

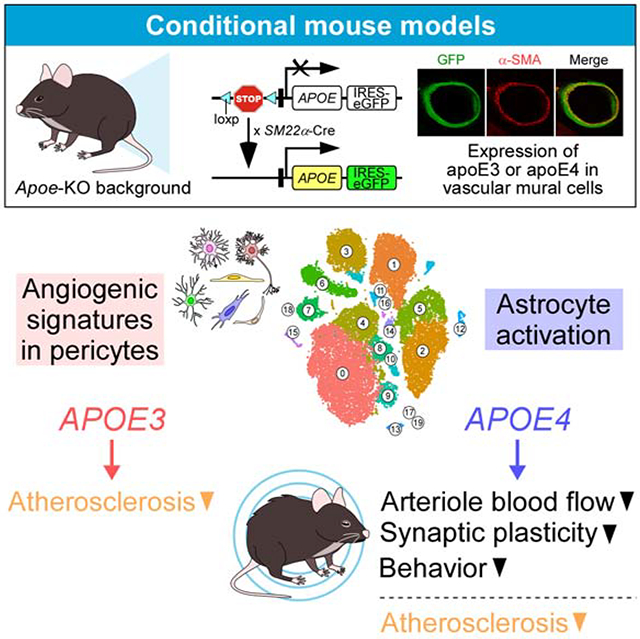
Summary
The ε4 allele of the apolipoprotein E gene (APOE4) is a strong genetic risk factor for Alzheimer’s disease (AD) and multiple vascular conditions. ApoE is abundantly expressed in multiple brain cell types including astrocytes, microglia, and vascular mural cells (VMCs). Here, we show that VMC-specific expression of apoE4 in mice impairs behavior and cerebrovascular functions. Expression of either apoE3 or apoE4 in VMCs was sufficient to rescue the hypercholesterolemia and atherosclerosis phenotypes seen in Apoe knockout mice. Intriguingly, vascular expression of apoE4, but not apoE3, reduced arteriole blood flow, impaired spatial learning, and increased anxiety-like phenotypes. Single cell RNA sequencing of vascular and glial cells revealed that apoE4 in VMCs was associated with astrocyte activation while apoE3 was linked to angiogenic signature in pericytes. Together, our data support cell-autonomous effects of vascular apoE on brain homeostasis in an isoform-dependent manner, suggesting a critical contribution of vascular apoE to AD pathogenesis.
Keywords: APOE, vascular mural cells, single cell RNA sequencing, Alzheimer’s disease, gliovascular function
Graphical Abstract
In Brief
Yamazaki et al. generated conditional mouse models expressing apoE isoforms uniquely in vascular mural cells on a murine Apoe knockout background. They show that vascular mural cell-derived apoE4 impairs vascular function and behavior. Single cell RNA-sequencing reveals apoE4-associated astrogliosis along the vasculature and apoE3-related angiogenesis.
Introduction
In humans, the apolipoprotein E gene (APOE) has three major polymorphic allelic variants with the resulting apoE isoforms (apoE2, apoE3 and apoE4) differ from one another by a single amino acid. However, these small differences have profound effects on their biochemical properties and functions (Liu et al., 2013; Yamazaki et al., 2019b). Consistent with an important role of apoE in brain homeostasis and injury repair, the APOE polymorphism is a strong risk determinant for several brain disorders including Alzheimer’s disease (AD) (Genin et al., 2011) and age-related cognitive decline (Caselli et al., 2009; Shinohara et al., 2016), where APOE4 increases these risks compared to APOE3.
Cerebrovascular alteration represents a major pathological hallmark for both AD (Yamazaki et al., 2019a) and age-related cognitive decline (Vemuri et al., 2015). In addition, APOE4 is associated with increased risk for small vessel diseases (Luo et al., 2017) and vascular cognitive impairment (VCI) (Rasmussen et al., 2018; Sun et al., 2015). Individuals with the APOE4 genotype have a significantly higher risk for white matter damage independent of vascular risk factors and AD pathology (Rojas et al., 2018; Schilling et al., 2013; Sudre et al., 2017). This suggests that the pathobiology of apoE4 may directly cause vascular dysfunctions, thereby contributing to the development of cognitive impairment. As such, dissecting the mechanism by which apoE4 compromises cerebrovascular functions would aid in better understanding of apoE pathobiology and discern the potential mechanism underlying APOE4-related conditions (Tai et al., 2016).
ApoE is expressed and secreted by multiple cell types in both brain and periphery including astrocytes, microglia, vascular mural cells (VMCs) referring to vascular smooth muscle cells and pericytes, hepatocytes, and macrophages (Casey et al., 2015; Xu et al., 2006; Yamazaki et al., 2020). ApoE produced by different cell types might synergistically or independently contribute to its isoform-specific effects. As such, disambiguating the in vivo effects of cell type-specific apoE isoforms represents a challenge and unique opportunity to better define the pathogenic mechanisms of apoE4 in AD and vascular conditions. In this study, by using novel conditional mouse models expressing human apoE isoforms only in VMCs, we attempt to address the effects of vascular apoE on cerebrovascular function and brain cognition. We found that expression of apoE4, but not apoE3, in VMCs leads to impairments in arteriole blood flow and behavior. Transcriptome profiling at the single cell level revealed apoE isoform-specific pathways that likely underlie the observed phenotypes. Given the phenotypes mediated by VMC-derived apoE4 substantially overlap with those globally expressing apoE4, our findings identify VMCs as a critical cell type contributing to the detrimental effects of apoE4 in AD and vascular conditions.
Results
Vascular expression of apoE rescues hypercholesterolemia and atherosclerosis phenotypes in Apoe-KO mice
To define the impact of VMC-derived apoE on brain and vascular functions, we developed mouse models where human apoE3 or apoE4 is conditionally expressed in VMCs on a murine Apoe knockout background. We used our recently-developed apoE conditional mouse models (Liu et al., 2017) where a transgene containing a loxP-flanked STOP cassette and human APOE3 or APOE4 sequence was integrated into the mouse ROSA-26 locus (Figure 1A; Figure S1A). We first bred these mice with SM22alpha-driven Cre recombinase mice to generate sm/E3 and sm/E4 mice. Upon further breeding with murine Apoe knockout mice (Apoe-KO mice), we generated mouse models where human APOE3 or APOE4 is conditionally expressed in VMCs on an Apoe knockout background. Hereinafter these models are referred to as iE3/Cre+ or iE4/Cre+ mice (“i” means inducible, Figure 1A; Figure S1A). To define the in vivo consequences of apoE isoform expression in VMCs, phenotypes in apoE conditional mice (iE3/Cre+ or iE4/Cre+) were compared with their Cre-negative littermates (hereinafter referred to as iE3/Cre− or iE4/Cre− mice; equivalent to Apoe-KO mice).
Figure 1. Generation and characterization of conditional mouse models expressing human apoE isoforms in VMCs.
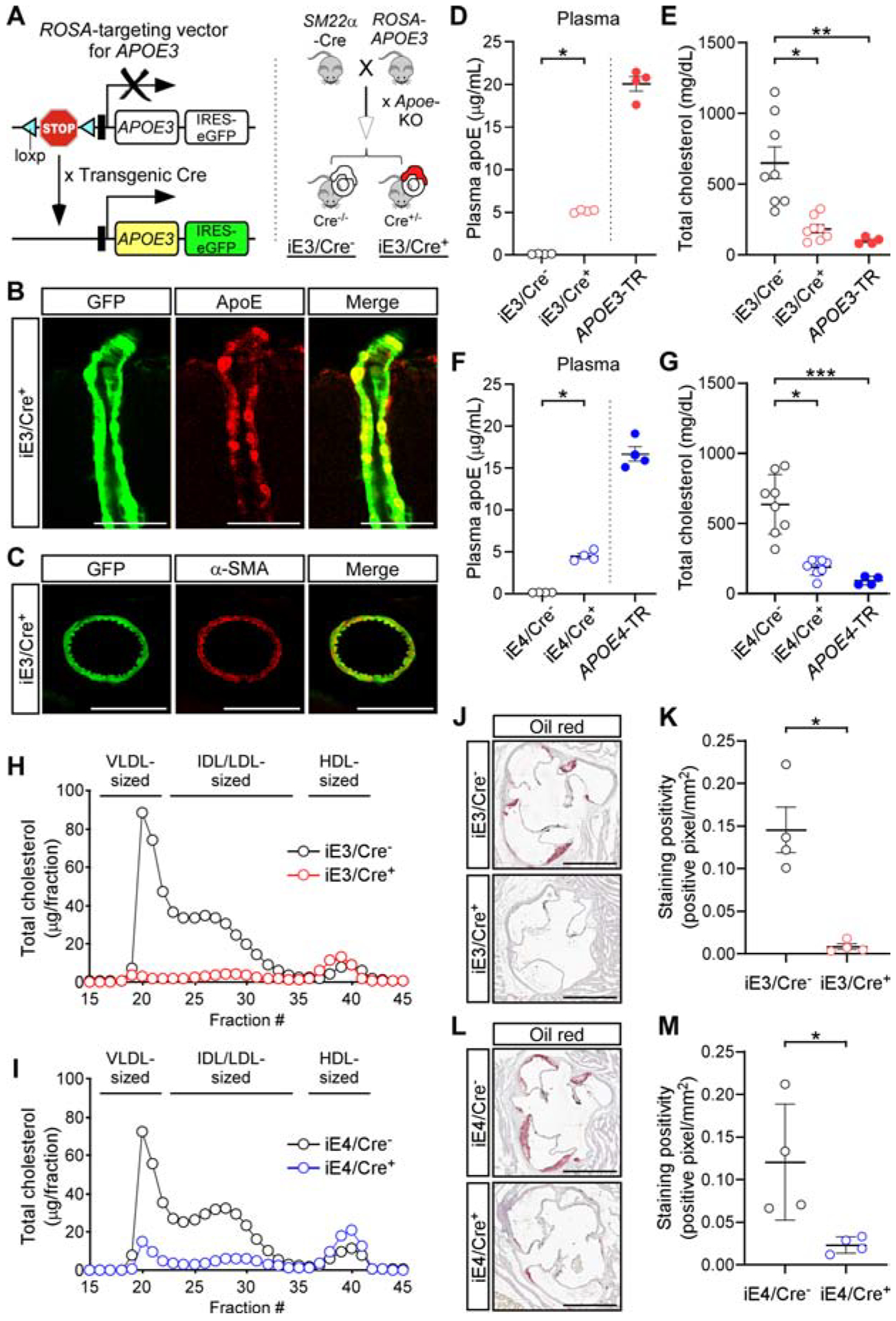
(A) Strategy for generating vascular APOE conditional mouse models.
(B and C) Brain sections from iE3/Cre+ mice immunostained for apoE (B), and smooth muscle cell marker (C, α-smooth muscle actin, α-SMA). Representative images are shown. Scale bar, 50 μm (B), 100 μm (C).
(D) The amount of apoE in plasma was determined by ELISA in iE3/Cre− (n=4, 2 males, 2 females) and iE3/Cre+ mice (n=4, 2 males, 2 females) with APOE3-TR mice (n=4, 2 males, 2 females) as controls. *P<0.05, iE3/Cre− vs. iE3/Cre+, Mann-Whitney test. Data in this and subsequent figures represent mean ± SEM.
(E) Total amount of plasma cholesterol was determined in iE3/Cre− (n=8, 4 males, 4 females), iE3/Cre+ (n=8, 4 males, 4 females), and APOE3-TR mice (n=4, 2 males, 2 females). **P<0.01, iE3/Cre− vs. APOE3-TR; *P<0.05, iE3/Cre− vs. iE3/Cre+, Kruskal-Wallis test followed by Dunn’s multiple comparison tests.
(F) The amount of apoE in plasma was determined by ELISA in iE4/Cre− (n=4, 2 males, 2 females), iE4/Cre+ mice (n=4, 2 males, 2 females), and APOE4-TR mice (n=4, 2 males, 2 females). *P<0.05, iE4/Cre− vs. iE4/Cre+, Mann-Whitney test.
(G) Total amount of plasma cholesterol was determined in iE4/Cre− (n=8, 4 males, 4 females), iE4/Cre+ (n=8, 4 males, 4 females), and APOE4-TR mice (n=4, 2 males, 2 females). ***P<0.001, iE4/Cre− vs. APOE4-TR; *P<0.05, iE4/Cre− vs. iE4/Cre+, Kruskal-Wallis test followed by Dunn’s multiple comparison tests.
(H) Size distribution profile of plasma cholesterol in iE3/Cre− (n=4, 2 males, 2 females) and iE3/Cre+ mice (n=4, 2 males, 2 females).
(I) Size distribution profile of plasma cholesterol in iE4/Cre− (n=4, 2 males, 2 females) and iE4/Cre+ mice (n=4, 2 males, 2 females).
(J and K) Atherosclerotic lesions in the aortic root of iE3/Cre− (n=4, 2 males, 2 females) and iE3/Cre+ mice (n=4, 2 males, 2 females) were visualized by oil-red staining (J) and quantified (K). Scale bar, 1,000 μm (J). *P<0.05, iE3/Cre− vs. iE3/Cre+, Student t-test.
(L and M) Atherosclerotic lesions in the aortic root of iE4/Cre− (n=4, 2 males, 2 females) and iE4/Cre+ mice (n=4, 2 males, 2 females). Scale bar, 1,000 μm (L). *P<0.05, iE4/Cre− vs. iE4/Cre+, Student t-test.
See also Figure S1.
The expression of APOE3 or APOE4 in perivascular cells of iE/Cre+ mice was confirmed with green fluorescent protein (GFP), a surrogate for the target gene expression (Liu et al., 2017) and through apoE immunostaining (Figure 1B; Figure S1B). Further, GFP signals were co-localized with α-smooth muscle actin confirming VMC-specific transgene expression (Figure 1C; Figure S1C). The total amount of plasma apoE and cortical apoE (RIPA-soluble fraction) in iE/Cre+ mice corresponded to 20–30% and 5–15% of apoE-targeted replacement (APOE-TR) mice (Knouff et al., 1999; Sullivan et al., 1997), respectively (Figure 1D and 1F; Figure S1D and S1F). In contrast, the amount of apoE associated with isolated brain micro-vessels in iE/Cre+ mice was comparable to or higher than that in APOE-TR mice (Figure S1E and S1G). As expected, there was no apoE detected in control iE/Cre− mice (Figure 1D and 1F; Figure S1D–S1G). The patterns of GFP signal across cortex and hippocampus were comparable between iE3/Cre+ and iE4/Cre+ mice (Figure S1H and S1I). Collectively, these results validated the cell type-specific expression of apoE in VMCs in our iE/Cre+ mice.
As apoE plays critical roles in plasma cholesterol homeostasis primarily by mediating the hepatic clearance of lipoprotein particles, apoE-deficient mice develop severe hypercholesterolemia and atherosclerosis (Chistiakov et al., 2017; Getz and Reardon, 2016). Consistent with this, the plasma cholesterol concentration of iE/Cre− mice was significantly higher compared to that of APOE-TR mice (Figure 1E and 1G). Importantly, we found that the hypercholesterolemia phenotype due to apoE deficiency was rescued by the conditional expression of apoE3 or apoE4 in VMCs (Figure 1E and 1G). We further analyzed the plasma lipid profile by fractionating plasma from iE/Cre− and iE/Cre+ mice through size-exclusion chromatography. The amount of cholesterol associated with very low-density lipoprotein (VLDL) particles was reduced upon apoE expression in VMCs (Figure 1H and 1I). Further, the size of atherosclerotic lesions at the aortic sinus was significantly lower in iE/Cre+ mice compared to iE/Cre− mice (Figure 1J–1M). Collectively, these data suggest that VMC-derived apoE is sufficient to rescue the atherogenic plasma lipid profiles and atherosclerotic phenotypes due to apoE deficiency regardless of isoforms.
Conditional expression of apoE4 in VMCs leads to increased anxiety-like behavior, impaired spatial learning, and compromised synaptic plasticity
To determine the impact of VMC-derived apoE on behavior, iE/Cre+ and iE/Cre− mice were subjected to a series of behavioral tests. The anxiety-like behaviors were not seen in iE3/Cre+ mice when compared with iE3/Cre− mice (Figure 2A–2C; Figure S2A; Figure S5A–S5C). Interestingly, in open-field assay (OFA), increased anxiety-like behavior was observed in iE4/Cre+ mice compared to iE4/Cre− mice (Figure 2D–2F; S5A–S5C). iE4/Cre+ mice also had a tendency to show a higher anxiety-like behavior compared to iE4/Cre− mice when examined by elevated plus maze (EPM) (Figure S2E). In contextual and cued fear conditioning (CFC), similar memory retention was observed between iE/Cre− and iE/Cre+ mice in both the hippocampus-dependent context test and the hippocampus/amygdala-dependent cued test (Figure S2B, S2C, S2F and S2G). In Morris water maze (MWM) tests, while there was no difference in the latency to find the visible platform (Figure S2D and S2H), iE4/Cre+ mice travelled farther to reach the platform during hidden platform training compared to iE4/Cre− mice (Figure 2I; Figure S5D). In contrast, the number of platform crossings during the probe trials did not differ between the groups (Figure S2J), suggesting a deficiency in iE4/Cre+ mice in spatial learning but not memory retention. Notably, deficiency in spatial learning and memory retention was not seen in iE3/Cre+ mice when compared with iE3/Cre− mice (Figure 2G; Figure S2I; Figure S5D). Motor coordination and balance were not affected by VMC-derived apoE when analyzed through the rotarod test (Figure S2K and S2L). Consistent with the detrimental effect of VMC-derived apoE4 on behavior, synaptic plasticity measured by long-term potentiation (LTP) was suppressed in iE4/Cre+ mice (Figure 2J) but not in iE3/Cre+ mice (Figure 2H). Together, these data indicate that vascular apoE4 expression is associated with increased anxiety-like behavior, impaired spatial learning, and compromised synaptic plasticity.
Figure 2. VMC-derived apoE4 increases anxiety-like behavior, and impairs spatial learning and synaptic plasticity.
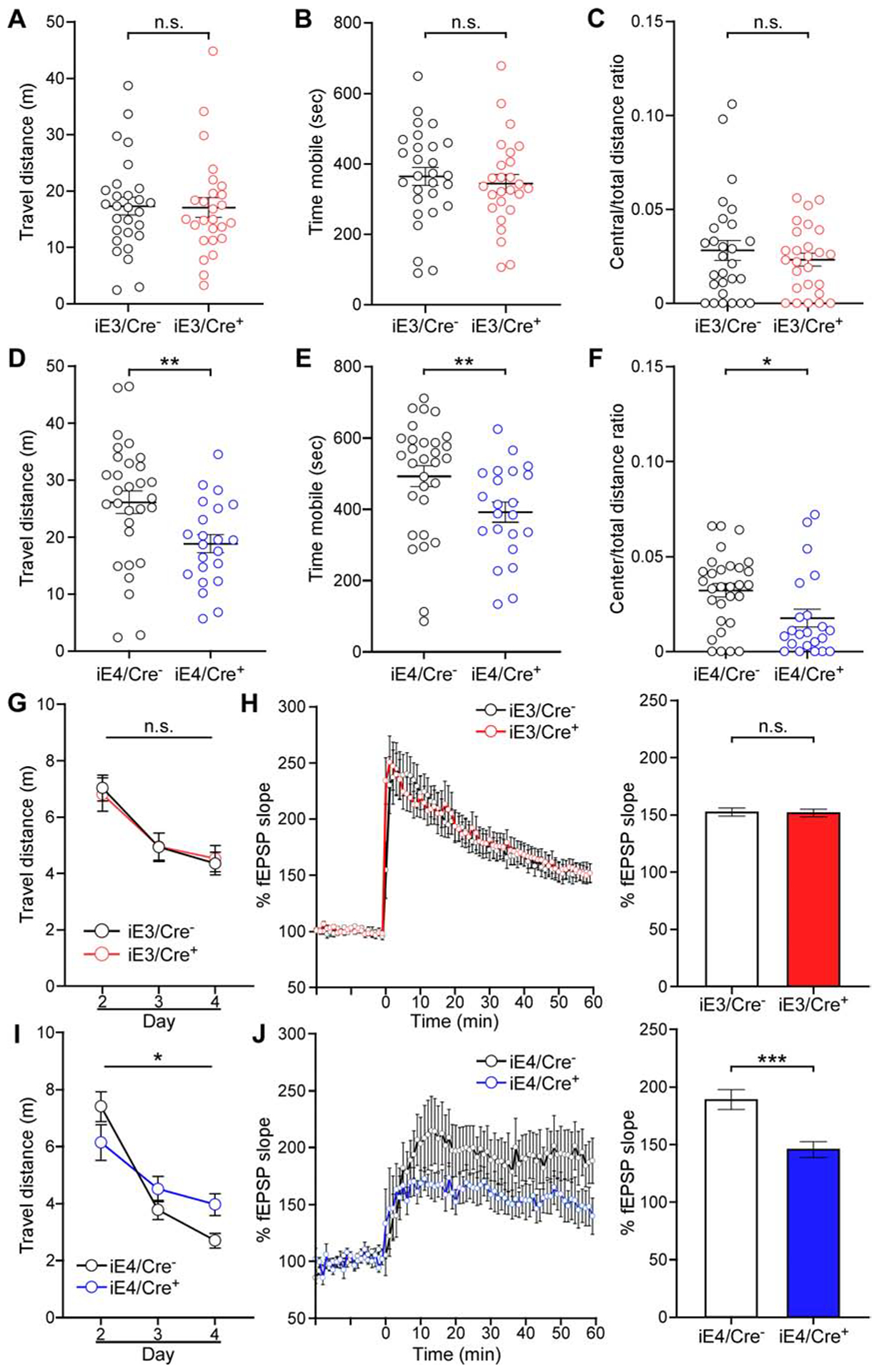
(A-C) Total distance travelled (A), time spent mobile (B), and center to total distance ratio (C) of iE3/Cre− and iE3/Cre+ mice in the OFA tests. n.s., not significant, iE3/Cre− vs. iE3/Cre+, Student t-test (A) and Mann-Whitney test (B and C). Data in this and subsequent figures represent mean ± SEM.
(D-F) Same analyses as in A, B and C except for comparing iE4/Cre− and iE4/Cre+ mice. **P<0.01, iE4/Cre− vs. iE4/Cre+, Student t-test (D). **P<0.01, *P<0.05, iE4/Cre− vs. iE4/Cre+, Mann-Whitney test (E and F).
(G) Travelled distance to visible platform of iE3/Cre− and iE3/Cre+ mice in the MWM test plotted against the training days. n.s., not significant, iE3/Cre− vs. iE3/Cre+, repeated measures ANOVA.
(H) Left, normalized fEPSP responses to field stimulation in the CA1 region of hippocampal slices from iE3/Cre− (n=5, all males) and iE3/Cre+ mice (n=6, all males). Right, averages of the last 5 min of fEPSP recording were quantified. n.s., not significant, iE3/Cre− vs. iE3/Cre+, Mann-Whitney test.
(I and J) Same analyses as in G and H except for comparing iE4/Cre− and iE4/Cre+ mice. *P<0.05, iE4/Cre− vs. iE4/Cre+, repeated measures ANOVA (I). ***P<0.001, iE4/Cre− vs. iE4/Cre+, Mann-Whitney test (J).
Conditional expression of apoE4 in VMCs leads to reduced cerebral arteriole blood flow
Impaired cerebral blood flow (CBF), dysfunction of the glio-vascular unit, and disruption of the blood-brain barrier (BBB) represent key pathological events that underlie vascular contributions to cognitive impairment (Gorelick et al., 2011; Toth et al., 2017). To gain insights into potential mechanisms by which VMC-derived apoE4 impairs behaviors, we evaluated vascular integrity and function in vascular-specific apoE mice. By employing in vivo two-photon microscopy, we found that the velocity of CBF in iE4/Cre+ mice was significantly lower than that in iE4/Cre− mice (Figure 3B; Figure S5E). In contrast, vascular expression of apoE4 had no evident effect on BBB integrity measured by the degree of extravasation of peripherally-administered tracers (Figure S3C and S3D). Consistent with these results, immunohistochemical analysis showed no difference between iE4/Cre− and iE4/Cre+ mice in the expression of several key molecules essential for BBB integrity, including cerebral endothelial cell marker glucose transporter type 1 (GLUT1) (Figure S3I and S3J), tight junction protein claudin-5 (Figure S3I and S3K), and pericyte marker CD13 (Figure S3I and S3L). There was no difference in the velocity of CBF or BBB integrity between iE3/Cre− and iE3/Cre+ mice (Figure 3A; Figure S3A and S3B; Figure S3E–S3H; Figure S5E). Collectively, these results indicate that vascular expression of apoE4 is associated with reduced CBF, likely contributing to the detrimental effect of vascular apoE4 on brain homeostasis and behavior.
Figure 3. VMC-derived apoE4 impairs cerebral blood flow.
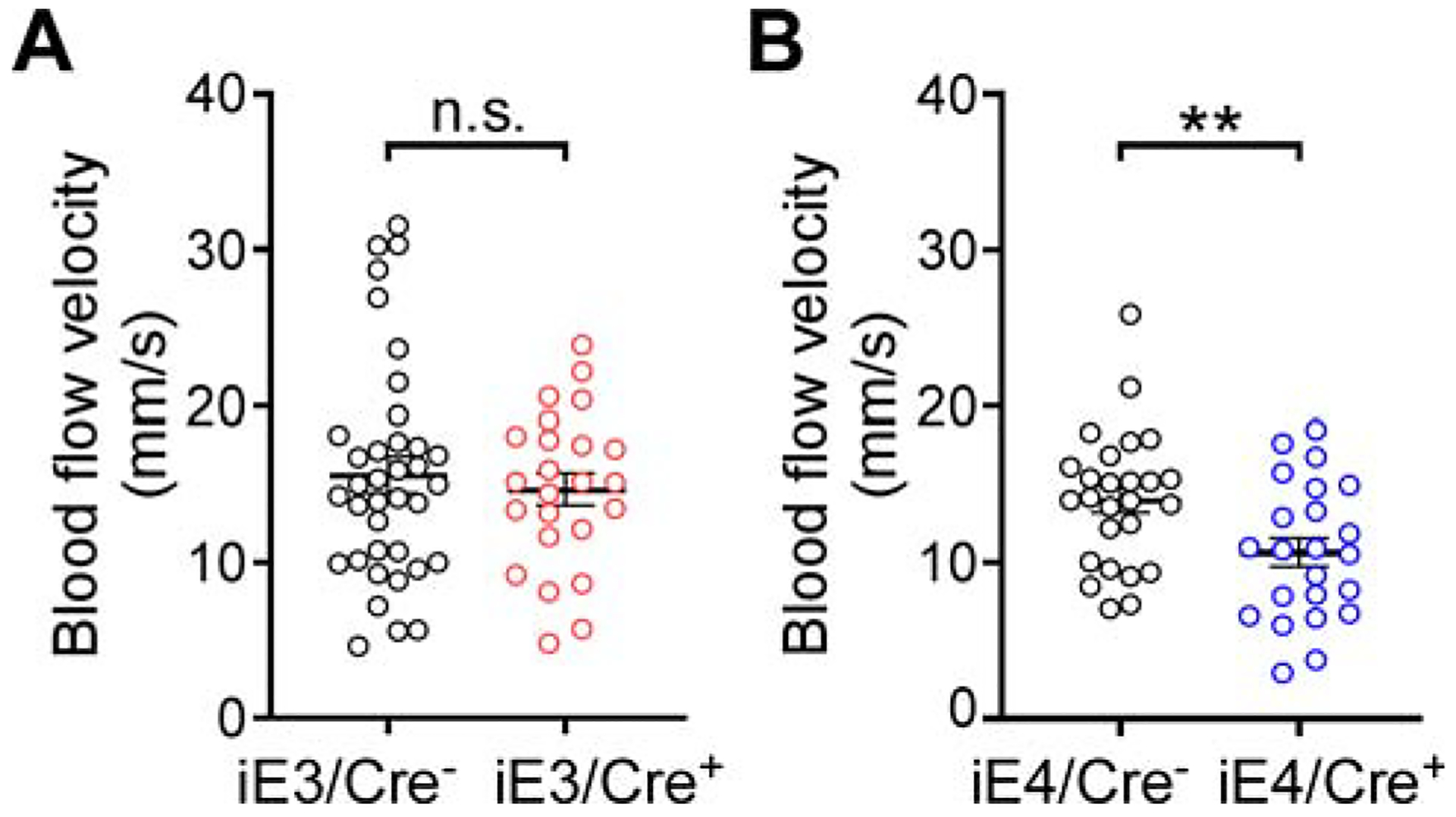
(A) Mean arteriole blood flow was measured in iE3/Cre− mice (n=3, all males, total 37 arterioles) and iE3/Cre+ mice (n=3, all males, total 24 arterioles). n.s., not significant, iE3/Cre− vs. iE3/Cre+, Mann-Whitney test. Data in this and subsequent figures represent mean ± SEM.
(B) Mean arteriole blood flow was measured in iE4/Cre− (n=3, all males, total 26 arterioles) and iE4/Cre+ mice (n=3, all males, total 23 arterioles). **P<0.01, iE4/Cre− vs. iE4/Cre+, Student t-test.
Conditional expression of apoE4 in VMCs modulates transcriptomic signatures in multiple cell types within the glio-vascular unit
To examine how VMC-derived apoE isoforms impact the molecular phenotypes within the glio-vascular unit, we performed single cell RNA-sequencing (scRNA-seq) of vascular and glial cells from iE3/Cre−, iE3/Cre+, iE4/Cre− and iE4/Cre+ mice (Figure 4A). Over 15 cell clusters were identified in the combined datasets, and annotation of these cell clusters by cell type-specific canonical markers confirmed that the majority of glial and vascular cell types were captured (Figure 4B and 4C; Figure S4A and S4B). These included astrocytes, pericytes, smooth muscle cells, choroid plexus cells, endothelial cells, fibroblasts and other types of cells (see also Table S1 and Table S2 for the list of averaged gene expressions in all cell clusters). Upregulation of Gt(ROSA)26Sor in smooth muscle cell clusters of iE/Cre+ mice (with arrowhead in Figure 4D and 4E; Figure S4C and S4D; Table S3; Table S4) was consistent with Cre-mediated conditional activation of transgene in VMCs and thus verified our mouse models.
Figure 4. Conditional expression of apoE4 in VMCs modulates transcriptomic signatures in a variety of cell types within glio-vascular unit.
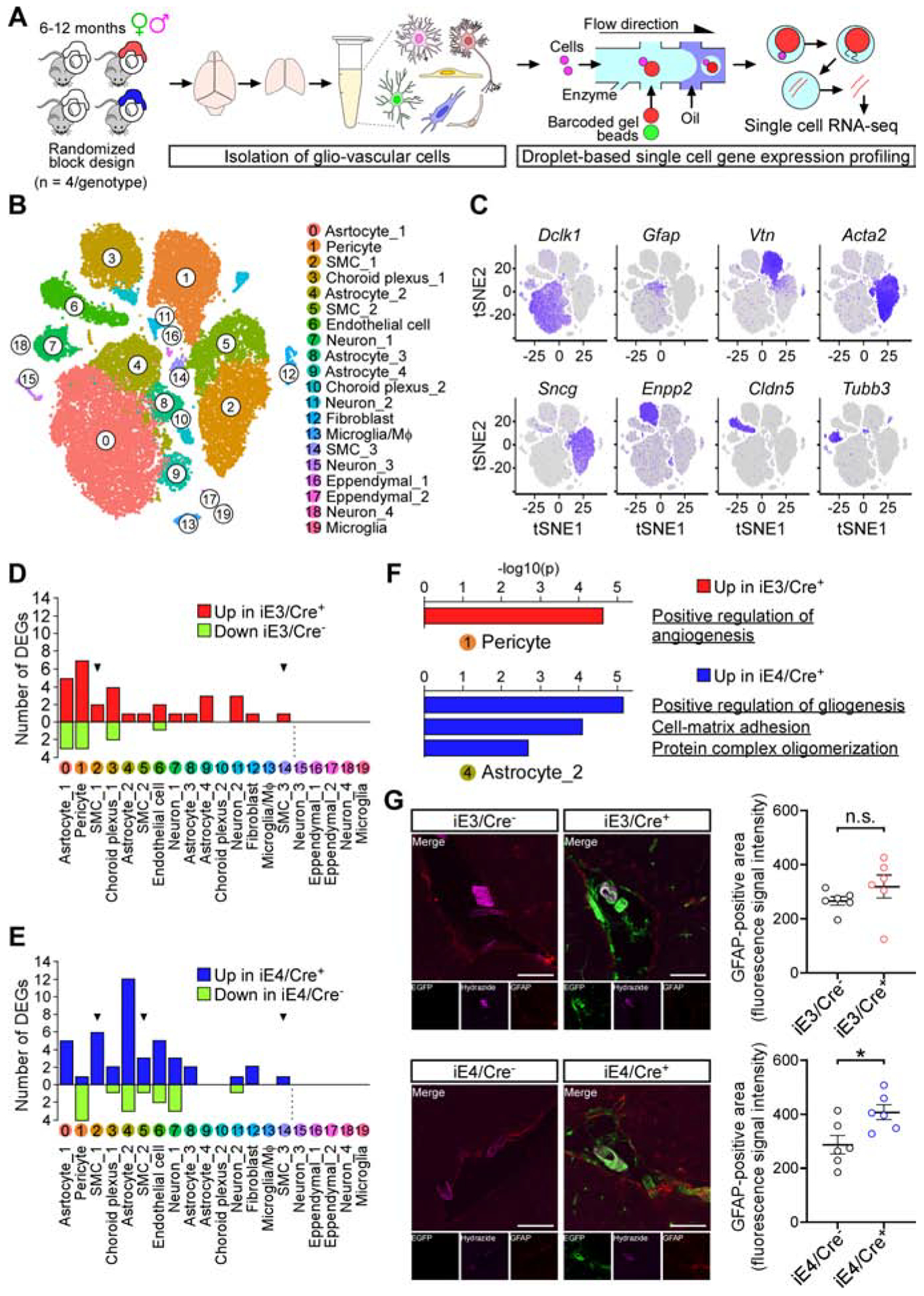
(A) Diagram showing an overview of the scRNA-seq experiment.
(B) t-SNE plots showing the diversity of single cell events captured in scRNA-seq datasets. Events from iE3/Cre−, iE3/Cre+, iE4/Cre−, and iE4/Cre+ mice were combined. SMC, smooth muscle cell; MΦ, macrophage.
(C) Feature plot of canonical markers defining major cell types.
(D) Bar graph depicting the number of genes affected by VMC-derived apoE3 across cell types. Red and green bars represent the number up- and downregulated genes in iE3/Cre+ mice, respectively. Arrowhead, cell population in which Gt(ROSA)26Sor was upregulated.
(E) Same as in D except for iE4/Cre+ mice.
(F) Top, Metascape analysis of upregulated genes (with adjusted p value < 0.01) in “Pericyte” cluster in iE3/Cre+ mice. Bottom, Metascape analysis of upregulated genes (with adjusted p value < 0.01) in “Astrocyte_2” cluster in iE4/Cre+ mice.
(G) Left, coronal brain sections of iE3/Cre−, iE3/Cre+, iE4/Cre−, and iE4/Cre+ mice were immunostained with GFAP antibody (red). EGFP signal (green) depicts the surrogate apoE3 or apoE4 expression in iE3/Cre+ or iE4/Cre+ mice, respectively. Arteriole-specific lamina propria was visualized by Alexa Fluor 633 Hydrazide (purple). Representative images are shown. Scale bar, 100 μm. Right, fluorescence intensity of GFAP signals was quantified in coronal brain sections containing the posterior cerebral artery (n=6, 3 males, 3 females, for each genotype group). n.s., not significant, iE3/Cre− vs. iE3/Cre+; *P<0.05, iE4/Cre− vs. iE4/Cre+, Mann-Whitney test. Data represent mean ± SEM.
The number of differentially expressed genes (DEGs) was highest in the pericyte clusters in the E3-scRNA-seq dataset (depicted as “Pericyte” in Figure 4D), whereas DEGs were highest in a subset of astrocyte clusters in the E4-scRNA-seq dataset (depicted as “Astrocyte_2” in Figure 4E). Metascape analysis of DEGs in “Pericyte” in the E3-scRNA-seq dataset identified pathways associated with a positive regulation of angiogenesis (Figure 4F; Table S3), suggesting the increased angiogenic potentials in pericytes in iE3/Cre+ mice. In contrast, “Astrocyte_2” DEGs in the E4-scRNA-seq dataset were associated with glial activation, including positive regulation of gliogenesis and cell-matrix adhesion (Figure 4F; Table S4). Consistently, we demonstrated that glial fibrillary acidic protein (GFAP) immunoreactivity around the penetrating arterioles, but not capillaries, was significantly higher in iE4/Cre+ mice compared to iE4/Cre− mice (Figure 4G; Figure S4E; Figure S5F). As such, vascular apoE could impact gene expression not only cell-autonomously in the VMCs but also non-cell-autonomously in other cell types including astrocytes and endothelial cells (Figure 4D and 4E; Table S3; Table S4). Moreover, distinct molecular signatures between iE3/Cre+ and iE4/Cre+ mice were detected in the glial and vascular cells, which may contribute to the differential brain and vascular functions modulated by apoE isoforms (Figure 2; Figure 3; Figure 4; Figure S5).
The number of DEGs in each cell cluster identified in E3-scRNA-seq dataset was lower than those in the E4-scRNA-seq dataset (Figure 4D and 4E). Among DEGs in both E3-scRNA-seq and E4-scRNA-seq datasets, Cpe in “Astrocyte_1”, Ttr in “Endothelial cell”, and Xist in “Choroid plexus_1 were upregulated in both iE3/Cre+ and iE4/Cre+ mice (Table S3; Table S4). We also identified cell type-specific DEGs that were upregulated in iE3/Cre+ mice but downregulated in iE4/Cre+ mice (i.e., Alad in “Pericyte”, Igkc in “Astrocyte_2”, and Uba52 in “Neuron_2”). The small number of DEGs might limit the identification of VMC-derived apoE isoform effect on biological pathways in vascular and glial cells. Nonetheless, these results provide another layer of evidence that the biological impact of VMC-derived apoE4 on vascular and glial cells is different from VMC-derived apoE3.
Discussion
Herein, using novel conditional mouse models, we showed that VMC-derived apoE differentially modulates behavior and vascular phenotypes in an isoform-dependent manner. Transcriptomic analyses at the single cell level further revealed distinct molecular signatures of vascular and glial cells in the presence of VMC-derived apoE4 compared to apoE3. These studies provide novel insights into the pathobiology of apoE4 by demonstrating that vascular apoE4 is sufficient to compromise behavior and synaptic function, supporting an overall vascular contribution to AD and dementia.
Considering that the dysregulation of cerebral blood supply represents a key component of vascular contribution to cognitive decline (Gorelick et al., 2011; Toth et al., 2017), reduced CBF in the apoE4 vascular mice likely contributes to the disrupted neuronal homeostasis. Further, increased GFAP immunoreactivity in apoE4 vascular mice might represent reactive astrocytes in the perivascular niche. These astrocytes might alter the communication between the glial limitans and arterioles (Pelligrino et al., 2011), exacerbating the dysregulation of CBF to the brain. Furthermore, as apoE is a secreted protein, it is possible that vascular apoE4 can be taken up by neuronal cells, triggering neuronal stress induced by apoE4 aggregation or fragmentation (Mahley, 2016; Najm et al., 2019). Thus, it is likely that vascular produced apoE4 compromises brain function in both cerebrovascular function dependent and independent manner.
In clear contrast to the differential modulation of brain and vascular functions, both VMC-derived apoE3 and apoE4 rescued atherogenic plasma lipid profiles and atherosclerotic phenotypes due to apoE deficiency. It is likely that VMC-derived apoE populated in plasma facilitates the hepatic clearance of lipoprotein remnant, thereby rescuing the atherogenic plasma lipid profiles in iE/Cre+ mice (Chistiakov et al., 2017; Getz and Reardon, 2016). The rescuing effect of VMC-derived apoE on aortic atherosclerosis in iE/Cre+ mice might be attributed to the correction of hypercholesterolemia. However, it should also be noted that tissue-specific expression of apoE, either in macrophage (Bellosta et al., 1995) or adrenal gland (Thorngate et al., 2000), at levels that were not sufficient to correct hypercholesterolemia, was sufficient to reduce atherosclerosis in apoE-deficient mice. Accordingly, the athero-protective action of VMC-derived apoE might also be mediated by the increased cholesterol efflux from macrophages or VMCs via apoE, which prevents the formation of foam cells in the arterial wall (Chistiakov et al., 2017). In either scenario, our results highlight a functional effect of VMC-derived apoE in promoting lipoprotein metabolism and athero-protection.
Several limitations need to be acknowledged. First, the phenotypes related to VMC-derived apoE were determined by comparing those in iE/Cre+ with those in iE/Cre− mice (Figure 1–4; Figure S1–S5), both of which have the background of murine apoE deficiency. Several phenotypes including cognitive decline, synaptic degeneration (Masliah et al., 1995), BBB dysfunction (Bell et al., 2012; Fullerton et al., 2001), and hypercholesterolemia are reported in the absence of murine apoE. Thus, there is a possibility that subtle phenotypes mediated by VMC-derived apoE were not captured in the presence of phenotypes related to murine Apoe knockout. Second, the expression of APOE in our models is not regulated by the physiological Apoe promoter. Thus, the relevance of phenotypes in our models to apoE pathobiology needs to be carefully interpreted. Nonetheless, the gain-of-toxic effects of VMC-derived apoE4 are well captured in our study. Third, although our findings provide a powerful experimental framework for defining brain homeostatic pathways mediated by VMC-derived apoE4, the exact cause-effect relationships between VMC-derived apoE4 expression, gliovascular dysfunctions, and behavioral phenotypes are not fully clear. Therefore, future studies aimed at examining the spatiotemporal dynamics of these phenotypes in mice at different ages would aid in determining their relationships, and defining potential mechanisms for reduced CBF and behavioral phenotypes.
In conclusion, we demonstrated that VMC-derived apoE isoforms differentially modulate glio-vascular functions and behavior. Given that phenotypes induced by VMC-derived apoE4 considerably recapitulate those globally expressing apoE4, our results suggest a critical contribution of vascular apoE4 to the pathogenesis of AD and VCI.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Guojun Bu (bu.guojun@mayo.edu).
Materials Availability
There are restrictions to the availability of iE3 and iE4: B6.Gt(ROSA)26Sortm1(rtTA-APOE) mice. We will consider distribution under an MTA subject to restrictions from commercial source.
Data and Code Availability
Single Cell RNA Sequencing data are available via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP-AD) Target Discovery Program and other National Institute on Aging (NIA)-supported programs to enable open-science practices and accelerate translational learning. The data, analyses and tools are shared early in the research cycle without a publication embargo on secondary use. Data is available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.synapse.org/DataAccess/Instructions). For access to content described in this manuscript see: https://doi.org/10.7303/syn22313650.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
An inducible APOE3-eGFP or APOE4-eGFP construct containing a floxed STOP cassette was knocked into the ROSA-26 locus of our mouse models (Figure 1A and Figure S1A) (Liu et al., 2017). Animals were bred with SM22alpha-driven Cre recombinase mice (Boucher et al., 2003), and further with murine Apoe knockout mice (obtained from the Jackson Laboratory). We used littermates for all experiments with Cre− mice equivalent of Apoe knockout as controls (hereinafter referred to as iE3/Cre− or iE4/Cre−) and Cre+ mice conditionally expressing apoE3 or apoE4 in VMCs as the experimental mice (hereinafter referred to as iE3/Cre+ or iE4/Cre+). Both male and female mice were used in all experiments except for extracellular field recording with hippocampal slices, and intravital two photon imaging study. For extracellular field recording with hippocampal slices, and intravital two photon imaging study, only male mice were used as we did not observe any sex-dependent differences in other critical readouts in this study. Mice were housed in a temperature-controlled environment with a 12-h light–dark cycle and free access to food and water. 6–8 months old animals were used for all functional, biochemical and histological experiments. For the single cell RNA sequencing experiments, 6–12 months old animals were used (n=4/genotype; age-matched male (n=2) and female (n=2) mice). All animal procedures were approved by the Animal Study Committee at Mayo Clinic and performed in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care.
METHOD DETAILS
Collection of Mouse Tissues and Sample Preparation
For plasma preparation, blood was collected using EDTA as anticoagulant from the inferior vena cava. EDTA-plasma was isolated via centrifugation at 1,000 g for 10 min at 4°C and was stored at −80°C. For tissue preparation, mice were trans-cardially perfused with PBS. Brains were dissected and were stored at −80°C. For biochemical analysis, brain tissues were homogenized at a ratio of 20 ml/g wet-weight brain in a mixture of ice-cold RIPA buffer (Cat. #20–188, Millipore), Complete protease inhibitors (Cat. #11697498001, Roche), and PhosSTOP phosphatase inhibitors (Cat. #4906845001, Roche). After centrifugation at 100,000 g for 1 hour at 4°C, the supernatant was collected and used for biochemical analysis. For histological analysis, brain and corresponding heart tissue were fixed in 4% paraformaldehyde for 24 hours. Samples were moved to 30% sucrose for cryostat sectioning. Some brain tissues were embedded in O.C.T. compound (Cat. #50-363-579, Fisher) without fixation and snap-frozen for cryostat sectioning. For preparation of samples for single cell RNA-seq, refer to ‘Preparation of Single Cell Suspensions Enriched in Brain Vascular Cells’.
Isolation of Brain Microvessels
Brain microvessels were isolated as described (Kanekiyo et al., 2012; Yamazaki et al., 2016). Mouse brains were homogenized in Dulbecco’s modified Eagle’s medium (DMEM, Cat. #11965175, Gibco) and re-suspended in 17% dextran. Sample was centrifuged again at 10,000 g for 15 min at 4°C, and pellets were suspended in DMEM containing 10% fetal bovine serum (FBS) and filtered through sterilized glass beads (425–600 μm, acid-washed; Cat. #G8772, Sigma-Aldrich) on a 70-μm nylon cell strainer (Cat. #352350, Corning). After transferring glass beads with captured microvessels onto a plastic dish with DMEM containing 10% FBS, microvessels were dissociated from glass beads by gentle agitation, collected, and lysed in RIPA buffer.
ApoE ELISA
ApoE ELISA was performed as described (Casey et al., 2015; Yamazaki et al., 2020). In brief, a 96-well plate was coated overnight with WUE4 antibody (Cat. #NB110–60531, Novus Biologicals). After blocking with 1% nonfat milk in PBS, samples were applied at the appropriate dilution and incubated with a detection antibody (Cat. #K74180B, Meridian Life Science) then incubated with streptavidin-poly-HRP (Cat. #85R-200, Fitzgerald). Tetramethylbenzidine (Cat. #T5569, Sigma-Aldrich) was then applied to each well, and the substrate-peroxidase reaction was stopped with sulfuric acid stop solution. The absorbance was read at 450 nm using a BioTek 600 plate reader. ApoE concentration of each sample was calculated against a standard curve derived from serial dilutions of recombinant human apoE3 or apoE4 protein (Cat. #30R-2381, #30R-2382) purchased from Fitzgerald.
Cholesterol Analysis in Plasma
Plasma samples were fractionated by gel filtration-FPLC using an AKTA system with a Superose 6, 10/300 GL column (Cat. #17517201, GE Healthcare). The amount of cholesterol in the plasma samples and subsequent fractions was determined with the Amplex Red cholesterol assay kit (Cat. #A12216, Invitrogen) according to the manufacturer’s instructions. Samples were pipetted into an opaque 96-well plate with a transparent bottom. Standards and samples were incubated with the Amplex Red reagent at 37°C for 30 min. Fluorescence was measured using excitation of 530–560 nm and emission detection at 590 nm. Commercially available cholesterol was used to determine the fractions that correspond to the size range of VLDL, IDL/LDL and HDL particles. We defined cholesterol separated in fractions 16–22 as VLDL-sized, fractions 23–33 as IDL/LDL-sized, and fractions 35–41 as HDL-sized in this study.
Immunofluorescence Staining
Free floating brain sections (40 μm thickness; coronal) were immunolabeled for apoE, smooth muscle cell marker and glial fibrillary acidic protein (GFAP). The tissue was blocked in 5% goat serum for 1 hour and incubated with the primary antibodies overnight. We used rabbit anti-human specific anti-apoE (1:200, Cat. #ab52607, abcam), rabbit anti-α smooth muscle actin (1:200, Cat. #ab5694, abcam), and mouse anti-GFAP (1:800, Cat. #MAB360, Millipore) primary antibodies. Sections were then incubated with Alexa Fluor-conjugated secondary antibodies for 1 hour at room temperature (1:1000, Cat. #A-11011, #A-11004, Invitrogen). To visualize arteriole-specific lamina propria, the sections stained with GFAP were further incubated with Alexa Fluor 633 Hydrazide for 1 hour at room temperature (1:1000, Cat. #A30634, Invitrogen) (Shen et al., 2012).
In another set of experiments, acetone/ethanol-fixed frozen brain sections (25 μm thickness; coronal) were immunolabeled for glucose transporter type 1 (GLUT1), claudin-5, and CD13. The tissue was blocked in 5% goat serum for 1 hour and incubated with the primary antibodies overnight. We used rabbit anti-GLUT1 antibody (1:200, Cat. #07–1401, Millipore), mouse anti-claudin-5 antibody (1:200, Cat. #35–2500, Invitrogen), and rat anti-CD13 (1:50, Cat. #558745, BD Biosciences) primary antibodies. Sections were then incubated with Alexa Fluor-conjugated secondary antibodies for 1 hour at room temperature (1:1000, Cat. #A-11034, Cat. #A-11001, Cat. #A-11006, Invitrogen). The images were captured by confocal laser-scanning fluorescence microscopy (LSM880, Carl Zeiss).
Confocal Image Analysis
Fluorescence Intensity Analysis of GLUT1, claudin-5 and CD13 immunoreactivities
Fifteen-micron maximum intensity projection z-stacks (420 × 420 μm field) within cortical areas were reconstructed. The fluorescence signal intensity of the dye used to visualize GLUT1, claudin-5 or CD13 immunoreactivity was quantified by Zen software (Black edition, Carl Zeiss). At least two different sections containing both cortex and hippocampus were randomly chosen and used for image acquisition per mouse. The signal intensity per section was averaged per mouse. Confocal images were acquired and analyzed by an experimentally blind investigator.
Fluorescence Intensity Analysis of GFAP Immunoreactivity Around Arterioles
Fifteen-micron maximum projection z-stacks (420 × 420 μm field) were reconstructed. The fluorescence signal intensity of the dye used to visualize GFAP immunoreactivity was quantified by Zen software (Black edition, Carl Zeiss). At least two different sections that contain posterior cerebral artery or its branches (visualized by Alexa Fluor 633 Hydrazide, Cat. #A30634, Invitrogen) were randomly chosen and used for image acquisition per mouse. The GFAP signal intensity per section was averaged per mouse. Confocal images were acquired and analyzed by an experimentally blind investigator.
Fluorescence Intensity Analysis of GFP in Cortex and Hippocampus
To quantify the fluorescence intensity for GFP signals in brain sections from iE3/Cre+ and iE4/Cre+ mice, Z-stack images of the free floating brain sections (40 μm thickness; coronal) were acquired by Keyence fluorescence microscopy (model BZ-X, Keyence) and converted into maximum z-stack projection images. At least two different brain slides per mouse were used for image acquisition. The intensity of GFP signals in cortex and hippocampus were quantified by Image J software (ImageJ bundled with 64-bit Java 1.8.0_112, NIH). Fluorescence images were acquired and analyzed by an experimentally blinded investigator.
Fluorescence Intensity Analysis of GFAP Immunoreactivity Around Capillaries
Acetone/ethanol-fixed frozen brain sections (25 μm thickness; coronal) were immunolabeled for GLUT1 and GFAP. The tissue was blocked in 5% goat serum for 1 hour and incubated with the primary antibodies overnight. We used rabbit anti-GLUT1 (1:200, Cat. #07–1401, Millipore), and mouse anti-GFAP (1:800, Cat. #MAB360, Millipore) primary antibodies. Sections were then incubated with Alexa Fluor-conjugated secondary antibodies for 1 hour at room temperature (1:1000, Cat. #A-11034, #A-11004, Invitrogen). Z-stack images of the stained sections were acquired by Keyence fluorescence microscopy (model BZ-X, Keyence) and converted into maximum z-stack projection images.
At least two different brain slides per mouse were used for image acquisition. The intensity of GFAP signals surrounding the capillaries was quantified and normalized to the GLUT1 signals. In brief, the GLUT1 or GFAP immunoreactivities were quantified by Image J software (ImageJ bundled with 64-bit Java 1.8.0_112, NIH). To quantify the GFAP immunoreactivities around capillaries, continuous capillaries clearly labeled by anti-GLUT1 antibody (>100 μm) were randomly selected as the region of interests (ROIs). Fluorescence images were acquired and analyzed by an experimentally blinded investigator.
Analysis of Atherosclerotic Lesions in Aortic Root
Serial 10-μm sections of formalin-fixed hearts were prepared using a cryostat. Sections were immersed in Oil Red O (Cat. #O1391, Sigma-Aldrich) and hematoxylin were used for nuclear counterstaining. Sections where three valve bases were visible were analyzed. The sections were captured using the Aperio AT2 image scanner (Leica Biosystems). The volume of the aortic lesion was determined by measuring the maximal size of an atherosclerotic lesion positive for Oil Red O. The positivity of Oil Red O staining in the aorta was calculated using the Positive Pixel Count Algorithm available with the ImageScope software (12.3.3, Leica Biosystems). Quantification was performed by an experimentally blind investigator.
Cognitive and Behavioral Tests
Contextual and cued fear conditioning (CFC), Morris water maze (MWM), open-field assay (OFA) and elevated plus maze (EPM) tests were performed as described (Kang et al., 2018; Sakae et al., 2016; Zhao et al., 2018). In CFC, mice were trained in a sound attenuating chamber with a grid floor capable of delivering an electric shock as described (Kang et al., 2018; Zhao et al., 2018). Freezing behavior was monitored with an overhead camera using FreezeFrame software (Actimetrics). On the following day, mice were returned to the testing chamber and freezing behavior was recorded for 5 min (context test). Mice were then returned to their home cage, moved to a different room, and allowed to re-acclimate for a minimum of 60 minutes. To examine cued learning/memory (cued test) to the auditory conditioned stimulus (CS) independent of the previous contextual cues, several parameters of the environment including visual and olfactory contextual cues were altered as described (Kang et al., 2018). Subsequently, mice were placed in the chamber for 3 min without tone 3 min of the auditory CS during which freezing time was recorded.
For Morris water maze (MWM), on Day 1 mice underwent one session of six pre-training trials with a visible marker placed on the platform (visible platform test). For hidden platform training on Days 2 to 4, the platform was submerged 1.5 cm below the surface located at the northwest quadrant of the pool. Mice underwent 6 trials per day sessions during hidden platform training (Days 2 to 4). For the probe test on Day 5, the platform was removed, and mice were allowed to swim for 60 seconds. The starting location for the probe test was at the southeast edge of the pool 180° from the northwest platform location used during training sessions (Days 2 to 4). Multiple measures, including escape latencies, distance traveled, and platform entries were analyzed.
In OFA, mice were placed in the center of an open-field arena (40 × 40 × 30 cm, W × L × H), and analyzed for multiple measures, including total distance traveled, average speed, time mobile, and time spent in the “center” (digitally defined by an 20 × 20 cm region).
In EPM, the maze was elevated 50 cm from the floor and consisted of four arms (50 × 10 cm) with two of the arms enclosed with roofless gray walls (35 × 15 cm, L × H). Mice were placed in the center of the maze facing an open arm, and were tracked for 5 min. In all of these tests, an overhead camera with ANY-Maze software (Stoelting Co.) was used to monitor and track their behaviors.
Rotarod test was performed as described (Delenclos et al., 2017). Each mouse was placed on an accelerating spindle (4–40 rpm) for 5 min (Med Associates, Inc) for four consecutive trials with at least 40 min of rest in between trials. The latency to fall time was recorded in each trial from Day 1 to 4.
Extracellular Field Recording with Hippocampal Slices
To examine the LTP in the hippocampus of experimental mice, extracellular recordings were performed as described with modification (Rogers et al., 2017). Mice were sacrificed and transverse slices were prepared for electrophysiology in ice-cold cutting solution containing 110 mM sucrose, 60 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 28 mM NaHCO3, 0.6 mM sodium ascorbate, 5 mM glucose, 7 mM MgCl2 and 0.5 mM CaCl2. Field excitatory post-synaptic potentials (fEPSPs) were obtained from the CA1 stratum radiatum using a glass microelectrode (2–4 mΩ) filled with artificial cerebrospinal fluid (aCSF) composed of 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM glucose, 1 mM MgCl2 and 2 mM CaCl2. fEPSPs were evoked through Schaffer collateral stimulation using a 0.1 msec biphasic pulse. To measure basic synaptic transmission (Input/Output curve), stimulation was strengthened incrementally every 0.5–1 mV until the maximum amplitude of the fEPSP was reached. All other following stimulation paradigms were induced at the same voltage, defined as the stimulus voltage which evokes 50–60% of the maximum fEPSP amplitude, for each individual slice. To examine the presynaptic function, paired-pulse facilitation (PPF) was performed using series of 15 paired-pulses with an initial 20ms interval and an incremental step of 20 msec. Baseline fEPSP responses were recorded for 20 min. Tetanus used to evoke LTP was a theta-burst stimulation (TBS) protocol consisting of 5 trains of 4 pulse bursts at 200 Hz separated by 200 msec, repeated 6 times with an inter-train interval of 10 sec. Following TBS induction, fEPSPs were recorded for 60 min. Potentiation was measured as the increase of the mean fEPSP descending slope following TBS normalized to the mean fEPSP descending slope of baseline recordings. All the recordings were analyzed using Matlab (9.6.0.1072779 R2019a, Mathworks).
Blood Flow Velocity and Permeability Measurement with Intravital Two Photon Imaging
Mice were anesthetized with isoflurane (4% for induction; 2% for surgery, and 0.5 to 1% for imaging) and immobilized in a custom-made stereotactic apparatus. After anesthesia, dexamethasone (2 mg/kg) and buprenorphine (0.2 mg/kg) were subcutaneously administered to reduce inflammation and pain. Body temperature was monitored by a rectal probe and maintained at 37°C using a heating blanket (Homeothermic blanket systems, Harvard Apparatus). Hair was removed to expose the parietal skull. A craniotomy of 3–4 mm in diameter was performed with a surgical drill above the somato-sensory and motor cortex. Experiments were performed only if the physiological variables remained within normal limits.
An upright laser scanning microscope (BX61WI, Olympus) was attached to a Ti:sapphire pulsed laser system (80 MHz repetition rate, <100 fs pulse width, Spectra Physics, Santa Clara, California) and software (Prairie View 5.3, Bruker) were used for two-photon fluorescence imaging. 20x water immersion (NA, 1.00; WD, 2 mm, Olympus), and 40x water-immersion objectives (NA 0.80, WD; 3.3 mm, Olympus) were selectively chosen for fluorescence imaging in vivo, Dextran conjugated with Texas Red (40 kDa, 100 mg/kg, Sigma) was intravenously administered to visualize the vasculature and blood flow in the cerebral cortex immediately before the imaging. 830-nm irradiation wavelength was used to excite Dextran conjugated with Texas Red, and light emission was differentiated and detected with 525/50 and 615/50 filters, respectively. Average laser power for imaging was less than 50 mW.
Time-lapse imaging of small cortical sub-volumes (30–40 image planes with 1–2 μm axial spacing), was performed for at least 40 minutes to track permeability in the cerebral cortex. The interval duration between stack sequences was 2–3 min; PMT settings and laser excitation power were kept constant during time-lapse imaging.
Line scan was performed along the central axis in the single vessel and perpendicular to the single vessel to measure blood flow velocity and vessel diameter, respectively. Arterioles, vein and capillary were discriminated by blood flow direction from the pial surface, line scan pattern, and vessel diameter. Capillaries were identified by their diameter (<5 μm).
Images were processed using open source software Fiji (NIH) and commercial software Matlab (8.5.0 R2015a, Mathworks). Software was used to perform intensity-based alignment of images at the different time points in Fiji. In addition, a custom-written Matlab-based script with the Radon transform algorithm was used to measure blood flow velocity in the cerebral cortex (Drew et al., 2010). Maximum intensity projection was used to calculate vessel diameter and full width at half maximum intensity was defined as vessel diameter.
To measure vascular permeability, the region of interest (ROI) was manually circumscribed within the brain parenchyma and a corresponding vessel. The mean fluorescence intensity (MFI) of each ROI was calculated minute by minute. The relative intensity of fluorescence in the brain parenchyma was defined as , where Fn and F0 were fluorescence intensity at any given time point and the initial time point, respectively.
Sodium Fluorescein Extravasation Assay
Sodium fluorescein (NaF), a relatively lower mass molecule (376 Da), was also used to evaluate BBB permeability as described with some modifications (Yamazaki et al., 2016). 100 μl of sodium fluorescein (100 mg/ml, Cat. #F6377, Sigma-Aldrich) was injected intraperitoneally. 45 minutes later, mice were anesthetized, and perfused with ice-cold PBS. Brains were removed and the cortical tissues were dissected. Dissected tissues were weighed and homogenized in PBS, and centrifuged at 10,000 × g for 15 min at 4°C to obtain supernatant. The supernatant was diluted with PBS to measure sodium fluorescein within a linear range of standards of known concentrations. The concentrations of NaF in brain samples were determined by measuring fluorescence intensity with a microplate reader (BioTek Synergy H1) at an excitation of 493 nm and an emission of 538 nm. The results are presented as (μg of NaF in tissue)/(mg of protein). Results were expressed as fold change with respect to control.
Preparation of Single Cell Suspensions Enriched in Brain Vascular Cells
Brain mural cells, brain endothelial cells and glial cells and other vascular-related cells were isolated as described (Vanlandewijck et al., 2018) with some modifications. After trans-cardial perfusion with PBS, cortical tissues were dissected from mouse brains. Cortical tissues were enzymatically digested using the Neural Tissue Dissociation Kit (P) (Cat. #130-092-628, Miltenyi Biotec), and myelin was removed using magnetic bead separation (Myelin Removal Beads II, Cat. #130-096-733, Miltenyi Biotec). After myelin removal, microglia and debris were removed by magnetic separation using anti-CD11b+ microbeads (Cat. #130-049-601, Miltenyi Biotec) and a Dead Cell Removal Kit (Cat. #130-090-101, Miltenyi Biotec), respectively, according to the manufacturer’s instruction. The final cell pellet was suspended in Ca/Mg-free PBS supplemented with 0.5% FBS for downstream analysis. In all samples, more than 90% of the cells were viable based on trypan blue examination.
Single Cell RNA Sequencing
Cells were prepared from cortical tissues of four different mice (male to female ratio = 1) for each genotype (i.e., 2 males and 2 females for iE3/Cre−, iE3/Cre+, iE4/Cre−, and iE4/Cre+ mice, respectively). Approximately 3,000 glio-vascular cells from one animal were loaded into each sample well on a chip and combined with Gel Beads containing barcoded oligonucleotides using a 10x Chromium Controller (10x Genomics). Single cell libraries were constructed according to the manufacturer’s instructions and were sequenced at the 3’ end by an Illumina HiSeq 4000 Sequencing Systems at an average depth of 60,000 reads per cell.
Single Cell Data Analysis
10X Genomics Cell Ranger Single Cell Software Suite (3.0.0) was used to de-multiplex raw base call (BCL) files generated from the sequencer into FASTQ files, perform alignment to mouse genome mm10, filter, barcode count, and UMI count. The gene expression matrix files were used for the subsequent analyses. Pre-processing, cell clustering, and differential expression analyses were performed using standard Seurat package procedures (2.3.4) (Satija et al., 2015). Briefly, the raw count matrices of the 4 samples in each genotype were merged into a new matrix. For each new matrix, we examined the distributions of genes, UMIs and reads mapped to mitochondrial genome for quality assurance. Cells that that had unique gene counts (over 4,000 or less than 200), and >25% mitochondrial counts were removed. Canonical correlation analysis (CCA) was performed to identify common sources of variation between cases and controls in the E3- and E4-scRNA-seq datasets (dataset composed of event from iE3/Cre− and iE3/Cre+ mice, and that composed of evens from iE4/Cre− and iE4/Cre+ mice, respectively). Based on the measurements of correlation strength, the first 20 canonical correlation vectors were used for dimension reduction and clustering analysis. Clusters were visualized using t-distributed stochastic nearest neighbor embedding (t-SNE). Function “FindConservedMarkers” was used to identify cluster-specific genes that were conserved across conditions. Function “SplitDotPlotGG” was used to visualize the conserved cell type markers in each cell cluster across conditions. Differentially expressed genes (DEGs) in each pairwise comparison of cell clusters were determined by the “FindMarkers” function with default parameters. Due to the nature of experimental design (Figure 1A and Figure S1A) pairwise comparison for DEG identification was performed between iE3/Cre− and iE3/Cre+ events, and iE4/Cre− and iE4/Cre+ events, respectively.
Annotation of Cell Clusters for Specific Cell Type
The annotation of each cluster was manually performed using the datasets for murine cell taxonomy (Saunders et al., 2018; Vanlandewijck et al., 2018).
Gene Ontology Analysis
Gene ontology (GO) analysis of upregulated or downregulated marker genes in specific cell clusters in iE3/Cre+ and iE4/Cre+ mice was performed using Metascape (http://metascape.org/) (Zhou et al., 2019).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Normal distribution of all data was evaluated by Shapiro-Wilk normality test. The assumption that data was sampled from Gaussian populations with equal variances was evaluated by F test. On passing both tests, data was analyzed using a two-tailed unpaired Student t-test (2 groups). Nonparametric datasets were analyzed by Mann-Whitney U test (2 groups) or Kruskal-Wallis one-way ANOVA test (3 groups). The type of tests used is indicated in the figure legends. P values <0.05 were considered significant. When comparing the effect of genotype on latency to find the platform (Figure S2D and S2H) and total distance travelled (Figure 2G and 2I) in MWM test, repeated measures ANOVA was used; trial (1–6) or day (2–4) served as the within-subject factors, and genotype served as the between-subject factor. When comparing the effect of genotype on latency to fall (Figure S2K and S2L) in the rotarod test, repeated measures ANOVA was used with trial (1–16) serving as the within-subject factors, and genotype serving as the between-subject factor. Similarly, when comparing the effect of genotype on are of extravascular rhodamine in two-photon experiments (Figure S3A and S3C), repeated measures ANOVA was used with time (0–30) serving as the within-subject factors, and genotype serving as the between-subject factor. Each dot in the figures represents readout from one animal unless otherwise noted in figure legends. All statistical analyses were conducted using GraphPad Prism 8.
Supplementary Material
Table S1. List of averaged gene expressions in all cell clusters identified in E3-scRNA-seq datasets, Related to Figure 4
Table S2. List of averaged gene expressions in all cell clusters identified in E4-scRNA-seq datasets, Related to Figure 4
Table S3. List of differentially expressed genes in each cell cluster identified in E3-scRNA-seq datasets, Related to Figure 4
Table S4. List of differentially expressed genes in each cell cluster identified in E4-scRNA-seq datasets, Related to Figure 4
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Apolipoprotein E Antibody (WUE-4) | Novus Biologicals | Cat#NB110-60531; RRID: AB_920623 |
| Goat Anti-Apolipoprotein E Polyclonal Antibody,Biotin Conjugated | Meridian Life Science | Cat#K74180B; RRID: AB_150544 |
| Rabbit Anti-Apolipoprotein E Monoclonal Antibody | Abcam | Cat#ab52607; RRID: AB_867704 |
| Rabbit Anti-Alpha Smooth Muscle Actin Polyclonal Antibody | Abcam | Cat#ab5694; RRID: AB_2223021 |
| Mouse Anti-Glial Fibrillary Acidic Protein (GFAP) Monoclonal Antibody | Millipore | Cat#MAB360; RRID: AB_2109815 |
| Rabbit Anti-GLUT-1 Polyclonal Antibody | Millipore | Cat#07-1401; RRID: AB_11212210 |
| Mouse Anti-Claudin-5 Monoclonal Antibody (4C3C2) | Invitrogen | Cat#35-2500; RRID: AB_2533200 |
| Rat Anti-CD13 Monoclonal Antibody | BD Biosciences | Cat#558744; RRID: AB_397101 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human apoE3 (for ELISA) | Fitzgerald Industries | Cat#30R-2381 |
| Recombinant human apoE4 (for ELISA) | Fitzgerald Industries | Cat#30R-2382 |
| Critical Commercial Assays | ||
| Amplex™ Red Cholesterol Assay Kit | Invitrogen | Cat#A12216 |
| Neural Tissue Dissociation Kit (P) | Miltenyi Biotec | Cat#130-092-628 |
| Myelin Removal Beads II | Miltenyi Biotec | Cat#130-096-733 |
| Anti-CD11b MicroBeads | Miltenyi Biotec | Cat#130-049-601 |
| Dead Cell Removal Kit | Miltenyi Biotec | Cat#130-090-101 |
| Chromium Single Cell 30Library & Gel BeadKit v3 | 10x Genomics | Cat#PN-1000075 |
| Deposited Data | ||
| scRNA-seq raw data | This paper | AMP-AD Knowledge Portal_VMC |
| Experimental Models: Organisms/Strains | ||
| Mouse: iE3 and iE4: B6.Gt(ROSA)26Sortm1(rtTA-APOE) | Guojun Bu, lead contact | N/A |
| Mouse: SM22alpha-driven Cre recombinase: B6.Cg-Tg(Tagln-cre)1Her/J | The Jackson Laboratory | Jax 017491 |
| Mouse: Apoe knockout: B6.129P2-Apoetm1Unc/J | The Jackson Laboratory | Jax 002052 |
| Software and Algorithms | ||
| Aperio ImageScope | Leica Biosystems | Version: 12.3.3 |
| FreezeFrame | Actimetrics | N/A |
| ANY-Maze software | Stoelting Co. | N/A |
| Matlab | Mathworks | Version: 9.6.0.1072779 R2019a Version: 8.5.0 R2015a |
| Fiji (ImageJ) | Fiji contributors | Version: 2.0.0 |
| Cell Ranger | 10x Genomics | Version: 3.0.0 |
| Seurat | Sajita Lab | Version: 2.3.4 |
| Metascape | https://metascape.org/ | N/A |
| GraphPad Prism | GraphPad | Version: 8 |
| ImageJ bundled with 64-bit Java | NIH | Version: 1.8.0_112 |
| Prairie View | Bruker | Version: 5.3 |
| Other | ||
| Superose 6, 10/300 GL | GE Healthcare | Cat#17517201 |
Highlights:
Conditional mouse models expressing apoE isoforms in vascular mural cells.
Vascular apoE4 is associated with impaired arteriole blood flow and behaviors.
Transcriptomic analysis suggests apoE4-related astrogliosis at the vasculature.
ApoE3 expressed in vasculature is linked to angiogenic signature in pericytes.
Acknowledgements:
The authors thank Dr. Laura Lewis-Tuffin for technical support. This work was supported by grants from the National Institutes of Health (NIH) (R37AG027924, RF1AG051504, RF1AG057181, P50AG016574, and RF1AG046205 to G.B., R01AG051574 to T.K., and R01AG062110 to C.-C.L.); a Cure Alzheimer’s Fund (to G.B.), an American Heart Association grant (15SDG22460003 to T.K.) and a fellowship (to Y.Y.); and Florida Department of Health Ed and Ethel Moore Alzheimer’s Disease Research Program grants (7AZ22 to T.K. and 8AZ07 to C.-C.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
The authors declare no competing interests.
References
- Alata W, Ye Y, St-Amour I, Vandal M, and Calon F (2015). Human apolipoprotein E varepsilon4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J Cereb Blood Flow Metab 35, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellosta S, Mahley RW, Sanan DA, Murata J, Newland DL, Taylor JM, and Pitas RE (1995). Macrophage-specific expression of human apolipoprotein E reduces atherosclerosis in hypercholesterolemic apolipoprotein E-null mice. J Clin Invest 96, 2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher P, Gotthardt M, Li WP, Anderson RG, and Herz J (2003). LRP: role in vascular wall integrity and protection from atherosclerosis. Science 300, 329–332. [DOI] [PubMed] [Google Scholar]
- Caselli RJ, Dueck AC, Osborne D, Sabbagh MN, Connor DJ, Ahern GL, Baxter LC, Rapcsak SZ, Shi J, Woodruff BK, et al. (2009). Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N Engl J Med 361, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey CS, Atagi Y, Yamazaki Y, Shinohara M, Tachibana M, Fu Y, Bu G, and Kanekiyo T (2015). Apolipoprotein E Inhibits Cerebrovascular Pericyte Mobility through a RhoA Protein-mediated Pathway. J Biol Chem 290, 14208–14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, and Bobryshev YV (2017). The phenomenon of atherosclerosis reversal and regression: Lessons from animal models. Exp Mol Pathol 102, 138–145. [DOI] [PubMed] [Google Scholar]
- Cruz Hernandez JC, Bracko O, Kersbergen CJ, Muse V, Haft-Javaherian M, Berg M, Park L, Vinarcsik LK, Ivasyk I, Rivera DA, et al. (2019). Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat Neurosci 22, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delenclos M, Faroqi AH, Yue M, Kurti A, Castanedes-Casey M, Rousseau L, Phillips V, Dickson DW, Fryer JD, and McLean PJ (2017). Neonatal AAV delivery of alpha-synuclein induces pathology in the adult mouse brain. Acta Neuropathol Commun 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Battista AM, Heinsinger NM, and Rebeck GW (2016). Alzheimer’s Disease Genetic Risk Factor APOE-epsilon4 Also Affects Normal Brain Function. Curr Alzheimer Res 13, 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PJ, Blinder P, Cauwenberghs G, Shih AY, and Kleinfeld D (2010). Rapid determination of particle velocity from space-time images using the Radon transform. J Comput Neurosci 29, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Shirman GA, Strittmatter WJ, and Matthew WD (2001). Impairment of the blood-nerve and blood-brain barriers in apolipoprotein e knockout mice. Exp Neurol 169, 13–22. [DOI] [PubMed] [Google Scholar]
- Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, Bullido MJ, Engelborghs S, De Deyn P, Berr C, et al. (2011). APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry 16, 903–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz GS, and Reardon CA (2016). Do the Apoe−/− and Ldlr−/− Mice Yield the Same Insight on Atherogenesis? Arterioscler Thromb Vasc Biol 36, 1734–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, et al. (2011). Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke 42, 2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday MR, Pomara N, Sagare AP, Mack WJ, Frangione B, and Zlokovic BV (2013). Relationship between cyclophilin a levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein e4 carriers and blood-brain barrier breakdown. JAMA Neurol 70, 1198–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Liu CC, Shinohara M, Li J, and Bu G (2012). LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer’s amyloid-beta. J Neurosci 32, 16458–16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SS, Kurti A, Baker KE, Liu CC, Colonna M, Ulrich JD, Holtzman DM, Bu G, and Fryer JD (2018). Behavioral and transcriptomic analysis of Trem2-null mice: not all knockout mice are created equal. Hum Mol Genet 27, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, Sullivan PM, and Maeda N (1999). Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J Clin Invest 103, 1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Kanekiyo T, Xu H, and Bu G (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9, 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW, and Bu G (2017). ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 96, 1024–1032 e1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Jiaerken Y, Yu X, Huang P, Qiu T, Jia Y, Li K, Xu X, Shen Z, Guan X, et al. (2017). Associations between APOE genotype and cerebral small-vessel disease: a longitudinal study. Oncotarget 8, 44477–44489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW (2016). Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler Thromb Vasc Biol 36, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, and Roses AD (1995). Neurodegeneration in the central nervous system of apoE-deficient mice. Exp Neurol 136, 107–122. [DOI] [PubMed] [Google Scholar]
- Najm R, Jones EA, and Huang Y (2019). Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer’s disease. Mol Neurodegener 14, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelligrino DA, Vetri F, and Xu HL (2011). Purinergic mechanisms in gliovascular coupling. Semin Cell Dev Biol 22, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, and Frikke-Schmidt R (2018). Absolute 10-year risk of dementia by age, sex and APOE genotype: a population-based cohort study. CMAJ 190, E1033–E1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GA, Burns MP, Weeber EJ, and Rebeck GW (2013). Young APOE4 targeted replacement mice exhibit poor spatial learning and memory, with reduced dendritic spine density in the medial entorhinal cortex. Learn Mem 20, 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JT, Liu CC, Zhao N, Wang J, Putzke T, Yang L, Shinohara M, Fryer JD, Kanekiyo T, and Bu G (2017). Subacute ibuprofen treatment rescues the synaptic and cognitive deficits in advanced-aged mice. Neurobiol Aging 53, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas S, Brugulat-Serrat A, Bargallo N, Minguillon C, Tucholka A, Falcon C, Carvalho A, Moran S, Esteller M, Gramunt N, et al. (2018). Higher prevalence of cerebral white matter hyperintensities in homozygous APOE-varepsilon4 allele carriers aged 45–75: Results from the ALFA study. J Cereb Blood Flow Metab 38, 250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakae N, Liu CC, Shinohara M, Frisch-Daiello J, Ma L, Yamazaki Y, Tachibana M, Younkin L, Kurti A, Carrasquillo MM, et al. (2016). ABCA7 Deficiency Accelerates Amyloid-beta Generation and Alzheimer’s Neuronal Pathology. J Neurosci 36, 3848–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Zimri S, Boehm-Cagan A, Liraz O, and Michaelson DM (2014). Hippocampus-related cognitive impairments in young apoE4 targeted replacement mice. Neurodegener Dis 13, 86–92. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030 e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling S, DeStefano AL, Sachdev PS, Choi SH, Mather KA, DeCarli CD, Wen W, Hogh P, Raz N, Au R, et al. (2013). APOE genotype and MRI markers of cerebrovascular disease: systematic review and meta-analysis. Neurology 81, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Lu Z, Chhatbar PY, O’Herron P, and Kara P (2012). An artery-specific fluorescent dye for studying neurovascular coupling. Nat Methods 9, 273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Kanekiyo T, Yang L, Linthicum D, Shinohara M, Fu Y, Price L, Frisch-Daiello JL, Han X, Fryer JD, and Bu G (2016). APOE2 eases cognitive decline during Aging: Clinical and preclinical evaluations. Ann Neurol 79, 758–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JA, Haley GE, and Raber J (2012). Apolipoprotein E isoform-dependent effects on anxiety and cognition in female TR mice. Neurobiol Aging 33, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudre CH, Cardoso MJ, Frost C, Barnes J, Barkhof F, Fox N, Ourselin S, and Alzheimer’s Disease Neuroimaging I (2017). APOE epsilon4 status is associated with white matter hyperintensities volume accumulation rate independent of AD diagnosis. Neurobiol Aging 53, 67–75. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, and Maeda N (1997). Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem 272, 17972–17980. [DOI] [PubMed] [Google Scholar]
- Sun JH, Tan L, Wang HF, Tan MS, Tan L, Li JQ, Xu W, Zhu XC, Jiang T, and Yu JT (2015). Genetics of Vascular Dementia: Systematic Review and Meta-Analysis. J Alzheimers Dis 46, 611–629. [DOI] [PubMed] [Google Scholar]
- Tai LM, Thomas R, Marottoli FM, Koster KP, Kanekiyo T, Morris AW, and Bu G (2016). The role of APOE in cerebrovascular dysfunction. Acta Neuropathol 131, 709–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambisetty M, Beason-Held L, An Y, Kraut MA, and Resnick SM (2010). APOE epsilon4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch Neurol 67, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorngate FE, Rudel LL, Walzem RL, and Williams DL (2000). Low levels of extrahepatic nonmacrophage ApoE inhibit atherosclerosis without correcting hypercholesterolemia in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 20, 1939–1945. [DOI] [PubMed] [Google Scholar]
- Toth P, Tarantini S, Csiszar A, and Ungvari Z (2017). Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol 312, H1–H20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Preboske GM, Kantarci K, Raman MR, Machulda MM, Mielke MM, Lowe VJ, et al. (2015). Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain 138, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, and Huang Y (2006). Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci 26, 4985–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG, Bu G, and Kanekiyo T (2016). Vascular Cell Senescence Contributes to Blood-Brain Barrier Breakdown. Stroke 47, 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Shinohara M, Shinohara M, Yamazaki A, Murray ME, Liesinger AM, Heckman MG, Lesser ER, Parisi JE, Petersen RC, et al. (2019a). Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain 142, 1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Shinohara M, Yamazaki A, Ren Y, Asmann YW, Kanekiyo T, and Bu G (2020). ApoE (Apolipoprotein E) in Brain Pericytes Regulates Endothelial Function in an Isoform-Dependent Manner by Modulating Basement Membrane Components. Arterioscler Thromb Vasc Biol 40, 128–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu CC, and Bu G (2019b). Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol 15, 501–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Liu CC, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, Heckman MG, Diehl NN, Shinohara M, Martens YA, et al. (2018). APOE epsilon2 is associated with increased tau pathology in primary tauopathy. Nat Commun 9, 4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of averaged gene expressions in all cell clusters identified in E3-scRNA-seq datasets, Related to Figure 4
Table S2. List of averaged gene expressions in all cell clusters identified in E4-scRNA-seq datasets, Related to Figure 4
Table S3. List of differentially expressed genes in each cell cluster identified in E3-scRNA-seq datasets, Related to Figure 4
Table S4. List of differentially expressed genes in each cell cluster identified in E4-scRNA-seq datasets, Related to Figure 4
Data Availability Statement
Single Cell RNA Sequencing data are available via the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the Accelerating Medicines Partnership (AMP-AD) Target Discovery Program and other National Institute on Aging (NIA)-supported programs to enable open-science practices and accelerate translational learning. The data, analyses and tools are shared early in the research cycle without a publication embargo on secondary use. Data is available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.synapse.org/DataAccess/Instructions). For access to content described in this manuscript see: https://doi.org/10.7303/syn22313650.