

SUMMARY
NMDA receptors (NMDAR) are glutamate-gated ion channels that mediate fast excitatory synaptic transmission in the nervous system. Applying glutamate to outside-out patches containing a single NMDAR, we find that agonist-bound receptors transition to the open state via two conformations, an ‘unconstrained pre-active’ state that contributes to fast synaptic events, and a ‘constrained pre-active’ state that does not. To define how glutamate drives these conformations, we decoupled the ligand-binding domains from specific transmembrane segments for GluN1 and GluN2A. Displacements of the pore-forming M3 segments define the energy of fast opening. However, to enter the unconstrained conformation and contribute to fast signaling, the GluN2 pre-M1 helix must be displaced before the M3 segments move. This pre-M1 displacement is facilitated by the flexibility of the S2-M4 of GluN1 and GluN2A. Thus, outer structures - pre-M1 and S2-M4 - work in concert to remove constraints and prime the channel for rapid opening, facilitating fast synaptic transmission.
Graphical Abstract
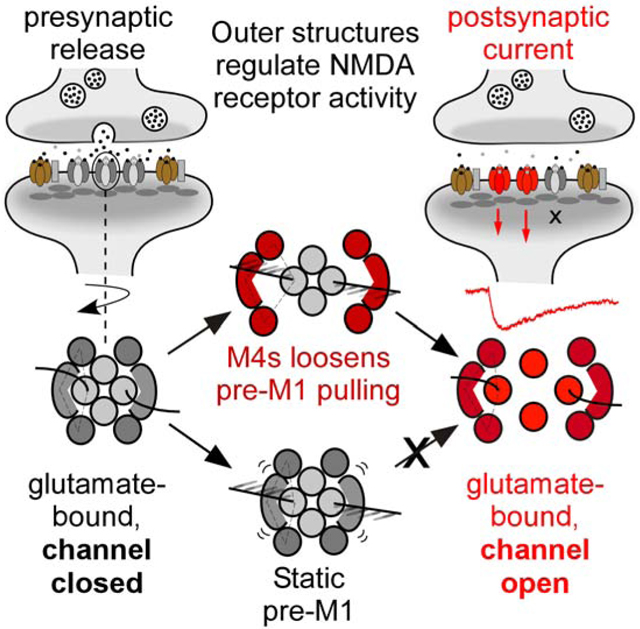
eTOC.
NMDA receptors are neurotransmitter-gated ion channels that regulate higher brain function. Using a unique technique to assay neurotransmitter-induced opening of the ion channel and available high-resolution structures, Amin et al. provides a framework to understand the structural underpinnings that regulate the activity of NMDA receptor activation at synapses.
INTRODUCTION
The transition from the closed, non-conducting conformation to the open, conducting conformation is the central physiological function of ion channels. The preferred low-energy conformation is generally the closed state, and channels require the input of energy to transition to the open state (Hille, 2001). For voltage-gated channels, displacements of the voltage-sensing domains generate this energy (Bezanilla, 2008, Toombes and Swartz, 2014, Ahern et al., 2016), whereas for ligand-gated channels, it arises from agonist-induced conformational changes (Auerbach, 2013, daCosta and Baenziger, 2013, Plested, 2016). High-resolution structures of the closed and open conformations provide the endpoints for ion channel opening, but do not reveal how energy is communicated between structural elements nor potential structural intermediates, which are typically extremely transient and energetically unstable. Indeed, almost all channels transition from the closed to the open state via multiple structural intermediates (Aldrich et al., 1983, Howe et al., 1991, Banke and Traynelis, 2003, Popescu and Auerbach, 2003, Colquhoun and Lape, 2012, Goldschen-Ohm et al., 2013). These structural or conformational intermediates are physiologically critical since they dictate the kinetics and probability of pore opening, and hence how an ion channel contributes to membrane physiology.
Ionotropic glutamate receptors (iGluRs), including AMPA (AMPAR), kainate, and NMDA (NMDAR) receptors, are ligand-gated ion channels that mediate the majority of fast synaptic transmission in the brain (Henley and Wilkinson, 2016, Hansen et al., 2017). iGluRs are highly modular, multi-layered proteins (Figure 1A). To open the ion channel pore, the ligand-binding domain (LBD) must energetically communicate with the pore-forming transmembrane-domain (TMD). While this might arise from direct physical interactions between domains, the most likely pathway for energetic coupling is via a set of short polypeptide linkers, the LBD-TMD linkers, which connect the LBD to specific transmembrane segments (Figures 1B & 1C) (Schmid et al., 2007, Sobolevsky et al., 2009, Talukder et al., 2010, Yelshanskaya et al., 2017, Hansen et al., 2018).
Figure 1. Structure and topology of NMDARs.
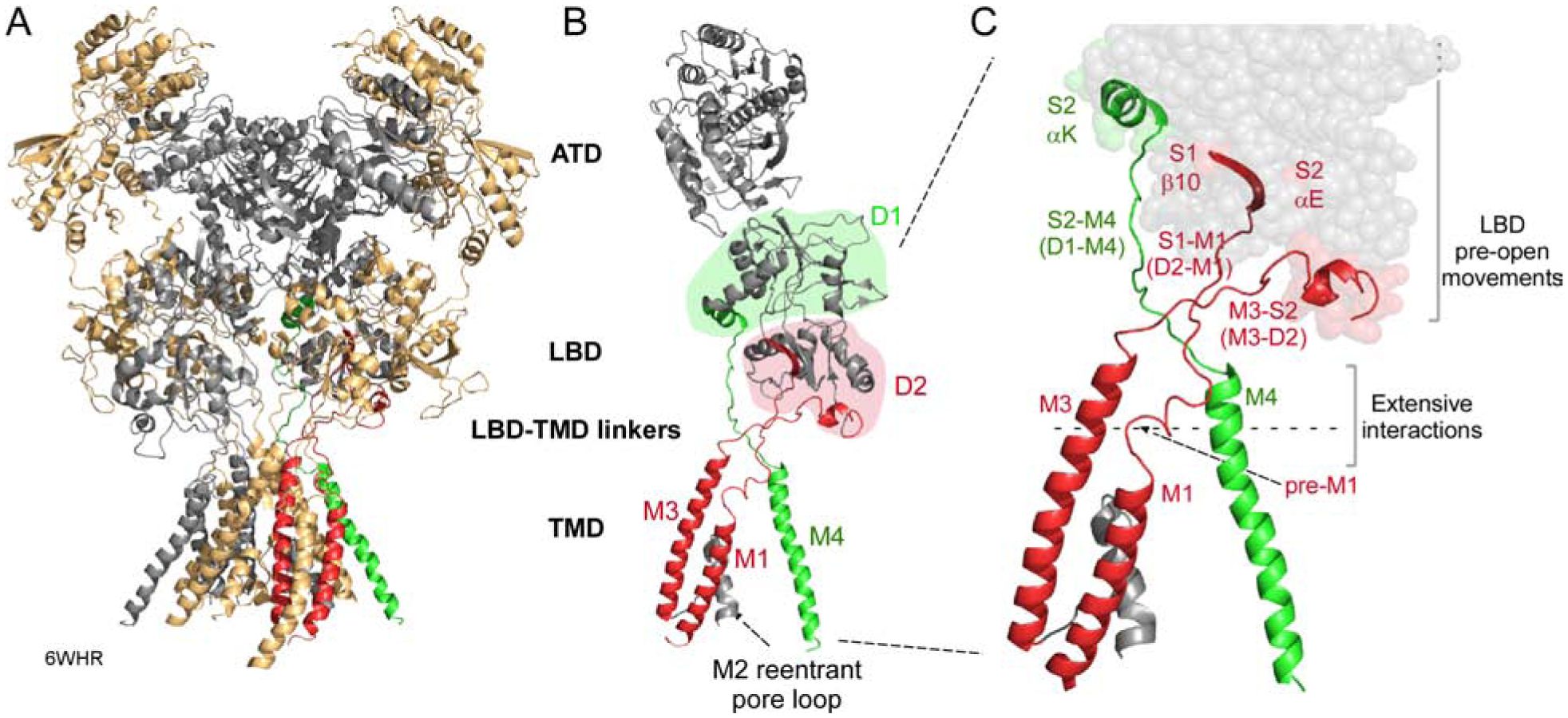
(A) NMDARs consist of four modular domains: extracellular amino-terminal (ATD) and ligand-binding (LBD) domains; the transmembrane domain (TMD) forming the ion channel; and an intracellular C-terminal domain (CTD). Subunits are colored light orange (GluN1) and gray (GluN2B). 6WHR (Chou et al., 2020).
(B) Topology of an individual subunit. LBD lobes are colored green (D1) or red (D2). The ion channel is formed by 3 transmembrane segments, M1, M3, and M4, and an intracellular M2 pore loop. Transmembrane segments are colored according to the LBD lobe to which they are connected.
(C) Bridging the LBD and TMD in each subunit are three LBD-TMD linkers: S1-M1 (D2-M1), M3-S2 (D2-M3), and S2-M4 (D1-M4). The most membrane proximal secondary structures in the LBD are: β10 (S1-M1); αE (M3-S2); and αK (S2-M4). With agonist binding these structures undergo movements that precede channel opening (Zhu et al., 2016, Tajima et al., 2016, Twomey et al., 2017, Chen et al., 2017a, Chou et al., 2020). Pre-M1 is a short helix in S1-M1 that surrounds the corresponding M3 segment (Sobolevsky et al., 2009).
The iGluRs LBD is comprised of two separate polypeptides, S1 and S2, which form a clamshell-like configuration comprised of two lobes, an upper D1 (mainly S1) and a membrane-proximal D2 (mainly S2) (Figure 1B)(Armstrong et al., 1998, Furukawa et al., 2005, Mayer, 2005). The core of the ion channel is the pore domain, consisting of transmembrane segments M1 and M3 and a cytoplasmic re-entrant M2 pore loop (Figure 1B). This pore domain is evolutionarily related to an inverted K+ channel (Wo and Oswald, 1995, Wood et al., 1995). Notably, the linkers coupling M1 (S1-M1) and M3 (M3-S2) to the LBD are D2-linked (Figure 1C). In contrast, the eukaryotic-specific M4 transmembrane segments, which surround the pore domain, are D1-linked (S2-M4).
In iGluRs, agonist binding induces “clamshell closure”, in which the D2 lobe presumably pulls away from the lipid bilayer (Armstrong and Gouaux, 2000, Twomey et al., 2017, Twomey and Sobolevsky, 2018). The M3 segments are the major pore-lining element, with their bundle helical crossing forming a gate (Chang and Kuo, 2008, Sobolevsky et al., 2009). The basic mechanism opening this M3 gate presumably entails mechanical pulling (Armstrong and Gouaux, 2000), with the agonist-induced displacements of the D2 lobe away from the membrane generating tension in the M3-S2 linker, providing the energy necessary for pore opening (Kazi et al., 2014, Ladislav et al., 2018, Twomey and Sobolevsky, 2018, Greger and Mayer, 2019).
The non-pore forming transmembrane segments, M1 and M4, are also displaced during channel opening (Kazi et al., 2013, Twomey et al., 2017, Chen et al., 2017a, Dolino et al., 2017). The S1-M1 linker includes a pre-M1 helix, which surrounds the inner M3 segments and regulates pore opening (Ogden et al., 2017, Gibb et al., 2018, McDaniel et al., 2020, Amin et al., 2020, Perszyk et al., 2020). The S2-M4 linker/M4 segments also regulate gating (Ren et al., 2003, Talukder et al., 2010, Yuan et al., 2014, Hughes and Woodward, 2016, Amin et al., 2017, Chen et al., 2017b, Shi et al., 2019). Still, the contribution of these non-pore forming elements to the energetics and timing of receptor gating remains unknown.
Here, we address the gating mechanism in NMDARs. At most synapses, NMDARs are obligate hetero-tetramers composed of two GluN1 and two GluN2 (A-D) subunits. To define conformational changes between agonist binding and pore opening, we applied glutamate to outside-out patches containing single NMDARs and assessed the time between when glutamate was first applied to when the channel first opened (‘latency to 1st opening’)(Aldrich et al., 1983). With this approach, we identified that wild type GluN1/GluN2A NMDARs, in the transition from agonist-bound closed to the open state, transitions through one of two pre-active conformations: an ‘unconstrained’ state that can rapidly transition to the open state and contributes to fast synaptic signaling, and a ‘constrained’ state that slowly transitions to the open state, presumably due to higher energy requirements, and does not contribute to fast signaling. By decoupling the LBD from the TMD for each specific LBD-TMD linker (S1-M1, M3-S2, and S2-M4, Figures 1B & 1C), we show that the non-pore forming linkers uniquely influence pore-opening by altering the stability of these pre-active conformations. Thus, our results highlight the temporal and coordinated movements from agonist binding to ion channel opening in NMDARs under non-equilibrium conditions, closely mimicking NMDAR activation at excitatory synapses.
RESULTS
To address the coupling between agonist-induced displacements of the LBD and ion channel opening, we recorded from outside-out patches containing a single NMDAR and rapidly applied glutamate (Figure 2A). Patches were continuously bathed in glycine which presumably occurs at synapses. We focused on the time interval between when glutamate was initially applied to the first identifiable opening of the ion channel. We refer to this period as ‘latency to 1st opening’ (Aldrich et al., 1983). This latency has physiological significance since it reflects how efficiently a glutamate signal is translated into channel opening. Further, it is the most direct assay of the coupling between LBD movements and those of the transmembrane segments leading to pore opening since it requires no assumptions about idealization and simultaneously captures non-equilibrium conditions for a single channel.
Figure 2. Agonist applications to outside-out patches containing a single wild-type GluN1/GluN2A.
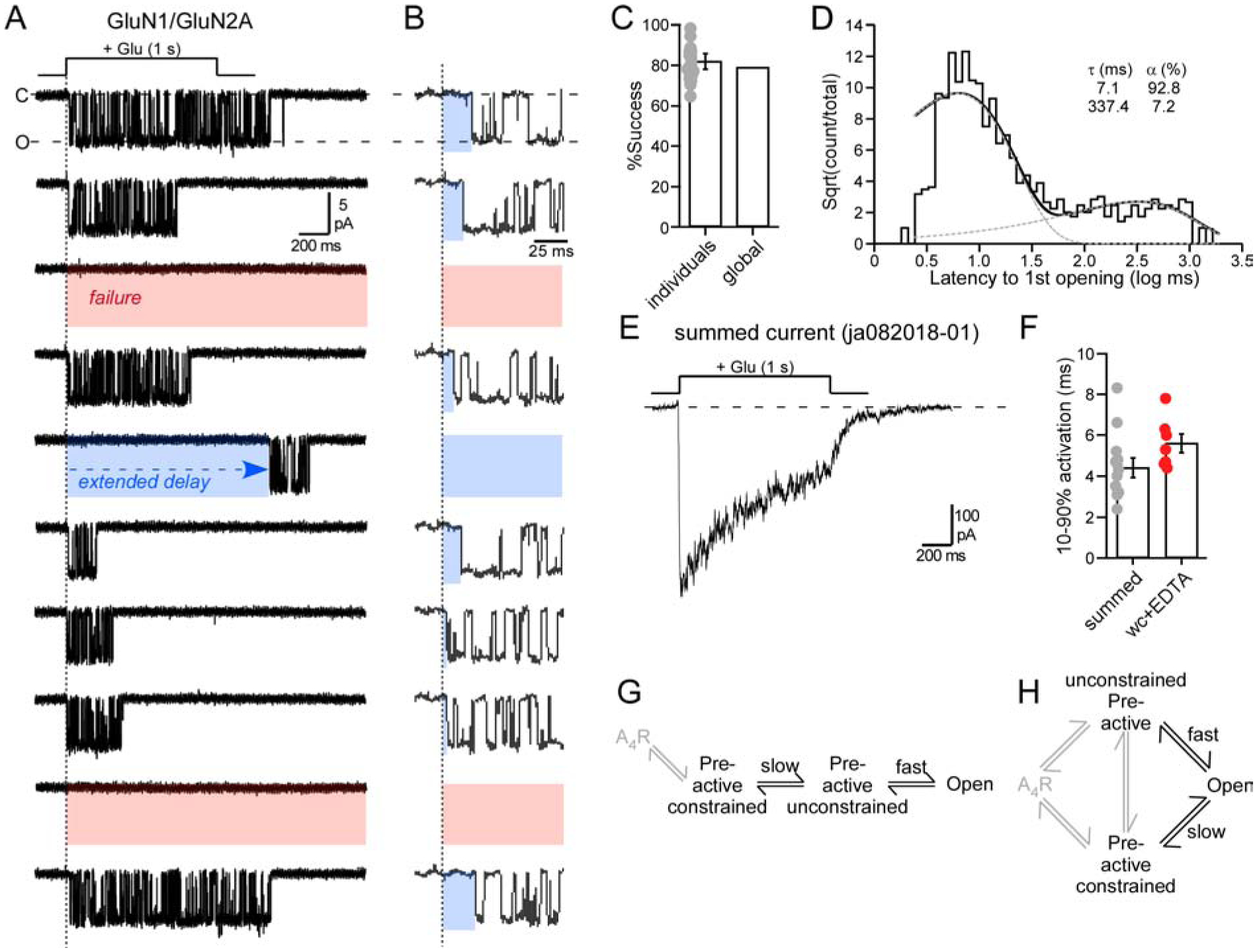
(A & B) Membrane currents from an outside-out patch (10 consecutive traces from same recording) containing a single wild-type GluN1/GluN2A channel. Traces in (B) are those in (A) but on an expanded time scale. The patch was exposed to pulses (1 s) of glutamate (1 mM) in the continuous presence of glycine (0.1 mM). Current responses reflected either successes (channel opening) or failures (red highlight), with successes showing variations to 1st opening (blue highlights). Currents were sampled at 50 kHz (displayed at ~ 1 kHz). Holding potential, −70 mV.
(C) Success rate (1 - failure rate) for the average of all patches (18 total patches, dots represent individual data points) or the global success rate (1011 successes out of 1279 total trials).
(D) Dwell time histogram of the latency to 1st opening. Histogram was best fit by 2 exponentials (dashed lines).
(E) Example summed current. Patch had 140 (123 successes) applications.
(F) Bar graph (mean ± SEM) showing activation rates (10–90% rise time) for summed currents (13 patches) from outside-out patches or from currents in the whole-cell (wc) mode (Table S1). Values were not significantly different (p = 0.069, t-test).
(G & H) Potential pathways for the transition to the open state. A4R is the receptor fully bound with agonist, which occurs rapidly after the start of the glutamate application because we used supersaturating concentrations.
Latency to 1st openings in wild-type GluN1/GluN2A
Wild-type GluN1/GluN2A channels typically activated shortly after glutamate exposure (Figure 2A, trace 1 & 2), but with variations (Figure 2B), including in some instances extended delays (trace 5). There were also instances of failures to open (3rd & 9th traces) (Erreger et al., 2005). For GluN1/GluN2A, we recorded from 18 different outside-out patches with the number of applications varying from 10 to 325 per patch. The latency to 1st opening and success rate (=1 - failures) showed no time dependence (Figure S1), indicating that they could be combined across patches. We made a total of 1279 applications to 18 patches of which 268 were failures. The success rate of the 18 individual patches was 82.0 ± 2% (n = 18) (mean ± SEM, n = number single channel patches), whereas the global success rate was 79% (n = 1011, where n = number of successful events out of 1279 applications) (Figure 2C).
For successes, the mean latency to 1st opening was 48.0 ± 7.9 ms (n = 18) (global mean latency, 47 ms, n = 1011). To characterize these latencies, we generated dwell time histograms (Figure 2D), which were best by 2 exponentials: a fast (τ = 7.1 ms, 92.8%) and a slow (τ = 337.4 ms, 7.2%) component. Thus, of the total successful events (1011), approximately 938 events corresponded to the fast component, while 73 events corresponded to the slow component. As a fraction of total trials (1279), 73.3% corresponded to the fast component, while 5.7% corresponded to the slow component with the remaining events failures.
Fast component events would contribute to synaptic events (Erreger et al., 2005), whereas slow component events (and failures) would not. Hence, for wild-type GluN1/GluN2A, synaptic efficiency - the fast component events out of the total number of trials - would be around 73%. Energetically, the activation energy through the fast component requires 1.96 kT (see STAR Methods).
Properties of summed currents
To further define currents arising in these single channel patches, we summed currents for each individual patch (e.g., Figure 2E) and characterized their properties (Table S1). The activation rate of these summed currents (10–90% rise time, 4.4 ± 0.5 ms, n = 13) was comparable albeit somewhat faster than whole-cell recordings under similar conditions (5.8 ± 0.5, n = 6) (Figure 2F; Table S1), supporting the idea that fast events would contribute to synaptic signaling.
Failures may reflect that receptors are already in a ‘desensitized’ state when glutamate is applied, either because the receptors have not recovered from desensitization or because of intrinsic structural changes (Vance et al., 2013, Yao et al., 2013). Interestingly, there is no relationship (r2 = 0.09) between failures and extent of desensitization for individual patches (Figure S2), highlighting the diverse structural factors affecting NMDAR function. While failures are important physiologically, we will not focus on them in detail.
Given the two kinetic components, we assume there are at least two major agonist-bound closed states between glutamate binding and ion channel opening. We will refer to these states as ‘unconstrained pre-active’ associated with the fast component, and ‘constrained pre-active’ associated with the slow component. These two states could be arranged linearly (Figure 2G) or in parallel (Figure 2H). Subsequent experiments will favor the parallel pathway (Figure 2H) (see Discussion). In this context and in terms of NMDAR physiology, there are two key considerations: (i) The availability of receptors in the ‘unconstrained pre-active’ state, that permits receptors to transition rapidly into an open conformation and contribute to synaptic signaling. We refer to this feature as ‘efficiency’. (ii) The energy required to open the channel in the ‘fast’ pathway. Hence, 73% of the receptors exist in the more efficient ‘unconstrained pre-active’ state and it takes 1.96 kT to open the channel.
Altering the linkage between the LBD and the TMD
Conformational changes in the LBD are coupled to changes in the TMD leading to pore opening via the LBD-TMD linkers (Figure 1C)(Twomey and Sobolevsky, 2018, Chou et al., 2020). To address the energetic coupling between agonist-induced displacements and those of the transmembrane elements leading to pore opening, we inserted glycine residues at different points in the LBD-TMD linkers in the GluN1 and GluN2A subunits and subsequently quantified channel function (Kazi et al., 2014). Our goal with these insertions was to increase the distance and/or flexibility in the LBD-TMD linkers, which would presumably alter the coupling and potentially the efficiency of energy transfer between the pre-open movements of the last secondary structure in the LBD to its associated transmembrane element (Figure 1C). Insertions, when tested, have no notable effect on LBD dynamics (Kazi et al., 2014).
LBD coupling to the M3 segments determines the energy of fast pore opening
The M3s are the main pore lining transmembrane segments and form an activation gate for channel opening (Chang and Kuo, 2008, Sobolevsky et al., 2009). To analyze pore opening, we used AMPAR closed (5WEK) and open (5WEO) structures (Twomey et al., 2017), because the ion channel in 5WEO is in a clear ‘open-like’ structure (Figure S3A). In contrast, for the most high-resolution NMDAR structures (Chou et al., 2020), the ion channel of ‘active’ conformations is not distinct from a ‘closed’ state (Figure S3B). The M3-S2 linkers couple αE in the D2 lobe of the LBD to the M3 segments (Figures 1C, 3A & 3B). These linkers show distinct subunit-specific orientations, but for both A/C (=GluN1) (Figure 3A) and B/D (=GluN2A) (Figure 3B), αE shows a strong displacement away from the approximate M3 activation gate (circle) with pore opening, with this displacement greatest for B/D subunits. To characterize how these M3-S2 linkers energetically contribute to pore opening, we assayed latency to 1st opening for glycine insertions (Table S2)(Kazi et al., 2014), either GluN1 (G666+1G) or GluN2A (G664+1G) (Figures 3C–3J).
Figure 3. M3-S2 insertions in GluN1 or GluN2A alter the energy for fast opening.
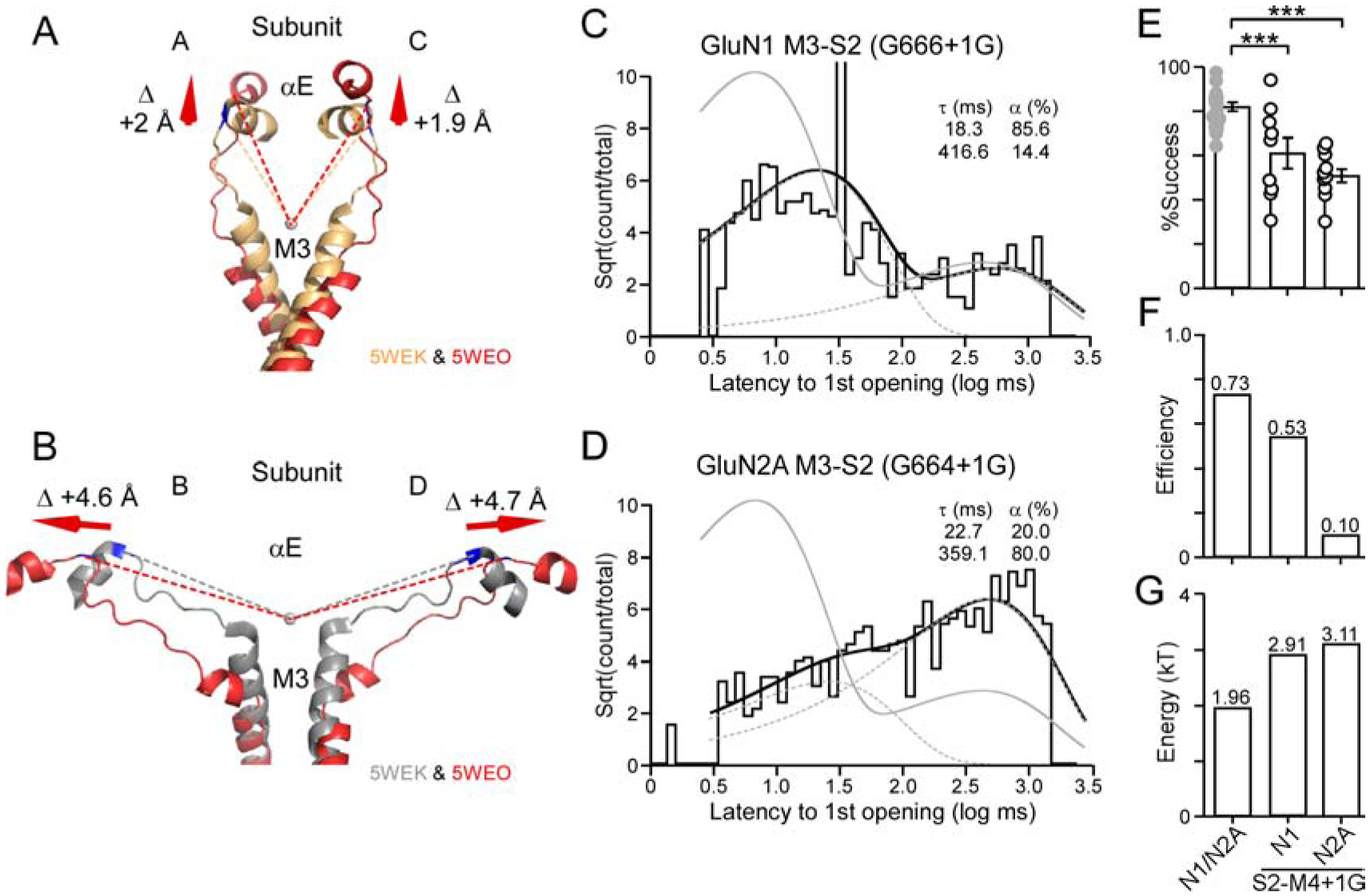
(A & B) Overlaid AMPAR structures of αE, M3-S2, and the top of M3 in the closed (5WEK) or open (5WEO) conformations (Twomey et al., 2017). Closed state is light gold (A/C)(~GluN1) (A) or gray (B/D)(~GluN2A) (B). Open state is red (D2-associated). Values shown are the difference between the closed and open states measured from the circle at the mouth of the pore to the last αC in αE (see STAR Methods).
(C & D) Dwell time histogram for insertions in GluN1 M3-S2 (G666+1G) (C) or in GluN2A M3-S2 (G664+1G) (D). Histograms were best fit by 2 exponentials (dashed lines). Gray line is wild type.
(E) Success rate (1 - failure rate) for the average of all patches for wild type, GluN1 M3-S2 +1G (9 total patches) (global success rate, 0.62; 315 out of 505 trials), or GluN2A M3-S2 +1G (10 total patches) (global success rate, 0.51; 415 out of 808 trials). ***p < 0.001, t-test.
(F) Efficiency (=successes × fraction fast component)/total number of trials) of fast component.
(G) Energy required for fast component is derived from a log transformation of the forward rate constant for activation (see STAR Methods).
Notably, fits to the dwell time histogram for the latency to 1st opening for M3-S2 insertions in either GluN1 (Figure 3C) or GluN2A (Figure 3D) required two kinetic components. Hence, transitions to a ‘fast’ and a ‘slow’ pathway still exist as in wild type. Nevertheless, these components were shifted relative to wild type. In addition, these M3-S2 insertions showed significantly reduced successes (Figure 3E).
For the GluN1 M3-S2 insertion, the efficiency of the fast component for GluN1 was reduced from 0.73 to 0.53 (Figure 3F), reflecting a reduced occupancy of the fast component as well as increased failures. Hence, receptors could transition ‘rapidly’ into an open state with about 53% of the glutamate applications. On the other hand, 2.91 kT (versus 1.96 kT for wild type) of energy was needed to open the channel in this ‘fast’ pathway (Figure 3G), presumably reflecting inefficient energy transfer from the LBD to M3 helix. In GluN2A, M3-S2 insertions severely reduced efficiency to 0.10 (Figure 3F) and the energy required for the rapid transition was 3.11 kT (Figure 3G).
In summary, the reduced entry into the fast component could arise because receptors when glutamate bound occupy the ‘unconstrained pre-active’ state only about 53% (GluN1) or 10% (GluN2A) of the time; alternatively, given the reduced energy transfer to the M3 segment, the energy generated in the M3 segments is insufficient to overcome other structural constraints. Importantly, a greater amount of energy is required to open the channel even when in the ‘fast’ pathway (Figure 3G). Hence, the mechanically pulling entailing the M3-S2 linkers is directly involved in opening the ion channel, consistent with its central role in iGluR gating (Kazi et al., 2014, Twomey et al., 2017, Chou et al., 2020).
Insertions in the GluN1 or GluN2A S1-M1 linkers attenuate receptor gating
In terms of S1-M1 linkers, the most proximal secondary structure in the LBD is the D2-associated β10 (Figures 1C, 4A & 4D)(Figures S4 & S5). A notable feature of the S1-M1 linkers are the pre-M1 helices that form a ring around the inner M3 segments (Sobolevsky et al., 2009, Karakas and Furukawa, 2014, Lee et al., 2014).
Figure 4. S1-M1 insertions in GluN1 or GluN2A reduce NMDAR gating.
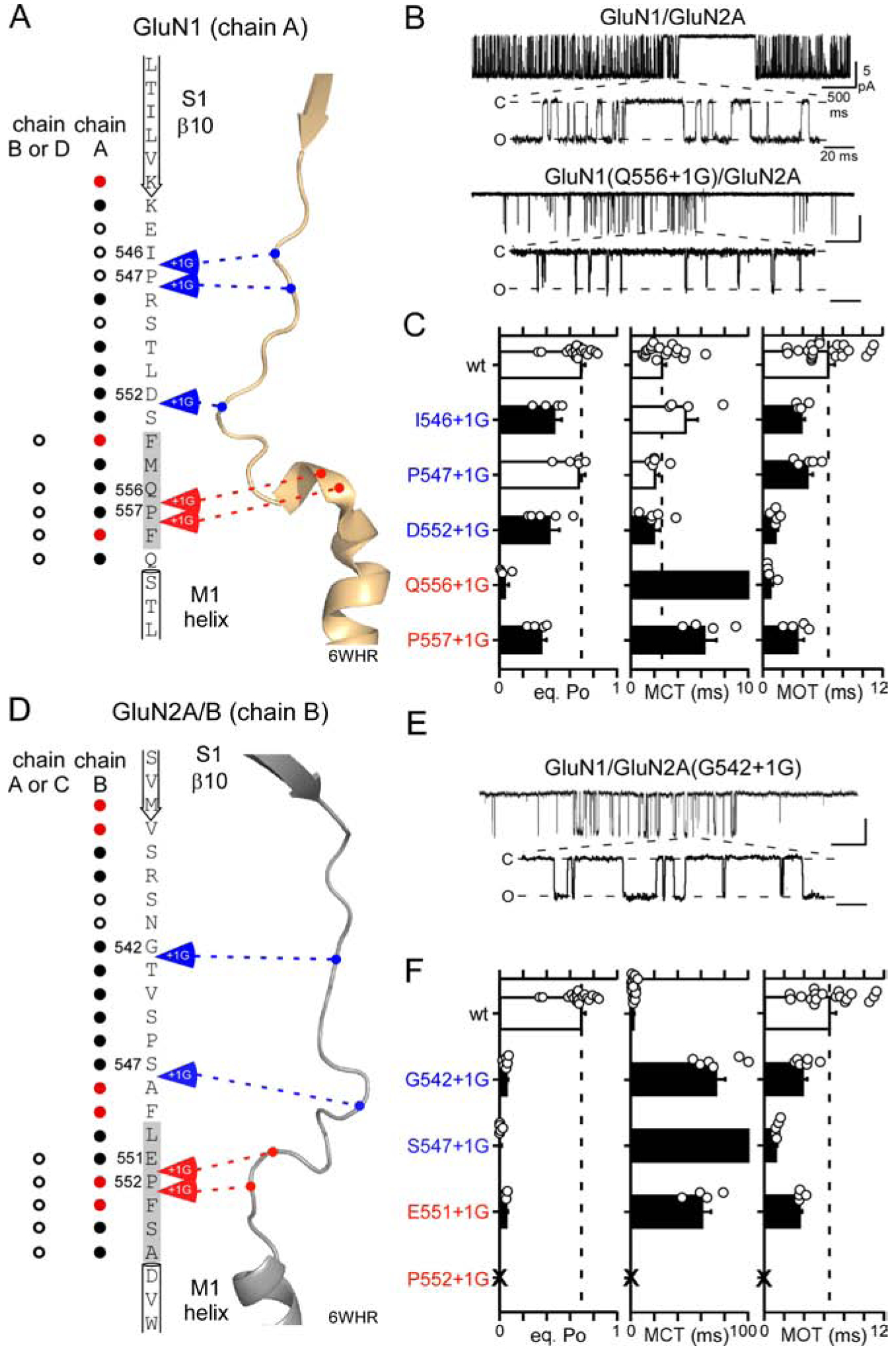
(A & D) Primary amino acid sequences and tertiary structures of the GluN1 (A) and GluN2B (D) S1-M1 linkers and surrounding regions. Gray shading in primary sequence highlight pre-M1 helices which is often not well-defined in GluN2B subunits (Figure S5). Circles indicate number of side chains in the closed state (6WHR) around that position [open, <6 Å; solid, 6–10 Å; red, >10 Å]. Arrows indicate insertions sites, with those in blue indicating few local interactions, whereas those in red indicate more interactions.
(B & E) Representative traces from on-cell single-channel patches at −100 mVs. Experiments were for wild-type and constructs containing single insertions either in GluN1 (B) or GluN2A (E) S1-M1. In each panel, upper trace shows 5 seconds of recording (1 kHz filtering), while lower trace is an expanded section (4 kHz). Closed (C) and open (O) states are indicated.
(C & F) Single channel eq. Po, MCT, and MOT (mean ± SEM) for wild-type and S1-M1 insertion constructs either in GluN1 (C) or GluN2A (F) (see Table S3). Dashed lines indicate mean wild-type values. Solid bars indicate values significantly different from wild type (p < 0.05, t-test). ‘X’ indicates no single channel currents could be detected.
We inserted glycines, individually, at largely equivalent points in the GluN1 (Figure 4A) and GluN2A (Figure 4D) S1-M1 linkers. The membrane-proximal residues of S1-M1 in GluN1 and GluN2A, encompassing the pre-M1 helices (highlighted in gray in primary sequence), interact with other residues in the closed state (red arrows), whereas the remainder of the S1-M1 have fewer local interactions (blue arrows). Notably, there is a membrane-proximal proline (P) conserved across all mammalian iGluR subunits (Figures S4B & S5B) [GluN1(P557) & GluN2A(P552)] as well as an additional proline in GluN1 (P547) (Alsaloum et al., 2016). Given the rigid secondary structure of proline, we inserted glycines on either side of these prolines. Numerous disease-associated variants have been identified at the membrane-proximal prolines (Ogden et al., 2017, Soto et al., 2019). To assay receptor function, we initially recorded the activity of single-channel patches in the on-cell mode because it is much easier to obtain recordings than outside-out patches.
Wild-type GluN1/GluN2A NMDARs (Figure 4B) show an equilibrium open probability (eq. Po) of around 0.70 (0.70 ± 0.03, n = 18)(mean ± SEM, n = number of independent on-cell patches)(Figure 4C)(Table S3). With one exception, GluN1(P552+1G), all S1-M1 insertions in the GluN1 (Figure 4C, left panel) and GluN2A (Figure 4F, left panel) significantly attenuated (black bars) single channel activity (Table S3). These effects occurred whether insertions were made membrane-proximal or membrane-distal.
There are four key observations from these S1-M1 insertions. First, in general, insertions in the GluN2A S1-M1 linkers have a much stronger effect on gating than those in GluN1. Indeed, the eq. Po of the insertions in GluN2A were considerably less than 0.1 whereas this occurred only for GluN1(Q556+1G) (Figures 4C & 4F, left panel; Table S3). Second, the most consistent effect of the insertions was a significant reduction in mean open time (MOT) (Figures 4C & 4F, right panel). Third, while the insertions in GluN1 and GluN2A had about the same effect on reducing MOT, the stronger effect on eq. Po in GluN2A reflects that they had a much stronger effect on increasing mean closed time (MCT) (Figures 4C & 4F, middle panel; note the changed scale in Figure 4F). Finally, insertions around the membrane-proximal proline, as with disease-associated variants at this proline (Ogden et al., 2017), strongly inhibited gating.
In summary, a presumed decoupling of the LBD (β10) from the pre-M1 helices strongly inhibits receptor gating with these effects stronger in GluN2A than in GluN1. In general, these results parallel those for M3-S2 insertions (Table S2)(Kazi et al., 2014).
The pre-M1 helices regulate the efficiency of ion channel opening
The S1-M1 linkers couple β10 in the D2 lobe of the LBD to the pre-M1 helices/M1 segments (Figure 1C)(see Figure S6). With the transition to the open state, β10 shows a strong displacement away from the M3 activation gate (circle) for both A/C (=GluN1) (Figure 5A) and B/D (=GluN2A) (Figure 5B), with this displacement greatest for B/D subunits. Hence, S1-M1 linker-mediated processes are also driven by the D2 moving away from the activation gate upon agonist binding, paralleling those in M3-S2 linkers.
Figure 5. Insertions in S1-M1 linkers slow receptor activation.
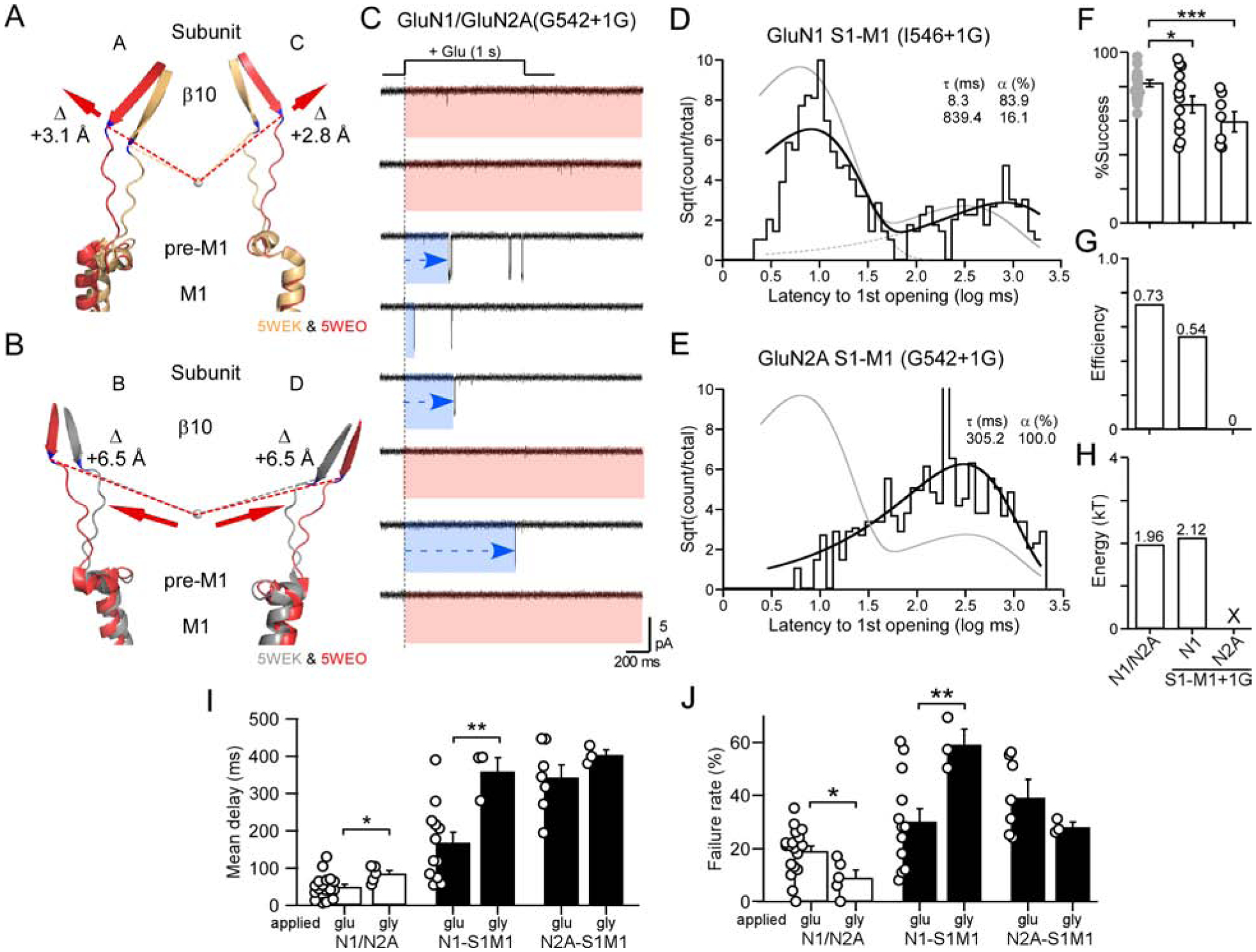
(A & B) Overlaid AMPAR structures of β10, S1-M1, pre-M1, and the top of M1 (see Figures 3A & 3B for details).
(C) Current traces from an outside-out patch of a single NMDAR containing an insertion in GluN2A S1-M1 (G542+1G). Note frequent extended delays to opening (blue).
(D & E) Dwell time histograms for GluN1 S1-M1 (I546+1G) (D) (best fit by 2 exponentials, dashed lines) and GluN2A S1-M1 (G642+1G) (E) (single exponential).
(F-H) Data analyzed and displayed as in Figures 3E–3G.
(F) Success rates for wild type, GluN1 S1-M1+1G (12 total patches) (global success rate, 0.65; 414 out of 639 trials), and GluN2A S1-M1+1G (7 total patches) (global success rate, 0.55; 311 out of 561 trials). *p < 0.05, ***p < 0.001, t-test.
(I & J) Glycine applications, in continuous glutamate, to single channel patches. Mean delay to 1st opening (I) and failures (J) (mean ± SEM) for wild-type and insertions in S1-M1s. Solid bars indicate values significantly different from wild type; asterisks indicate those different from corresponding glutamate application (*p < 0.05,*p<0.01, ANOVA, Tukey pairwise). For glycine applications: wild-type, n = 5; GluN1(I546+1G), n = 3; and GluN2A(G542+1G), n = 3.
To address the role of β10 agonist-induced displacements in channel opening, we rapidly applied glutamate to single channel patches containing S1-M1 insertions in either GluN1 or GluN2A. For these experiments, we selected insertions, I546+1G in GluN1 and G542+1G in GluN2A (Figure 5C), that were membrane-distal and less likely to have extensive local interactions. The major effect of these insertions is to presumably remove the mechanical coupling between β10 and displacement of the pre-M1 helices and hence reduce the efficiency of energy transfer as in M3-S2 insertions.
For GluN1 S1-M1 insertions, the dwell time histogram was best fit by 2 exponentials (Figure 5D), and there was a weak albeit significant decrease in successes (Figure 5F). Correspondingly, efficiency was reduced from 0.73 to 0.54 (Figure 5G), similar to what is observed in GluN1 M3-S2 insertions (Figure 3). However, in contrast to what is observed for GluN1 M3-S2 insertions, decoupling the GluN1 pre-M1 helix had limited effects on the energy required for the ‘fast’ pathway (1.96 kT versus 2.12 kT) (Figure 5H). Hence, decoupling the GluN1 pre-M1 helices from agonist-induced displacements reduces the occupancy of the ‘unconstrained pre-active’ state, but does not alter the energy required for fast opening.
On the other hand, decoupling the GluN2A pre-M1 helices has a dramatic effect on the dwell time histogram, which now only has a single slow component (Figure 5E). There is a significant reduction in successes (Figure 5F). However, given the lack of a fast component, there is no efficiency (Figure 5G). Hence, when the GluN2A pre-M1 helix is decoupled from the LBD, the receptor is no longer able to enter into the ‘unconstrained pre-active’ state.
In summary, while the on-cell data superficially suggest a similarity between decoupling S1-M1 and M3-S2 from the LBD, there are notable differences: decoupling the pre-M1 helices from the LBD either inefficiently (GluN1 pre-M1) or are completely unable (GluN2A pre-M1) to enter the fast pathway. Most likely (see Discussion), entry into the fast pathway via the ‘unconstrained pre-active’ state reflects where at least one GluN2 pre-M1 is displaced and presumably at least one GluN1 pre-M1, though energetically the GluN1 pre-M1 is less critical. In contrast, the ‘constrained pre-active’ state reflects that neither GluN2 pre-M1 helices are displaced. This interpretation is consistent with recent results suggesting that the GluN2 pre-M1 helices regulate NMDAR pore opening (Gibb et al., 2018, McDaniel et al., 2020).
Non-equilibrium gating for glycine binding
For our outside-out experiments, glutamate was applied in the continuous presence of glycine. To test non-equilibrium gating for glycine binding to GluN1, we rapidly applied glycine in the continuous presence of glutamate (Figures 5I & 5J). Given the challenge of these experiments, we collected a limited number of patches and characterized the mean latency to 1st opening.
For decoupling the GluN1 pre-M1 helices, glycine activation caused the mean latency for GluN1 to be significantly slowed compared to glutamate activation (Figure 5I) as well as an increase in failure rate (Figure 5J), highlighting the role of non-equilibrium gating for the glycine-binding subunit. Notably, for decoupling the GluN2A pre-M1, flipping the agonists did not have any significant effect on either the mean latency (Figure 5I) or the failure rate (Figure 5J). Hence, these experiments highlight two important points. Non-equilibrium conditions are important for defining kinetics. Second, independent of agonist, displacement of GluN2 pre-M1 is always the rate-limiting step (Gibb et al., 2018).
Insertions in the GluN1 or GluN2A S2-M4 linkers enhance receptor gating
For S2-M4 linkers, the most proximal secondary structure in the LBD is αK in the D1 lobe (Figures 1C, 6A & 6D) (Figures S7 & S8). We inserted glycines (arrows) at largely equivalent positions in GluN1 (Figure 6A) and GluN2A (Figure 6D) S2-M4s. There is also a membrane-proximal leucine (L) that in GluN2A (L812) has disease-associated variants (Yuan et al., 2014). We therefore inserted glycines around this leucine in GluN2A (Q811+1G & L812+1G) and in the homologous leucine in GluN1 (T807+1G & L808+1G).
Figure 6. Insertions in the GluN1 or GluN2A S2-M4 linkers potentiate gating.
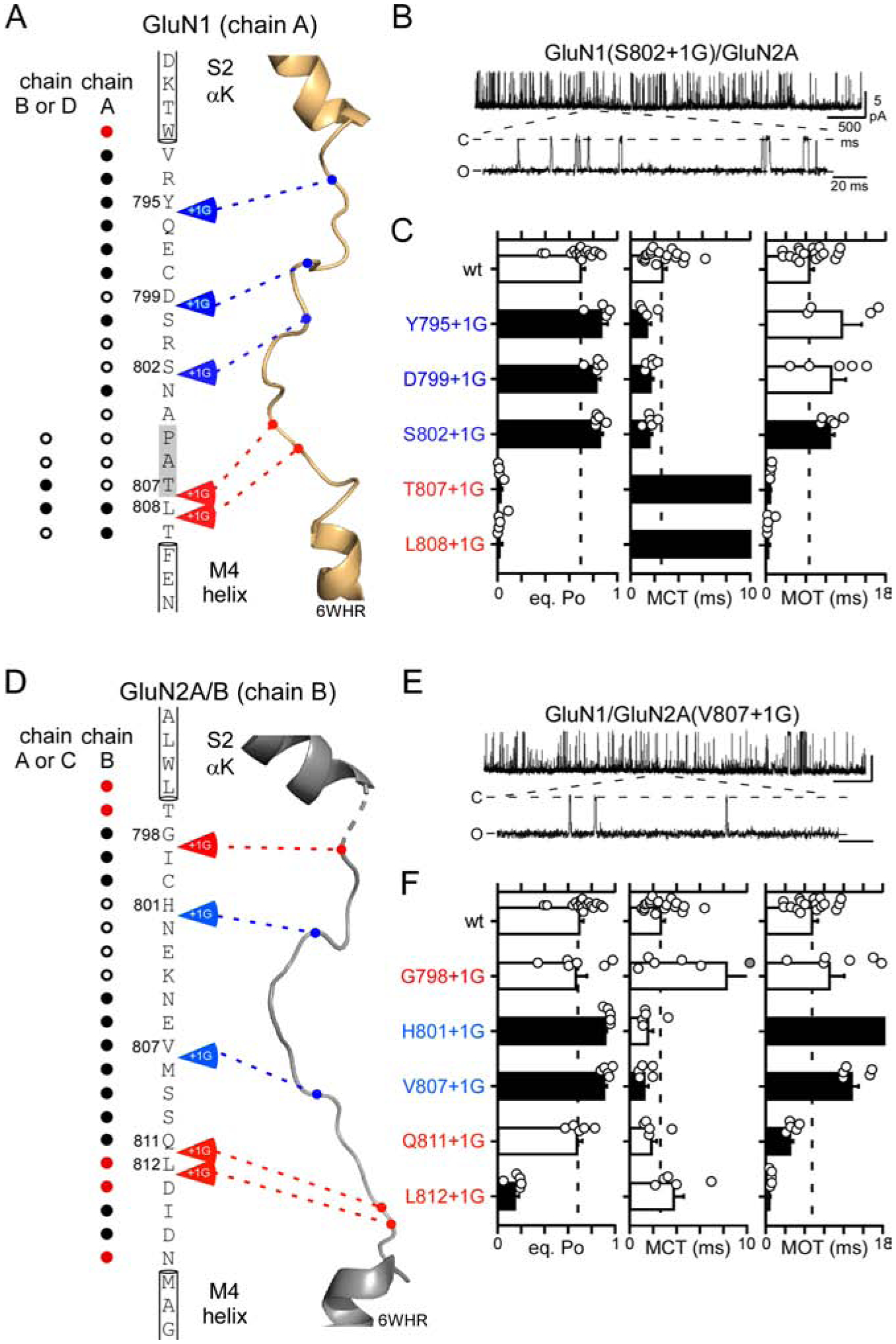
(A & D) Primary amino acid sequences from the GluN1 and GluN2A S2-M4 linkers and surrounding regions.
(B & E) Representative traces of S2-M4 insertion constructs.
(C & F) Single channel eq. Po, MCT, and MOT (mean ± SEM) for wild-type and S2-M4 insertions either in GluN1 (C) or GluN2A (F) (Table S4).
In contrast to insertions in M3-S2 (Table S2) or S1-M1 (Figure 4; Table S3), S2-M4 insertions had more diverse effects. Certain insertions had no effect on eq. Po (Figures 6C & 6F, open bars), and a subset of insertions inhibited gating (T807+1G & L808+1G in GluN1 & L812+1G in GluN2A) (Figures 6C & 6F, black bars). However, these effects occurred only at insertions around the membrane-proximal regions in GluN1 (T807+1G & L808+1G) and GluN2A (L812+1G) or at a position proximal to αK in GluN2A (G798). Notably, all of these insertions occurred at positions proximal to other structural elements (Figures 6A & 6D).
The most distinctive and consistent phenotype of S2-M4 insertions was a potentiation of receptor gating (Figures 6). Indeed, despite a relatively high wild-type eq. PO (about 0.7), S2-M4 insertions in either GluN1 (Y795+1G, D799+1G, & S802+1G) or GluN2A (H801+1G & V807+1G) significantly increased eq. Po (Figures 6C & 6F). These increases in eq. PO generally arose through decreases in MCT in GluN1 (Figure 6C) or increases in MOT in GluN2A (Figure 6F).
In summary, the most consistent phenotype for S2-M4 insertions was receptor potentiation. Notably, this potentiation phenotype was associated with insertions located away from sites of local interactions (blue arrows, Figures 6A & 6D), suggesting that these insertions were mainly affecting the properties of S2-M4 rather than local interactions. Hence, GluN1 or GluN2A S2-M4 insertions, which add length or flexibility to S2-M4 linkers, enhance receptor gating.
S2-M4 insertions in GluN1 and GluN2A enhance gating efficiency
The S2-M4 linkers couple αK in the D1 lobe to the M4 segments (Figures 1C, 7A & 7B)(see also Figure S9). In the transition to the open state, the D1-associated αK shows a displacement towards the M3 activation gate (circle) for both A/C (=GluN1) (Figure 7A) and B/D (=GluN2A) (Figure 7B). Hence, in contrast to D2-associated linkers, S2-M4 linker-mediated processes are driven by D1 moving towards the activation gate upon agonist binding.
Figure 7. NMDARs containing insertions in GluN1 or GluN2A S2-M4 linkers only activate through the fast pathway.
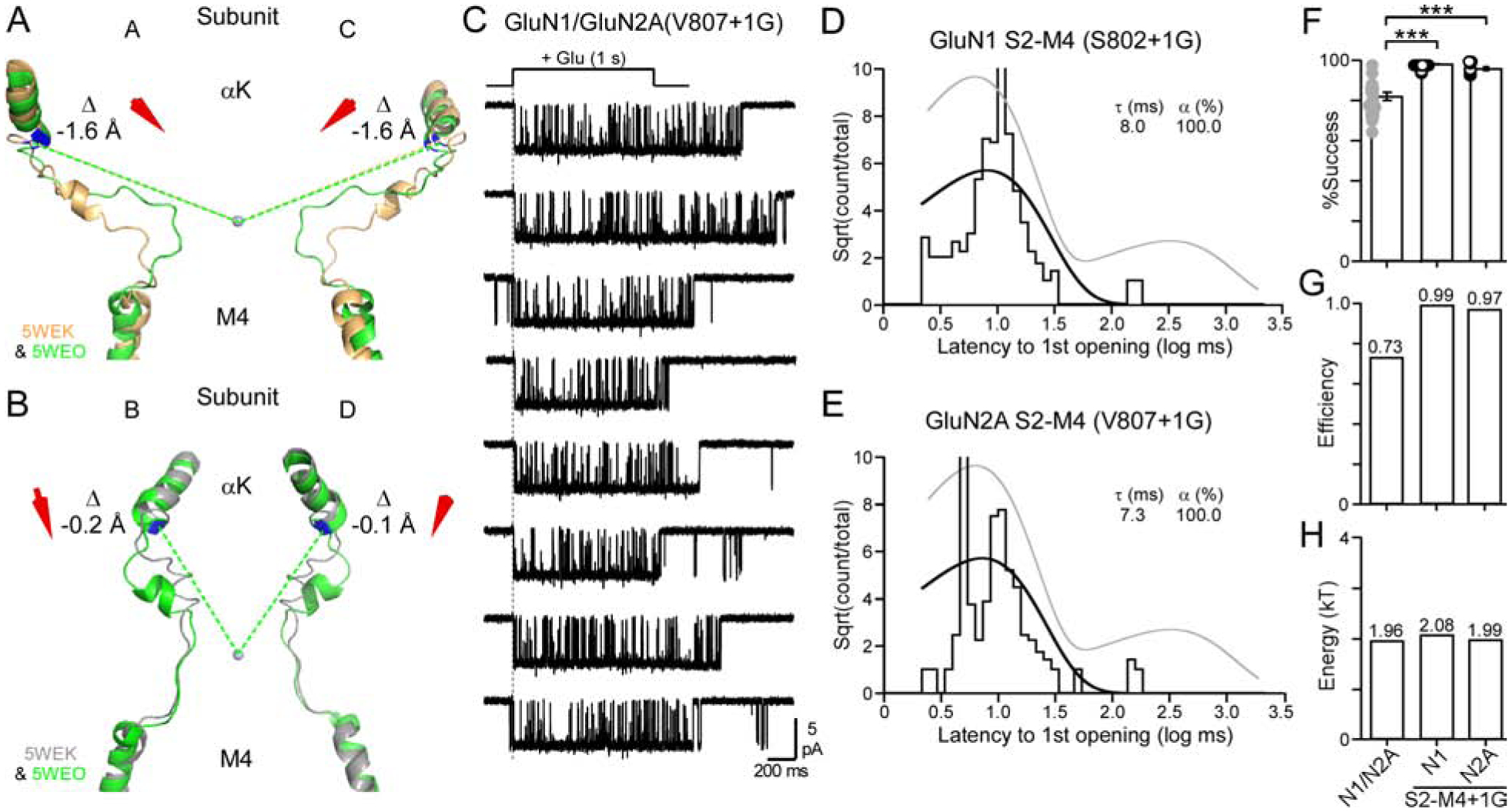
(A & B) Overlaid AMPAR structures of αK, S2-M4, and the top of M4, displayed as in Figures 3A & 3B except that the open state is green (D1-associated).
(C) Current traces from an outside-out patch of a single NMDAR containing an insertion in the GluN2A S2-M4 (V807+1G). See Figure 2A for details.
(D & E) Dwell time histograms for GluN1 S2-M4 (S802+1G) (D) or GluN2A S2-M4 (V807+1G) (E). Histograms best fit by single exponentials.
(F-H) Data displayed and analyzed as in Figures 3E–3G.
(F) Success rates for wild type, GluN1 S2-M4+1G (10 total patches) (global success rate, 0.99; 304 out of 304 trials), and GluN2A S2-M4+1G (7 total patches) (global success rate, 0.97; 213 out of 219 trials). ***p < 0.001, t-test.
To address the role of αK and S2-M4 linkers in agonist-induced displacements to open the channel, we applied glutamate to single channel patches containing S2-M4 insertions in either GluN1 or GluN2A (Figures 7C–7H). Notably, for either GluN1 (Figure 7D) or GluN2A (Figures 7C & 7E) insertions, the dwell time histogram was best fit by a single exponential, and in both cases, there was only a ‘fast’ component. Indeed, for both constructs, failures where largely non-existent (Figures 7F), leading to nearly 100% efficiency (Figure 7G). The energy for the fast component was notably indistinguishable from wild type GluN1/GluN2A (Figure 7H).
In summary, S2-M4 insertions in either GluN1 or GluN2A had largely comparable effects on receptor function, dramatically enhancing receptor activation. Importantly, these S2-M4 insertions did not reduce the energy of the fast transition and hence did not enhance receptor activation by easing the transition to the open state. Rather, given the near 100% efficiency, they enhance gating by allowing receptors to always enter the ‘unconstrained pre-active’ state. Given that the ‘unconstrained pre-active’ state reflects the displacement of the GluN2A pre-M1 helices, we assume that insertions in GluN1 or GluN2A S2-M4, and correspondingly the presumed downward movement of D1, facilitates displacement of the GluN2A pre-M1 helices.
DISCUSSION
To assay NMDAR activation, we applied glutamate to single NMDAR patches and measured the ‘latency to 1st opening’, the time between when glutamate was initially applied to when the channel first opened. This latency assays events occurring between agonist binding and ion channel opening. Using this approach, we found that GluN1/GluN2A NMDARs transition to the open state through, at minimum, two different intermediate pathways: a fast pathway referred to as the ‘unconstrained pre-active’ that can contribute to fast synaptic events, and a slow pathway referred to as the ‘constrained pre-active’ that does not (Figure 2). Given that we can make manipulations to the receptors such that it visits only the slow (Figure 5) or fast (Figure 7) pathways indicates that entry into these states is an either/or process and hence occurs in a parallel fashion (Figures 2H & 8).
Figure 8. Outer structures must rearrange for efficient pore opening.
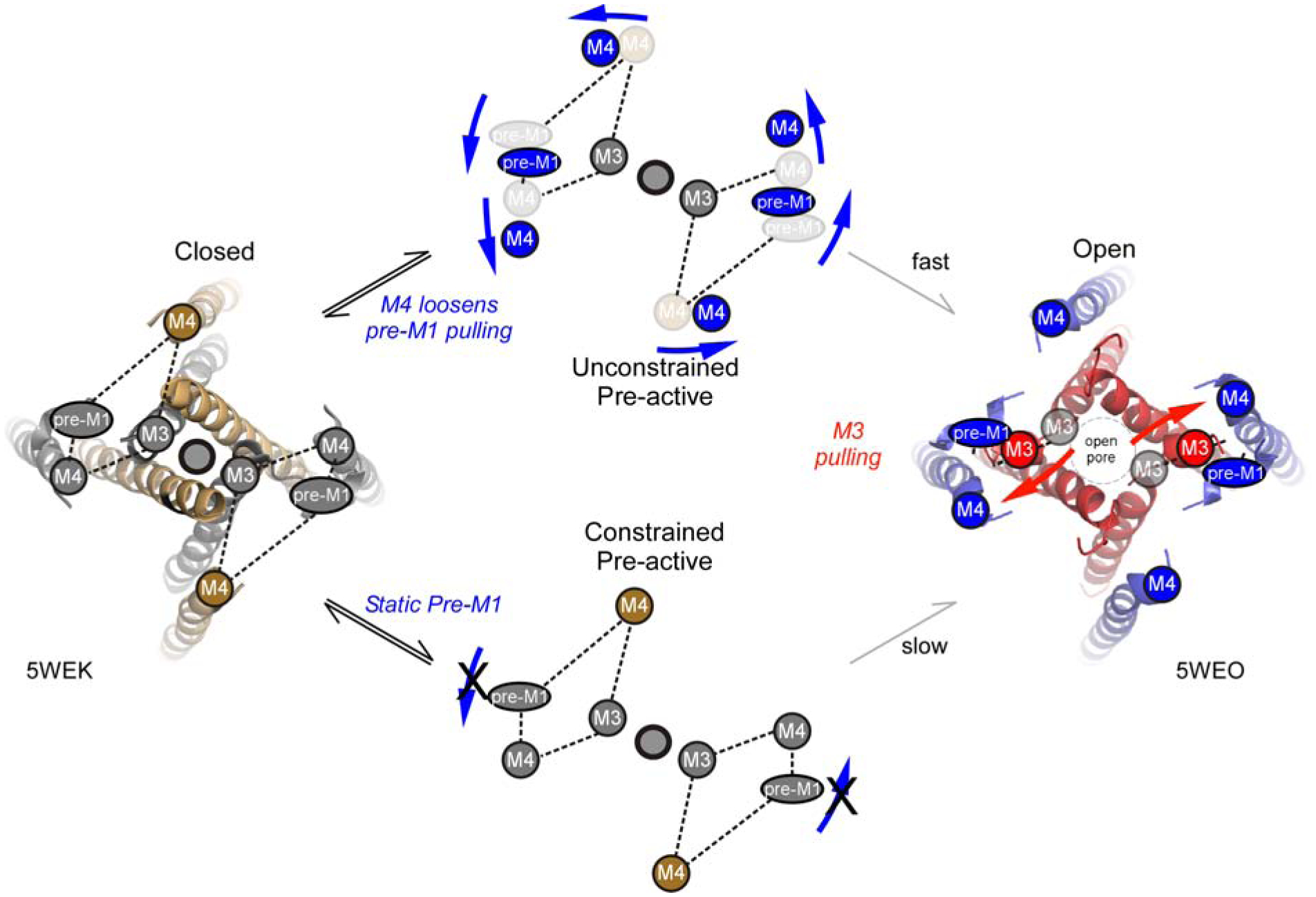
Model of NMDAR activation. Movements of the outer structures - GluN1 M4 (gold circle) and GluN2A pre-M1 and M4 (inner gray circles) - regulate entry from agonist-bound closed (A4R) (left) to either the unconstrained (upper) or constrained (lower) pre-active states. To efficiently enter the unconstrained state, the pre-M1 helices as well as the S2-M4/M4 must be displaced (upper). In contrast, if the GluN2A pre-M1 helix is not displaced, the receptor enters the constrained state, where it still can open but requires more energy to do so (lower). Pre-activation movements are shown in blue, whereas pore opening movements are shown in red.
The entry into these intermediate pathways will determine how strongly NMDARs contribute to synaptic activity. Notably, decoupling the GluN2A pre-M1 helix from the D2-lobe displacement completely removes the ability of receptors to enter the ‘fast’ pathway (Figure 5), highlighting the importance GluN2 pre-M1 displacement to channel opening (Ogden et al., 2017, McDaniel et al., 2020, Perszyk et al., 2020). In addition, insertions in the GluN1 and GluN2A S2-M4 linkers, possibly by mimicking the D1-lobes downward displacement of αK (Figures 7A & 7B)(Twomey et al., 2017), drives receptors into the ‘unconstrained pre-active’ state (Figure 7). Thus, the outer linkers - GluN2A pre-M1 and GluN1 and GluN2A S2-M4/M4 - play critical roles in regulating how NMDARs contribute to synaptic signaling.
Our experiments identify that all LBD-TMD linkers - S1-M1, M3-S2, and S2-M4 - contribute to the fast process of NMDAR activation, but they do so at different points in this process. Notably, the rapid opening of the ion channel requires 1.96 kT of energy (Figure 2). Predictably, the energetic requirement for the fast opening pathway is increased when the central GluN1 (2.91 kT) or GluN2 (3.11 kT) M3 segments are decoupled from the LBD. In contrast, decoupling the outer linkers have no effect on the energetics of the fast component, but rather altered the probability of entering either the ‘unconstrained’ or ‘constrained’ pre-active states (Figures 5 & 7). Hence, S1-M1/pre-M1 and S2-M4/M4 movements are not strongly involved in the final step of pore opening, but rather determine pre-channel opening conformational changes (Figure 8).
D1-versus D2-lobes in opening the ion channel
In terms of channel opening, LBD movements can be divided into two general categories (Twomey et al., 2017): D2-lobe movements, which entails β10 (S1-M1/pre-M1) and αE (M3-S2/M3), and D1-lobe movements, which entails αK (S2-M4/M4). D2-associated movements for both A/C (~GluN1) and B/D (~GluN2) show strong displacement away from the activation gate (Figures 3A, 3B, 5A, & 5B). As would be expected with mechanical coupling, decoupling pre-M1 (insertions in S1-M1) or M3 (insertions in M3-S2) from D2 in either GluN1 or GluN2A strongly attenuated gating. Nevertheless, as assayed by glutamate applications to outside-out patches, the pre-M1 helices and the M3 segments contribute differently to the pore opening process.
Previous experiments have highlighted a gating triad between the GluN2A pre-M1 helices and the M3 segments and GluN1 S2-M4/M4s (Ogden et al., 2017, Gibb et al., 2018, McDaniel et al., 2020, Perszyk et al., 2020). Our results strongly support the role of the GluN2A pre-M1 helix in regulating NMDAR channel opening. Indeed, if the GluN2A pre-M1 helices are decoupled, that is they can no longer receive sufficient mechanical displacement from the LBD, the receptor can no longer enter the ‘unconstrained pre-active’ state (Figure 5E) and makes no contribution to fast synaptic activity (Figure 5G).
In contrast, the D1-associated linker show a net displacement towards the M3 activation gate (Figures 7A & 7B). Insertions in either GluN1 or GluN2A S2-M4 potentiated receptor gating (Figure 5). These insertions may be mimicking the downward displacement of D1 by reliving tension in the S2-M4, though additional experiments are needed to verify this alternative. It is unlikely to reflect S2-M4 unwinding (Twomey et al., 2017), since this is fairly A/C subunit specific (Figures 7A & 7B). In either case, these results indicate that the D1 lobe is not passive in channel activation, but rather plays an active role in modulating ion channel opening. Interestingly, allosteric modulators at the extracellular ATD act by inducing a rolling movement in the LBD (Tajima et al., 2016, Esmenjaud et al., 2019, Chou et al., 2020) that may act on the αK helix, to either ‘loosen’ S2-M4 with positive allosteric modulators or ‘tighten’ S2-M4 with negative allosteric modulators. Hence, in addition to its active role, the S2-M4 linkers may represent a pathway for allosteric regulation of gating (Talukder et al., 2010, Yuan et al., 2014, Chen et al., 2017b). Still, this view of the S2-M4/upper M4s is certainly simplistic and does not capture their full role in regulating iGluR function (Poulsen et al., 2019).
A surprising result was the gating potentiation associated with the S2-M4 was not subunit dependent, occurring in both GluN1 and GluN2A (Figures 6 & 7). We assume that the action of S2-M4/M4 is to facilitate displacements of the GluN2A pre-M1 helices, allowing all receptors to enter into the ‘unconstrained’ pre-active state. Nevertheless, future experiments will be needed to fully test the specific action of the GluN1 and GluN2A S2-M4/M4 on receptor gating including the GluN2A centered gating triad.
Using latency to 1st opening to assay receptor function
Assaying latency to 1st opening is a powerful tool to study ion channel function (Aldrich et al., 1983, Goldschen-Ohm et al., 2013). We assume that the fast pathway would contribute to synaptic events whereas the slow pathway, as well as failures, would not. However, the slow component could contribute to synaptic events during high levels of activity when the glutamate concentration might be increased at the synapse. Hence, under our ionic conditions and because of these intermediate conformations, GluN1/GluN2A NMDARs would contribute to synaptic events about 73% of the time. For the present set of experiments, we used an external solution at pH 8 containing no Ca2+ and added EDTA. We preferred this solution since it removed uncertainty as to the effects of short- and long-term effects of Ca2+, Zn2+ and pH on receptor function (see STAR Methods). Presumably similar conformations also exist in more physiological solutions, but the relative ratios may differ.
Model of NMDAR activation
Our model of NMDAR activation (Figure 8) does not substitute for available kinetic models since it does not capture extremely fast events critical to the gating process (Banke and Traynelis, 2003, Popescu and Auerbach, 2003, Erreger et al., 2005, Gibb et al., 2018). To capture details of the fast intermediate states between agonist binding and channel opening using latency to 1st opening will require an extremely large number of applications, far beyond the present pool of samples. In addition, the model does not consider events after the 1st channel opening, which play key roles in NMDAR physiology (Popescu et al., 2004, Zhang et al., 2008).
Conclusion
Our experiments highlight the critical role of LBD-TMD linkers in regulating NMDAR activity. Notable, the S2-M4/M4 linkers display numerous disease-associated variants (Yuan et al., 2014, Amin et al., 2018, Vyklicky et al., 2018, Amin et al., 2020, Perszyk et al., 2020). Further, the extracellular position of S2-M4 linkers compared to the membrane-proximal positions of M4 provides a strategic advantage for designing new drug therapies (Ogden et al., 2014, Hackos and Hanson, 2017, Perszyk et al., 2018). Importantly, knowing what role these linkers play in the gating process allows us to fine tune NMDAR activity.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Dr. Lonnie Wollmuth (lonnie.wollmuth@stonybrook.edu).
Materials Availability
All constructs generated in this study are available upon request.
Data and Code Availability
The datasets generated and analyzed during the current study are available from the lead contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Transfections
For all experiments, we used mammalian human embryonic kidney 293 (HEK293) cells. We obtained from ATCC® a stock of HEK 293 cells (CRL-1573™) in 2016. Multiple aliquots of this stock were made and frozen down in liquid nitrogen. Approximately once per month, we unfreeze one of these aliquots and use for a subsequent time period. Cells derived from these aliquots have displayed highly consistent morphology and growth patterns. If we have concerns about our stocks of HEK 293 cells, we will obtain a new stock of HEK 293 cells from ATCC®.
We transiently co-transfected cDNA constructs of GluN1 and GluN2A into mammalian human embryonic kidney 293 (HEK293) with a separate pEGFP-Cl vector (Clontech) at a ratio of 4.5:4.5:1 (GluN1/GluN2A/EGFP) using X-treme GENE HP (Roche). Cells were grown at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) for 24 h before transfection. To increase cell viability via limiting Ca2+ influx, the media bathing transfected cells also contained the NMDAR competitive antagonist APV (100 μM) and non-competitive antagonist Mg2+ (100 μM). Patch clamp recordings were performed 16–48 h post-transfection.
METHOD DETAILS
Mutagenesis and expression
Site-directed insertions were made in GluN1 (GluN1–1a) (NCBI Protein database accession no. P35439) or GluN2A (Q00959) subunits with the QuikChange site-directed mutagenesis kit (Agilent) with XL1-Blue super-competent cells. All constructs were built in rat orthologues. To ensure that site-directed mutagenesis is done correctly, i.e., that the mutation is what we intend and is introduced where we desire, we sequenced the DNA over the region, typically 400 base pairs, where the mutation is introduced. Residue numbering included the signal peptide (GluN1, 18 residues; GluN2A, 19 residues). In previous publications, we typically used numbering for the mature protein.
Structural analysis of closed and open states
For this analysis, we used the AMPAR closed (5WEK) and open (5WEO) state structures (Twomey et al., 2017), because the end states are better defined and the relationship of the LBD-TMD linkers could be visualized. Although potentially comparable conformations exist for NMDARs (Tajima et al., 2016, Zhu et al., 2016, Chou et al., 2020), there is no associated open ion channel in these structures (Figures S3A & S3B). Analysis of structures was done using PyMOL (http://pymol.org). To define a reference point for each set of structures, we identified the center of mass for the structure and would adjust slightly this reference point such that it was positioned near the center of the mouth for the ion channel pore, approximately just above the M3 helices bundle helical crossing which forms the activation gate. Using the measuring function in PyMOL, we measured from this reference point to the last αC in the designated secondary structure either in the closed (5WEK) or open (5WEO) state.
Outside-out single channel recordings
Outside-out single channel recordings were collected at room temperature (21–24°C) using an EPC-10 patch clamp amplifier interfaced with PatchMaster (HEKA). We used thick-walled, borosilicate glass (Sutter Instrument) for patch pipettes, which were pulled and fire-polished yielding resistances between 5 and 20 MΩ when measured in the bath solution (with applied positive pressure of ~150 mbar). Patch pipettes contained 140 mM KCl, 10 mM HEPES, and 1 mM BAPTA (pH 7.2, NaOH).
For all of the outside-out patch experiments, we used an external solution containing (in mM): 150 NaCl, 10 HEPES, 0.05 EDTA, pH 8.0 (NaOH). Although this external solution does not mimic what occurs at synapses, we used it since it removes the complications of proton (high pH) and divalent (Zn2+) inhibitory effects (Popescu and Auerbach, 2003) and the short- (Maki and Popescu, 2014) and long- (Legendre et al., 1993) term effects of Ca2+ on NMDARs. We applied 1 s pulses of either 1 mM glutamate or 0.1 mM glycine through a piezo-driven double-barrel application pipette system (10–90% rise time of 400–600 μs). For both glutamate and glycine application experiments, the baseline barrel contained our standard Na-based external and either 0.1 mM glycine (for glutamate test applications) or 1 mM glutamate (for glycine test applications) while the test barrel contained the identical solution except for added 1 mM glutamate or 0.1 mM glycine, respectively. There were 4 s intervals in between pulses to allow recovery from desensitization (tau of recovery from desensitization for wild type GluN1/GluN2A is around 1 sec (Alsaloum et al., 2016)). Currents were recorded at −70 mV.
For these experiments, we were mainly interested in the latency between when glutamate was first applied and the 1st opening of the ion channel (latency to 1st opening). Given the high single channel current amplitudes of GluN1/GluN2A NMDARs which greatly facilitated detection of the 1st opening, we did not rigorously control for noise. This allowed for a large number of patches/events to be recorded, greatly improving statistical analysis.
On-cell single channel recordings
On-cell single channel recordings were collected at room temperature (21–24°C) on-cell single channel recordings of NMDARs at steady state, using an Axopatch 200B (Molecular Devices) integrating patch clamp amplifier. All on-cell patches were held at −100 mV. Currents were analog filtered at 10 kHz (lowpass Bessel filter) and digitized at 50 kHz (ITC-16 interfaced with PatchMaster, HEKA). The recordings were ~4–60 min in duration to provide a substantial number of events for analysis. In general, we did not include on-cell patches in analysis unless it contained a minimum of 10,000 (typically >50,000) events, except for some of the GluN2A S1-M1 constructs that had extremely low open probability.
The bath (patch pipette) solution contained the same external solution as used for outside-out patches plus 1 mM glutamate and 0.1 mM glycine. Patch pipettes were the same as used in outside-out cell recordings and had resistances between 5 and 35 MΩ. Seal resistances were between 1 and 15 GΩ.
Macroscopic or whole-cell currents
Whole-cell currents were recorded at room temperature (21–24°C) using an EPC-10 amplifier with Patchmaster software, digitized at 10 kHz and low-pass filtered at 2.9 kHz (−3 dB) using an 8 pole low pass Bessel filter (Yelshansky et al., 2004). Patch microelectrodes were filled with an intracellular solution (in mM): 140 KCl, 10 HEPES, 1 BAPTA, pH 7.2 (KOH). Our standard extracellular solution consisted of (in mM): 140 NaCl, 1 CaCl2, 10 HEPES, pH 7.2 (NaOH). We recorded currents with or without 0.01 mM EDTA which was added to our standard extracellular solution to approximate conditions used in single channels. Pipettes had resistances of 2–6 MΩ when filled with the pipette solution and measured in the Na+ external solution. We did not use series resistance compensation, nor did we correct for junction potentials. Currents were measured within 15 minutes of going whole-cell.
Analysis of outside-out patches
Outside-out patches were exposed to super-saturating concentrations of glutamate (1 mM) and glycine (100 μM) (Traynelis et al., 2010) to ensure receptor saturation and that the agonist binding steps occurred as rapidly as possible. At synapses, presynaptic-released glutamate appears rapidly, but is also present only transiently for ~1 to 2 ms (Clements et al., 1992). For our experiments, we applied glutamate for 1 second, which allowed us to readily determine whether patches contained just a single receptor, facilitating obtaining a high number of events. Given the high open probability and relatively long open time of most of the constructs tested, we are quite confident that patches contained a single receptor; many patches were rejected because of 2 or more channels. In contrast, several constructs, most notably glycine insertions in GluN2A S1-M1 and M3-S2, had low open probability and brief open times, making it less certain that patches contained a single channel.
To analyze outside-out patches, we exported data from PatchMaster to Igor, where they were analyzed to detect for failures and for successes when the channel first opened after the intial application of glutamate (or glycine) (latency to 1st opening). Applications displaying significant amounts of noise were removed (~5–10% of applications). Nevertheless, noise was not rigorously controlled for since it was extremely straight-forward to detect the large NMDAR current amplitudes.
Latency to 1st opening
Durations from the start of an application to the first open event (latency or latency to 1st opening) were reported (Aldrich et al., 1983, Goldschen-Ohm et al., 2013). For each individual record, we estimated single channel amplitudes and used a threshold crossing to assist in identifying the 1st opening. Each individual application was subsequently visually inspected to verify amplitude and latency time.
Latency to 1st opening times were pooled and imported into ChannelLab (Synaptosoft). Latencies were binned at ~ 60 μs intervals and histograms displaying number of events as a function of latency to 1st opening were generated. The cumulative histogram was fitted by exponential components, until the log likelihood score could not be further improved.
Failure to open.
In some instances, agonist application showed no discernible NMDAR-mediated currents either during or after agonist removal (2 seconds total). These instances are referred to as failures. In the text, successes = 1 - failure rate.
Analysis of on-cell recordings
Analysis of single-channel records was similar to Talukder and Wollmuth, 2011 (Talukder and Wollmuth, 2011). Briefly, PatchMaster data were exported to QuB. Each recording was visually inspected in its entirety for multiple simultaneous openings, signal-to-noise fluctuations, high-frequency artifacts, and baseline drifts. Further processing was done to eliminate occasional obvious brief noise spikes (e.g., high amplitude, stereotypical exponential-like decay, etc) by matching them to the level of adjacent events using the ‘erase’ function in QuB. Long periods of high noise were deleted with the remaining flanking segments separated as discontinuous segments. Baseline drift was corrected by resetting baseline to zero current levels. Processed data were idealized using the segmental k-means (SKM) algorithm in QuB with a dead time of 40 μs (Qin, 2004). Single-channel amplitude (pA), equilibrium open probability (Eq. PO), mean closed time (MCT), mean open time (MOT), and number of events were extracted. Eq. PO is the fractional occupancy of the open states in the entire single-channel recording, including long lived closed states.
Identification of single channel patches.
For wild type GluN1/GluN2A and S2-M4 insertion constructs, the high Po allowed us to distinguish single-channels quite easily, as recordings with multiple channels had apparent multiple conductance states (Colquhoun and Hawkes, 1990). For low Eq. Po S1-M1 and M3-S2 glycine insertion constructs, we ensured that recordings were of long duration (~10,000–150,000 events) to maximize the likelihood of observing multiple conductance states. Nevertheless, certain constructs showed an extremely low Eq. Po (<0.07). Although we did not detect multiple conductance states, we cannot rule out that these patches contained more than one channel.
Analysis of macroscopic currents
To determine the extent and rate of desensitization in the whole-cell mode, we applied glutamate at −70 mV for 2.5 secs. Percent desensitization (% des) was calculated from the ratio of peak (Ipeak) and steady-state (Iss) current amplitudes: %des = 100 × (1 − Iss/Ipeak). Time constants of desensitization (weighted τ) were determined by fitting the decaying phase of currents to a double exponential function. In some instances when current amplitudes were small, we averaged 3–8 records.
To determine the rates of activation and deactivation in the whole-cell mode, we applied glutamate for 2 ms at −70 mV. For activation times, we used the 10–90% rise time. Deactivation times (weighted τs) were derived by fitting the decay phase of currents with a double-exponential function.
Where possible, currents were summed for outside-out patches. These summed currents were analyzed for peak and steady state current (to derive %des), rate of desensitization (weighted τdes), activation rate (10–90% rise time), and deactivation rate (fitted with double exponential function to derive weighted τdeact). In some instances, we were not able to analyze a particular feature of summed currents either because of too few events and/or too strong of desensitization.
Energetic analysis
We quantified the activation energy (Ea) energy for the transition rate to channel activation:
where kf (= 1/τ) is the forward rate constant for activation. tau is derived from the fitted exponential.
For our experiments, we identified two kinetic components for the latency to 1st opening. However, we focused quantitatively only on the energetics and properties of the fast component, which we assume entails only a single process. We did not focus on the slow component because its delay may entail a complex array of different processes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis was performed using IgorPro, QuB, Excel, and ChannelLab. Results from QuB or Igor analysis for each recording were organized in a Microsoft Excel sheet. All average values are presented as mean ± S.E.M. For statistical analysis, we used Excel or MiniTab 18. In instances where we were only interested in whether outcomes were statistically different from those of wild type, we used an unpaired two-tailed Student’s t-test to test for significant differences. Statistical significance was typically set at p < 0.05. In instances where we were interested in how constructs varied from each other, we used an ANOVA and followed with Tukey’s test (p < 0.05).
We did not run a statistical test to determine sample size a priori. Sample sizes resemble those from previous publications. Statistical significance is shown in the Figure legends as well as in associated tables shown in the Supplemental Information. Figures were generated using Canvas.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| N/A | ||
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| APV, NMDAR antagonist | Tocris | Cat#: 0106; RRID: SCR_003689 |
| Critical Commercial Assays | ||
| N/A | ||
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| HEK 293 cells | American Type Culture Collection, Manassas, Virginia |
https://www.atcc.org/ Cat#: CRL-1573™ |
| Experimental Models: Organisms/Strains | ||
| N/A | ||
| Oligonucleotides | ||
| N/A | ||
| Recombinant DNA | ||
| Rat GluN1–1a | Dr. P. Seeburg, Max Planck Institute for Medical Research, Heidelberg, Germany | P35439 |
| Rat GluN2A | Dr. P. Seeburg, Max Planck Institute for Medical Research, Heidelberg, Germany | Q00959 |
| Software and Algorithms | ||
| Office Package 2016 | Microsoft Corporation, Redmond, WA | https://www.microsoftsoftwares.com/collections/office-2016?gclid=CjwKCAjwiOv7BRBREiwAXHbv3G2xLLOeWdzUjrCZkIeWvn3nZWpiYN7Ht0sFKY4VckiiBi8bOlTXTBoCgIAQAvD_BwE |
| PatchMaster | HEKA Elektronik, Ludwigshafen, Germany | https://heka.com/ |
| QuB | QuB, Buffalo, NY | http://www.sachslab.buffalo.edu/Auerbach/info/ind_qub.htm |
| Igor Pro | WaveMetrics, Portland, OR | https://www.wavemetrics.com/ |
| Canvas 12 | Canvas GFX Inc, Boston, MA | https://www.canvasgfx.com/ |
| MiniTAB | Minitab, State College, PA | https://www.minitab.com/en-us/ |
| Channel Lab | Synaptosoft, Decatur, GA | http://www.synaptosoft.com/Channels |
Highlights.
NMDARs transition through two distinct structural conformations before opening.
The efficiency of NMDAR opening depends on which intermediate pathway is entered.
Fast NMDAR opening requires glutamate-induced displacement of the GluN2 pre-M1 helix.
Glutamate-induced displacement of the S2-M4/M4 axis also facilitates rapid channel gating.
Acknowledgments
We thank Donna Schmidt for technical assistance; Dr. Rashek Kazi for comments on the manuscript. This manuscript is dedicated to the memory of Dr. James Howe (RIP), who assisted in developing key early ideas in it.
This work was supported by NIH Grant R01 NS088479 (LPW), including a minority supplement (JA), and an SBU-BNL SEED Grant (LPW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Ahern CA, Payandeh J, Bosmans F & Chanda B 2016. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J Gen Physiol, 147, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich RW, Corey DP & Stevens CF 1983. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature, 306, 436–41. [DOI] [PubMed] [Google Scholar]
- Alsaloum M, Kazi R, Gan Q, Amin J & Wollmuth LP 2016. A Molecular Determinant of Subtype-Specific Desensitization in Ionotropic Glutamate Receptors. J Neurosci, 36, 2617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin JB, Leng X, Gochman A, Zhou H-X & Wollmuth LP 2018. A conserved glycine harboring disease-associated mutations permits NMDA receptor slow deactivation and high Ca2+ permeability. Nature Communications, 9, 3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin JB, Moody GR & Wollmuth LP 2020. From bedside-to-bench: What disease-associated variants are teaching us about the NMDA receptor. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin JB, Salussolia CL, Chan K, Regan MC, Dai J, Zhou HX, Furukawa H, Bowen ME & Wollmuth LP 2017. Divergent roles of a peripheral transmembrane segment in AMPA and NMDA receptors. J Gen Physiol, 149, 661–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong N & Gouaux E 2000. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron, 28, 165–81. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Sun Y, Chen GQ & Gouaux E 1998. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature, 395, 913–7. [DOI] [PubMed] [Google Scholar]
- Auerbach A 2013. The energy and work of a ligand-gated ion channel. J Mol Biol, 425, 1461–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG & Traynelis SF 2003. Activation of NR1/NR2B NMDA receptors. Nat Neurosci, 6, 144–52. [DOI] [PubMed] [Google Scholar]
- Bezanilla F 2008. How membrane proteins sense voltage. Nat Rev Mol Cell Biol, 9, 323–32. [DOI] [PubMed] [Google Scholar]
- Chang HR & Kuo CC 2008. The activation gate and gating mechanism of the NMDA receptor. J Neurosci, 28, 1546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhao Y, Wang Y, Shekhar M, Tajkhorshid E & Gouaux E 2017a. Activation and Desensitization Mechanism of AMPA Receptor-TARP Complex by Cryo-EM. Cell, 170, 1234–1246 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Tankovic A, Burger PB, Kusumoto H, Traynelis SF & Yuan H 2017b. Functional Evaluation of a De Novo GRIN2A Mutation Identified in a Patient with Profound Global Developmental Delay and Refractory Epilepsy. Mol Pharmacol, 91, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TH, Tajima N, Romero-Hernandez A & Furukawa H 2020. Structural Basis of Functional Transitions in Mammalian NMDA Receptors. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE & Westbrook GL 1992. The time course of glutamate in the synaptic cleft. Science, 258, 1498–501. [DOI] [PubMed] [Google Scholar]
- Colquhoun D & Hawkes AG 1990. Stochastic properties of ion channel openings and bursts in a membrane patch that contains two channels: evidence concerning the number of channels present when a record containing only single openings is observed. Proceedings of the Royal Society of London. Series B, Containing papers of a Biological character. Royal Society, 240, 453–77. [DOI] [PubMed] [Google Scholar]
- Colquhoun D & Lape R 2012. Perspectives on: conformational coupling in ion channels: allosteric coupling in ligand-gated ion channels. J Gen Physiol, 140, 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacosta CJ & Baenziger JE 2013. Gating of pentameric ligand-gated ion channels: structural insights and ambiguities. Structure, 21, 1271–83. [DOI] [PubMed] [Google Scholar]
- Dolino DM, Chatterjee S, Maclean DM, Flatebo C, Bishop LDC, Shaikh SA, Landes CF & Jayaraman V 2017. The structure-energy landscape of NMDA receptor gating. Nat Chem Biol, 13, 1232–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erreger K, Dravid SM, Banke TG, Wyllie DJ & Traynelis SF 2005. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J Physiol, 563, 345–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmenjaud JB, Stroebel D, Chan K, Grand T, David M, Wollmuth LP, Taly A & Paoletti P 2019. An inter-dimer allosteric switch controls NMDA receptor activity. EMBO J, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H, Singh SK, Mancusso R & Gouaux E 2005. Subunit arrangement and function in NMDA receptors. Nature, 438, 185–92. [DOI] [PubMed] [Google Scholar]
- Gibb AJ, Ogden KK, Mcdaniel MJ, Vance KM, Kell SA, Butch C, Burger P, Liotta DC & Traynelis SF 2018. A structurally derived model of subunit-dependent NMDA receptor function. J Physiol, 596, 4057–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschen-Ohm MP, Capes DL, Oelstrom KM & Chanda B 2013. Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nat Commun, 4, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH & Mayer ML 2019. Structural biology of glutamate receptor ion channels: towards an understanding of mechanism. Curr Opin Struct Biol, 57, 185–195. [DOI] [PubMed] [Google Scholar]
- Hackos DH & Hanson JE 2017. Diverse modes of NMDA receptor positive allosteric modulation: Mechanisms and consequences. Neuropharmacology, 112, 34–45. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Yi F, Perszyk RE, Furukawa H, Wollmuth LP, Gibb AJ & Traynelis SF 2018. Structure, function, and allosteric modulation of NMDA receptors. J Gen Physiol, 150, 1081–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KB, Yi F, Perszyk RE, Menniti FS & Traynelis SF 2017. NMDA Receptors in the Central Nervous System. Methods Mol Biol, 1677, 1–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JM & Wilkinson KA 2016. Synaptic AMPA receptor composition in development, plasticity and disease. Nat Rev Neurosci, 17, 337–50. [DOI] [PubMed] [Google Scholar]
- Hille B 2001. Ion channels of excitable membranes, MA, Sinauer Associates, Inc. [Google Scholar]
- Howe JR, Cull-Candy SG & Colquhoun D 1991. Currents through single glutamate receptor channels in outside-out patches from rat cerebellar granule cells. J Physiol, 432, 143–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes BA & Woodward JJ 2016. Disruption of S2-M4 linker coupling reveals novel subunit-specific contributions to N-methyl-d-aspartate receptor function and ethanol sensitivity. Neuropharmacology, 105, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalali-Yazdi F, Chowdhury S, Yoshioka C & Gouaux E 2018. Mechanisms for Zinc and Proton Inhibition of the GluN1/GluN2A NMDA Receptor. Cell, 175, 1520–1532 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas E & Furukawa H 2014. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science, 344, 992–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi R, Dai J, Sweeney C, Zhou HX & Wollmuth LP 2014. Mechanical coupling maintains the fidelity of NMDA receptor-mediated currents. Nat Neurosci, 17, 914–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazi R, Gan Q, Talukder I, Markowitz M, Salussolia CL & Wollmuth LP 2013. Asynchronous Movements Prior to Pore Opening in NMDA Receptors. J Neurosci, 33, 12052–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladislav M, Cerny J, Krusek J, Horak M, Balik A & Vyklicky L 2018. The LILI Motif of M3-S2 Linkers Is a Component of the NMDA Receptor Channel Gate. Front Mol Neurosci, 11, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Lu W, Michel JC, Goehring A, Du J, Song X & Gouaux E 2014. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature, 511, 191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P, Rosenmund C & Westbrook GL 1993. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J. Neurosci, 13, 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki BA & Popescu GK 2014. Extracellular Ca(2+) ions reduce NMDA receptor conductance and gating. J Gen Physiol, 144, 379–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML 2005. Crystal structures of the GluR5 and GluR6 ligand binding cores: molecular mechanisms underlying kainate receptor selectivity. Neuron, 45, 539–52. [DOI] [PubMed] [Google Scholar]
- Mcdaniel MJ, Ogden KK, Kell SA, Burger PB, Liotta DC & Traynelis SF 2020. NMDA receptor channel gating control by the pre-M1 helix. J Gen Physiol, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JA, Stein IS, Lau CG, Peixoto RT, Aman TK, Kaneko N, Aromolaran K, Saulnier JL, Popescu GK, Sabatini BL, Hell JW & Zukin RS 2014. Phosphorylation of Ser1166 on GluN2B by PKA is critical to synaptic NMDA receptor function and Ca2+ signaling in spines. J Neurosci, 34, 869–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Chen W, Swanger SA, Mcdaniel MJ, Fan LZ, Hu C, Tankovic A, Kusumoto H, Kosobucki GJ, Schulien AJ, Su Z, Pecha J, Bhattacharya S, Petrovski S, Cohen AE, Aizenman E, Traynelis SF & Yuan H 2017. Molecular Mechanism of Disease-Associated Mutations in the Pre-M1 Helix of NMDA Receptors and Potential Rescue Pharmacology. PLoS Genet, 13, e1006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden KK, Khatri A, Traynelis SF & Heldt SA 2014. Potentiation of GluN2C/D NMDA receptor subtypes in the amygdala facilitates the retention of fear and extinction learning in mice. Neuropsychopharmacology, 39, 625–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perszyk R, Katzman BM, Kusumoto H, Kell SA, Epplin MP, Tahirovic YA, Moore RL, Menaldino D, Burger P, Liotta DC & Traynelis SF 2018. An NMDAR positive and negative allosteric modulator series share a binding site and are interconverted by methyl groups. Elife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perszyk RE, Myers SJ, Yuan H, Gibb AJ, Furukawa H, Sobolevsky AI & Traynelis SF 2020. Hodgkin-Huxley-Katz Prize Lecture: Genetic and pharmacological control of glutamate receptor channel through a highly conserved gating motif. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plested AJ 2016. Structural mechanisms of activation and desensitization in neurotransmitter-gated ion channels. Nat Struct Mol Biol, 23, 494–502. [DOI] [PubMed] [Google Scholar]
- Popescu G & Auerbach A 2003. Modal gating of NMDA receptors and the shape of their synaptic response. Nat Neurosci, 6, 476–83. [DOI] [PubMed] [Google Scholar]
- Popescu G, Robert A, Howe JR & Auerbach A 2004. Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature, 430, 790–3. [DOI] [PubMed] [Google Scholar]
- Poulsen MH, Poshtiban A, Klippenstein V, Ghisi V & Plested AJR 2019. Gating modules of the AMPA receptor pore domain revealed by unnatural amino acid mutagenesis. Proc Natl Acad Sci U S A, 116, 13358–13367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F 2004. Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophys J, 86, 1488–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Honse Y, Karp BJ, Lipsky RH & Peoples RW 2003. A site in the fourth membrane-associated domain of the N-methyl-D-aspartate receptor regulates desensitization and ion channel gating. J Biol Chem, 278, 276–83. [DOI] [PubMed] [Google Scholar]
- Schmid SM, Korber C, Herrmann S, Werner M & Hollmann M 2007. A domain linking the AMPA receptor agonist binding site to the ion pore controls gating and causes lurcher properties when mutated. J Neurosci, 27, 12230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi EY, Yuan CL, Sipple MT, Srinivasan J, Ptak CP, Oswald RE & Nowak LM 2019. Noncompetitive antagonists induce cooperative AMPA receptor channel gating. J Gen Physiol, 151, 156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP & Gouaux E 2009. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature, 462, 745–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto D, Olivella M, Grau C, Armstrong J, Alcon C, Gasull X, Santos-Gomez A, Locubiche S, Gomez De Salazar M, Garcia-Diaz R, Gratacos-Batlle E, Ramos-Vicente D, Chu-Van E, Colsch B, Fernandez-Duenas V, Ciruela F, Bayes A, Sindreu C, Lopez-Sala A, Garcia-Cazorla A & Altafaj X 2019. l-Serine dietary supplementation is associated with clinical improvement of loss-of-function GRIN2B-related pediatric encephalopathy. Sci Signal, 12. [DOI] [PubMed] [Google Scholar]
- Tajima N, Karakas E, Grant T, Simorowski N, Diaz-Avalos R, Grigorieff N & Furukawa H 2016. Activation of NMDA receptors and the mechanism of inhibition by ifenprodil. Nature, 534, 63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukder I, Borker P & Wollmuth LP 2010. Specific sites within the ligand-binding domain and ion channel linkers modulate NMDA receptor gating. J Neurosci, 30, 11792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukder I & Wollmuth LP 2011. Local constraints in either the GluN1 or GluN2 subunit equally impair NMDA receptor pore opening. J Gen Physiol, 138, 179–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toombes GE & Swartz KJ 2014. Divining the design principles of voltage sensors. J Gen Physiol, 143, 139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, Mcbain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ & Dingledine R 2010. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev, 62, 405–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC & Sobolevsky AI 2018. Structural Mechanisms of Gating in Ionotropic Glutamate Receptors. Biochemistry, 57, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twomey EC, Yelshanskaya MV, Grassucci RA, Frank J & Sobolevsky AI 2017. Channel opening and gating mechanism in AMPA-subtype glutamate receptors. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance KM, Hansen KB & Traynelis SF 2013. Modal gating of GluN1/GluN2D NMDA receptors. Neuropharmacology, 71, 184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky V, Krausova B, Cerny J, Ladislav M, Smejkalova T, Kysilov B, Korinek M, Danacikova S, Horak M, Chodounska H, Kudova E & Vyklicky L 2018. Surface Expression, Function, and Pharmacology of Disease-Associated Mutations in the Membrane Domain of the Human GluN2B Subunit. Front Mol Neurosci, 11, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wo ZG & Oswald RE 1995. Unraveling the modular design of glutamate-gated ion channels. Trends Neurosci, 18, 161–168. [DOI] [PubMed] [Google Scholar]
- Wood MW, Vandongen HMA & Vandongen AMJ 1995. Structural conservation of ion conduction pathways in K channels and glutamate receptors. Proc. Nat. Acad. Sci. (USA), 92, 4882–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Belcher J, Berger AJ, Mayer ML & Lau AY 2013. Conformational analysis of NMDA receptor GluN1, GluN2, and GluN3 ligand-binding domains reveals subtype-specific characteristics. Structure, 21, 1788–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelshanskaya MV, Mesbahi-Vasey S, Kurnikova MG & Sobolevsky AI 2017. Role of the Ion Channel Extracellular Collar in AMPA Receptor Gating. Sci Rep, 7, 1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelshansky MV, Sobolevsky AI, Jatzke C & Wollmuth LP 2004. Block of AMPA receptor desensitization by a point mutation outside the ligand-binding domain. J Neurosci, 24, 4728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Hansen KB, Zhang J, Pierson TM, Markello TC, Fajardo KV, Holloman CM, Golas G, Adams DR, Boerkoel CF, Gahl WA & Traynelis SF 2014. Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy. Nat Commun, 5, 3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Howe JR & Popescu GK 2008. Distinct gating modes determine the biphasic relaxation of NMDA receptor currents. Nat Neurosci, 11, 1373–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stein RA, Yoshioka C, Lee CH, Goehring A, Mchaourab HS & Gouaux E 2016. Mechanism of NMDA Receptor Inhibition and Activation. Cell, 165, 704–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available from the lead contact upon reasonable request.