

Abstract
Rationale:
The cardiac sodium channel Nav1.5 has a fundamental role in excitability and conduction. Previous studies have shown that sodium channels cluster together in specific cellular subdomains. Their association with intracellular organelles in defined regions of the myocytes, and the functional consequences of that association, remain to be defined.
Objective:
To characterize a subcellular domain formed by sodium channel clusters in the crest region of the myocytes, and the subjacent subsarcolemmal mitochondria (SSM).
Methods and Results:
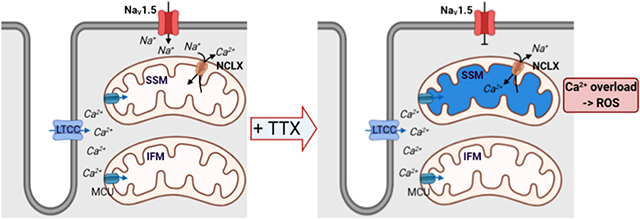
Through a combination of imaging approaches including super-resolution microscopy and electron microscopy we identified, in adult cardiac myocytes, a Nav1.5 subpopulation in close proximity to SSM; we further found that SSM preferentially host the mitochondrial Na+/Ca2+ exchanger (NCLX). This anatomical proximity led us to investigate functional changes in mitochondria resulting from sodium channel activity. Upon TTX exposure, mitochondria near Nav1.5 channels accumulated more Ca2+ and showed increased ROS production when compared to interfibrillar mitochondria. Finally, crosstalk between Nav1.5 channels and mitochondria was analyzed at a transcriptional level. We found that SCN5A and SLC8B1 (which encode Nav1.5 and NCLX, respectively) are negatively correlated both in a human transcriptome dataset (GTEx) and in human-induced pluripotent stem cell-derived cardiac myocytes deficient in SCN5A.
Conclusions:
We describe an anatomical hub (a couplon) formed by sodium channel clusters and SSM. Preferential localization of NCLX to this domain allows for functional coupling where the extrusion of Ca2+ from the mitochondria is powered, at least in part, by the entry of sodium through Nav1.5 channels. These results provide a novel entry-point into a mechanistic understanding of the intersection between electrical and structural functions of the heart.
Keywords: Nav1.5/ subsarcolemmal mitochondria/ NCLX/ mitochondrial homeostasis, sodium channels, mitochondria, Na+/Ca2+ exchange
Subject Terms: Basic Science Research, Cell Biology/Structural Biology, Ion Channels/Membrane Transport, Metabolism
Graphical Abstract
INTRODUCTION
The voltage-gated sodium channel (Na-channel) is central to cardiac electrogenesis. Its dysfunction can lead to lethal arrhythmias both in acquired (ischemia;1, 2 heart failure2), and genetic disorders.3-5 Rare and common genetic variants in SCN5A, encoding the pore-forming Na-channel subunit Nav1.5, are strongly associated with life-threatening arrhythmias4, 6 and with structural heart disease.7-9 Yet, insight into Na-channel function, particularly as it extends beyond electrogenesis, is limited.
Na-channels form macromolecular complexes that cluster at specific subcellular domains (or “pools”).10-12 An emerging concept is that certain Na-channel partners localize only to one subcellular domain, thus endowing the functional complex with region-specific properties that, if disrupted, facilitate arrhythmias.13-17
Subcellular organelles such as mitochondria also organize in region-specific subdomains. Mitochondria in myocytes are highly organized, spatially separated into at least three subpopulations: one just beneath the surface sarcolemma (subsarcolemmal mitochondria, SSM), a second one (the largest fraction) between myofibrils in longitudinal chains (interfibrillar mitochondria, IFM), and a third one, occupying the perinuclear subdomain (PNM). Data show that these subpopulations possess distinct biochemical properties, morphology, Ca2+ handling, and that they may interact differently with other intracellular structures, causing functional specificity.18,19
The relationship between Na-channel and mitochondrial function is well established in one direction only: mitochondrial function and/or molecular composition can alter sodium current (INa).20,21 Recently, however, Zhang et al demonstrated that reduced levels of Scn5a expression lead to accumulation of myocardial reactive oxygen species (ROS),22 and Wan et al documented that expression of Nav1.5 mutant F1759A leads to mitochondrial injury.23 The mechanism by which sodium channel expression or activity can affect mitochondrial function or integrity is not known.
We investigated the anatomical and functional relation between mitochondria and Nav1.5 in adult cardiac myocytes. Advanced imaging methods allowed us to observe that a subpopulation of membrane-resident Nav1.5 clusters sits in close apposition to subjacent SSM, and that it is in this mitochondrial subpopulation where the mitochondrial Na+/Ca2+ exchanger (NCLX; encoded by SLC8B1) is primarily located. This anatomical proximity led us to investigate functional changes in mitochondria resulting from Na-channel activity. In particular, exposing adult cardiac myocytes to the Na-channel blocker tetrodotoxin (TTX) caused accumulation of mitochondrial Ca2+ and ROS production, predominantly in SSM. We speculate that proximity to functional Nav1.5 clusters facilitates sodium-dependent activity of NCLX to extrude mitochondrial Ca2+, whereas reduced sodium entry impairs Ca2+ extrusion. In addition, we observed that the relation between Nav1.5 and NCLX is not only local. Indeed, human transcriptome analysis shows that SCN5A negatively correlates with SLC8B1. Experimental confirmation was obtained from human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM) in which SCN5A was knocked down. We conclude that not only mitochondrial metabolism can affect INa but Na-channels can, in turn, positively affect mitochondrial function. Our data lead us to propose the existence of a Na-channel subpopulation that clusters with subjacent mitochondria to create an anatomical couplon for electro-metabolic coupling.
METHODS
Data Availability.
The authors declare that all supporting data are available within the article and its Online supplement files.
Expanded Methods available in Online Data Supplement.
Mice.
Wild-type black mice C57BL/6N strain 027 (Charles River Laboratories) were treated in accordance to the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health. Procedures were approved by the NYU IACUC committee, protocol 160726. Both genders were included and animals were between 3-6 months of age.
Focused Ion Beam-Scanning Electron Microscopy (FIB-SEM).
Protocol modified from Wilke et al. 24 Tissue was processed and en bloc lead staining performed to enhance membrane contrast.25 Acquisition was done using Auto Slice and View G3 software.26
Inmunostaining.
For immunolocalization, HL-1 cells were incubated with Mitotracker and NCLX antibody, as described in Online Supplement Data.
Cardiomyocyte dissociation.
Adult mouse ventricular myocytes were obtained by enzymatic dissociation, as described in Online Supplement Data.
Single-molecule localization microscopy (SMLM) by stochastic optical reconstruction microscopy (STORM).
Ventricular cardiomyocytes were incubated with Mitotracker before fixation and immunostained with NCLX, Nav1.5 and α-actinin antibodies. Interaction factor analysis (details in Supplemental Data) provided a measure of the degree to which mitochondria and Nav1.5 proximity can be the result of random (vs deterministic) distribution.
Mitochondrial Ca2+ dynamics.
As described,27 mitochondrial Ca2+ was measured with Rhod-2-AM dye. Analysis and quantification were done using LASX and ImageJ software. To evaluate mitochondrial Ca2+ dynamics in response to drugs, we monitored Rhod-2 fluorescence signal before (F0) and after (F) adding: TTX, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) + oligomycin A and CGP-37157.
Mitochondrial superoxide detection.
Superoxide production was measured using MitoSOX, per manufacturer's instructions. To evaluate mitochondrial superoxide changes in response to TTX, we measured MitoSOX fluorescence signal before (F0) and after (F) TTX.
SCN5A correlation analysis in human left ventricle.
RNAseq raw data from human left ventricular tissue were downloaded from the Genotype-Tissue Expression v8 (GTEx) consortium (https://www.gtexportal.org/home/datasets). Analysis done using Rstudio.
hiPSC-CM model of SCN5A knockout.
hiPSCs were maintained as previously described.28 CRISPR/Cas9 was used to generate the SCN5A knock-out line. Differentiation into hiPSC-CMs was modified from a previously described protocol.29,30
Statistics.
Numerical results are given as means±standard error of the mean (S.E.M.). Statistical tests were used as stated. Differences were considered significant at p<0.05.
RESULTS
Mitochondria are in close apposition to the sarcolemma.
Figure 1A shows an electron microscopy image of mouse left ventricle revealing the alignment of mitochondria along the interfibrillar space. A complete volumetric image of cardiac tissue acquired using FIB-SEM (Fig. 1B) showed mitochondria “nested” immediately under the sarcolemma in the dome-shaped surface spanning the distance between z-disks (the “crest”31), with both structures (sarcolemma and SSM) reaching points of close apposition (red arrows; small panels show different planes of the same preparation). A zoom-in of Figure 1B-A, with detailed description of structures observed in the left end of the image, is presented in Supplemental Fig.I. Segmentation analysis of FIB-SEM images facilitated visualization (Fig. 1C) and revealed multiple connections of SSM with deeper mitochondria both immediately subjacent and, through mitochondria that bridge across sarcomeres, to other interfibrillar chains in the cell, hence expanding into a complete mitochondrial reticulum31 (Fig. 1C and Supplemental Fig.II). Approximately 13% of the mitochondrial surface in the subsarcolemmal space was found <20 nm apart from the membrane (Supplemental Fig.III).
Figure 1. Mitochondria in close apposition to sarcolemma.

A. Focused Ion Beam-Scanning Electron Microscopy (FIB-SEM) image of ventricular myocytes. Notice the presence of mitochondria between z-disks (red box). Scale bar 2 μm. B. FIB-SEM single plane of complete 3D-volumetric image of cell membrane and subjacent mitochondria. Notice mitochondria under the crest. Enlarged images in bottom frames: sequential z-sections showing proximity of membranes and points where membranes come in close apposition (red arrows). C. Segmentation analysis of subsarcolemmal mitochondria connecting to mitochondrial reticulum. Top left: Mitochondria in bottom images of panel B, highlighted in blue. Top right: Segmentation of layers below shows subsarcolemmal mitochondria coupling to first chain of interfibrillar mitochondria. Dotted circles identify points of proximity. Lower panel in left shows subsarcolemmal mitochondria (purple) that bifurcates to reach two interfibrillar chains. Bottom right: bifurcating mitochondrial from a different angle. Through these connections, mitochondria form a complete reticulum, as documented by Glancy et al.37,38
Nav1.5 clusters adjacent to SSM in a non-random organization.
In separate experiments, we used dual-color STORM to localize Nav1.5 in the membrane surface of adult ventricular myocytes. α-Actinin was used to mark sarcomeric edges (z-disks). As shown in Figure 2A, we observed large Nav1.5 clusters between z-disks, in agreement with previous functional data.14 Negative controls are shown in Supplemental Fig.IV. Nav1.5 distribution led us to speculate that a subpopulation of Na-channels may localize in close proximity to SSM, creating a macromolecular complex.
Figure 2. Nav1.5 is found between z-disks close to mitochondria.

A. Stochastic optical reconstruction microscopy (STORM) image of adult ventricular myocyte. Blue: α-actinin as marker of z-disk. Green: Nav1.5. White-boxed area enlarged in bottom panel. Scale bars 5 μm. Inset scale bar 1 μm. Apparent misalignment of α-actinin lines is due to cell surface curvature. Notice that large clusters of Nav1.5 fall not on α-actinin (the expectation for costameric localization), but in-between, corresponding to the crest. B-C. STORM-acquired images of Mitotracker (red), Nav1.5 (green) and α-Actinin (only in panel C, blue) in single myocytes dissociated from wild-type mice. White-boxed areas enlarged in respective bottom panels. Scale bars 5 μm. Inset scale bar 2 μm. Notice alignment of Nav1.5 with SSM at cell surface, spaced by marker of z-disk.
Figure 2B presents an image of a single, isolated adult cardiac myocyte labeled for Nav1.5 (green) and Mitotracker (red) to localize mitochondria. At the cell lateral membrane (distinguishable as the edge framing the long axis of the cell) a seemingly preferential proximity of mitochondria and Nav1.5 was noted (white rectangles indicating area enlarged in the bottom of each panel). For some experiments, a third color was added (blue) to mark α-actinin (Fig. 2C). Data are consistent with the localization of Nav1.5 and of mitochondria separately detected by FIB-SEM and by STORM (Figs. 1 and 2A, respectively). Moreover, the organization of both resembles the shape of the crest, with a Nav1.5 cluster resting on top of the mitochondria, and between z-disks. Nav1.5 cluster properties are indicated in Supplemental Fig.V.
We developed an analytical tool to address the question of whether proximity is random or deterministic. A line traced over the aligned SSM was used as reference (Fig. 3A). Each Nav1.5 cluster was defined as an ellipse. Two parameters were defined: the distance between the centroid of the ellipse and the reference line, and the angle formed between the long axis of the ellipse, and the reference line (details on online supplement). Parameters were correlated for each cluster on an xy plot (Fig. 3B). Experimental data were compared to a simulated image where mitochondria were kept as in the experimental data but Nav1.5 clusters were distributed randomly, though constrained by the edge of the cell (Fig. 3C-D). As shown in Figure 3B, most experimental Nav1.5 clusters assembled in a small window of 10 to 300 nm in distance and a 0-60° angle to the reference line. However, in simulated images, green clusters were scattered along the plot (Fig. 3D). The method was further validated when comparing the relation between SSM and a line formed by immunostaining of α-actinin, which traces the z-disk perpendicular to the lateral surface of the membrane. In this case, almost all α-actinin clusters were gathered in a very small window, with a 500 to 2000 nm distance and a 70-90° angle to the mitochondria long axis, as expected (Fig. 3E-H). Overall, our results support the concept that the Na-channel complex and mitochondria form a structural macromolecular complex. Whether the association has a functional correlate was investigated next.
Figure 3. Nav1.5 localizes on top of SSM in a deterministic matter.

A-B: STORM-acquired image of Mitotracker (red) and Nav1.5 (green) from the lateral membrane of an adult murine myocyte (“Experimental image”; A) and data analysis (B). Reference line was traced over the first string of mitochondria under the cell membrane (SSM). This line was used as reference to measure distance to centroid of the Nav1.5 cluster (abscisae in B) and its inclination, namely, the angle of the long axis of the ellipse defining the cluster (ordinates in B). A window of 300 nm and up to 60° in inclination captured 80% of the clusters. Distribution was compared to that obtained by modeling a random distribution of green clusters over the same region of interest (C; “randomized image”). In that condition, only 15% of clusters fell within the same window (D). To validate the model, results were compared to those obtained for a domain established by clusters of known architecture, namely, α-actinin (E: “Experimental image” of Mitotracker (red) and α-Actinin (blue). Analysis in F. In this case, to enhance visualization, the angle of inclination is reported as 90-angle (so perpendicular lines cluster around zero). A similar experiment in the “randomized image” is presented in G-H. As predicted, α-actinin is tightly organized in relation to mitochondria, and such organization is lost when clusters distribute randomly. Distance to reference line measured in nm. Similar results were obtained in n=28 cells from N=4 mice.
Mitochondrial NCLX is predominantly found in SSM near Nav1.5 clusters.
NCLX plays an important role in maintaining mitochondrial function. Sodium concentration, in turn, is key to the ability of NCLX to extrude Ca2+ from the mitochondrial space. Given the found relation between Nav1.5 and mitochondria, we examined whether NCLX had a preferential localization in the SSM space.
Figure 4 shows that the immunofluorescent signal for NCLX (red) was more abundant in the SSM than in mitochondria localized in the intracellular space; we also show significant co-localization of NCLX with Nav1.5-positive clusters (green). Indeed, ~80% of Nav1.5-NCLX clusters colocalized (i.e., were located within 20 nm of each other) at the lateral membrane vs ≈30% in the cell interior (Supplemental Fig. VI). To further validate NCLX antibody specificity for mitochondrial NCLX, we immunolocalized mitochondria with NCLX in HL-1 cells. Supplemental Fig.VII shows colocalization of the two structures and a round-punctuate pattern, characteristic of mitochondrial structures.
Figure 4. Mitochondrial NCLX is mainly found in SSM near Nav1.5 clusters.

Dual-color stochastic optical reconstruction microscopy (STORM)-acquired images of NCLX (red) and Nav1.5 (green) in single myocytes dissociated from wild-type mice. White-boxed area enlarged in right image. Notice abundance of NCLX signal at lateral membrane and proximity to Nav1.5 clusters. Scale bar 5 μm. Inset scale bar 1 μm.
Given the proximity of NCLX to Nav1.5, we postulated that the presence or absence of INa may impact on NCLX function and as such, on the extent of Ca2+ accumulation in the mitochondrial space.
Mitochondrial Ca2+ dynamics: differences between SSM and IFM.
To assess the relation between NCLX and Na-channel function we used a mitochondrial Ca2+ indicator (Rhod2-AM; Fig. 5A; see also Supplemental Fig.VIII.A, “Methods” and27). Supplemental Fig.IX shows preservation of striated cell morphology after treatment. Rhod2-AM intensity was measured before (F0) and after drug treatment (F) and measurements of the ratio F/F0 in SSM were compared to those obtained in IFM. Measurements in control conditions (no drugs added) are shown as the first two bars of the graph in Figure 5B, corresponding to data acquired from SSM (red) and IFM (gray), respectively. Since cytoplasmic Na+ concentration is a major driver of NCLX forward movement, we first tested whether blocking INa with TTX would affect mitochondrial Ca2+. Rhod2-AM fluorescence was first recorded in a solution containing 148 mmol/L Na+; then, TTX 10 μmol/L was added. The bar graph (bars with vertical lines) in Figure 5B shows that, upon TTX exposure, SSM accumulated significantly more Ca2+ (detected as increase in Rhod2 fluorescence) than IFM (F/Fo 1.1±0.03 and 0.99±0.03 for SSM and IFM, respectively; p<0.05). This was consistent with the notion that reduced entry of sodium via Nav1.5 limits NCLX activity, and that this effect is more noticeable in the area where the Nav1.5/NCLX complex is abundant (see Supplemental Fig.VIII.B). Of note, when low concentrations of TTX (100 nmol/L) were used, we observed a non-statistically significant tendency for increased Rhod2 intensity in SSM (Supplemental Fig.X).
Figure 5. Mitochondrial Ca2+ in relation to sodium channel activity.

A. Confocal 2D images of cardiomyocytes loaded with Rhod 2-AM at 4°C and then incubated at 37°C for 3-5 hours (see Methods). Two regions were defined, SSM (first string of mitochondria immediately below cell surface) and IFM (in the interior of the cell). B. Quantitative analysis of F/F0 (F corresponds to Rhod2-AM intensity at rest, and F0 to Rhod2-AM intensity after treatment) ratio under conditions described in the table below. Notice increased fluorescence emission units (mitochondria) after tetrodotoxin (TTX), TTX+FCCP+Oligomycin and CGP-37157 treatment, particularly in SSM. The ratio F/F0 of Rhod-2 fluorescence intensities was compared between SSM and IFM: higher ratio reflects more mitochondrial Ca2+ accumulation. Scale bar 10 μm. +TTX: 15 cells from 8 mice; +TTX+FCCP+Oligomycin: 7 cells from 4 mice; +TTX+FCCP+Oligomycin+CGP-37157: 7 cells from 3 mice; CGP-37157: 6 cells from 3 mice; Negative control: 17 cells from 5 mice. Student’s t-test: between SSM and IFM for each treatment: *p<0.05 vs SSM, **p<0.01 vs SSM, ***p<0.001 vs SSM; between treatment vs negative control: #p<0.05 vs negative control, ###p<0.001 vs negative control.
We next compared Ca2+ dynamics in SSM vs IFM in the presence of TTX and after mitochondrial depolarization with 1 μmol/L FCCP + 2 μmol/L oligomycin. FCCP alone can induce mitochondrial ATP depletion via the reverse of F0/F1-ATPase, which is prevented by adding the F0/F1-ATPase inhibitor oligomycin. FCCP induces a mitochondrial depolarization of around 100 mV, enough for NCLX to function in reverse mode (Ca2+in/ Na+out of the mitochondria).32 Consistent with this notion, FCCP+oligomycin augmented Rhod2 fluorescence exclusively at the SSM (Fig. 5B, bars with horizontal lines). Most importantly, adding the NCLX inhibitor CGP-371257 attenuated the mitochondrial Ca2+ accumulation in SSM and no significant difference in Rhod2 fluorescence was observed between SSM and IFM (Fig. 5B, bar with dots).
We analyzed Rhod2 fluorescence in cells treated only with 2 μmol/L CGP-37157, a selective NCLX inhibitor. As expected, after adding CGP-37157, Rhod2 fluorescence increased in SSM (ratio 1.10±0.024 vs IFM ratio 0.98±0.01, p<0.001, Fig. 5B bars with crosses) indicating that Ca2+ influx through the mitochondrial calcium uniporter (MCU) cannot be compensated by extrusion since NCLX is inhibited. Rhod2 fluorescence was practically unchanged after CGP-37157 addition in IFM, suggesting that IFM have another way of extruding Ca2+ that is NCLX- and Na+-independent.
Finally, we measured mitochondrial Ca2+ dynamics at different pacing rates (3 and 5Hz) alone or in the presence of either TTX or CGP-37157. Rapid pacing led to mitochondrial Ca2+ accumulation in SSM and IFM (vs rest #p<0.05). When TTX was added (10 μmol/L), there was a tendency for SSM to accumulate more Ca2+ in response to pacing (3Hz vs 3Hz+ TTX p=0.06; 5Hz vs 5Hz+ TTX p=0.07) and when comparing the two mitochondrial subpopulations, mitochondrial Ca2+ increased significantly more in SSM than in IFM, *p<0.05). Similar results were obtained with CGP-37157. Details in Supplemental Fig.XI. Altogether, the data suggest functional coupling between Nav1.5 and NCLX in the sarcolemmal-subsarcolemmal space in the crest region of adult cardiac myocytes, with functional implications to the adaptation necessary to meet demand at increased rates.
Increased superoxide production after TTX addition.
There is a reciprocal interaction between Ca2+ and ROS which, in physiological conditions, plays an important role in regulating cellular signaling networks.33,34 Pathological changes in one can affect the other, leading to more damage. Since TTX increases mitochondrial Ca2+, we wondered if this could lead to increased ROS production.
We used the fluorescent dye MitoSOX Red, which detects mitochondrial superoxide, to measure ROS in live myocytes before and after TTX. When in mitochondria, MitoSOX exhibits red fluorescence. As seen in Figure 6, at resting conditions (F0) ventricular myocytes showed low MitoSOX fluorescence, higher in SSM, as previously reported.35 Co-staining with MitoTracker confirmed MitoSox mitochondrial localization (Supplemental Fig.XII).
Figure 6. Increased superoxide production after TTX.

A. Confocal 2D images of cardiomyocytes loaded with MitoSOX (see Methods). Two images from same cell are shown: at rest (F0; left), and after 10 μmol/L addition of TTX (F; right). Notice brighter fluorescence at SSM at resting conditions compared to IFM. Scale bar 10 μm. B. Ratio FTTX/F0 of MitoSOX fluorescence intensities was compared between SSM and IFM: higher ratio reflects more ROS produced. Average was then plotted. N=3 mice, n=12 cells. No statistical differences (Student’s t-test).
After exposure to TTX 10 μmol/L (pacing at 0.5Hz), MitoSOX fluorescence increased in both SSM and IFM, the latter being less pronounced (Paired Student’s t-test: p<0.001 +TTX vs rest in SSM; p<0.05 +TTX vs rest in IFM). This result indicates that blocking Na+ entry with TTX leads to increased mitochondrial ROS production, though in this case there was no predominant effect in the SSM, suggesting that the role of INa on mitochondrial function may go beyond its local interaction with NCLX.
Correlation analysis of human left ventricle transcriptome in relation to SCN5A expression.
We used the Genotype-Tissue Expression repository (GTEx; v8) of 386 samples of left ventricle tissue from deceased human donors to compare the abundance of SCN5A transcript against SLC8B1 and SLC8A1, which encode the mitochondrial NCLX and the plasma membrane NCX, respectively. Resulting plots are shown in Figure 7A-B. Data show that SLC8A1 is positively correlated with SCN5A (regression coefficient β= 0.62, log-P-value= 4E-58) whereas SLC8B1 is negatively correlated with SCN5A (regression coefficient β= −0.17, log-P-value= 1.66 E-30). This observation is only correlational. We therefore tested whether reduced abundance of one transcript would affect the abundance of the other. To this aim, we used an hiPSC-CM line deficient in SCN5A (see “Methods” and Supplemental Figs.XIII and XIV). As shown in Figure 7C the abundance of SLC8B1 was significantly increased in cells where SCN5A was knocked down, supporting the notion of a transcriptional relation between the two gene products.
Figure 7. SLC8B1 (encoding NCLX), negatively correlates with SCN5A.

A-B. Association analysis of human left ventricle transcriptome in relation to SCN5A expression: correlation between expression of SLC8B1 (panel A) or SLC8A1 (panel B) vs SCN5A expression. Each point represents the normalized level (VST, variance stabilizing transformation) of SCN5A vs that of SLC8B1 (A) or SLC8A1 (B) for one individual in the dataset. Notice the positive slope (=positive correlation) between SCN5A and SLC8A1 (plasma membrane NCX) and negative correlation with SLC8B1 (mitochondrial NCLX), suggesting that SCN5A transcript levels correlate with expression of both in human heart. C. qPCR for SCN5A, SLC8A1 and SLC8B1 in hiPSC-CMs wild-type (WT) and SCN5A-KO. Notice drastic reduction in SCN5A and increase in SLC8B1 in hiPSC-CM SCN5A-KO. N=7. Statistical test: Mann Whitney U-test between SCN5A-KO and WT for each gene. ***p<0.001 vs WT.
DISCUSSION
Our data show that SSM and a subpopulation of Nav1.5 at the lateral membrane are in close apposition and that in ventricular cardiomyocytes, not only mitochondria can regulate INa,20,21 but also INa can regulate NCLX activity. NCLX preferentially localized at SSM near Nav1.5 clusters. We speculate that such distribution creates a high-Na+ microdomain that is energy-efficient, comparable to the high-Ca2+ microdomain formed between MCU and the sarcoplasmic reticulum (SR). Many diseases have been linked to changes in intracellular Na+ concentration36 which, in turn, could affect NCLX function, therefore affecting ATP production and ROS generation. Overall, we show the presence of a Na-channel-mitochondria complex and suggest that Nav1.5-NCLX proximity may have functional consequences to metabolic homeostasis via crosstalk with the mitochondrial reticulum.
Cardiomyocytes are rich in mitochondria, which occupy a third of total cell volume.18 Although mitochondria form a network reaching throughout the cell37,38, they are divided into at least three subpopulations with distinct morphology, composition, biochemical properties and function, contributing to their capacity for adaptation.18,19,35 Here, we focus on SSM. As others,39 we found SSM nested in-between z-disks. A nanometric-resolution cell-surface scan shows a pattern of crests and valleys (or “grooves”) with ~2 μm periodicity, that is, the length of a sarcomere.31 In this pattern, grooves match the position of the z-disk (prompting the name “z-grooves”), with crests being dome-shaped surfaces spanning the distance between two z-grooves.31 Previously, using scanning patch clamp, we measured larger INa in the crest compared to grooves.14 This matches our data showing Nav1.5 clusters preferentially aligned with SSM at the crest. We also show that the cell membrane in the crest region reaches close proximity with the outer membrane of the mitochondria, in a manner reminiscent of the T-tubule-junctional SR dyad (see, e.g.40,41) or between cell membranes in the perinexus.42 In these structures, the narrow intermembrane space allows functional coupling between channels residing in opposing membranes. The molecular mechanisms of formation of this Nav1.5-mitochondrial complex remain undefined. Yet, we show that the relation between mitochondria and Nav1.5 is deterministic and of a particular topology.
We identify NCLX, a molecule key for Na+ and Ca2+ homeostasis, as resident of the Nav1.5-mitochondria hub. NCLX (encoded by SLC8B1) is proposed to be the main pathway for mitochondrial Ca2+ extrusion in excitable cells, while MCU is responsible for Ca2+ transport into mitochondria.43,44 NCLX transport is voltage-dependent and electrogenic, with stoichiometry of 3-4 Na+ for 1 Ca2+.45 Unlike plasma membrane NCX, unique residues confer NCLX Li+ selectivity.46 The Ca2+ extrusion rate of NCLX is ≈100 times slower than MCU-Ca2+ influx rate. Therefore, NCLX activity is the rate-limiting step of mitochondrial Ca2+ cycling.44,47,48
NCLX is fundamental to life. A conditional knockout of NCLX quickly leads to heart failure associated with mitochondrial Ca2+ overload and consequent increase in ROS generation.43 In contrast, NCLX overexpression in mouse heart increases mitochondrial Ca2+ extrusion preventing pathogenic Ca2+ accumulation and ROS generation.43 Whether loss or over-abundance of NCLX affects INa or subcellular localization of Nav1.5 remains unclear.
NCLX is very sensitive to changes in cytoplasmic [Na+],32,48,49 given that the Km of association is similar to resting [Na+]i (Km ~7-10 mmol/L). Moreover, studies from Skogestad et al show that [Na+]i in an active, excitable myocyte, is not homogeneous. Rather, it is highest in the subsarcolemma and distributes in pools, likely, corresponding to the location of Na+-channel clusters.50 We propose that NCLX is close to Nav1.5 channels to rapidly sense Na+ signals and properly extrude Ca2+ out of mitochondria, in a way similar to how MCU’s low affinity for Ca2+ is compensated by proximity to the SR (diagram in Supplemental Fig.VIII).51,52 Several studies describe an interaction of NCLX with plasma membrane transporters: a) In pancreatic β-cells, glucose-dependent depolarization leads to cytosolic Na+ influx via voltage-gated Na+ channels, which promotes NCLX activity, maintaining ATP production and controlling insulin secretion.53,54 b) The nociceptive noxious heat-activated receptor (TRPV1) triggers a Na+-dependent depolarization that controls NCLX Ca2+ extrusion, which at the same time regulates TRPV1 conductance.55,56 c) In macrophages and melanoma cells, activation of a splice variant of Nav1.6 causes release of Na+ from vesicular compartments and Na+-uptake-Ca2+release by NCLX.57
Through human transcriptome analysis, we found SLC8B1 negatively correlating with SCN5A. Though the common denominator of this relation remains unknown, its presence may have important functional consequences. Indeed, reduced expression of SCN5A may be compensated by increased expression of SCL8B1 to increase the ability to capture Na+ entering the cell. The reverse would also have functional effects: an excess Na+ entry could be compensated by reduced expression of the exchanger, maintaining mitochondrial homeostasis. This is consistent with the notion that NCLX activity is strongly regulated by even slight changes in cytosolic Na+.48,58-63 Their physical proximity (shown here) and transcriptional inverse relation would facilitate maintenance of this delicate equilibrium.
In resting Na+ and Ca2+ conditions and with an inner mitochondrial membrane potential of ~−180 mV, NCLX operates in forward mode, exporting Ca2+ out of the mitochondrial matrix in exchange for cytosolic Na+. However, in depolarized mitochondria, NCLX functions in reverse mode, transporting Ca2+ into the mitochondria and extruding Na+.64 Studies have shown that increasing cytosolic Na+ accelerates mitochondrial Ca2+ decay due to increased NCLX forward mode activity,63,64 but these studies did not compare SSM to IFM. Our mitochondrial Ca2+ dynamic experiments show that NCLX is predominantly located at SSM, confirming our SMLM experiments, and TTX leads to mitochondrial Ca2+ accumulation more prominently in SSM, connecting Nav1.5 and NCLX function. The Ca2+ accumulation after TTX+FCCP+Oligomycin exposure in SSM seen in our experiments could be explained by 1) active uptake of calcium by the reverse NCLX, or 2) increased mitochondrial uptake via MCU. However, MCU open probability decreases with mitochondrial depolarization (i.e. with FCCP).64 In fact, IFM show a slight decrease in Rhod2 fluorescence after TTX+FCCP+Oligomycin which could be accounted for an inhibited MCU and lack of reverse mode NCLX-Ca2+ uptake in IFM. We used the benzothiazepine compound CGP-37157 to selectively block NCLX (IC50= 0.36 μM).59,62 At least a 10-fold higher concentration of CGP-37157 is needed to affect plasma membrane NCX, SERCA and RyR2.59 It is known that cytosolic Na+ is required for powering NCLX to activate mitochondrial Ca2+ efflux. Therefore, our mitochondrial Ca2+ dynamics data suggest that a healthy INa is needed to maintain mitochondrial Ca2+ homeostasis. However, and compared to Ca2+ imaging, studying cellular Na+ dynamics is challenging, particularly due to the relatively small changes of intracellular Na+ concentration.53,58
There is mutual interaction between ROS and Ca2+, where ROS can regulate Ca2+ signaling and Ca2+ signaling is essential for ROS production.33,43 Physiologically, ROS are generated as by-product of mitochondrial respiratory chain activity or by specialized ROS-generating enzymes to regulate signaling functions such as cell proliferation, migration, and vascular tone; but toxic levels associate with pathology. Mitochondrial Ca2+ accumulation leads to ROS production. Our results show increased ROS production after TTX exposure in both SSM and (less pronounced) IFM, even though only SSM showed increased mitochondrial Ca2+ after TTX. It is worth noting that, even though they are part of distinct mitochondrial subpopulations, SSM and IFM are connected, forming a mitochondrial reticulum in which ROS can propagate.37,38,65 Most importantly, ROS production leads to decreased INa.66 ROS can alter the oxidative state of PKA, PKC, CaMKII and NAD(H), which in turn regulates INa.33,67 Genetically engineered Scn5a heterozygous mice show increased ROS accumulation.68 This, together with our results, suggests that there could be an amplification loop: increased ROS decreases INa, leading to less Na+ entering the cytoplasm, which in turn affects NLCX activity which leads to mitochondrial Ca2+ accumulation, leading to more ROS production.
One question that arises is how do IFM extrude calcium if NCLX is mainly found in SSM? Even though NCLX is the major transporter for Ca2+ extrusion, mitochondria contain both Na+-dependent and Na+-independent mechanisms for Ca2+ extrusion.45,64 This may include leucine zipper EF-hand-containing transmembrane protein 1 (LETM1;45,64,69). Somehow controversial, it has been proposed that transient mitochondrial permeability transition pore openings could also help discharge mitochondrial Ca2+ load.45,47,70 Other studies show Cx43 hemichannels in mitochondria71 and particularly in SSM72 which could act as mitochondrial Ca2+ gateway. Overall, our data are consistent with previous descriptions of proteins that distribute and/or work differently in various sub-populations of mitochondria (see, e.g.73,74).
Our data suggest the need of healthy INa for (proper) mitochondrial Ca2+ balance. Many diseases associate with cytoplasmic unbalanced Na+ concentration. A pathogenic increase in cytoplasmic Na+ accelerates mitochondrial Ca2+ efflux trough NCLX, which inhibits mitochondrial metabolic rate and ATP production, as seen in heart failure, ischemia, and seizure-like events.48,59,63,75,76 In melanoma cells, an increase in Na+ mediated by vesicular Nav1.6 activates NCLX, which accelerates invasiveness of these tumor cells.57 Conversely, hypoxia is known to reverse NCLX mode, which leads to mitochondrial Ca2+ overload and ROS production, in turn accelerating cytoplasmic Na+ accumulation.77 Future studies need to be done to analyze NCLX activity in pathologies where INa is reduced (e.g. Brugada syndrome).
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The voltage-gated sodium channels, formed by the protein Nav1.5, carry the excitatory inward current (the sodium current) that is responsible for the action potential upstroke in adult cardiac ventricular myocytes.
Nav1.5 channels localize to different subdomains within the myocyte, associating with other molecules in a domain-specific manner.
Mitochondrial function can alter the magnitude and properties of the sodium current.
What New Information Does This Article Contribute?
Nav1.5 clusters that localize to the membrane region between two z-lines (the “crest” of the sarcomere) are in close apposition to subsarcolemmal mitochondria and to the mitochondrial Na+/Ca2+ exchanger.
This anatomical proximity leads to a functional relation between Nav1.5 and the mitochondrial Na+/Ca2+ exchanger.
Nav1.5 channel malfunction leads to unbalanced mitochondrial calcium dynamics, potentially impacting cell metabolism.
Na-channels form macromolecular complexes that, in adult ventricular myocytes, cluster at specific subdomains. Dysfunction of the channel itself, or of its partners, can lead to arrhythmias that cause sudden cardiac death. The relation between Na-channels and mitochondrial function has been described in one direction only: mitochondrial byproducts (such as ROS) affect sodium current. Here we show that this relation is reciprocal, namely, Na-channel activity influences mitochondrial function. Using imaging techniques, we describe the deterministic localization of Na-channels and subsarcolemmal mitochondria, where we primarily locate the mitochondrial Na+/Ca2+ exchanger. Functionally, the blockade of Na-channels leads to a local mitochondrial Ca2+ accumulation and ROS production. We propose that NCLX is powered by the entry of Na+ through Na-channels to properly extrude Ca2+. This proposed model for electro-metabolic coupling redefines Nav1.5 as a key regulator of cell metabolism and opens a new window to understand electrical and structural disorders of the heart.
ACKNOWLEDGMENTS
We thank Chris Petzold for EM sample preparation and image acquisition. GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
SOURCES OF FUNDING
Supported by NIH RO1-HL134328, RO1-HL136179 and RO1-HL145911 (MD), the Wilton W. Webster Fellowship in Pediatric Electrophysiology from the Heart Rhythm Society (MP-H), a Leducq Foundation Transatlantic network of Excellence (MD, PB) and the Swiss National Science Foundation P1BEP3_172237 (SV). Microscopy Laboratory is supported by: NIH/NCI P30CA016087 and S10 ODO019974-01A1.
Nonstandard Abbreviations and Acronyms:
- FIB-SEM
Focused Ion Beam-Scanning Electron Microscopy
- GTEx
Genotype-Tissue Expression
- IFM
InterFibrillar Mitochondria
- SSM
SubSarcolemmal Mitochondria
- NCLX
Mitochondrial Na+/Ca2+/Li+ Exchanger
- ROS
Reactive Oxygen Species
- SMLM
Single-Molecule Localization Microscopy
- STORM
Stochastic Optical Reconstruction Microscopy
- TTX
Tetrodotoxin
Footnotes
DISCLOSURES
None.
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
REFERENCES
- 1.Janse MJ and Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiological reviews. 1989;69:1049–169. [DOI] [PubMed] [Google Scholar]
- 2.Bankston JR and Kass RS. Fading sodium channels in failing hearts. Circ Res. 2007;101:1073–4. [DOI] [PubMed] [Google Scholar]
- 3.Wilde AA and Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res. 2011;108:884–97. [DOI] [PubMed] [Google Scholar]
- 4.Wilde AA, Moss AJ, Kaufman ES, Shimizu W, Peterson DR, Benhorin J, Lopes C, Towbin JA, Spazzolini C, Crotti L, Zareba W, Goldenberg I, Kanters JK, Robinson JL, Qi M, Hofman N, Tester DJ, Bezzina C, Alders M, Aiba T, Kamakura S, Miyamoto Y, Andrews ML, McNitt S, Polonsky B, Schwartz PJ, Ackerman MJ. Clinical Aspects of Type 3 Long QT Syndrome: An International Multicenter Study. Circulation. 2016;134(12):872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.George AL. Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res. 2015;116:1919–36. [DOI] [PubMed] [Google Scholar]
- 7.te Riele AA-PE, James CA, Leo-Macias A, Cerrone M, Zhang M, Lin X, Lin B, Sobreira NL, Amat-Alarcon N, Narsman RF, Murray B, Tichnell C, can der Heijden JF, Dooijes D, van Veen TA, Tandri H, Fowleer SJ, Hauer RN, Tomaselli G, van den Berg MP, Taylor Mr, Brun F, Sinagra G, Wilde AA, Mestroni L, Bezzina CR, Calkins H, van Tintelen PJ, Bu L, Delmar M, Judge DP. Multilevel analyses of SCN5A mutations in ARVD/C suggest noncanonical mechanisms of disease pathogenesis. Cardiovasc Res. 2017;113:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu M, Yang KC, Dudley SC Jr. Cardiac sodium channel mutations: why so many phenotypes? Nature reviews Cardiology. 2014;11:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu M, Yang KC, Dudley SC Jr. Cardiac Sodium Channel Mutations: Why so Many Phenotypes? Curr Top Membr. 2016;78:513–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abriel H, Rougier JS, Jalife J. Ion channel macromolecular complexes in cardiomyocytes: roles in sudden cardiac death. Circ Res. 2015;116:1971–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. SAP97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels Nav1.5 in cardiomyocytes. Circ Res. 2011;108:294–304. [DOI] [PubMed] [Google Scholar]
- 12.Shy D, Gillet L, Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta. 2013;1833:886–94. [DOI] [PubMed] [Google Scholar]
- 13.Lin X, Liu N, Lu J, Zhang J, Anumonwo JM, Isom LL, Fishman GI, Delmar M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 2011;8:1923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhargava A, Lin X, Novak P, Mehta K, Korchev Y, Delmar M, Gorelik J. Super-resolution scanning patch clamp reveals clustering of functional ion channels in adult ventricular myocyte. Circ Res. 2013;112:1112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marsman RF, Bezzina CR, Freiberg F, Verkerk AO, Adriaens ME, Podliesna S, Chen C, Purfurst B, Spallek B, Koopmann TT, Baczko I, Dos Remedios CG, George AL Jr, Bishopric NH, Lodder EM, de Bakker JM, Fischer R, Coronel R, Wilde AA, Gotthardt M, Remme CA. Coxsackie and adenovirus receptor is a modifier of cardiac conduction and arrhythmia vulnerability in the setting of myocardial ischemia. J Am Coll Cardiol. 2014;63:549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillet L, Rougier JS, Shy D, Sonntag S, Mougenot N, Essers M, Shmerling D, Balse E, Hatem SN, Abriel H. Cardiac-specific ablation of synapse-associated protein SAP97 in mice decreases potassium currents but not sodium current. Heart Rhythm. 2015;12(1):181–92. [DOI] [PubMed] [Google Scholar]
- 17.Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, Taffet SM, Delmar M. Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, Ventura-Clapier R, Joubert F. Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol. 2013;4:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu X, Thai PN, Lu S, Pu J, Bers DM. Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes. J Mol Cell Cardiol. 2019;136:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu M, Sanyal S, Gao G, Gurung IS, Zhu X, Gaconnet G, Kerchner LJ, Shang LL, Huang CL, Grace A, London B, Dudley SC Jr. Cardiac Na+ current regulation by pyridine nucleotides. Circ Res. 2009;105:737–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC Jr. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang X, Yoon JY, Morley M, McLendon JM, Mapuskar KA, Gutmann R, Mehdi H, Bloom HL, Dudley SC, Ellinor PT, Shalaby AA, Weiss R, Tang WHW, Moravec CS, Singh M, Taylor AL, Yancy CW, Feldman AM, McNamara DM, Irani K, Spitz DR, Breheny P, Margulies KB, London B, Boudreau RL. A common variant alters SCN5A-miR-24 interaction and associates with heart failure mortality. J Clin Invest. 2018;128:1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, Zakharov SI, Yang L, Morrow JP, Garan H, Marx SO. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest. 2016;126:112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilke SA, Antonios JK, Bushong EA, Badkoobehi A, Malek E, Hwang M, Terada M, Ellisman MH, Ghosh A. Deconstructing complexity: serial block-face electron microscopic analysis of the hippocampal mossy fiber synapse. J Neurosci 2013;33,507–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walton J Lead asparate, an en bloc contrast stain particularly useful for ultrastructural enzymology. J Histochem Cytochem 1979; 27, 1337–1342. [DOI] [PubMed] [Google Scholar]
- 26.Leo-Macias A, Agullo-Pascual E, Sanchez-Alonso JL, Keegan S, Lin X, Arcos T, Feng-Xia-Liang, Korchev YE, Gorelik J, Fenyö D, Rothenberg E, Rothenberg E, Delmar M. Nanoscale visualization of functional adhesion/excitability nodes at the intercalated disc. Nat Commun 2016;7:10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JC, Pérez-Hernández M, Alvarado FJ, Maurya SR, Montnach J, Yin Y, Zhang M, Lin X, Vasquez C, Heguy A, Liang FX, Woo SH, Morley GE, Rothenberg E, Lundby A, Valdivia HH, Cerrone M, Delmar M. Disruption of Ca2+i Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019;140(12):1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuo H-H, Gao X, DeKeyser J-M, Fetterman KA, Pinheiro EA, Weddle CJ, Fonoudi H, Orman MV, Romero-Tejeda M, Jouni M, Blancard M, Magdy T, Epting CL, George AL, Burridge PW. Negligible-Cost and Weekend-Free Chemically Defined Human iPSC Culture. Stem Cell Reports. 2020;14(2):256–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burridge PW, Holmström A, Wu JC. Chemically Defined Culture and Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. Current protocols in human genetics. 2015;87:21.3.1–21.3.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC. Chemically defined generation of human cardiomyocytes. Nature methods. 2014;11:855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gu Y, Gorelik J, Spohr HA, Shevchuk A, Lab MJ, Harding SE, Vodyanoy I, Klenerman D, Korchev YE. High-resolution scanning patch-clamp: new insights into cell function. FASEB J. 2002;16:748–50. [DOI] [PubMed] [Google Scholar]
- 32.Kim B, Matsuoka S. Cytoplasmic Na+-dependent modulation of mitochondrial Ca2+ via electrogenic mitochondrial Na+-Ca2+ exchange. J Physiol. 2008;586(6):1683–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sag CM, Wagner S, Maier LS. Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radic Biol Med. 2013;63:338–49. [DOI] [PubMed] [Google Scholar]
- 34.Görlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol. 2015;6:260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adhihetty PJ, Ljubicic V, Menzies KJ, Hood DA. Differential susceptibility of subsarcolemmal and intermyofibrillar mitochondria to apoptotic stimuli. Am J Physiol Cell Physiol. 2005;289(4):C994–C1001. [DOI] [PubMed] [Google Scholar]
- 36.Murphy E, Eisner DA. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res. 2009;104(3):292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glancy B, Hartnell LM, Malide D, Yu ZX, Combs CA, Connelly PS, Subramaniam S, Balaban RS. Mitochondrial reticulum for cellular energy distribution in muscle. Nature. 2015;523:617–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glancy B, Hartnell LM, Combs CA, Fenmou A, Sun J, Murphy E, Subramaniam S, Balaban RS. Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell reports. 2017;19:487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miragoli M, Sanchez-Alonso JL, Bhargava A, Wright PT, Sikkel M, Schobesberger S, Diakonov I, Novak P, Castaldi A, Cattaneo P, Lyon AR, Lab MJ, Gorelik J. Microtubule-Dependent Mitochondria Alignment Regulates Calcium Release in Response to Nanomechanical Stimulus in Heart Myocytes. Cell Rep. 2016;14(1):140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong T and Shaw RM. Cardiac T-Tubule Microanatomy and Function. Physiological reviews. 2017;97:227–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lavorato M, Huang TQ, Iyer VR, Perni S, Meissner G and Franzini-Armstrong C. Dyad content is reduced in cardiac myocytes of mice with impaired calmodulin regulation of RyR2. J Muscle Res Cell Motil. 2015;36:205–14. [DOI] [PubMed] [Google Scholar]
- 42.Veeraraghavan R, Lin J, Hoeker GS, Keener JP, Gourdie RG, Poelzing S. Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: an experimental and modeling study. Pflugers Arch. 2015;467(10):2093–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR, Elrod JW. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature. 2017;545(7652):93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA. 2010;107:436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giorgi C, Marchi S, Pinton P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol. 2018;19(11):713–730. [DOI] [PubMed] [Google Scholar]
- 46.Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, Silverman WF, Argaman M, Sekler I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem. 2004; 279(24):25234–40. [DOI] [PubMed] [Google Scholar]
- 47.Bernardi P, von Stockum S. The permeability transition pore as a Ca2+ release channel: new answers to an old question. Cell Calcium 2012; 52(1):22–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verkhratsky A, Trebak M, Perocchi F, Khananshvili D, Sekler I. Crosslink between calcium and sodium signalling. Exp Physiol. 2018;103(2):157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Despa S. Myocyte [Na+]i Dysregulation in Heart Failure and Diabetic Cardiomyopathy. Front Physiol. 2018;9:1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skogestad J, Lines GT, Louch WE, Sejersted OM, Sjaastad I, Aronsen JM. Evidence for heterogeneous subsarcolemmal Na(+) levels in rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2019;316(5):H941–H957. [DOI] [PubMed] [Google Scholar]
- 51.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13(9):566–78. [DOI] [PubMed] [Google Scholar]
- 52.De La Fuente S, Lambert JP, Nichtova Z, Fernandez Sanz C, Elrod JW, Sheu SS, Csordás G. Spatial Separation of Mitochondrial Calcium Uptake and Extrusion for Energy-Efficient Mitochondrial Calcium Signaling in the Heart. Cell Rep. 2018;24(12):3099–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sekler I, Hershfinkel M, Nita II. The mitochondrial Na+/Ca2+ exchanger-NCLX is an integrating hub for glucose dependent Na+ and Ca2+ signaling in pancreatic β cells. FASEB J. 2013;27:xxx. [Google Scholar]
- 54.Nita II, Hershfinkel M, Kantor C, Rutter GA, Lewis EC, Sekler I. Pancreatic β-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. FASEB J. 2014;28:3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim MS and Usachev YM. Mitochondrial Ca2+ cycling facilitates activation of the transcription factor NFAT in sensory neurons. J Neurosci. 2009;29:12101–12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nita II, Caspi Y, Gudes S, Fishman D, Lev S, Hersfinkel M, Sekler I, Binshtok AM. Privileged crosstalk between TRPV1 channels and mitochondrial calcium shuttling machinery controls nociception. Biochim Biophys Acta. 2016;1863:2868–2880. [DOI] [PubMed] [Google Scholar]
- 57.Carrithers MD, Chatterjee G, Carrithers LM, Offoha R, Iheagwara U, Rahner C, Graham M, Waxman SG. Regulation of podosome formation in macrophages by a splice variant of the sodium channel SCN8A. J Biol Chem. 2009;284(12):8114–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kostic M and Sekler I. Function, regulation and physiological role of the mitochondrial Na+/Ca2+ exchanger, NCLX. Current Opinion in Physiology. 2018;3:63–70. [Google Scholar]
- 59.Palty R, Hershfinkel M, Sekler I. Molecular identity and functional properties of the mitochondrial Na+/Ca2+ exchanger. J Biol Chem. 2012;287(38):31650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Griffiths EJ. Mitochondrial calcium transport in the heart: physiological and pathological roles. J. Mol. Cell Cardiol 2009;46:789–803. [DOI] [PubMed] [Google Scholar]
- 61.Boyman L, Williams GS, Khananshvili D, Sekler I, Lederer WJ. NCLX: the mitochondrial sodium calcium exchanger. J. Mol. Cell Cardiol. 2013;59:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cox DA, Conforti L, Sperelakis N, Matlib MA. Selectivity of inhibition of Na(+)-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J Cardiovasc Pharmacol. 1993;21:595–599. [DOI] [PubMed] [Google Scholar]
- 63.Maack C, Cortassa S, Aon M, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res 2006;99:172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samanta K, Mirams GR, Parekh AB. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat Commun. 2018;9(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aon MA, Cortassa S, Marbán E, O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278(45):44735–44. [DOI] [PubMed] [Google Scholar]
- 66.Wang HJ, Li YL, Zhang LB, Zucker IH, Gao L, Zimmerman MC, Wang W. Endogenous reactive oxygen species modulates voltage-gated sodium channels in dorsal root ganglia of rats. J Appl Physiol. 1985;2011;110(5):1439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu M, Liu H, Dudley SC Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ Res. 2010;107(8):967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park DS, Fishman GI. SCN5A: the greatest HITS collection. J Clin Invest. 2018;128(3):913–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009;326(5949):144–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu X, Kwong JQ, Molkentin JD, Bers DM. Individual cardiac mitochondria undergo rare transient permeability transition pore openings. Circ. Res 2016;118: 834–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gadicherla AK, Wang N, Bulic M, Agullo-Pascual E, Lissoni A, De Smet M, Delmar M, Bultynck G, Krysko DV, Camara A, Schlüter KD, Schulz R, Kwok WM7, Leybaert L. Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res Cardiol. 2017;112(3):27. [DOI] [PubMed] [Google Scholar]
- 72.Boengler K, Stahlhofen S, van de Sand A, Gres P, Ruiz-Meana M, Garcia-Dorado D, Heusch G, Schulz R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res Cardiol. 2009;104(2):141–7. [DOI] [PubMed] [Google Scholar]
- 73.Takahashi M, Hood DA. Protein import into subsarcolemmal and intermyofibrillar skeletal muscle mitochondria. Differential import regulation in distinct subcellular regions. J Biol Chem. 1996;271(44):27285–91. [DOI] [PubMed] [Google Scholar]
- 74.Holmuhamedov EL, Oberlin A, Short K, Terzic A, Jahangir A. Cardiac subsarcolemmal and interfibrillar mitochondria display distinct responsiveness to protection by diazoxide. PLoS One. 2012;7(9):e44667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fekete A, Franklin L, Ikemoto T, Rózsa B, Lendvai B, Sylvester Vizi E, Zelles T. Mechanism of the persistent sodium current activator veratridine-evoked calcium elevation: implication for epilepsy. J. Neurochem 2009;111:745–756. [DOI] [PubMed] [Google Scholar]
- 76.Kovács R, Kardos J, Heinemann U, Kann O. Mitochondrial calcium ion and membrane potential transients follow the pattern of epileptiform discharges in hippocampal slice cultures. J. Neurosci 2005;25:4260–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Griffiths EJ. Reversal of mitochondrial sodium/calcium exchange during metabolic inhibition in rat cardiomyocytes. FEBS Lett. 1999;453:400–404. [DOI] [PubMed] [Google Scholar]
- 78.Bermudez-Hernandez K, Keegan S, Whelan DR, Reid DA, Zagelbaum J, Yin Y, Ma S, Rothenberg E, Fenyö D. A Method for Quantifying Molecular Interactions Using Stochastic Modelling and Super-Resolution Microscopy. Sci Rep. 2017;7(1):14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Montnach J, Agullo-Pascual E, Tadros R, Bezzina CR, Delmar M. Bioinformatic analysis of a plakophilin-2-dependent transcription network: implications for the mechanisms of arrhythmogenic right ventricular cardiomyopathy in humans and in boxer dogs. Europace. 2018;20:iii125–iii132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the article and its Online supplement files.
Expanded Methods available in Online Data Supplement.