

Abstract
Elevated uptake of saturated fatty acid palmitate is associated with metastatic progression of cancer cells; however, the precise signaling mechanism behind the phenomenon is unclear. The loss of cell adhesion proteins, such as desmoplakin (DSP), is a key driving event in the transformation of cancer cells to more aggressive phenotypes. Here we investigated the mechanism by which palmitate induces the loss of DSP in liver and breast cancer cells. We propose that palmitate activates the IRE1-XBP1 branch of the endoplasmic reticulum (ER) stress pathway to upregulate the ZEB transcription factor, leading to transcriptional repression of DSP. Using liver and breast cancer cells treated with palmitate, we found loss of DSP leads to increased cell migration independent of E-cadherin. We report that the ZEB family of transcription factors function as direct transcriptional repressors of DSP. CRISPR-mediated knockdown of IRE1 confirmed that the transcription of ZEB, loss of DSP, and enhanced migration in the presence of palmitate is dependent on the IRE1-XBP1 pathway. Additionally, by analyzing the somatic expression and copy number variation profiles of over 11,000 tumor samples, we corroborate our hypothesis and establish the clinical relevance of DSP loss via ZEB in human cancers.
Keywords: Palmitate, ER-stress, unfolded protein response (UPR), epithelial to mesenchymal transition (EMT), HepG2, Hep3B, MDA-MB-231, TCGA
Introduction
The loss of cell adhesion junctions has been recognized as a hallmark of cancer and an important first step towards the metastatic progression of cancer cells (1). Mounting evidence suggests the epithelial to mesenchymal transition (EMT) program is responsible for the transcriptional repression and breakdown of the components of cell adhesion junctions driving tumor towards malignancy (2). This program is controlled by a number of key transcription factor families including SNAIL, TWIST and ZEB (3,4). These transcription factors suppress the expression of key cell adhesion components, like E-cadherin (CDH1), that are required for the proper assembly of cell adhesion junctions (5). Recent studies have established that the EMT program can be invoked in cancer cells in the presence of elevated palmitate (PA) levels due to increased expression of fatty acid uptake molecules like CD36 (6,7). In vitro studies have shown that treating cancer cell lines with PA results in the loss of desmoplakin (DSP) expression (8,9). DSP serves as an obligate component of desmosomes that function as intercellular adhesion junctions and sites for intermediate filament attachment (10). Besides maintaining the integrity of desmosomes, DSP also plays an important role as tumor suppressor (11–13) by regulating various signaling pathways in cancer cells (14–16). However, the precise mechanism that lead to the loss of DSP expression in PA treated cells is unknown.
In the presence of elevated PA, accumulation of unfolded proteins and other cellular stresses trigger the endoplasmic reticulum (ER) stress response (17). Three transmembrane ER signaling proteins PERK (EIF2AK3) (protein kinase R (PKR)-like ER kinase), IRE1α (ERN1) (inositol-requiring enzyme) and ATF6 (activating transcription factor 6) function as critical sensors of ER stress leading to the activation of unfolded protein response (UPR) (18). A number of signaling pathways, including apoptosis and cell death, are controlled by the UPR that affect the survival outcomes of cells in a dose-dependent manner (19,20). Research from our group and others established that PA induces ER stress in mammalian and yeast cells via the IRE1 branch (21–24). It is well-established that the IRE1 branch of UPR response detects unfolded proteins and chaperones via the IRE1α luminal domain, while it senses membrane lipid saturation via its transmembrane domain (21–24). The self-association and dimerization of IRE1α subsequently activates the RNase domain (RD) (25). This active form of IRE1α splices the mRNA of the X-Box binding protein-1 (XBP1) to produce the spliced XBP1 transcription factor (XBP1S) that upregulates genes involved in enhancing ER protein-folding capacity (26,27). Besides regulating expression of stress-response related transcripts, XBP1 can also bind to the promoters of EMT transcription factors like SNAIL1, ZEB2 and TCF3 (28).
Based on this link, we hypothesized that the activation of the IRE1-XBP1 pathway triggers the loss of DSP in cancer cells via the ZEB transcription factors. We corroborated the hypothesis by analyzing the somatic expression and copy-number variations profile of over 11,000 pan-cancer tumor samples, and discussed their clinical impact based on survival outcomes. We showed that elevated PA levels lead to loss of DSP and promote migration in liver cancer (HepG2 and Hep3B) and breast cancer (MDA-MB-231) cell lines. Using a luciferase DSP-promoter assay and ChIP-qPCR, we validate that the transcriptional repression of DSP is mediated by the ZEB transcription factors downstream of the IRE1-XBP1 pathway. CRISPR-mediated knockout (KO) of IRE1 abrogated ZEB-mediated DSP repression and cell migration. Thus, our results demonstrate the mechanism of DSP loss in cancer cells driven by elevated PA uptake depends on the IRE1-XBP1-ZEB pathway.
Materials and methods
Cell lines, culture medium and treatment
HepG2, Hep3B, SNU-387, MCF10A, MDA-MB-231 and HCT116 cells were purchased from ATCC. Cell line authentication/certification was performed by ATCC. The cell lines were tested for mycoplasma using a PCR-based method (ATCC’s Universal Mycoplasma Detection Kit). HepG2, Hep3B and MDA-MB-231 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). SNU-387 cells were cultured in RPMI1640 medium supplemented with 10% FBS. HCT116 cells were cultured in McCoy’s 5A medium supplemented with 10% FBS. MCF10A cells were cultured in DMEM/F12 medium supplemented with 5% horse serum, 0.5μg/ml hydrocortisone, 100ng/ml cholera toxin, 10μg/ml insulin and 20ng/ml epidermal growth factor. All cell lines were maintained in a humid incubator with 5% CO2 at 37°C and were used between P3-P15 passage numbers.
For DSP knockdown, Hep3B and HepG2 cells were seeded at a density of 5×105 cells/ml and transfected with DSP siRNAs (Thermo Fisher, cat # s4333, s4335) or scramble siRNA (control, Thermo Fisher, cat # AM4611) using Lipofectamine RNAiMAX (Thermo Fisher, cat # 13778030) following manufacturers protocol. A dose-response experiment was performed to determine the concentration of siRNA required for >70% knockdown of DSP expression. Concentration of 100 nM and 80 nM were used to knockdown ZEB1 and ZEB2, respectively. These concentrations were used for all further experiments. For ZEB1/2 knockdown, MDA-MB-231 and Hep3B cells were seeded at a density of 5×105 cells/ml and transfected with ZEB1/2 siRNAs (ORIGENE, cat # SR304746A-C for ZEB1,Thermo Fisher, cat # AM16708 for ZEB2) or scramble siRNA (control: ORIGENE cat # SR30004 for ZEB1, Thermo Fisher, cat # AM4611 for ZEB2) using Lipofectamine RNAiMAX following manufacturers protocol. For Western blot analysis, 48 or 72 hours of RNAiMAX transfections were performed. For qRT-PCR, 24 hours of RNAiMAX transfections were performed.
IRE1−/− CRISPR knockout (KO) cell lines
The pSpCas9(BB)-2A-GFP (PX458) and pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmids were a gift from Dr. Feng Zhang (Addgene plasmid # 48138 and # 62988). The pX333 plasmid was a gift from Dr. Andrea Ventura (Addgene plasmid # 64073). To target the Cas9 enzyme to the IRE1 gene, we designed sgRNAs using several CRISPR design servers, e.g. crispr.mit.edu and crispr.dbcls.jp, and identified two sgRNAs (gRNA-A and gRNA-B) with high scores on exon 1, which were evaluated by COSMID and Cas OFFinder for off-target effects. Additionally, we also identified a sgRNA on exon 17 that target the kinase domain of IRE1 (gRNA-C) with high score and verified by COSMID, to further ensure KO of IRE1 function. Three high scoring single guide RNAs (sgRNAs) were synthesized and inserted into the plasmids. gRNA-A (TCACCGCCTCGCTGTCGTCGCGG) and gRNA-B (CCGTACCGCCCCCGGAGCCAGGG) is inserted in the pX333 plasmid, while gRNA-C (CCAGAGGGAGGCCGCCGAATGGG) was inserted in the pSpCas9(BB)-2A-Puro (PX459) V2.0 plasmid. Subsequently, gRNA-A and gRNA-C were inserted within the Bbs1 cut sites and gRNA-B was inserted between the AflII and KpnI sites. All inserted constructs were confirmed by DNA sequencing. The plasmids were transfected into MDA-MD-231, Hep3B and HepG2 cells using Lipofectamine 3000 (Thermo Fisher, cat # L3000001). After using the appropriate selection agents, GFP and puromycin, individual clonal populations were expanded and analyzed for loss of IRE1 expression using Western blotting. Clones with no detectable IRE1 were selected for further studies.
Boyden’s chamber assay
Trans-well (Boyden’s chamber) migration assays were performed using cell culture inserts (8μm pore size) (Sigma-Aldrich, cat # CLS3464 and CELLTREAT #230639). 5×105 cells in serum-free media supplemented with 1μg/ml mitomycin C (Sigma Aldrich, cat # M4287) were seeded in the upper chamber of the inserts. To assess migration upon DSP silencing, lipofectamine RNAiMAX (Thermo Fisher, cat #13778030) was used for transfection following manufacturers protocol. After 24 hours of transfection at 37°C, the media was replaced with serum-free media supplemented with 1μg/ml mitomycin C. The inserts were placed in wells with 500 uL of regular medium containing 10% FBS as chemoattractant. To assess migration with PA treatment, cells were seeded with 200 uL of serum-free media supplemented with 1μg/ml mitomycin C in the upper chamber of the insert for 24 hours. The media in the insert was replaced with 200 uL PA media without FBS but contains 1μg/ml mitomycin C. The insert was then placed in an outer well (using a 24 well plate) containing either BSA or PA media with 10% FBS as a chemoattractant for 24 hours. After 24 hours of incubation at 37°C, the inserts (of the DSP silencing or PA treatment) were removed, washed with PBS, fixed in 70% ethanol for 10 minutes. The inserts were then dried for 15 minutes, stain with 0.2% (w/v) crystal violet (Sigma Aldrich, cat# C0775) in methanol for 15 minutes and washed with PBS twice. After 15 minutes of drying, the number of cells that migrated across the membrane were counted across 3 fields of view using a fluorescence microscope at 10x or 20x magnification. Each migration assay was repeated three times.
XBP1 splicing assay
Total mRNA was isolated from Hep3B, HepG2 and MDA-MB-231 wild-type (WT) and IRE1−/− KO cells. A one-step RT-PCR kit was used to amplify XBP1 cDNA (Qiagen, cat # 210210). XBP1-for 5’-TTACGAGAGAAAACTCATGGCC-3’ and XBP1-rev- 5’- GGGTCCAAGTTGTCCAGAATGC-3’ primers were designed to flank the 26bp intron sequence and the products were resolved on a 2.5% agarose gel made in 1X Tris/Borate/EDTA (TBE).
qRT-PCR
Total mRNA was isolated from MDA-MB-231, Hep3B, and HepG2 WT and IRE1−/− KO cells with an RNeasy Plus mRNA extraction kit (Qiagen cat # 74134). cDNA was prepared (Thermo, cat # 4368814) and equal amounts (100ng) used for qRT-PCR with the iQ SyBr Green supermix (Bio-Rad, cat # 1708882) using the following parameters for 40 cycles: 95oC for 30s, 58oC for 30s and 72oC for 45s. The primer sequences for ZEB1 were 5’-ATGCAGCTGACTGTGAAGGT-3’ (forward) and 5’- GAAAATGCATCTGGTGTTCC-3’ (reverse), ZEB2 were 5’-TATGGCCTACACCTACCCAAC-3’ (forward) and 5’-AGGCCTGACATGTAGTCTTGTG-3’ (reverse), for DSP were 5’-TGTACGACTGGAGCGACAAG-3’ (forward) and 5’- GCGTCTGCAGAGTGTCCATA-3’ (reverse), and the housekeeping gene GAPDH were 5’- CAGCCGCATCTTCTTTTGCG −3’ (forward) and 5’- TGGAATTTGCCATGGGTGGA-3’ (reverse).
Western Blots
MDA-MB-231, Hep3B, and HepG2 cell lysates were collected using RIPA buffer containing a protease inhibitor cocktail (Roche, cat # 4693159001). 30–50ug total protein was loaded onto an SDS-PAGE gel run in a Tris/glycine/SDS buffer system. The proteins were transferred onto a nitrocellulose membrane and blocked with either 5% BSA or 5% non-fat dried milk for 3 hours or 1 hour, respectively, at room temperature. Primary antibody incubation was done overnight at 4°C with antibodies against IRE1 (Cell Signaling Tech, cat # 3294), DSP I + II (Abcam, cat # ab71690), E-cadherin (Cell Signaling Tech, cat # 3195), Vimentin (Cell Signaling Tech, cat # 3932), ZEB1 (ORIGENE, cat # TA802298), ZEB2 (ORIGENE, cat # TA802113) and GAPDH (GeneTex, cat # GTX100118).
Luciferase assay
The LightSwitch Luciferase Assay system (Active Motif) was used to insert the DSP promoter sequence upstream of an engineered luciferase gene (RenSP). A 96 well plate was seeded with 5000 cells/well. 150ng of reporter plasmid containing the DSP promoter was then transfected into Hep3B, HepG2 cells and MDA-MB-231 cells. siRNA designed against ZEB1, ZEB2 or a scrambled sequence was co-transfected along with the plasmid at a concentration of 200nM per well. The luciferase assay was carried out according to the LightSwitch Luciferase Assay Kit instructions after 24 hrs (Active Motif, cat # 32031).
Chromatin Immunoprecipitation
Human cancer cells (Hep3B, MDA-MB-231, HepG2) were fixed with 1% formaldehyde for 15 min and quenched with 0.125 M glycine. Chromatin was isolated by the addition of lysis buffer, followed by disruption with a Dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300–500 bp. Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heat for de-crosslinking, followed by ethanol precipitation. Pellets were resuspended and the resulting DNA was quantified on a ClarioStar spectrophotometer. Extrapolation to the original chromatin volume allowed quantitation of the total chromatin yield.
An aliquot of chromatin (30 μg) was precleared with protein G agarose beads (Invitrogen). Genomic DNA regions of interest were isolated using 4 μg of antibody against ZEB1 (Santa Cruz Biotechnologies, cat # sc-10572, lot # K0702). Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65 C, and ChIP DNA was purified by phenol-chloroform extraction and ethanol precipitation.
Quantitative PCR (qPCR) reactions were carried out in triplicate on specific genomic regions using SYBR Green Supermix (Bio-Rad, cat # 170–8882) on a CFX Connect™ Real Time PCR system. The qPCR were performed using one positive control primer pair for CDH1 5’-GTGAACCCTCAGCCAATCAG-3’ (forward) and 5’-TCACAGGTGCTTTGCAGTTC-3’ (reverse), a negative control primer pair that amplifies a region in a gene desert on chromosome 12 (Untr12; Active Motif Cat# 71001) and primers for four regions of interest along the DSP promoter region containing putative ZEB1/2 binding sites: three putative ZEB binding regions upstream of DSP transcription start site (−1k, −462, +/− 500p from TSS) and one region located in the first intron. These regions were selected based on promoter analysis for putative binding sites and ZEB peaks observed in the ENCODE project ChIP-seq data. Primer sequences for target sequenes were: DSP −1k 5’-TTTTCATGGATGGTGTGACTG-3’ (forward), 5’-GGGCCACTCCCAGTTAAGAG-3’ (reverse); DSP −462 5’-CCCCAAACAAAACTCTATCATC-3’ (forward), 5’-GCTACCTACCACGCATTGAC-3’ (reverse); DSP Pro 5’-CTAAAGCGTTCCCTCCTCAC-3’ (forward), 5’-CGATTGGAGACTTACTGGTCAA-3’ (reverse); DSP In1 5’-GCCTCCGCAAGTAAGAGCTA-3’ (forward), 5’-GGAGCTTCTGCCTCTGTGAC-3’. The resulting signals were normalized for primer efficiency by carrying out qPCR for each primer pair using input DNA from each cell line.
Confocal microscopy
Confocal microscopy with HepG2 cells transfected with DSP siRNA was performed as described previously (7). HepG2 cells were fixed 48 hours post siRNA transfection and incubated overnight with primary antibodies against DSP, JUP, and CDH1. The cells were then washed and incubated with AlexaFluor 488 secondary antibody, followed by nuclear counter-staining with DAPI. Images were acquired at 40X magnification using an Olympus FluoView 1000 inverted microscope with identical exposure and PMT settings for each image.
DSP promoter, TCGA and ICGC data analysis
Promoter sequence (1000bp upstream of transcription start site) for the human DSP gene was retrieved from the UCSC genome browser (hg38) and transcription factor binding analysis for putative ZEB binding sites was performed using the JASPAR gene regulatory database (29). Gene expression, methylation, copy number variation (CNV) and survival data for the Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) cohorts were retrieved from the genomics data commons hub at the UCSC Xena portal for cancer genomics (30). The RNAseq gene expression profiles (FPKM) were upper-quantile normalized and standardized (mean = 0, standard deviation = 1) prior to further analysis. The mean beta values of the DSP gene were obtained from Illumina methylation 450 array and log2 transformed. For ERN1 CNV analysis, masked copy number segmentation values from the GISTIC2 pipeline were averaged for the ERN1 gene. Kaplan-Meier survival analyses were performed by segregating the TCGA and ICGC samples into two groups. The samples were clustered based on standardized expression or average masked copy number segmentation values using K-means clustering (k=2) yielding groups with high or low average expression or CNV represented as cluster centers.
Statistical analyses
Statistical analyses for differences in mean were performed using Student’s T-test in GraphPad Prism 7 with P < 0.05 considered significant. Kaplan-Meier survival analysis, linear regression and correlation analyses were performed in R Studio (R version 3.5.1) with P < 0.05 considered significant. Statistical analysis for differences in mean in experiments with PA and BSA treated WT and IRE1−/− cells were performed using two-way ANOVA with P < 0.05 considered significant.
Results
Palmitate-induced desmoplakin loss and enhanced migration depends on the IRE1-XBP1 pathway
To evaluate the impact of PA on DSP expression and migration in cancer cells, we compared the Hep3B liver cancer and MDA-MB-231 breast cancer cell lines treated with 0.3mM PA or 2% BSA (control). In each cell line, we observed a significant reduction in DSP expression by 24–48 hours post-treatment (Figure 1A and 1B). Next, we determined whether the reduction in DSP expression in PA treated cells depended on the IRE1-XBP1 pathway. Using CRISPR gene editing, we generate IRE1−/− KO HepG2, Hep3B, and MDA-MB-231 cell lines (Supplementary Figure S1A, and S2–S4). While PA treatment resulted in accumulation of XBP1 splice product (XBP1s) in the parental lines, indicating activation of the IRE1-XBP1 pathway, the IRE1−/− cells did not accumulate XBP1s upon PA treatment (Supplementary Figure S1B). In the IRE−/− cells, PA treatment did not result in a decrease in DSP expression levels, as compared to WT cells (Figure 1A and 1B).
Figure 1:

A. Western blots showing expression levels of DSP I + II in WT or IRE1−/− KO Hep3B and MDA-MB-231 cells. The cells were cultured in the presence of either 0.3mM palmitate (PA) or 2% BSA. The Hep3B cell lysates were collected after 48 hours post-treatment, whereas MDA-MB-231 cell lysates were collected at 24 and 48-hour time points B. Quantification of the western blots comparing DSP I + II expression levels in WT and IRE1−/− KO Hep3B and MDA-MB-231 cells. Data shown here is mean ±SE of three independent experiments, with * indicating a Student’s T-test P < 0.05. C-E. Quantification of the number of migrating cells Boyden’s chamber assay in HepG2 (C), Hep3B (D) and MDA-MB-231 (E) cells treated with DSP siRNA or scramble siRNA (control). The bars here represent mean ±SE of three independent experiments, with * indicating a Student’s T-test P < 0.05. F-H. Quantification of the number of migrating cells determined using Boyden’s chamber assay in WT or IRE1−/− KO HepG2 (F), Hep3B (G) and MDA-MB-231 (H) cells. The cells were additionally incubated with either BSA or PA. The bars here represent mean ±SE of three independent experiments, with * indicating two-way ANOVA with P < 0.05 and ** indicating P <0.01. Two-ANOVA analysis shows both PA treatment and IRE1 KO status significantly affected cell migration. PA treatment resulted in an increase (P < 0.05) while IRE1 KO resulted in a decrease in cell migration (P < 0.05) in all three cell lines.
Next, we evaluated the impact of DSP knockdown and PA treatment on rates of migration in Hep3B, HepG2, and MDA-MB-231 cells. As expected, the rates of migration were elevated in cells with siRNA mediated DSP knockdown (Figure 1C–E, Supplementary Figure S5A, S6). In comparison, cells treated with PA exhibited higher migration rates comparable to the rates observed in the cells with DSP knockdown (Figures 1F–H, Supplementary Figure S5B, S7). However, this increase in migration rates was not observed in IRE1−/− cells, suggesting the activation of the IRE1-XBP1 pathway was critical in mediating the effects of PA on cell migration (Figures 1F–H).
Loss of DSP promotes migration of cancer cells independent of classic EMT markers
Previous studies have linked loss of cell adhesion molecules like DSP with the activation of EMT program, a complex set of cellular alterations that drive multiple metastatic processes including cell migration. To determine whether loss of DSP triggered EMT activation or was sufficient in driving cancer cell migration on its own, we assessed the impact of loss of DSP expression on the established EMT markers, including E-cadherin (CDH1) and vimentin (VIM). While cells transfected with DSP siRNA showed a decrease in DSP expression (Figures 2A and 2B), neither E-cadherin (Figure 2C) or vimentin (Figure 2D) were impacted by the loss of DSP expression in Hep3B, HepG2, and MDA-MB-231 cells. Next, we assessed the expression and localization of the desmosomal components DSP and junction plakoglobin (JUP) in HepG2 cells compared to E-cadherin. Upon siRNA-mediated DSP knockdown, we observed both a decrease in DSP and JUP expression, as well loss of localization at the cell membrane (Figure 2E). This suggested DSP knockdown resulted in loss of desmosomes. However, neither the expression nor the cell membrane localization of E-cadherin was impacted by DSP knockdown (Figure 2E). Taken together with earlier results showing enhanced migration in cells with DSP knockdown, these results suggest that the loss of DSP itself may be sufficient to promote cell migration.
Figure 2.

A: Western Blots showing expression of DSP I + II, CDH1, VIM and GAPDH in HepG2, Hep3B, and MDA-MB-231 cells treated with two individual or pooled DSP siRNAs. B-D. Quantification of the western blots for DSP siRNA-treated HepG2, Hep3B, and MDA-MB-231 cells, comparing the expression levels of DSP, CDH1, and VIM. Bars represent mean ±SE across 3 independent replicates with * indicating Student’s T-test P < 0.05 and ** indicating P < 0.01. E. Confocal images showing the expression and localization of the desmosomal component DSP and JUP, along with CDH1, in HepG2 cells treated with DSP siRNA or scramble siRNA (control). Each image was acquired using uniform image acquisition parameters and the scale bar represents a length of 30μm.
ZEB regulates DSP expression downstream of the IRE1-XBP1 pathway
Considering recent evidence linking XBP1 with transcriptional activation of the ZEB family of transcriptional repressors (28), we analyzed the promoter sequence of DSP and found three putative ZEB binding sites (Figure 3A). To corroborate the hypothesis that ZEB transcription factors function as transcriptional repressors of DSP, we analyzed gene expression data from the TCGA PANCAN cohort. Correlation analysis revealed that ZEB1 and ZEB2 showed strong negative correlation with DSP expression (both P < 2.2×10−16) (Figure 3B). These correlative patterns were not just limited to liver or breast cancer, but consistent across different cancer types (Supplementary Figure S8). Additionally, average DSP methylation levels were negatively correlated with DSP expression while being positively correlated with ZEB1 and ZEB2 expression (Figure 3C). We evaluated the correlation between DSP methylation levels and the expression of DSP, ZEB1, and ZEB2 across liver, breast and colorectal cancer tumors and found consistent patterns within these individual cancer types (Supplementary Figure S9). These results suggest that ZEB transcription factors may be responsible for transcriptional repression of DSP.
Figure 3:

A. Schematic diagram of the DSP promoter (−1000bp upstream of transcription start site) displaying putative ZEB binding sites B. Scatterplots showing the correlation between DSP, ZEB1 and ZEB2 expression levels across 10,022 samples in the TCGA PANCAN (pan-cancer) dataset C. Scatterplots showing the correlation between DSP methylation levels with DSP, ZEB1 and ZEB2 expression in the TCGA PANCAN dataset. PCC indicates Pearson’s correlation coefficient and the P values indicates significance of correlation
We next measured the basal expression levels of DSP, ZEB1, and ZEB2 in six cancer cell lines to validate the correlation patterns observed in the TCGA analysis. Similar to the human tumor data, we found a strong negative correlation between DSP and ZEB expression across the six cell lines (Figure 4A). Since MDA-MB-231 cells have the highest endogenous levels of ZEB and lowest endogenous levels of DSP, we knocked-down ZEB1/2 expression using siRNAs and found that DSP levels increased significantly (Supplementary Figure S10). Likewise, we found similar results with the Hep3B cells (Supplementary Figure S11). To functionally validate the role of ZEB in the regulation of DSP expression, we cloned the DSP promoter sequence in a luciferase reporter gene with the three putative ZEB binding sites upstream of the transcription start site. Upon co-transfecting Hep3B and MDA-MB-231 cells with the DSP-promoter luciferase plasmid and siRNAs targeting ZEB1 or ZEB2, we found that DSP promoter activity was significantly enhanced upon knockdown of ZEB1 and ZEB2 (Figures 4B and 4C). Additionally, for further validation we performed chromatin immunoprecipitation (ChIP)-qPCR to detect enrichment of ZEB1 binding on various predicted sites along the DSP promoter (Figure 4D–F, Supplementary Figure S12). In each cell line, we observed a significantly greater number of ZEB1 binding events at different putative binding sites as compared to negative control.
Figure 4.
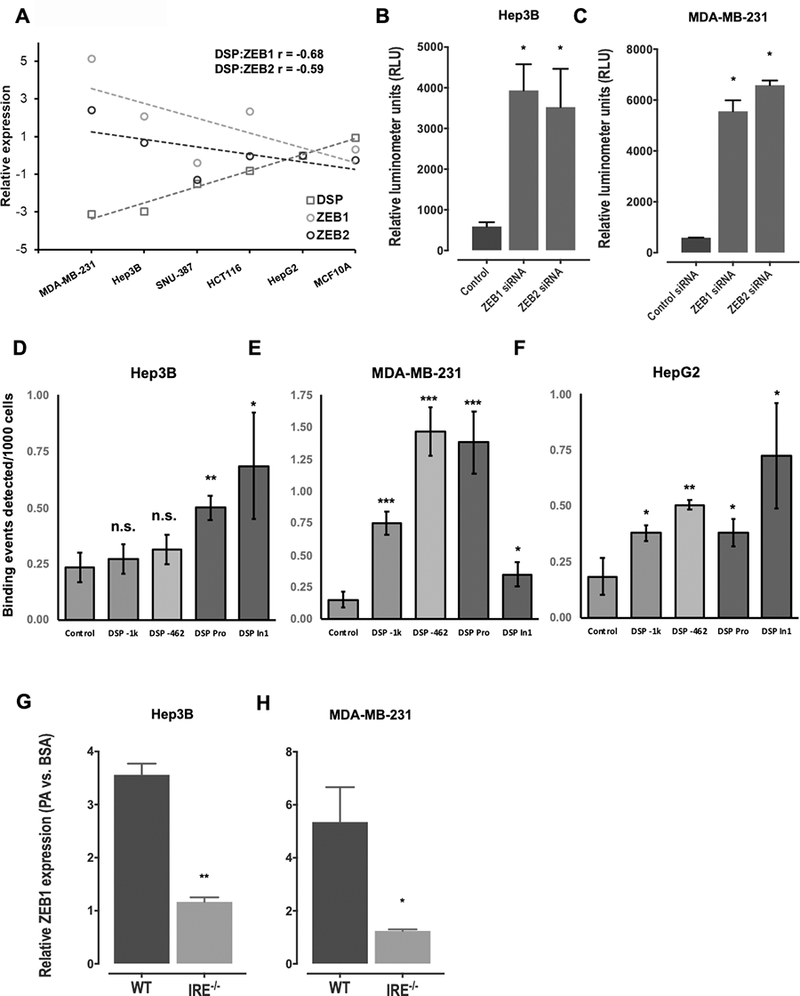
A: Scatterplots showing correlation in basal expression levels between DSP, ZEB1, and ZEB2 across six cancer cell lines. The expression levels (Y-axis) of the three genes were normalized to GAPDH in each cell line and represented relative to the levels in HepG2 cells. B-C. Barplots showing results of the DSP promoter luciferase assay in response to siRNA-mediated knockdown of ZEB1 or ZEB2 in Hep3B (C) or MDA-MB-231 cells (C). D-F. Barplots displaying results of ZEB1 ChIP-qPCR assay in Hep3B (D), MDA-MB-231 (E) or HepG2 (F) cells. The Y-axis represents number of binding events detected per 1000 input cells. The binding events were quantified by qPCR for various segments along the DSP promoter and first DSP intron that contained putative ZEB binding sites, and compared with negative control. G, H. Loss of ZEB1 gene expression in IRE1−/− KO Hep3B (G) or MDA-MB-231 (H) cells. The barplots in each panel represent mean ±SE of three independent replicates with * indicating Student’s T-test P < 0.05, ** indicating P < 0.01 and ns indicating not significant.
Next, we investigated the direct impact of the IRE1-XBP1 pathway in the activation of ZEB transcription factors in the presence of elevated PA. We observed a significant increase in the expression levels of ZEB1 upon PA treatment in WT Hep3B and MDA-MB-231 cells (Figure 4G–H). However, the ZEB1 expression was significantly lower in the PA treated IRE−/− cells, suggesting ZEB expression was dependent on the IRE1-XBP1 pathway (Figure 4G–H, Supplementary Figures S13 and S14).
Relevance of DSP and IRE1 in human cancers
The in vitro experiments confirmed that DSP loss via PA depended on the activation of the IRE1-XBP1-ZEB pathway and led to enhanced migration of cancer cells. As increased migration rates are often considered a hallmark of advanced malignancy, we explored the clinical relevance of somatic alterations of DSP and IRE1 in cancer patients. We first analyzed the relative impact of DSP expression on patient survival outcomes in the TCGA PANCAN and ICGC cohorts. We grouped tumor samples from the two cohorts using K-means clustering, and determined the impact of samples clustered on the basis of low or high expression of survival outcomes. As shown in Figure 5A, the group of samples with low DSP expression were associated with worse overall survival outcomes compared to high DSP expression. In addition, we compared DSP expression in TCGA samples based on pathological assessment of metastatic status (where available). In this analysis, DSP expression levels were significantly lower in tumors with evidence of distant metastasis (M1) compared to tumors without any metastases (M0) (Figure 5B). Next, we evaluated the impact of IRE1 (ERN1) on survival outcomes. While IRE1 expression levels did not impact survival outcomes, we found increased copy number of the ERN1 gene were associated with poor overall survival outcomes in both the TCGA and ICGC cohorts (Figure 5C). While preliminary, this evidence suggest both loss of DSP and gain of IRE1/ERN1 may be important factors in determining worse patient prognosis, demanding further investigations.
Figure 5.

A: Kaplan-Meier (survival) curves of TCGA PANCAN (n=10022) or ICGC (n=4429) patients grouped according to expression levels of DSP. The samples in each cohort were clustered using K-means (K=2). Barplots on the right indicate centers of the expression clusters in each study. P values indicate significance of the log-rank (Mantel-Cox) test. B. Boxplots comparing the expression levels of DSP in the TCGA PANCAN cohort based on distant metastasis status. M0 indicates no reported indications of metastasis whereas M1 indicates presence of one or more distant metastases. C. Kaplan-Meier (survival) curves of TCGA PANCAN patients grouped according to copy number variation status of ERN1 (IRE1). Samples were clustered using K-means clustering (K=2) based on mean segment copy number, with P values indicating significance of the log-rank (Mantel-Cox) test. D. A proposed PA-induced pathway driving migration of the cancer cells through the loss of DSP mediated by activation of the IRE1-XBP1 pathway and ZEB transcription factors.
Discussion
Along with the intrinsic cellular factors that drive transformation to advanced metastatic state, the tumor microenvironment is increasingly being recognized as a crucial mediator in the survival and growth of cancer cells (31,32). We now recognize fatty acid metabolism plays an important role in cancer progression and response to therapeutic intervention (33,34). Here we report that elevated palmitate levels can promote migration of cancer cells by depleting desmoplakin expression. Owing to the critical role of desmoplakin in the structural integrity of desmosomes, previous studies have speculated a tumor suppressor role of desmoplakin in cancers (10,13). These reports highlighted loss of desmosomal adhesion as prerequisite for the induction of EMT program, along with the unidirectional dependency of desmosome formation on adherence junctions (35). However, our results suggest that loss of desmoplakin itself is sufficient to enhance migration of cancer cells. Interestingly, this effect seems to be independent of the loss of E-cadherin or the induction of the classic EMT program. Thus, our study suggests an important and independent mechanism of cancer cell migration that depends on the loss of desmoplakin.
At the cellular level, elevated fatty acid levels have been reported to influence various signaling pathways, including ER-stress and the UPR. The activation of IRE1-XBP1 axis in the UPR is a well-established consequence of elevated palmitate levels in cancer cells (36). In our study, we show that the loss of desmoplakin actually depends on the activation of the IRE1-XBP1 pathway. In the presence of palmitate, cancer cell lines undergo loss of desmoplakin and enhanced migration. However, these effects of elevated palmitate levels were abrogated in the IRE−/− KO cells. Thus, our results provide direct evidence linking the IRE1-XBP1 pathway with loss of DSP and enhanced cancer cell migration in the presence of palmitate.
We further explored the precise mechanism by which IRE-XBP1 pathway could regulate DSP expression. Based on systematic analysis of the DSP promoter, we identified multiple ZEB binding sites that could suggest a transcriptional repression mechanism triggered by IRE1-XBP1. This hypothesis derives from a recent study suggesting the spliced and active form of XBP1 (XBP1s) is a direct transcriptional activator of SNAI1, SNAI2, ZEB2 and TCF3 transcription factors (28). We corroborated the correlative expression patterns between DSP expression and methylation levels with ZEB expression across a large cohort of human tumor samples from the TCGA and ICGC project. Moreover, we demonstrated a direct link between ZEB expression and DSP promoter activity, thereby confirming the regulatory link between ZEB and DSP. In addition, we demonstrate that ZEB expression levels were elevated in cells treated with PA, while this effect was suppressed in the IRE−/− KO cells. Based on these results, we confirmed our hypotheses of DSP regulation via the IRE1-XBP1-ZEB axis and proposed the functional model in Figure 5D.
As we continue to gain further evidence supporting the molecular mechanisms that govern cancer cell progression in the presence of elevated palmitate, we must also scrutinize the phenotypic impact of these alterations in patient tumors. We investigated the relevance of loss of DSP expression and gain of IRE1 genomic copy number in patient prognosis. Our analyses revealed a significant impact of both alterations on poor patient outcomes, as would be expected based on enhanced migration of the cancer cells observed in our experiments. While these are preliminary and speculative connections, they highlight the need to study the role of these pathways in further details.
Supplementary Material
Implications:
Provides mechanistic link on palmitate-induced activation of IRE1α to cancer cell migration.
Acknowledgments
The authors would like to thank Dr. Melinda Frame at the Center for Advanced Microscopy at Michigan State University for assistance with the confocal microscopy experiments. The authors are also grateful for the excellent service and support received from Maddy Craske’s team at Active Motif for the ChIP-qPCR experiments. This work was supported in part by the National Science Foundation (CBET 1547518, 1510895, 1802992, 2029319) and the National Institute of Health (R21NS116496).
Financial support: National Science Foundation (CBET 1510895, CBET 1547518, and CBET 1802992).
Footnotes
Conflict of interest: The authors declare no conflicts of interest
References
- 1.Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell 2011;144:646–74 [DOI] [PubMed] [Google Scholar]
- 2.Ye X, Weinberg RA. Epithelial–Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends in Cell Biology 2015;25:675–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009;139:871–90 [DOI] [PubMed] [Google Scholar]
- 4.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nature Reviews Molecular Cell Biology 2014;15:178–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Molecular Cancer 2016;15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS-O, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2016;541:41–5 [DOI] [PubMed] [Google Scholar]
- 7.Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Scientific Reports 2015;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Nath A, Yang X, Portis A, Walton SP, Chan C. Synergy Analysis Reveals Association between Insulin Signaling and Desmoplakin Expression in Palmitate Treated HepG2 Cells. PLoS ONE 2011;6:e28138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swagell CD, Henly DC, Morris CP. Expression analysis of a human hepatic cell line in response to palmitate. Biochemical and Biophysical Research Communications 2005;328:432–41 [DOI] [PubMed] [Google Scholar]
- 10.Chidgey M, Dawson C. Desmosomes: a role in cancer? British Journal of Cancer 2007;96:1783–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou G, Yang L, Gray A, Srivastava AK, Li C, Zhang G, et al. The role of desmosomes in carcinogenesis. OncoTargets and Therapy 2017;Volume 10:4059–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clurman BE, Chun MGH, Hanahan D. Genetic Deletion of the Desmosomal Component Desmoplakin Promotes Tumor Microinvasion in a Mouse Model of Pancreatic Neuroendocrine Carcinogenesis. PLoS Genetics 2010;6:e1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dusek RL, Attardi LD. Desmosomes: new perpetrators in tumour suppression. Nature Reviews Cancer 2011;11:317–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Najor NA. Desmosomes in Human Disease. Annual Review of Pathology: Mechanisms of Disease 2018;13:51–70 [DOI] [PubMed] [Google Scholar]
- 15.Yang L, Chen Y, Cui T, Knösel T, Zhang Q, Albring KF, et al. Desmoplakin acts as a tumor suppressor by inhibition of the Wnt/β-catenin signaling pathway in human lung cancer. Carcinogenesis 2012;33:1863–70 [DOI] [PubMed] [Google Scholar]
- 16.Kam CY, Dubash AD, Magistrati E, Polo S, Satchell KJF, Sheikh F, et al. Desmoplakin maintains gap junctions by inhibiting Ras/MAPK and lysosomal degradation of connexin-43. The Journal of Cell Biology 2018;217:3219–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic Palmitate But Not Oleate Exposure Induces Endoplasmic Reticulum Stress, Which May Contribute to INS-1 Pancreatic β-Cell Apoptosis. Endocrinology 2006;147:3398–407 [DOI] [PubMed] [Google Scholar]
- 18.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 1999;13:1211–33 [DOI] [PubMed] [Google Scholar]
- 19.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2013;1833:3460–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Achard CS, Laybutt DR. Lipid-Induced Endoplasmic Reticulum Stress in Liver Cells Results in Two Distinct Outcomes: Adaptation with Enhanced Insulin Signaling or Insulin Resistance. Endocrinology 2012;153:2164–77 [DOI] [PubMed] [Google Scholar]
- 21.Halbleib K, Pesek K, Covino R, Hofbauer HF, Wunnicke D, Hanelt I, et al. Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol Cell 2017;67:673–84 e8 [DOI] [PubMed] [Google Scholar]
- 22.Kono N, Amin-Wetzel N, Ron D. Generic membrane-spanning features endow IRE1alpha with responsiveness to membrane aberrancy. Mol Biol Cell 2017;28:2318–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A 2013;110:4628–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho H, Stanzione F, Oak A, Kim GH, Yerneni S, Qi L, et al. Intrinsic Structural Features of the Human IRE1alpha Transmembrane Domain Sense Membrane Lipid Saturation. Cell Rep 2019;27:307–20 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J 2011;30:894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001;107:881–91 [DOI] [PubMed] [Google Scholar]
- 27.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 2003;23:7448–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cuevas EP, Eraso P, Mazón MJ, Santos V, Moreno-Bueno G, Cano A, et al. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Scientific Reports 2017;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Research 2018;46:D260–D6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldman M, Craft B, Brooks AN, Zhu J, Haussler D. The UCSC Xena Platform for cancer genomics data visualization and interpretation. bioRxiv 2018:326470 [Google Scholar]
- 31.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nature Medicine 2013;19:1423–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nature Cell Biology 2014;16:717–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nature Reviews Cancer 2016;16:732–49 [DOI] [PubMed] [Google Scholar]
- 34.Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging roles of lipid metabolism in cancer metastasis. Molecular Cancer 2017;16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis JE, Wahl JK, Sass KM, Jensen PJ, Johnson KR, Wheelock MJ. Cross-Talk between Adherens Junctions and Desmosomes Depends on Plakoglobin. The Journal of Cell Biology 1997;136:919–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends in Cell Biology 2014;24:472–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.