

Summary
Lesions on DNA uncouple DNA synthesis from the replisome, generating stretches of unreplicated single-stranded DNA (ssDNA) behind the replication fork. These ssDNA gaps need to be filled in to complete DNA duplication. Gap-filling synthesis involves either translesion DNA synthesis (TLS) or template switching (TS). Controlling these processes, ubiquitylated PCNA recruits many proteins that dictate pathway choice, but the enzymes regulating PCNA ubiquitylation in vertebrates remain poorly defined. Here we report that the E3 ubiquitin ligase RFWD3 promotes ubiquitylation of proteins on ssDNA. The absence of RFWD3 leads to a profound defect in recruitment of key repair and signaling factors to damaged chromatin. As a result, PCNA ubiquitylation is inhibited without RFWD3, and TLS across different DNA lesions is drastically impaired. We propose that RFWD3 is an essential coordinator of the response to ssDNA gaps, where it promotes ubiquitylation to drive recruitment of effectors of PCNA ubiquitylation and DNA damage bypass.
Keywords: anemia, TLS, ubiquitylation, PCNA, RFWD3, ICL repair, DPC repair, HMCES, CHROMASS, UV lesions
Graphical Abstract
Highlights
-
•
RFWD3 promotes ubiquitylation of proteins on ssDNA
-
•
RFWD3 regulates DNA damage-induced PCNA ubiquitylation
-
•
RFWD3 stimulates gap-filling DNA synthesis across different DNA lesions
Using Xenopus egg extracts, Gallina et al. describe a new role for the ubiquitin ligase RFWD3 in stimulating PCNA ubiquitylation and the bypass of different polymerase-blocking DNA lesions, providing an understanding of RFWD3’s essential function in safeguarding the genome.
Introduction
DNA replication encounters a plethora of lesions on its template that stall DNA synthesis. While helicase-stalling lesions inhibit the translocation of the CMG (Cdc45/Mcm2-7/GINS) helicase and therefore block the entire replisome, polymerase-stalling lesions involve small modifications on template DNA that stall replicative polymerases behind the replication fork. If encountered on the leading strand template, on which CMG translocates (Fu et al., 2011), these lesions uncouple CMG unwinding from leading strand synthesis and thereby generate stretches of unreplicated single-stranded DNA (ssDNA) that activate the S phase replication checkpoint (Cortez, 2019).
Among helicase-stalling lesions are DNA-protein crosslinks (DPCs). Because of their bulky nature, DPCs inhibit CMG translocation if encountered on the leading strand template (Duxin et al., 2014; Fu et al., 2011; Kose et al., 2019). However, CMG can ultimately bypass intact DPCs (Sparks et al., 2019), and the protein adduct constitutes a further block to replicative polymerases (Duxin et al., 2014; Sparks et al., 2019). The DPC is subsequently degraded to a short peptide-DNA adduct by the protease SPRTN and/or the proteasome, allowing bypass by translesion DNA synthesis (TLS) polymerases (Duxin et al., 2014; Larsen et al., 2019). In contrast to DPCs, most base lesions on the leading strand template, such as ultraviolet (UV) light-induced thymidine dimers, base oxidation, or small peptide-DNA adducts, do not stall CMG translocation and therefore trigger rapid uncoupling (Cortez, 2019).
Once uncoupled from the helicase, nascent leading strand synthesis can recouple with the replisome via different mechanisms. First, a new primer on the leading strand template can be synthesized past the lesion to reinitiate DNA synthesis. This mechanism has been well characterized in bacteria (Yeeles and Marians, 2011), and is believed to operate in eukaryotes via PrimPol (Bianchi et al., 2013; García-Gómez et al., 2013; Mourón et al., 2013). Repriming generates DNA gaps behind the replication fork that can be filled in via two alternative mechanisms, collectively called DNA damage tolerance (DDT). In one mechanism, error-prone TLS polymerases are recruited to synthesize DNA across the damage, and the nature of the lesion most likely dictates TLS polymerase choice. Alternatively, a recombination-mediated mechanism known as template switching (TS) can be employed to bypass the lesion in an error-free manner, using the undamaged sister chromatid as a template. Both TLS and TS are stimulated by the ubiquitylation of the polymerase processivity factor PCNA (Hoege et al., 2002; Stelter and Ulrich, 2003).
The central role of PCNA in orchestrating DDT has been well characterized in yeast. Upon DNA damage, PCNA first becomes mono-ubiquitylated by the E3 ubiquitin ligase Rad18 (Hoege et al., 2002; Stelter and Ulrich, 2003). Rad18 is recruited to stalled polymerases by RPA, which binds and protects the ssDNA generated during uncoupling (Davies et al., 2008; Huttner and Ulrich, 2008). Mono-ubiquitylated PCNA then promotes switching from a replicative to a TLS polymerase that replicates across the damage. This is because most TLS polymerases harbor ubiquitin binding motifs, in addition to their PCNA-interacting peptide (PIP) domain, enhancing their affinity for ubiquitylated PCNA (Bienko et al., 2005; Guo et al., 2006; Kannouche et al., 2004). PCNA can be further poly-ubiquitylated via K63-linked ubiquitin chains by the E3 ligase Rad5 (Hoege et al., 2002; Parker and Ulrich, 2009). PCNA poly-ubiquitylation channels repair toward TS.
In vertebrates, RAD18-dependent PCNA mono-ubiquitylation is also observed upon treatments that stall DNA replication (Kannouche et al., 2004; Watanabe et al., 2004). In contrast, the poly-ubiquitylation of PCNA has been challenging to monitor in cells. The Rad5 orthologs HLTF and SHPRH can both catalyze the addition of ubiquitin to mono-ubiquitylated PCNA in vitro, and depletion of these proteins leads to the reduction of some poly-ubiquitylated PCNA species in cells (Lin et al., 2011; Motegi et al., 2006, 2008; Unk et al., 2006, 2008). However, mouse embryonic fibroblasts (MEFs) lacking both SHPRH and HLTF are not sensitive to DNA damaging agents, and these cells can still promote PCNA poly-ubiquitylation, suggesting the involvement of another ubiquitin ligase (Krijger et al., 2011). Moreover, TS has not been accurately monitored in vertebrates, and whether PCNA mono- and poly-ubiquitylation dictate TLS and TS, respectively, remains unclear. In summary, although the regulation and outcomes of PCNA ubiquitylation are well defined in yeast, whether the same mechanisms apply to vertebrates remains poorly defined.
Here, we identify a critical role for the E3 ubiquitin ligase RFWD3 in promoting DNA synthesis across polymerase-stalling lesions. We show that in the absence of RFWD3, ubiquitin signaling and protein accumulation at damaged chromatin are severely compromised. As a result, PCNA ubiquitylation is drastically reduced and TLS across different types of DNA lesion impaired. Our results indicate that RFWD3 is one of the earliest responders and an essential regulator of gap-filling synthesis and repair.
Results
RFWD3 is required for replication-coupled bypass of DPCs
RFWD3 was previously shown to physically interact with RPA and to participate in the restart of stalled replication forks and the repair of double-stranded breaks (DSBs) via homologous recombination (HR) (Elia et al., 2015; Feeney et al., 2017; Gong and Chen, 2011; Inano et al., 2017; Liu et al., 2011). Accordingly, during DNA replication of DPC-containing plasmids, the recruitment of RFWD3 strongly correlated with RPA recruitment, which matched with the generation of DNA gaps (Figure 1A) (Figure 2 in Larsen et al., 2019). To investigate whether RFWD3 regulates replication-coupled DPC repair, we generated two specific antibodies against RFWD3 that efficiently immunodeplete the protein from Xenopus egg extracts (Figure S1A). We first replicated pMH, a plasmid containing a single crosslinked M.HpaII (Duxin et al., 2014), in the presence of radiolabeled [α-32P]dATP in mock- or RFWD3-depleted extracts. During replication of pMH, converging forks transiently stall at the DPC, after which resolved daughter molecules appear. Daughter molecules containing the DPC first accumulate as open circular (OC) molecules and are then converted to supercoiled (SC) molecules following proteolysis of the DPC and bypass of the peptide adduct by TLS (Figures 1B and 1C, lanes 1–5) (Duxin et al., 2014). RFWD3 depletion severely inhibited the conversion of the damaged plasmid from OC to SC, without affecting replication kinetics, suggesting a defect in replication of the adducted strand (Figure 1C, lanes 6–10, red arrowheads and quantification). To determine how RFWD3 depletion affects replication across the DPC, we analyzed nascent leading strands on a denaturing polyacrylamide gel (Figure 1D). In mock-depleted extracts, synthesis of the nascent leading strand first stalls ~30–40 nt upstream of the DPC because of the footprint of the CMG helicase colliding with the DPC. Following CMG bypass of the DPC, the nascent strand is extended up to the lesion (−1, 0, +1), before TLS polymerases can synthesize across the DNA-peptide adduct (Figure 1E, lanes 1–5). In the absence of RFWD3, nascent strands reached the crosslink with normal kinetics but then permanently stalled at the −1 position and never extended past the lesion, indicating a severe TLS defect (Figure 1E, lanes 6–10). Depleting RFWD3 with antibody F mimicked RFWD3 depletion with antibody N, eliminating the possibility of an off-target effect (Figure S1B). Despite many attempts, we were unable to generate functional recombinant RFWD3 and could not rescue RFWD3 depletion in egg extracts (see Discussion). Nevertheless, we confirmed specificity of the depletion by analyzing the proteome of RFWD3-depleted extracts using mass spectrometry (MS). RFWD3 was the only protein significantly depleted from these extracts (Figure S1C; Table S1), reducing the possibility that the effects are caused by co-depletion of an interacting partner. Furthermore, we confirmed that none of the known TLS factors participating in DPC bypass were co-depleted with RFWD3 (Figure S1D). To address whether RFWD3 controls DNA synthesis across other DPCs, we generated a DPC plasmid containing a bacterial glycosylase (Fpg) covalently linked to an abasic site (Figure S1E) (Gilboa et al., 2002). RFWD3 depletion also blocked TLS across crosslinked Fpg, indicating that RFWD3 is essential for replication across protein adducts (Figure S1E).
Figure 1.

RFWD3 is essential for TLS across peptide adducts
(A) Heatmap depicting the mean of the Z score, log2, label-free quantification (LFQ) intensity from four biochemical replicates of pCTRL and pDPCLeads (originally published in Larsen et al., 2019). Red arrow indicates RFWD3. Geminin was added to block DNA replication. Ubiquitin-vinyl sulfone (UbVS) was added to deplete the pool of free ubiquitin.
(B) Replication intermediates generated during replication of pMH (Duxin et al., 2014).
(C) pMH was replicated in egg extracts in the presence of [α-32P]dATP in mock- or RFWD3-depleted extracts. Reaction samples were analyzed by native agarose gel electrophoresis. RI, replication intermediate; OC, open circular; SC, supercoiled. Red arrowheads indicate OC molecules. Lower graph: quantification of replication efficiencies from three independent experiments. Error bars represent the standard error of the mean (SEM). To deplete RFWD3, the RFWD3-N antibody was used in all experiments unless otherwise indicated.
(D) Nascent leading strand and extension products generated upon FspI and AatII digest of pMH. Double digestion generates shorter damaged and longer undamaged extension products, which can be resolved on a denaturing polyacrylamide gel (see E, top panel). The CMG helicase is depicted in green, and the crosslinked M.HpaII is depicted in gray.
(E) Samples from (C) were digested with FspI and AatII (upper panel) or AatII (lower panel) and separated on a denaturing polyacrylamide gel. Location of the stalling points in relation to the DPC is indicated on the right of the gel.
(F) Generation of pMHPK.
(G) pMHPK was replicated in mock- or RFWD3-depleted extracts and analyzed as in (C).
(H) Samples from (G) were analyzed as in (E).
(I) pMHPK-Lead or pMHPK-Lag were replicated in egg extracts in the presence of LacI. Samples were digested with FspI and AatII (upper panel) or either AatII (pMHPK-Lead, lower panel) or BssHII (pMHPK-Lag, lower panel) and analyzed as in (E).
Next, we asked whether RFWD3 is required for TLS because of a role in DPC proteolysis. To this end, we replicated pMHPK, a DPC plasmid pretreated with proteinase K, which generates a 4-amino acid adduct that overcomes the need for proteolysis during replication (Figure 1F) (Figures S4L and S4M in Larsen et al., 2019). RFWD3 depletion also blocked bypass across this short peptide adduct, and synthesis of nascent leading strands again permanently stalled at −1 (Figures 1G and 1H, lanes 6–10). Thus, RFWD3 is essential for replication across peptide-DNA adducts regardless of their size, and this function is independent of DPC proteolysis.
Because DNA lesions can form on either the leading or the lagging strand template, we addressed whether the function of RFWD3 is strand specific. To this end, we replicated a plasmid containing a lac repressor array that is flanked on one side by a M.HpaII peptide adduct on either the top or the bottom strand (Figure 1I) (Dewar et al., 2015; Duxin et al., 2014). In the presence of LacI, the rightward fork stalls at the array, allowing the leftward fork to encounter the peptide adduct on either the leading or the lagging strand template. As shown in Figure 1I, RFWD3 depletion blocked nascent strand bypass of the peptide adduct on either template, indicating that RFWD3 is essential for both leading and lagging strand TLS.
Finally, because TLS can occur directly at the replication fork or at ssDNA gaps during gap-filling DNA synthesis (Daigaku et al., 2010), we addressed whether RFWD3 also regulates TLS in the absence of a replication fork. For this purpose, M.HpaII was crosslinked across from a 29 nt gap (Figure S1F) (Larsen et al., 2019), and the resulting plasmids, pMHssDNA or pMHssDNA-PK, were incubated in mock- or RFWD3-depleted non-licensing egg extracts that do not support loading of the replicative helicase MCM2-7 but are proficient in gap-filling DNA synthesis (Larsen et al., 2019). In these conditions, RFWD3 depletion again blocked TLS across both substrates (Figure S1F). We conclude that RFWD3 is essential for replication across peptide-DNA adducts not only during DNA replication, but also during gap-filling DNA synthesis, which can occur throughout the cell cycle.
RFWD3-mediated TLS across M.HpaII crosslinks is a two-step mutagenic process
Our data suggest a fundamental role for RFWD3 in regulating TLS across DPCs. The Polζ TLS polymerase is a multi-subunit complex that interacts tightly with the TLS regulator REV1 (Martin and Wood, 2019). Because of this tight interaction, depletion of Polζ subunits in egg extracts leads to co-depletion of REV1, and vice versa (Budzowska et al., 2015), allowing only investigation of the REV1-Polζ complex. Accordingly, we have previously shown that upon immunodepletion of REV7 or REV1, nascent strands permanently stall at the −1, 0, and +1 positions during replication of pMH, suggesting an essential role for REV1-Polζ extending past the peptide adduct (Figures 2A and 2B lanes 9–12; Figure S2A) (Duxin et al., 2014). This contrasts with an RFWD3 depletion, in which nascent strands predominantly stall at −1 (Figure 1E). Thus, a TLS polymerase other than REV1-Polζ performs the insertion across the adducted base, and this process requires RFWD3. Because DNA Polη is known as an insertion polymerase (Cruet-Hennequart et al., 2010), we immunodepleted it from egg extracts and replicated pMH. Although Polη depletion did not affect the appearance of SC molecules (Figure 2A, compare lanes 1–4 and 5–8), it abolished the insertion signature across the adducted base (Figure 2B, lanes 6–7), which was restored by reintroducing recombinant Polη (Figure S2B). Consistent with the generation of replicated SC molecules, Polη depletion minimally affected the kinetics of bypass and generation of full-length extension products (Figure 2B, upper panel, lanes 5–8). Similar results were obtained during gap-filling synthesis of pMHssDNA (Figure S2C). When Polη and REV1 were immunodepleted together (Figure S2A), nascent strands permanently stalled at −1 (Figure 2B, lanes 13–16). Altogether, our data show that replication across crosslinked M.HpaII is a regulated two-step TLS process involving Polη insertion and REV1-Polζ extension. However, when Polη is absent, REV1-Polζ appears to both insert and rapidly extend past the peptide adduct. This two-step mechanism is reminiscent of DNA synthesis across UV-induced 6-4 photoproducts or cisplatin-induced DNA intra-strand crosslinks (Pt-GG) (Hirota et al., 2010; Johnson et al., 2001; Shachar et al., 2009; Zhao et al., 2012).
Figure 2.

Replication of pMH is a two-step mutagenic process
(A) pMH was replicated in mock-, Polη-, REV1-, or Polη- and REV1-depleted extracts. Samples were analyzed as in Figure 1C.
(B) Samples from (A) were digested and analyzed as in Figure 1E. U, undamaged strand; D, damaged strand.
(C) pMH was replicated in mock-, Polη-, RFWD3-, or Polη- and RFWD3-depleted extracts. Samples were analyzed as in Figure 1E.
(D) Quantification of mutation frequencies measured after replication of pCTRL or pMH. Replication samples were amplified by PCR and analyzed by next-generation sequencing (see STAR methods). The 0 position corresponds to the location of the protein adduct. One of three independent experiments is shown.
(E) Distribution of nucleotide misincorporation from the data generated in (D).
(F) pMH was replicated in either mock- or Polη-depleted extracts, and samples were amplified and analyzed as in (D). One of three independent experiments is shown.
(G) Distribution of nucleotide misincorporation from the data generated in (F).
The observation that RFWD3 depletion blocked nascent strand synthesis at −1 suggested that not only Polη insertion but also downstream extension by REV1-Polζ are inhibited in the absence of RFWD3. To test this possibility, we immunodepleted RFWD3 in Polη-depleted extracts, which depend on REV1-Polζ for bypass. As seen in Figure 2C, in the absence of both Polη and RFWD3, nascent strands permanently stalled at −1 (lanes 13–16), indicating that the requirement of REV1-Polζ for lesion bypass in the absence of Polη (lanes 5–8) is RFWD3 dependent. Consistently, replication across the Fpg crosslink, which depended on REV1-Polζ, but not on Polη (Figures S2D–S2F), also required RFWD3 (Figure S1E). Thus, RFWD3 is essential for different TLS polymerases to replicate across peptide-DNA adducts.
To ensure that we are monitoring TLS, we sequenced the repair products and measured mutagenesis following replication-coupled DPC repair. When comparing pCTRL and pMH, we found increased mutagenesis across the crosslinked cytosine (0.45% mutation frequency) (Figure 2D). Guanine to adenine substitutions were the most common mutations (Figure 2E). Consistent with our conclusion that Polη inserts across the crosslinked base, in the absence of Polη, we observed a ~3-fold increase in mutagenesis at the insertion nucleotide (Figures 2F and 2G). In contrast, the absence of Polη did not affect the mutagenesis signature of pFpg (Figures S2G and S2H), consistent with this process depending exclusively on REV1-Polζ. Altogether, our data show that DPCs are mutagenic lesions that exhibit different mutation signatures depending on the nature of the protein crosslink and the TLS polymerase used to bypass the lesion. RFWD3 affects TLS polymerases at multiple steps of these bypass processes.
RFWD3 promotes TLS across different polymerase-stalling lesions
Given that RFWD3 depletion constitutes an absolute block to the bypass of small peptide adducts, we asked whether RFWD3 also regulates TLS across other types of polymerase-stalling lesions. To this end, we generated a plasmid carrying a site-specific cyclobutane pyrimidine dimer (CPD). Because CPD lesions were unstable in egg extracts, likely due to repair by the CPD photolyase during the 30 min licensing incubation (data not shown), we placed the CPD across from a 29 nt DNA gap to generate pCPDssDNA, which allowed nascent strands to reach the lesion by 30 s, before repair had occurred (Figure 3A). During gap-filling synthesis of pCPDssDNA, the nascent strand stalled at the −1 position before rapidly extending past the lesion (Figure 3A, lanes 6–10). Polη depletion severely inhibited bypass, consistent with the primary role of Polη in replicating across CPDs (Figure 3B, lanes 5–8 and quantification) (Johnson et al., 1999; Masutani et al., 2000; McCulloch et al., 2004), whereas REV1 depletion had no impact on the reaction (Figure 3B, lanes 9–12). Remarkably, in the absence of RFWD3, lesion bypass was strongly impaired, and nascent leading strands accumulated at the −1 position for up to 30 min (Figure 3B, lanes 13–16). We conclude that RFWD3 is also required for TLS across CPDs. However, RFWD3 depletion does not completely block bypass of CPDs, in contrast to the absolute block observed across DPC lesions. This might reflect that unlike REV1-Polζ, Polη can still partially operate without RFWD3.
Figure 3.

RFWD3 is essential for DNA synthesis across different polymerase-stalling DNA lesions
(A) pCTRLssDNA or pCPDssDNA were incubated in non-licensing egg extracts in the presence of [α-32P]dATP. Samples were analyzed on a denaturing polyacrylamide gel following AatII-ApoI digest. Top scheme: extension products generated by AatII-ApoI digest.
(B) pCPDssDNA was incubated in mock-, Polη-, REV1-, or RFWD3-depleted non-licensing extracts. Samples were analyzed as in (A). Quantification of the relative intensity of the −1 product for three independent experiments is shown in the lower panel. Intensity of the −1 product was quantified for each lane and normalized to the maximum value. Error bars represent SEM.
(C) pICLpt was replicated in mock- or RFWD3-depleted extracts (with either the RFWD3-N or the RFWD3-F antibody), in the presence of pQuant. Reaction samples were analyzed by native agarose gel electrophoresis.
(D) Intermediates generated by AflIII digest on pICLpt.
(E) Samples from (C) were digested with AflIII and analyzed on a denaturing polyacrylamide gel.
Because biallelic mutations in RFWD3 cause Fanconi anemia (FA) (Knies et al., 2017), we also addressed the role of RFWD3 during replication of a cisplatin interstrand crosslinks (ICL), which also requires TLS (Räschle et al., 2008). During replication-coupled repair of a cisplatin ICL-cointaining plasmid (pICLPt), replication forks converge on either side of the ICL, after which one of the parental strands is incised to unhook the ICL (Knipscheer et al., 2009; Räschle et al., 2008). Replication of the daughter molecule containing the remaining adduct depends on REV1-Polζ (Budzowska et al., 2015), whereas the incised strands are repaired by HR (Figure S3A) (Long et al., 2011). As previously reported in cells, RFWD3 depletion in egg extracts had no effect on the mono-ubiquitylation of FANCD2, which regulates nuclease recruitment during ICL repair (Figure S3B) (Feeney et al., 2017; Klein Douwel et al., 2014; Knipscheer et al., 2009). Consistent with no defect in FANCD2 ubiquitylation, incisions around the ICL occurred with normal kinetics in the absence of RFWD3 (Figure 3C; Figures S3C and S3D). However, without RFWD3, nascent strands persisted at the −1 and 0 positions up to 4 h, consistent with a severe TLS defect (Figure 3D; Figure 3E, lanes 8–10 and 13–15). Faithful repair by HR was also abolished without RFWD3 (Figures S3C–S3E). Thus, the essential role of RFWD3 in TLS is not limited to DPCs but can be extended to different types of polymerase-stalling lesions.
RFWD3 regulates PCNA ubiquitylation
Next, we addressed how RFWD3 regulates TLS. Previous reports indicated that RFWD3 controls the activation of the replication checkpoint (Gong and Chen, 2011), although this has been disputed (Liu et al., 2011). To address whether RFWD3 affects checkpoint signaling in egg extracts, we monitored the phosphorylation of CHK1 during replication of a DPC plasmid. As seen in Figure 4A, RFWD3-depleted extracts triggered robust and persistent phosphorylation of CHK1 (lanes 6–10), consistent with the accumulation of DNA gaps on the plasmid. Similarly, RFWD3 depletion did not inhibit CHK1 phosphorylation during pICLPt replication (Figure S4A). Therefore, the requirement for RFWD3 in TLS is unlikely to reflect a defect in ATR checkpoint activation.
Figure 4.

RFWD3 regulates PCNA ubiquitylation
(A) pMHLeads was replicated in mock- or RFWD3-depleted extracts and analyzed as in Figure 1C (upper panel) or blotted with the indicated antibodies (bottom three panels). Asterisks indicate unspecific bands.
(B) pMH or pMHPK were replicated in the presence or absence of ubiquitin E1 inhibitor. Reaction samples were digested and analyzed as in Figure 1E.
(C) Sperm chromatin was either untreated or treated with 20 J/m2 UV-C and then replicated in egg extracts. A CDC7 inhibitor was added to block origin firing and DNA replication. Proteins associated with isolated chromatin were blotted with the indicated antibodies. Red dots correspond to PCNA ubiquitylation (likely mono-, di-, and tri-ubiquitin). The black dot corresponds to mono-sumoylated PCNA (see Figure S4K for bands assignment).
(D) Sperm chromatin was either untreated or treated with 20 J/m2 UV-C and then replicated in mock- or RFWD3-depleted extracts. Chromatin was isolated, and the proteins associated were blotted with the indicated antibodies. The percentage of ubiquitylated PCNA over unmodified PCNA is indicated underneath the PCNA blot.
(E) pCTRL or pMHLeads were replicated in egg extracts and analyzed as in Figure 1C.
(F) Reactions from (E) were subjected to plasmid pull-down, and samples were blotted with the indicated antibodies.
(G) pMHLeads was replicated in mock-, RFWD3-, or REV1-depleted extracts. Reactions were processed as in (F). The percentage of ubiquitylated PCNA over unmodified PCNA is indicated.
RFWD3 was shown to ubiquitylate RPA, which was suggested to stimulate the exchange of RPA for RAD51 on chromatin to promote HR (Elia et al., 2015; Inano et al., 2017). This exchange is mediated by p97, which recognizes and extracts ubiquitylated RPA from chromatin (Inano et al., 2017). As seen in Figures S4B and S4C, p97 inhibition had a negligible impact on replication of pMHPK, in contrast to the severe defect observed by p97 inactivation during pICLPt replication (Fullbright et al., 2016). Thus, a defect in p97-mediated RPA removal is unlikely causative of the TLS defect observed in the absence of RFWD3.
To interrogate the role of ubiquitin signaling in replication across a peptide DNA adduct, pMH and pMHPK were replicated in the presence of an inhibitor of the E1 ubiquitin-activating enzyme, which efficiently blocks de novo ubiquitylation and proteasome-mediated CDT1 destruction (Figure S4D). Consistent with DNA replication initiation and elongation being independent of ubiquitylation, E1 inhibition did not affect replication kinetics (Figure 4B; Figure S4E). However, in the presence of the E1 inhibitor, nascent strands permanently arrested at −1 (Figure 4B, lanes 6–10 and 16–20), mimicking the effect of RFWD3 depletion. We also monitored whether bypass requires ubiquitin chain extension by adding an excess of different ubiquitin mutants to the extracts. Although addition of ubiquitin K48R had no effect on insertion and extension past the protein adduct, addition of ubiquitin K63R specifically blocked the extension step regulated by REV1-Polζ (Figure S4F). Similar results were obtained upon depletion of the K63-specific E2 ubiquitin-conjugating enzyme UBC13, which is essential for PCNA poly-ubiquitylation (Figures S4G–S4J) (Hoege et al., 2002; Stelter and Ulrich, 2003). Thus, replication across a M.HpaII crosslink adduct requires de novo ubiquitylation for insertion and K63-linked ubiquitin chains for extension.
Because PCNA ubiquitylation regulates TLS, we next investigated PCNA status in the absence of RFWD3. First, we confirmed that PCNA is mono- and K63 ubiquitylated in egg extracts during replication of UV-damaged sperm chromatin (Figure 4C, lanes 6 to 9; Figure S4K) (Göhler et al., 2008; Leach and Michael, 2005). Furthermore, when UV-treated chromatin was replicated in the absence of RFWD3 (Figure S4L), we observed a marked reduction in PCNA mono- and poly-ubiquitylation, which correlated with a significant decrease of Polζ (REV1 and REV7) and Polη recruitment to chromatin (Figure 4D, compare lanes 9–12 to 13–16). In contrast, small amounts of PCNA mono-ubiquitylation were observed during replication of undamaged chromatin, consistent with a previous report (Leach and Michael, 2005), and this mono-ubiquitylation was mostly independent of RFWD3 (Figure 4D, compare lanes 1–4 to 5–8). Similarly, replication of an undamaged plasmid induced PCNA mono-ubiquitylation (Figures 4E and 4F, lanes 2–3), which was also independent of RFWD3 (Figure S4M). However, replication of DPC-containing plasmids stabilized this initial mono-ubiquitylation and triggered subsequent PCNA poly-ubiquitylation at 20 and 30 min, when TLS occurs on the plasmid (Figures 4E and 4F, lanes 6–10). In this scenario, RFWD3 depletion did not affect the initial mono-ubiquitylation of PCNA (Figure 4G, compare lanes 2 and 6), which is likely damage independent. However, in the absence of RFWD3, mono-ubiquitylated PCNA rapidly disappeared and no PCNA poly-ubiquitylation was observed at the time the mock reaction underwent TLS (Figure 4G, lanes 8–10). This loss of PCNA ubiquitylation correlated with a decrease and delay in REV1 and REV7 recruitment (Figure 4G, lanes 6–9). We conclude that the absence of RFWD3 leads to a defect in damaged-induced PCNA ubiquitylation, which likely explains the failure to recruit TLS polymerases to perform lesion bypass.
RFWD3 regulates PCNA ubiquitylation in human cells
We next addressed whether RFWD3 also regulates PCNA ubiquitylation in human cells. To this end, U2OS cells expressing Strep-hemagglutinin (HA)-PCNA were treated with UV in the presence of different small interfering RNAs (siRNAs) targeted against RFWD3, and PCNA was pulled down using Strep-Tactin Sepharose beads under denaturing conditions. As expected, UV treatment induced mono- and poly-ubiquitylation of PCNA (Figure 5A, lane 3), which was severely reduced by 3 independent siRNAs that depleted RFWD3 (Figure 5A, lanes 4–7; siRNA 3 does not deplete RFWD3). The PCNA ubiquitylation defect in RFWD3-depleted cells could be restored in a cell line stably expressing ectopic wild-type (WT) RFWD3 resistant to the siRNA (Figure 5B, compare lanes 4 and 6), but not a point mutant in the RING domain devoid of E3 ligase activity (Figure 5B, lane 8) (Feeney et al., 2017). RFWD3 overexpression also triggered PCNA ubiquitylation in the absence of UV damage (Figures 5C, lane 3, and 5D). This again depended on its E3 ligase activity (Figure 5C, lane 5), which occurred exclusively on the canonical PCNA K164 ubiquitylation site (Figure 5C, lane 4) and depended on RAD18 (Figure 5E, compare lanes 4 and 5). RFWD3-mediated PCNA ubiquitylation was greatly impaired by the I639K mutation that causes FA and abrogates RFWD3 interaction with RPA (Figure 5C, lane 6) (Feeney et al., 2017). We conclude that PCNA ubiquitylation in cells is also regulated by RFWD3 and depended on an intact RPA-RFWD3 interaction.
Figure 5.

RFWD3 ubiquitin ligase activity regulates PCNA ubiquitylation in human cells
(A) U2OS cells or U2OS cells expressing Strep-HA-PCNA were transfected with siCtrl or 4 different siRNAs against RFWD3 and either left untreated or treated with UV (30 J/m2). PCNA was recovered under denaturing conditions via Strep-Tactin pull-down and analyzed by immunoblotting with the indicated antibodies.
(B) U2OS cells or U2OS/FRT GFP-RFWD3 WT or catalytic inactive (C315A) cells were treated with the indicated siRNAs, transfected with either empty vector (EV) or Strep-HA-PCNA plasmids, and treated with doxycycline to induce expression of RFWD3. UV treatment and protein pull-down were performed as in (A).
(C) U2OS cells were transfected with the indicated plasmids and then subjected to Strep-Tactin pull-down as in (A).
(D) U2OS/FRT GFP-RFWD3 WT cells were treated with doxycycline and transfected with EV or Strep-HA-PCNA 24 h before lysis in denaturing buffer. Lysates were subjected to Strep-Tactin pull-down in denaturing conditions, washed, and incubated with USP2 (ubiquitin protease) and/or UPL1 (SUMO protease), as indicated.
(E) U2OS cells or U2OS/FRT GFP-RFWD3 WT cells were transfected with either control (siCtrl) or RAD18 siRNAs. After 48 h, cells were transfected with either EV or Strep-HA-PCNA plasmids and treated with doxycycline to induce the expression RFWD3 WT. Then, cells were treated with UV for 4 h and processed for Strep-Tactin pull-down as described in (A). The asterisk denotes a non-specific band.
RFWD3 is required for ubiquitylation and protein accumulation at ssDNA
Next, we performed CHROMASS (chromatin mass spectrometry) to identify proteins whose recruitment depends on RFWD3 (Räschle et al., 2015). Sperm chromatin was either left untreated or treated with a high dose of UV, incubated in mock- or RFWD3-depleted non-licensing egg extracts, and analyzed by label-free MS. As a control, REV1-depleted extracts were used to monitor changes in protein recruitment to chromatin that are specific to defects in TLS. As seen in Figure 6A, REV1 depletion led to the specific downregulation on chromatin of REV1, REV7, and REV3 (subunits of Polζ), as well as Polκ. Although REV7 and REV3 are already co-depleted from extracts by REV1 depletion, loss of Polκ, which is not co-depleted by REV1, supports the critical role for REV1 in recruiting Polκ (Figure S6B) (Guo et al., 2003; Ohashi et al., 2004) and demonstrates that CHROMASS reliably monitors TLS-dependent protein recruitment to UV-damaged chromatin. Conversely, 14 proteins were enriched in the absence of REV1. These included Polη and its interactor protein WRNIP1 (Yoshimura et al., 2014; Yuasa et al., 2006) and proteins participating in DSB repair (RNF168, 53BP1, PTIP, PTIP-associated protein 1, and the BRCA1-A complex) (Figure 6A; Table S2). We reason that in the absence of REV1-Polζ, gap-filling synthesis across some UV lesions (e.g., 6-4 photoproducts) is impaired, leading to the accumulation of Polη on chromatin and the generation of DNA DSBs.
Figure 6.

RFWD3 regulates ubiquitin levels and protein recruitment to UV damaged chromatin
(A) MS analysis of protein recruitment to UV-treated sperm chromatin in mock- or REV1-depleted extracts. The volcano plot shows the difference in abundance of proteins between the two sample conditions (x axis), plotted against the p value resulting from two-tailed Student’s t testing (y axis). Proteins significantly down- or upregulated (false discovery rate [FDR] < 5%) in REV1-depleted reactions are represented in red or blue, respectively. n = 4 biochemical replicates. FDR < 5% corresponds to permutation-based, FDR-adjusted q < 0.05. Different isoforms of the same protein can be detected (e.g., REV1).
(B) Same experiment as in (A) but comparing mock- to RFWD3-depleted extracts. Small red dots, 1% < FDR < 5%; large red dots, FDR < 1%.
(C) STRING network of proteins highly significantly upregulated (FDR < 1%) on UV-treated sperm chromatin compared with mock-treated chromatin and highly significantly downregulated (FDR < 1%) on UV-treated sperm chromatin with RFWD3 depletion compared with mock depletion.
(D) An independent experiment was analyzed by immunoblot using the indicated antibodies.
(E) Average PCNA and PCNA K164-ubiquitin abundance on sperm chromatin identified by tandem mass spectrometry (MS/MS), quantified in a label-free manner, and plotted as a ratio over untreated control. n = 4 biochemical replicates. Error bars represent SEM. ∗∗p < 0.01, via two-tailed Student’s t testing.
(F) Quantification of ubiquitin linkages on sperm chromatin directly identified by MS/MS via diglycine-modified lysine residues in the corresponding peptide sequences and quantified in a label-free manner. n = 4 biochemical replicates. Error bars represent SEM. ∗p < 0.05, ∗∗p < 0.01, via two-tailed Student’s t testing.
We next compared MS profiles with or without RFWD3. Strikingly, the absence of RFWD3 led to profound impairment of protein recruitment to UV-treated chromatin, affecting more than 200 proteins, which are recruited to chromatin in response to UV damage (Figure 6B; Figure S6A). Among the most de-enriched proteins were the TLS polymerases REV1-Polζ, Polη, and Polκ; the PCNA-ubiquitylating enzyme RAD18 and its associated complex SLF1-SLF2-SMC5/6; PCNA ubiquitylation-interacting proteins such as ZRANB3 and WRNIP1; and DSB repair factors, such as RNF168, 53BP1, PTIP, PTIP-associated protein 1, and the BRCA1-A complex (Figures 6B–6D). We also noticed a profound defect in recruitment of ubiquitin to damaged chromatin (Figures 6B and 6C). When quantifying ubiquitylated lysine residues, we confirmed that PCNA ubiquitylation on K164, but not total levels of PCNA, was specifically reduced without RFWD3, but not REV1 (Figure 6E). Ubiquitin K63 and K48 linkages were also dramatically reduced in the absence of RFWD3 (Figure 6F). In contrast to ubiquitin, both SUMO2 and SUMO3 levels were unaffected without RFWD3 (Figure S6C). We conclude that lack of RFWD3 leads to a profound reduction of ssDNA-associated ubiquitylation, abolishing recruitment of multiple components of pathways involved in PCNA ubiquitylation, post replicative gap-filling DNA synthesis, and DSB repair.
RFWD3 stimulates ubiquitylation of different proteins on ssDNA
The reduction in ubiquitylation and repair factors on damaged chromatin described earlier suggests that RFWD3 may promote ubiquitin modifications at polymerase-stalling lesions that stimulate protein recruitment, PCNA ubiquitylation, and ultimately gap-filling repair. RFWD3 was previously shown to ubiquitylate the ssDNA binding proteins RPA and RAD51 on multiple lysines upon DNA damage (Elia et al., 2015; Inano et al., 2017). Altogether, these observations raise the possibility that RFWD3 may stimulate ubiquitylation of multiple proteins on ssDNA to promote protein accumulation and bypass/repair at DNA gaps. To directly test whether RFWD3 stimulates protein ubiquitylation in the context of ssDNA, we monitored a protein exogenous to egg extracts on ssDNA. To this end, the bacterial glycosyase Fpg was either crosslinked to duplex (pFpg) or ssDNA (pFpgssDNA) and added to non-replicating egg extracts immunodepleted of SPRTN to avoid potential DPC degradation. In this setting, pFpgssDNA triggered modifications of Fpg, whereas pFpg did not (Figure 7A), as we have previously shown for M.HpaII crosslinks (Larsen et al., 2019). Upon inhibition of de novo ubiquitylation, Fpg persisted mostly unmodified, indicating that these modifications are indeed ubiquitin (Figure 7B, lanes 5–8). RFWD3 was specifically recruited to pFpgssDNA (Figure S7A), and RFWD3 depletion abolished Fpg ubiquitylation (Figure 7C, lanes 5–8). Moreover, Fpg ubiquitin linkages appeared to be formed from a mixture of K63- and K48-linked ubiquitin chains (Figure S7B), consistent with the RFWD3-mediated ubiquitylation signature observed on RPA (Elia et al., 2015). Similar results were observed on M.HpaII, although here an additional ligase likely contributes to the residual ubiquitylation activity observed without RFWD3 (Figure S7C). We confirmed the specificity of RFWD3 depletion using antibody F (Figures S7D and S7E) and cross-complemented RFWD3-F depletion with a peptide-eluted extract generated from an RFWD3-N immunoprecipitate.
Figure 7.

RFWD3 simulates ubiquitylation of proteins on ssDNA
(A) Fpg bacterial glycosylase was crosslinked to either double-stranded (pFpg) or single-stranded DNA (pFpgssDNA) and added to SPRTN-depleted non-licensing egg extracts. DPC pull-down under stringent conditions was performed at the indicated time points, and samples were blotted against crosslinked Fpg. Slow mobility bands represent ubiquitylated Fpg species (see B).
(B) pFpgssDNA was incubated in SPRTN-depleted non-licensing extracts, and ubiquitin E1 inhibitor was added where indicated. Plasmids were recovered, and samples were blotted against Fpg as in (A).
(C) pFpgssDNA was incubated in mock- or RFWD3-depleted non-licensing extracts (also depleted of SPRTN) for the indicated time points and samples processed as in (A).
(D) Generation of an AP site on ssDNA (pAPssDNA) to induce HMCES crosslinking.
(E) pAPssDNA was incubated in SPRTN-depleted non-licensing extracts, and ubiquitin E1 inhibitor was added where indicated. Plasmids were recovered, and proteins were blotted against HMCES. The black dot indicates sumoylated HMCES (see F).
(F) pAPssDNA was incubated in mock- or RFWD3-depleted non-licensing extracts (depleted of SPRTN), and ubiquitin E1 inhibitor or SUMO E1 inhibitor was added where indicated. Plasmids were recovered and analyzed as in (D).
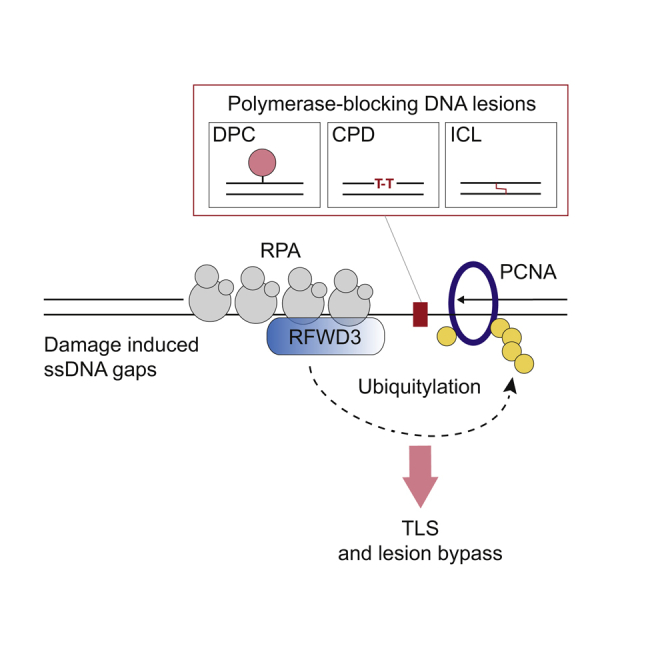
(G) Model illustrating the role of RFWD3 in gap-filling DNA synthesis (see Discussion).
Finally, we addressed whether an endogenous protein fixed on ssDNA could also be targeted by RFWD3. To this end, we took advantage of the recently identified suicide enzyme HMCES, which links and shields abasic (AP) sites on ssDNA (Mohni et al., 2019). We observed rapid crosslinking of HMCES to the plasmid when the AP site was located on ssDNA (Figure 7E, lane 1). Following crosslinking, HMCES underwent rapid modifications that were largely suppressed upon addition of the ubiquitin E1 inhibitor (Figure 7E, compare lanes 1–3 and 4–6). In this setting, RFWD3 depletion again abolished HMCES ubiquitylation (Figure 7F, lanes 5–8). Altogether, these data indicate that RFWD3 promotes ubiquitylation of various proteins residing at RPA-coated ssDNA, thereby playing a central role in regulating ubiquitin-dependent protein accumulation and repair processes at these genomic regions.
Discussion
RFWD3 was previously shown to interact with and ubiquitylate RPA and to participate in the restart of stalled replication forks via HR, possibly by promoting the exchange of RPA from ssDNA (Elia et al., 2015; Feeney et al., 2017; Inano et al., 2017). Here, we show that RFWD3 has an additional and essential function safeguarding the integrity of replicating genomes by promoting DNA synthesis across a spectrum of polymerase-stalling lesions. We show that RFWD3 stimulates ubiquitylation of proteins when these are located on ssDNA. Consequently, in the absence of RFWD3, ubiquitin signaling at ssDNA gaps is severely compromised, which in turn undermines key events required for timely and productive DNA damage bypass. Most notably, proteins known to stimulate PCNA ubiquitylation and to participate in DDT are no longer recruited to these sites, which leads to a profound defect in PCNA ubiquitylation and a nearly complete absence of DNA damage bypass. We propose that RFWD3 is one of the upstream responders to ssDNA gaps and stimulates ubiquitin chain formation to create a recruitment platform to amplify PCNA ubiquitylation and promote gap-filling repair (Figure 7G).
RFWD3 promotes PCNA ubiquitylation and gap-filling DNA synthesis
The drastic loss of PCNA ubiquitylation observed in the absence of RFWD3 likely explains the severe TLS defects observed in egg extracts. We show that PCNA ubiquitylation in human cells is also regulated by RFWD3 and depended on RFWD3’s E3 ligase activity. However, we can envision different models to explain how RFWD3 affects PCNA ubiquitylation and downstream gap-filling DNA synthesis. One possibility is that RFWD3 directly ubiquitylates PCNA when RFWD3 is bound to RPA beyond the lesion. Accordingly, RFWD3-mediated PCNA ubiquitylation in cells also depended on an intact RPA-RFWD3 interaction. Moreover, although the Rad5 orthologs, HLTF and SHPRH, have been shown to poly-ubiquitylate PCNA in vertebrates (Lin et al., 2011; Motegi et al., 2006; Unk et al., 2006, 2008), combined depletion of these two ubiquitin ligases in cells did not affect PCNA ubiquitylation upon UV damage in our hands (Figures S5A and S5B). In agreement with these findings, hltf/shprh double-knockout MEFs still exhibit PCNA poly-ubiquitylation and are not sensitive to UV treatment (Krijger et al., 2011). Thus, another ubiquitin ligase likely compensates when the Rad5 orthologs are absent from vertebrates. Could RFWD3 provide this missing activity? Although our recombinant RFWD3 preparations, which are needed to test this model, exhibited robust ubiquitin ligase activity in vitro, these proteins underwent rapid auto-ubiquitylation and degradation when added to extracts (data not shown). We believe that this is caused by an unfolded RFWD3-WD40 propeller domain, which is critical for interaction with RPA (Feeney et al., 2017) and could restrain RFWD3 ligase activity. RPA depletion from egg extracts co-depletes RFWD3 (data not shown). This shows that RPA-RFWD3 interaction is not restricted to ssDNA but is likely constitutive in solution and could restrain RFWD3 ligase activity until RPA is loaded onto ssDNA.
Although a model in which RFWD3 poly-ubiquitylates PCNA remains to be tested, the following evidence favors an indirect stimulation model. First, previous studies in cells have shown that upon DNA damage, RFWD3 promotes RPA ubiquitylation on different subunits and on at least 18 lysine residues (Elia et al., 2015). RAD51, another ssDNA binding protein, was also shown to be ubiquitylated by RFWD3 (Inano et al., 2017). Moreover, we show here that three additional proteins become ubiquitylated in an RFWD3-dependent manner in egg extracts, but only when these are linked to ssDNA. Altogether, these data indicate that RFWD3 is able to stimulate the ubiquitylation of a diverse range of substrates on ssDNA, rendering specific PCNA ubiquitylation on K164 by RFWD3 an unlikely scenario. Instead, we favor a model in which RFWD3 is recruited to ssDNA gaps by RPA, where it is activated to initiate ubiquitylation of RPA and proteins in its vicinity, giving rise to an accumulation of ubiquitin conjugates at ssDNA regions (Figures 7Gi and 7Gii). These initial ubiquitylation events likely stimulate the recruitment and retention of factors participating in gap-filling repair to the vicinity of the lesions (Figure 7Giii). Most proteins involved in DDT contain one or more ubiquitin binding motifs (e.g., RAD18, REV1-Polζ, Polη, Polk, WRNIP1, ZRANB3, SLX4, FAN1, USP1, SMN1A, and SPRTN), and most of these factors are significantly depleted from damaged chromatin in the absence of RFWD3. Therefore, RFWD3-dependent ubiquitylation would, on the one hand, enrich DDT factors in the vicinity of the lesion and, on the other hand, promote PCNA mono-and poly-ubiquitylation via RAD18 to dictate pathway choice (Figures 7Giii and 7Giv). Thus, our model predicts that in addition to TLS, TS and other post-replicative repair pathways, such as SPRTN-mediated proteolysis (Larsen et al., 2019), should be regulated by RFWD3. Consistent with this, the recruitment of ZRANB3, the enzyme responsible for reversing replication forks (which is a way to initiate TS) (Ciccia et al., 2012; Vujanovic et al., 2017), is drastically reduced in the absence of RFWD3.
PCNA ubiquitylation and TLS regulation
We show that RFWD3-mediated bypass across a M.HpaII crosslink is a mutagenic process that most likely involves Polη insertion followed by REV1-Polζ extension. However, in the absence of Polη, REV1-Polζ appears to both insert and extend with comparable kinetics to mock reactions but with increased mutagenesis at the insertion step, which is consistent with studies on other DNA lesions, such as CPDs and (6-4) photoproducts (Gibbs et al., 2005; Szüts et al., 2008). But how is Polη favored to perform insertion when Polη and REV1-Polζ are both available? The current dogma is that PCNA mono-ubiquitylation targets all TLS polymerases, whereas RAD5-mediated poly-ubiquitylation of PCNA triggers TS. Although this model is well defined in Saccharomyces cerevisiae, we have gathered evidence to suggest an alternative model in which Polη depends on PCNA mono-ubiquitylation and REV1-Polζ activity is regulated by PCNA poly-ubiquitylation. First, during replication of a DPC plasmid, REV1-Polζ recruitment to the lesion did not correlate with the mono-ubiquitylation of PCNA but instead appeared late in the reaction, when a large fraction of PCNA is poly-ubiquitylated on chromatin (Figure 4F, lanes 8–10). Moreover, the exogenous addition of lysine-deficient ubiquitin or ubiquitin K63R mutants, which blocked poly-ubiquitylation of PCNA, but not mono-ubiquitylation of PCNA, allowed insertion by Polη but inhibited extension by REV1-Polζ. In agreement, depletion of UBC13, which stabilizes PCNA in its mono-ubiquitylated form, also specifically blocked the extension step. A previous report in fission yeast indicated that Polζ-dependent error-free bypass of UV-induced 6-4 photoproducts depends on the RAD8Rad5/Ubc13-Mms2 ubiquitin ligase complex, highlighting a clear difference in the regulation of TLS between fission and budding yeast (Coulon et al., 2010). Although we cannot exclude that a substrate other than PCNA could be ubiquitylated by UBC13, our results are consistent with a model in which the mono-ubiquitylation of PCNA targets the first round of insertion polymerases (e.g., Polη), whereas the sequential poly-ubiquitylation would promote the REV1-Polζ complex. Because we observe residual levels of PCNA mono-ubiquitylation in the absence of RFWD3, this model is also consistent with RFWD3 depletion delaying, but not completely blocking, the bypass of CPD lesions, which depends on Polη.
In addition to damaged-induced ubiquitylation, PCNA was shown to be mono-ubiquitylated during replication of undamaged sperm chromatin (Leach and Michael, 2005). A recent report suggests that this is also likely the case in human cells (Thakar et al., 2020), and our data generated during replication of undamaged plasmids agree with these findings. Although the significance of damage-independent PCNA mono-ubiquitylation remains unclear, RFWD3 depletion did not affect it, suggesting independent mechanisms for inducing PCNA ubiquitylation at the fork during unstressed replication versus at ssDNA gaps upon DNA damage and RFWD3 regulating only the latter of the two.
RFWD3 function in human cells and FA
How do our results in Xenopus egg extracts account for observations in mammalian systems? First, the severe defect observed in bypassing DNA polymerase-stalling lesions in the absence of RFWD3 would most likely be incompatible with cellular proliferation. In agreement, RFWD3 was identified as an essential gene in a genome-wide screen (Wang et al., 2015), and full knockout of RFWD3 in human cells was not possible (Feeney et al., 2017), although surprisingly, RFWD3 knockout mice have been generated (Knies et al., 2017). Importantly, heterozygous mutations in RFWD3 have been identified in a patient with classical FA symptoms (Knies et al., 2017). In this patient, an I639K abrogates the interaction of RFWD3 with RPA2 (Feeney et al., 2017), compromising RFWD3 recruitment to ICLs and inhibiting ICL repair. In Figure 3, we show how RFWD3 depletion in egg extracts similarly abolished the repair of an ICL-containing plasmid. Consistent with cellular work, FANCD2 was activated normally in the absence of RFWD3, suggesting that RFWD3 acts downstream of incisions (Feeney et al., 2017). However, our work provides a new layer of understanding to the ICL repair defect resulting from loss of RFWD3 function, because we show that the TLS step, which is required for HR, is inhibited in the absence of RFWD3. Although we could not reconstitute RFWD3 with the FA mutation in our system, we predict that both TLS and HR would be affected by the mutation. In accordance, our experiments in human cells show that RFWD3-dependent PCNA ubiquitylation is conserved and hindered by the FA mutation.
Importantly, our work highlights that the function of RFWD3 in replication-coupled repair is unlikely to be restricted to ICLs, because we show that the response to multiple DNA lesions (and likely all polymerase-stalling lesions) depends on its function. Accordingly, a genome-wide CRISPR-Cas9 screen against 27 genotoxic agents clustered RFWD3 next to REV1-Polζ and RAD18, away from the other Fanconi/HR genes (Olivieri et al., 2020). Moreover, RFWD3-mediated ubiquitylation of DPCs located on ssDNA, such as HMCES, might also stimulate their destruction. In analogy to the recently identified role for TRAIP in ubiquitylating proteins ahead of the replication fork to stimulate clearance or repair of obstructions that hinder CMG translocation (Larsen et al., 2019; Wu et al., 2019), we propose that RFWD3 has an analogous function at polymerase-stalling lesions behind the replication fork to promote gap-filling synthesis and repair.
Limitations of study
As stated earlier, we were unable to rescue RFWD3 depletion in egg extracts, which does not allow us to fully exclude the possibility that the effects attributed to RFWD3 are caused by the co-depletion of another critical factor interacting with RFWD3. Nevertheless, important evidence supports that our observations are specific to RFWD3 loss. First, the effects caused by RFWD3 depletion in egg extracts are recapitulated with independent antibodies, and we show via whole proteome analysis that RFWD3 is the only protein significantly depleted from these extracts. Moreover, the defect in damaged-induced PCNA ubiquitylation, which likely impairs damage bypass by TLS, is recapitulated in human cells using different siRNAs against RFWD3 and rescued with WT, but not with a point mutant, in the RING domain of RFWD3. Altogether, these observations support that our results are most likely specific to RFWD3 loss and dependent on RFWD3 ubiquitin ligase activity.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Rev1-N | Budzowska et al., 2015 | N/A |
| Rabbit polyclonal anti-Rev1-C | Budzowska et al., 2015 | N/A |
| Rabbit polyclonal anti-Rev7 | Räschle et al., 2008 | N/A |
| Rabbit polyclonal anti-PCNA | Kochaniak et al., 2009 | N/A |
| Rabbit polyclonal anti-Fancd2 | Räschle et al., 2008 | N/A |
| Rabbit polyclonal anti-Cdt1 | Arias and Walter, 2005 | N/A |
| Rabbit polyclonal anti-MCM6 | Larsen et al., 2019 | 2926 |
| Rabbit polyclonal anti-RPA | Walter and Newport, 2000 | N/A |
| Rabbit polyclonal anti-TraiP | Larsen et al., 2019 | 3472 |
| Rabbit polyclonal anti-BRCA1 | Joukov et al., 2006 | N/A |
| Rabbit polyclonal anti-Sprtn | Larsen et al., 2019 | 3703 |
| Rabbit polyclonal anti-M.HpaII | Duxin et al., 2014 | N/A |
| Rabbit polyclonal anti-phospho-Chk1 (S345) | Cell Signaling Technology | Cat#2341; RRID: AB_330023 |
| Rabbit polyclonal anti-histone H3 | Cell Signaling Technology | Cat#9715; RRID: AB_331563 |
| Rabbit monoclonal antibody anti-ubiquityl-PCNA (K164) (D5C7P) | Cell Signaling Technology | Cat#13439; RRID: AB_2798219 |
| Rabbit polyclonal anti-Polη | This paper | 3923 |
| Rabbit polyclonal anti-HMCES | This paper | 4639 |
| Rabbit polyclonal anti-Rfwd3-N | This paper | 3575 and 4056 |
| Rabbit polyclonal anti-Ubc13 | This paper | 4262 |
| Rabbit polyclonal anti-PolD1 | This paper | 4000 |
| Rabbit polyclonal anti-PolD2 | This paper | 4516 |
| Rabbit polyclonal anti-PolD3 | This paper | 4517 |
| Rabbit polyclonal anti-Rad6 | This paper | 4266 |
| Rabbit polyclonal anti-Polκ | This paper | 3924 |
| Rabbit polyclonal anti-Zranb3 | This paper | 4057 |
| Rabbit polyclonal anti-Nse2 | This paper | 3484 |
| Rabbit polyclonal anti-Rad18 | This paper | 4265 |
| Rabbit polyclonal anti-RPA-70 | This paper | 4495 |
| Rabbit polyclonal anti-RPA-14 | This paper | 4493 |
| Rabbit polyclonal anti-Wrnip1 | This paper | 4082 |
| Rabbit polyclonal anti-Rfwd3-F | This paper | N/A |
| Mouse monoclonal anti-GFP | Roche | Cat#11814460001; RRID: AB_390913 |
| Rabbit polyclonal anti-GFP | Santa Cruz Biotachnology | Cat#sc-8334; RRID: AB_641123 |
| Rabbit polyclonal anti-HA | Santa Cruz Biotachnology | Cat#sc-805; RRID: AB_631618 |
| Goat polyclonal antibody anti MCM6 | Santa Cruz Biotachnology | Cat#sc-9843; RRID: AB_2142543 |
| Rabbit polyclonal antibody anti RAD18 | Bethyl Laboratories | Cat#A301-340A; RRID: AB_937974 |
| Goat polyclonal antibody anti HLTF | Santa Cruz Biotachnology | Cat#sc-27542; RRID: AB_2117306 |
| Rabbit polyclonal antibody anti SHPRH | abcam | Cat#ab80129; RRID: AB_2042909 |
| Rabbit polyclonal antibody anti RFWD3 | abcam | Cat#ab138030; RRID: AB_2687568 |
| Rabbit polyclonal antibody anti UBC13 | Cell Signaling Technology | Cat#4919; RRID: AB_2211168 |
| Mouse monoclonal antibody anti ubiquitin | Santa Cruz Biotachnology | Cat#sc-8017; RRID: AB_2762364 |
| Mouse monoclonal antibody anti vinculin | Sigma-Aldrich | Cat#V9131; RRID: AB_477629 |
| Chemicals, peptides, and recombinant proteins | ||
| MLN-7243 Ubiquitin E1 inhibitor | Active Biochem | Cat#A-1384; CAS:1450833-55-2 |
| ML-792 SUMO E1 inhibitor | Medkoo Biosciences | Cat#407886; CAS:1644342-14-2 |
| NMS-873 p97 inhibitor | Sigma-Aldrich | Cat#SML1128; CAS:1418013-75-8 |
| PHA 767491 Cdc7 inhibitor | Sigma-Aldrich | Cat#PZ0178; CAS:845538-12-7 |
| LacI-biotin | Duxin et al., 2014 | N/A |
| M.HpaII | Duxin et al., 2014 | N/A |
| xlPOLη | This paper | N/A |
| Fpg | New England BioLabs | Cat#M0240L |
| UDG | New England BioLabs | Cat#M0280S |
| Human recombinant ubiquitin No Lys | Boston Biochem | Cat#UB-NOK |
| Human recombinant ubiquitin mutant K48R | Boston Biochem | Cat#UM-K48R |
| Human recombinant ubiquitin mutant K63R | Boston Biochem | Cat#UM-K63R |
| Human recombinant ubiquitin mutant K63 only | Boston Biochem | Cat#UM-K630 |
| Human chorionic gonadotropin | Sigma-Aldrich | Cat#CG10-10VL |
| Alpha-32-deoxyadenosinetriphosphate | Perkin Elmer | Cat#BLU512H250UC |
| Gamma-32-adenosinetriphosphate | Perkin Elmer | Cat#BLU502A100UC |
| Gel Loading Buffer II | Thermo Fischer | Cat#AM8547 |
| Lambda exonuclease | New England BioLabs | Cat#M0262S |
| Exonuclease I | New England BioLabs | Cat#M0293S |
| Acc65I restriction enzyme | New England BioLabs | Cat#R0599L |
| HincII restriction enzyme | New England BioLabs | Cat#R0103S |
| SapI restriction enzyme | New England BioLabs | Cat#R0596S |
| Proteinase K, recombinant | Roche | Cat#3115879001 |
| SYBR® Gold Nucleic Acid Gel Stain | Thermo Fisher | Cat#S11494 |
| Phusion® High-Fidelity DNA Polymerase | New England BioLabs | Cat#M0530S |
| Protein A Sepharose Fast Flow | GE Heath Care | Cat#17-1279-01 |
| RNase A | Thermo Fischer | Cat#EN0531 |
| Streptavidin-coupled magnetic beads M-280 | Invitrogen | Cat#11205D |
| Benzonase | Novagen | Cat#70746-3 |
| LysC | Life Technologies | Cat#90051 |
| Trypsin | Life Technologies | Cat#90305 |
| FuGENE 6 Transfection Reagent | Promega | Cat#E2692 |
| Lipofectamine RNAiMAX | Invitrogen | Cat#13778075 |
| Doxycyclin | Sigma-Aldrich | Cat#D9891; CAS: 23390-14-5 |
| Strep-Tactin Sepharose resin | IBA BioTAGnology | Cat#2-1201-010 |
| Blasticidin S | Invitrogen | Cat#ant-bl-1 |
| Hygromycin | Thermo Fisher | Cat#10687010 |
| Critical commercial assays | ||
| TnT® Sp6 Quick Coupled Transcription/Translation System | Promega | Cat#L2080 |
| QuickChange II Site directed mutagenesis kit | Agilent | Cat#200524 |
| Deposited data | ||
| CHROMASS | This paper; PRIDE partner repository | PRIDE: PXD018217 |
| Total proteome | This paper; PRIDE partner repository | PRIDE: PXD021445 |
| DNA deep sequencing | This paper; European nucleotide archive | ENA: PRJEB39253 |
| Experimental models: cell lines | ||
| Human: U2OS cell line | ATCC | HTB-96 |
| U2OS/Strep-HA-PCNA WT | Mosbech et al., 2012 | N/A |
| U2OS FRT Flp-In TRex | Jakob Nilsson Lab | N/A |
| U2OS/FRT GFP RFWD3 WT siR2 | This paper | N/A |
| U2OS/FRT GFP RFWD3 C315A siR2 | This paper | N/A |
| Experimental models: organisms/strains | ||
| Xenopus laevis (females) | Nasco | Cat#LM0053MX |
| Xenopus laevis (males) | Nasco | Cat#LM00715MX |
| Oligonucleotides | ||
| For pFpg: TCA GCA TCC GGT AGC TAC TCA ATC C(8oxoG)G TAC C | This paper | Tag Copenhagen |
| For pCPD: TGA GGT ACC GGA (T-T)G AGT AGC TAC CGG ATG C | This paper | TriLink, USA |
| For pAP: TCA GCT AGT TAU AAT AGC CC | This paper | Tag Copenhagen |
| Strand-specific primer DNA library: GGATCCATGTCGCAGTTGCGCA GCCTGAATGG |
This paper | Tag Copenhagen |
| Fw primer DNA library: TAGGATCCATCACGCAGTTG | This paper | Tag Copenhagen |
| Fw primer DNA library: TANGGATCCATCACGCAGTTG | This paper | Tag Copenhagen |
| Fw primer DNA library: TANNGGATCCATCACGCAGTTG | This paper | Tag Copenhagen |
| Rv primer DNA library: GTCGGGGCTGGCTTAACTATGC | This paper | Tag Copenhagen |
| siCtrl: GGGAUACCUAGACGUUCUA | (Thorslund et al., 2015) | N/A |
| siRFWD3#2: GGAAACAGGCCGAGUUAGA | Elia et al., 2015 | N/A |
| siRFWD3#3: GUUAAGAUGUUGAGUACU | Elia et al., 2015 | N/A |
| siRFWD3#4: GGACCUACUUGCAAACUAU | Elia et al., 2015 | N/A |
| siRFWD3#601: AACUCCUGCACAUGACUGC | This paper | N/A |
| siRAD18: ACUCAGUGUCCAACUUGCU | Mosbech et al., 2012 | N/A |
| siTRAIP: CCGUGAUGAUAUUGAUCUCAA | (Hoffmann et al., 2016) | N/A |
| siGENOME Human SHPRH siRNA | Dharmacon | N/A |
| siGENOME Human HLTF siRNA | Dharmacon | N/A |
| Recombinant DNA | ||
| pJLS2 | Larsen et al., 2019 | N/A |
| pNBL101 | Kose et al., 2019 | N/A |
| pFpg | This paper | N/A |
| pCPD | This paper | N/A |
| pAP | This paper | N/A |
| pCMV-Sport-xlPolη | This paper | N/A |
| pcDNA4/TO Strep HA PCNA WT | Mosbech et al., 2012 | N/A |
| pcDNA4/TO Strep HA PCNA K164R | Mosbech et al., 2012 | N/A |
| pcDNA5 FRT/TO GFP-RFWD3 WT | Feeney et al., 2017 | N/A |
| pcDNA5 FRT/TO GFP-RFWD3 C315A | Feeney et al., 2017 | N/A |
| pcDNA5 FRT/TO GFP-RFWD3 I639K | Feeney et al., 2017 | N/A |
| pcDNA5 FRT/TO GFP-RFWD3 WT siR2 | This paper | N/A |
| pcDNA5 FRT/TO GFP-RFWD3 C315A siR2 | This paper | N/A |
| pcDNA5 FRT/TO GFP-RFWD3 I639K siR2 | This paper | N/A |
| pOG44 | Invitrogen | Cat#V600520 |
| pQuant | (Knipscheer et al., 2012) | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| AdapterRemoval | Schubert et al., 2016 | https://adapterremoval.readthedocs.io/en/latest/ |
| samtools | Li et al., 2009 | http://www.htslib.org |
| Picard | Broad Institute, GitHub repository. | http://broadinstitute.github.io/picard/ |
| MaxQuant | (Cox and Mann, 2008) | http://www.coxdocs.org/doku.php |
| Perseus | (Tyanova et al., 2016) | http://www.coxdocs.org/doku.php |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Julien P. Duxin (julien.duxin@cpr.ku.dk).
Materials availability
Plasmids generated in this study are available upon request.
Data and code availability
The accession number for the CHROMASS proteomics data reported in this paper is ProteomeXchange: PXD018217. The accession number for the total proteome proteomics data reported in this paper is ProteomeXchange: PXD021445.
The accession number for the deep sequencing of repair products reported in this paper is ENA: PRJEB39253. Original data have been deposited to Mendeley Data: DOI:10.17632/9mbb8sp7b7.1
Experimental model and subject details
Egg extracts were prepared using Xenopus laevis (Nasco Cat #LM0053MX, LM00715MX). All experiments involving animals were approved by the Danish Animal Experiments Inspectorate, and are conform to relevant regulatory standards and European guidelines.
Method details
Xenopus egg extracts and DNA replication reactions
Preparation of Xenopus egg extracts was performed as described previously (Lebofsky et al., 2009). For replication of plasmid DNA, plasmids were first licensed in high-speed supernatant (HSS) for 20-30 min at RT at a final concentration of 7.5 ng/μL. Replication was initiated by addition of two volumes of nucleoplasmic egg extract (NPE). For gap filling reactions in non-licensing extracts, one volume of HSS and two volumes of NPE were premixed prior to the addition of plasmid DNA (final concentration of 10 ng/μL). For replication in the presence of LacI, plasmid DNA (150 ng/μL) was incubated with an equal volume of 12 μM LacI for 1 hr prior to licensing (Duxin et al., 2014). Ubiquitin E1 inhibitor (MLN7243; Active Biochem) was supplemented to egg extracts at the final concentration of 200 μM, 10 min prior to initiating the reaction. SUMO E1 inhibitor (ML-792; Medkoo Biosciences) was supplemented to egg extracts at the final concentration of 50 μM. To inhibit p97, NMS-873 (Sigma) was supplemented to NPE at the final concentration of 300 μM. For visualization of DNA replication intermediates, replication reactions were supplemented with [α-32P]dATP (Perkin Elmer) and 1 μL for each time point was added to 5 μL of stop buffer (5% SDS, 80 mM Tris pH 8.0, 0.13% phosphoric acid, 10% Ficoll) supplemented with 1 μL of Proteinase K (20 mg/mL) (Roche). Samples were incubated at 37°C for 1 h, followed by separation by 0.9% native agarose gel electrophoresis and visualization using a phosphorimager. Radioactive signal was quantified using ImageJ (NIH, USA).
Preparation of DNA constructs
pMH, pMHLeads, and pMHssDNA were previously described in Larsen et al. (2019) as pDPC, pDPC2xLead and pDPCssDNA, respectively. To generate pMHPK or pMHssDNA-PK, pMH or pMHssDNA were treated with Proteinase K (37°C overnight in presence of 0.5% SDS) to degrade the crosslinked M.HpaII to a 4 amino acids peptide adduct. The plasmids were subsequently recovered by phenol/chloroform extraction and ethanol precipitation. To generate pFpg, pJLS2 (Larsen et al., 2019) was nicked with Nt.BbvCI and annealed with the following oligo: 5′-TCA GCA TCC GGT AGC TAC TCA ATC C(8oxoG)G TAC C-3′. The resulting plasmid was ligated and purified. 3U/μL Fpg (New England BioLabs) was crosslinked to the 8oxoG containing plasmid (90ng/μL) in: 25mM HEPES pH 6.8, 1mM EDTA, 1mM DTT, 50mM NaBH4 for 2 h at 37°C. Subsequently the reaction buffer was quenched with 400mM glucose on ice overnight. To generate pCPDssDNA, first pJLS2 (Larsen et al., 2019) was modified to create pNBL101 (Kose et al., 2019). To generate a plasmid containing a site-specific cyclobutene thymidine dimer (pCPD), pNBL101 was nicked with Nt.BbvCI and ligated with the following oligo containing a thymidine dimer lesion 5′-TGA GGT ACC GGA (T-T)G AGT AGC TAC CGG ATG C 3′ (TriLink, USA). To generate pCPDssDNA, pCPD was nicked with Nb.BbvCI and the resulting ssDNA fragment was melted off and captured by annealing to an excess complementary oligo. Excess oligos were degraded by Exonuclease I treatment (New England BioLabs). The plasmid containing a gap across the CPD lesion was then recovered by phenol/chloroform extraction and ethanol precipitation. pICLPt was prepared as previously described (Räschle et al., 2008). To generate pAPdsDNA, pNBL101 was digested with Nb.BbvCI and ligated with the following oligo containing an uracil (5′-TCA GCT AGT TAU AAT AGC CC-3′). To generate the abasic site, 600 ng of plasmid were incubated with 5 U of uracil-DNA glycosylase (UDG; New England BioLabs) for 30 min at 37°C prior to incubation in egg extracts. To generate pAPssDNA, pAPdsDNA was nicked with Nt.BbvCI and the resulting ssDNA fragment was melted off and captured by annealing to an excess complementary oligo. Excess oligos were degraded by Exonuclease I treatment (New England BioLabs). The plasmid containing a gap across the AP site was then recovered by phenol/chloroform extraction and ethanol precipitation.
Antibodies and Immunodepletions
Antibodies against REV1 (REV1-N and REV1-C (Budzowska et al., 2015), REV7 (Räschle et al., 2008), PCNA (Kochaniak et al., 2009), FANCD2 (Räschle et al., 2008), CDT1 (Arias and Walter, 2005), MCM6 (Larsen et al., 2019), RPA (Walter and Newport, 2000), TRAIP (Larsen et al., 2019), BRCA1 (Joukov et al., 2006), SPRTN (Larsen et al., 2019), M.HpaII (Larsen et al., 2019) were described previously. Antibodies against phosphorylated CHK1 (S345) (Cell Signaling, #2341), Histone H3 (Cell Signaling, #9715) and Ubiquityl-PCNA (K164) (Cell Signaling, #13439) are commercially available. The following antibodies were raised against the indicated peptides (New England Peptide) POLη (Ac-CDGNRTLDAFFKSPK-OH), HMCES (Ac-CQWLKKEDGEPSPKRAKK-OH), RFWD3-N (H2N-MAQEEMEVDLPNLC-amide), UBC13 (H2N-MAGLPRRIIKETQRLC-amide), POLD1 (Ac-SSQTKKLRGDWDDDC-amide), POLD2 (Ac-CQPISFSGFGADDELGD-OH), POLD3 (H2N-MDELYLENIDELVTDQNKC-amide), RAD6 (Ac-CKREYEKRVSAIVEQSWNDS-OH), Polκ (Ac-CPASKKSKPNSSKNTIDRFFK-OH), ZRANB3 (Ac-CSKYGSDISKFFSKM-OH), NSE2 (Ac-CKRTIDILNKQKGRH-OH), RAD18 (Ac-CRPKTSSGEIKPRSKRHRK-OH), RPA-70 (Ac-CHEFPQYYPIGHSSSE-OH), RPA-14 (Ac-CSRRLIMNIRKMATQGV-OH), WRNIP1 (Ac-CEQDYMPEEMKGVTFV-OH). The RFWD3-F antibody was raised against a protein fragment of Xenopus laevis RFWD3 spanning from amino acid 252 to 406 (BioGenes). The fragment was tagged on the N terminus with His6 and purified from bacteria under denaturing conditions.
To immunodeplete Xenopus egg extracts of REV1, one volume of Protein A Sepharose Fast Flow (PAS) (GE Health Care) beads was bound to one volumes of REV1-N or REV1-C antibodies overnight at 4°C. The beads were then washed twice with 500 μL PBS, once with ELB (10 mM HEPES pH 7.7, 50 mM KCl, 2.5 mM MgCl2, and 250 mM sucrose), twice with ELB supplemented with 0.5 M NaCl, and twice with ELB. One volume of precleared HSS or NPE was then depleted by mixing with 0.2 volumes of antibody-bound beads and then incubated at room temperature for 15 min, before being harvested. For HSS, the depletion procedure was performed once with REV1-N coupled beads and once with REV1-C coupled beads. For NPE the depletion procedure was performed twice with REV1-N coupled beads and once with REV1-C coupled beads. To immunodeplete Xenopus egg extracts of Polη, one volume of PAS beads was bound to five volumes of affinity purified POLη antibody (1 mg/mL). The beads were washed as described above, and one volume of precleared HSS or NPE was then depleted by mixing with 0.2 volumes of antibody-bound beads for 15 min at room temperature. The depletion procedure was performed once for HSS and three times for NPE. For Rev1 and Polη combined depletion, one volume of beads was first pre-bound to five volumes of POLη antibody for 1 h at room temperature, followed by addition of one volume of REV1-N or REV1-C antibodies overnight at 4°C. The beads were washed, and the depletion was performed as described above, twice for HSS and three times for NPE. To immunodeplete Xenopus egg extracts of RFWD3, one volume of PAS beads was bound to four volumes of affinity purified RFWD3-N antibody (1 mg/mL) or to three volumes of RFWD3-F antibody. The beads were washed as described above, and one volume of precleared HSS or NPE was then depleted by mixing with 0.2 volumes of antibody-bound beads for 15 min at room temperature. The depletion procedure was performed once for HSS and two times for NPE. To immunodeplete Xenopus egg extracts of UBC13, one volume of PAS beads was bound to twenty volumes of affinity purified UBC13 antibody (1 mg/mL). The beads were washed as described above, and one volume of precleared HSS or NPE was then depleted by mixing with 0.2 volumes of antibody-bound beads for 15 min at room temperature. The depletion procedure was performed three times for HSS and three times for NPE.
Protein purification
Xenopus laevis POLη (S. homeolog, Horizon) was cloned into the pCMV-Sport vector under the Sp6 promoter. The protein was then expressed by in vitro translation in rabbit reticulocytes lysate. More specifically, 2 μg of pCMV-Sport- POLη were incubated with 100 μL of TnT® Sp6 Quick Master Mix (Promega) supplemented with 4 μL of 1 mM methionine for 90 min at 30°C. Afterward, the reaction volume was adjusted to 400 μL with PBS and DNA was precipitated by addition of 0.06% polymin-P and incubation for 30 min at 4°C with rotation. The mixture was then centrifuged at 14000 g for 30 min and the proteins in the supernatant were precipitated with saturated ammonium sulfite to a final concentration of 55% for 30 min at 4°C with rotation, followed by centrifugation at 16000 g for 30 min. The protein pellet was resuspended in 15 μL of ELB buffer, dialyzed for 3 h at 4°C in ELB buffer. As a negative control for rescue experiments, a reaction without DNA was performed.
Nascent leading strand analysis
For nascent leading strand analysis, 3-4 μL of replication reaction were added to 10 volumes of transparent stop buffer (50 mM Tris-HCl, pH 7.5, 0.5% SDS, 25 mM EDTA), and replication intermediates were purified as previously described (Knipscheer et al., 2009; Räschle et al., 2008). DNA was digested with the indicated restriction enzymes and supplemented with 0.5 volumes of denaturing PAGE Gel Loading Buffer II (Life technologies). The digested DNA products were resolved on either a 6 or 7% polyacrylamide sequencing gel.
pICLPt repair assay
Timing of dual incisions on pICLpt was monitored as described before (Knipscheer et al., 2012). Briefly, replication intermediates were purified as described above for the nascent leading strand analysis, followed by digestion with the HincII restriction enzyme (New England BioLabs) for 3 h and separation of the repair intermediates on a 0.8% native agarose gel. As exemplified in Figure S3C the appearance over time of long arm (3.3 kb), short arm (2.3 kb) and linear fragments is indicative of successful dual incision of the ICL lesion. To monitor the repair of the ICL replication intermediates were digested with HincII and SapI. Percentage repair was calculated as described previously and normalized using pQuant plasmid (Knipscheer et al., 2012).
Plasmid pull-down
Plasmid pull-downs were performed as described previously (Budzowska et al., 2015). Briefly, 6 μL Streptavidin-coupled magnetic beads (Dynabead M-280, Invitrogen) per pull-down reaction, were equilibrated with wash buffer 1 (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1mM EDTA pH 8, 0.02% tween-20) and then incubated with 12 pmol of biotinylated LacI at RT for 40 min. The beads were washed four times with pull-down buffer 1 (10 mM HEPES pH 7.7, 50 mM KCl, 2.5 mM MgCl2, 250 mM sucrose, 0.25 mg/mL BSA, 0.02% Tween-20) and resuspended in 40 μL and stored on ice. At the indicated time points, 8 μL of reaction was added to the beads and rotated for 30 min at 4°C. The beads were subsequently washed twice in wash buffer 2 (10 mM HEPES pH 7.7, 50 mM KCl, 2.5 mM MgCl2, 0.25 mg/mL BSA, 0.03% Tween 20) and resuspended in 2x Laemmli sample buffer.
DPC pull-down
DPC pull-downs were performed as described previously (Larsen et al., 2019). Streptavidin-coupled magnetic beads (Dynabeads M-280, Invitrogen; 5uL per pull-down) were washed twice with 50 mM Tris pH 7.5, 150mM NaCl, 1mM EDTA pH 8, 0.02% Tween-20. Biotinylated LacI was added to the beads (1 pmol per 5 μL of beads) and incubated at room temperature for 40 min. The beads were then washed four times with DPC pull-down buffer (20 mM Tris pH 7.5, 150 mM NaCl, 2 mM EDTA pH 8, 0.5% IPEGAL-CA630) and then stored in the same buffer on ice until needed. At the indicated times, equal volumes (2-10 μL) of reaction were withdrawn and stopped in 300 μL of DPC pull-down buffer on ice. After all of the time points were taken, 5 μL of LacI-coated streptavidin Dynabeads were added to each sample and allowed to bind for 30-60 min at 4°C rotating. 20 μL of pull-down supernatant was reserved in 20 μL of 2X Laemmli sample buffer for input. The beads were subsequently washed four times with DPC pull-down buffer and then twice with Benzonase buffer (20mM Tris pH 7.5, 150mM NaCl, 2mM MgCl2, 0.02% Tween-20) before being resuspended in 15 μL Benzonase buffer containing 1 μL Benzonase (Novagen). Samples were incubated for 1hr at 37°C to allow for DNA digestion and DPC elution, after which the beads were pelleted and the supernatant eluate was mixed with 2X Laemmli sample buffer for subsequent western blotting analysis.
Chromatin spin-down
Demembranated Xenopus sperm chromatin was prepared as described (Sparks and Walter, 2019) and stored at −80°C at a concentration of 100000 sperm chromatin/μL (320 ng/μL). For analysis of UV-damaged chromatin, sperm chromatin was diluted to either 25000 or 50000 sperm chromatin/μL in ELB buffer (10 mM HEPES pH 7.7, 50 mM KCl, 2.5 mM MgCl2, and 250 mM sucrose), deposited on parafilm and irradiated with the indicated dose of UV-C. For replication reactions, undamaged or UV-damaged sperm chromatin was added to HSS at a final concentration of 16 ng/ μL and licensed for 30 min, followed by addition of two volumes of NPE. For analysis of replication-independent repair in non-licensing extracts, one volume of HSS and two volumes of NPE were premixed prior to the addition of sperm chromatin (final concentration of 8 ng/μL). PHA 767491 (Cdc7 inhibitor; Sigma-Aldrich) was added to NPE at the final concentration of 100 μM 10 min prior to initiating the reaction to block origin firing. Where indicated, recombinant ubiquitin mutants (Boston Biochem) were added to NPE at the final concentration of 1mg/mL. At the indicated time points, 8 μL of replication reaction was stopped with 60 μL of ELB buffer supplemented with 0.2% Triton-X. The mixture was carefully layered on top on a sucrose cushion (10 mM HEPES pH 7.7, 50 mM KCl, 2.5 mM MgCl2, and 500 mM sucrose) and spun for 1 min at 6800 x g in a swing bucket centrifuge at 4°C. The chromatin pellet was carefully washed twice with 200 μL of ice-cold ELB buffer and resuspended in 2X Laemmli buffer.
Sequencing of replication products
pCTRL (pJLS2-FdC) or pMH were replicated in Xenopus egg extract and DNA samples (8 μL) were collected at the end of the replication reaction (240 min). Samples were diluted in transparent stop buffer (50 mM Tris-HCl, pH 7.5, 0.5% SDS, 10 mM EDTA), supplemented with 4 μL RNase A (4 mg/mL) at 37°C for 30 min, followed by incubation with 2 μL Proteinase K (20 mg/mL) overnight at RT. DNA was phenol/chloroform-extracted, ethanol-precipitated and resuspended in 8 μL 10 mM Tris-HCl, pH 8. Because in pMH, M.HpaII is crosslinked only on one strand, the damaged and undamaged daughter DNA molecules can be discriminated by the presence or absence of the DPC peptide adduct (Duxin et al., 2014). To exclude the undamaged daughter DNA molecules from the sequencing analysis, DNA extracted from replication of pMH was first digested with Acc65I, which linearizes the undamaged DNA molecules but is blocked by the presence of the DPC peptide adduct. The linearized molecules were subsequently degraded by combined addition of Lambda exonuclease and Exonuclease I (New England BioLabs). Reactions were stopped in transparent stop buffer and DNA was purified as described above and resuspended in 8 μL 10 mM Tris-HCl, pH 8. pCtrl and pMH samples were subsequently linearized with the AatII restriction enzyme and the newly replicated strand was specifically amplified by 30 cycles of annealing and extension using a single strand-specific primer (5′-GGATCCATGTCGCAGTTGCGCAGCCTGAATGG-3′). Amplified ssDNA products were separated on a 6% denaturing Urea-PAGE and the gel was stained in 1x SYBR Gold solution (Thermo Fisher). The DNA band corresponding to the amplified repair products was visualized with a blue lamp, excised, transferred to a 1.5 mL tube and minced in small pieces by pressing with a plastic stick to be bottom of the tube. To elute the DNA from the gel pieces, 500 μL of PAGE elution buffer (0.5 M NaOAc, pH 7, 1 mM EDTA, 0.1% SDS) were added, and the mixture was incubated for 2 h at 37°C shaking at 1200 rpm. Eluted DNA was separated from the gel pieces through a homemade nitex column, purified by phenol/chloroform extraction followed by ethanol precipitation and resuspended in 12 μL of 10 mM Tris-HCl, pH 8 (approximately 10 ng/ μL of DNA were recovered). 10 ng of ssDNA template were amplified with Phusion High Fidelity DNA polymerase (New England BioLabs) for 18 cycles using a mix of primers (Forward primers: 5′-TAGGATCCATCACGCAGTTG; TANGGATCCATCACGCAGTTG-3′; 5′-TANNGGATCCATCACGCAGTTG-3′. Reverse primer: 5′-GTCGGGGCTGGCTTAACTATGC-3′), where N represents a random nucleotide, to optimize the sequencing procedure. PCR products were separated on an 8% TBE polyacrylamide gel, which was stained in 1x SYBR Gold solution and visualized with a blue lamp. Product of approximately 170 bp (mix of 169 bp, 170 bp and 171 bp products) were excised from the gel, transferred into a 1.5 mL tube and minced in small pieces by pressing with a plastic stick to the bottom of the tube. DNA was eluted from the gel pieces by addition of 300 μL elution buffer (10 mM Tris-HCl, pH 8, 1 mM EDTA, 300 mM NaCl), followed by overnight agitation at 1200 rpm at 30°C. Eluted DNA was separated from the gel pieces through a homemade nitex column, ethanol precipitated and resuspended in 12 μL of 10 mM Tris-HCl, pH 8 (approximately 100 ng/ μL of DNA were recovered). DNA fragments were ligated to barcoded Illumina adaptors according to the manufacturer’s instructions and sequenced with a MiSeq sequencer (150 bp read length, paired ends) at the Danish National High-Throughput Sequencing Center. Read data are available at the European Nucleotide Archive with the accession number PRJEB39253.
Sequencing data analysis
Sequencing data were demultiplexed and exported to paired FASTQ files. The paired FASTQ files were adaptor-trimmed and quality controlled using AdapterRemoval (v. 2.2.2. –minlength 30,–mm 3–trimns–trimqualities–minquality 2–mm 3) (Schubert et al., 2016). Each read pair was collapsed into a single consensus sequence with a minimum overlapping length of 11 bases. In order to compute the base-specific variation on the region of interest, quality controlled reads were then mapped to amplicon sequence using bwa mem alignment (Li and Durbin, 2009). BAM files were then filtered for high quality mapping reads using samtools (v. 1.9. -q30 –F4) (Li et al., 2009) and sorted by mapping region with Picard (v. 2.9.1; http://broadinstitute.github.io/picard/). The position-wise read coverage and base distribution could then be computed by processing the output of samtools mpileup (v. 1.9), using an in-house python script for each sample. To adjust for sample differences in sequencing depth, the relative base frequency was used for the downstream analysis. Subsequent statistical analysis and plotting were conducted using Rstudio (version 3.5.2 - code available by request).
CHROMASS
CHROMASS experiments were performed as previously described (Räschle et al., 2015). Briefly, isolated sperm chromatin was either untreated or treated with 2000 J/m2 of UV-C. Each reaction was performed in quadruplicate. The sperm chromatin was then incubated at a final concentration of 16 ng/μL in non-licensing extracts that were either mock-, REV1- or RFWD3-depleted. Reactions were stopped after 45 min. Specifically, 10 μL of replication reaction was stopped with 60 μL of ELB buffer supplemented with 0.2% Triton-X, and chromatin spin down performed as described above. The chromatin pellet was then resuspended in 50 μl denaturation buffer (8 M urea, 100 mM Tris-HCl, pH 8), and transferred to a new low binding tube. Cysteines were reduced (1 mM DTT for 15 min at RT) and alkylated (0.55 M chloroacetamide for 40 min at RT protected from light). Proteins were first digested with 0.5 μg LysC (2.5 h at RT) and then with 0.5 μg tripsin at 30°C overnight. Peptides were acidified with 10% trifluoroacetic acid (pH < 4), followed by addition of 400 mM NaCl, and purified by stage tipping (C18 material). For this, stage tips were first activated in 100% methanol, then equilibrated in 80% acetonitrile/ 10% formic acid, and finally washed twice in 0.1% formic acid. Samples were loaded on the equilibrated stage tips and washed twice with 50 μL 0.1% formic acid. StageTip elution was performed with 80 μL of 25% acetonitrile in 0.1% formic acid, eluted samples were dried to completion in a SpeedVac at 60°C, dissolved in 10 μL 0.1% formic acid, and stored at −20°C until MS analysis.
Whole proteome analysis
Nucleoplasmic egg extracts (NPE) where either mock- or RFWD3- depleted (either with the N or F antibodies) as described above. Depleted samples were recovered and diluted 10-fold with 50 mM ABC and digested using modified sequencing grade Trypsin (1:100 w/w; Sigma), for 30 min at 30°C. Subsequently, TCEP and CAA were added to final concentrations of 10 mM, and digestion was allowed to continue for a total of 3 h. Tryptic peptides were fractionated on-StageTip at high-pH essentially as described previously (Hendriks et al., 2018)). Peptides were eluted from StageTips as eight fractions (F1-8) using 80 μL of 2, 4, 7, 10, 13, 17, 25, and 40% ACN in 50 mM ammonium. All fractions were dried to completion in LoBind tubes, using a SpeedVac for 3 h at 60°C, after which the dried peptides were dissolved using 20 μL of 0.1% formic acid. Between 3 μL and 10 μL were used from each fraction, depending on peptide content, to equalize protein load for analysis by MS.
Mass spectrometry
MS data acquisition
MS samples were analyzed on an EASY-nLC 1200 system (Thermo) coupled to either a Q Exactive HF-X Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo) for CHROMASS samples, or an Orbitrap Exploris 480 mass spectrometer (Thermo) for total proteome samples. Separation of peptides was performed using 15-cm columns (75 μm internal diameter) packed in-house with ReproSil-Pur 120 C18-AQ 1.9 μm beads (Dr. Maisch). Elution of peptides from the column was achieved using a gradient ranging from buffer A (0.1% formic acid) to buffer B (80% acetonitrile in 0.1% formic acid), at a flow rate of 250 nL/min. For CHROMASS samples, gradient length was 80 min per sample, including ramp-up and wash-out, with an analytical gradient of 57.5 min ranging from 8% to 28% buffer B. For total proteome samples, gradient length was 80 min per sample, including ramp-up and wash-out, with an analytical gradient of 60 min ranging in buffer B from 5%–25% for F1, 5%–29% for F2, 5%–33% for F3, 6%–36% for F4, 8%–38% for F5, 10%–40% for F6, 12%–42% for F7, and 14%–44% for F8. The columns were heated to 40°C using a column oven, and ionization was achieved using either a NanoSpray Flex ion source (Thermo) for CHROMASS, or a NanoSpray Flex NG ion source (Thermo) for total proteome samples. Spray voltage set at 2 kV, ion transfer tube temperature to 275°C, and RF funnel level to 40%. CHROMASS samples were measured as two technical replicates, with 5 μL of the sample per injection, and with different technical settings (“Normal” and “Fast”) to balance speed versus sensitivity. Raw files with a “b” appended to the file name correspond to those analyzed using “Fast” settings. CHROMASS measurements were performed with a full scan range of 300-1,750 m/z, MS1 resolution of 60,000, MS1 AGC target of 3,000,000, and MS1 maximum injection time of 60 ms. Precursors with charges 2-6 were selected for fragmentation using an isolation width of 1.3 m/z and fragmented using higher-energy collision disassociation (HCD) with a normalized collision energy of 25. Precursors were excluded from re-sequencing by setting a dynamic exclusion of 45 s. MS2 AGC target was set to 200,000 and minimum MS2 AGC target to 20,000. For “Normal” runs, MS2 maximum injection time was 55 ms, MS2 resolution was 30,000, and loop count was 14. For “Fast” runs, MS2 maximum injection time was 25 ms, MS2 resolution was 15,000, and loop count was 20. For total proteome measurements, full scan range was set to 300-1,750 m/z, MS1 resolution to 120,000, MS1 AGC target to “200” (2,000,000 charges), and MS1 maximum injection time to “Auto.” Precursors with charges 2-6 were selected for fragmentation using an isolation width of 1.3 m/z and fragmented using higher-energy collision disassociation (HCD) with normalized collision energy of 25. Monoisotopic Precursor Selection (MIPS) was enabled in “Peptide” mode. Precursors were excluded from re-sequencing by setting a dynamic exclusion of 60 s, with an exclusion mass tolerance of 15 ppm, exclusion of isotopes, and exclusion of alternate charge states for the same precursor. MS2 resolution was set to 15,000, MS2 AGC target to “200” (200,000 charges), MS2 intensity threshold to 430,000, MS2 maximum injection time to “Auto,” and TopN to 18.
MS data analysis
All MS RAW data were analyzed using the freely available MaxQuant software (Cox and Mann, 2008), version 1.5.3.30. Default MaxQuant settings were used, with exceptions specified below. For generation of theoretical spectral libraries, the Xenopus laevis FASTA database was downloaded from Uniprot on the 10th of February 2018. In silico digestion of proteins to generate theoretical peptides was performed with trypsin, allowing up to 3 or 4 missed cleavages for the CHROMASS and total proteome experiment, respectively. Allowed variable modifications were oxidation of methionine (default), protein N-terminal acetylation (default) for all samples. For total proteome analysis, deamination of asparagine and peptide N-terminal pyroglutamate formation from glutamine and glutamic acid were additionally allowed. Maximum variable modifications per peptide were reduced to 3. Label-free quantification (LFQ) was enabled (Cox et al., 2014), with “Fast LFQ” disabled. For total proteome analysis, “LFQ min. ratio count” set to 3, and iBAQ was enabled. Stringent MaxQuant 1% FDR data filtering at the PSM- and protein-levels was applied (default). Second peptide search was enabled. Matching between runs was enabled, with an alignment window of 20 min and a match time window of 1 min, and for total proteome analysis matching was only allowed within the same fractions.
MS data annotation and quantification
The Xenopus laevis FASTA database downloaded from Uniprot lacked comprehensive gene name annotation. Missing or uninformative gene names were, when possible, semi-automatically curated by drawing informative gene names from Uniprot, otherwise Xenbase, otherwise the Session et al. database (Session et al., 2016), otherwise RefSeq (via Xenbase), and otherwise InterPro annotations were used. Quantification of the MaxQuant output files was performed using Perseus software (Tyanova et al., 2016). For quantification purposes, all protein LFQ intensity values were log2 transformed, and filtered for presence in 4 out of 4 replicates (n = 4/4) in at least one experimental condition. Missing values were imputed below the global experimental detection limit at a downshift of 1.8 and a randomized width of 0.3 (in log2 space; Perseus default). Statistical significance of differences was tested using two-tailed Student’s t testing, with permutation-based FDR-control applied at an s0 value of 0.5. Both p values and FDR-adjusted q-values are reported in Table S1 (total proteome) or Table S2 (CHROMASS).
Human cell experiments
Human cell plasmids and siRNAs
pcDNA4/TO Strep HA PCNA WT and K164R plasmids were described previously (Mosbech et al., 2012). pcDNA5 FRT/TO GFP-RFWD3 WT, C315A and I639K plasmids were gifts from John Rouse (Feeney et al., 2017). RFWD3 plasmids were rendered insensitive to RFWD3 siRNA#2 by introducing silent mutations into the siRNA target sequences using site-directed mutagenesis.
Plasmid DNA and siRNA transfections were performed using FuGENE 6 Transfection Reagent (Promega) and Lipofectamine RNAiMAX (Invitrogen), respectively, according to the manufacturers’ protocols. All siRNAs were used at a final concentration of 50 nM. The following siRNA oligonucleotides were used: Non-targeting control (Ctrl): 5′-GGGAUACCUAGACGUUCUA-3′; RFWD3#2: 5′-GGAAACAGGCCGAGUUAGA-3′; RFWD3#3: 5′-GUUAAGAUGUUGAGUACUG-3′; RFWD3#4: 5′-GGACCUACUUGCAAACUAU-3′; RFWD3#601: 5′- AACUCCUGCACAUGACUGC-3′; RAD18: 5′- ACUCAGUGUCCAACUUGCU-3′. siRNA duplexes specifically targeting SHPRH and HLTF (SMARTpool) were purchased from Dharmacon.
Cell culture
Human U2OS cells were obtained from ATCC. Cell lines stably expressing Strep-HA-PCNA WT were described previously (Mosbech et al., 2012). To generate a cell line expressing GFP-tagged, siRNA-resistant RFWD3 WT or C315A in a tet-on system, RFWD3 plasmids were co-transfected with Flp recombinase (pOG44, Invitrogen) into U2OS FRT Flp-In TRex cells and selected in 100 μg/mL hygromycin and 5 μg/mL blasticidin. The expression of GFP-RFWD3 was induced by doxycycline (1 μg/mL, Sigma Aldrich) for 24 h. All cell lines used in this study were cultured in DMEM containing 10% FBS at 37°C and 5% CO2 and were regularly tested for mycoplasma infection. Cells were exposed to UV light (30 J/m2) for 4 h unless otherwise stated.
Immunochemical methods
For Strep-Tactin pull-downs under denaturing conditions, cells were lysed in denaturing buffer (20 mM Tris, pH 7.5, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.5% NP-40, 0.5% sodium deoxycholate, and 0.5% SDS) supplemented with protease and phosphatase inhibitors and incubated on ice for 10 min. Lysates were sonicated and cleared by centrifugation for 10 min at 20,000 rpm. Then, lysates were incubated with Strep-Tactin Sepharose (IBA BioTAGnology) for 2 h on an end-over-end rotator at 4°C and washed five times with denaturing buffer. For USP2 and ULP1 treatment, beads were subsequently washed three times in reaction buffer A (50 mM Tris, pH 7.5; 50 mM NaCl; 5 mM DTT) and incubated with USP2/ULP1 for 30 min at 37°C. Then, beads were resuspended in 2X Laemmli sample buffer and the proteins resolved by SDS–PAGE and analyzed by immunoblotting.
Antibodies used in this study included: GFP (11814460001, Roche (1:500)) and sc-8334, Santa Cruz (1:1,000)), HA (sc-805, Santa Cruz (1:1,000)), MCM6 (sc-9843, Santa Cruz (1:1,000)), RAD18 (A301-340A, Bethyl (1:1,000)), HLTF (sc-27542, Santa Cruz (1:500)), SHPRH (ab80129, abcam (1:1,000)), RFWD3 (138030, abcam (1:1,000)), UBC13 (4919, Cell Signaling (1:1,000)), Ubiquitin (sc-8017, Santa Cruz (1:1,000)), Vinculin (V9131, Sigma (1:10,000)).
Quantification and statistical analysis
Bioinformatic analysis of mass spectrometry data were carried out with the Perseus software. Statistical significance of differences was tested using two-tailed Student’s t testing, with permutation-based FDR-control applied at an s0 value of 0.5. Autoradiographs and western blots were quantified using ImageJ. The error bars represent standard error.
Acknowledgments
We thank Jiri Lukas, Robert Fuchs, Johannes C. Walter, Andres Lopez-Contreras, and members of the Duxin laboratory for feedback on the manuscript. We thank Magdalena Budzowska and Johannes C. Walter for sharing pICLPt. The Novo Nordisk Foundation Center for Protein Research is supported financially by the Novo Nordisk Foundation (grant agreement NNF14CC0001). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement 715975), the Lundbeck Foundation (grant R303-2018-3212), and the Novo Nordisk Foundation (grant NNF16CC0020906 and NNF18OC0030752).
Author contributions
I.G. performed the experiments presented in Figures 1, 2, 3, 4, and 6 unless stated otherwise. S.H. performed the experiments presented in Figure 5 and Figure S5, and N.B.L. performed the experiments presented in Figure 7 and Figures S4D, S4M, and S7. I.A.H. performed MS analysis on CHROMASS samples (Figure 6; Figure S6). I.A.H. and C.S.C.-C. performed MS analysis on total proteome samples (Figure S1C). The MS run and analysis were done under the supervision of M.L.N. J.J. analyzed the sequencing data presented in Figure 2 under the supervision of S.R. L.S. established the conditions for the UV-damaged CHROMASS and performed the experiments in Figures 3C–3E and Figures S3D and S3E. Z.F. performed the experiments in Figures S1C and S1D. U.K. generated the abasic site DNA substrate, and S.S.-B. performed the experiment in Figure S2B. M.R. prepared Figure 1A, and A.O.G. performed preliminary experiments for this project. I.G., N.B.L., and J.P.D. designed and analyzed the experiments performed in Xenopus egg extracts. S.H., N.M., and J.P.D. designed and analyzed the experiments in human cells. I.G. and J.P.D. prepared the manuscript with feedback and input from all authors of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: December 14, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.11.029.
Supporting citations
The following references appear in the supplemental information: Hashimoto et al. (2010).
Supplemental information
C-C, mock-depleted extracts incubated in undamaged chromatin; C-UV, mock-depleted extracts incubated with UV-treated chromatin; RFWD3Δ-UV, RFWD3-depleted extracts incubated with UV-treated chromatin; REV1Δ-UV, REV1-depleted extracts incubated with UV-treated chromatin.
References
- Arias E.E., Walter J.C. Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev. 2005;19:114–126. doi: 10.1101/gad.1255805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi J., Rudd S.G., Jozwiakowski S.K., Bailey L.J., Soura V., Taylor E., Stevanovic I., Green A.J., Stracker T.H., Lindsay H.D., Doherty A.J. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell. 2013;52:566–573. doi: 10.1016/j.molcel.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienko M., Green C.M., Crosetto N., Rudolf F., Zapart G., Coull B., Kannouche P., Wider G., Peter M., Lehmann A.R. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science. 2005;310:1821–1824. doi: 10.1126/science.1120615. [DOI] [PubMed] [Google Scholar]
- Budzowska M., Graham T.G.W., Sobeck A., Waga S., Walter J.C. Regulation of the Rev1-pol ζ complex during bypass of a DNA interstrand cross-link. EMBO J. 2015;34:1971–1985. doi: 10.15252/embj.201490878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A., Nimonkar A.V., Hu Y., Hajdu I., Achar Y.J., Izhar L., Petit S.A., Adamson B., Yoon J.C., Kowalczykowski S.C. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D. Replication-Coupled DNA Repair. Mol. Cell. 2019;74:866–876. doi: 10.1016/j.molcel.2019.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon S., Ramasubramanyan S., Alies C., Philippin G., Lehmann A., Fuchs R.P. Rad8Rad5/Mms2-Ubc13 ubiquitin ligase complex controls translesion synthesis in fission yeast. EMBO J. 2010;29:2048–2058. doi: 10.1038/emboj.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cruet-Hennequart S., Gallagher K., Sokòl A.M., Villalan S., Prendergast A.M., Carty M.P. DNA polymerase eta, a key protein in translesion synthesis in human cells. Subcell. Biochem. 2010;50:189–209. doi: 10.1007/978-90-481-3471-7_10. [DOI] [PubMed] [Google Scholar]
- Daigaku Y., Davies A.A., Ulrich H.D. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–955. doi: 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A.A., Huttner D., Daigaku Y., Chen S., Ulrich H.D. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol. Cell. 2008;29:625–636. doi: 10.1016/j.molcel.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar J.M., Budzowska M., Walter J.C. The mechanism of DNA replication termination in vertebrates. Nature. 2015;525:345–350. doi: 10.1038/nature14887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxin J.P., Dewar J.M., Yardimci H., Walter J.C. Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell. 2014;159:346–357. doi: 10.1016/j.cell.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia A.E.H., Wang D.C., Willis N.A., Boardman A.P., Hajdu I., Adeyemi R.O., Lowry E., Gygi S.P., Scully R., Elledge S.J. RFWD3-Dependent Ubiquitination of RPA Regulates Repair at Stalled Replication Forks. Mol. Cell. 2015;60:280–293. doi: 10.1016/j.molcel.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney L., Muñoz I.M., Lachaud C., Toth R., Appleton P.L., Schindler D., Rouse J. RPA-Mediated Recruitment of the E3 Ligase RFWD3 Is Vital for Interstrand Crosslink Repair and Human Health. Mol. Cell. 2017;66:610–621.e4. doi: 10.1016/j.molcel.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.V., Yardimci H., Long D.T., Ho T.V., Guainazzi A., Bermudez V.P., Hurwitz J., van Oijen A., Schärer O.D., Walter J.C. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011;146:931–941. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullbright G., Rycenga H.B., Gruber J.D., Long D.T. p97 Promotes a Conserved Mechanism of Helicase Unloading during DNA Cross-Link Repair. Mol. Cell. Biol. 2016;36:2983–2994. doi: 10.1128/MCB.00434-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Gómez S., Reyes A., Martínez-Jiménez M.I., Chocrón E.S., Mourón S., Terrados G., Powell C., Salido E., Méndez J., Holt I.J., Blanco L. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell. 2013;52:541–553. doi: 10.1016/j.molcel.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs P.E.M., McDonald J., Woodgate R., Lawrence C.W. The relative roles in vivo of Saccharomyces cerevisiae Pol eta, Pol zeta, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6-4) photoadduct and T-T cis-syn cyclobutane dimer. Genetics. 2005;169:575–582. doi: 10.1534/genetics.104.034611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa R., Zharkov D.O., Golan G., Fernandes A.S., Gerchman S.E., Matz E., Kycia J.H., Grollman A.P., Shoham G. Structure of formamidopyrimidine-DNA glycosylase covalently complexed to DNA. J. Biol. Chem. 2002;277:19811–19816. doi: 10.1074/jbc.M202058200. [DOI] [PubMed] [Google Scholar]
- Göhler T., Muñoz I.M., Rouse J., Blow J.J. PTIP/Swift is required for efficient PCNA ubiquitination in response to DNA damage. DNA Repair (Amst.) 2008;7:775–787. doi: 10.1016/j.dnarep.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Gong Z., Chen J. E3 ligase RFWD3 participates in replication checkpoint control. J. Biol. Chem. 2011;286:22308–22313. doi: 10.1074/jbc.M111.222869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Fischhaber P.L., Luk-Paszyc M.J., Masuda Y., Zhou J., Kamiya K., Kisker C., Friedberg E.C. Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003;22:6621–6630. doi: 10.1093/emboj/cdg626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Tang T.-S., Bienko M., Parker J.L., Bielen A.B., Sonoda E., Takeda S., Ulrich H.D., Dikic I., Friedberg E.C. Ubiquitin-binding motifs in REV1 protein are required for its role in the tolerance of DNA damage. Mol. Cell. Biol. 2006;26:8892–8900. doi: 10.1128/MCB.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y., Ray Chaudhuri A., Lopes M., Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks I.A., Lyon D., Su D., Skotte N.H., Daniel J.A., Jensen L.J., Nielsen M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat Commun. 2018;0 doi: 10.1038/s41467-018-04957-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K., Sonoda E., Kawamoto T., Motegi A., Masutani C., Hanaoka F., Szüts D., Iwai S., Sale J.E., Lehmann A., Takeda S. Simultaneous disruption of two DNA polymerases, Polη and Polζ, in Avian DT40 cells unmasks the role of Polη in cellular response to various DNA lesions. PLoS Genet. 2010;6:e1001151. doi: 10.1371/journal.pgen.1001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C., Pfander B., Moldovan G.L., Pyrowolakis G., Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- Hoffmann S., Smedegaard S., Nakamura K., Mortuza G.B., Räschle M., Ibañez de Opakua A., Oka Y., Feng Y., Blanco F.J. TRAIP is a PCNA-binding ubiquitin ligase that protects genome stability after replication stress. J Cell Biol. 2016;212:63–75. doi: 10.1083/jcb.201506071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner D., Ulrich H.D. Cooperation of replication protein A with the ubiquitin ligase Rad18 in DNA damage bypass. Cell Cycle. 2008;7:3629–3633. doi: 10.4161/cc.7.23.7166. [DOI] [PubMed] [Google Scholar]
- Inano S., Sato K., Katsuki Y., Kobayashi W., Tanaka H., Nakajima K., Nakada S., Miyoshi H., Knies K., Takaori-Kondo A. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination. Mol. Cell. 2017;66:622–634.e8. doi: 10.1016/j.molcel.2017.04.022. [DOI] [PubMed] [Google Scholar]
- Johnson R.E., Prakash S., Prakash L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Poleta. Science. 1999;283:1001–1004. doi: 10.1126/science.283.5404.1001. [DOI] [PubMed] [Google Scholar]
- Johnson R.E., Haracska L., Prakash S., Prakash L. Role of DNA polymerase eta in the bypass of a (6-4) TT photoproduct. Mol. Cell. Biol. 2001;21:3558–3563. doi: 10.1128/MCB.21.10.3558-3563.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joukov V., Groen A.C., Prokhorova T., Gerson R., White E., Rodriguez A., Walter J.C., Livingston D.M. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell. 2006;127:539–552. doi: 10.1016/j.cell.2006.08.053. [DOI] [PubMed] [Google Scholar]
- Kannouche P.L., Wing J., Lehmann A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- Klein Douwel D., Boonen R.A.C.M., Long D.T., Szypowska A.A., Räschle M., Walter J.C., Knipscheer P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell. 2014;54:460–471. doi: 10.1016/j.molcel.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knies K., Inano S., Ramírez M.J., Ishiai M., Surrallés J., Takata M., Schindler D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J. Clin. Invest. 2017;127:3013–3027. doi: 10.1172/JCI92069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P., Räschle M., Schärer O.D., Walter J.C. Replication-coupled DNA interstrand cross-link repair in Xenopus egg extracts. Methods Mol Biol. 2012;920:221–243. doi: 10.1007/978-1-61779-998-3_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P., Räschle M., Smogorzewska A., Enoiu M., Ho T.V., Schärer O.D., Elledge S.J., Walter J.C. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochaniak A.B., Habuchi S., Loparo J.J., Chang D.J., Cimprich K.A., Walter J.C., van Oijen A.M. Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J. Biol. Chem. 2009;284:17700–17710. doi: 10.1074/jbc.M109.008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kose H.B., Larsen N.B., Duxin J.P., Yardimci H. Dynamics of the Eukaryotic Replicative Helicase at Lagging-Strand Protein Barriers Support the Steric Exclusion Model. Cell Rep. 2019;26:2113–2125.e6. doi: 10.1016/j.celrep.2019.01.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger P.H.L., Lee K.-Y., Wit N., van den Berk P.C.M., Wu X., Roest H.P., Maas A., Ding H., Hoeijmakers J.H.J., Myung K., Jacobs H. HLTF and SHPRH are not essential for PCNA polyubiquitination, survival and somatic hypermutation: existence of an alternative E3 ligase. DNA Repair (Amst.) 2011;10:438–444. doi: 10.1016/j.dnarep.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen N.B., Gao A.O., Sparks J.L., Gallina I., Wu R.A., Mann M., Räschle M., Walter J.C., Duxin J.P. Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol. Cell. 2019;73:574–588.e7. doi: 10.1016/j.molcel.2018.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach C.A., Michael W.M. Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J. Cell Biol. 2005;171:947–954. doi: 10.1083/jcb.200508100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebofsky R., Takahashi T., Walter J.C. DNA replication in nucleus-free Xenopus egg extracts. Methods Mol. Biol. 2009;521:229–252. doi: 10.1007/978-1-60327-815-7_13. [DOI] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.-R., Zeman M.K., Chen J.-Y., Yee M.-C., Cimprich K.A. SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell. 2011;42:237–249. doi: 10.1016/j.molcel.2011.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Chu J., Yucer N., Leng M., Wang S.-Y., Chen B.P.C., Hittelman W.N., Wang Y. RING finger and WD repeat domain 3 (RFWD3) associates with replication protein A (RPA) and facilitates RPA-mediated DNA damage response. J. Biol. Chem. 2011;286:22314–22322. doi: 10.1074/jbc.M111.222802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long D.T., Räschle M., Joukov V., Walter J.C. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333:84–87. doi: 10.1126/science.1204258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.K., Wood R.D. DNA polymerase ζ in DNA replication and repair. Nucleic Acids Res. 2019;47:8348–8361. doi: 10.1093/nar/gkz705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masutani C., Kusumoto R., Iwai S., Hanaoka F. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. EMBO J. 2000;19:3100–3109. doi: 10.1093/emboj/19.12.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch S.D., Kokoska R.J., Masutani C., Iwai S., Hanaoka F., Kunkel T.A. Preferential cis-syn thymine dimer bypass by DNA polymerase eta occurs with biased fidelity. Nature. 2004;428:97–100. doi: 10.1038/nature02352. [DOI] [PubMed] [Google Scholar]
- Mohni K.N., Wessel S.R., Zhao R., Wojciechowski A.C., Luzwick J.W., Layden H., Eichman B.F., Thompson P.S., Mehta K.P.M., Cortez D. HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell. 2019;176:144–153.e13. doi: 10.1016/j.cell.2018.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosbech A., Gibbs-Seymour I., Kagias K., Thorslund T., Beli P., Povlsen L., Nielsen S.V., Smedegaard S., Sedgwick G., Lukas C. DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat. Struct. Mol. Biol. 2012;19:1084–1092. doi: 10.1038/nsmb.2395. [DOI] [PubMed] [Google Scholar]
- Motegi A., Sood R., Moinova H., Markowitz S.D., Liu P.P., Myung K. Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J. Cell Biol. 2006;175:703–708. doi: 10.1083/jcb.200606145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi A., Liaw H.-J., Lee K.-Y., Roest H.P., Maas A., Wu X., Moinova H., Markowitz S.D., Ding H., Hoeijmakers J.H.J., Myung K. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA. 2008;105:12411–12416. doi: 10.1073/pnas.0805685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourón S., Rodriguez-Acebes S., Martínez-Jiménez M.I., García-Gómez S., Chocrón S., Blanco L., Méndez J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013;20:1383–1389. doi: 10.1038/nsmb.2719. [DOI] [PubMed] [Google Scholar]
- Ohashi E., Murakumo Y., Kanjo N., Akagi J., Masutani C., Hanaoka F., Ohmori H. Interaction of hREV1 with three human Y-family DNA polymerases. Genes Cells. 2004;9:523–531. doi: 10.1111/j.1356-9597.2004.00747.x. [DOI] [PubMed] [Google Scholar]
- Olivieri M., Cho T., Álvarez-Quilón A., Li K., Schellenberg M.J., Zimmermann M., Hustedt N., Rossi S.E., Adam S., Melo H. A Genetic Map of the Response to DNA Damage in Human Cells. Cell. 2020;182:481–496.e21. doi: 10.1016/j.cell.2020.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.L., Ulrich H.D. Mechanistic analysis of PCNA poly-ubiquitylation by the ubiquitin protein ligases Rad18 and Rad5. EMBO J. 2009;28:3657–3666. doi: 10.1038/emboj.2009.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Räschle M., Knipscheer P., Enoiu M., Angelov T., Sun J., Griffith J.D., Ellenberger T.E., Schärer O.D., Walter J.C. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Räschle M., Smeenk G., Hansen R.K., Temu T., Oka Y., Hein M.Y., Nagaraj N., Long D.T., Walter J.C., Hofmann K. DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science. 2015;348:1253671. doi: 10.1126/science.1253671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M., Lindgreen S., Orlando L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes. 2016;9:88. doi: 10.1186/s13104-016-1900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Session A.M., Uno Y., Kwon T., Chapman J.A., Toyoda A., Takahashi S., Fukui A., Hikosaka A., Suzuki A. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. 2016;538:336–343. doi: 10.1038/nature19840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar S., Ziv O., Avkin S., Adar S., Wittschieben J., Reissner T., Chaney S., Friedberg E.C., Wang Z., Carell T. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28:383–393. doi: 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks J., Walter J.C. Extracts for Analysis of DNA Replication in a Nucleus-Free System. Cold Spring Harb. Protoc. 2019 doi: 10.1101/pdb.prot097154. Published online March 1, 2019. [DOI] [PubMed] [Google Scholar]
- Sparks J.L., Chistol G., Gao A.O., Räschle M., Larsen N.B., Mann M., Duxin J.P., Walter J.C. The CMG Helicase Bypasses DNA-Protein Cross-Links to Facilitate Their Repair. Cell. 2019;176:167–181.e21. doi: 10.1016/j.cell.2018.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelter P., Ulrich H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- Szüts D., Marcus A.P., Himoto M., Iwai S., Sale J.E. REV1 restrains DNA polymerase zeta to ensure frame fidelity during translesion synthesis of UV photoproducts in vivo. Nucleic Acids Res. 2008;36:6767–6780. doi: 10.1093/nar/gkn651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakar T., Leung W., Nicolae C.M., Clements K.E., Shen B., Bielinsky A.-K., Moldovan G.L. Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat. Commun. 2020;11:2147. doi: 10.1038/s41467-020-16096-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk I., Hajdú I., Fátyol K., Szakál B., Blastyák A., Bermudez V., Hurwitz J., Prakash L., Prakash S., Haracska L. Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA. 2006;103:18107–18112. doi: 10.1073/pnas.0608595103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T., Ripplinger A., Hoffmann S., Wild T., Uckelmann M., Villumsen B., Narita T., Sixma T.K., Choudhary C. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015;52:389–393. doi: 10.1038/nature15401. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Unk I., Hajdú I., Fátyol K., Hurwitz J., Yoon J.-H., Prakash L., Prakash S., Haracska L. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. USA. 2008;105:3768–3773. doi: 10.1073/pnas.0800563105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujanovic M., Krietsch J., Raso M.C., Terraneo N., Zellweger R., Schmid J.A., Taglialatela A., Huang J.-W., Holland C.L., Zwicky K. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell. 2017;67:882–890.e5. doi: 10.1016/j.molcel.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J., Newport J. Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha. Mol. Cell. 2000;5:617–627. doi: 10.1016/s1097-2765(00)80241-5. [DOI] [PubMed] [Google Scholar]
- Wang T., Birsoy K., Hughes N.W., Krupczak K.M., Post Y., Wei J.J., Lander E.S., Sabatini D.M. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K., Tateishi S., Kawasuji M., Tsurimoto T., Inoue H., Yamaizumi M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004;23:3886–3896. doi: 10.1038/sj.emboj.7600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.A., Semlow D.R., Kamimae-Lanning A.N., Kochenova O.V., Chistol G., Hodskinson M.R., Amunugama R., Sparks J.L., Wang M., Deng L. TRAIP is a master regulator of DNA interstrand crosslink repair. Nature. 2019;567:267–272. doi: 10.1038/s41586-019-1002-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles J.T.P., Marians K.J. The Escherichia coli replisome is inherently DNA damage tolerant. Science. 2011;334:235–238. doi: 10.1126/science.1209111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A., Kobayashi Y., Tada S., Seki M., Enomoto T. WRNIP1 functions upstream of DNA polymerase η in the UV-induced DNA damage response. Biochem. Biophys. Res. Commun. 2014;452:48–52. doi: 10.1016/j.bbrc.2014.08.043. [DOI] [PubMed] [Google Scholar]
- Yuasa M.S., Masutani C., Hirano A., Cohn M.A., Yamaizumi M., Nakatani Y., Hanaoka F. A human DNA polymerase eta complex containing Rad18, Rad6 and Rev1; proteomic analysis and targeting of the complex to the chromatin-bound fraction of cells undergoing replication fork arrest. Genes Cells. 2006;11:731–744. doi: 10.1111/j.1365-2443.2006.00974.x. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Biertümpfel C., Gregory M.T., Hua Y.-J., Hanaoka F., Yang W. Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc. Natl. Acad. Sci. USA. 2012;109:7269–7274. doi: 10.1073/pnas.1202681109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
C-C, mock-depleted extracts incubated in undamaged chromatin; C-UV, mock-depleted extracts incubated with UV-treated chromatin; RFWD3Δ-UV, RFWD3-depleted extracts incubated with UV-treated chromatin; REV1Δ-UV, REV1-depleted extracts incubated with UV-treated chromatin.
Data Availability Statement
The accession number for the CHROMASS proteomics data reported in this paper is ProteomeXchange: PXD018217. The accession number for the total proteome proteomics data reported in this paper is ProteomeXchange: PXD021445.
The accession number for the deep sequencing of repair products reported in this paper is ENA: PRJEB39253. Original data have been deposited to Mendeley Data: DOI:10.17632/9mbb8sp7b7.1