

Abstract
Metabolic constraints in the tumor microenvironment constitute a barrier to effective anti-tumor immunity and similarities in the metabolic properties of T cells and cancer cells impede the specific therapeutic targeting of metabolism in either population. To identify distinct metabolic vulnerabilities of CD8+ T cells and cancer cells, we developed a high-throughput in vitro pharmacologic screening platform and used it to measure the cell type–specific sensitivities of activated CD8+ T cells and B16 melanoma cells to a wide array of metabolic perturbations during antigen-specific killing of cancer cells by CD8+ T cells. We illustrated the applicability of this screening platform by showing that CD8+ T cells were more sensitive to ferroptosis than B16 and MC38 cancer cells. Overexpression of ferroptosis suppressor protein 1 (FSP1) or cytosolic GPX4 yielded ferroptosis-resistant CD8+ T cells without compromising their function, while genetic deletion of the ferroptosis sensitivity–promoting enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4) protected CD8+ T cells from ferroptosis, but impaired anti-tumor CD8+ T cell responses. Our screen also revealed high T cell–specific vulnerabilities for compounds targeting NAD+ metabolism or autophagy and ER stress pathways. We focused the current screening effort on metabolic agents. However, this in vitro screening platform may also be valuable for rapid testing of other types of compounds to identify regulators of anti-tumor CD8+ T-cell function and potential therapeutic targets.
Keywords: T cells, cancer, metabolism, screening, ferroptosis
Introduction
CD8+ T cells are critical effectors of the anti-tumor immune response. They can directly kill malignant cells through the secretion of cytolytic molecules like granzyme B (GzmB) and perforin (1). Although immune checkpoint blockade (ICB) and chimeric antigen receptor (CAR) T-cell strategies have harnessed the therapeutic potential of T cells for cancer immunotherapy (2–4), ICB response rates remain limited and CAR T cells have had little success in solid cancers (5–7). Therefore, there is a critical need to understand how to enhance the efficacy of cancer immunotherapies.
One barrier to successful anti-tumor immune responses, particularly in the setting of solid tumors, is the metabolic constraints of the tumor microenvironment (TME) (8–13). The combination of high metabolic activity of cells in the TME and limited blood supply to the tumor may result in a scarcity of important nutrients and oxygen, which cancer cells and immune cells then compete for (12,14,15). Metabolic pathways, including glycolysis, glutaminolysis and one carbon metabolism, are critical for effective T-cell activation and differentiation (16–19). However, the metabolic requirements for maintenance and execution of effector functions are less well-studied. These may be particularly important in the context of an anti-tumor immune response, where T cells are activated in a lymph node before encountering metabolic challenges and interacting with cancer cells in the TME (20).
Cancer cells and activated T cells are both anabolic and rely on many of the same metabolic pathways to survive in the TME (21). This overlap in metabolic dependencies complicates efforts to either target cancer metabolism therapeutically without also impairing anti-tumor immunity or to enhance T-cell metabolism without also promoting tumor growth. Thus, identifying unique metabolic vulnerabilities of T cells and cancer cells could inform more selective therapeutic strategies.
To identify specific metabolic vulnerabilities of CD8+ T cells and cancer cells, we developed a high-throughput pharmacologic screening platform and used it to screen a metabolic small molecule library. We measured antigen-specific killing of cancer cells by activated CD8+ T cells in vitro, while pharmacologically imposing metabolic perturbations on both co-cultured cell populations. We observed that CD8+ T cells were highly sensitive to glutathione peroxidase 4 (GPX4) inhibitors, which provoked the lipid peroxidation-induced cell death pathway ferroptosis in CD8+ T cells at concentrations that did not impact B16 melanoma or MC38 colorectal adenocarcinoma cell numbers. Using genetic manipulations, we demonstrated that GPX4 and ferroptosis suppressor protein 1 (FSP1) protected T cells from ferroptosis, whereas acyl-CoA synthetase long-chain family member 4 (ACSL4) promoted ferroptosis sensitivity in CD8+ T cells. In addition, we discovered that ACSL4 deficiency reduced CD8+ T-cell survival. The dependency of CD8+ T cells on ACSL4 for optimal effector responses may contribute to their sensitivity to ferroptosis induction. Our in vitro screening platform facilitated the discovery of cell-type specific metabolic vulnerabilities of CD8+ T cells and cancer cells during antigen-specific interaction, identifying pathways for in vivo validation studies. This platform may enable rapid testing of the effects of a variety of molecules during antigen-specific killing of cancer cells by activated CD8+ T cells.
Materials and Methods
Mice
6- to 23-week old female mice were used for all experiments. Wildtype C57BL/6J mice were purchased from the Jackson Laboratory (cat. #000664). OT-1 (C57BL/6J-Tg(Tcra/Tcrb)1100Mjb/J) mice were purchased from the Jackson Laboratory (cat. #003831) and bred in-house. These mice were used to generate congenically marked (CD45.1/2) OT-1 mice. Cas9-expressing mice were generated in-house as previously described (22,23). These mice were then used to generate Cas9-expressing congenically marked (CD45.1/2) OT-1 mice as previously described (22,23). Mice were housed in specific pathogen-free conditions and used in accordance with approvals from the Harvard Medical School Institutional Animal Care and Use Committee.
Cell lines
The B16.F10 (B16WT) cell line was a gift from G. Dranoff (Novartis Institutes for Biomedical Research; acquired in 2012). The MC38WT cell line was a gift from D. Vignali (University of Pittsburgh School of Medicine; acquired in 2012). These lines were authenticated (most recently in 2018) using whole-exome sequencing. B16 cells expressing wildtype ovalbumin (B16-OVAWT) and no fluorescent marker, which were used for in vivo tumor experiments, were generated as previously described (24). HEK293x cells were a gift from C. Kadoch (Dana-Farber Cancer Institute; acquired in 2017). Phoenix-ECO cells (ATCC; cat. #CRL-3214) and Platinum-E cells (Cell Biolabs; cat. #RV-101) were purchased in 2015 and 2019, respectively. All cell lines were cultured in DMEM (Thermo Fisher Scientific; cat. #11965-118) supplemented with 10% FBS (MilliporeSigma; cat. #F244; lot #7H115), 100 units/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific; cat #5140-122). B16-OVAWT cells without fluorescent marker were selected in 2 μg/mL puromycin (Thermo Fisher Scientific; cat. #D3861). Platinum-E cells were selected in 1 μg/mL puromycin and 10 μg/mL blasticidin (Thermo Fisher Scientific; cat. #A11139-03).
Variants of B16.F10 and MC38 expressing OVAWT or OVA without SIINFEKL (OVAΔ257-264) with fluorescent markers were generated by retroviral transduction. Plasmids for retroviral transduction were generated by replacing the puromycin resistance cassette in the MSCV-PIG plasmid (Addgene; cat. #18751) with OVAWT or OVAΔ257-264 and either leaving green fluorescent protein (GFP) in place or replacing GFP with red fluorescent protein (RFP) or violet-excited GFP (Vex) and cloned using One Shot Stbl3 chemically competent E. coli (Thermo Fisher Scientific, cat. #C7373-03). Phoenix-ECO cells were transfected with the various plasmids using a polyethylenimine (PEI) (Polysciences; cat. #24765-2) and DNA mixture at a 1:3 DNA:PEI mass ratio in Opti-MEM I reduced serum medium (Life Technologies; cat. #31985-062) for 24hr. Retroviral supernatants were collected 72hr after transfections and any cellular debris was removed by either filtrating the supernatants through 45 μm filters (Thermo Fisher Scientific; cat. #723-9945) or by centrifugation at 500 g for 10 min. B16.F10 and MC38 were transduced by culture in equal volumes of media containing polybrene (Santa Cruz Biotechnology; cat. #sc-134220) and retroviral supernatant, for a final polybrene concentration of 10 μg/mL for 24hr. Transduced cancer cells were selected by flow cytometric sorting (see Flow cytometry and flow sorting below) using a 100 μm nozzle of reporter protein-expressing cells ≥2 times to generate variants expressing OVAWT with GFP, RFP, or Vex, or OVAΔ257-264 with GFP, RFP, or Vex. Maintenance of construct expression was ensured by flow cytometry before each experiment. MC38WT, B16WT, B16-OVAWT, HEK293x, Phoenix and Platinum-E cells were all confirmed Mycoplasma negative in the year before submission of the manuscript (≤8 passages from most recent use) using PCR-based assays. The OVAWT-GFP/-RFP/-Vex- and OVAΔ257-264-GFP/-RFP/-Vex-expressing cell lines were thus generated using Mycoplasma-negative cells but were not tested themselves. All cells were used 3-14 days after thawing.
CD8+ T-cell activation and in vitro killing assay
Primary naïve CD8+ and CD4+ T cells were purified from spleens and inguinal lymph nodes of mice using a naïve CD8a+ T cell isolation kit (Miltenyi Biotec; cat. #130-096-543) or a naïve CD4+ T-cell isolation kit (Miltenyi Biotec; cat. #130-104-453). The cells were activated for 72hr at 37°C in plates coated with 1 μg/mL anti-CD3 (clone 145-2C11; BioXCell; cat. #BE0001-1) and 1 μg/mL anti-CD28 (clone 37.51; BioXCell; cat. #BE0015-1) in the presence of 100 units/mL IL-2 (R&D Systems; cat. #202-IL-050) and 10 ng/mL IL-12 (PeproTech; cat. #210-12-50ug), unless otherwise noted. Where indicated, cancer cells were pre-treated with IFNγ (PeproTech; cat. #315-05-500UG). For killing assays, 4,000 activated CD8+ OT-1 T cells and 24,000 cancer cells (12,000 OVAWT- and 12,000 OVAΔ257-264-expressing cells) (unless otherwise indicated) were plated together in 96-well plates after 72hr T-cell activation, and compounds were added at that time. T-cell cultures and killing assays were conducted in RPMI-1640 (Thermo Fisher Scientific, cat. #11875-119) supplemented with 10% FBS (MilliporeSigma; cat. #F244; lot #17H115), 100 units/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific; cat. #15140-122), 10 mM HEPES (Thermo Fisher Scientific; cat. #15630-130), 1 mM sodium pyruvate (Thermo Fisher Scientific; cat. #11360-070) and 55 μM 2-mercaptoethanol (Thermo Fisher Scientific; cat. #21985-023). After co-culture for 24hr (unless otherwise noted), plates were trypsinized, cells were resuspended in MACS buffer (consisting of phosphate-buffered saline [Life Technologies; cat. #14190-250] with 1% FBS and 2 mM EDTA [Invitrogen; cat. #15575-020]) and analyzed by flow cytometry to determine the numbers of cancer cells and T cells (as described below in Flow cytometry and flow sorting). The percentage of B16-/MC38-OVAWT was calculated by the following formula: number of B16-/MC38-OVAWT / (number of B16-/MC38-OVAWT + number of B16-/MC38-OVAΔ257-264) x 100%. For analysis of cancer cell numbers in wells without T cells, the sum of B16-/MC38-OVAWT and B16-/MC38-OVAΔ257-264 cell numbers was used. Absolute cell numbers were calculated by dividing the number of recorded cells by the portion of the well volume collected during flow cytometry. For T-cell numbers, the average number of background events in wells where no T cells were added was subtracted. Where indicated, IFNγ and/or TNFα were neutralized using blocking antibodies from BioLegend (LEAF purified anti-mouse IFNγ [clone XMG1.2; cat. #505812], LEAF purified anti-mouse TNFα [clone MP6-XT22; cat. #506310], LEAF purified rat IgG1, κ isotype control [clone RTK2071; cat. #400427]). Where indicated, 1S,3R-RSL3 (MilliporeSigma; cat. #SML2234-5mg), ML210 (MilliporeSigma; cat. #SML0521-5mg), ML162 (Cayman Chemical; cat. #20455), Ferrostatin-1 (MilliporeSigma; cat. #SML0583-5mg), α-Tocopherol (MilliporeSigma; cat. #T3251-25g), and rosiglitazone (MilliporeSigma; cat. #R2408-10mg) were used at the noted concentrations.
High-throughput in vitro killing assay
The workflow for the high-throughput killing assay, as used for the screen, was the same as that used in the low-throughput killing assay described above (see CD8+ T-cell activation and in vitro killing assay), except that Thermo Multidrop Combi machines were used for plating of cells and addition of media, trypsin, and MACS buffer to plates, library compounds were added using a Seiko D-TRAN XM3106-31 PN 4-axis cartesian Compound Transfer Robot, and read-out was performed using the Intellicyt iQue Screener PLUS high-throughput flow cytometer. Each of the 10 384-well compound libraries was screened in a separate run consisting of 16 96-well assay plates (4 quadrants, with and without T cells, in duplicate). Each assay plate contained control wells without drug (DMSO-only) or with 2 μM cyclosporin A (Tocis; cat. #1101). These assays were conducted in collaboration with the ICCB-Longwood screening facility.
Screen analysis
Flow cytometry data were analyzed using ForeCyt software to obtain numbers of T cells, Vex+ B16-OVAWT cells and GFP+ B16-OVAΔ257-264 cells. The percentage of B16-OVAWT was calculated for each well. This was used to generate MinMax-normalized specific killing values for T cell–containing wells by comparison to DMSO-only control wells with T cells (defined as specific killing of 1) and without T cells (defined as specific killing of 0). The following formula was used: specific killing = 1–(X–Y)/(Z–Y), where X = % B16-OVAWT in assay well, Y = average % B16-OVAWT in DMSO-only control wells with T cells, and Z = average % B16-OVAWT in corresponding DMSO-only control wells without T cells. T-cell toxicity was calculated for T cell–containing wells using the following formula: T-cell toxicity = 1 – (X/Y), where X is the number of T cells in the well, and Y is the average number of T cells in corresponding DMSO-only control wells with T cells. For wells without T cells, cancer-cell toxicity was calculated using the following formula: cancer-cell toxicity = 1 – (X/Z), where X is the number of B16-OVAWT + number of B16-OVAΔ257-264 cells in the well and Z is the average number of B16-OVAWT + number of B16-OVAΔ257-264 cells in DMSO-only control wells without T cells. Average values of the replicates were calculated for each compound concentration and used for downstream analyses. GraphPad Prism was used to generate dose-response curves by non-linear regression and calculate area under the curve (AUC) for each test compound. AUCs were calculated using the lowest nine of ten concentrations to allow more compounds to be included in the analysis. The specific killing inhibition area was calculated by subtracting the specific killing AUC from the total plot area of 1 x (Log(20,000/3) – Log(20,000/39)): the area between the lowest and highest compound concentrations. T cell–specific vulnerability was calculated for each compound by subtracting the cancer-cell toxicity AUC from the specific killing inhibition area.
For six of the 240 test compounds, one replicate was excluded because of technical collection issues at one or more concentration. These compounds were still included in the analysis using the remaining replicates. For six test compounds, fluorescence properties were impacted at the highest compound concentration such that accurate gating of GFP- and Vex-expressing populations was not possible. These compounds were still included in the analysis since AUC calculations were performed from the lowest nine concentrations for all compounds. Another 12 test compounds had to be excluded from the analysis because the impact on fluorescence properties prevented accurate gating at multiple drug concentrations. To ensure that a reliable specific killing score could be calculated from the percentage of B16-OVAWT cells, this parameter was included only for wells where >50 B16 cells were recorded. For six compounds, this was not the case at just the highest concentration (still included in AUC analysis), and for three compounds this was the case at multiple concentrations (excluded for analyses involving specific killing calculations). In all, 228 of 240 compounds were included in the analyses of T-cell and cancer-cell toxicities, and 225 of 240 compounds were included in analyses involving specific killing calculations.
C11-BODIPY581/591 staining
C11-BODIPY581/591 staining was conducted by incubating samples with 5 μM C11-BODIPY581/591 (Thermo Fisher Scientific; cat. #D3861) for 2-3hr at 37 °C. Fluorescence emission around 510 nm, indicative of oxidation, was read out in the GFP or FITC channel by flow cytometry.
Flow cytometry and flow sorting
Flow cytometry was performed on BD LSR II and BD FACSymphony machines. FACS sorting was conducted on a BD Aria II or MoFlo Astrios EQ. Flow analyses were performed in FlowJo 10.6.1. For histogram flow plots, modal Y axes were used to display relative cell numbers (normalized to mode). All flow antibodies were used at a 1:100 dilution unless indicated otherwise. The following antibodies were purchased from Biolegend: TruStain fcX (anti-mouse CD16/32; clone 93; cat. #101320), APC anti-mouse H-2Kb bound to SIINFEKL (clone 25-D1.16; cat. #141606), PE/Cy7 anti-mouse H-2Kb bound to SIINFEKL (clone 25-D1.16; cat. #141608), APC anti-mouse IFN-γ (clone XMG1.2; cat. #505810), PE/Cy7 anti-mouse IFN-γ (clone XMG1.2; cat. #505826), Pacific Blue anti-human/mouse Granzyme B (clone GB11; cat. #515408), FITC anti-human/mouse Granzyme B (clone GB11; cat. #515403), APC anti-mouse CD3ε (clone 145-2C11; cat. #100312), Brilliant Violet 421 anti-mouse CD8a (clone 53-6.7; cat. #100753), PE/Cy7 anti-mouse/human CD11b (clone M1/70; cat. #101216), FITC anti-mouse CD3ε (clone 145-2C11; cat. #100306), PE/Cy7 anti-mouse CD8b (clone YTS156.7.7, cat. #126616), Brilliant Violet 421 anti-mouse CD45.1 (clone A20; cat. #110732), FITC anti-mouse CD8b (Ly-3) (clone YTS156.7.7; cat. #126606), APC anti-mouse CD8b (Ly-3) (clone YTS156.7.7; cat. #126614), PE anti-mouse CD8b (Ly-3) (clone YTS156.7.7; cat. #126608), Brilliant Violet 605 anti-mouse CD279 (PD-1) (clone 29F.1A12; cat. #135220); PerCP/Cy5.5 anti-mouse/human CD44 (clone IM7; cat. #103032), Brilliant Violet 605 anti-mouse/human CD11b (clone M1/70; cat. #101257), PE anti-mouse TCR Vβ5.1, 5.2 (clone MR9-4; cat. #139504), PE anti-mouse TER-119 (clone TER-119; cat. #116208), PE anti-mouse/human CD45R/B220 (clone RA3-6B2; cat. #103208), PE anti-mouse Ly-6G/Ly-6C (Gr-1) (clone RB6-8C5; cat. #108408), Brilliant Violet 510 anti-mouse CD45.1 (clone A20; cat. #110741). The following antibodies were purchased from BD Biosciences: BUV395 rat anti-mouse CD8b (clone H35-17.2; cat. #740278), BUV395 mouse anti-mouse CD45.2 (clone 104; cat. #564616), BUV805 rat anti-mouse CD4 (clone GK1.5; cat. #612900), BUV737 rat anti-mouse/human CD11b (clone M1/70; cat. #564443), PerCP/Cy5.5 mouse anti-mouse Ki67 (clone B56; cat. #561284). LIVE/DEAD fixable near-IR dead cell stain kit (Thermo Fisher Scientific; cat. #L34976) was used to determine cell viability. The eBioscience Foxp3 / transcription factor staining buffer set (Thermo Fisher Scientific; cat. #00-5523-00) was used to stain for intracellular antigens. UltraComp beads (Thermo Fisher Scientific; cat. #01-2222-42) were used for compensation.
Tumor implantations
Mice were anesthetized with 2.5% 2,2,2-tribromoethanol (MilliporeSigma; cat. #T48402-25g) and 2.5 x 105 B16-OVAWT cells without fluorescent label were injected in the flank subcutaneously. Tumors were measured with a caliper every 2–3 days once palpable. Tumor volumes were calculated using the formula ½ x D x d2, where D is the longer diameter and d is the shorter diameter. Mice were sacrificed when tumors reached 2 cm3 or ulcerated or the mice had poor body condition score, unless they were harvested earlier for described analyses.
Tumor harvests
Tumors were excised and manually dissociated, followed by incubation in collagenase (Worthington Biochemical Corporation; cat. #LS004194) and DNase (MilliporeSigma; cat. #10104159001) for 20 min at 37 °C. Lymphocytes were enriched using a Percoll gradient (VWR; cat. #89428-526): cells were resuspended in 5 mL 40% salt-adjusted Percoll and 2 mL 70% salt-adjusted Percoll was underlaid, followed by centrifugation for 20 min at 800 g at room temperature and recovery of leukocytes from the interface of the 40% and 70% Percoll layers. The Percoll gradient step was not used when performing ex vivo C11-BODIPY581/591 staining. After cell isolations, cells were stained for flow cytometry.
T-cell transductions
Plasmids for expression of GPX4 and FSP1 were generated by replacing the OVA-IRES-Vex section of the MSCV plasmid generated above with Vex-IREX-GPX4, FSP1-IRES-Vex, or Vex-only as an empty vector control. For GPX4, entire transcript variants 4 (cytosolic; NM_001367995.1) and 1 (mitochondrial; NM_008162.4) of mouse Gpx4 (Gene ID: 625249) were expressed to include the 3’ UTR that contains a SECIS element necessary for selenocysteine incorporation. For FSP1, the coding region of transcript variant 2 (NM_178058.4) of mouse Aifm2 (Gene ID: 71361) was expressed, with a C-terminal HA-tag (TACCCATACGATGTTCCAGATTACGCT) inserted before the stop codon. Retrovirus was produced using Platinum-E cells using the same protocol as described for Phoenix-ECO cells above. Our T-cell transduction protocol was a modification of published protocols (18,25). Primary naïve CD8+ T cells were isolated using a naïve CD8α+ T-cell isolation kit (Miltenyi Biotec; cat. #130-096-543) and activated on plates coated with 2 μg/mL anti-CD3 and 2 μg/mL anti-CD28 in the presence of 200 units/mL IL-2 (R&D Systems; cat. #202-IL-050) for 24–27hr at 37 °C prior to transduction in the same RPMI-based media as described for T-cell culture and killing assays above. Transductions were performed in non-tissue culture-treated plates coated with 20 μg/mL retronectin (Takara Bio; cat. #T100B) by “spinfecting” the T cells at 726 g for 90 minutes at 37 °C. The plates were then put in an incubator (37 °C, 5% CO2) for 4hr before washing off the virus. The T cells were expanded for 72hr in media with 100 U/mL IL-2 (R&D Systems; cat. #202-IL-050) prior to flow cytometric sorting of transduced cells based on Vex expression. After sorting, the T cells were rested in the same RPMI-based media as described for T-cell culture and killing assays above with5 ng/mL IL7 (PeproTech; cat. #217-17-10ug) for 48hr and used in downstream experiments using the same methods described for naïve T cells (e.g., T-cell activation conditions for killing assays).
Western blotting
Transduced cells were collected for Western blotting immediately following sorting of Vex+ T cells. Cells were lysed in RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific; cat. #89901) supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific; cat. #78440) for 15 min on ice. Whole cell lysates were centrifuged at 20,817 g at 4 °C for 15 min. Supernatants were collected and transferred to new Eppendorf tubes and 10 μL of each lysate was taken for protein estimation using the Pierce BCA protein assay kit (Thermo Fisher Scientific; cat. #23225) to normalize protein loading. Cleared lysates were denatured with 4x Laemmli sample buffer (Bio-Rad; cat. #1610747) containing 2-mercaptoethanol and boiled for 5 min at 95 °C. 15 μg of each lysate was loaded and run on a NuPAGE 4–12% bis-tris protein gel (Thermo Fisher Scientific; cat. #NP0322BOX) and then transferred onto a nitrocellulose membrane (LI-COR Biosciences; cat. #926-31092). Membranes were blocked for 1hr in 5% milk (MilliporeSigma; cat. #M7409-1BTL) at room temperature and then incubated with primary antibodies, including anti-glutathione peroxidase 4 rabbit IgG monoclonal (1:1000; Abcam; clone EPNCIR144; cat. #ab125066), anti-HA high affinity rat IgG1 monoclonal (1:500; MilliporeSigma; clone 3F10; cat. #11867423001) and anti-β-actin mouse IgG2b monoclonal (1:1000; Cell Signaling Technologies; clone 8H10D10; cat. #3700S), overnight in 5% milk at 4 °C. Membranes were washed in TBS-T buffer and incubated with the respective HRP-linked secondary antibodies (HRP-linked anti-rabbit IgG, HRP-linked anti-mouse IgG and HRP-linked anti-rat IgG; 1:2000; Cell Signaling Technologies; cat. #7074P2, #7076P2 and #7077S, respectively) in 5% milk for 1hr at room temperature. Membranes were then treated with SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific; cat. #34580) and imaged using the Amersham Imager 600. When necessary, membranes were stripped with Restore PLUS Western Blot Stripping Buffer (Thermo Fisher Scientific; cat. #46430) for 15 min at room temperature, blocked and reprobed with primary and secondary antibodies as described above. Expression levels were analyzed using Image Studio software and quantified in pixels.
CHimeric IMmune Editing (CHIME)
CHimeric IMmune Editing was performed as previously described (22,23). Briefly, gRNAs were designed using the Broad Institute sgRNA designer software and cloned into our lentiviral pXPR_053 vector (Addgene; cat. #113591). gRNA sequences were GCGAGGTATTCGGCTCCGCG (Scramble), GTCCAGGGATACGTTCACAC (ACSL4 gRNA-1) and CAATAGAGCAGAGTACCCTG (ACSL4-2). Bone marrow cells were isolated from femurs, tibias, hip bones and spines of Cas9+ CD45.1 single-positive (SP) and CD45.1/2 double-positive (DP) OT-1 mice. The CD45.1/2 congenic markers were used to allow for the detection of adoptively transferred cells during downstream adoptive T-cell transfer experiments (see below). LSK (lineage− Sca-1+ Kit+) cells were magnetically enriched using CD117 MicroBeads (Miltenyi Biotec; cat. #130-091-224) and flow sorted for purity. Lentivirus was generated by PEI-mediated transfection of HEK293x cells with gRNA plasmid DNA as well as psPAX2 (Addgene; cat. # 12260) and pMD2.G (Addgene; cat. # 12259) packaging and envelope plasmids using a 1:3:4:24 pMD2.G : psPAX2 : gRNA vector : PEI ratio in Opti-MEM, following the same transfection and supernatant harvest protocol as described for retrovirus with Phoenix-ECO cells above. LSK cells were spin transduced in plates coated with 100 μg/mL retronectin at a multiplicity of infection of approximately 30, followed by adoptive transfer into irradiated CD45.2+ wildtype C57BL/6J recipients.
Adoptive T-cell transfer
For transfer of GPX4 or FSP1 overexpressing OT-1 CD8+ T cells, transduced CD45.1 SP and CD45.1/2 DP cells were collected following a 48hr rest in IL7, as described above (see T-cell transductions). For transfer of naïve OT-1 CD8+ T cells from CHIME bone marrow chimeras, naïve CD8+ T cells were isolated from spleens and inguinal lymph nodes of chimeras ≥8 weeks after immune reconstitution using a naïve CD8a+ T cell isolation kit (Miltenyi Biotec; cat. #130-096-543) followed by sorting for Vex-positivity, absence of lineage markers (TER-119, B220, Gr-1), and viability (7-AAD-negativity; BD Biosciences; cat. #559925). Mixes of 5,000 CD45.1 SP and 5,000 CD45.1/2 DP cells were adoptively transferred into wildtype or Cas9-expressing recipients (to avoid rejection of Cas9-expressing OT-1 cells) one day prior to B16-OVAWT tumor implantations. Tumors were harvested between day 15 and 18 post-implantation. For calculations of CD45.1 SP to CD45.1/2 DP ratios upon tumor harvest (as above; see Tumor harvests), samples with ≥50 recorded Vex+ CD8+ T cells were included to ensure that reliable ratios were obtained. Ratio fold changes were calculated using the formula (DP:SP)TIL / (DP:SP)input when DP cells were modified and SP cells were controls, or (SP:DP)TIL / (SP:DP)input when SP cells were modified and DP cells were controls. To analyze expression of markers within the SP or DP populations (e.g., PD-1, Ki-67, and GzmB), samples with ≥100 recorded cells in each group were included to ensure that reliable percentages were obtained.
MiSeq for indel quantification
For indel quantification, DNA was isolated from Vex+ CD8+ T cells using a DNeasy blood & tissue kit (Qiagen; cat. #69506). The guide RNA target region was amplified by PCR, purified using the QIAquick PCR purification kit (Qiagen; cat. #28106), and sequenced by MiSeq at the MGH DNA Sequencing Core using the following primers: ACSL4 gRNA-1 forward CAAGTAGACCAACCCCTTCAGACAT and reverse ATCCTACAGCCATAGGTAAAGCATGA; ACSL4 gRNA-2 forward AGTGTGACAAATTGAATAGCTGGCTT and reverse TCTGTCATGTGCAGTCTTGATTACTT. The percentage indels in the guide RNA target region was quantified using Basepair software.
Cell-trace violet proliferation assay
To quantify T-cell proliferation, naïve CD8+ T cells were labelled with cell-trace violet (CTV) (Thermo Fisher Scientific; cat. #C34557) per the manufacturer’s instructions and activated in the same RPMI-based media used for the T-cell culture and killing assays described above in plates coated with 4 μg/mL anti-CD3 and 4 μg/mL anti-CD28 in the presence of 100 units/mL IL-2 for 72hr. The cells were then collected, stained, and analyzed by flow cytometry as described above (see Flow cytometry and flow sorting). Proliferation indices were determined in FlowJo software.
Statistical analyses
Statistical analyses were conducted in GraphPad Prism 8.3.1. A two-sided paired Student’s t-test was used for comparison of two groups with paired samples. A two-sided unpaired Student’s t-test was used for comparison of two unpaired groups. One-way analysis of variance (ANOVA) was used for single comparisons with >2 groups. Two-way ANOVA was used for multiple comparisons within groups. Pearson’s correlation coefficients were used to analyze correlations. Graphs display mean +/− standard deviation (SD). p values are denoted as *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Results
Development of a Platform for Pharmacologic Screening of Anti-Tumor CD8+ T-Cell Function
To develop a platform suitable for in vitro pharmacologic screening of tumor-specific CD8+ T cell function, we built upon an in vitro killing assay (26), in which naïve T-cell receptor (TCR)-transgenic CD8+ T cells are activated and then co-cultured with antigen-expressing cancer cells. To quantify antigen-specific cancer cell killing by CD8+ T cells, we generated B16 and MC38 cancer cells expressing either OVAWT, which contains the SIINFEKL peptide, a potent MHC-I–presented epitope recognized by OT-1 CD8+ T cells (27), or OVAΔ257-264, which lacks SIINFEKL. These tumor cells also expressed a fluorescent marker—either GFP, RFP or Vex —to enable us to distinguish among these populations by flow cytometry. The presence of the SIINFEKL peptide in H-2Kb MHC-I on the OVAWT but not the OVAΔ257-264-expressing cell lines was confirmed by flow cytometry (Supplementary Fig. S1A and S1B). We cultured a 1:1 ratio of cancer cells with (OVAWT) and without (OVAΔ257-264) the SIINFEKL epitope with activated OT-1 T cells, and measured antigen-specific killing by a reduction in the percentage of OVAWT-expressing cells (Fig. 1A and Supplementary Fig. S1C). Cancer cells expressing OVAWT were specifically killed by OT-1 T cells, as reflected by a change in the percentage of OVAWT-expressing among all (OVAWT- or OVAΔ257-264-expressing) B16 cells, regardless of which fluorescent protein was co-expressed (Supplementary Fig. S1D).
Figure 1. Development of a Platform for Pharmacologic Screening of Anti-Tumor CD8+ T-Cell Function.
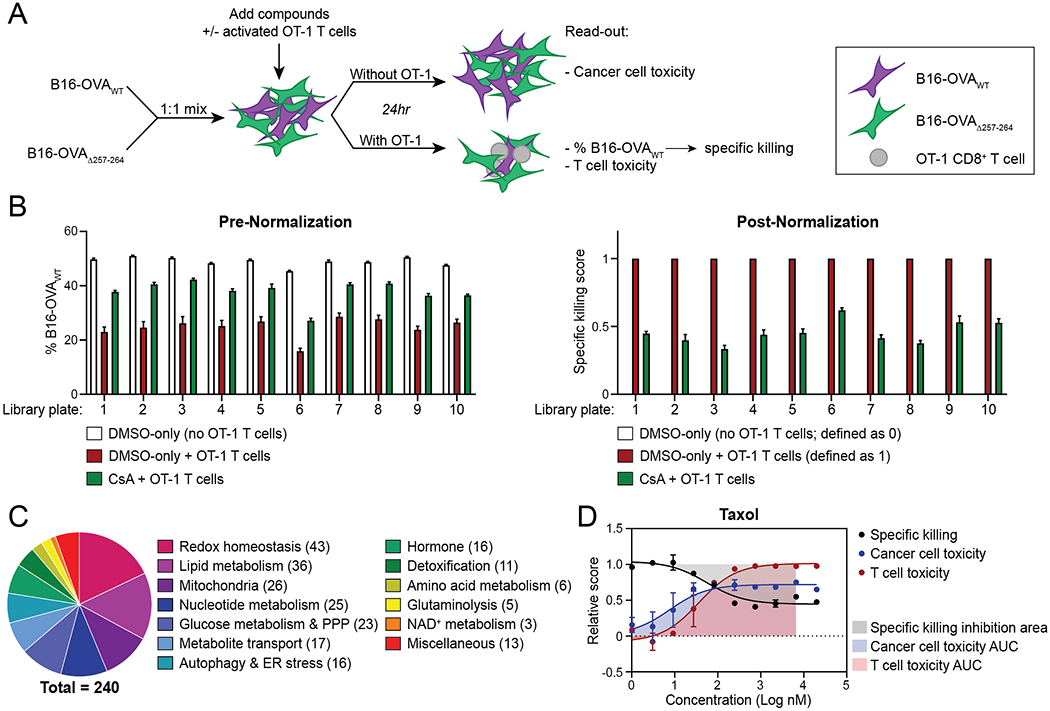
A, Schematic depicting experimental setup used for pharmacologic screening.
B, Average read-outs for T cell–mediated specific killing before and after normalization, as indicated by average % B16-OVAWT and average specific killing score among control wells of each assay plate respectively.
C, Numbers of compounds in the Ludwig Metabolic Library targeting indicated metabolic pathways.
D, Dose-response curve displaying T cell–mediated specific killing, cancer-cell toxicity and T-cell toxicity of taxol at the indicated concentrations.
Graphs display mean +/− SD. n=8 (B,C) or n=2 (D) technical replicates per condition. CsA, Cyclosporin A. PPP, pentose-phosphate pathway. ER, endoplasmic reticulum.
To ensure that the T-cell activation protocol yielded potently cytotoxic OT-1 CD8+ T cells, we measured antigen-specific killing by OT-1 CD8+ T cells activated in the presence or absence of IL-12, a cytokine known to enhance CD8+ T-cell cytotoxicity and proliferation (28). OT-1 T cells activated without IL-12 displayed little specific killing, while those activated with IL-12 had robust specific cytotoxicity, both in the presence and absence of antibodies neutralizing IFNγ and TNFα during co-culture (Supplementary Fig. S1E). We therefore activated CD8+ T cells with IL-12 in our screening experiments.
To determine the optimal killing assay conditions for pharmacologic screening, we conducted killing assays with either B16 or MC38 as target cells (with and without pre-treatment with IFNγ to upregulate antigen presentation), two T cell – cancer cell co-culture durations (24hr and 48hr), and multiple T cell to cancer cell ratios (Supplementary Fig. S1F and G). We observed robust antigen-specific killing with both B16 and MC38 cells, independent of IFNγ pre-treatment (Supplementary Fig. S1F and G). Based on these results, we selected a 24hr assay period using B16 cells without IFNγ pre-stimulation at a 1:6 T cell to cancer cell ratio for our screening effort, aiming for a change in percentage of OVAWT-expressing cells from 50% to ~20-30% to allow detection of both enhanced and reduced antigen-specific killing. Thus, by optimizing CD8+ T-cell activation and T cell – cancer cell co-culture killing assay conditions we established a reliable and sensitive screening platform for measurement of anti-tumor CD8+ T cell function.
Pharmacologic Screening Identifies Vulnerability of CD8+ T Cells to GPX4 Inhibitors
Having optimized the general killing assay workflow, we employed high-throughput plating and flow cytometry methods for pharmacologic screening. Each assay plate included control wells without drug (DMSO-only) and wells with 2 μM cyclosporin A (CsA), a known T-cell inhibitor (29). At the end of the 24hr co-culture period, we used flow cytometry to read out the numbers of B16-OVAWT, B16-OVAΔ257-264 and T cells in each well. Using these numbers, we calculated the percentage B16-OVAWT (of all B16-OVAWT and B16-OVAΔ257-264 cells) and T-cell toxicity for wells with T cells, and cancer-cell toxicity for wells without T cells (Fig. 1A). The percentage B16-OVAWT was converted to a normalized specific killing score to allow for comparison of findings across 10 screen runs (Fig. 1B). The average percentage B16-OVAWT in DMSO-only control wells without T cells was defined as a specific killing score of 0, and the average percentage B16-OVAWT DMSO-only wells with T cells was defined as a specific killing score of 1.
We screened the Ludwig Metabolic Library, which consists of 240 compounds at 10 concentrations each (dose range 1 nM to 20 μM), broadly targeting metabolic pathways (30) (Fig. 1C and Supplementary Table S1). The compounds were added at the start of T cell – cancer cell co-culture to mimic the encounter of metabolic challenges when activated T cells enter the TME (Fig. 1A). The library was screened in duplicate both with and without T cells. Replicate values for these parameters were well-correlated, especially for specific killing score, which does not rely on absolute cell numbers (Supplementary Fig. S2A–C).
To analyze the screen data, we generated dose-response curves for each of the 240 compounds in the library, displaying specific killing score, cancer-cell toxicity and T-cell toxicity at each of the concentrations tested (Fig. 1D, Supplementary Table S2, and Supplemental Data File S1). To quantify the overall effects of each drug, we calculated the AUC for cancer-cell toxicity and T-cell toxicity and plotted the area minus the specific killing AUC as a measure of inhibition of T cell–mediated specific killing of cancer cells (Fig. 1D and Supplementary Table S3). The highest concentration was not included in AUC calculations to allow inclusion of compounds for which the top concentration resulted in significant skewing of flow cytometric properties or a very high toxicity such that no reliable B16-OVAWT to B16-OVAΔ257-264 ratio could be determined. Using these parameters, we ranked the compounds by specific killing inhibition, cancer-cell toxicity and T-cell toxicity (Supplementary Fig. S2D–F). Most metabolic compounds in the library decreased T cell–mediated specific killing rates, and very few drugs enhanced specific killing (Supplementary Fig. S2D). Moreover, those compounds that did increase specific killing did so only marginally, as exemplified by the dose-response curve of the vitamin A derivative isotretinoin, which yielded the highest increase in specific killing of all library compounds (Supplementary Fig. S2G). Similarly, most compounds were toxic to cancer cells and T cells, with only few compounds resulting in small increases in cell numbers (Supplementary Fig. S2E and F).
To assess the comparative sensitivities of cancer cells versus T cells to the tested metabolic compounds, we compared T-cell toxicity AUC to cancer-cell toxicity AUC. This showed that T cells were more sensitive than cancer cells to most of the compounds in the library, suggesting that effector CD8+ T cells are generally more sensitive to metabolic perturbations (Fig. 2A). To identify which compounds most specifically affected T cells, in terms of either cell numbers or function, we plotted the T cell–specific vulnerability of the compounds, defined as the difference between inhibition of T cell–mediated specific killing of cancer cells and cancer-cell toxicity AUC (Fig. 2B and C). Here, a high score indicates high inhibition of specific killing and/or low cancer-cell toxicity, while a low score indicates low inhibition of specific killing and/or high cancer-cell toxicity. The highest T cell–specific vulnerabilities were obtained for compounds targeting GPX4, NAD+ metabolism or autophagy and endoplasmic reticulum (ER) stress pathways (Fig. 2B). Strikingly, all three GPX4 inhibitors in the library, 1S,3R-RSL3 (RSL3), ML162 and ML210, were among the 10 compounds with the highest scores, indicating a high vulnerability of T cells compared to B16 cancer cells to these compounds (Fig. 2C). Moreover, two of the three library compounds targeting NAD+ metabolism, which were nicotinamide phosphoribosyltransferase (NAMPT) inhibitors such as the top hit FK866, were in the top 10, while most of the remaining top hits targeted autophagy and ER stress (Fig. 2B and C). Among the compounds with the lowest scores were several modulators of nucleotide and mitochondrial metabolism (Fig. 2C). These low scores were mostly explained by a relatively intact specific killing score with high cancer-cell toxicity, even when T-cell toxicity was as high or even higher than cancer-cell toxicity, as illustrated by the dose-response curves of the top two hits methotrexate and antimycin A (Supplementary Fig. S2H).
Figure 2. Pharmacologic Screening Identifies Vulnerability of CD8+ T Cells to GPX4 Inhibitors.
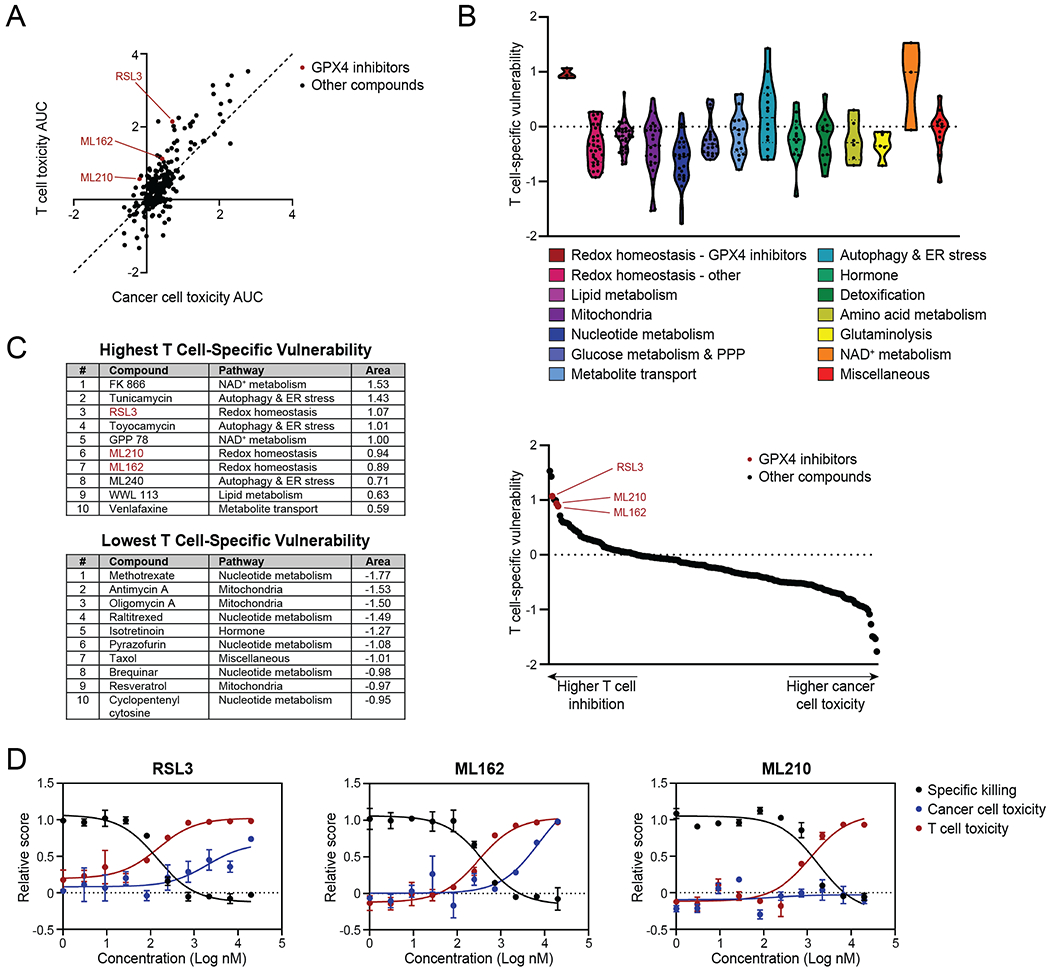
A, Plot showing T-cell toxicity AUC and cancer-cell toxicity AUC, highlighting GPX4 inhibitors. Dashed line represents y=x function. Each dot represents one compound.
B, Violin plots of T cell–specific vulnerability for each compound, organized by pathway. Each dot represents one compound, dashed lines indicate medians, dotted lines indicate quartiles.
C, Tables showing the top 10 highest and lowest library compounds by T cell–specific vulnerability (left) and plot showing library compounds ranked by this parameter (right).
D, Dose-response curves displaying specific killing, cancer-cell toxicity and T-cell toxicity of RSL3, ML162 and ML210 at the indicated concentrations. Graphs display mean +/− SD. n=2 technical replicates per condition.
PPP, pentose phosphate pathway. ER, endoplasmic reticulum.
T cells that genetically lack GPX4 were previously shown to undergo ferroptosis, leading to defective immunity to infection (31). Since GPX4 inhibitors were among the top hits and GPX4 has not been examined in T cells in the context of anti-tumor immunity, we selected the GPX4 inhibitors for further validation and follow-up. Dose-response curves of these compounds showed that T-cell numbers and specific killing were reduced at concentrations that were not toxic to cancer cells (Fig. 2D). Thus, the in vitro pharmacologic screen identified compounds targeting metabolic pathways to which T cells were more vulnerable than B16 cancer cells, including GPX4 inhibitors.
GPX4 Inhibitors Selectively Induce Ferroptosis in CD8+ T Cells
GPX4 is the only glutathione peroxidase enzyme that has the ability to reduce hydroperoxides in membranes, thereby protecting cells from the iron-dependent cell death pathway ferroptosis (Fig. 3A) (32,33). Induction of this pathway in cancer cells is being pursued as a potential novel therapeutic approach (34,35). To validate the vulnerability of CD8+ T cells to GPX4 inhibition, we conducted low-throughput killing assays with GPX4 inhibitors RSL3, ML162 and ML210 at concentrations that selectively impacted CD8+ T cells in the screen. Confirming the screen results, OT-1 T cells were sensitive to GPX4 inhibitors, as shown by reduced specific killing rates and T-cell numbers, at concentrations that did not affect the numbers of B16 cancer cells in the absence of T cells (Fig. 3B–D). Since CD8+ T cell-derived IFNγ increases ferroptosis sensitivity in cancer cells (36), we hypothesized that GPX4 inhibition might impact cancer cells more when co-cultured with OT-1 T cells than when cultured alone. We therefore also assessed B16 and MC38 cell numbers in the wells with OT-1 T cells, and found that the numbers of these cancer cells increased with the tested concentrations of GPX4 inhibitors (Fig. 3E), indicating that exposure of T cells and cancer cells to GPX4 inhibition in the same environment negatively impacted CD8+ T cells more than B16 or MC38 cancer cells, providing the cancer cells a net advantage.
Figure 3. GPX4 Inhibitors Selectively Induce Ferroptosis in CD8+ T Cells.
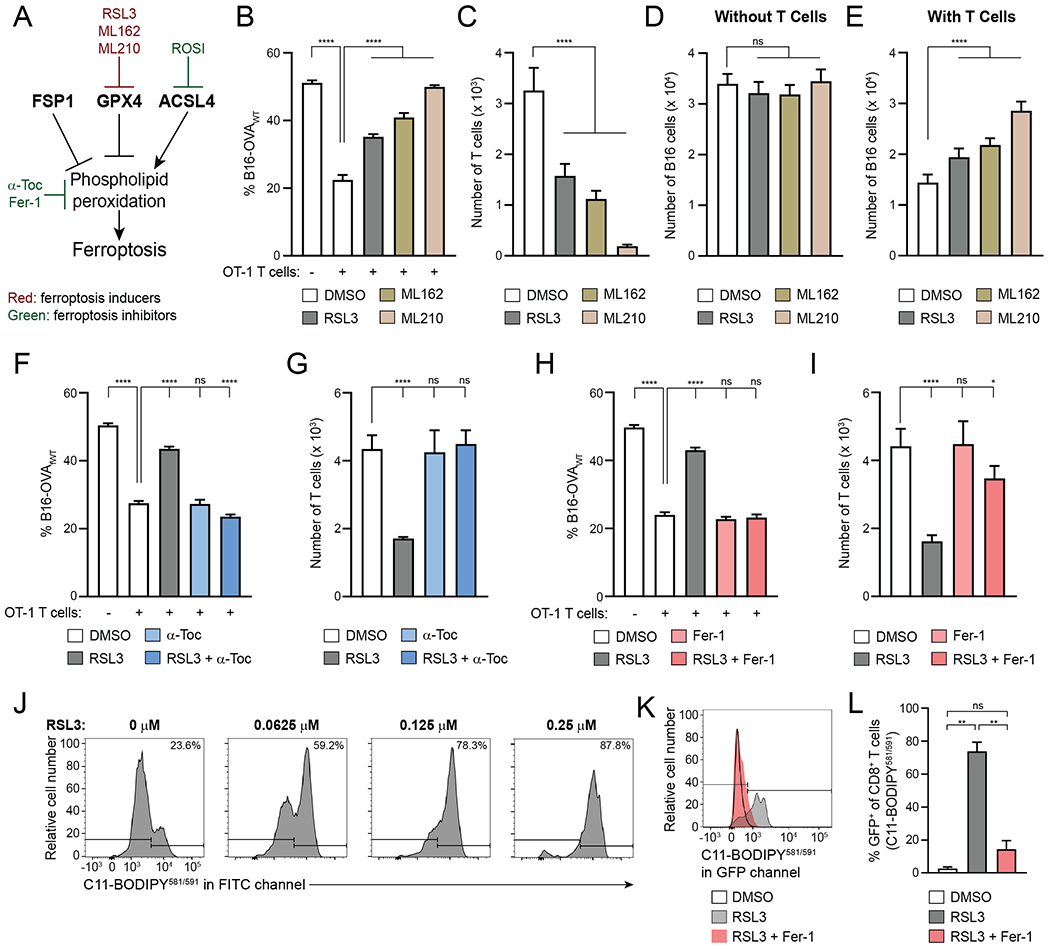
A, Schematic displaying (inhibitors of) enzymes that regulate lipid peroxidation and ferroptosis.
B–E, Killing assay with B16-OVAWT and B16-OVAΔ257-264 cells in the presence of 0.25 μM RSL3, 0.74 μM ML162, 2.2 μM ML210 or DMSO control, showing % B16-OVAWT (B), number of T cells (C), and total number of B16 cells in wells without (D) or with (E) T cells at end of assay. n=5-10 technical replicates per condition. Statistical significance determined by one-way ANOVA.
F–I, Killing assay with B16-OVAWT and B16-OVAΔ257-264 cells with or without 0.25 μM RSL3, 100 μM α-Toc (F,G) and/or 1 μM Fer-1 (H,I), showing % B16-OVAWT (F,H) and number of T cells at end of assay (G,I). n=4 technical replicates per condition. Statistical significance determined by one-way ANOVA.
J, C11-BODIPY581/591 staining of CD8+ T cells that were activated for 4 days and then treated with indicated concentrations of RSL3 for 24hr.
K,L, Representative examples (K) and quantification (L) of C11-BODIPY581/591 staining of CD8+ T cells that were activated for three days and then treated for 24hr with 0.25 μM RSL3 with or without 1 μM Fer-1. n=2 technical replicates per condition. Statistical significance determined by one-way ANOVA.
*p<0.05 **p<0.01 ***p<0.001 ****p<0.0001. ns, not significant. Graphs display mean +/− SD. α-Toc, α-tocopherol vitamin E. Fer-1, ferrostatin-1. ROSI, rosiglitazone. Data representative of ≥2 independent experiments.
To test whether our findings with GPX4 inhibitors were specific to the B16 cancer model, we conducted the same in vitro validation experiments with MC38 cells, and found that CD8+ T cells were also more sensitive to GPX4 inhibition than these cancer cells (Supplementary Fig. S3A–D). When culturing these cell types individually, numbers of both B16 and MC38 cancer cells, even after IFNγ treatment, were less affected by a 24hr treatment with the GPX4 inhibitor RSL3 than activated CD8+ T cells (Supplementary Fig. S3E–G). Finally, we found that CD8+ and conventional CD4+ T cells (Tconvs) were similarly sensitive to GPX4 inhibition after activation in the presence of IL-12 (Supplementary Fig. S3H).
Given that GPX4 inhibition can lead to cell death through ferroptosis (33), we examined whether the reduction in numbers of RSL3-treated CD8+ T cells was due to ferroptosis. The lipophilic antioxidant α-tocopherol vitamin E (α-Toc) and ferroptosis inhibitor ferrostatin-1 (Fer-1) can inhibit ferroptosis (31,37–39). We therefore tested whether these agents could prevent the effects of GPX4 inhibitors on CD8+ T cells in the context of anti-tumor function. Indeed, α-Toc and Fer-1 rescued T cell–mediated specific killing of cancer cells as well as T-cell numbers in the presence of RSL3, supporting the notion that GPX4 inhibition causes ferroptosis in CD8+ T cells (Fig. 3F–I). We did not observe a direct effect of α-Toc and Fer-1 on B16 cell numbers in the absence of OT-1 T cells (Supplementary Fig. S3I and J). GPX4 inhibition also resulted in a small reduction in GzmB expression, which was rescued by addition of α-Toc and Fer-1, while IFNγ expression was unchanged (Supplementary Fig. S3K–N).
Excess phospholipid peroxidation is a hallmark of ferroptosis (33,39,40). Accordingly, increasing lipid peroxidation was observed in T cells treated with increasing concentrations of RSL3, as measured by fluorescent emission in the GFP/FITC channel upon C11-BODIPY581/591 staining, and this was reversed by co-treatment with Fer-1 during RSL3 treatment (Fig. 3J–L). In summary, in vitro validations confirmed that GPX4 inhibitors induce ferroptosis in activated CD8+ T cells at concentrations that do not affect B16 and MC38 cancer cell numbers.
In addition to GPX4 inhibitors, we conducted in vitro validation experiments with FK866 (Supplementary Fig. S4A–D) and isotretinoin (Supplementary Fig. S4E–H), which also confirmed the screen results for these compounds. This further demonstrates the robustness of the screen platform.
CD8+ Tumor-Infiltrating Lymphocytes Display Lipid Peroxidation Ex Vivo
Since high levels of reactive oxygen species are common in tumors (41), we investigated whether lipid peroxidation occurs in T cells in the TME. We implanted B16-OVAWT tumors in mice subcutaneously and harvested tumors and draining lymph nodes (dLNs) two weeks later, followed by C11-BODIPY581/591 and flow cytometry staining (Supplementary Fig. S5A). As a control, ex vivo C11-BODIPY581/591 staining was conducted in the absence and presence of Fer-1. Tumor-derived, but not dLN-derived, CD8+ T cells displayed significant amounts of lipid peroxidation (Supplementary Fig. S5B–F). In CD4+ T cells, a substantial portion of which are regulatory T cells (Tregs) in B16-OVA tumors (42), we did not observe the same effect (Supplementary Fig. S5G–I). These data indicate that lipid peroxidation occurs in CD8+ tumor-infiltrating lymphocytes (TILs).
Overexpression of FSP1 or Cytosolic GPX4 Reduces Ferroptosis Sensitivity in CD8+ T Cells but Does Not Affect Their Anti-Tumor Function In Vivo.
GPX4 and FSP1 can protect other cell types from ferroptosis induction (33,38,43,44) (Fig. 3A). To investigate their roles in modulating ferroptosis sensitivity in CD8+ T cells, we retrovirally transduced OT-1 CD8+ T cells with plasmids encoding Vex as well as GPX4 or HA-tagged FSP1 or a Vex-only empty vector (EV) control (Fig. 4A and Supplementary Fig. S6A and B). For GPX4, both the cytosolic (cGPX4) and mitochondrial (mGPX4) isoforms were expressed.
Figure 4. Overexpression of FSP1 or Cytosolic GPX4 Reduces Ferroptosis Sensitivity in CD8+ T Cells.
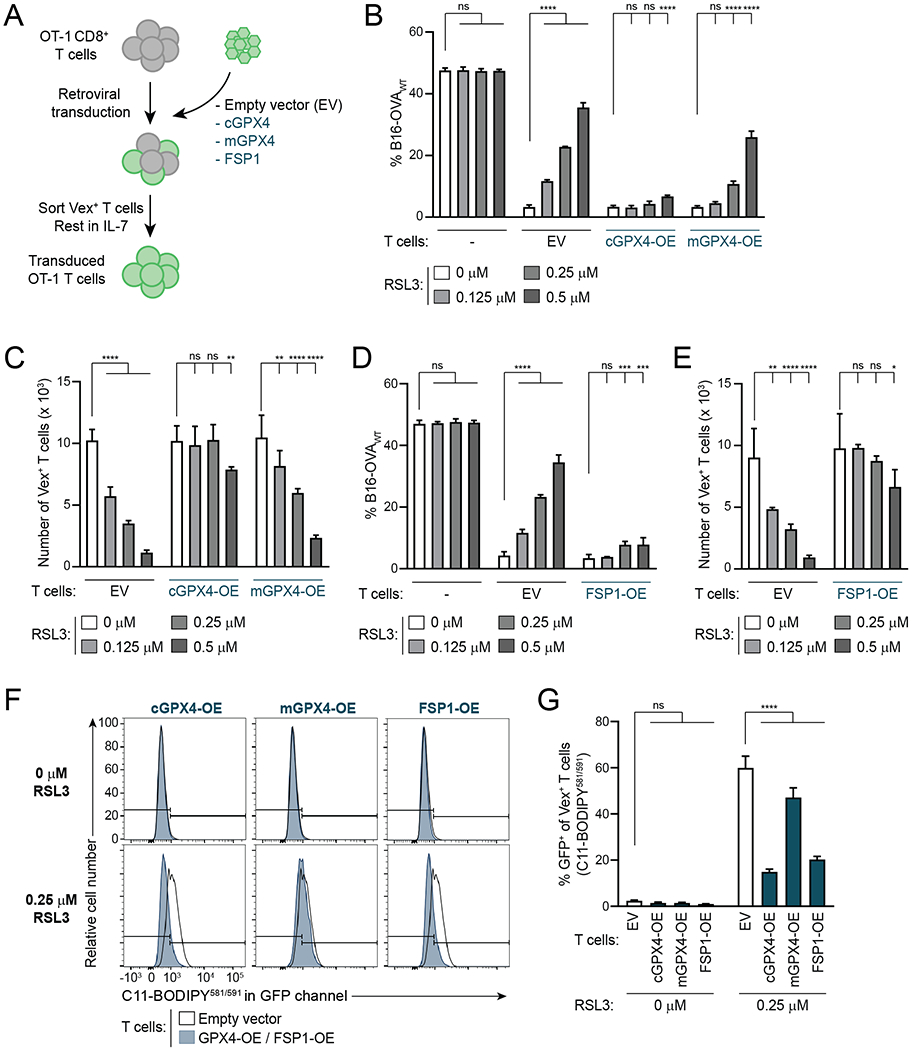
A, Schematic depicting generation of GPX4- and FSP1-OE OT-1 CD8+ T cells.
B–E, Specific killing (B,D) and T cell numbers (C,E) at the end of killing assays with GPX4-OE (B,C) or FSP1-OE (D,E) OT-1 T cells and EV control OT-1 T cells. n=2-6 technical replicates per condition. Statistical significance determined by two-way ANOVA.
F,G, Representative examples (F) and quantification (G) of C11-BODIPY581/591 staining of OT-1 EV, GPX4-OE and FSP1-OE T cells treated with indicated concentrations of RSL3 for 24hr after 72hr activation. Flow plots in F compare each condition to the same EV control sample. n=3-4 technical replicates per condition. Statistical significance determined by two-way ANOVA.
*p<0.05 **p<0.01 ***p<0.001 ****p<0.0001. ns, not significant. Graphs display mean +/− SD. Data representative of ≥2 independent experiments.
In killing assays, OT-1 T cells overexpressing cytosolic GPX4 (cGPX4-OE) displayed a markedly reduced sensitivity to ferroptosis inducer RSL3, as measured by T cell–mediated specific killing rates as well as number of T cells at the end of the assay with increasing concentrations of RSL3 (Fig. 4B and C). This effect was much less pronounced with overexpression of mGPX4 (Fig. 4B and C). FSP1-OE T cells also showed resistance to ferroptosis induction, similar to that seen with cGPX4-OE (Fig. 4D and E). Accordingly, activated cGPX4-OE and FSP1-OE CD8+ T cells displayed lower levels of lipid peroxidation upon treatment with RSL3, as measured by C11-BODIPY581/591 staining, whereas mGPX4-OE T cells had only a small reduction in lipid peroxidation compared to EV control cells (Fig. 4F and G). In the absence of RSL3, specific killing and T cell numbers were not compromised by overexpression of GPX4 or FSP1 (Fig. 4B–E). GzmB and IFNγ expression were not altered with GPX4 or FSP1 overexpression in the absence of GPX4 inhibition (Supplementary Fig. S6C and D). B16 cell numbers were not affected by even the highest RSL3 concentration of 0.5 μM in the absence of T cells (Supplementary Fig. S6E). These data demonstrate that FSP1 and cytosolic GPX4 can mediate resistance to ferroptosis induction in CD8+ T cells.
To investigate whether cGPX4 or FSP1 overexpression promotes anti-tumor CD8+ T cell responses in vivo, we transferred a 1:1 mix of congenically labeled cGPX4- or FSP1-OE and EV control OT-1 CD8+ T cells to mice in which B16-OVAWT tumors were implanted one day later (Fig. 5A). Upon tumor harvest (day 15 to 18 after implantation), the ratios of cGPX4- or FSP1-OE (CD45.1.2 double-positive (DP)) to control (CD45.1 single-positive (SP)) intratumoral Vex+ CD8+ T cells were assessed (Fig. 5B and Supplementary Fig. S7A). cGPX4- and FSP1-OE T cells did not have a competitive advantage compared to control T cells, as measured by the ratio of CD45.1.2 DP to CD45.1 SP Vex+ T cells in tumors compared to input (Fig. 5C). Moreover, markers of T-cell function (GzmB) and proliferation (Ki-67) were not different between these populations (Fig. 5D and E and Supplementary Fig. S7B). These results suggest that, despite displaying lipid peroxidation, CD8+ TILs do not die from ferroptosis at high rates. Notably, induction of ferroptosis resistance by overexpressing cGPX4 or FSP1 does not compromise anti-tumor CD8+ T cell function in vivo.
Figure 5. Overexpression of cGPX4 or FSP1 Does Not Affect CD8+ T cell Anti-Tumor Function In Vivo.
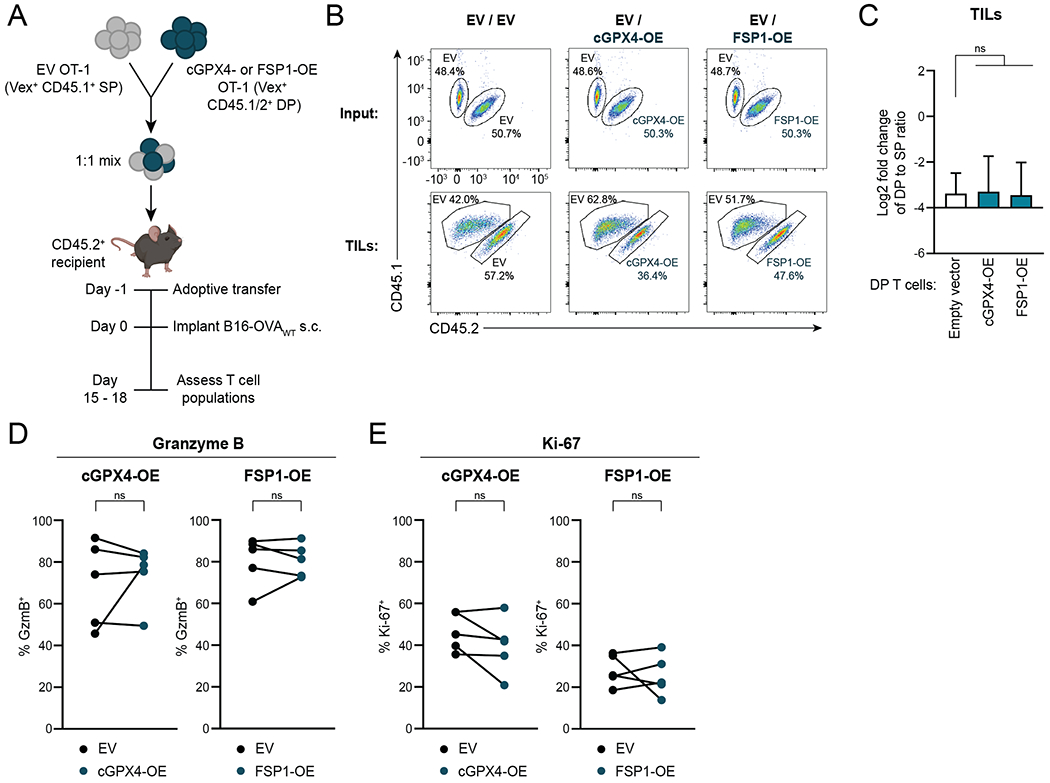
A, Schematic depicting adoptive transfer experiment. The mouse image was created using BioRender.com.
B,C, Representative plots (B) and quantification (C) of flow cytometric measurement of the change in ratio of CD45.1/2 DP to CD45.1 SP among Vex+ CD8+ T cells in B16-OVAWT tumors compared to input. Tumors were harvested for analysis on day 15 to 18 after implantation. Ratio fold-change calculated as (DP:SP)TIL / (DP:SP)input. n=5-8 animals per group. Statistical significance determined by one-way ANOVA.
D,E, Percentages of Vex+ CD8+ T cells expressing GzmB (D) and Ki-67 (E) as measured by flow cytometry after tumor harvest. n=5 mice per group. Statistical significance determined by paired t tests.
SP, CD45.1 single-positive. DP, CD45.1/2 double-positive. *p<0.05 **p<0.01 ***p<0.001 ****p<0.0001. ns, not significant. Graphs display mean +/− SD. Data representative of ≥2 independent experiments. TIL, tumor-infiltrating lymphocyte.
ACSL4 Promotes Ferroptosis Sensitivity in CD8+ T Cells
Next, we tested the role of ACSL4, which promotes ferroptosis sensitivity in other cell types by facilitating the incorporation of oxidation-prone polyunsaturated fatty acids (PUFAs) into membranes (Fig. 3A) (45–48). First, we tested whether inhibition of ACSL4 would protect CD8+ T cells from ferroptosis induction by GPX4 inhibition by using thiazolidinedione rosiglitazone (ROSI) to inhibit ACSL4 (49). When activated CD8+ T cells were cultured with the GPX4 inhibitor RSL3 in the presence or absence of ROSI, ROSI rescued RSL3-treated CD8+ T cell numbers in a dose-dependent manner (Supplementary Fig. S8A). These findings indicate that inhibition of ACSL4 reduces ferroptosis sensitivity in CD8+ T cells.
Next, we used CRISPR/Cas9–mediated CHIME to generate Vex-expressing ACSL4-knockout (KO) OT-1 CD8+ T cells with two distinct ACSL4-targeting guide RNAs (gRNAs) (Fig. 6A and Supplementary Fig. S8B) (22). ACSL4 deficiency effectively protected CD8+ T cells from ferroptosis induction by GPX4 inhibition in killing assays, resulting in higher T cell-mediated specific killing and T-cell numbers than scramble gRNA-expressing control T cells at RSL3 concentrations ≥0.25 μM (Fig. 6B and C). However, in the absence of GPX4 inhibition, ACSL4-KO CD8+ T cells displayed reduced specific killing and T-cell numbers at the end of the assay (Fig. 6B and C). This decrease in specific killing by ACSL4-KO CD8+ T cells was not caused by a defect in the expression of effector molecules GzmB and IFNγ, as nearly all cells expressed GzmB in both groups, and IFNγ was even increased in ACSL4-KO cells (Supplementary Fig. S8C and D). In line with a reduced sensitivity to GPX4 inhibition, ACSL4-KO CD8+ T cells displayed less lipid peroxidation upon RSL3 treatment compared to scramble control T cells (Fig. 6D and E).
Figure 6. ACSL4 Promotes Ferroptosis Sensitivity in CD8+ T Cells.
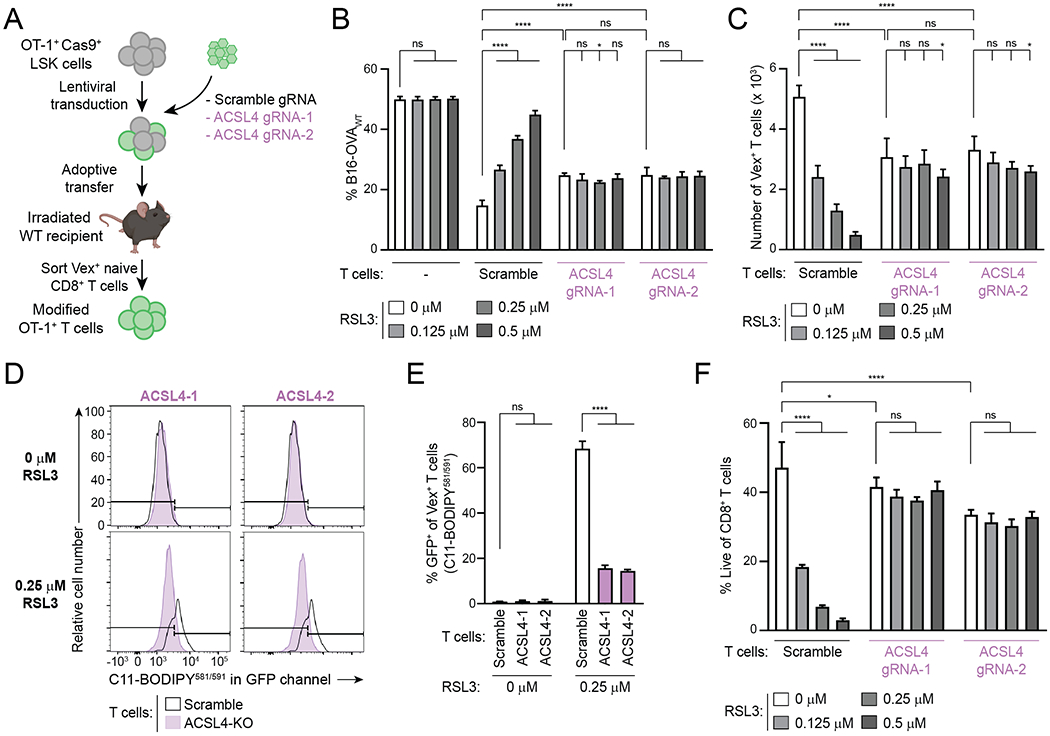
A, Schematic depicting generation of ACSL4-deficient OT-1 CD8+ T cells using CHIME. The mouse image was created using BioRender.com.
B,C, Specific killing (B) and T cell numbers (C) at the end of a killing assay with ACSL4-targeting gRNA-transduced OT-1 T cells and scramble gRNA control OT-1 T cells. n=4 technical replicates per condition. Statistical significance determined by two-way ANOVA.
D,E, Representative examples (D) and quantification (E) of C11-BODIPY581/591 staining of OT-1 T cells transduced with ACSL4 gRNA-1 (ACSL4-1), ACSL4 gRNA-2 (ACSL4-2) or scramble gRNAs and treated with indicated concentrations of RSL3 for 24hr after 72hr activation. Flow plots in (D) compare each condition to the same scramble control sample. n=2–3 technical replicates per condition. Statistical significance determined by two-way ANOVA.
F, Viability of OT-1 CD8+ T cells after 24hr culture with the indicated concentrations of RSL3, following a 72hr activation, as determined by flow cytometry. n=3 technical replicates per condition. Statistical significance determined by two-way ANOVA.
*p<0.05 **p<0.01 ***p<0.001 ****p<0.0001. ns, not significant. Graphs display mean +/− SD. Data representative of ≥2 independent experiments. LSK, Lineage− Sca-1+ Kit+. WT, wildtype. gRNA, guide RNA. KO, knockout.
Next, we investigated whether ACSL4-KO cells displayed reduced proliferation or survival. Using a CTV proliferation assay, we found that ACSL4-deficient CD8+ T cells proliferate similarly to control cells during a 72hr activation period (Supplementary Fig. S8E and F). We determined the viability of activated ACSL4-KO and control T cells after 24hr of culture in the presence or absence of RSL3, and observed that in the absence of RSL3, the viability of ACSL4-KO T cells is reduced (Fig. 6F). These data show that ACSL4 deficiency protects CD8+ T cells from ferroptosis induction by GPX4 inhibition, but ACSL4 is required for an optimal anti-tumor CD8+ T cell response in vitro by preserving T-cell survival.
ACSL4 Deficiency Impairs Anti-Tumor CD8+ T Cells In Vivo
We next investigated how ACSL4 deficiency affects CD8+ T cells during an in vivo anti-tumor response. We transferred a 1:1 mix of congenically marked ACSL4-KO and scramble control OT-1 T cells to mice that received B16-OVAWT tumors one day later (Fig. 7A). After harvesting tumors (day 15 to 18 after implantation), we determined the ratios of ASCL4-KO (CD45.1 SP) to control (CD45.1/2 DP) intratumoral Vex+ CD8+ T cells (Fig. 7B and Supplementary Fig. S9A). ACSL4-deficient cells had a competitive disadvantage compared with control T cells, as measured by a reduction in the ratio of ACSL4-KO CD45.1 to control CD45.1/2 Vex+ T cells in tumors compared to input (Fig. 7C). The expression of PD-1, GzmB and Ki-67 was not different between ACSL4-KO and control OT-1 T cells (Fig. 7D–F and Supplementary Fig. S9B), consistent with our in vitro finding that ACSL4 deficiency reduced T-cell numbers but not functionality on a per cell basis (Fig. 6B and C and Supplementary Fig. S8C and D). ACSL4 deficiency thus leads to a cell-intrinsic defect in CD8+ T cells, indicating that CD8+ T cells require ACSL4 for optimal anti-tumor immunity and dependency on ACSL4 contributes to the observed ferroptosis sensitivity of CD8+ T cells.
Figure 7. ACSL4 Deficiency Impairs Anti-Tumor CD8+ T Cells In Vivo.
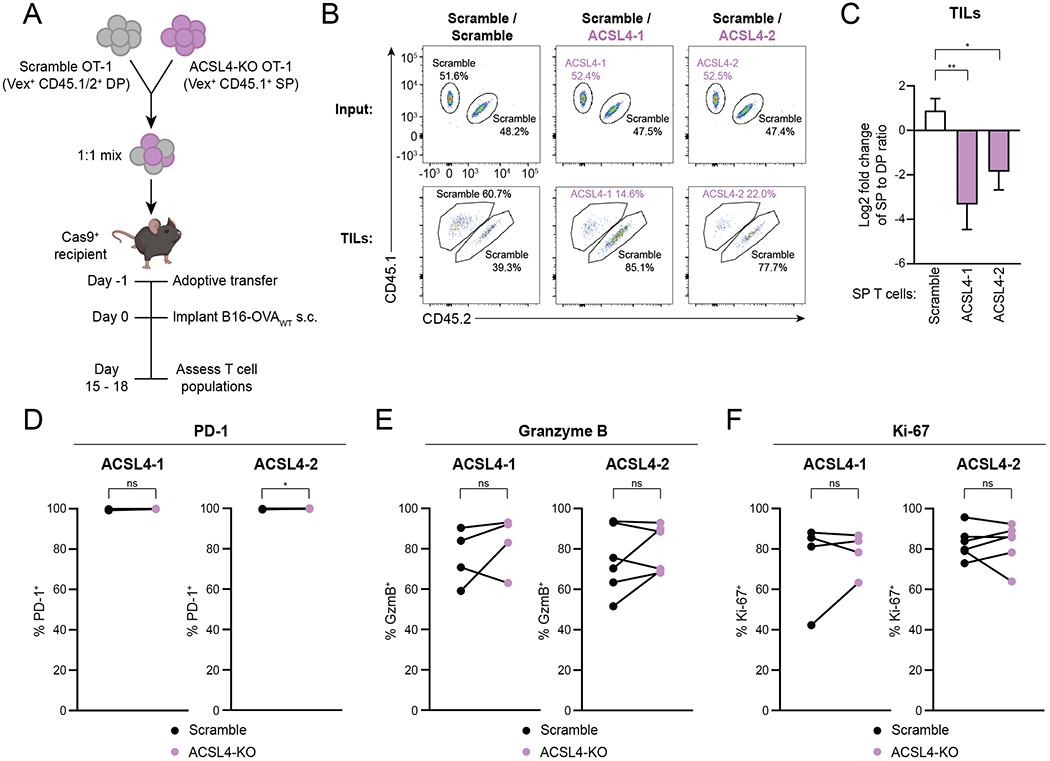
A, Schematic depicting adoptive transfer experiment. The mouse image was created using BioRender.com.
B,C, Representative plots (B) and quantification (C) of flow cytometric measurement of the change in ratio of CD45.1 SP to CD45.1/2 DP among Vex+ CD8+ T cells in B16-OVAWT tumors compared to input. Tumors were harvested for analysis on day 15 to 18 after implantation. Ratio fold-change calculated as (SP:DP)TIL / (SP:DP)input. n=2 animals for scramble control and n=5 animals for each ACSL4-KO group. Statistical significance determined by one-way ANOVA. Data representative of 2 independent experiments.
D–F, Percentages of Vex+ CD8+ T cells expressing PD-1 (D), GzmB (E) and Ki-67 (F) as measured by flow cytometry after tumor harvest. n=4–6 mice per group. Statistical significance determined by paired t tests. Data pooled from 2 independent experiments.
SP, CD45.1 single-positive. DP, CD45.1/2 double-positive. *p<0.05 **p<0.01 ***p<0.001 ****p<0.0001. ns, not significant. Graphs display mean +/− SD. TIL, tumor-infiltrating lymphocyte.
Discussion
In this study, we developed an in vitro pharmacologic screening platform to compare metabolic vulnerabilities between CD8+ T cells and B16 cancer cells. We designed this platform to address questions relevant to the in vivo anti-tumor immune response: T cells were activated prior to encountering metabolic challenges and interacting with cancer cells to simulate T cells entering the TME after activation in a lymph node (20) and we investigated metabolic requirements for execution rather than acquisition of effector functions. We used pharmacologic agents to mimic the exposure of both T cells and cancer cells to the same metabolic conditions in the TME and determined the net effects of these metabolic perturbations on T cells, cancer cells and their interactions. While we focused our current screening effort on metabolic agents, the platform described here may also be valuable for screening other compound libraries to study anti-cancer CD8+ T cell function and identify potential therapeutic targets.
GPX4 inhibitors, which induce ferroptosis, emerged as a top hit of our screen. The term ferroptosis was coined in 2012 to describe an iron-dependent cell death pathway, distinct from other known forms of cell death (37). Shortly thereafter, it was determined that GPX4, a selenoprotein with the ability to reduce lipid peroxidation in membranes (50), is a key ferroptosis regulator, and the absence of its activity results in excess lipid peroxidation (33,38). ACSL4 expression is a key determinant of ferroptosis sensitivity, as this enzyme facilitates the incorporation of oxidation-prone PUFAs, such as arachidonic acid and adrenic acid, into membranes (45,46,48). Most of these and other ferroptosis studies have focused on ferroptosis sensitivity of various cancer types, suggesting the exciting possibility of exploiting this liability therapeutically in cancer (34,35). This is a particularly appealing approach given the observation that cancer cells that exist in a high mesenchymal state, which is associated with resistance to multiple treatment modalities, including chemo- and immunotherapies, are often sensitive to GPX4 inhibition (51–53).
The importance of ferroptosis in immune cells in the tumor microenvironment is less understood. It has been reported that T cells lacking GPX4 fail to expand and function upon activation and undergo ferroptosis (31). However, the sensitivity of T cells to pharmacologic inhibition of GPX4 in the post-activation effector phase and in anti-tumor immune function have not been explored. Our assay enabled a side-by-side comparison of ferroptosis sensitivity of CD8+ T cells and cancer cells during the same perturbation in the same environment, and demonstrated exquisite sensitivity of effector CD8+ T cells to pharmacologic inhibition of GPX4: all three GPX4 inhibitors in our library emerged in the top 10 compounds with the highest T cell–specific vulnerability of 240 metabolic compounds screened. Moreover, activated CD8+ T cells were substantially more sensitive to GPX4 inhibition than both cancer cell lines tested, B16 melanoma and MC38 colorectal adenocarcinoma, even in the presence of IFNγ. B16 cells were previously found to be sensitive to ferroptosis, and this sensitivity was promoted by CD8+ T cell–derived IFNγ, suggesting that increasing ferroptosis sensitivity in cancer cells is one of the mechanisms by which CD8+ T cells exert their anti-tumor effects (36). At the same time, since cancer cells contribute to accumulation of reactive oxygen species in the TME (54), they could conceivably promote lipid peroxidation in local T cells and other cell types. Complex intercellular interactions in the TME may thus impact the respective ferroptosis sensitivities of T cells, cancer cells and other immune and stromal cells. Unfortunately, currently available GPX4 inhibitors are not suitable for systemic administration in vivo (44,51), so it is not yet possible to evaluate the systemic effects of GPX4 inhibition.
We found that, as in other cell types, ACSL4 promoted ferroptosis sensitivity in CD8+ T cells. ACSL4 deficiency thus protected CD8+ T cells from ferroptosis induction, resulting in a benefit of ACSL4-deficient CD8+ T cells compared to control T cells during treatment with high concentrations of GPX4 inhibitor RSL3. However, ACSL4 deficiency also caused a defect in CD8+ T cells that reduced specific killing in vitro in the absence of GPX4 inhibition by reducing CD8+ T-cell viability. Similarly, ACSL4 deficiency resulted in reduced numbers with intact expression of functional markers in an in vivo anti-tumor immune response. These findings suggest that a dependency of CD8+ T cells on ACSL4 for optimal effector responses is an important factor contributing to their sensitivity to ferroptosis induction. The mechanisms underlying the requirement for ACSL4 for optimal CD8+ T-cell function are an interesting topic for future investigation. Potential mechanisms include the requirement of membrane PUFAs for appropriate membrane fluidity (55), which could affect T-cell signaling and other aspects of T-cell biology, and synthesis of eicosanoids, which have been ascribed a variety of effects on T cells (56).
CD8+ T cells displayed a higher vulnerability than B16 cancer cells to most metabolic compounds in the screen, suggesting that T cells have less metabolic flexibility. For example, CD8+ T cells are more sensitive to NAMPT inhibitors as well as several compounds targeting autophagy and ER stress pathways than B16 tumor cells. Our findings with NAMPT, the enzyme that catalyzes the rate-limiting step of NAD+ synthesis, are consistent with previous work showing that NAMPT is important for the maintenance of NAD+ levels, proliferation and function of T cells, and interventions that increase intracellular NAD+ levels can enhance anti-tumor function of CD4+ T cells (57,58). Similarly, manipulations of ER stress pathways can modulate anti-tumor T-cell function (59,60). Future studies that examine whether the TME represents a niche that imposes constraints on NAD+ metabolism and/or ER stress pathways may provide further insights into the metabolic state of the TME. Moreover, our screening approach opens an avenue to discover metabolic liabilities that may be physiologically relevant in other in vivo settings, such as infection and autoimmunity, as well.
In summary, we developed a pharmacologic screening approach to identify metabolic vulnerabilities of CD8+ T cells. Studying the metabolic regulation of T-cell survival and function in vivo, both in cancer and other contexts, has been complicated by both biological (e.g., the high dependency of metabolic pathways on contextual factors) and technical (e.g., how rapidly metabolite abundances can change during sample processing such as flow cytometric sorting) challenges. Our in vitro screening platform enables the study of metabolic pathways in a high-throughput manner and the identification of specific metabolic pathways for further interrogation. In vivo validation of in vitro findings is crucial, as important differences between the metabolic properties of T cells in vitro and in vivo have been observed (61). Although we used our screening platform to study metabolic compounds, this platform enables high-throughput testing of various types of molecules and may inform the design of new cancer (immuno)therapies.
Supplementary Material
Synopsis:
It is challenging to develop metabolism-targeted therapeutics because T cells and cancer cells have similar metabolic properties. The authors develop an in vitro pharmacologic screening platform and highlight ferroptosis as a metabolic vulnerability of CD8+ T cells.
Acknowledgments
We would like to thank the entire Sharpe and Haigis labs for productive discussions. We also thank Dr. Jennifer Smith and the entire ICCB-Longwood Screening Facility for their assistance with the screen. We would like to thank Alos Diallo and Artem Sokolov for their advice on data analysis.
Financial support: This study was supported by a grant from the Ludwig Center at Harvard (M.C.H. and A.H.S.) and NIH grant U54-CA225088 (M.C.H. and A.H.S.). J.M.D. was supported by a predoctoral F31 NIH fellowship (5F31CA224601). A.E.R was supported by a postdoctoral fellowship from the American Cancer Society (130373-PF-17-132-01-CCG). M.W.L. was supported by a predoctoral T32 NIH fellowship (T32CA207021). K.K. is a Gilead Sciences Fellow of the Life Sciences Research Foundation.
Disclosures of Potential Conflicts of Interest: A.H.S. has patents on the PD-1 pathway licensed by Roche/Genentech and Novartis, consults for Novartis, is on the scientific advisory boards for Surface Oncology, Sqz Biotech, Elstar Therapeutics, Elpiscience, Selecta and Monopteros and has research funding from Merck, Novartis, Roche, and Quark Ventures. M.C.H. is a co-founder and on the scientific advisory board for Pori Therapeutics and has research funding from Roche. J.M.D. has consulted for ElevateBio and Third Rock Ventures. The remaining authors declare no competing interests.
References
- 1.Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol. 2019;234:8509–21. [DOI] [PubMed] [Google Scholar]
- 2.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LaFleur MW, Muroyama Y, Drake CG, Sharpe AH. Inhibitors of the PD-1 Pathway in Tumor Therapy. J Immunol. 2018;200:375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–5. [DOI] [PubMed] [Google Scholar]
- 5.Dougan M, Dranoff G, Dougan SK. Cancer Immunotherapy: Beyond Checkpoint Blockade. Annu Rev Cancer Biol. 2019;3:55–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newick K, O’Brien S, Moon E, Albelda SM. CAR T Cell Therapy for Solid Tumors. Annu Rev Med. 2017;68:139–52. [DOI] [PubMed] [Google Scholar]
- 7.Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16:372–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res. 2017;5:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction Immunity. Elsevier Inc.; 2016;45:374–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scharping NE, Delgoffe GM. Tumor Microenvironment Metabolism: A New Checkpoint for Anti-Tumor Immunity. Vaccines. 2016;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugiura A, Rathmell JC. Metabolic Barriers to T Cell Function in Tumors. J Immunol. 2018;200:400–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drijvers JM, Sharpe AH, Haigis MC. The effects of age and systemic metabolism on anti-tumor T cell responses. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho PC, Bihuniak JD, MacIntyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses Cell. Elsevier Inc.; 2015;162:1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV., O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis Cell. Elsevier Inc.; 2013;153:1239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation Immunity. Elsevier Inc.; 2011;35:871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation Cell Metab. Elsevier Inc.; 2016;24:104–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell. 2017;169:570–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen DS, Mellman I. Oncology meets immunology: The cancer-immunity cycle. Immunity. 2013;39:1–10. [DOI] [PubMed] [Google Scholar]
- 21.Andrejeva G, Rathmell JC. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017;26:49–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.LaFleur MW, Nguyen TH, Coxe MA, Yates KB, Trombley JD, Weiss SA, et al. A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nat Commun. 2019;10:1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LaFleur MW, Nguyen TH, Coxe MA, Miller BC, Yates KB, Gillis JE, et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat Immunol. 2019;20:1335–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20:326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurachi M, Kurachi J, Chen Z, Johnson J, Khan O, Bengsch B, et al. Optimized retroviral transduction of mouse T cells for in vivo assessment of gene function. Nat Protoc. 2017;12:1980–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78:110–7. [DOI] [PubMed] [Google Scholar]
- 28.Mehrotra PT, Wu D, Crim JA, Mostowski HS, Siegel JP. Effects of IL-12 on the generation of cytotoxic activity in human CD8+ T lymphocytes. J Immunol. 1993;151:2444–52. [PubMed] [Google Scholar]
- 29.Havele C, Paetkau V. Cyclosporine blocks the activation of antigen-dependent cytotoxic T lymphocytes directly by an IL-2-independent mechanism. J Immunol. 1988;140:3303–8. [PubMed] [Google Scholar]
- 30.Harris IS, Endress JE, Coloff JL, Selfors LM, McBrayer SK, Rosenbluth JM, et al. Deubiquitinases Maintain Protein Homeostasis and Survival of Cancer Cells upon Glutathione Depletion. Cell Metab. 2019;29:1166–1181. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–303. [DOI] [PubMed] [Google Scholar]
- 33.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell. 2019;35:830–49. [DOI] [PubMed] [Google Scholar]
- 35.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee H-J, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137–47. [DOI] [PubMed] [Google Scholar]
- 40.Dixon SJ, Stockwell BR. The Hallmarks of Ferroptosis. Annu Rev Cancer Biol. 2019;3:35–54. [Google Scholar]
- 41.Weinberg F, Ramnath N, Nagrath D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers (Basel). 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MWL, Ngiow SF, et al. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. 2010;70:7788–99. [DOI] [PubMed] [Google Scholar]
- 43.Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8. [DOI] [PubMed] [Google Scholar]
- 44.Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338–43. [DOI] [PubMed] [Google Scholar]
- 48.Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol. 2015;10:1604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Horn CG, Caviglia JM, Li LO, Wang S, Granger DA, Coleman RA. Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: identification of a novel variant of isoform 6. Biochemistry. 2005;44:1635–42. [DOI] [PubMed] [Google Scholar]
- 50.Ursini F, Maiorino M, Gregolin C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta. 1985;839:62–70. [DOI] [PubMed] [Google Scholar]
- 51.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell. 2019;49:361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Costa A, Scholer-Dahirel A, Mechta-Grigoriou F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol. 2014;25:23–32. [DOI] [PubMed] [Google Scholar]
- 55.Agmon E, Stockwell BR. Lipid homeostasis and regulated cell death. Curr Opin Chem Biol. 2017;39:83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lone AM, Taskén K. Proinflammatory and immunoregulatory roles of eicosanoids in T cells. Front Immunol. 2013;4:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bruzzone S, Fruscione F, Morando S, Ferrando T, Poggi A, Garuti A, et al. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS One. 2009;4:e7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, et al. CD38-NAD+Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018;27:85–100. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song M, Cubillos-Ruiz JR. Endoplasmic Reticulum Stress Responses in Intratumoral Immune Cells: Implications for Cancer Immunotherapy. Trends Immunol. 2019;40:128–41. [DOI] [PubMed] [Google Scholar]
- 60.Song M, Sandoval TA, Chae C-S, Chopra S, Tan C, Rutkowski MR, et al. IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. 2018;562:423–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S, et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8+ T Cells. Immunity. 2019;51:856–870. e5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.