (D) Heatmap demonstrating Z-scored, log2-transformed expression of NOTCH1 target genes in pAVICs in the presence and absence of NO donor.
To confirm that pAVICs were representative of VICs in vivo, we compared our in vitro scRNA-seq data from pAVICs to published scRNA-seq data of 1-month-old [postnatal day 30 (P30)] mouse AoV cells (21). We observed significant correlation between the expression of mouse AVIC marker genes, Col1a1, Col3a1, and Serpinf1, with pAVICs and the weak expression of endothelial (Cdh5, Nos3, and Emcn), melanocyte (Dct, Mitf, and Tyrp1), or immune cell (Tyrobp, Ptprc, and Cx3cr1) genes in pAVICs (fig. S2A). This observation indicates that the pAVIC culture is homogeneous and does not contain a significant percentage of other cell types found within the adult aortic valve.
Differential expression analysis of scRNA-seq data of pAVICs revealed that ACTA2 (α-SMA) expression is strongly suppressed in pAVICs cultured in the presence of NO donor (Fig. 2B and fig. S2B). As ACTA2 is a characteristic marker of myofibroblast differentiation, this decrease demonstrates that NO suppresses conversion of AVICs into myofibroblast-like cells. In addition, other myofibroblast markers, such as TAGLN (transgelin), VIM (vimentin), and CNN1 (calponin) (22), were also reduced, further supporting the idea that NO inhibits myofibroblast activation of pAVICs (Fig. 2B and fig. S2B). To test whether this NO-mediated inhibition of myofibroblast activation is transient, we maintained pAVICs in the presence of NO donor for the first 3 days followed by culturing cells without NO donor for an additional 2 days and performed scRNA-seq. RNA expression profile after NO donor withdrawal is similar to that of the pAVICs cultured in the absence of NO donor (Fig. 2C). This suggests that the effect of NO on gene expression is not permanent, and cellular markers of a myofibroblast-like state, including ACTA2 and VIM, rapidly reappear in these cells (Fig. 2C and data file S1). To examine gene expression after S-nitrosylation, we cultured pAVICs for 5 days in the presence of GSNO followed by scRNA-seq. We observed that the gene expression profile after GSNO exposure is similar to NO donor exposure, implying that NO-mediated changes to gene expression in pAVICs are in large part due to S-nitrosylation in this in vitro system (Fig. 2C). Consistent with this mechanism, expression of GUCY1A3 and GUCY1B3, which encodes sGC, was not significantly affected after addition of NO donor (fig. S2B). This observation further indicates that the sGC/cGMP pathway is not involved in NO-mediated inhibition of myofibroblast activation and calcification of pAVICs, and the effect of NO on calcification of cultured pAVICs is due to S-nitrosylation.
Our previous studies have shown that NOTCH signaling is increased by NO treatment, as demonstrated by increased nuclear localization of the NOTCH1 intracellular domain (NICD) (10). Examination of scRNA-seq data demonstrated that NOTCH1 mRNA expression was not increased after NO treatment (fig. S2B). This prompted us to examine whether NOTCH1 signaling targets were affected by NO treatment. We compared pAVIC scRNA-seq data with published NOTCH1 target genes that were identified by combining chromatin immunoprecipitation sequencing and RNA-seq datasets from NOTCH1 small interfering RNA (siRNA)–treated human aortic VECs (AVECs) subjected to shear stress (23). We observed changes of NOTCH1 target genes in pAVICs after NO exposure similar to that observed in human AVECs in response to shear stress (Fig. 2D and fig. S2C). This similarity of gene expression confirms that NOTCH signaling is affected by NO treatment in pAVICs. The absence of NOTCH1 mRNA changes at the transcriptional level, but alterations in NOTCH1 signaling targets supported a posttranslational mechanism for the regulation of NOTCH1 signaling by NO. On the basis of the transcriptomic changes observed with GSNO treatment, S-nitrosylation was the most likely posttranslational mechanism for this regulation.
USP9X, an activator of NOTCH1 signaling, is S-nitrosylated in pAVICs
To identify proteins that were S-nitrosylated after exposure to NO donor in AVICs, we used a modified biotin switch technique (BST) followed by MS [liquid chromatography–tandem MS (LC/MS-MS)]. Rat AVICs were used for the LC/MS-MS identification of S-nitrosylated proteins as the porcine protein sequences are not well annotated. Similar to pAVICs, rat AVICs undergo calcification and the addition of NO donor inhibited their osteogenic differentiation, as shown by RUNX2 expression (fig. S3A). Modified BST followed by LC/MS-MS identified a total of 579 S-nitrosylated proteins in rat AVICs (untreated control and NO donor–treated samples), and among them, 217 proteins were overrepresented after NO treatment (fig. S3, B and C, and data file S2). Enrichment analysis of S-nitrosylated proteins using WebGestalt 2019 (WEB-based Gene SeT AnaLysis Toolkit) (24) revealed that ubiquitin-specific processing proteases were the most enriched pathway after NO treatment. In addition, the deubiquitination pathway, representing 12 similar proteins as identified for ubiquitin-specific processing proteases, was also overrepresented (Fig. 3A).
Fig. 3. Mass spectrometric analysis of S-nitrosylated proteins in rat AVICs exposed to NO donor demonstrates an enrichment of ubiquitin-specific processing proteases and identifies USP9X.
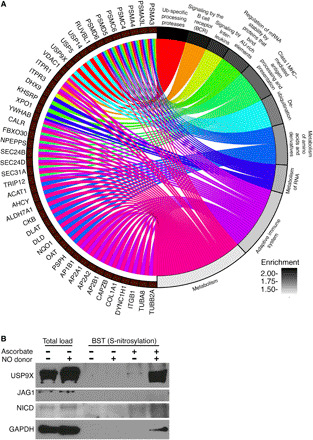
(A) Chord plot illustrates overrepresentation of NO-enriched S-nitrosylated proteins. Top 10 Gene Ontology pathways (GO Slim) and gene sets are shown. (B) Immunoblot shows S-nitrosylation of USP9X in the presence of NO donor, where negative ascorbate serves as a technical negative control. NICD and JAG1 immunoblots with ascorbate serve as biological negative controls. GAPDH, a known S-nitrosylated protein, serves as a biological positive control. Total load represents the initial cell lysates used to purify S-nitrosylated proteins. AU-rich element, adenylate/uridylate-rich elements.
Among the 12 S-nitrosylated proteins, we were specifically interested in USP9X, given its previous link to NOTCH signaling (25). USP9X is an evolutionarily conserved deubiquitinase, and its Drosophila homolog, faf, regulates NOTCH signaling during eye development (26). Further, in human breast cancer cell lines, USP9X deubiquitinates and stabilizes MIB1, an activator of ligand-dependent canonical NOTCH1 signaling that is important for cardiac development (25, 27). Therefore, we postulated that nitrosylated USP9X deubiquitinates and stabilizes MIB1 in valvular interstitial cells, which potentiates the ability of a ligand-producing cell to activate NOTCH on a neighboring cell. First, we confirmed that USP9X undergoes posttranslational modification by S-nitrosylation in pAVICs after NO treatment using modified BST followed by immunoblotting for USP9X (Fig. 3B). In similar conditions, JAG1 and activated NICD, which are two important components of NOTCH1 signaling, did not show evidence of S-nitrosylation and served as negative controls. Glyceraldehyde phosphate dehydrogenase (GAPDH), a well-known S-nitrosylated protein (28), was used as a positive control (Fig. 3B).
NO activates USP9X to deubiquitinate MIB1
To investigate whether the deubiquitination activity of S-nitrosylated USP9X regulates NOTCH1, we examined the ubiquitination status of MIB1, a known activator of NOTCH1 signaling. For these studies, we generated stable human embryonic kidney (HEK) 293 cells expressing both MIB1-FLAG and hemagglutinin (HA)–ubiquitin (fig. S4A). This cell line expressing tagged MIB1 and ubiquitin (Ub) was treated with NO donor, GSNO, or WP1130, a cell-permeable small molecule reported as an inhibitor of deubiquitinases, including USP9X (29) for 48 hours, and proteasomal degradation was blocked by the addition of N-carbobenzyloxy-l-leucyl-l-leucyl-l-leucinal (MG132) before the preparation of cell lysate. Immunoprecipitation of MIB1 using an anti-FLAG antibody was followed by measurement of HA-Ub-MIB1-FLAG using anti-HA antibody (fig. S4A). For this experiment, S-nitrosylation of USP9X was confirmed after treatment with NO donor or GSNO while remaining at basal level in untreated control and after WP1130 treatment (fig. S4B).
Ubiquitin is an 8.6-kDa protein, and varying degrees of ubiquitination are possible for a target protein (mono-, multimono-, and polyubiquitination). Therefore, bands of increasing molecular weights found by immunoblot after the proteasome block represent ubiquitination (fig. S4A). With the addition of NO donor and GSNO, we found decreased ubiquitination of MIB1 when compared to treatment with the deubiquitinase inhibitor WP1130 or untreated control (Fig. 4, A to D). No significant difference in the expression of endogenous USP9X or MIB1-FLAG was observed, indicating that up-regulated deubiquitinase activity is due to S-nitrosylation (Fig. 4, A to D).
Fig. 4. S-nitrosylation activates USP9X to deubiquitinate MIB1.
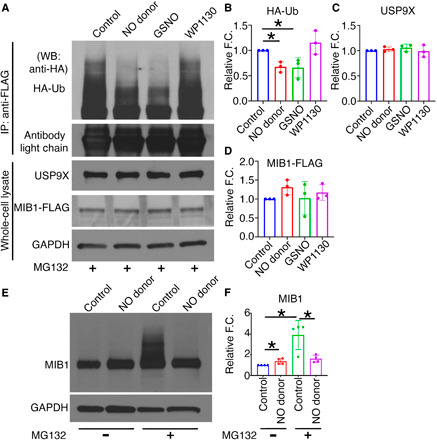
HEK293 cells expressing MIB1-FLAG and HA-Ub were cultured in the presence of DETA NONOate (NO donor), GSNO (S-nitrosylating agent), or WP1130 (USP9X inhibitor). (A) After addition of MG132 and immunoprecipitation (IP) with anti-FLAG antibody, immunoblot with anti-HA antibody shows HA-Ub-MIB1-FLAG of various molecular weights in untreated condition (control) and with WP1130, indicating the presence of poly-Ub-MIB1. This is decreased with NO donor and GSNO and is quantified in (B). (C and D) Quantification of USP9X and MIB1-FLAG expression after normalization against GAPDH. (E) pAVICs were cultured in the presence and absence of NO donor for 5 days, and immunoblot for MIB1 is shown with and without the addition of MG132. (F) Quantification of MIB1 is shown after normalization against GAPDH. *P ≤ 0.05. For Western blots (WB), two-tailed unpaired t test was performed. F.C., fold change.
To examine the effect of S-nitrosylation of USP9X on the deubiquitination of MIB1, we cultured pAVICs for 5 days in the presence and absence of NO donor. To measure ubiquitination, we inhibited proteasomal degradation by adding MG132, and ubiquitinated MIB1 was examined by immunoblot. We found decreased ubiquitination of MIB1 after NO donor treatment compared to the untreated condition, which confirms the NO-dependent activation USP9X in pAVICs (Fig. 4, E and F).
Inhibition of USP9X reduces expression of MIB1 and NOTCH1 to promote calcification
To examine the effect of USP9X inhibition on NOTCH1 signaling and calcification of pAVICs, we used both pharmacological inhibition (with WP1130) and siRNA-mediated knockdown of USP9X (Fig. 5). The addition of WP1130 to pAVICs decreased levels of MIB1 and NICD, while treatment with NO donor was associated with increased MIB1 and NICD (Fig. 5, A and B, and fig. S5, B and C). Accordingly, we also found increased RUNX2 expression with WP1130 treatment compared to untreated cells while RUNX2 expression decreased with NO donor (Fig. 5, C and D). Notably, culturing pAVICs with NO donor or WP1130 did not alter USP9X expression (fig. S5A). In addition, our scRNA-seq data indicated no significant alteration of MIB1 expression at the transcriptional level after exposure to NO donor (fig. S2B). Overall, these data indicate that S-nitrosylated USP9X stabilizes MIB1, which is involved in NOTCH1 activation and inhibition of calcification.
Fig. 5. Inhibition of USP9X causes reduced expression of MIB1 and NICD and increased RUNX2 expression in pAVICs.
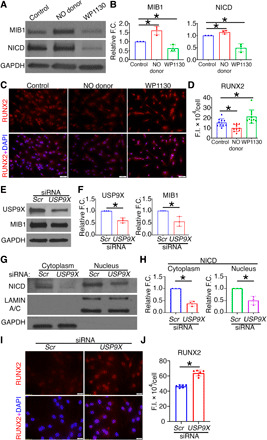
(A) Immunoblot shows decreased levels of MIB1 and NICD in pAVICs cultured with WP1130 for 5 days as compared to untreated cells (Control), while NO donor results in increased MIB1 and NICD. (B) Quantification of MIB1 and NICD expression is shown. (C) RUNX2 expression (red) is increased with WP1130, while RUNX2 is decreased with NO donor when compared to untreated cells (Control). Costained with nuclear DAPI (blue). (D) Quantification of RUNX2 expression is shown. (E) Addition of USP9X-siRNA to pAVICs reduced protein expression of USP9X and MIB1 as compared to scramble (Scr) control, which is quantified in (F). (G) With USP9x knockdown, nuclear NICD is reduced, and this is quantified in (H) after normalization with LAMIN A/C (nuclear). Cytoplasmic NICD was normalized with GAPDH (G and H). (I) After treatment with USP9X-siRNA, RUNX2 expression (red) is increased when compared to scramble (Scr) control cells, costained with nuclear DAPI (blue). (J) Quantification of RUNX2 expression is shown. Scale bars, 50 μm (C) and 20 μm (I). *P ≤ 0.05 (two-tailed). For immunostaining and WBs, Mann-Whitney test and unpaired t test were performed, respectively.
In addition to USP9X, WP1130 can target other deubiquitinases (29) such as USP5 and USP14, which are S-nitrosylated after NO donor treatment (Fig. 3A). USP5 and USP14 are also reported to regulate Notch and Wnt signaling pathways in cancer and in Drosophila eye development (30, 31). As WP1130 can target other deubiquitinases besides USP9X, we used siRNA to knock down USP9X for these studies. After optimization, we used 30 nM concentration of pooled siRNA against USP9X, which resulted in a 60% reduction in USP9X mRNA and 40% reduction in USP9X protein (Fig. 5, E and F, and fig. S5D). With USP9X knockdown in pAVICs, we found reduced levels of MIB1 and NICD (Fig. 5, E to H, and fig. S5, E to G). In addition, we found evidence of osteogenic differentiation with up-regulation of RUNX2 (Fig. 5, I and J, and fig. S5, H and I). Together, these data indicate that NO activates USP9X by S-nitrosylation, which deubiquitinates and stabilizes MIB1, and this is followed by the activation of NOTCH1 signaling to prevent calcification.
Mice harboring endothelial cell deletion of Usp9x display aortic valve stenosis and regurgitation
To determine whether USP9X deficiency leads to AoV disease in vivo, we used mice in which Usp9x was genetically deleted. As germline deletion of Usp9x results in early embryonic lethality, mice harboring a floxed Usp9x allele (Usp9xfl/fl) were used, which allowed conditional deletion of the gene (32, 33). Usp9x resides on the X chromosome; therefore, only male mice were used to test the effect of Usp9x deletion. Usp9xfl/fl females were bred with males harboring Tie2Cre, which is expressed in the endothelial/endocardial cell lineage along with a subset of hematopoietic cells (34) (Fig. 6A). The progenies were genotyped at P10, and then only male mice were switched to a high-fat Western diet at 6 weeks of age. These mice subsequently underwent echocardiography at 6, 16, and 24 weeks of age followed by euthanasia to perform gross and cardiac histological examination (Fig. 6B). We observed a partial lethality before P10 in Usp9xfl/Y; Tie2Cre male mice, and this is presumed to occur during embryogenesis or soon after birth (Fig. 6C). Upon examination, the adult male survivors showed an increase in heart size within the Cre+ animals and confirmed reduction of USP9X protein expression in the AoVs of Cre+ mice as compared to Cre− mice (fig. S6, A and B). Echocardiographic analysis of the Cre+ mice revealed a significantly increased velocity across the AoV at 6, 16, and 24 weeks of age compared to Cre− controls (Usp9xfl/Y; Tie2Cre-), indicative of functional AoV stenosis (Fig. 6D). In addition, an increase in left ventricular end-systolic volume was noted at 16 and 24 weeks of age, consistent with increased afterload associated with AoV stenosis (fig. S6C). While not statistically significant, there was a trend toward decreased left ventricular ejection fraction and increased left ventricular end diastolic volume, suggestive of ventricular dysfunction (fig. S6C).
Fig. 6. Aortic valve disease in mice with Usp9x deletion in endothelial cell lineage.
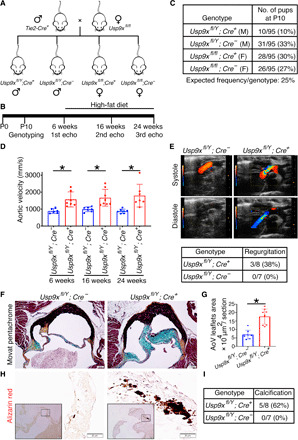
(A) Breeding scheme to generate mice harboring endothelial and endothelial-derived cell deletion of Usp9x (Usp9xfl/fl; Tie2Cre). (B) Experimental scheme for Usp9xfl/Y; Cre+ males and Usp9xfl/Y; Cre− controls. (C) Survival in Usp9xfl/Y; Cre+ male mice at P10. (D) Aortic velocity measured by echocardiogram in Cre+ males at 6, 16, and 24 weeks of age compared to Cre− controls. (E) Doppler image shows aortic valve regurgitation in Cre+ male at 24 weeks of age as compared to Cre− control. Table shows incidence of aortic valve regurgitation in Cre+ and Cre− males. (F) Histological section of aortic valve stained with Russell-Movat pentachrome shows increased proteoglycan deposition in Cre+ males compared to Cre− controls. (G) Graph shows quantification of aortic valve area in Cre+ and Cre− males. (H) Alizarin red staining demonstrates calcification in Cre+ males. (I) Table shows incidence of calcification in Cre+ and Cre− males. Scale bars, 100 μm (pentachrome staining) and 20 μm (Alizarin red staining). *P ≤ 0.05 (two-tailed Mann-Whitney test).
Three of eight Cre+ mice (38%) demonstrated AoV regurgitation, indicative of incomplete coaptation of the AoV leaflets compared to Cre− (zero of seven) controls (Fig. 6E and movies S1 and S2). Histological analysis using Russell-Movat pentachrome staining of AoVs collected from 6-month-old mice revealed proteoglycan accumulation throughout the thickened leaflets of Cre+ mice as compared to Cre− control (Fig. 6, F and G). Evidence of calcification was detected by positive Alizarin red staining in five of eight Cre+ mice (62%) as compared to the lack of staining in the valves of Cre− control mice (zero of seven) at 6 months of age (Fig. 6, H and I). Expression of RUNX2 was increased, while MIB1 and NICD were decreased in Cre+ as compared to Cre− control mice (fig. S6, D to H).
Calcified human aortic valves display reduced S-nitrosylation of USP9X
To determine whether the S-nitrosylation of USP9X was altered in CAVD, we obtained explanted AoV tissue from patients at the time of surgical valve replacement. Aortic stenosis was the indication for surgery, and all of the patients had tricuspid aortic valves. The disease was categorized as moderate (n = 2) or moderate-severe (n = 2) valve calcification (fig. S7A). In all calcified valve leaflets, RUNX2 expression was increased, as expected (Fig. 7A). We also found a reduction in the amount of S-nitrosylated USP9X, which was inversely correlated with increased disease severity (Fig. 7A). We also found an overall reduction of USP9X protein expression in calcified human valves (Fig. 7A). Further quantification of S-nitrosylated USP9X normalized against total USP9X demonstrates reduced USP9X-S-nitrosylation in all of the moderate-severely calcified valves (fig. S7B). These data suggest that in addition to S-nitrosylation, expression of USP9X may also contribute to CAVD in humans. Furthermore, there was reduced expression of MIB1, NICD, and the Notch ligand, JAG1, when compared to a control noncalcified aortic valve (Fig. 7A and fig. S7C).
Fig. 7. Calcified human aortic valves display reduced S-nitrosylation of USP9X along with decreased expression of MIB1 and NICD.
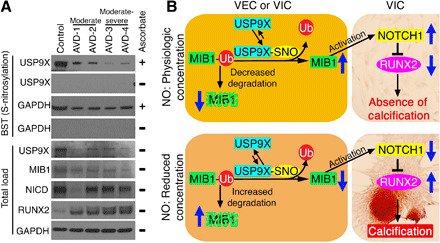
(A) Immunoblot for USP9X shows significantly reduced S-nitrosylation in aortic valves from adults with moderate (AVD-1 and AVD-2) and moderate-severe (AVD-3 and AVD-4) CAVD when compared to uncalcified aortic valve (Control). S-nitrosylation of GAPDH is also shown. Absence of a band in the ascorbate-negative experiment served as the negative control. Total load shows protein expression in the cell lysates before purification of S-nitrosylated proteins where USP9X, MIB1, and NICD expression is reduced in calcified valves while RUNX2 is increased. GAPDH serves as a loading control. (B) Schematic showing the proposed model where S-nitrosylation of USP9X leads to MIB1 stabilization via deubiquitination, which increases ligand-mediated NOTCH1 activation in neighboring cells and the inhibition of calcification in physiological concentrations of NO. This pathway is abrogated under low concentrations of NO.
Overall, these data demonstrate NO-mediated inhibition of AoV calcification via activation of a USP9X-MIB1-NOTCH1 signaling axis. We propose that under physiological NO concentrations, S-nitrosylation of USP9X results in stabilization of MIB1 by its deubiquitination, which prevents proteasomal degradation (Fig. 7B). The stable MIB1 subsequently triggers ligand-dependent NOTCH1 activation in neighboring cells, which inhibits RUNX2 expression and the calcification gene program. At low NO concentrations, USP9X is not activated by S-nitrosylation allowing for degradation of MIB1 through the ubiquitin-proteasome pathway. The loss of MIB1 prevents ligand-dependent NOTCH1 activation, thereby promoting calcification (Fig. 7B).
DISCUSSION
In this study, we describe a previously unknown molecular mechanism where NO regulates NOTCH1 signaling via the deubuiquitinase, USP9X, in the setting of CAVD. Here, we show that S-nitrosylation, a NO-dependent posttranslational modification, is important for the prevention of CAVD through inhibition of myofibroblast activation of AVICs. Our proteomic analysis revealed the role of S-nitrosylation in regulation of the ubiquitin-proteasome pathway. We also discovered that S-nitrosylation of USP9X is critical for its stabilization of MIB1, which potentiates the ligand-mediated activation of NOTCH1 signaling. Furthermore, deletion of Usp9x in the endothelial cell lineage in mice resulted in aortic valve stenosis and calcification. Last, translational studies using calcified human AoVs showed alterations in levels of S-nitrosylated USP9X, MIB1, and NICD that were consistent with our proposed mechanism (Fig. 7B).
Our group along with others have reported the protective role of a healthy valve endothelium in CAVD and, in our previous publication, showed that this protection is partly achieved by endothelial-derived NO regulation of NOTCH signaling (10, 18, 35). Using an ex vivo approach, a protective effect from endothelial NO that was mediated by an sGC/cGMP-dependent pathway was also reported by Richards et al. (18). An sGC/cGMP-dependent mechanism has also been supported by the investigation of mechanisms of vascular calcification where NO was shown to regulate transforming growth factor–β (TGF-β) signaling (36). Further, C-type natriuretic peptide inhibits myofibroblast activation and osteogenic differentiation of VICs via activation of cGMP-dependent protein kinase in mice (37, 38). In total, these studies indicate that sGC/GMP-dependent mechanisms play a role in vivo. While we did not find that activation of sGC/cGMP was sufficient to protect pAVICs from calcification in our in vitro system, we cannot exclude the possibility that elevated NO production by increased laminar blood flow or shear stress with subsequent activation of the sGC/cGMP pathway is protective against CAVD.
Apart from the NOTCH pathway, USP9X can also regulate the Wnt, TGF-β, and Hippo signaling pathways, which are also important for AoV development and disease (32, 39). The impact of S-nitrosylation of USP9X on these pathways is not known. In addition to USP9X, we also have identified S-nitrosylation of other ubiquitin pathway members in our proteomic analysis of AVICs treated with NO donor, and they include USP5 and USP14 (Fig. 3A). Similar to USP9X, USP5 and USP14 are also reported to regulate NOTCH and Wnt signaling pathways in a variety of cancers and also in Drosophila eye development (30, 31). USP5 regulates EMT by stabilizing SLUG, while USP14 controls ciliogenesis by regulating Hedgehog signaling (40, 41). Both of these pathways have well-established roles in valve morphogenesis but have not been investigated in valve calcification (42, 43). Intriguingly, the ubiquitin-proteasome pathway was the topmost, and the deubiquitination pathway was in the top 10 pathways in our proteomic analysis (Fig. 3A). The ubiquitin-proteasome pathway, including deubiquitination, has already drawn attention as a potential drug target for other diseases, such as cancer, neurodegenerative diseases, and immunological disorders as it is involved in numerous important regulatory processes. The small number (<100) of deubiquitinases are likely subject to multiple layers of regulation that modulate both their activity and their specificity (44). However, the role of the ubiquitin-dependent pathway in valve development and disease, including calcification, has not been studied. To the best of our knowledge, this is the first report demonstrating the association between the ubiquitin pathway and valve calcification.
Our data suggest that the S-nitrosylated USP9X deubiquitinates to stabilize MIB1, which then ubiquitinates the NOTCH ligands, a crucial step for ligand internalization and NOTCH1 activation in neighboring cells (45, 46). The importance of MIB1 and JAG1 for AoV development and calcification has been shown. Deletion of Mib1 in cardiac progenitor cells using Nkx2.5-Cre leads to neonatal lethality due to valve dysfunction, and deletion of Jag1 in endothelial cells using Cdh5-Cre results in a spectrum of cardiac phenotypes from congenital heart defects, reminiscent of tetralogy of Fallot, to adult valve disease characterized by thickened semilunar valves and calcification (27, 47). We used Tie2-Cre mice to delete Usp9x in endothelial and endothelial-derived cells and observed an embryonic/perinatal lethality of ~58% (Fig. 6C). The etiology of this lethality is under investigation.
As both Cdh5 and Tie2 express before EMT, these Cre lines can delete target genes in endothelial and endothelial-derived cells, such as VICs (34). Therefore, we are unable to determine whether USP9X function is important in either VECs or VICs, or both. In an attempt to address this question in vivo, we deleted Usp9x using Twist2-Cre, which can delete target genes in only VICs but we were unsuccessful in generating live adult mice with deletion of Usp9x in VICs (48). We also performed in vitro experiments using porcine AVECs (pAVECs) and pAVICs. Here, USP9X knockdown was performed using siRNA against USP9X in pAVECs, pAVICs, or both. When pAVECs or pAVICs were cultured alone, we found increased expression of RUNX2 in pAVICs at baseline, and USP9X knockdown in either cell type resulted in increased expression, although it was more dramatic in pAVICs (fig. S8, A to D and I). When pAVICs and pAVECs were cocultured, we observed decreased RUNX2 expression, as expected (fig. S8, C, E, and I). In this coculture system, USP9X knockdown in either pAVICs or pAVECs also resulted in increased RUNX2 expression, but here, it was more dramatic in pAVECs (fig. S8, E to I). These in vitro experiments support roles for USP9X in both valve endothelial and interstitial cells in CAVD.
Our scRNA-seq analysis of pAVICs, which spontaneously calcify in vitro, and reports from other laboratories demonstrate that VICs in culture conditions are already activated toward myofibroblast differentiation with the expression of the stress fiber gene ACTA2 (22, 49). This myofibroblast activation can be inhibited by culturing VICs in three-dimensional hydrogels or by using a fibroblast media formulation and a collagen-coated surface that maintains a quiescent phenotype of VICs (22, 49). Here, we observed that S-nitrosylation can reverse myofibroblast VICs toward an inactivated/quiescent VIC by reducing expression of ACTA2, VIM, TAGLN, and CNN1 (Fig. 2B). However, this phenotypic reversal is temporary and always requires elevated NO concentrations in culture, which indicates that S-nitrosylation is required for maintenance of the quiescent state of VICs (Fig. 2C). The in vivo significance of this observation for CAVD pathogenesis requires further study.
In a healthy valve, NO is primarily produced by NOS3, which is expressed by endothelial cells (21). Expression of the NOS3 gene is regulated by the transcription factors Krüppel-like factor 2 (KLF2) and KLF4. KLF2 and KLF4 are known to be regulated in endothelial cells by blood flow via the mechanosensor PLEXIND1 (50). Flow-dependent endothelial cell–derived NO is a freely diffusible, but short-lived molecule. Its diffusion coefficient in tissue is nearly fourfold smaller than in solution (51). This low diffusion rate may restrict the ability of NO to travel from VECs of the ventricular side to the VICs populating the aortic side of the leaflet. We observed increased S-nitrosylation on the flow side of the aortic and mitral valves in mice (fig. S8J). Therefore, an increased amount of activated USP9X and NOTCH1 signaling are expected in the ventricular side of the aortic valve, which is consistent with the observation that calcific nodules primarily occur on the fibrosa/aortic side of the human aortic valve (52). Notably, mouse heart valves are much thinner compared to human heart valves, and therefore, the bioavailability of diffusible NO is more in mice as compared to humans. This may explain the relative difficulty in developing models of CAVD in mice and potentially explains why additional genetic manipulation that results in thickened leaflets is required to see the development of CAVD in Notch1 heterozygous mice (10, 53). Last, epidemiologic studies have demonstrated gender differences in CAVD with males being more affected than females (35). While this issue was not investigated here, further experiments are required to determine whether USP9X, which is located on the X chromosome, may contribute to these observed differences (54).
In conclusion, we have discovered a previously unknown molecular mechanism by which endothelial NO regulates the NOTCH1 signaling pathway in CAVD that involves the ubiquitin-proteasome system. These findings open the door to investigate regulators of the ubiquitin-proteasome pathway as potential therapeutic targets in CAVD.
METHODS
AVEC and AVIC culture and treatments
pAVICs and pAVECs were collected from juvenile pig valve cusps and isolated as previously described (55). Briefly, valve leaflets were subjected to 5-min collagenase (Worthington Biochemical, #LS004176) digestion at 37°C, and the endothelial layer was gently sheared by a sterile swab onto the leaflet. Dislodged pAVECs were collected from the cell suspension and cultured in endothelial media as described earlier (55). At near confluency, the pAVECs were passaged with TrypLE Express (Thermo Fisher Scientific, #12-604-021) and further cultured in endothelial cell growth medium-2 (EGM-2) media (Lonza, #CC-3162). pAVECs used in this study were between passages 2 and 4. After removal of the endothelial layer, valve leaflets were subjected to a 15-hour collagenase digestion at 37°C, and the pAVICs were cultured in an interstitial porcine medium as previously described (55). At near confluency, pAVICs were passaged with trypsin-EDTA. For all experiments, pAVICs were cultured on stiff surface (plastic tissue culture plates) in Media 199 (Thermo Fisher Scientific, #11150059) supplemented with 10% fetal bovine serum. Only cells between passages 3 and 7 were used. Rat aortic VICs (rat AVICs) were similarly isolated from Sprague-Dawley adult rats by a collagenase digestion and cultured in Media 199. To identify S-nitrosylated proteins, we used the modified BST followed by MS. Rat AVICs were cultured in osteogenic media, by supplementing Media 199 with ascorbate-2-phosphate (50 μg/ml; Sigma-Aldrich, #49752), 10 nM dexamethasone (Sigma-Aldrich, #D4902), and 10 μM β-glycerol phosphate (Sigma-Aldrich, #G9422). For each cell culture experiment performed in the presence of exogenous reagents, media were supplemented with either DETA NONOate (NO donor: 150 μM) (Thermo Fisher Scientific, #AC328651000), GSNO (S-nitrosylating agent: 200 μM) (MilliporeSigma, #N4148), 8-Br-PET-cGMP (cGMP analog: 0.025 μM) (Axxora, #BLG-P003-10), or WP1130 (USP9X inhibitor: 1 μM for HEK293 and 0.05 μM for pAVICs) (Cayman, #15227) and were refreshed daily. To stop proteasomal degradation, 10 μM MG132 (MilliporeSigma, #474787) was added to the culture media 10 hours before preparation of the cell lysate.
Alizarin red staining
To stain calcific nodules, pAVICs were fixed in 2.5% paraformaldehyde (PFA) for 15 min at 4°C, incubated with Alizarin red solution (Millipore, #2003999) for 5 to 7 min, followed by washing with distilled water. The number of nodules per well was counted, and images were captured using an Olympus BX51 microscope attached with an Olympus DP71 camera.
Immunofluorescence staining of S-nitrosylated proteins
To visualize the S-nitrosylated proteins in pAVICs (n ≥ 3) and in a 3-month-old murine valve tissue section, BST was performed using the S-Nitrosylated Protein Detection Kit (Cayman Chemical, #10006518). Immunofluorescence staining was performed following the manufacturer’s protocol, and images were captured using an Olympus IX51 microscope attached with an Olympus DP72 camera. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI).
DAR4M staining
To estimate endogenous levels of NO, unfixed pAVICs were incubated with 10 μM DAR-4M AM (Sigma-Aldrich, #251765) for 15 min at room temperature, followed by washing with phosphate-buffered saline (PBS). Nuclei were stained with DAPI. Images were captured using an Olympus IX51 microscope attached with an Olympus DP72 camera.
scRNA-seq and analyses
pAVICs were dissociated with TrypLE Express, followed by filtration through a 40-μm cell strainer (BD Falcon, #352340) and resuspension in 0.04% bovine serum albumin (BSA). Cell viabilities (>85%) were measured using the Automated Cell Counter (Thermo Fisher Scientific, #AMQAF1000). Single-cell droplet libraries from this suspension were generated in the 10X Genomics Chromium controller (~2000 target cell recovery per group) according to the manufacturer’s instructions. For each sample, the quality and integrity of complementary DNA (cDNA) and cDNA libraries were quantitated using the High Sensitivity D5000 and D1000 ScreenTape (Agilent, #5067-5592 and 5067-5584) on the Agilent 2200 TapeStation. All libraries were sequenced (150 base pairs paired-end) in an Illumina HiSeq 4000 platform at The Steve and Cindy Rasmussen Institute for Genomic Medicine in Nationwide Children’s Hospital.
We used the 10X Genomics’ Cell Ranger pipeline to demultiplex and convert Illumina bcl output into fastq files. We mapped the fastq reads to the pig genome Sscrofa11 with Y sequences from WTSI_X_Y_pig V2 (GCF_000003025.6_Sscrofa11.1_genomic.fna) and gene annotation (GCF_000003025.6_Sscrofa11.1_genomic_genes.filtered.gtf) downloaded from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/assembly/GCF_000003025.6/). scRNA-seq data with different treatments (control, GSNO, NO donor, and NO donor withdrawal after 3 days) were combined using the cell ranger aggregate function yielding a total of 7037 cells (control, 1683; GSNO, 1966; NO donor, 1485; and NO donor withdrawal, 1903) with an average of 3397 genes per cell. These data were processed using Seurat (version 3.0) (56) to perform normalization and identification of genes with the highest variability. The top 100 genes that showed significant differential expression (adjusted P ≤ 0.05) between control and NO-treated samples were used to plot the heatmap. Z-score–transformed scaled expression of genes between the four samples was plotted using the DoHeatmap function of Seurat (3.0) package in R.
Genes up-regulated (green) and down-regulated (pink) are shown in a volcano plot based on the negative log10P value (2 cutoff) between control and NO-treated samples (fig. S2). The volcano plot was created using the ggplot2 (3.2.1) package of R.
In parallel, we imported P30 mouse aortic valve data (21) from the Gene Expression Omnibus (GEO) database (GSE117011). To compare the mouse data to our porcine data, we replaced the mouse gene names with corresponding porcine homolog’s gene names downloaded from BioMart (Ensemble; https://useast.ensembl.org/info/data/biomart/index.html). Mouse gene names without a clear porcine homolog were retained as mouse gene names. These data were also processed using Seurat similar to the porcine single-cell data. The mouse and the porcine data were then integrated and scaled, and dimensionality reduction was performed using Seurat. Violin plots were created to show marker gene expression in different clusters and drawn using a modified VlnPlot from Seurat (3.0) package in R.
Differential gene expression analysis was performed by splitting the pAVICs of one treatment into three sample groups of equal number of cells. These in silico replicates were then analyzed using DESeq2 (57) to identify differentially expressed genes between conditions. We compared the gene expression changes in the pAVICs to the expression changes observed in human AVECs subjected to shear stress (23) by comparing the relative gene expression changes in each sample. For the pAVICs, we used relative changes with respect to control cells, and for the human AVECs, we used control cells treated with shear stress as our baseline. Shear-responsive genes with a significant impact (P ≤ 0.05) in their expression in endothelial cells after NOTCH1 knockdown using siRNA were used to identify homologs in the porcine genome. Genes that had clear homologs in porcine genome (downloaded from BioMart), and those that were expressed in pAVICs (at least one read count in any of the samples), were evaluated for changes in expression following NO treatment.
BST and MS
BST was performed as previously described with minor modifications (58). Until the purification of the S-nitrosylated proteins, all of the procedures were performed in the dark. Briefly, cultured cells or frozen aortic valve tissues that were crushed under liquid nitrogen were harvested in HEN (100 mM Hepes, 1 mM EDTA, and 0.1 mM neocuproine) lysis buffer containing 50 mM NaCl, 0.2% methyl methanethiosulfonate (MMTS), 1 mM phenylmethylsulfonyl fluoride, 1% NP-40, and protease inhibitor cocktail (1 tablet/10 ml; MilliporeSigma, #11836170001). Protein concentrations in the cell lysates were estimated using the Quick Start Bradford Dye Reagent (Bio-Rad, #5000205) followed by blocking of free thiol groups in HEN buffer containing 0.2% MMTS and 2.5% SDS at 50°C for 30 min. After blocking, the proteins were purified by acetone precipitation and further resuspended in HENS (HEN + 1% SDS) buffer. These proteins were designated as “total load.” Further purification of S-nitrosylated proteins was done using Thiopropyl Sepharose 6B beads (GE Healthcare, #170420001) in the presence of 50 mM Na-ascorbate as a mild reducing agent, which will reduce ─SNO to ─SH, keeping the disulfide bond (─S─S─) intact. For the negative control, Na-ascorbate was replaced by HENS buffer. Purified proteins were eluted in SDS–polyacrylamide gel electrophoresis sample loading buffer and separated in 4 to 20% Mini-PROTEAN TGX Precast Gels (Bio-Rad, #4561094 and 4561096) for Western blot (WB) or stained with Imperial Protein Stain (Thermo Fisher Scientific, #24615). No band in the negative control lane demonstrated specificity of the purified S-nitrosylated proteins (fig. S3B). Stained lanes were excised from the gel and sent for MS (LC/MS-MS) at the Center for Proteomics and Bioinformatics, Case Western Reserve University. MassMatrix bioinformatics software (59) was used to search the acquired MS data against the rat database from UniProt. Identified S-nitrosylated proteins with 0% decoy were selected for analysis. We performed a total of six MS runs, triplicate for both with and without NO donor. S-nitrosylated proteins were designated if present in at least two of six samples. Any protein identified in more than two samples in the presence of NO donor compared to samples in the absence of NO donor were designated as NO-enriched. NO-enriched proteins were analyzed using WebGestalt 2019 software (24) to identify overrepresented pathways and associated proteins. The top 10 GO (Gene Ontology) Slim pathways and associated proteins were used to create the chord plot using a modified GOChord script in R.
Generation of stable cell lines
HEK293 cells were transfected with pMIB1-FLAG (Addgene, #37116) and pHA-Ub (Addgene, #18712) plasmids using Lipofectamine 3000 (Thermo Fisher Scientific, #L3000015) following the standard protocol. Stable cells were selected by culturing transfected cells in Dulbecco’s modified Eagle’s medium (DMEM) (American Type Culture Collection, #30-2002) in the presence of geneticin (500 μg/ml; Thermo Fisher Scientific, #10131035) and puromycin (10 μg/ml; Thermo Fisher Scientific, #A1113803). For MIB1 ubiquitination assay, transfected HEK293 cells were cultured in DMEM without antibiotics for 3 days.
USP9X knockdown by siRNA
A pool of three target-specific (19- to 25-nucleotide) siRNAs was used to silence USP9X expression (table S2). The siRNAs targeting porcine USP9X (ENSSSCG00000012251) transcript were designed using BLOCK-iT RNAi Designer (Thermo Fisher Scientific) and synthesized from Eurofins Genomics. A control siRNA was used as a scramble (Scr) siRNA, targeting no known gene. pAVICs maintained at 60 to 70% confluency were transfected with 30 nM USP9X or Scr siRNA using Lipofectamine 3000 Transfection Reagent (#L3000015, Thermo Fisher Scientific). Cell lysates were collected and examined for USP9X protein expression 72 hours after transfection.
For coculture experiments, pAVECs and pAVICs were transfected separately. After 12 hours of transfection, cells were detached using TrypLE Express and cocultured in EGM-2 media (Lonza, #CC-3162) in different combinations for an additional 60 hours as described in fig. S8 (A to H).
RNA purification and quantitative real-time polymerase chain reaction
RNA was extracted from pAVICs using TRIzol Reagent (Thermo Fisher Scientific, #15596018) following the manufacturer’s instructions. RNA (0.5 to 1.0 μg) was used for reverse transcription using the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific, #11754-050). SYBR Green–based quantitative real-time polymerase chain reaction (PCR) was performed using the Applied Biosystems 7500 real-time PCR. Mean relative gene expression was calculated after normalization of Ct values to GAPDH using the ΔΔCt method (table S1).
Immunoprecipitation and WB analysis
Cell lysates were prepared from cultured cells in an assay specific buffer. For immunoprecipitation, the cell lysate was prepared in Pierce IP Lysis Buffer (Thermo Fisher Scientific, #87787), and for WB, it was prepared in RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, #89900) supplemented with Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific, #87785). Cell lysates were centrifuged at 15,871g for 15 min at 4°C, and the supernatants were collected. Protein concentrations were estimated using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, #23227). Protein (400 to 500 μg) from the stably transfected HEK293 cells was used to immunoprecipitate FLAG-tagged MIB1 using EZview Red ANTI-FLAG M2 affinity gel (MilliporeSigma, #F2426) following the manufacturer’s protocol at 4°C overnight.
For nuclear-cytoplasmic fractionation, NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, #78833) were used following the manufacturer’s protocol. To test the purity of nuclear and cytoplasmic extracts, a WB against LAMIN A/C (Santa Cruz Biotechnology, #sc-7292) and a WB against GAPDH (Novus Biologicals, #NB300-221) were performed, respectively.
For the WBs, 10 to 25 μg of the protein samples was boiled for 5 min with 6X Laemmli SDS-Sample Buffer (Boston BioProducts, #BP-111R) containing β-mercaptoethanol and separated in 4 to 20% Mini-PROTEAN TGX Precast Gels (Bio-Rad, #4561094 and 4561096). After separation, the proteins were transferred into a polyvinylidene difluoride membrane (Bio-Rad, #1620177) and blocked with 5% nonfat milk in tris-buffered saline (TBS) containing 0.1% Tween 20 (TBST). Membranes were probed with primary antibodies against USP9X (Cell Signaling Technology, #5751), MIB1 (Santa Cruz Biotechnology, #sc-393811), NICD (Abcam, #ab8925 and Cell Signaling Technology, #4147), JAG1 (Santa Cruz Biotechnology, #sc8303 and #sc390177), RUNX2 (Cell Signaling Technology, #8486), HA-tag (Sigma-Aldrich, #H-9658), FLAG (Sigma-Aldrich, #F1804), pVASP (Cell Signaling Technology, #3114), and SMA (Abcam, #ab18147). After probing with primary antibodies, membranes were further probed with horseradish peroxidase–conjugated anti-rabbit and anti-mouse secondary antibodies (Vector Laboratories, #PI-1000 and PI-2000). Immunoblots were developed using either Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, #32106) or SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific, #34075). Densitometric analysis was performed to normalize the protein levels with respect to GAPDH or otherwise mentioned as loading control using ImageJ software. For reprobing with different primary antibodies, membranes were stripped with Restore Western Blot Stripping Buffer (Thermo Fisher Scientific, #21059) following the manufacturer’s protocol.
Immunofluorescence and immunohistochemistry in vitro and in vivo
For immunofluorescence staining, cultured pAVICs were fixed in 2.5% PFA. Cells were permeabilized with PBST (PBS containing 0.1% Triton X-100), and nonspecific immunoreactions were blocked using 1% BSA in PBST for 1 hour at room temperature. pAVICs were incubated overnight with primary antibodies against RUNX2 (Abcam, #ab76956) and activated NICD (Abcam, #ab8925), followed by washing with PBST and incubating with Alexa Fluor 594–conjugated anti-rabbit secondary antibody (Thermo Fisher Scientific, #A21207) for 1 hour in the dark. Nuclei were stained with DAPI (1.5 μg/ml; Sigma-Aldrich, #D9542). Images were captured using an Olympus IX51 microscope attached with an Olympus DP72 camera.
A similar protocol was performed to stain the tissue sections. The section underwent deparaffinization, using xylene and grades of ethanol, followed by antigen retrieval using citrate-based Antigen Unmasking solution (Vector Laboratories, #H-3300) following the manufacturer’s protocol. After permeabilization and blocking with 1% BSA for 1 hour, tissue sections were incubated with primary antibodies against MIB1 (Abcam, #ab124929), USP9X (Cell Signaling Technology, #14898), and RUNX2 (Abcam, #ab76956) overnight at 4°C. Following PBST wash, sections were incubated with anti-rabbit and anti-mouse secondary antibodies that were conjugated to Alexa Fluor 594/568/488 (Thermo Fisher Scientific, #A21207, A10037, A11001, and A11008) for 1 hour at room temperature. Nuclei were stained with VECTASHIELD HardSet Antifade Mounting Medium with DAPI (Vector Laboratories, #H-1500). Images were visualized using an Olympus BX51 microscope attached with an Olympus DP71 camera.
For immunohistochemical (IHC) staining, tissue sections were deparaffinized in xylene and rehydrated in grades of ethanol and PBS, followed by an antigen retrieval. Sections were incubated at room temperature with 3% H2O2 for 10 min to quench endogenous peroxidase activity and blocked by 5% normal goat serum (Vector Laboratories, #S-1000) in TBST for 1 hour. After blocking, sections were incubated with a primary antibody against NICD (Abcam, #ab8925) overnight at 4°C. Following primary antibody incubation, sections were further incubated with SignalStain Boost anti-Rabbit IHC Detection Reagent (Cell Signaling Technology, #8114) at room temperature for 30 min. Sections were visualized using the SignalStain DAB Substrate Kit (Cell Signaling Technology, #8059) and imaged using Zeiss Axio Imager A2 attached with an AxioCam MRc camera.
Experimental mouse model
Animal experiments were approved by the Institutional Animal Care and Use Committee at the Research Institute at Nationwide Children’s Hospital. Usp9xfl/fl mice were obtained from Charles River Laboratories, Italy, originally made by Ozgene Pty Ltd, Australia, with permission of S. Wood, Griffith University, Australia (33). Usp9xfl/fl females were bred withTie2Cre males (Jax, #008863), to obtain experimental Cre+ and control Cre− male (Usp9xfl/Y; Tie2Cre) mice. Generated pups were genotyped at P10 for Cre, and echocardiography was performed to assess cardiac function at 6, 16, and 24 weeks. After the baseline echocardiography at 6 weeks, mice were fed a high-fat Western diet (Envigo, #TD.88137) until 24 weeks of age and then euthanized to collect hearts for histological analyses. Transthoracic echocardiographic studies were performed after anesthetization with isoflurane (2% in 100% oxygen at a flow rate of 1.5 liters/min) using a Vevo 2100 device equipped with 18- to 38-MHz linear-array transducer with a digital ultrasound system (VisualSonics Inc., Canada). Body temperature was monitored throughout the procedure using a rectal probe thermometer. Pulsed-wave Doppler analysis of three consecutive cardiac cycles across the aortic valve was performed and averaged along the parasternal long axis to obtain maximal transvalvular velocity. All echocardiographic experiments were performed by an experienced investigator blinded to the study design and animal genotypes.
After euthanasia, hearts were perfused with PBS, removed, and fixed in 4% PFA at 4°C overnight. After fixation, the hearts were embedded in paraffin and sectioned. For gross histological analysis, tissue sections were stained with Russell-Movat pentachrome stain (American MasterTech, #KTRMP) to visualize collagen, elastin, muscle, mucin, and fibrin and with Alizarin red (Acros Organics, #400480250) to visualize calcium deposition. For both staining, tissue sections were deparaffinized in xylene and rehydrated in grades of ethanol and in water. Russell-Movat pentachrome staining was performed following the manufacturer’s protocol. For calcium deposition staining, tissue sections were incubated with 2% Alizarin red in water for 5 to 7 min followed by washing in water. All images were visualized using an Olympus BX51 microscope attached with an Olympus DP71 camera. Valve leaflet area was quantified using ImageJ software at least on three sections per aortic valve.
Collection of aortic valve leaflets from human patients
Aortic valve leaflets were excised by the surgeons during aortic valve replacement surgery (primary surgical indication: calcific aortic stenosis) at Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, and processed as described before (60). After excision, leaflets were placed in saline at 4°C for approximately 30 min and transported to the laboratory quickly (approximately within 15 min) in DMEM (Thermo Fisher Scientific, #10569044) without serum or antibiotics. Leaflets were then rinsed with cold PBS, and intact leaflets were placed into dry, 1.5-ml Eppendorf tubes, followed by a slow freezing in −80°C. Noncalcified aortic valve was obtained from the Heart Center Biorepository at Nationwide Children’s Hospital. Protein was extracted from aortic valve leaflets and used for BST and WB as described above.
Statistics
All experiments were performed at least in triplicate per treatment condition. Data are presented as means ± SD. Two-tailed Student’s t test and Mann-Whitney test were performed to determine statistical significance using the GraphPad Prism 8 software package. P ≤ 0.05 was considered statistically significant.
Study approval
This study was approved by the Institutional Animal Care and Use Committee at Nationwide Children’s Hospital (protocol AR16-00053) and conducted in accordance with the National Institutes of Health’s guidelines. Human valve studies were approved by the Institutional Review Board (IRB) at Nationwide Children’s Hospital (IRB, #07-00298) and Brigham and Women’s Hospital (IRB, #2011P001703). Informed consent was obtained from study participants or parents of participants less than 18 years of age. These aortic valves were received after surgery, without identification.
Acknowledgments
We thank members of the Biomorphology Core and A. P. Matos Nieves at Nationwide Children’s Hospital for histology support, M. Lockhart for assistance during preparation of pAVIC primary culture, and the Heart Center Biorepository at Nationwide Children’s Hospital. We also thank L. Wang, Center for Proteomics and Bioinformatics, Case Western Reserve University, for LC/MS-MS analysis. Funding: This work was supported by funding from National Heart, Lung, and Blood Institute grants: R01 HL132801 (V.G., J.L., and B.L.), R01 HL121797 (V.G.), and R01 HL136431, R01 HL147095, and R01 HL141917 (E.A.); NHLBI Postdoctoral Fellowship T32 HL098039 (S.M.); and AHA/Children’s Heart Foundation grant 18CDA34110330 (M.B.). Author contributions: V.G. and U.M. conceived the project, and experiments were designed by U.M. and V.G. with input from B.L., S.E.C., and J.L. U.M. performed and analyzed the experiments. The murine studies were performed by U.M. and E.C. M.B. performed scRNA-seq library preparation and siRNA-mediated knockdown experiments. Y.U. and M.R.M. performed and analyzed the mouse echocardiograms. Bioinformatic analyses were performed by S.M. M.C.B. and E.A. collected, graded, and provided human tissues. S.W. generated and provided Usp9xloxp/loxp mice. U.M., V.G., B.L., S.E.C., and J.L. interpreted the data. U.M. and V.G. wrote the manuscript with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The authors declare that all supporting data are available within the article and its Supplementary Materials. RNA-seq data have been deposited in GEO under accession number GSE161123. The plasmid constructs are commercially available from Addgene. The mouse models are commercially available from the Jackson Laboratory, USA, and Charles River Laboratories, Italy. Any other relevant data are available from the authors upon reasonable request. Source data are provided with this paper.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabe3706/DC1
REFERENCES AND NOTES
- 1.Yadgir S., Johnson C. O., Aboyans V., Adebayo O. M., Adedoyin R. A., Afarideh M., Alahdab F., Alashi A., Alipour V., Arabloo J., Azari S., Barthelemy C. M., Benziger C. P., Berman A. E., Bijani A., Carrero J. J., Carvalho F., Daryani A., Duraes A. R., Esteghamati A., Farid T. A., Farzadfar F., Fernandes E., Filip I., Gad M. M., Hamidi S., Hay S. I., Ilesanmi O. S., Naghibi Irvani S. S., Jurisson M., Kasaeian A., Kengne A. P., Khan A. R., Kisa A., Kisa S., Kolte D., Manafi N., Manafi A., Mensah G. A., Mirrakhimov E. M., Mohammad Y., Mokdad A. H., Negoi R. I., Thi Nguyen H. L., Nguyen T. H., Nixon M. R., Otto C. M., Patel S., Pilgrim T., Radfar A., Rawaf D. L., Rawaf S., Rawasia W. F., Rezapour A., Roever L., Saad A. M., Saadatagah S., Senthilkumaran S., Sliwa K., Tesfay B. E., Tran B. X., Ullah I., Vaduganathan M., Vasankari T. J., Wolfe C. D. A., Yonemoto N., Roth G. A.; Global Burden of Disease Study 2017 Nonrheumatic Valve Disease Collaborators , Global, regional, and national burden of calcific aortic valve and degenerative mitral valve diseases, 1990–2017. Circulation 141, 1670–1680 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Otto C. M., Prendergast B., Aortic-valve stenosis—From patients at risk to severe valve obstruction. N. Engl. J. Med. 371, 744–756 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Hutcheson J. D., Aikawa E., Merryman W. D., Potential drug targets for calcific aortic valve disease. Nat. Rev. Cardiol. 11, 218–231 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers M. A., Aikawa E., Cardiovascular calcification: Artificial intelligence and big data accelerate mechanistic discovery. Nat. Rev. Cardiol. 16, 261–274 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Lincoln J., Garg V., Etiology of valvular heart disease-genetic and developmental origins. Circ. J. 78, 1801–1807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hjortnaes J., Shapero K., Goettsch C., Hutcheson J. D., Keegan J., Kluin J., Mayer J. E., Bischoff J., Aikawa E., Valvular interstitial cells suppress calcification of valvular endothelial cells. Atherosclerosis 242, 251–260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rabkin-Aikawa E., Farber M., Aikawa M., Schoen F. J., Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J. Heart Valve Dis. 13, 841–847 (2004). [PubMed] [Google Scholar]
- 8.Chen J. H., Simmons C. A., Cell-matrix interactions in the pathobiology of calcific aortic valve disease: Critical roles for matricellular, matricrine, and matrix mechanics cues. Circ. Res. 108, 1510–1524 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Shapero K., Wylie-Sears J., Levine R. A., Mayer J. E. Jr., Bischoff J., Reciprocal interactions between mitral valve endothelial and interstitial cells reduce endothelial-to-mesenchymal transition and myofibroblastic activation. J. Mol. Cell. Cardiol. 80, 175–185 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosse K., Hans C. P., Zhao N., Koenig S. N., Huang N., Guggilam A., LaHaye S., Tao G., Lucchesi P. A., Lincoln J., Lilly B., Garg V., Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell. Cardiol. 60, 27–35 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garg V., Muth A. N., Ransom J. F., Schluterman M. K., Barnes R., King I. N., Grossfeld P. D., Srivastava D., Mutations in NOTCH1 cause aortic valve disease. Nature 437, 270–274 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Gould N., Doulias P. T., Tenopoulou M., Raju K., Ischiropoulos H., Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J. Biol. Chem. 288, 26473–26479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lima B., Forrester M. T., Hess D. T., Stamler J. S., S-nitrosylation in cardiovascular signaling. Circ. Res. 106, 633–646 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou H. L., Stomberski C. T., Stamler J. S., Cross talk between S-nitrosylation and phosphorylation involving kinases and nitrosylases. Circ. Res. 122, 1485–1487 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swatek K. N., Komander D., Ubiquitin modifications. Cell Res. 26, 399–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim R., Sugino T., Nolte H., Andrade J., Zimmermann B., Shi C., Doddaballapur A., Ong Y. T., Wilhelm K., Fasse J. W. D., Ernst A., Kaulich M., Husnjak K., Boettger T., Guenther S., Braun T., Kruger M., Benedito R., Dikic I., Potente M., Deubiquitinase USP10 regulates Notch signaling in the endothelium. Science 364, 188–193 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Shin S., Kim K., Kim H. R., Ylaya K., Do S. I., Hewitt S. M., Park H. S., Roe J. S., Chung J. Y., Song J., Deubiquitylation and stabilization of Notch1 intracellular domain by ubiquitin-specific protease 8 enhance tumorigenesis in breast cancer. Cell Death Differ. 27, 1341–1354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards J., El-Hamamsy I., Chen S., Sarang Z., Sarathchandra P., Yacoub M. H., Chester A. H., Butcher J. T., Side-specific endothelial-dependent regulation of aortic valve calcification: Interplay of hemodynamics and nitric oxide signaling. Am. J. Pathol. 182, 1922–1931 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oelze M., Mollnau H., Hoffmann N., Warnholtz A., Bodenschatz M., Smolenski A., Walter U., Skatchkov M., Meinertz T., Munzel T., Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ. Res. 87, 999–1005 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Majumdar U., Biswas P., Subhra Sarkar T., Maiti D., Ghosh S., Regulation of cell cycle and stress responses under nitrosative stress in Schizosaccharomyces pombe. Free Radic. Biol. Med. 52, 2186–2200 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Hulin A., Hortells L., Gomez-Stallons M. V., O’Donnell A., Chetal K., Adam M., Lancellotti P., Oury C., Potter S. S., Salomonis N., Yutzey K. E., Maturation of heart valve cell populations during postnatal remodeling. Development 146, dev173047 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porras A. M., van Engeland N. C., Marchbanks E., McCormack A., Bouten C. V., Yacoub M. H., Latif N., Masters K. S., Robust generation of quiescent porcine valvular interstitial cell cultures. J. Am. Heart Assoc. 6, e005041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White M. P., Theodoris C. V., Liu L., Collins W. J., Blue K. W., Lee J. H., Meng X., Robbins R. C., Ivey K. N., Srivastava D., NOTCH1 regulates matrix gla protein and calcification gene networks in human valve endothelium. J. Mol. Cell. Cardiol. 84, 13–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao Y., Wang J., Jaehnig E. J., Shi Z., Zhang B., WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Izrailit J., Jaiswal A., Zheng W., Moran M. F., Reedijk M., Cellular stress induces TRB3/USP9x-dependent Notch activation in cancer. Oncogene 36, 1048–1057 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Overstreet E., Fitch E., Fischer J. A., Fat facets and Liquid facets promote Delta endocytosis and Delta signaling in the signaling cells. Development 131, 5355–5366 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Luxan G., Casanova J. C., Martinez-Poveda B., Prados B., D’Amato G., MacGrogan D., Gonzalez-Rajal A., Dobarro D., Torroja C., Martinez F., Izquierdo-Garcia J. L., Fernandez-Friera L., Sabater-Molina M., Kong Y. Y., Pizarro G., Ibanez B., Medrano C., Garcia-Pavia P., Gimeno J. R., Monserrat L., Jimenez-Borreguero L. J., de la Pompa J. L., Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 19, 193–201 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Kornberg M. D., Sen N., Hara M. R., Juluri K. R., Nguyen J. V., Snowman A. M., Law L., Hester L. D., Snyder S. H., GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 12, 1094–1100 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kapuria V., Peterson L. F., Fang D., Bornmann W. G., Talpaz M., Donato N. J., Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 70, 9265–9276 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Ling X., Huang Q., Xu Y., Jin Y., Feng Y., Shi W., Ye X., Lin Y., Hou L., Lin X., The deubiquitinating enzyme Usp5 regulates Notch and RTK signaling during Drosophila eye development. FEBS Lett. 591, 875–888 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Jung H., Kim B.-G., Han W. H., Lee J. H., Cho J.-Y., Park W. S., Maurice M. M., Han J.-K., Lee M. J., Finley D., Jho E.-H., Deubiquitination of Dishevelled by Usp14 is required for Wnt signaling. Oncogenesis 2, e64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murtaza M., Jolly L. A., Gecz J., Wood S. A., La FAM fatale: USP9X in development and disease. Cell. Mol. Life Sci. 72, 2075–2089 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stegeman S., Jolly L. A., Premarathne S., Gecz J., Richards L. J., Mackay-Sim A., Wood S. A., Loss of Usp9x disrupts cortical architecture, hippocampal development and TGFβ-mediated axonogenesis. PLOS ONE 8, e68287 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kisanuki Y. Y., Hammer R. E., Miyazaki J., Williams S. C., Richardson J. A., Yanagisawa M., Tie2-Cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230–242 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Rajamannan N. M., Evans F. J., Aikawa E., Grande-Allen K. J., Demer L. L., Heistad D. D., Simmons C. A., Masters K. S., Mathieu P., O’Brien K. D., Schoen F. J., Towler D. A., Yoganathan A. P., Otto C. M., Calcific aortic valve disease: Not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease—2011 update. Circulation 124, 1783–1791 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanno Y., Into T., Lowenstein C. J., Matsushita K., Nitric oxide regulates vascular calcification by interfering with TGF- signalling. Cardiovasc. Res. 77, 221–230 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Yip C. Y., Blaser M. C., Mirzaei Z., Zhong X., Simmons C. A., Inhibition of pathological differentiation of valvular interstitial cells by C-type natriuretic peptide. Arterioscler. Thromb. Vasc. Biol. 31, 1881–1889 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Blaser M. C., Wei K., Adams R. L. E., Zhou Y. Q., Caruso L. L., Mirzaei Z., Lam A. Y., Tam R. K. K., Zhang H., Heximer S. P., Henkelman R. M., Simmons C. A., Deficiency of natriuretic peptide receptor 2 promotes bicuspid aortic valves, aortic valve disease, left ventricular dysfunction, and ascending aortic dilatations in mice. Circ. Res. 122, 405–416 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Thanh Nguyen H., Andrejeva D., Gupta R., Choudhary C., Hong X., Eichhorn P. J., Loya A. C., Cohen S. M., Deubiquitylating enzyme USP9x regulates hippo pathway activity by controlling angiomotin protein turnover. Cell Discov. 2, 16001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meng J., Ai X., Lei Y., Zhong W., Qian B., Qiao K., Wang X., Zhou B., Wang H., Huai L., Zhang X., Han J., Xue Y., Liang Y., Zhou H., Chen S., Sun T., Yang C., USP5 promotes epithelial-mesenchymal transition by stabilizing SLUG in hepatocellular carcinoma. Theranostics 9, 573–587 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Massa F., Tammaro R., Prado M. A., Cesana M., Lee B. H., Finley D., Franco B., Morleo M., The deubiquitinating enzyme Usp14 controls ciliogenesis and Hedgehog signaling. Hum. Mol. Genet. 28, 764–777 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niessen K., Fu Y., Chang L., Hoodless P. A., McFadden D., Karsan A., Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 182, 315–325 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fulmer D., Toomer K., Guo L., Moore K., Glover J., Moore R., Stairley R., Lobo G., Zuo X., Dang Y., Su Y., Fogelgren B., Gerard P., Chung D., Heydarpour M., Mukherjee R., Body S. C., Norris R. A., Lipschutz J. H., Defects in the exocyst-cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation 140, 1331–1341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komander D., Clague M. J., Urbe S., Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Gordon W. R., Zimmerman B., He L., Miles L. J., Huang J., Tiyanont K., McArthur D. G., Aster J. C., Perrimon N., Loparo J. J., Blacklow S. C., Mechanical allostery: Evidence for a force requirement in the proteolytic activation of notch. Dev. Cell 33, 729–736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langridge P. D., Struhl G., Epsin-dependent ligand endocytosis activates notch by force. Cell 171, 1383–1396.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hofmann J. J., Briot A., Enciso J., Zovein A. C., Ren S., Zhang Z. W., Radtke F., Simons M., Wang Y., Iruela-Arispe M. L., Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development 139, 4449–4460 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goddard L. M., Duchemin A. L., Ramalingan H., Wu B., Chen M., Bamezai S., Yang J., Li L., Morley M. P., Wang T., Scherrer-Crosbie M., Frank D. B., Engleka K. A., Jameson S. C., Morrisey E. E., Carroll T. J., Zhou B., Vermot J., Kahn M. L., Hemodynamic forces sculpt developing heart valves through a KLF2-WNT9B paracrine signaling axis. Dev. Cell 43, 274–289.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goto S., Rogers M. A., Blaser M. C., Higashi H., Lee L. H., Schlotter F., Body S. C., Aikawa M., Singh S. A., Aikawa E., Standardization of human calcific aortic valve disease in vitro modeling reveals passage-dependent calcification. Front. Cardiovasc. Med. 6, 49 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mehta V., Pang K. L., Rozbesky D., Nather K., Keen A., Lachowski D., Kong Y., Karia D., Ameismeier M., Huang J., Fang Y., Del Rio Hernandez A., Reader J. S., Jones E. Y., Tzima E., The guidance receptor plexin D1 is a mechanosensor in endothelial cells. Nature 578, 290–295 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X., Srinivasan P., Collard E., Grajdeanu P., Zweier J. L., Friedman A., Nitric oxide diffusion rate is reduced in the aortic wall. Biophys. J. 94, 1880–1889 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Otto C. M., Kuusisto J., Reichenbach D. D., Gown A. M., O’Brien K. D., Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 90, 844–853 (1994). [DOI] [PubMed] [Google Scholar]
- 53.Theodoris C. V., Mourkioti F., Huang Y., Ranade S. S., Liu L., Blau H. M., Srivastava D., Long telomeres protect against age-dependent cardiac disease caused by NOTCH1 haploinsufficiency. J. Clin. Invest. 127, 1683–1688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Porras A. M., McCoy C. M., Masters K. S., Calcific aortic valve disease: A battle of the sexes. Circ. Res. 120, 604–606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gould R. A., Butcher J. T., Isolation of valvular endothelial cells. J. Vis. Exp. 2010, 2158 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R., Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forrester M. T., Thompson J. W., Foster M. W., Nogueira L., Moseley M. A., Stamler J. S., Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 27, 557–559 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu H., Freitas M. A., A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics 8, 133 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlotter F., Halu A., Goto S., Blaser M. C., Body S. C., Lee L. H., Higashi H., DeLaughter D. M., Hutcheson J. D., Vyas P., Pham T., Rogers M. A., Sharma A., Seidman C. E., Loscalzo J., Seidman J. G., Aikawa M., Singh S. A., Aikawa E., Spatiotemporal multi-omics mapping generates a molecular atlas of the aortic valve and reveals networks driving disease. Circulation 138, 377–393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabe3706/DC1