

Abstract
Recruitment of myeloid-derived suppressor cells (MDSCs) into the tumor microenvironment (TME) contributes to cancer immune evasion. MDSCs express the chemokine receptor CXCR2, and inhibiting CXCR2 suppresses the recruitment of MDSCs into the tumor and the pre-metastatic niche. Here we compared the growth and metastasis of melanoma and breast cancer xenografts in mice exhibiting or not exhibiting targeted deletion of Cxcr2 in myeloid cells (CXCR2myeΔ/Δ vs. CXCR2 myeWT). Detailed analysis of leukocyte populations in peripheral blood and in tumors from CXCR2myeΔ/Δ mice revealed that loss of CXCR2 signaling in myeloid cells resulted in reduced intratumoral MDSCs and increased intratumoral CXCL11. The increase in intratumoral CXCL11 was derived in part from tumor-infiltrating B1b cells. The reduction in intratumoral MDSCs coupled with an increase in intratumoral B1b cells expressing CXCL11 resulted in enhanced infiltration and activation of effector CD8+ T cells in the TME of CXCR2myeΔ/Δ mice, accompanied by inhibition of tumor growth in CXCR2myeΔ/Δ mice compared to CXCR2myeWT littermates. Treatment of tumor-bearing mice with a CXCR2 antagonist (SX-682) also inhibited tumor growth, reduced intratumoral MDSCs, and increased intratumoral B1b cells expressing CXCL11, leading to an increase in activated CD8+ T cells in the tumor. Depletion of B220+ cells or depletion of CD8+ T cells reversed the tumor inhibitory properties in CXCR2myeΔ/Δ mice. These data revealed a mechanism by which loss of CXCR2 signaling in myeloid cells modulates antitumor immunity through decreasing MDSCs and enriching CXCL11-producing B1b cells in the TME, which in turn increases CD8+ T-cell recruitment and activation in tumors.
Keywords: CXCL11-producing B cells, MDSC, Myeloid CXCR2, Breast cancer, Melanoma
Introduction
Myeloid cells are important in adaptive immunity, and some myeloid cells are immunosuppressive (1). Although myeloid progenitor cells differentiate into monocytes, neutrophils, and dendritic cells (DCs), when tumors are present, a subpopulation of these cells does not differentiate along these pathways and become myeloid-derived suppressor cells (MDSCs)(1,2), which can be granulocytic (G-MDSCs) or monocytic (M-MDSCs)(3,4). Both G- and M-MDSCs exhibit a robust suppressive function towards T cell-mediated immune function, resulting in tumor progression and metastasis (3,5). Therefore, targeting these two populations together may be beneficial for overcoming immune suppression in the TME (6).
In cancer patients, growing tumors secrete a variety of cytokines that are key signals involved in the recruitment of MDSCs. The CXC chemokine receptor 2 (CXCR2) is expressed by MDSCs, and its ligands, including CXCL1, are produced by cancer cells (7). CXCR2 not only regulates neutrophil trafficking from the bone marrow to peripheral circulation or inflammation sites (8), but also plays a role in tumor progression by facilitating the migration of tumor-associated myeloid cells into the TME (9). Therefore, targeting CXCR2 could alter tumor-associated myeloid cells and MDSC accumulation in the TME, favoring a more immune-suppressive TME.
In contrast to the favorable prognosis associated with elevated tumor–infiltrating T cells in tumors, the prognosis associated with elevations in tumor-infiltrating B cells has been controversial. B cells can represent up to 30% of all recruited immune cells, and are reported to be associated with improved prognosis in primary melanoma (10,11) or poorer prognosis in other tumor types (12–15). Several studies have linked B-cell accumulation in the TME with activation of myeloid cells by B cell-secreted immunoglobulins (12,13,16). In prostate carcinogenesis, B cell-derived lymphotoxin, in addition to a variety of other cytokines and cytokines, promotes chemo-resistant carcinomas (17).
These compelling studies from solid tumors in either mice or humans prompted us to investigate the functional significance and clinical importance of CXCR2, in reference to MDSCs in metastatic breast cancer and melanoma. In the present study, we investigated how loss of CXCR2 in myeloid cells affects the MDSC and CD8+ T-cell populations in tumors. We uncovered a critical mechanism by which disruption of CXCR2 in myeloid cells resulted in reduced intratumoral myeloid cells, increased intratumoral B cells, and elevated CXCL11. We also identified the B1b population of B cells as the primary source of intratumoral CXCL11. Depletion of the intratumoral B-cell population reduced CXCL11 in the TME, decreased the recruitment of CD8+ T cells into the tumor, and diminished the tumor growth inhibition observed in the mice with myeloid depletion of CXCR2. We identified a positive correlation among B-cell markers (B220 and/or CD19), CXCL11, and CD8α in TCGA and GEO-NCBI databases of breast cancer and melanoma patients.
Materials and Methods
CXCR2 knockout models
All animal experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committee. CXCR2fl/fl mice (Jackson Labs) were backcrossed five generations on C57BL/6 mice (Jackson Labs). To selectively delete CXCR2 in myeloid cells in the CXCR2fl/fl C57BL/6 mice, CXCR2fl/fl mice were crossed to LysM-Cre-recombinase C57Bl/6 mice (Jackson Laboratories)(18). The resulting mice were then bred to C57Bl/6 mice harboring the loxP-flanked tdTomato (mT) following the EGFP (mG) cassette. These mT/mG mice served as a Cre-reporter strain, and after Cre-mediated recombination, myeloid cells that are CXCR2-null are green. Mice with CXCR2-null myeloid cells are designated as CXCR2myeΔ/Δ mice. Littermate LysMCre::mT/mG mice without CXCR2f/f alleles were used as CXCR2myeWT controls (referred to as CXCR2WT hereafter).
Cell lines and metastatic tumor models
C57Bl/6 PyMT cell lines established from PyMT breast tumors were provided by the Hal Moses laboratory, Vanderbilt University. The cells have been previously characterized (19,20) and were maintained in DMEM/F-12 medium (Catalog number 11330–032, Gibco) containing 5% FBS. The stable PyMT reporter cell line was generated by infection with pRBow_Flue-GFP2 lentiviral particles (Zijlstra laboratory, Vanderbilt University). The Rich1.1 melanoma cell line with BrafV637E PtenR74X, Q396X has been characterized in our laboratory. All cell cultures were mycoplasma free. Cultures were tested monthly for mycoplasma using a sensitive PCR technique (e-MycoTM Plus, LiliF Diagnostics). Any mycoplasma positive cultures were discarded. Cell phenotypes were closely examined for authenticity by cytological and histologic characterization of the cells in culture and in mouse models.
In order to establish a lung metastatic model, the PyMT breast cancer cells (1×106), derived from C57BL/6 mice, were intravenously injected into C57BL/6 CXCR2myeΔ/Δ or littermate control CXCR2WT mice to generate a metastatic lung tumor model. Two weeks after injection, lungs were harvested, and lung weight was determined and normalized to tumor-free lung. In some instances, lungs were harvested at other timepoints if mice with lung tumors developed breathing difficulty.
For some experiments, the effect of the CXCR2 antagonist, SX-682 (Syntrix Pharmaceuticals), on metastatic tumor progression in vivo was assessed. Immunocompetent C57BL/6 mice were subcutaneously implanted with syngeneic Rich 1.1 cells. C57BL/6 mice were fed with chow containing SX-682 (0.756g/kg) ± intraperitoneal (IP) delivery of anti-PD-1 (100μg/mouse, #BE0273, BioXcell) or control IgG (#BE0089, BioXcell), or vehicle control chow without SX-682 + IP delivery of anti-PD-1 or isotype IgG every other day. After three weeks of treatment, lungs were harvested, and lung weight was determined and normalized to tumor-free lungs. Immune cells were isolated from the lungs of mice as described below, then analyzed by flow cytometry as described below.
Depletion of immune cells in vivo
For depletion of CD8+ T cells, mice were intraperitoneally injected with 200 μg of CD8α monoclonal antibody (mAb)(#BE0117, BioXCell) or the same amount IgG2b isotype control mAb (#B0090, BioXCell) daily for three days and then maintained by injection of 100 μg/mouse every other day (21). For depletion of B cells, mice were intraperitoneally injected with 200 μg of B220 mAb (#BE0067, BioXCell) or the same amount IgG2a isotype control mAb (#BP0089, BioXCell) daily for three days and then maintained with 200 μg/mouse every other day. The antibody injection for specific immune cell depletion started three days prior to tumor cell implantation and ended a day before the collection of tumor samples.
Purification of tissue immune cells
The Mojosort mouse CD8+ T-cell isolation kit (#480008, Biolegend) was used to negatively isolate CD8+ T-cells per the manufacture’s instruction from digested fresh lung tumor tissues of CXCR2myeΔ/Δ mice or littermate control CXCR2WT mice one week after 1×106 PyMT cells were intravenously injected. Lung tissues were digested with collagenase (320 U/mL), dispase (0.26 U/mL), and DNAase (5 mU/mL) at 37°C for one hour and processed using the “m_impTumor_01.01” program on a Miltenyi gentleMACS Dissociator. We obtained 87% purity of CD8+ T-cell with this protocol. The isolated CD8+ T cells were cultured in RPMI 1640 medium (Catalog number 22400–089, Gibco) containing 0.5% FBS. In the same way, B cells were also negatively isolated from the tumor tissues using the Mojosort mouse pan B-cell isolation kit (#480051, Biolegend). We obtained 85% purity of B-cells, and the B cells were cultured in Excellebrate B-cell media (#CCM031, R&D Systems).
CTL assay
MDSCs and CD8+T cells were isolated from lung tumors of CXCR2myeΔ/Δ mice and littermate CXCR2myeWT mice one week after 1×106 PyMT cells were intravenously injected. A cell isolation kit (Miltenyi Biotec) was used to isolate MDSCs (Myeloid-Derived Suppressor Cell Isolation Kit #130-094-538), and CD8+ T cells were isolated using EasySep Mouse CD8+ T-cell Enrichment Cocktail (Cat#19753, Stemcell Technologies). The target PyMT cells were genetically engineered with a luciferase reporter construct, as described above. When indicated, these cells were mixed at different ratios as indicated in the specific figure legend and cocultured in the RPMI 1640 medium containing 2%-10% FBS, IL7 (0.5 μg/mL, #577802, Biolegend), IL2 (30 U/mL, 575402, Biolegend), and CD3/CD28 Dynabeads (#11456D, Thermo Fisher) at a bead-to-cell ratio 2:1 for various times based on the experimental design. Subsequently, dead cells were removed by washing, and the remaining live cells were lysed using 2× Cell Lysis Buffer (#AA-Lys16ml, Raybiotec, Inc) for luciferase activity quantitation to reflect the CTL killing capacity. Percent (%) CD8+ T-cell killing ability = (the luciferase activity of target cells alone – the luciferase activity of target cells plus MDSCs and/or CD8+ T cells) divided by the luciferase activity of target cells X100.
Chemotaxis assay
For determining CD8+ T-cell migration, CD8+ T cells isolated from the tumor tissues were cultured overnight in 0.5% FBS RPMI 1640 medium. The migration of these CD8+ T cells was evaluated with the Cell Migration/Chemotaxis Assay Kit (96-well, 5 μm) (#ab235693, Abcam) by loading 150 μL supernatant from culture medium of 150,000 B cells isolated from the same tumor tissues into the bottom chamber and 50,000 CD8+ T cells/100μl on the top chamber. The negative control utilized addition of Excellebrate B-cell media not cultured with B cells. To characterize the role of CXCR3 in CD8+ T-cell migration, 20 nM of the CXCR3 antagonist, SCH546738, (HY-10017, MedChemExpress) was added to the top chamber, or recombinant mouse CXCL11 (#578302, Biolegend) was added in the bottom chamber containing medium without B cells. Boyden chamber assays were performed by incubating plates at 37°C, in a 5% CO2 atmosphere. After an 18-hour incubation, cells in the bottom chambers were collected, and the number of CD8+ T cells was determined by flow cytometry.
For determining B-cell migration, B cells were isolated by negative selection from the tumor tissues using the Mojosort mouse pan B-cell isolation kit (#480051, Biolegend). The migration of these B cells was evaluated with the Cell Migration/Chemotaxis Assay Kit (96-well, 3 μm)(#ab235692, Abcam) by loading 50,000 B cells in 100 μL Excellebrate B-cell media with/without the CXCR3 antagonist, SCH46738, 10 ng/mL (20 nM) to the top chamber and 100 μL Excellebrate B-cell media containing recombinant (r)CXCL11 (1 μg/mL; #578302, Biolegend) to the bottom chamber. Boyden chamber assays were performed by incubating plates at 37°C, in a 5% CO2 atmosphere. After a 24-hour incubation, cells in the bottom chambers were collected, and the B cells were stained with CD45, CD19, B220 antibodies (as described below), and B-cell number was determined by flow cytometry.
Cytokine assessment
Cytokine arrays were performed using RayBio mouse cytokine antibody array G-series 3 kit (Cat# AAM-CYT-G3) to screen for expression of 62 cytokines (Supplementary Table S1) and to screen for expression of 40 inflammation cytokines using the RayBio mouse inflammation antibody array G-series (Cat# AAM-INF-G1). CXCL11 and CXCL13 ELISAs were performed using the Mouse CXCL11/I-TAC DuoSet ELISA kit (Cat# DY572, R&D Systems) and the LEGEND MAX mouse CXCL13 (BLC) ELISA Kit (Cat#41907, Biolegend). Samples included serum and tumor tissue lysates prepared from tumor-bearing CXCR2myeΔ/Δ mice or littermate CXCR2WT mice at the end of the experiments. Supernatants of PYMT and Rich1.1 cells were collected after 48 hours of culture in 6-well plates with 2 × 105 cells. All ELISAs followed the manufacture’s procedures. For cytokine array analysis, the glass chips were scanned at excitation frequency of 532 nm using a GenoPix 4000B Scanner (Molecular Devices, Sunnyvale, CA). For each spot, the net density was determined by subtracting the background. The relative fold-difference in cytokine amount was determined in reference to the amount present on the control samples.
Flow cytometry analysis and antibodies
For flow cytometric analysis, tissues were minced on a programmable gentleMACS dissociator and digested with an enzyme solution of collagenase 1 (1500 CDU, CAT#234153, Calbiochem), dispase II (1mg/mL, CAT#13689500, Roche), and DNase 1 (0.1 mg/mL, CAT#260913, Calbiochem). Digested lungs were passed through a 70 μm strainer to obtain a single-cell suspension. Mouse spleens were pressed through 40 μm strainer using a syringe plunger to obtain a single-cell suspension. Red blood cells (RBCs) present in whole blood or cell preparations were removed using ACK Lysing Buffer (CAT#RGF-3015, KD Medical) prior to the staining. Staining and analyses protocols were according to our previously published methodology (22). Cells were incubated with Ghost Dye TM Violet 510 (Tonbo Biosciences), an amine-reactive viability dye used to discriminate live/dead cells, then washed with FACS buffer (PBS containing 2% v/v FBS). After blocking Fc receptors with anti-mouse CD16/CD32 (Biolegend) in FACS buffer for 15 minutes, cells were incubated with fluorescence-conjugated monoclonal antibodies specific to mouse immune cell surface or intracellular (BD Cytofix/Cytoperm Plus Kit#554715), markers indicated below for additional 30 minutes on ice. Cells were washed twice in FACS buffer (23), and data were acquired with FACSCanto II (Becton Dickenson), and data (fcs files) were analyzed using FlowJo software (Version 10.1). For cell counting, absolute numbers of lymphocytes were counted using AccuCheck counting beads (Thermo Fisher).
For cell-surface markers, the following monoclonal antibodies from eBioscience (San Diego, CA) were used: CD11b-FITC (clone M1/70), CD3-FITC (clone 17A2), CD3-PECy5 (clone 17A2), CD103-Brilliant Violet 421 (clone 2E7), CD4-Pacific Blue (clone RM4–4), CD4-APC/Cy7 (clone RM4–4), CD8-PECy7 (clone 53–5.8), CD11c-APC (clone N418), CD95-APC (clone SA367H8), CD19-PECy7 (clone N418), B220-APC (clone RA3–6B2), CD45-PerCp/Cy5.5 (clone 30-F11), CD45-APC/Cy7 (clone 30-F11), CD44-APC/Cy7 (clone IM7), NK1.1-APC/Cy7 (clone PK136), F4/80-Pacific Blue (clone BM8), CD69-APC (clone H1.2F3), CD107b-Alexa Fluor 647 (clone M3/84), CD62L-Alexa Fluor 647 (clone MEL-14), Ly6G-APC (clone 1A8), Ly6C-Alexa Fluor 647 (clone HK1.4), CD25-PerCp/Cy5.5 (clone 3C7), CD44-APC (clone IM7), CD43-PECy7 (clone S11), CD5-PE Cy7 (53–7.3), MHC II-Alexa Fluor 647 (clone AF6–120.1), CD206-PE (clone C068C2), PD-1-APC/Cy7 (clone 29F.1A12), PD-L1-APC (clone 10F.9G2).
For intracellular markers, monoclonal antibodies from Biolegend (San Diego, CA, USA) were used as follows: Foxp3-Alexa Fluor 647 (clone 150D), Ki67-Pacific Blue (clone 16A8), CXCL11 (578302). We also used the following antibodies: IFNγ-Alexa Fluor 700 (clone XMG1.2, from BD Bioscience, San Diego, CA, USA); Gost Dye violet 450, Gost Dye violet 510 Gost Dye violet 780 (from Tonbo Biosciences, San Diego, CA, USA).
Cellular immunofluorescence
For visualizing intracellular CXCL11 expressed by B cells, B cells were isolated from collagenase-digested tumor tissues using the MojoSort Mouse Pan B-cell Isolation Kit (Cat# 480051, Biolegend). The enriched B cells were fixed in cold methanol overnight and then in blocking buffer (1X PBS / 5% normal serum (#13551 Stemcell) / 0.3% Triton™ X-100, CAT#T8787, Sigma) for 60 minutes. Cells were co-stained with FITC-anti-mouse CXCL11 (aa22–100, Cat# LS-C693458, LSBio) or its FITC-IgG Isotype Control (Cat# LS-C149376, LSBio) and PE-anti-mouse B220 (Cat# 50–0452-U100, Biolegend) at a 1:200 dilution in buffer (1X PBS / 1% BSA / 0.3% Triton™ X-100) and incubated overnight at 4°C. The stained cells were then washed with PBS buffer (Cat# 46–013-CM, Corning) and mounted on slides. PE-labeled B220+ cells co-expressing CXCL11 (based upon co-staining for FITC) were visualized by LSM 510 Meta confocal microscopy and photographed. Images were analyzed in ImageJ using the corrected total cell fluorescence (CTCF) method and the provided region of interest (ROI) tool. CTCF of CXCL11 was calculated for the cytosol by the following formula: CTCF = Integrated Density − (Area of Region of Interest * Fluorescence of background reading).
Analysis of publicly available datasets
Whole-genome copy number alteration (CNA) analysis, microarray, and RNA sequencing-based gene expression profiling in TCGA databases (previously known as TCGA Provisional. Source data from GDAC Firehose), including Breast Invasive Carcinoma and Skin Cutaneous Melanoma, were obtained from cBioPortal for Cancer Genomics (www.cbioportal.org). Kaplan-Meier survival curves were used to show the expression of target genes on the outcomes of cancer patients. Breast cancer datasets published in GEO-NCBI (www.ncbi.nlm.nih.gov/geo) with available gene expression profiles of CD19, CXCL11, and CD8A were retrieved to validate cBioPortal results (datasets and information are in Supplementary Table S2). Downloaded raw data were sorted in Excel (Microsoft 365) and analyzed using GraphPad Prism software version 8.3.0.
Statistical analysis
Statistical analyses were performed using R 3.6.0 or GraphPad Prism software version 8.3.0. Data were summarized in figures expressing as mean±SD. Treatment effects in standard two-group experiments were compared using a two-sample t-test with unequal variances or the Wilcoxon rank-sum test. For more than two-group experiments, a one-way analysis of variance (ANOVA) with post hoc test was used to compare the differences between treatments. The experimental differences between two groups by variables, such as the difference between CXCR2WT and CXCR2myeΔ/Δ mice, was assessed in the context of a two-way ANOVA. Pairwise differences between two groups were compared using model-based mean comparisons. The mixed-effects model with post-hoc tests was used to compare the difference in tumor growth between treatments. The Benjamini and Hochberg (BH) was used to adjust p-value for multiple comparisons as noted (denoted by asterisks) in the text and figure legends to control the within experiment false discovery rate to less than 5%. Spearman correlation coefficient or a quadratic polynomial regression were used to evaluate the association between two continuous variables. Survival curves are estimated using the Kaplan-Meier method and compared statistically using the log-rank test. Where indicated, *p<0.05, **p<0.01, and ***p<0.001.
Results
CXCR2 deficiency in myeloid cells results in a switch to an antitumor phenotype
C57Bl/6 mice with deletion of Cxcr2 in myeloid cells (CXCR2myeΔ/Δ) were generated as indicated in the Methods. CXCR2myeΔ/Δ mice exhibited an extensively reduced expression of both Cxcr2 mRNA transcript and CXCR2 protein in myeloid cells (Fig. 1A–B). To determine how CXCR2 expression in myeloid cells impacted immunity in an experimental breast cancer metastasis model, we evaluated the lung colonization of syngeneic PyMT cells intravenously injected into CXCR2myeΔ/Δ mice or littermate CXCR2WT mice. Two weeks later, CXCR2WT mice exhibited difficulty in breathing, and mice were euthanized. Tumor weight in CXCR2WT mice reached 468±81 mg. In CXCR2myeΔ/Δ tumor-bearing mice, the tumor weight was 52% lower than in the CXCR2WT tumor-bearing mice (226±107 mg, p<0.001, Fig. 1C–D). A similar result was achieved when these mice were implanted with Rich 1.1 melanoma cells (Supplementary Fig. S1A).
Figure 1. Myeloid Cxcr2-knockout reduces tumor burden.
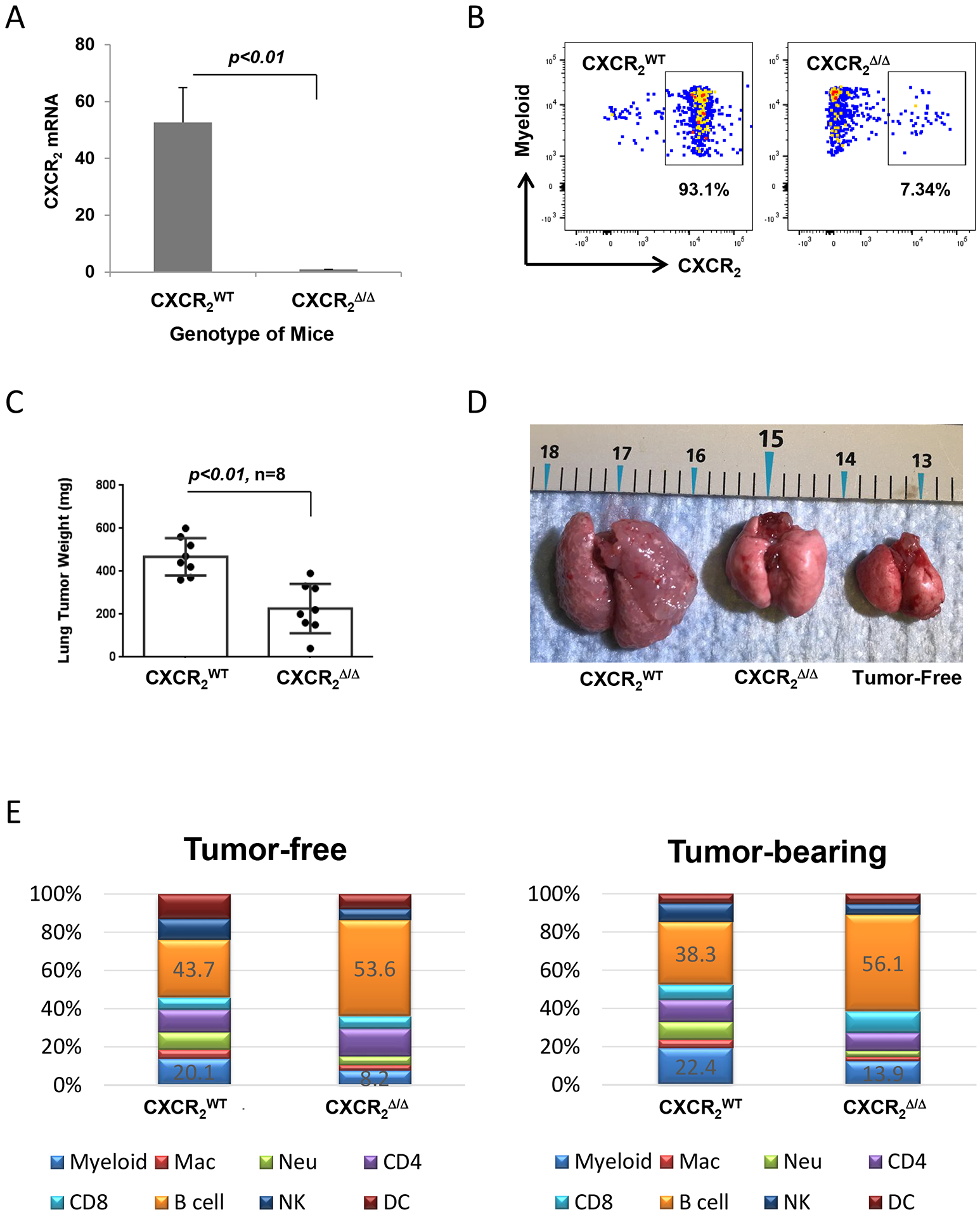
A, qRT-PCR for Cxcr2 mRNA expression in myeloid cells from CXCR2myeΔ/Δ and CXCR2WT mice (n=3). B, Representative plots showing the expression of myeloid CXCR2 protein on myeloid cells by flow cytometry. C, Tumor burden in mice with loss of Cxcr2 in myeloid cells. The metastatic lung tumors were intravenously implanted with PyMT breast cancer cells in CXCR2myeΔ/Δ or littermate CXCR2WT mice (n=8) for two weeks. D, Representative photo of tumor-free or tumor-bearing lungs from CXCR2WT and littermate CXCR2myeΔΔ mice. E, Immune cell profile of peripheral blood from CXCR2myeΔ/Δ and littermate CXCR2WT mice (n=6) that were tumor-free (left) or tumor-bearing (right). Mac: macrophage; Neu: neutrophil. Data were analyzed by two-sample t test with unequal variances for panels (A) and (C); p values indicated. Experiments were repeated, and values represent mean±SD.
To examine whether disruption of CXCR2 signaling altered the immune cell population in blood, we analyzed immune cells from peripheral blood by flow cytometry. Cells with tissue-specific LysM-Cre-mediated deletion of myeloid Cxcr2 were identified based on GFP expression. This analysis showed that the peripheral myeloid cell population was significantly decreased in CXCR2MyeΔ/Δ mice in comparison to that of CXCR2WT mice (Fig. 1E, p<0.001), where both neutrophils (CD11b+Ly6C−Ly6G+) and macrophages were significantly decreased in the CXCR2MyeΔ/Δ mice by 61% (p<0.001) and 52% (p<0.05), respectively. Specifically, B cells were increased in CXCR2MyeΔ/Δ mice by 23% (p<0.001), which coincided with a higher expression of CXCL13 in both serum and tumor tissues (Supplementary Fig. S1B). However, the total number of peripheral CD45+ cells remained unchanged between CXCR2mye Δ/Δ mice and littermate CXCR2WT mice, in both tumor-free and tumor-bearing conditions (Supplementary Fig. S1C). These data suggested a functional link between immune cell alterations and the antitumor phenotype in the CXCR2myeΔ/Δ mouse.
Effects of CXCR2MyeΔ/Δ on PBMCs in PyMT lung tumor-bearing mice
To determine how loss of Cxcr2 expression in myeloid cells would affect the overall immune profile of tumor-bearing mice, PyMT breast cancer cells were intravenously injected into CXCR2MyeΔ/Δ or littermate CXCR2WT mice. One week after injection, the peripheral blood leukocytes were collected and analyzed by flow cytometry as previously performed (23). We observed that the M1 macrophage population (CD11b+F4/80+MHCII+CD206−) in tumor-bearing CXCR2myeΔ/Δ mice was increased by 2.2-fold, in contrast to that of the tumor-bearing CXCR2WT mice (Supplementary Fig. S2A). Compared to CXCR2WT mice, the CXCR2myeΔ/Δ mice showed both Ly6C−Ly6G+ and Ly6G−Ly6C+ myeloid cells decreased by 26% and 22%, respectively (Supplementary Fig. S2B), although CXCR2 expression on M-MDSCs was barely detected (Supplementary Fig. S2C). CD8+ T cells were increased by 21%, and CD4+ T cells were decreased by 29% (Supplementary Fig. S2D). However, the population of effector memory CD4+ T cells (CD3+CD4+CD62L−CD44+) was increased by 66%, although the naïve CD4+ T-cell population frequency was not altered in the CXCR2myeΔ/Δ mice compared to CXCR2WT tumor-bearing mice (Supplementary Fig. S2E). Similar observations were observed in the Rich1.1 melanoma mouse model (Supplementary Fig. S2F–K). Altogether, these data indicated that knockout of Cxcr2 in myeloid cells reprogramed the circulating immune cells toward an antitumor phenotype.
Antitumor microenvironment immunity gained from myeloid Cxcr2 deletion
To examine the effects of Cxcr2 loss in myeloid cells on tumor-infiltrating leukocytes (TILs) in the lungs of CXCR2WT and CXCR2myeΔ/Δ mice, we evaluated the TIL population in the lungs of mice two weeks after they were intravenously implanted with PyMT cells. CD45+ leukocytes in lung tumors were isolated and analyzed by flow cytometry. We demonstrated that compared with CXCR2WT mice, CXCR2myeΔ/Δ mice exhibited a 46% decrease in tumor-infiltrating M2-like (CD206+) macrophages, whereas M1-like macrophages increased by 35% (Fig. 2A). Based on the gating strategy for MDSCs (Supplementary Fig. S1D), G- or M-MDSCs decreased by 36% and 29%, respectively, in CXCR2myeΔ/Δ mice (Fig. 2B). No significant alteration in natural killer (NK) cells expressing CD107a and CD178 was seen (Fig. 2C). Using the gating strategy for the CD4+ T-cell subpopulation (Supplementary Fig. S1E), effector memory CD4+ T cells increased by 29% and naïve or regulatory T cells decreased 22% in CXCR2myeΔ/Δ mice (p<0.01, Fig. 2D). CXCR2myeΔ/Δ mice also had CD11c+MHCII+CD103+ DCs increased by 40% (Fig. 2E). CD107a+CD8+ T cells increased by 49% (p<0.01), and CD69+CD8+ T cells increased by 44% in CXCR2myeΔ/Δ mice (p<0.05, Fig. 2F). However, the total number of CD45+ cells per 100mg lung tumor tissue was not significantly changed between CXCR2mye Δ/Δmice and the littermate CXCR2WT mice (Supplementary Fig. S2L). The similar immune profiles were observed in the Rich1.1 melanoma mouse model (Supplementary Fig. S3A–F). These data suggested that the targeted deletion of CXCR2 in myeloid cells produces a tumor immune environment that is antitumor.
Figure 2. Loss of CXCR2 in myeloid cells alters the immune cell profile of tumor-infiltrating immune cells.
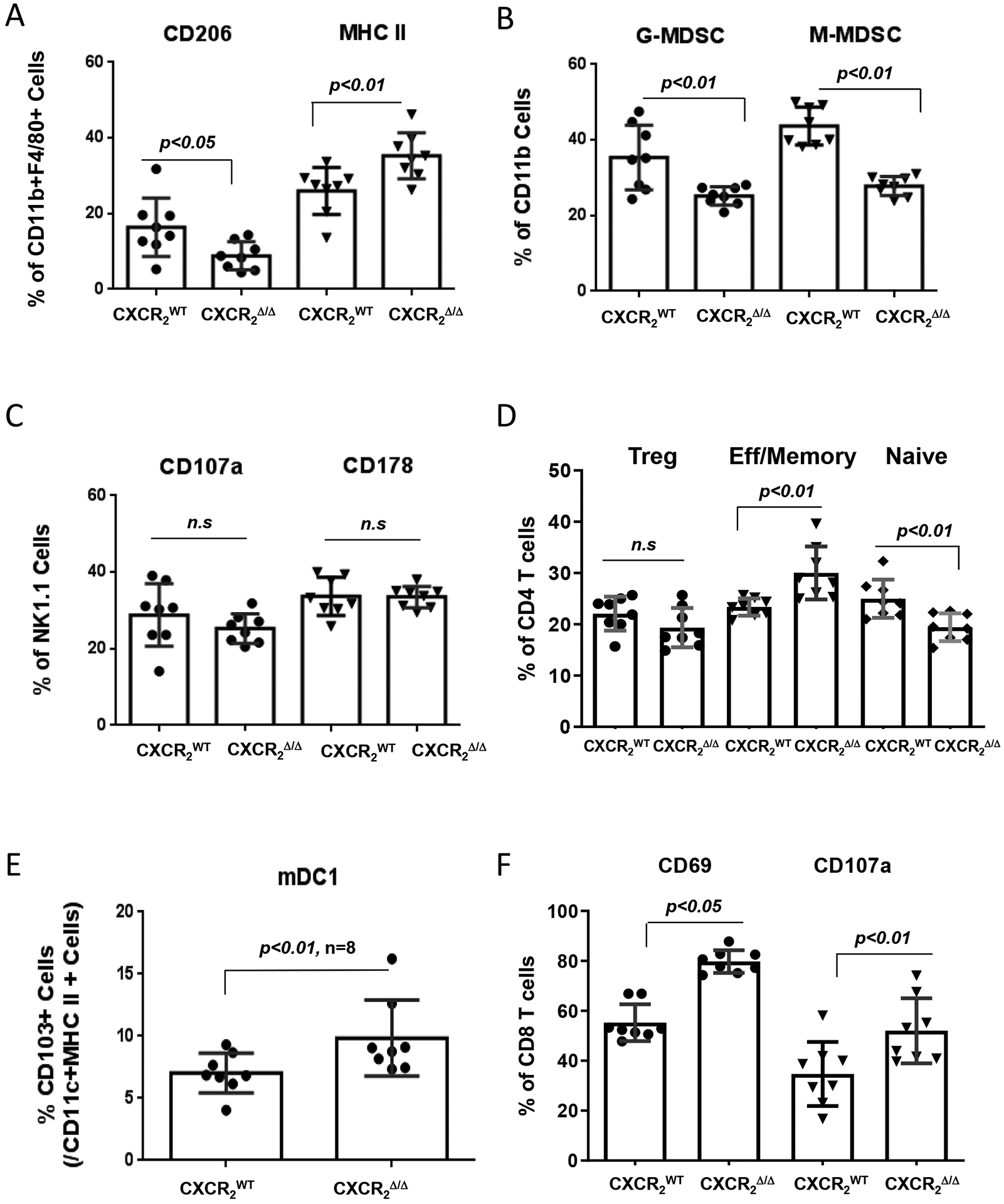
1×106 PyMT breast cancer cells were intravenously injected into CXCR2myeΔ/Δ (n=8) or littermate CXCR2WT mice (n=8). Two weeks after injection, TILs in the lung tumors were stained with markers specific for subsets of immune cells and analyzed by flow cytometry to identify: A, M1 macrophages (MHC IIhi) or M2 (CD206hi); B, neutrophils or granulocytic (G-)MDSCs (CD11b+Ly6C−Ly6G+) or monocytic (M-)MDSCs (CD11b+Ly6G−Ly6C+); C, NK cells (CD45+CD3−NK1.1+); D, CD4+ T cells of effector/memory (CD3+CD62L−CD44+), naïve (CD3+CD44−CD62L+), or Treg (CD3+CD4+CD25hi) lineage; E, mDC1 (CD45+/GFP−/CD11c+/MHC II+/CD103+); and F, alteration of CD8+ T-cell activation markers. Data were analyzed using the two-sample t-test with unequal variances; p values indicated; n.s.: not significant. Experiments were repeated, and values represent mean±SD.
MDSC-mediated CD8+ T-cell cytotoxicity is regulated by CXCR2 signaling
Firstly, to verify whether CD8+T cells were essential for the enhanced antitumor immunity in CXCR2myeΔ/Δ mice, CD8+ T cells were depleted in our model with anti-CD8α (controls given an IgG isotype) prior to implantation of PyMT cells. We observed that mice lacking CD8+ T cells exhibited a 2.1-fold increase in tumor burden, in contrast to the mice with intact CD8+ T cells (tumor weight 896±158 mg vs. 428±155 mg, p<0.01, Fig. 3A–B). However, depletion of CD8+ T cells in tumor-bearing CXCR2WT mice had no significant effect on lung tumor burden (Supplementary Fig. S4A), which is consistent with our previous study showing that CD8+ T-cell depletion in CXCR2WT mice bearing PyMT tumors does not significantly affect tumor growth compared with isotype IgG control mice (20).
Figure 3. Loss of CXCR2 expression in myeloid cells inhibits tumor growth through reduced MDSC activity and enhanced activation of CD8+ T cells.
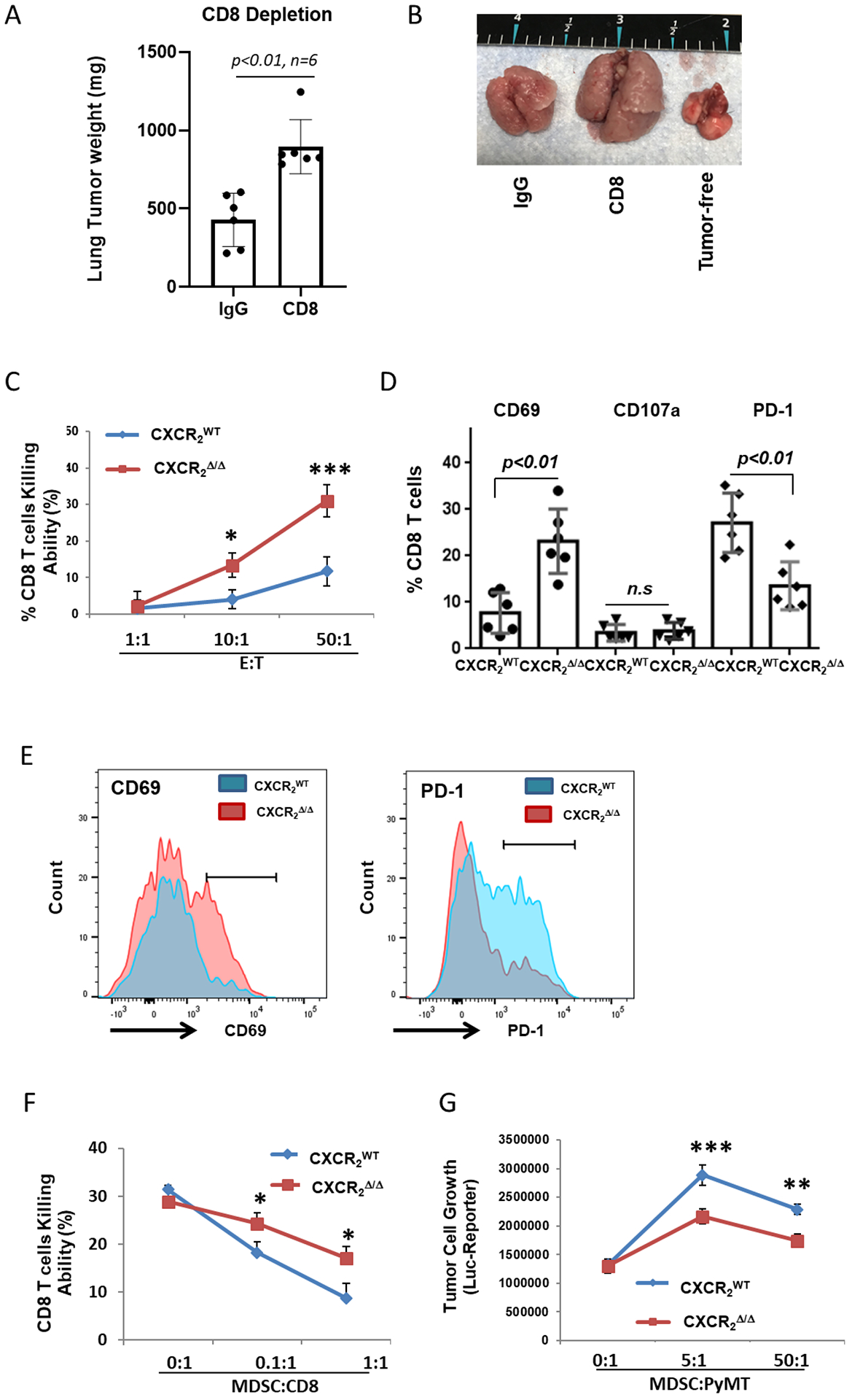
A, Tumor growth in CXCR2myeΔ/Δ mice. CXCR2myeΔ/Δ mice peritoneally treated with CD8a mAb (CD8) (n=6) or control IgG (n=6) daily for the first three days then maintained twice weekly. Three days after antibody treatment, mice were injected intravenously with 1×106 PyMT cells. Three weeks after, the mice were sacrificed, and the weight of the tumor-bearing lung was assessed. B, Representative photo of lungs from tumor-bearing mice treated with CD8 mAb or IgG control. Tumor-free lungs also shown. C, CD8+ T-cell cytotoxicity ex vivo. CD8+ T cells isolated from tumor-bearing lungs of CXCR2myeΔ/Δ and CXCR2WT mice (n=3) as effector cells (E) cocultured with firefly luciferase (luc)-expressing target tumor cells (T) at the indicated E:T ratios. After 18 hours, luciferase activity in the remaining PyMT-luc cells was determined to estimate viable tumor cells and %CD8+ T-cell killing was calculated. D, MDSCs isolated from tumor-bearing lungs of CXCR2myeΔ/Δ or CXCR2WT mice (n=3) were cocultured with CD8+ T cells from lung tumor of CXCR2myeΔ/Δ or CXCR2WT mice at ratio 1:1 for 3 days and then CD8+ T cells were analyzed by flow cytometry. E, Representative histograms of CD8+ T-cell surface markers (cells from D). F, MDSCs from tumor-bearing lungs of CXCR2myeΔ/Δ or CXCR2WT mice and CD8+ T cells from lung tumor of CXCR2myeΔ/Δ mice (n=3) at the indicated ratios were cocultured with PyMT-luc cells. After 3 days, luciferase activity was measured, and %CD8+ T-cell killing was calculated. G, MDSCs from tumor-bearing lungs of CXCR2myeΔ/Δ or CXCR2WT mice were cocultured with PyMT-luc cells at the indicated ratios. After 18 hours, luciferase activity in PyMT-luc cells was determined. Data were analyzed using the two-sample t-test with unequal variances for panels (A) and (D)(p values indicated); two-way ANOVA with a post-hoc test for panels (C), (F), and (G); *p<0.05, **p<0.01, ***p<0.001. Experiments were repeated, and values represent mean±SD.
To evaluate whether myeloid CXCR2/CXCL1 signaling was crucial for CD8+ T-cell toxicity, we isolated CD8+ T cells from the lungs of tumor-bearing mice one week-post PyMT cell implantation, and the ex vivo toxicity of the CD8+ T cells targeting PyMT cells was evaluated by incubating the tumor-derived CD8+ T cells with PyMT luciferase-expressing tumor cells. CD8+ T cells from tumor-bearing CXCR2myeΔ/Δ mice exhibited increased the killing compared to that of the CD8+ T cells from CXCR2WT mice (Fig. 3C). There was an increase in CD8+ T cells expressing the activation marker CD69 and a decrease in CD8+ T cells expressing PD-1. However, no detectable increase in CD8+CD107a+ T cells from CXCR2myeΔ/Δ mice was seen compared to CXCR2WT mice (Fig. 3D–E). To illustrate the interaction between MDSCs and CD8+ T cells, we isolated MDSCs from lung tumors then incubated with CD8+ T cells isolated from the CXCR2myeΔ/Δ bearing metastatic PyMT tumors. After three days of cocultured MDSCs and CD8+T cells at the indicated ratio, the CD8+ T-cell killing capacity was significantly rescued by the MDSCs from CXCR2myeΔ/Δ mice compared to MDSCs from CXCR2WT mice (Fig. 3F). Although CD8+ T cells isolated from CXCR2WT mice bearing metastatic PyMT tumors, we observed only a very limited MDSC inhibitory effect on CD8+ T-cell killing (Supplementary Fig. S4B). Thus, MDSC inhibitory effects on the CD8+ T cells was CXCR2 signal-dependent. To determine the direct effect of Gr1+ MDSCs on PyMT cells, we isolated MDSCs from lungs bearing metastatic PyMT tumors in either CXCR2myeΔ/Δ or littermate CXCR2WT mice. The MDSC cells were cocultured with the target PyMT cells that had been genetically engineered with a luciferase reporter. After overnight coculture, MDSCs from CXCR2WT mice promoted PyMT growth more than MDSCs from CXCR2myeΔ/Δ mice (Fig. 3G). These data suggested that the myeloid CXCR2 signaling axis regulates both MDSC-mediated suppression of CD8+ T-cell responses and promotes tumor cell proliferation.
B cell-derived CXCL11 is involved in CXCR2myeΔ/Δ-mediated CD8+ T-cell activity
To explore what cytokines were present in serum of CXCR2myeΔ/Δ and littermate CXCR2WT mice, we performed a 62-cytokine array assay (Supplementary Table S1). Serum CXCL11 expression was 72-fold higher in the CXCR2myeΔ/Δ mice than in CXCR2WT mice (Fig. 4A, Supplementary Fig. S5A–B). Also, CXCL9 was increased 6-fold in the serum of CXCR2myeΔ/Δ mice. To pursue the source of CXCL11 in the serum of tumor-bearing mice, CXCL11 concentration in the serum, spleen, and tumor tissue lysates of the tumor-bearing mice of the respective genotypes were determined by ELISA. We observed that tumor-bearing lungs were the major source of CXCL11 (Fig. 4B), whereas the spleen and peripheral blood provided minor quantities of CXCL11 (Fig. 4C–D). However, CXCL11 expression was minimal in the lung, spleen, and peripheral blood of tumor-free mice (Supplementary Fig. S5C–E). CXCL11 expression was low in cultured breast cancer and melanoma cell lines (Supplementary Fig. S5F). To further identify the specific immune cells producing CXCL11 in the TME, intracellular CXCL11 expression in TILs was analyzed by flow cytometry. This revealed that B cells had high expression of CXCL11, and B cells from the lungs of tumor-bearing CXCR2myeΔ/Δ mice exhibited a significant increase in CXCL11 compared to lungs from tumor-bearing CXCR2WT mice (Fig. 4E). Thus, tumor-infiltrating B cells were the major CXCL11-producing cell type in the TME.
Figure 4. Disruption of myeloid CXCR2 promotes CXCL11 production.
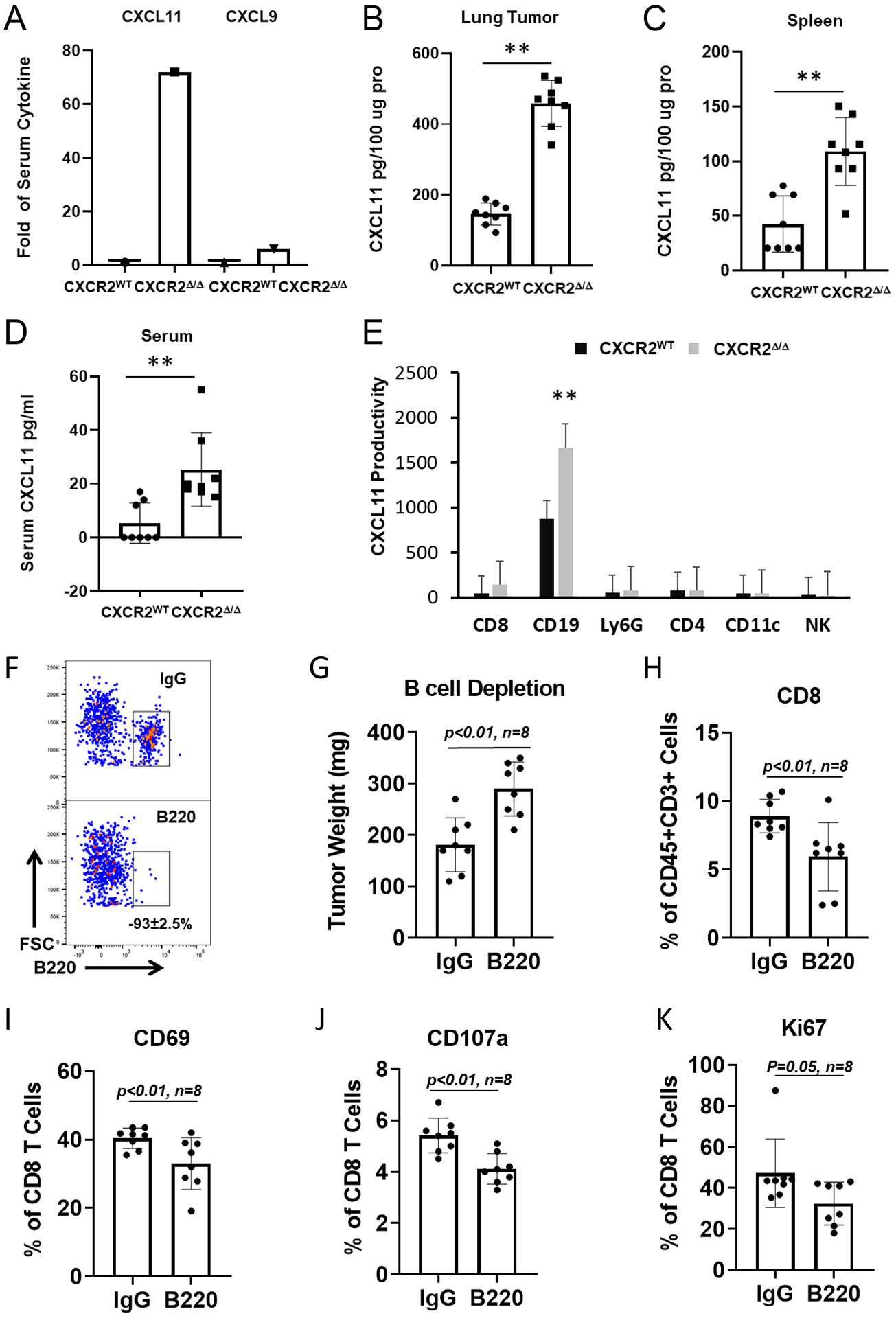
A, 1×106 PyMT breast cancer cells were intravenously injected into CXCR2myeΔ/Δ (n=8) or littermate CXCR2WT mice (n=8). Two weeks after injection, tissue samples were collected. The expression of CXCL11 was quantified using ELISAs to determine CXCL11 in (B) lungs (n=8), (C) spleen (n=8), (D) serum (n=8), and (E) in the indicated immune cells (n=8). F, B-cell depletion in mice (n=8). CXCR2myeΔ/Δ mice (n=8) pre-treated with 200μg B220 mAb (or isotype control IgG) daily for 3 days were intravenously injected with 1×106 PyMT breast cancer cells. The status of peripheral B-cell depletion was monitored for B220+ B cells by flow cytometry. G, 17 days after tumor implantation, tumor lung (n=8) were weighted and subtracted from tumor-free lungs in mice. H, Tumor-infiltrating CD8+ T-cell expression of surface activating markers (I) CD69, (J) CD107a, and (K) proliferation marker Ki67, analyzed by flow cytometry. Data were analyzed by the two-sample t-test with unequal variances; panels (G-K), p values indicated; **p<0.01. Experiments were repeated, and values represent mean±SD.
To verify whether CXCL11-producing B cells were required for CD8+ T-cell antitumor immunity, over 93% of B cells were depleted with a B220 mAb in tumor-bearing mice (Fig. 4F). We observed that growth of metastatic tumors in the lung was significantly increased by 38% in the CXCR2myeΔ/Δ mice depleted of B cells compared to IgG control-treated mice (p<0.01, Fig. 4G). CXCL11 expression in tumor tissue was reduced by 67% in B cell-depleted mice, compared with that of control mice treated with an IgG isotype mAb (Supplementary Fig. S5G). We also observed that depletion of B cells resulted in reduced CD8+ T-cell infiltration into tumors (Fig. 4H), reduced CD8+ T-cell activation (CD69+) and cytotoxicity (CD107a+)(Fig. 4I–J), and reduced CD8+ T-cell proliferation based on Ki67 staining (Fig. 4K). Altogether, these data indicated that tumor-infiltrating B cells may play an essential role in CD8+ T-cell activation within the TME of CXCR2myeΔ/Δ mice.
Myeloid Cxcr2 deletion promotes B1b cell proliferation and CXCL11 production
We observed that 77% of the CXCL11 production in the lungs of tumor-bearing mice was produced by B220+ B cells based on analysis of intracellular CXCL11+ cells in the TME (Fig. 5A, Supplementary Fig. S6A). To obtain an immune profile of the specific B-cell subpopulation, we examined B-cell surface markers (CD19, B220, CD43 or CD5) to identify B-cell subsets such as pre-B, B1, B2 or B1a, B1b (Supplementary Fig. S6B). We observed that in tumor-bearing CXCR2Δ/Δ mice, the infiltrating B cells, particularly the B1 and B1b subpopulations, were significantly increased in the TME (p<0.01, Fig. 5B), as well as in peripheral circulation (Supplementary Fig. S6C). The intracellular CXCL11 expression in subpopulations of B cells was investigated (Supplementary Fig. S6D). Myeloid Cxcr2 deletion resulted in significantly increased CXCL11 expression in the CD5−CD43highB220highCD19+ B1b cell population in the TME (Fig. 5C) and in the peripheral blood of these mice (Supplementary Fig. S6D). Using immunofluorescence confocal microscopy, we verified that the B220 cells expressed CXCL11 (Fig. 5D). CD19+B220−CD43− pre-B cells, as a minor population in the peripheral blood (Supplementary Fig. S6D) but not in the TME (Fig. 5C), had high expression of CXCL11. Altogether, our data suggested that the CXCR2/CXCL1 axis in myeloid cells plays an inhibitory function on either recruitment or expansion of intratumoral CXCL11-producing B cells, and therefore, disruption of CXCR2 in myeloid cells promoted B-cell response and CXCL11 secretion, particularly in the B1b subpopulation.
Figure 5. CXCL11 is differentially produced by B-cell subsets.
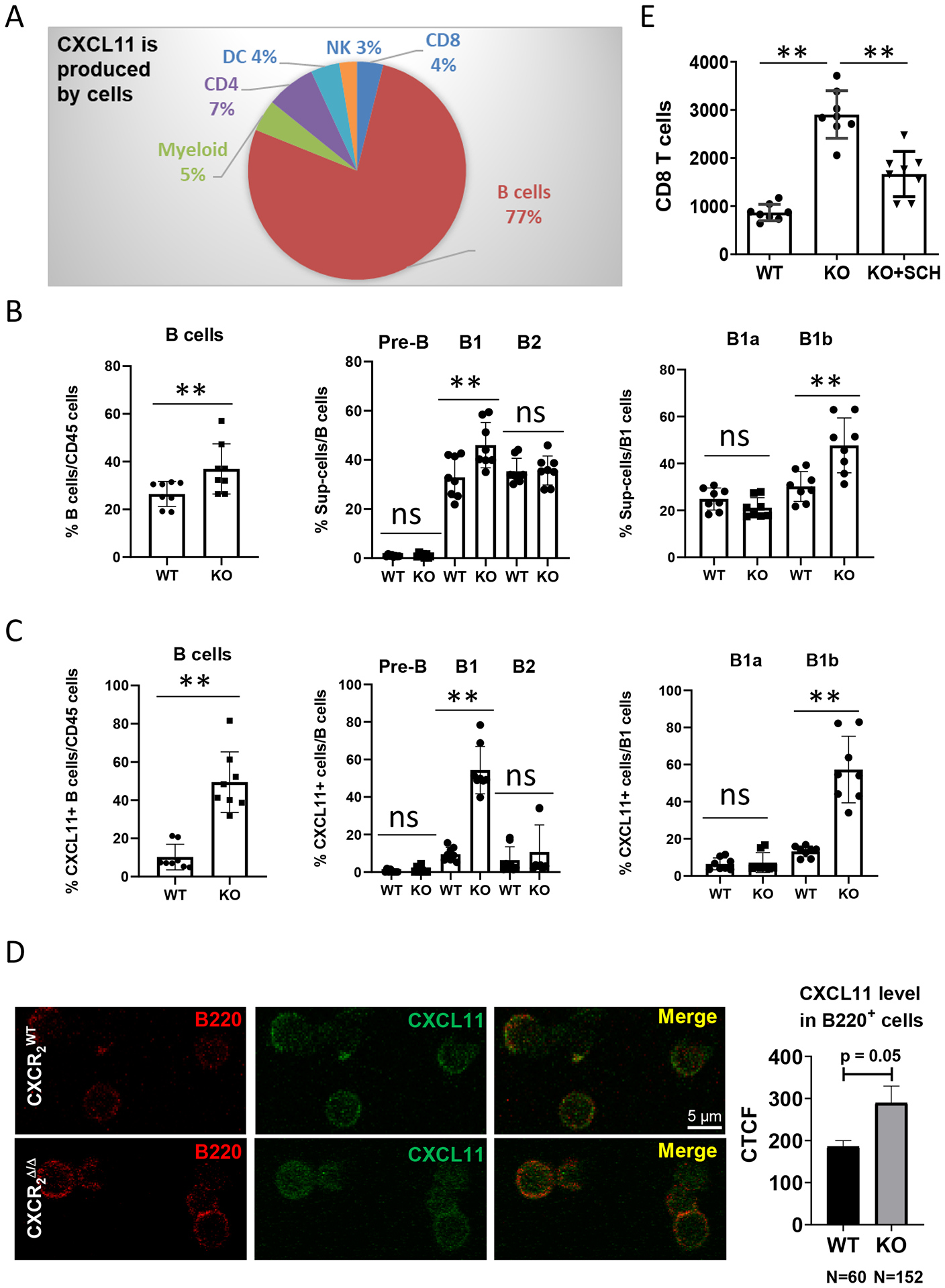
1×106 PyMT breast cancer cells were intravenously injected into CXCR2myeΔ/Δ (n=8) or littermate CXCR2WT mice (n=8). Two weeks after injection, (A) intracellular CXCL11 expression in the indicated intratumoral CD45+ cell subsets was analyzed by flow cytometry. B, Frequencies of B-cell subsets in TME: B cells (CD11b-CD19+B220+); B1 cells (CD19+B220+CD43+); B2 cells (CD19+B220+CD43−); Pre-B cells (CD19+B220−CD43−); B1a cells (CD19+B220+CD5+); and B1b cells (CD19+B220+CD5−). n=8. C, Percentage of CXCL11-expressing subpopulations of B cells. n=8. D, Localization of CXCL11 in B220+ B cells. B cells were isolated from tumor tissues of either CXCR2WT or CXCR2myeΔ/Δ mice and stained for B220 and CXCL11. Expression was visualized through confocal microscopy. Representative micrographs of tumor B cells (red) and intracellular CXCL11 (green). Quantification as corrected total cell fluorescence (CTCF) was performed and are presented as the mean of CXCL11-expressing B220+ B cells. E, 1×106 PyMT breast cancer cells were intravenously injected into CXCR2myeΔ/Δ (KO, n=8) or littermate CXCR2WT mice (WT, n=8). One week after injection, CD8+ T cells and pan B cells were isolated from tumor-bearing lungs, cultured short-term (18h), and conditioned medium was collected. Isolated CD8+ T cells were analyzed for migration toward the B cell-conditioned medium using a Boyden Chamber chemotaxis assay as described in Methods. Shown: number of CD8+ T cells in response to B cell-conditioned medium, or from B cell-conditioned medium from CXCR2myeΔ/Δ mice treated with 20 nM of the CXCR3 antagonist, SCH546738. Data were analyzed using the two-sample t-test with unequal variances for panels (B-C) and using one-way ANOVA with post-hoc test for panel (E); **p<0.01. Experiments were repeated, and values represent mean±SD.
Because CXCL13, a chemoattractant for B cells, was increased in tumors of CXCR2myeΔ/Δ mice (Supplementary Fig. S1B), it is likely that CXCL13 could be recruiting B cells into the TME. To examine whether the secretion of CXCL11 by tumor-infiltrating B cells was responsible for the increased tumor-infiltrating CD8+ T cells (Supplementary Fig. S2D), CD8+ T cells and pan B cells were isolated from lung tumors. After overnight culture of CD8+ T cells in low serum-containing medium, the cells were analyzed for chemotactic response to the supernatant of cultured B cells over an 18-hour incubation period. We observed that the number of migrated CD8+ T cells was significantly increased in the tumor-bearing CXCR2myeΔ/Δ mice compared to the tumor-bearing CXCR2WT mice (p<0.01, Fig. 5E). In order to verify that this was CXCR3 receptor-dependent, 20 nM of the CXCR3 antagonist, SCH546738, was added in the top chamber with CD8+ T cells from the tumor-bearing CXCR2myeΔ/Δ mice. Treatment with the CXCR3 antagonist resulted in a decrease of CD8+ T-cell migration (p<0.01, Fig. 5E). To confirm that CXCL11 was chemotactic to CD8+ T cells, CD8+ T cells from spleens of tumor-bearing mice were exposed to increasing concentrations of CXCL11. A dose-dependent chemotactic response was seen and revealed that CXCL11 induced chemotaxis of CD8+ T cells (p<0.001, Supplementary Fig. S6E). Thus, data suggested that intratumoral B cells recruit CD8+ T cells in a CXCR3/CXCL11-dependent manner.
To learn whether tumor implantation influenced the B-cell ratio in the lungs, flow cytometry of the B-cell subpopulation was performed on cell suspensions of lung from tumor-free or -bearing mice. The B1a subpopulation was significantly increased by the tumor implantation (p<0.01) in both CXCR2WT (Supplementary Fig. S6F) and CXCR2myeΔ/Δ mice (Supplementary Fig. S6G). These data supported our model that Cxcr2 deletion in myeloid cells was specific to the regulation of B1b cells, whereas induction of B1a cells was related to tumor implantation/growth, but not CXCR2 deletion-dependent. Tumor-dependent induction of B1a cells is considered as a regulatory mechanism by which B cells (B1a cells) deliver IL10 to the TME and affect the CD8+ T-cell response in melanoma (24). We observed that B cells did not undergo chemotaxis in response to CXCL11, ruling out the possibility that B cells were being recruited into the tumor through a CXCL11 autocrine loop (Supplementary Fig. S6H–I).
Systemic CXCR2 antagonist enhances the antitumor activity of CD8+ T cells
A selective small molecule CXCR1 and CXCR2 antagonist, SX-682, is currently in clinical trials for melanoma treatment in combination with anti-PD-1 (NCT03161431). This targeted therapy is expected to inhibit MDSC accumulation in the TME, which is associated with a tolerant immune microenvironment, providing a growth advantage to the tumor (3,5). To investigate the potential effect of SX-682 on metastatic tumor progression in vivo, immunocompetent C57Bl/6 mice were subcutaneously implanted with syngeneic Rich 1.1 cells. Mice were fed with chow containing SX-682 (3.023g/kg) ± intraperitoneal (IP) delivery of anti-PD-1 (100μg/mouse) or control IgG, or vehicle control chow without SX-682 + IP delivery of anti-PD-1 (100μg/mouse) or isotype IgG every other day. After three weeks of treatment with the SX-682 or vehicle chow, tumor burden was reduced 63% in the mice fed SX-682 chow compared with vehicle control-treated animals (p<0.05, Fig. 6A), whereas tumor volume in the group treated with anti-PD-1 alone was not significantly different than the vehicle control. However, co-treatment with SX-682 and anti-PD-1 significantly reduced tumor burden compared to vehicle control (p<0.001) and to SX-682 alone (p<0.05, Fig. 6A). There was no evidence of toxicity or weight loss in association with treatments (Supplementary Fig. S6J). Analysis of the leukocytes in the tumor revealed that mice fed the CXCR2 antagonist exhibited an increase in the density of B cells, particularly the B1b population (p<0.01, Fig. 6B–C), enhanced CD8+T cells and CD8+CD69+ activated T effector-cell density (p<0.01, Fig. 6D–E), and decreased Gr1+MDSC frequency in the TME (p<0.01, Fig. 6F–G). These changes were similar to the changes induced by genetic knockout of Cxcr2 in myeloid cells (Fig. 2B, Fig. 5B). Both myeloid CXCR2 deletion and systemic CXCR2 antagonism resulted in an elevation of effector CD8+ T cells in the TME (p<0.01). However, the addition of anti-PD-1 did not elevate the number of effector CD8+ T cells in the TME over that observed with the SX-682 alone (Fig. 6D–E), although the combination treatment revealed significantly more CD8+ T cells than anti-PD-1 treatment alone (p<0.01, Fig. 6D–E). These data suggested that treatment with the CXCR2 antagonist, SX-682, results in enhanced CD8+ T-cell recruitment to the tumor and when combined with anti-PD-1, can enhance antitumor immunity. Thus, this combination therapy may prove to be promising for clinical application in advanced-stage melanoma patients.
Figure 6. CXCR2 antagonist, SX-682, inhibits tumor growth and activates CD8+ T cells.
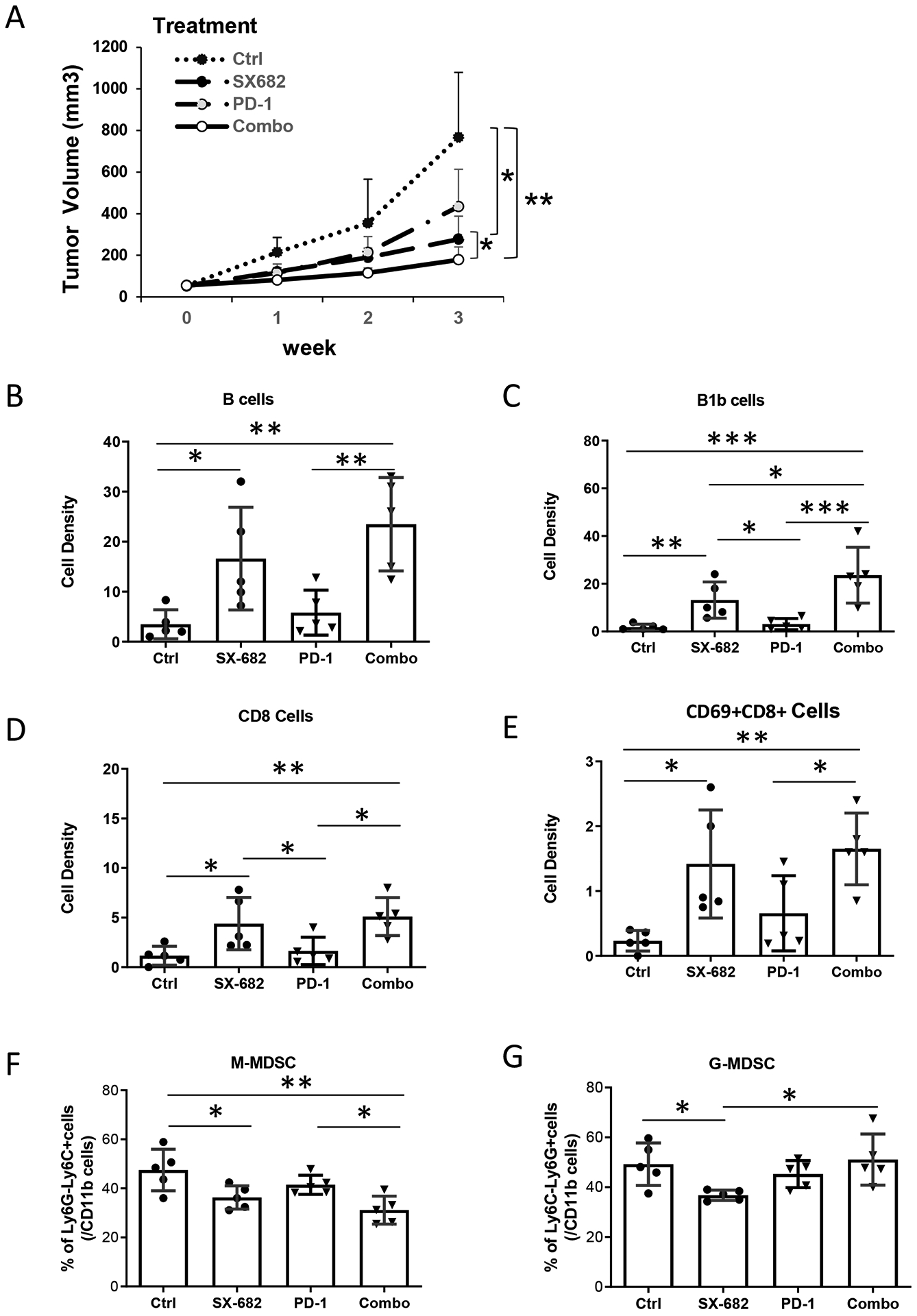
C57/BL6 mice (5 mice/group) were allowed free access to chow containing SX-682 or vehicle control from one week prior to subcutaneous implantation with Rich1.1 melanoma cells (1×106), and thereafter for the duration of the experiment. When tumor size reached 50 mm3, mice fed with SX-682 or vehicle chow were treated with or without IP injection of 100 μg anti-PD-1 or IgG isotype control every other day for a total of 26 days. A, Tumor growth was monitored weekly for three weeks. Mice were then euthanized and single-cell suspensions of tumors were prepared for flow cytometry. B, B cells (CD45+CD11b−CD19+B220+); C, B1b cells (CD19+B220+CD5−); D, CD8+ T cells (CD45+CD3+CD8+). E, Activated CD8+ T-cell (CD8+CD69+) density (cell number per mg tissue) in the TME. F, M-MDSCs or G, G-MDSC in the tumor as a percent of intratumoral CD11b+ cells. Data were plotted and analyzed by the mixed-effect model for panel (A) and by the Wilcoxon rank-sum test for panels (B-G); *p<0.05, **p<0.01, ***p<0.001. Data values represent mean±SD.
B220 and CXCL11 expression positively correlate with a better clinical outcome
To explore whether the B cell-CXCL11 axis has a potential role in the prediction of survival in breast cancer, we queried TCGA datasets. Upon examination of the somatic DNA copy number alterations (CNA) of B220 in a cohort of 1064 available patients (Fig. 7A), we observed that although B220-deletion was rare (~2%), gain (~64%) and amplification (~11%) of B220 were common. A clear trend of increased percent survival and median survival of breast cancer patients with increased B220 CNAs was observed. Although patients who were diploid for B220 had an estimated median survival of 120.53 months, patients with B220-amplification showed significantly better overall survival (OS), with >70% of patients surviving more than 255.49 months. Consistent with our murine data, B220 and CXCL11 mRNA expression were tightly associated in breast cancer patients (p<0.0001, Spearman r=0.6699). The changes of B220 and CXCL11 in breast tumors were positively associated with CD8α mRNA expression (p<0.0001, Spearman r=0.7988), suggesting a potential linkage between CXCL11-producing B cells with antitumoral T-cell responses. The observation was consistently observed in all 11 breast cancer studies published in GEO-NCBI databases (Supplementary Table S2, n=1678). To validate whether our observation was breast cancer-specific, we further analyzed the melanoma-TCGA dataset (Fig. 7B, n=358). We confirmed that over 55% of melanoma patients exhibited an increased (either gain or amplification) copy number of B220. Although melanoma patients’ diploid for B220 had an estimated median survival of 103.12 months, patients with B220-amplification and B220-deletion exhibited a median survival of 246.85 and 53.48 months, respectively. As expected, the mRNAs of B220, CXCL11, and CD8α positively correlated with one another in melanoma tumors. Altogether, these data suggested that CXCL11-producing B220+ B cells have a significant biological impact on the pathogenesis of breast cancer and melanoma patients.
Figure 7. Higher intratumoral expression of B220 is positively correlated with CXCL11 expression in cancer patients with a better clinical outcome.
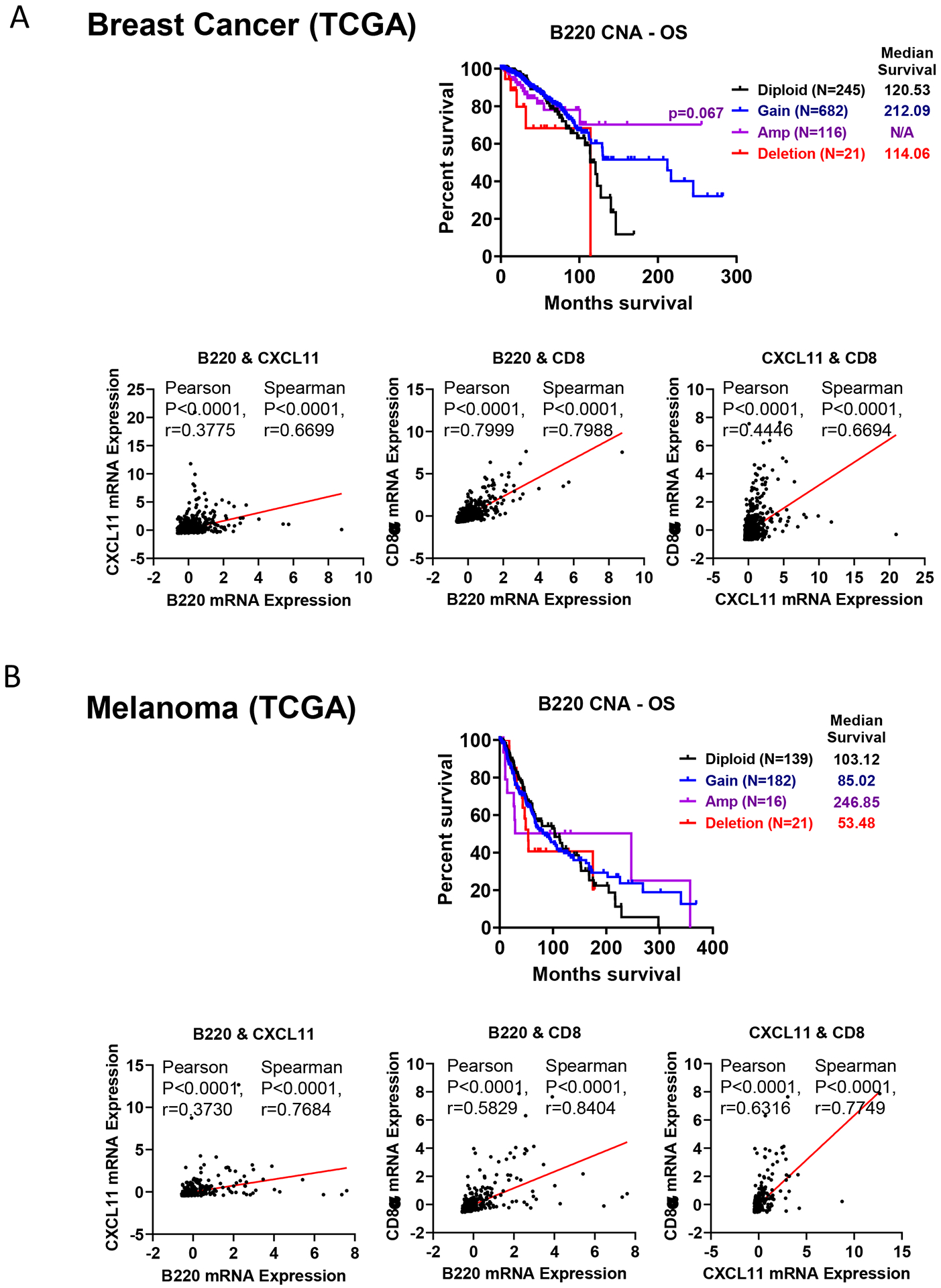
The somatic DNA copy number alterations (CNAs) of B220 were obtained through The Cancer Genome Atlas (TCGA) from cBioportal for Cancer Genomics. Microarray and RNA sequencing-based gene expression profiling of B220, CXCL11, and CD8α were obtained to further identify molecular signatures associated with overall survival of patients with (A) breast cancer (n=1064) and (B) melanoma (n=358). Log-rank (Mantel-Cox) test and the defined median survival months (MS) are indicated if determined. Spearman correlation analyses of mRNA expression of B220 with CXCL11 and CD8α are also shown.
Discussion
Survival of cancer cells in the host depends on escape from host immunosurveillance. A growing body of evidence supports the concept that MDSCs contribute to cancer immune evasion via suppressing functions of CD8+ T and NK cells (1). MDSCs in peripheral blood correlates well with clinical cancer stage and metastatic tumor burden in cancer patients (25). The factors that modulate the movement of MDSCs from the vasculature into the TME, and the mechanism by which MDSCs suppress CD8+ effector T-cell function have been examined (5,26). Our findings not only uncovered a role for CXCR2-mediated recruitment of MDSCs in the inhibition of CD8+ T-cell function, but also revealed a role for CXCR2-expressing myeloid cells in the regulation of recruitment of CXCL11-expressing B cells into the tumor, thus influencing the recruitment of cytotoxic CD8+ T cells.
In most experimental tumor models, MDSCs are expanded in the TME (3). The crucial immune function of CXCR2 is not only to regulate myeloid mobilization from bone marrow to peripheral circulation (8), but also to maintain the survival/self-renewal of hematopoietic stem/progenitor cells (27). In mice, CXCR2 is a shared cellular receptor for the murine CXC cytokine ligands KC (hCXCL1) and MIP-2 (hCXCL2/3), ENA-78/LIX (hCXCL5), NAP-2 (hCXCL7), and GCP-2 (hCXCL6)(28–31). CXCL1 is overexpressed by both human tumor cells (7) and murine tumor cells, including the PyMT breast cancer cells and melanoma cells used in this study. The tumor-derived chemokines that bind CXCR2 had a major role in the recruitment of bone marrow-derived myeloid cells to the tumor site. Even in tumor-free mice, obstructing CXCR2 expression in myeloid cells resulted in a decline in the peripheral blood myeloid population. In contrast, B cells were elevated in the both CXCR2myeΔ/Δ tumor-free and tumor-bearing mice, while in tumor-bearing mice, both the G- and M-MDSC subpopulations were significantly reduced in the peripheral blood and the TME. CXCR2 regulated the influx of M-MDSCs indirectly by inhibiting the differentiation of the hematopoietic stem cells, and macrophage and dendritic cell progenitor cells that give rise to M-MDSCs (27,32). We showed here that MDSC-associated inhibition of CD8+ T-cell activity was reduced in both breast cancer and melanoma models following targeted deletion of Cxcr2 in myeloid cells.
Crosstalk between immune and cancer cells takes place by direct cell-cell interactions and by cytokine regulation (33,34). Studies focused on MDSCs show that MDSCs promote tumor progression, metastasis, and immune escape by suppressing antitumor immune responses in the tumor stroma and in the pre-metastatic niche (1,11,16,35). Our studies indicated that MDSCs not only directly suppressed the tumor-killing function of CD8+ T cells, but also enhanced the growth of tumor cells. In the mouse models described in this report, tumor-produced ligands for CXCR2 were critical mediators in the activation of the myeloid CXCR2 signal cascade, which was associated with suppression of the function of tumor-infiltrating CD8+ T cells. Using a LysM-Cre-mediated targeted deletion of Cxcr2 in myeloid cells, we observed that CD8+ T cells isolated from tumors in CXCR2myeΔ/Δ mice exhibited enhanced tumor cell killing capacity when incubated with MDSCs from CXCR2myeΔ/Δ mice compared to MDSCs from CXCR2WT mice. In contrast, when CD8+ T cells were isolated from tumors of CXCR2WT mice, we observed only a 10% tumor cell killing capacity and no significant differences were observed when these CD8+ T cells were incubated with MDSCs isolated from CXCR2WT versus CXCR2myeΔ/Δ mice. Although we cannot rule out the possibility that there were some small differences in perforin, granzyme, and IFNγ expression in CD8+ T cells cocultured with MDSCs from CXCR2WT versus CXCR2myeΔ/Δ mice, these differences, where they existed, did not alter the killing capacity of the CD8+ T cells isolated from the tumors growing in CXCR2WT mice.
There was an increase in CXCL13 in blood and tumors from CXCR2myeΔ/Δ mice. This CXCL13 likely interacts with CXCR5 expressed on the B cells (36) to promote B-cell chemotaxis (37,38). This resulted in an increase in intratumoral B cells (CD19+B220+) in tumor-bearing CXCR2myeΔ/Δ mice. Specifically, we observed that the B1/ B1b subpopulation of B cells was significantly increased in peripheral blood and in the tumors of CXCR2myeΔ/Δ mice. These B1 and B1b cells expressed CXCL11, a chemokine expressed in multiple cell types that is predominantly induced by IFNγ (39).
In the mouse model of myeloid Cxcr2 knockout reported here, in naïve, non-tumor bearing mice, expression of G-CSF increased, and the expression of the CX3C chemokine fractalkine decreased, but IFNγ expression was not altered in peripheral blood. However, in tumor-bearing mice, CXCL11 and leptin had high expression in both peripheral blood and in tumors. The chemokine receptor for CXCL11, CXCR3, binds CXCL9, CXCL10, and CXCL11. The affinity of CXCL11 for CXCR3 is the highest of the three ligands, followed by CXCL10 and CXCL9 (40). Therefore, CXCL11 may play a major role in the recruitment of cytotoxic CXCR3-expressing CD8+ T cells into the TME in the models studied here. Our demonstration that disruption of CXCR2 signaling in myeloid cells resulted in alterations in B-cell subpopulations with enhanced expression of CXCL11. Analysis of TCGA datasets revealed a positive correlation between intratumoral B cells (based on B220 expression) and CXCL11 expression, and this correlated positively with overall survival for both breast cancer and melanoma patients. Evidence linking tumor-infiltrating B cells to clinical outcome have been reported previously, showing a correlation with improved prognosis in primary melanoma, but also enhanced metastasis in squamous sarcoma (10,11,41). Thus, the nature of the TME and the type of infiltrating B cells may be linked to survival outcomes.
In conclusion, the myeloid CXCR2 signaling axis had a pro-tumor role by controlling the accumulation of MDSCs in the TME. Targeting of CXCR2 in myeloid cells reduced the infiltration of MDSCs in the tumor, increased cytotoxic CD8+ T-cell activity in the tumor, and resulted in intratumoral elevations in B cell-derived CXCL11, and thus enhanced recruitment of CD8+ T cells into the TME. These data suggest that systemic inhibition of CXCR2 may contribute to antitumor activity in combination with therapies that target tumor cells but coincidently increase the recruitment of MDSCs into the tumor. Currently, three clinical trials are ongoing using CXCR2 or CXCR1/CXCR2 antagonists in cancer patients: (i) AZD5069 (CXCR2 antagonist) in patients with metastatic castration resistant prostate cancer (NCT03177187); (ii) SX-682 (CXCR1/CXCR2 inhibitor) in subjects with myelodysplastic syndrome with disease progression or intolerance to prior therapy (NCT04245397); and (iii) SX-682 plus anti-PD-1 (pembrolizumab) in stage III and stage IV melanoma patients (NCT03161431). It will be interesting to see how blocking CXCR2 affects tumor growth and survival in the three ongoing trials and to sort out mechanisms by which any observed effects occur.
Supplementary Material
Acknowledgements
We would like to acknowledge the contributions of Dr. Rebecca Shattuck-Brandt and Ms. Kerry W. Vazquez for their administrative support and Dean Maeda at Syntrix for supplying the CXCR2 antagonist. We are thankful for grant support from the NCI including: R01CA34590 (AR), R01CA243326 (AR), R01CA116021 (AR), P30CA06845 (CCSG Core Facilities Support), V1R37CA233770-01 (AEV); and funding from the Department of Veterans Affairs 101BX002301 (AR), VA SRCS Award (AR).
References
- 1.Gabrilovich DI, Nagaraj S: Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009;9:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S et al. : The terminology issue for myeloid-derived suppressor cells. Cancer Res 2007;67:425; author reply 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI: Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 2008;181:5791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI: Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol 2012;91:167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gabrilovich DI: Myeloid-derived suppressor cells. Cancer Immunol Res 2017;5:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S et al. : Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell 2017;32:654–68 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang J, Richmond A: Constitutive IkappaB kinase activity correlates with nuclear factor-kappaB activation in human melanoma cells. Cancer Res 2001;61:4901–9. [PubMed] [Google Scholar]
- 8.Eash KJ, Greenbaum AM, Gopalan PK, Link DC: CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest 2010;120:2423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma B, Nannuru KC, Varney ML, Singh RK: Host Cxcr2-dependent regulation of mammary tumor growth and metastasis. Clin Exp Metastasis 2015;32:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT et al. : Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res 2012;72:1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garg K, Maurer M, Griss J, Bruggen MC, Wolf IH, Wagner C et al. : Tumor-associated B cells in cutaneous primary melanoma and improved clinical outcome. Hum Pathol 2016;54:157–64. [DOI] [PubMed] [Google Scholar]
- 12.Affara NI, Ruffell B, Medler TR, Gunderson AJ, Johansson M, Bornstein S et al. : B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell 2014;25:809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D et al. : Hif1a deletion reveals pro-neoplastic function of B cells in pancreatic neoplasia. Cancer Discov 2016;6:256–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D et al. : Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015;521:94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woo JR, Liss MA, Muldong MT, Palazzi K, Strasner A, Ammirante M et al. : Tumor infiltrating B-cells are increased in prostate cancer tissue. J Transl Med 2014;12:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Visser KE, Korets LV, Coussens LM: De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell 2005;7:411–23. [DOI] [PubMed] [Google Scholar]
- 17.Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M: B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010;464:302–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Li M, Spangler LC, Spear C, Veenstra M, Darnall L et al. : Functional defect of peripheral neutrophils in mice with induced deletion of CXCR2. Genesis 2013;51:587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pickup MW, Hover LD, Polikowsky ER, Chytil A, Gorska AE, Novitskiy SV et al. : BMPR2 loss in fibroblasts promotes mammary carcinoma metastasis via increased inflammation. Mol Oncol 2015;9:179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sai J, Owens P, Novitskiy SV, Hawkins OE, Vilgelm AE, Yang J et al. : PI3K inhibition reduces mammary tumor growth and facilitates antitumor immunity and anti-PD1 responses. Clin Cancer Res 2017;23:3371–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Hawkins OE, Barham W, Gilchuk P, Boothby M, Ayers GD et al. : Myeloid IKKbeta promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Res 2014;74:7274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Splittgerber R, Yull FE, Kantrow S, Ayers GD, Karin M et al. : Conditional ablation of Ikkb inhibits melanoma tumor development in mice. J Clin Invest 2010;120:2563–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Kumar A, Vilgelm AE, Chen SC, Ayers GD, Novitskiy SV et al. : Loss of CXCR4 in myeloid cells enhances antitumor immunity and reduces melanoma growth through NK cell and FASL mechanisms. Cancer Immunol Res 2018;6:1186–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi T, Oishi K, Okamura A, Maeda S, Komuro A, Hamaguchi Y et al. : Regulatory B1a cells suppress melanoma tumor immunity via IL-10 production and inhibiting T helper type 1 cytokine production in tumor-infiltrating CD8(+) T cells. J Invest Dermatol 2019;139:1535–44 e1. [DOI] [PubMed] [Google Scholar]
- 25.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ: Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother 2009;58:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V: Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012;12:253–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sinclair A, Park L, Shah M, Drotar M, Calaminus S, Hopcroft LE et al. : CXCR2 and CXCL4 regulate survival and self-renewal of hematopoietic stem/progenitor cells. Blood 2016;128:371–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bozic CR, Gerard NP, von Uexkull-Guldenband C, Kolakowski LF Jr., Conklyn MJ, Breslow R et al. : The murine interleukin 8 type B receptor homologue and its ligands. Expression and biological characterization. J Biol Chem 1994;269:29355–8. [PubMed] [Google Scholar]
- 29.Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI: Chemokine binding and activities mediated by the mouse IL-8 receptor. J Immunol 1995;155:2158–64. [PubMed] [Google Scholar]
- 30.Lazennec G, Richmond A: Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol Med 2010;16:133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rossi D, Zlotnik A: The biology of chemokines and their receptors. Annu Rev Immunol 2000;18:217–42. [DOI] [PubMed] [Google Scholar]
- 32.Han X, Shi H, Sun Y, Shang C, Luan T, Wang D et al. : CXCR2 expression on granulocyte and macrophage progenitors under tumor conditions contributes to mo-MDSC generation via SAP18/ERK/STAT3. Cell Death Dis 2019;10:598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanahan D, Coussens LM: Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012;21:309–22. [DOI] [PubMed] [Google Scholar]
- 34.Korkaya H, Liu S, Wicha MS: Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest 2011;121:3804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M et al. : Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008;13:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Legler DF, Loetscher M, Roos RS, Clark-Lewis I, Baggiolini M, Moser B: B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J Exp Med 1998;187:655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ansel KM, Harris RB, Cyster JG: CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity 2002;16:67–76. [DOI] [PubMed] [Google Scholar]
- 38.Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD et al. : A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature 2000;406:309–14. [DOI] [PubMed] [Google Scholar]
- 39.Metzemaekers M, Vanheule V, Janssens R, Struyf S, Proost P: Overview of the mechanisms that may contribute to the non-redundant activities of interferon-inducible CXC chemokine receptor 3 ligands. Front Immunol 2017;8:1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weng Y, Siciliano SJ, Waldburger KE, Sirotina-Meisher A, Staruch MJ, Daugherty BL et al. : Binding and functional properties of recombinant and endogenous CXCR3 chemokine receptors. J Biol Chem 1998;273:18288–91. [DOI] [PubMed] [Google Scholar]
- 41.Martinez-Rodriguez M, Thompson AK, Monteagudo C: A significant percentage of CD20-positive TILs correlates with poor prognosis in patients with primary cutaneous malignant melanoma. Histopathology 2014;65:726–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.