

Abstract
Introduction-
A better understanding of the underlying molecular mechanism of diseases is critical for developing more effective diagnostic tools and therapeutics towards precision medicine. However, many challenges remain to unravel the complex nature of diseases.
Areas covered-
Changes in protein isoform expression and post-translation modifications (PTMs) have gained recognition for their role in underlying disease mechanisms. Top-down mass spectrometry (MS)-based proteomics is increasingly recognized as an important method for the comprehensive characterization of proteoforms that arise from alternative splicing events and/or PTMs for basic and clinical research. Here, we review the challenges, technological innovations, and recent studies that utilize top-down proteomics to elucidate changes in the proteome with an emphasis on its use to study the diseases of the heart.
Expert Opinion-
Proteoform-resolved information can substantially contribute to the understanding of the molecular mechanisms underlying various diseases and for the identification of novel proteoform targets for better therapeutic development toward precision medicine. Despite the challenges of sequencing intact proteins, top-down proteomics has enabled a wealth of information regarding protein isoform switching and changes in PTMs. Continuous developments in sample preparation, intact protein separation, and instrumentation for top-down MS have broadened its capabilities to characterize proteoforms from a range of samples on an increasingly global scale.
Keywords: Top-down Proteomics, Mass Spectrometry, Post-translational Modifications, Proteoforms, Heart Diseases
1.1. Introduction
Mutations, alternative splicing events, and post-translational modifications (PTMs) result in a host of proteoforms (a term referring to all the protein products that arise from a single gene) that greatly diversify the proteome [1–4]. To understand the functional roles of specific proteoforms in biological systems, it is critical to unambiguously determine how they regulate protein activity, localization, degradation, and their alteration in diseases [5,6]. Moreover, mutations and differential expression of protein isoforms have been shown to drive molecular mechanisms underlying disease phenotypes, necessitating their in-depth characterization [5,7].
Mass spectrometry (MS)-based proteomics has emerged as the most versatile and comprehensive strategy for protein characterization [8,9]. Typically, proteins are extracted from a biological sample of interest and enzymatically or chemically digested. The resulting peptides are separated by liquid chromatography (LC) coupled to online MS, fragmented, and the resulting data are processed by proteomics software to yield protein identifications, expression-level quantification, and PTM mapping [10–12]. This “bottom-up” proteomics approach is conventional as peptides, resulting from intact protein digestion, are biochemically more soluble, easier to separate, and there is numerous well-developed bioinformatics software available for protein identification and quantification [10]. However, the ‘peptide-to-protein inference’ problem in bottom-up proteomics leads to a loss in proteoform information because of many homologous sequence regions are shared between different protein forms and the generally low protein sequence coverage is observed [13,14]. For this reason, the “top-down” proteomics approach, which forgoes digestion to peptides and instead analyzes intact proteins, is the preferred method to achieve unambiguous, proteoform-resolved molecular detail (Figure 1) [15]. Top-down proteomics is a powerful tool for proteoform analysis and enables accurate protein identification, PTM localization including possible sequence variants, and permits the relative quantification of expression levels of various proteoforms [16–20]. Here, we review the challenges and innovations (particularly in the last 2 years) in top-down proteomics with a particular focus on sample preparation, proteoform separation and enrichment, instrumentation, data acquisition and analysis, and the significant applications of top-down proteomics for understanding the mechanisms underlying cardiac diseases.
Figure 1.
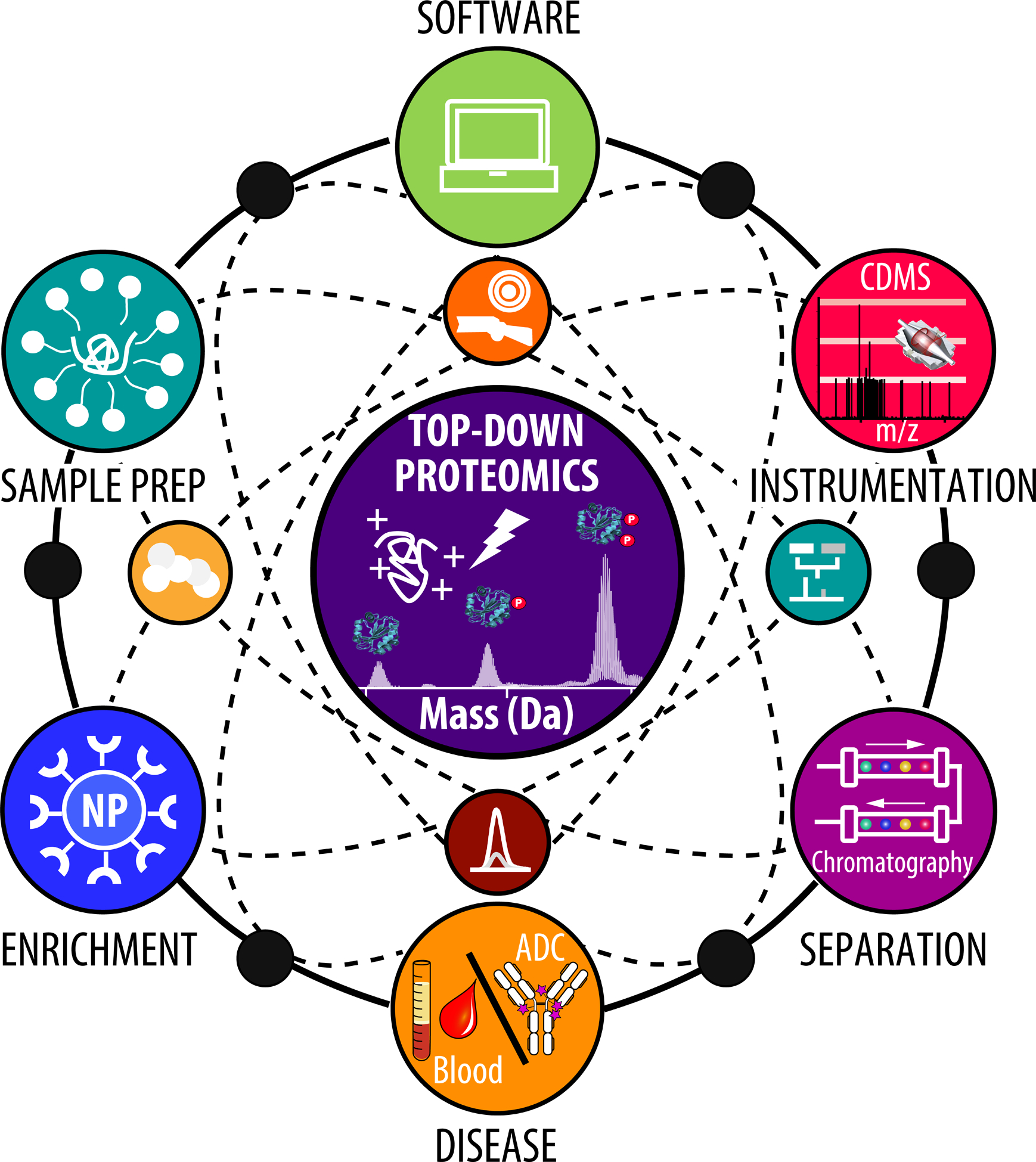
Illustration of the different components of top-down proteomics highlighting the major areas of recent development.
1.2. Methods for Top-down Proteomics
1.2.1. Sample Preparation for Top-down Proteomics
Sample preparation is critical for achieving reliable proteomics data. Commonly, protein extraction for biomedical research involves the use of Good’s buffers, high salt (>100 mM), protease and phosphatase inhibitors, and surfactants, like sodium dodecyl sulfate (SDS) or Triton X-100, for protein solubilization [15,21]. However, most salts and conventional surfactants are generally incompatible with MS analysis and must be removed before downstream analysis [22].
To standardize sample preparation, a “best practices and benchmarks” was reported by Agar and colleagues in the Consortium of Top-down Proteomics [23]. Using their outlined approach, incompatible salts and other components are removed from target proteins by ultrafiltration spin filters with specific molecular weight cutoffs to exchange MS-incompatible components for low concentration organic acid or volatile ammonium-based salts before analysis. On the other hand, protein precipitation can be used to eliminate contaminants for the analysis of complex biological mixtures. In particular, precipitation is often required when using surfactants, which are often employed during cell lysis to solubilize hydrophobic membrane proteins, because they significantly inhibit MS signal by outcompeting proteins for ionization or by forming harmful adducts [24,25]. However, precipitation can be time-consuming and may add unintended sample variability and sample loss, as some proteins are not readily re-solubilized [26,27].
Previously, acid-cleavable surfactants such as Rapigest [28], ProteaseMAX [29], and MS-compatible degradable surfactant MaSDeS [30] were developed as MS-compatible surfactants to enable protein solubilization for digestion and MS-analysis. However, it was found that these surfactants are incompatible with top-down proteomics [31]. Brown et al. have recently developed a photocleavable ionic surfactant, 4-hexylphenylazosulfonate (Azo) [31], which shows comparable protein extraction efficiency to common surfactants such as SDS and was compatible with top-down MS analysis after rapid UV-degradation (Figure 2) [31]. Azo was used to broadly solubilized proteins from cardiac tissues including important membrane proteins such as phospholamban [32], receptor expression enhancing protein 5 [33], and every subunit of the ATP synthase complex [34,35]. Additionally, Azo was shown to enable rapid enzymatic digestion by trypsin, demonstrating its unique ability as an “all-in-one” surfactant for integrated top-down and bottom-up proteomics [36].
Figure 2.
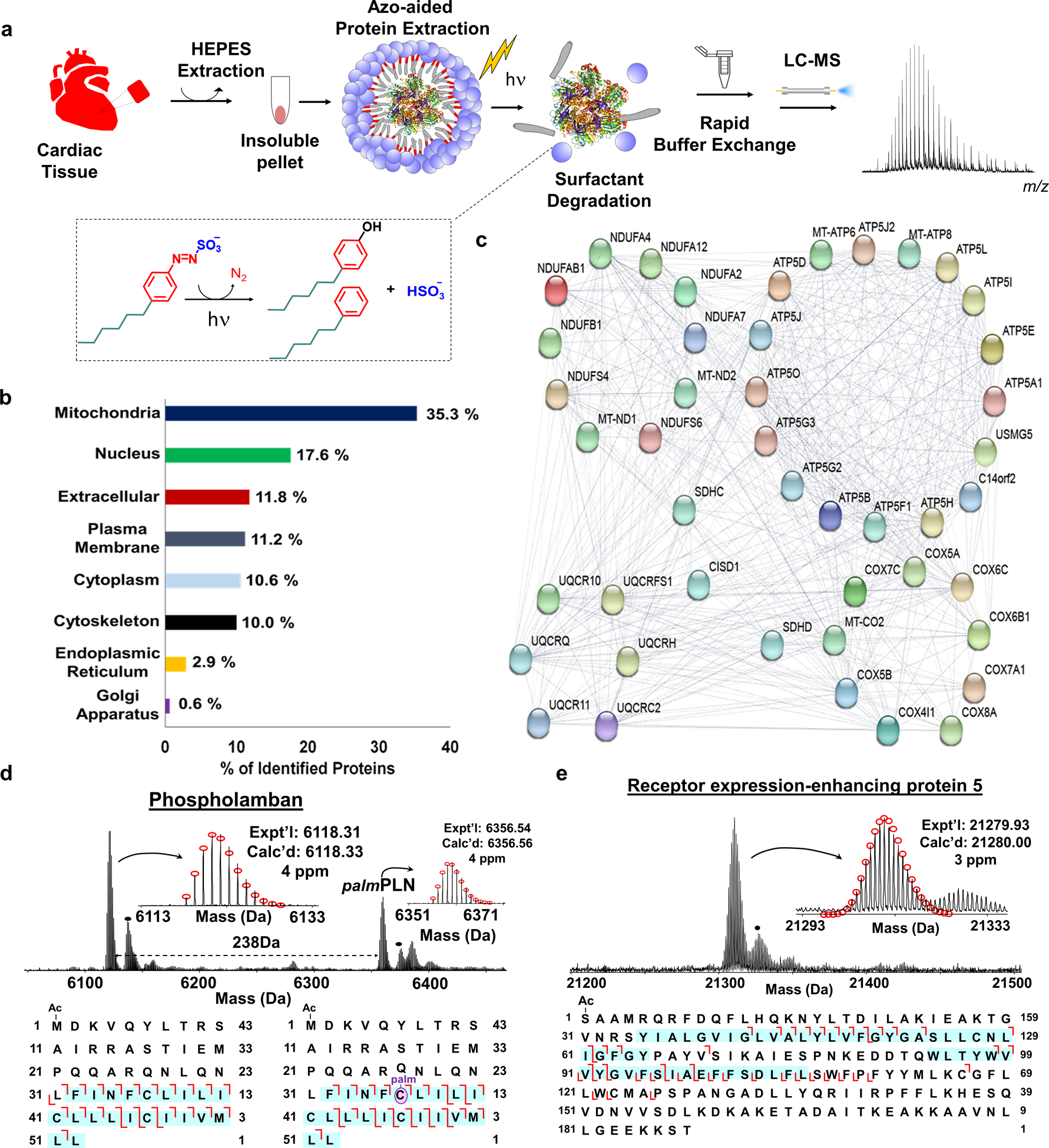
a, Scheme illustrating photocleavable surfactant (Azo)-enabled membrane proteomics. Proteins from cardiac tissue were extracted using HEPES buffer (2x) to deplete the cytosolic proteins, followed by an extraction using 0.5% Azo. The surfactant was rapid degradation (into 4-hexylphenol, 4-hexylbenzene, nitrogen, and hydrogen sulfate under UV irradiation) of the surfactant with UV irradiation (λmax = 305 nm) and buffer exchange before LC-MS analysis for proteoform characterization. Note that the molecules are not drawn to scale. b, Subcellular location of proteins identified in cardiac tissue in the Azo-aided top-down proteomics. c, Interactome map of the identified proteins with Azo that belong to the electron transport chain d-e, MS, and tandem MS analysis of representative membrane proteins. Phospholamban (PLN) and palmitoylated PLN (palmPLN) with palmitoylation localized at cysteine 36 residue (d), and receptor expression-enhancing protein 5 (e). The sequences below the spectra represent the fragmentation maps with sequence coverage and PTM localization based on online RPLC–MS/MS analysis. The regions representing the transmembrane domains are highlighted by blue shading. The dot represents +16 Da. Figure adapted from reference [31]. Copyright 2019 Springer Nature.
To improve the sensitivity and automation of top-down sample preparation, Zhu and coworkers have adapted the nanoPOTS (nanodroplet processing in one pot for trace samples) [37] technology for top-down proteomics. They found that a combination of dodecyl-β-D-maltopyranoside (DDM) with urea enabled the extraction and identification of ~170–620 proteoforms from as little as ~70–770 HeLa cells [38]. The nanoPOTs technology, although still highly specialized, shows enormous promise to enable highly sensitive top-down proteomics (potentially even single-cell top-down analysis), but the majority of protein identifications were limited to sizes <10 kDa.
Traditionally, proteomics is performed in the denatured mode to study the primary protein sequence [39]. More recently, native top-down MS has been utilized to study protein structure [40] and a more extensive summary of this emerging technology can be found in a recent review [41]. Briefly, native top-down MS involves using non-denaturing conditions and often employing 100–200 mM ammonium acetate as a volatile buffer for the analysis of intact proteins and protein complexes. Valuable information regarding protein complex subunit stoichiometry [42], metal-binding [43,44], protein interaction partners [45], and lipid binding [46] can be achieved using native MS. Due to the hydrophobic nature of membrane proteins, the use of nonionic surfactants, such as DDM or the recently developed oligoglycerol class of detergents (shown to improve purification and MS-based characterization of G-protein coupled receptors) [47], to solubilize and stabilize membrane proteins is generally required to enable native MS analysis [48–50]. In this case, the entire protein-micelle complex is ionized, and the protein is liberated from the surfactant micelle using high collisional energy in the mass spectrometer [49]. Robinson and coworkers have successfully demonstrated native MS analysis for a number of applications including most recently analyzing protein assemblies directly for native membranes bypassing the use of detergents [51].
1.2.2. Protein and Proteoform Enrichment for Top-down Proteomics
The human proteome has a high dynamic range; therefore, the analysis of certain protein classes, low abundance proteoforms, and target proteins often requires an enrichment step [4,52]. Proteoform-level enrichment is important for identifying and characterizing low-abundance species with consideration for their molecular variability. Enrichment strategies employing metal oxide materials such as titanium oxide [53] and immobilized metal affinity chromatography (IMAC) [54], or enrichment beads coated with sugar-binding lectin [55] are common for phosphopeptide and glycopeptide enrichment, respectively. On the other hand, intact proteoform enrichment, which is necessary for top-down proteomics, is relatively underdeveloped [56]. To address this gap in technology development, the Ge group has developed superparamagnetic cobalt ferrite nanoparticles (NPs) functionalized with GAPT-Zn ligands for phosphoprotein enrichment before LC-MS/MS analysis for confident identification and localization of phosphorylation sites [57]. More recently, the synthetic scalability, reproducibility, and enrichment efficacy of these NPs were improved using surface-silanized superparamagnetic magnetite (Fe3O4) particles coupled with the GAPT-Zn ligand [58]. The newly developed surface-functionalized NP platform could be further generalized to other enrichment targets, provided a suitable affinity reagent is available [58].
For targeted protein characterization, affinity purification (AP) is often employed to purify proteins and characterize their modifications [59–61]. Antibodies are the most common affinity reagents for protein purification. However, antibodies possess many inherent limitations: batch-to-batch variation, high cost of production, poor chemical stability, and lack of proteoform-specific epitope recognition that collectively contribute to reproducibility issues [62–64]. To address the limitation of the antibody-based approach, Tiambeng and Roberts led the development of a nanoproteomics strategy using functionalized magnetic NPs with a peptide affinity ligand to specifically target cardiac troponin I (cTnI) [65–67], an important and gold-standard cardiac biomarker [68]. The NP-peptide approach enabled the enrichment of cTnI with high specificity (< 1 ng/mL) and reproducibility, while at the same time depleted highly abundant proteins like human serum albumin. Overall, the NP-peptide approach outperformed conventional monoclonal antibody and top-down characterization of cTnI, enriched from serum, yielded proteoform-resolved molecular fingerprints of diverse cTnI proteoforms for evaluation of proteoform-pathophysiology relationships (Figure 3) [68]. These advancements demonstrate the great potential for the improved enrichment of target proteins and their proteoforms for top-down proteomics.
Figure 3. Nanoproteomics enables proteoform-resolved analysis of low-abundance cardiac troponin I in human serum.
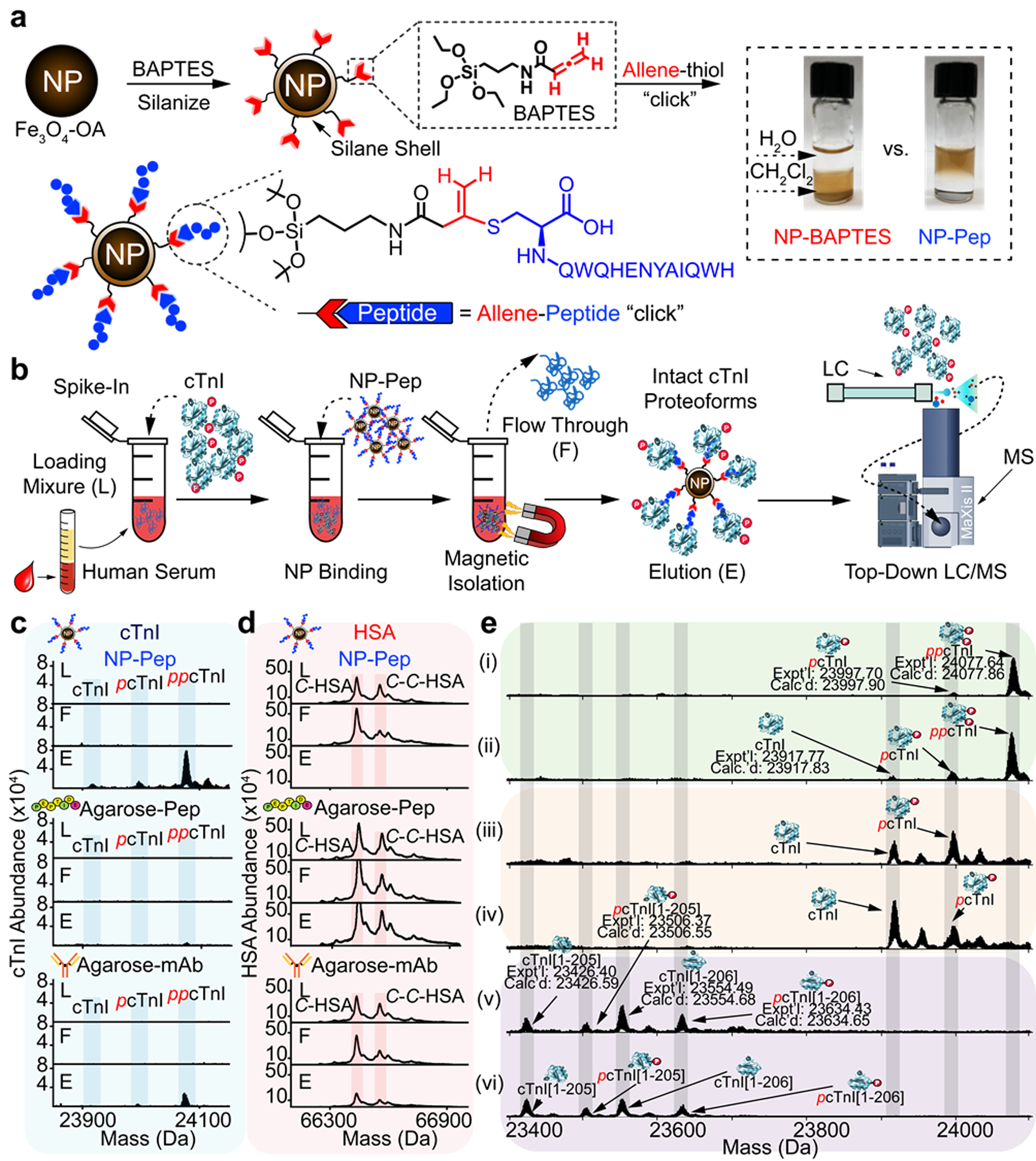
a, Silanization of Fe3O4 NPs using an allene carboxamide-based organosilane monomer (BAPTES) for cysteine thiol-specific bioconjugation. The rationally designed NPs are surface-functionalized with a 13-mer peptide that has a high affinity for cTnI (NP-Pep) for cTnI enrichment. The 13-mer peptide possesses a C-terminal cysteine that selectively reacts with the allene carboxamide moiety on the silanized NPs. The photograph shows functionalized NPs in a biphasic mixture of dichloromethane (CH2Cl2) and water (H2O), comparing the solvent compatibility of the NP-BAPTES and the NP-Pep. b, Nanoproteomics assay utilizing NP-Pep for specific enrichment of cTnI from serum and subsequent top-down MS analysis of cTnI proteoforms. cTnI is first spiked into human serum to prepare the loading mixture (L). The NPs are then incubated with the serum loading mixture, the cTnI-bound NPs are magnetically isolated, the unwanted and nonspecific proteins are removed as flow-through (F). The captured cTnI is then eluted and the final elution fraction after enrichment is analyzed by top-down LC/MS. c-d, Normalized deconvoluted mass spectra corresponding to enriched cTnI (c) and depleted HSA (d), illustrating the abundance of cTnI and HSA before and after enrichment using NP-Pep, high-affinity peptide-functionalized agarose (Agarose-Pep), and antibody (mAb M46) functionalized with agarose (Agarose-mAb). e, Deconvoluted MS corresponding to cTnI proteoforms enriched from human serum. The cTnI (~10–20 ng/mL) spiked in the human serum (10 mg) were extracted from various human hearts: (i) and (ii), donor hearts; (iii) and (iv), diseased hearts with dilated cardiomyopathy, (v) and (vi), post-mortem hearts. cTnI proteoforms were identified using accurate intact mass measurement, using the most abundant mass based on the amino acid sequence of entry name TNNI3_human from the UniProtKB sequence database. p, phosphorylation. pp, bisphosphorylation. Figure adapted from reference [68]. Copyright 2020 Springer Nature.
1.2.3. Intact Protein Separation
Global proteoform characterization necessitates high-resolution protein separation before top-down analysis. However, front-end separation of intact proteins represents a major challenge for in-depth proteome coverage in part because of dramatic variation in physicochemical properties of proteins (size, hydrophobicity, charge, etc.).
Reversed-phase liquid chromatography (RPLC) is the predominant method for online sample desalting and separation coupled to MS. Typically a single-dimension of RPLC can only enable the identification of ~100 intact proteoforms with limited coverage of larger species (>30 kDa) [69]. Although long columns (>1 m) were demonstrated to provide a peak capacity of >400 and confidently identified ~900 proteoforms, they operate at ultrahigh pressures (~14,000 psi) and are not always practical [70]. On the other hand, monolithic columns offer high-resolution protein separation with low backpressure [71]. Liang et al. demonstrated that bridged hybrid bis(triethoxysilyl)ethylene monolith with C8 functional groups afforded high-resolution separation of complex cardiac lysate including a large proteoform, α-actinin2 (103.77 kDa) [72].
Capillary zone electrophoresis (CZE) has recently shown great promise as an alternative or complementary front-end separation due to its inherent sensitivity and peak capacity [73]. The development of sheathflow and sheathless interfaces have enabled facile coupling of CZE to MS [73]. However, the application of CZE for intact protein separation has been limited by the low loading capacity and narrow separation window common to CZE systems. To address these major issues, the Sun group optimized the dynamic pH junction CZE method, using 10% acetic acid as the background electrolyte, to load ~1 μL of sample and they demonstrated the use of single-shot CZE-MS analysis to identify ~600 proteoforms from an Escherichia coli lysate [74].
Offline hydrophilic interaction chromatography (HILIC) was previously utilized to separate intact subunits of mitochondria membrane proteins offline before intact protein MS analysis [75]. More recently, Gargano et al. developed online capillary HILIC for top-down proteomics [76]. Notably, they utilized a C4 trap column for loading (bypassing the traditional use of high organic solvent for sample composition) and doped propionic acid in the electrospray interface to overcome the signal suppression of trifluoroacetic acid in the mobile phase [76]. However, the proteome coverage was limited and significant work will still be needed before the full potential of HILIC for top-down proteomics is recognized.
Online LC-MS under non-denaturing conditions for protein separation before top-down analysis is gaining popularity, particularly for the analysis of large biomolecule therapeutics. For example, online size exclusion chromatography (SEC) has been used for the rapid online desalting of antibodies and protein complexes [77]. Similarly, ion-exchange chromatography (IEX)-MS has been used to characterize protein conjugates [78,79]. Hydrophobic interaction chromatography (HIC)-MS shows great promise for characterizing intact protein directly coupled to top-down MS [80]. Chen et al. demonstrated online HIC-MS analysis for determination of the relative hydrophobicity, intact masses, and glycosylations of monoclonal antibodies (mAb) as well as gaining sequence and structural information using MS/MS [81].
While the continued development of online separation strategies is essential for enhancing the capabilities of top-down proteomics, no single separation mode can sufficiently handle the complexity of the whole proteome. Therefore, multidimensional separation approaches are commonly employed for deeper proteome coverage [82]. Often large proteins, which have an inherently lower signal-to-noise, are underrepresented in top-down studies [83]. For this reason, gel-based techniques are commonly used to separate proteins based on molecular weight. Gel-eluted liquid fraction entrapment electrophoresis (GELFrEE) is one of the most common pre-fractionation techniques owing to its high-resolution size separation. In 2012, the Kelleher group combined solution isoelectric focusing, GELFrEE, and RPLC-MS/MS to identify ~3,000 proteoforms from 1043 protein groups including proteins up to 105 kDa [84]. More recently, PEPPI (passively eluting protein from polyacrylamide gel as intact species)-MS was introduced as an alternative gel-based separation technique [85]. Using PEPPI, good protein recovery, and high-resolution separation was achieved for top-down proteomics of protein <50 kDa. Overall, gel-based strategies suffer from the use of SDS, which requires sample clean-up that may result in protein loss and variability [26].
To bypass the use of SDS for separation, serial SEC (sSEC) was developed by coupling columns with differing pore sizes and using an MS-compatible mobile phase (1% formic acid). sSEC was used to achieve high-resolution size-based separation of cardiac sarcomere proteins and, when used as for offline fractionation before RPLC-MS, enabled the detection of ~4,000 unique proteoforms up to 223 kDa [69]. SEC was also coupled to CZE for the identification of ~5700 proteoforms from Escherichia coli [86]. Yu et al. took a different approach using offline fractionation by high pH RPLC followed by online low pH RPLC coupled to MS to enable the identification of 2778 proteoforms from 628 proteins [87].
Multidimensional separation strategies have become increasingly adopted for top-down proteomics in non-denaturing mode (i.e., native top-down MS). For example, native SEC and CZE were coupled to identify 672 proteoforms, 144 proteins, and 23 protein complexes [88]. Using Native GELFrEE and IEX, Skinner et al. identified 125 intact endogenous complexes from mouse hearts and human cancer cell lines [89].
Despite the advancements in multidimensional separation strategies, performing offline protein fractionation remains labor-intensive, low-throughput, and requires relatively large sample quantities making it not suitable for sample limited applications. However, some recent online approaches have been developed which include using weak-cationic exchange HILIC and RPLC to enable the characterization of hundreds of histone proteoforms from ~1.5 μg of protein [90]. Additionally, online high pH RPLC coupled to low pH RPLC shows promise for intact protein separation before top-down MS [91]. Moving forward, the growth of online multidimensional separations will be crucial for in-depth proteome coverage and high-throughput top-down proteomics analysis.
1.2.4. Instrumentation and Fragmentation for Proteoform Identification and Characterization
Although several ionization techniques are available, ESI is still predominately utilized for top-down proteomics as it is easy to interface with common LC systems [92]. Although ESI enables the highly sensitive detection of analytes, protein detection by ESI suffers from signal competition with small-molecule contaminates that outcompete large molecules and results in the need for highly purified samples [23,83]. Recently, submicron ESI emitter tips, developed by the Williams group, have pushed the boundaries of ESI sensitivity while also showing improved salt tolerance up to physiological concentrations (25 mM Tris, 150 mM KCl) by virtue of creating smaller droplets [93]. Another emerging technique is liquid extraction surface analysis (LESA), which directly sampling proteins from a solid surface for analysis of protein and protein complexes [94,95].
A growing number of instrument platforms are now capable of top-down proteomic analysis. Traditionally, Fourier-transform ion cyclotron resonance (FTICR) mass spectrometers are the instrument of choice for top-down because of its capabilities to resolve very large species [96]; however, the slower scan speed can limit its capabilities for effective LC-MS/MS analysis and global protein characterization. Quadrupole time-of-flight (q-TOF) mass spectrometers are a common alternative instrument platform, as they are robust, affordable, and capable of analyzing relatively large protein species [69,97]. Currently, benchtop Orbitrap mass spectrometers are the most common instrument for proteomics as they bypass the need for cryogenic cooling (which is needed for the magnets of FTICR instruments) while still offering high-resolution and faster acquisition speeds [98,99]. Moreover, global top-down proteomics using the Orbitrap has enabled the identification and quantification of thousands of proteoforms [84,100]. However, top-down proteomics using an Orbitrap MS was limited to low-molecular-weight species as the signal of larger biomolecules rapidly decays due to collisions with the background gas [101]. Work by the Brodbelt and Kelleher group in tuning the Orbitrap instrument parameters has increased its scope for targeted and global analysis of larger proteoforms [98,102]. More recently, instrument hardware improvements have led to enhanced large ion transmission and detection, especially with the implementation of ultra-high mass range (UHMR) Orbitraps that are capable of analyzing species at 70,000 m/z and mass as large as 800 kDa [103,104].
Beyond new instrumentation, unique approaches have been implemented to improve the detection of intact proteins (particularly large proteins) by generally reducing the complexity of the resulting mass spectra. The simplest of these is using non-denaturing conditions, which inherently improves the detection of larger biomolecules by reducing the number of protein charge states to collapse MS signal into fewer isotopic features [105]. Similarly, proton transfer reaction (PTR) MS has been used to reduce the average number of charge states for improved signal-to-noise and reduced spectral complexity for the identification of protein complex mixtures [106]. Arguably, one of the most important recent developments is charge detection mass spectrometry (CDMS) (also referred to as individual ion MS) for top-down proteomics [107–110]. CDMS analysis of intact proteins is achieved by simultaneously measuring m/z and z for individual ions contained in an electrostatic ion trap from the frequency and amplitude of the resulting signal, which can then be converted into a mass measurement [111]. The major advantage of CDMS is the analysis of very large biomolecules including virus particles (2.47 MDa) and highly complex intact protein mixtures [108,109]. Additionally, Kafader et. al demonstrated increase sensitivity and sequence coverage when employing CDMS of intact protein analysis [107].
1.2.5. Protein Fragmentation for Sequence Determination
A significant benefit of top-down proteomics is the comprehensive characterization of the protein primary sequence. Effective ion activation for MS/MS analysis is essential for characterizing proteoforms [112]. Generally, data acquisition in top-down on an LC-MS timescale is performed using data-dependent acquisition (DDA) wherein the topmost intense peaks in the initial MS scan are selected for fragmentation [113]. Collisionally activated dissociation (CAD) and higher-energy collisional dissociation (HCD) are the most prevalent and robust techniques for protein fragmentation especially for global top-down proteomics studies [100,113]. Using CAD or HCD, ions of interest are accelerated and collide with inert gas (nitrogen) breaking the most labile bonds. The result is a series of b/y ions due to bond breakage between amides (the “peptide bond”) along the peptide backbone. However, because CAD and HCD often break the most labile bonds, it may cleave labile PTMs (e.g., phosphorylation) and cause unwanted neutral loss of water and ammonium [112].
Electron capture dissociation (ECD) [114] and electron transfer dissociation (ETD) [115], on the other hand, are nonergodic techniques for protein fragmentation. Fragmentation by ECD and ETD cleave between the secondary amine and α-carbon resulting in c/z ions. Breakages along the protein backbone occur more randomly enabling higher sequence coverage while preserving labile PTMs. However, the slower reaction kinetics, protein-to-protein variability, and charge state dependence of these techniques limit their application for global proteomic analysis[112]. Recently, electron ionization dissociation (EID), which uses high energy electrons for fragmentation, has shown promise for improved internal fragmentation for higher sequence coverage [116].
Ultraviolet photodissociation (UVPD) also preserves labile modification, but unlike ECD and ETD, it is charge state independent [20]. This UVPD approach uses high-energy photons that translates into an array of protein fragment ions. Predominately, UVPD breaks the bond between the alpha carbon and the carbonyl, resulting in a/x ions, but also can produce b/y and c/z ions. The diversity of bond breakages afforded by UVPD makes it ideal for deep sequence coverage, but higher numbers of protein identifications are still obtained using HCD for global top-down analysis by LC-MS [117].
Hybrid dissociation technique like electron-transfer dissociation/higher-energy collisional dissociation (EThcD) [118,119] and activated ion-election transfer dissociation (AI-ETD) [19,120] show great promise for intact protein characterization. Generally, these techniques involve activation of protein ions with collisional energy or photon bombardment before ion-ion reaction for improved fragmentation [112]. For example, Riley et al. demonstrated AI-ETD could identify 935 proteoforms while HCD, ETD, and EThcD identified 1014 proteoforms, 915, and 817, respectively [121]. Importantly, AI-ETD generally outperformed HCD, ETD, and EThCD in terms of sequence coverage and proteoform characterization scores.
Beyond primary sequence coverage, MS/MS analysis has been utilized to study protein complexes [122] and protein-ligand binding [123]. For example, Gault et al. demonstrated the use of MS4 analysis to identify both the proteoform state and the associated ligand structure [123]. Using the ‘Nativeomics’ approach, endogenous ligands such as lipids and even drug binding could be identified as demonstrated for several membrane proteins. Overall, the growing number of different fragmentation tools available has enabled high protein sequence coverage, improved global proteoform characterization, and characterization of higher-order protein structure [112].
1.2.6. Data Acquisition and Analysis
After acquisition, complex spectra are generated from the fragmentation of protein ions resulting in many isotopic (possibly overlapping) clusters. Thus, data analysis software is critical for interpreting spectra. Thorough High-Resolution Analysis of Spectra by Horn (THRASH) was the first software to automate the deconvolution of high-resolution mass spectra and is still widely used [124]. THRASH uses a theoretical isotopomer envelope based on the average elemental composition of amino acids (averagine) [125] to compare to the experimental envelopes. A statistical score is then assigned based on the overlap of the experimental and theoretical fit. On the other hand, MS-Deconv generates a large set of candidate isotopomer envelopes and scores sets of envelopes rather than a single fit [126]. UniDec was developed to handle highly heterogeneous spectra, which has been critical for the interpretation of native protein mass spectra [127]. Recently, FLASHDeconv used pattern matching to quickly deconvolute MS with speeds orders of magnitudes higher than conventional approaches [128]. Other deconvolution strategies have been developed including TopFD [129], pParseTD [130], and SNAP (a vendor-specific tool). Recently, combining deconvolution results from a single data set using a simple voting ensemble method or random forest machine learning-enabled improved fragment identification and confidence compared to a single algorithm [131]. After deconvolution is used to create a fragment mass list, there are several software tools for protein identification. Examples of searching software include ProSight PC [132], MS-Align [133], TopPIC [129], pTop [134], Mascot Top Down [135], MS-TopDown [136], Promex [137], and Proteoform Suite [138]. To take advantage of the unique aspects of several of these algorithms, recently the Ge group developed MASH Explorer, which integrates multiple spectral deconvolution and database searching algorithms into a single, freely-available universal platform that can process top-down proteomics data from various vendor formats [139].
1.3. Application in Disease Research
MS-based proteomics has become a staple in biomedical research for the discovery of novel biomarkers and elucidation of disease mechanisms [140]. Commonly, proteomics is used to examine changes in the protein expression levels or the relative abundance of various PTMs between healthy and diseased states. For example, in the heart, protein isoform switching [141], and PTMs are known to regulate cardiac contraction and relaxation, as well as play a role in the progression of heart failure [61,142,143]. Additionally, decreased level of cTnI phosphorylation was found in hearts with increased chronic heart failure [61] and circulating cTnI released into the blood upon myocardial infarction is a known cardiac protein biomarker [144–146]. Using the previously discussed NP-based method, Tiambeng and Roberts et al. demonstrated that different low-abundance cTnI proteoforms could be enriched directly from human serum and characterized by top-down MS, showing promise for improved clinical diagnosis [68]. Notably, the use of top-down proteomics to identify characteristic PTM or isoform profiles from low-abundance proteins shows promise as a “molecular fingerprint” with the potential to serve as the next-generation biomarkers.
Hypertrophic cardiomyopathy is a commonly heritable cardiovascular disease that results in the thickening of the ventricular septum, which results in obstructive heart failure if left untreated [147]. While around 1,400 mutations have been identified in 11 genes related to the sarcomere, the molecular events leading to disease phenotype remain largely misunderstood [148]. Tucholski et al. performed a large-scale quantitative top-down proteomics study of donor (n=16) and obstructive HCM (n=16) samples to compare the proteoform landscape between the two groups (Figure 4) [149]. Since this disease is genetically heterogeneous, the authors aim to determine what the effect of different HCM causing mutations had on the sarcomeric landscape of severe cases of HCM resulting in septal myectomy. Their results convincingly show that regardless of the HCM causing mutation, convergent sarcomeric proteoforms were observed in the HCM samples compared to the donor samples. Interestingly, the authors determined a strong linear correlation of decreased phosphorylation levels of cardiac troponin I and enigma homolog 2 in the HCM samples suggesting protein crosstalk or dysregulation of signaling by cAMP-dependent protein kinase A (Figure 4) [149]. In addition, Tucholski et al. also observed highly consistent changes in the phosphorylation levels of cardiac troponin T, tropomyosin isoform 1.1, and the ventricular isoform of myosin light chain 2 in the HCM samples. This study indicates a conserved pathway is associated with clinical phenotypes in patients diagnosed with obstructive HCM, providing a new opportunity for the development of interventions that target the HCM phenotype rather than the individual genotype [149].
Figure 4.
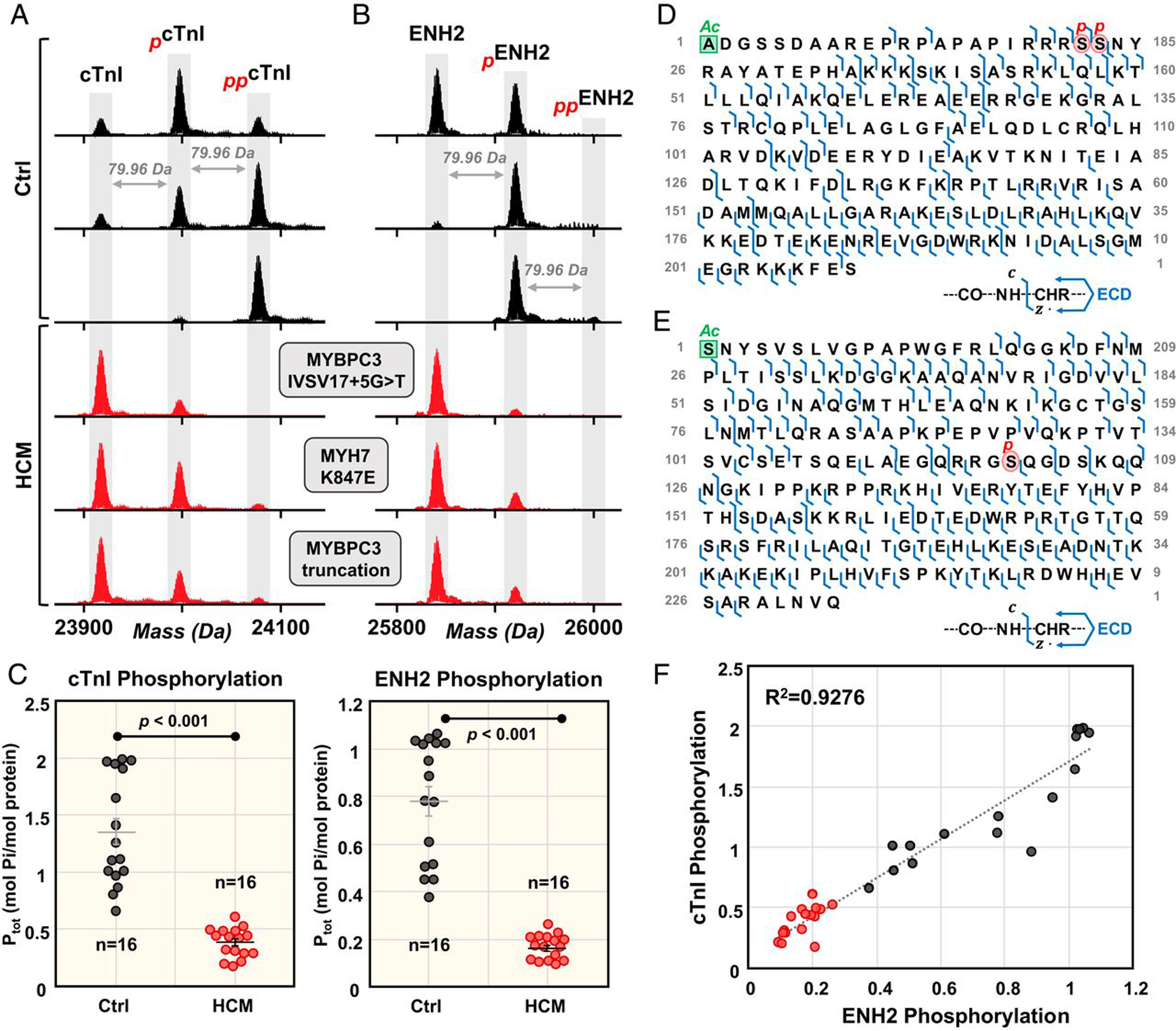
Top-down proteomics reveals a common proteoform profile in patients with hypertrophic cardiomyopathy (HCM). Coordinated decrease in cTnI and ENH2 phosphorylation in HCM tissues. Representative deconvoluted mass spectra for (A) cTnI and (B) ENH2 from donor hearts (black) and HCM tissues (red). Mono- and bis-phosphorylation are denoted by red p and pp, respectively. (C) Total protein phosphorylation (Ptot) calculated by mol Pi/mol protein for cTnI and ENH2 in Ctrl (n = 16) and HCM (n = 16). Horizontal bars represent the mean of the group and error bars represent SEM in gray for Ctrl and black for HCM. Groups were considered significantly different at P < 0.05. (D) Localization of cTnI phosphorylation to Ser22/23 and (E) localization of ENH2 phosphorylation to Ser118 by ECD. (F) Linear correlation between cTnI phosphorylation and ENH2 phosphorylation (R2 = 0.9276). Figure from reference [149]. Copyright 2020 National Academy of Sciences.
While patient tissue samples are the most relevant for disease studies, human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) offer unprecedented clinical opportunities to study heritable cardiovascular diseases, test various drug candidates, and enable cardiac regeneration and precision medicine [150]. However, a major limitation to hiPSC-CM resembles fetal CMs rather than fully matured CMs. In addition, there is a lack of comprehensive methods to measure the maturity of hiPSC-CMs. To address this challenge, Cai et al. developed an unbiased method for the assessment of hiPSC-CMs maturation by combining functional assessments and proteomics to assess CM maturation in 2-dimensional (2D) monolayer systems and 3-dimensional (3D) engineered cardiac tissues (ECTs) [151]. The top-down proteomics was employed to quantify contractile protein isoform and PTM changes between early and late-stage 2D hiPSC-CM monolayers and 3D hiPSC-ECTs (Figure 5) [151]. Distinct changes in protein isoform expression, such as a switch in expression from the atrial to the ventricular isoform of myosin light chain 1, were observed between the two maturation states. For the first time, contractile PTMs such as a decrease in alpha tropomyosin phosphorylation were discovered as promising new markers of hPSC-CMs maturation. This unbiased method enables benchmarking of hiPSC-CM maturation laying a strong foundation for uncovering the molecular pathways involved in cardiac development, drug responses and disease [151].
Figure 5.
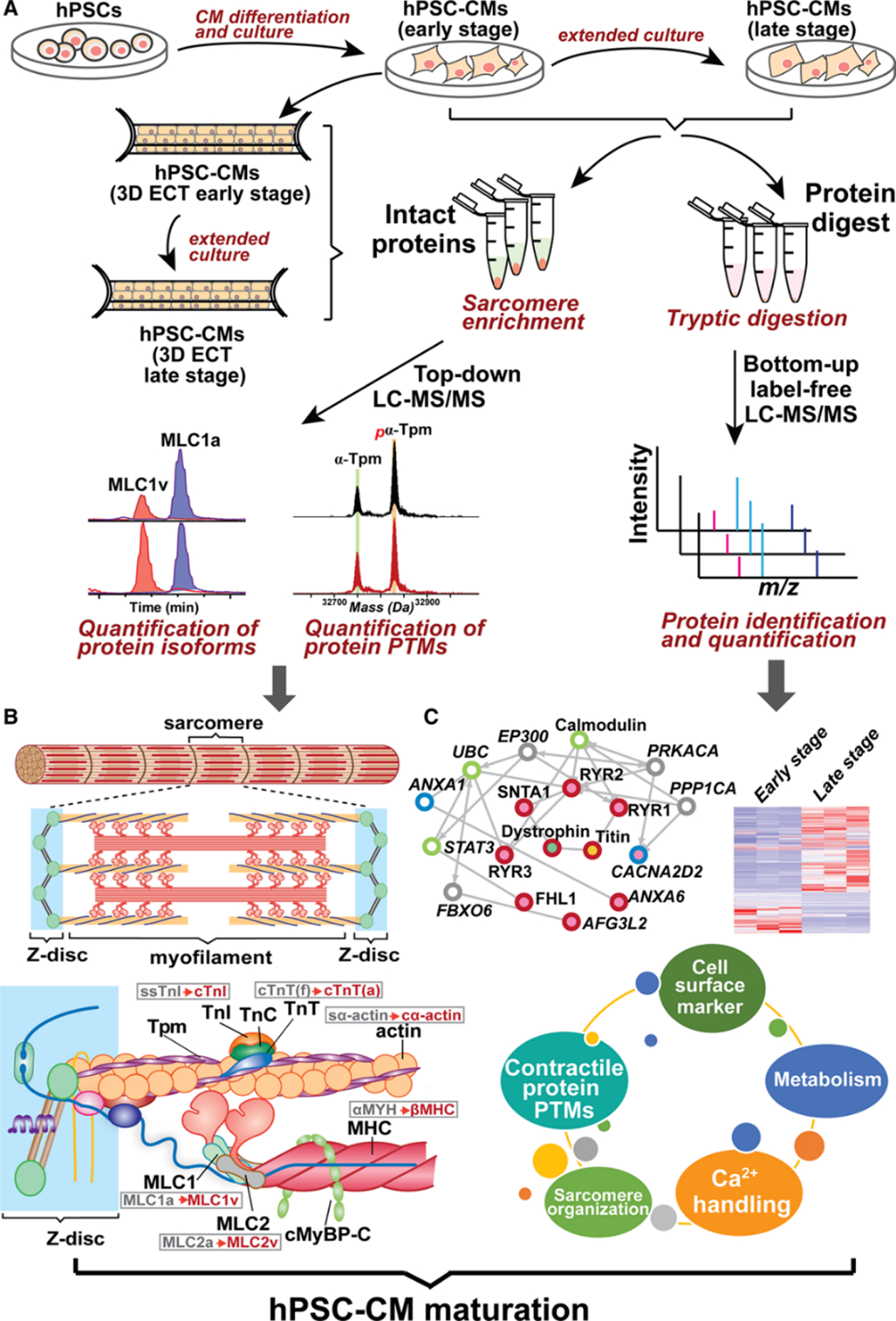
A comprehensive proteomics platform for the assessment of human pluripotent stem cell (hPSC)-cardiomyocyte (CM) maturation. A, Experimental scheme for integrated top-down and bottom-up proteomics analyses of hPSC-cardiomyocytes from early- and late-stage 2-dimensional monolayer culture and 2-dimensional (3D) engineered cardiac tissue (ECT). B, Top-down liquid chromatography (LC)-mass spectrometry (MS)–based proteomics for concurrent quantification of contractile protein isoform expression and post-translational modification (PTMs). Gray and red font represent fetal and adult isoforms, respectively, in the ventricular cardiomyocytes. C, Bottom-up label-free quantitative proteomics for determining global differential regulation of proteins. CM indicates cardiomyocyte; cMyBP-C, cardiac myosin–binding protein C; cTnI, cardiac troponin I; cTnT(f)/(a), cardiac troponin T fetal/adult isoform; cα-actin, cardiac α-actin; MHC, myosin heavy chain; MLC1a, myosin light chain 1 atrial isoform; MLC1v, myosin light chain 1 ventricular isoform; MLC2a, myosin light chain 2 atrial isoform; MLC2v, myosin light chain 2 ventricular isoform; ssTnI, slow skeletal troponin I; sα-actin, skeletal α-actin; TnC, troponin C; TnI, troponin I; TnT, troponin T; and Tpm, tropomyosin. Figure from reference [151]. Copyright © 2019, Wolters Kluwer Health
The effects of aging, or sarcopenia, has detrimental effects on skeletal muscle due to the deterioration of muscle mass [152]. However, there remains an incomplete understanding of the molecular mechanism underlying sarcopenia. Wei et. al applied a top-down proteomics strategy to study the sarcomere proteome of fast- and slow-twitch skeletal muscles of aging rats [153]. Importantly, quantitative top-down proteomics revealed age-related alterations, such as the phosphorylation levels of the Z-disk protein cypher, in the fast-twitch skeletal muscles of the aging rats [153]. This finding represented the first report of PTMs in Z-disk proteins and implies the importance of the Z-disk in the progression of sarcopenia. While this report was important to study the effects of sarcopenia, the study was limited to only two skeletal muscle tissues: gastrocnemius (fast-twitch) and soleus (slow-twitch). Melby et al. expanded upon this study by performing top-down proteomics on seven different rat skeletal muscle tissues: vastus lateralis (VL), vastus medialis (VM), vastus intermedius (VI), rectus femoris (RF), soleus (SOL), gastrocnemius (GAS), and plantaris (PLN) [154]. These skeletal muscle tissues represent a range of constituent fast-twitch (i.e., VL, GAS, RF), slow-twitch (i.e., SOL), and mix of fiber types (i.e., VI, VM, PLN) to determine if there are differences in the myofilament proteoforms found in the myriad muscle tissues. Quantitative top-down proteomics discovered that there were distinct differences in the myofilament proteoforms found across the seven various rat skeletal muscle tissues, which demonstrates the highly heterogeneous nature of skeletal muscle (Figure 6) [154]. This high throughput top-down proteomics platform is amenable for a larger scale study on the effects of sarcopenia across a large cohort of skeletal muscle tissues to further elucidate the molecular mechanisms of aging.
Figure 6.
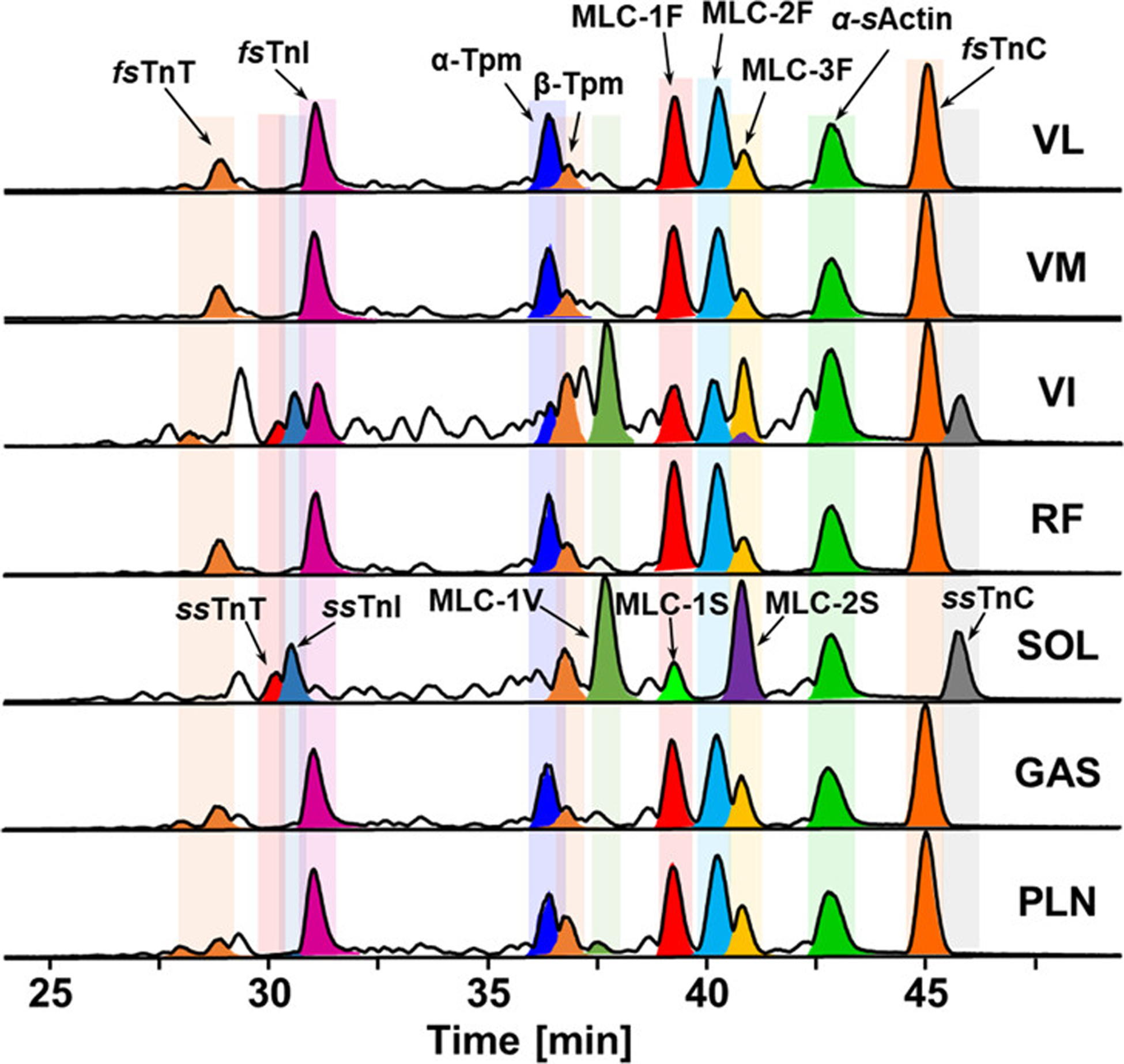
LC-MS profiling of myofilament proteins detected in different types of rat muscle. LC-MS of labeled myofilament protein base peak chromatograms (BPCs) of seven different rat skeletal muscles, VL, VM, VI, RF, SOL, GAS, and PLN. Reprinted (adapted) with permission from reference [154]. Melby JA, Jin Y, Lin Z et al. Top-Down Proteomics Reveals Myofilament Proteoform Heterogeneity among Various Rat Skeletal Muscle Tissues. Figure from reference [154] Journal of Proteome Research, 19(1), 446–454 (2020). Copyright 2019 American Chemical Society.
A range of clinical samples including serum, urine, or biopsy tissue, have been studied using top-down proteomics to identify protein profiles characteristic of a certain disease generating a more reliable biomarker [155–157]. Recently, Toby et al. developed a top-down proteomics-based protocol for the analysis of human peripheral blood mononuclear cells [157]. Important studies such as this provide standardized protocols for identifying proteoform changes in clinical samples, which provides a baseline for continued improvement, makes top-down proteomics more accessible, and increases its potential impact for biomarker discovery and clinical diagnosis. He et al. utilized ultrahigh-resolution MS on a 21 T FTICR enabling the diagnosis of hemoglobinopathy and β-thalassemia from blood with a 3 min assay [158]. Targeted top-down proteomics was employed by Ntai et al. to investigate mutation in KRAS [59]. They identified changes in PTMs (such as Cys118 nitrosylation and C-terminal methylation) using both isogenic KRAS colorectal cancer (CRC) cell lines and patient CRC tumors.
Undoubtedly, the advancement of top-down proteomics is stimulated by the commercial growth of large molecule therapeutics such as monoclonal antibodies and antibody-drug-conjugates as detailed and high-throughput methods for their characterization and quality control are required [81,159,160]. For example, analysis of antibodies by top-down can quickly determine the modification state [81], drug conjugate ratio [161], quality control screening [162], and more [163,164]. As the number of large-molecule therapeutics increases, top-down proteomics is expected to be increasingly utilized by the pharmaceutical industry.
Article highlights.
Top-down proteomics represents a powerful technology for comprehensively characterizing proteoforms for the discovery of novel biomarkers and the elucidation of disease mechanisms.
Recently, significant advancements have been made in the sample preparation for improved protein solubilization, sample clean-up, and protein/proteoform enrichment.
The development of intact protein separations has advanced the scope of top-down proteomics for global proteoform characterization.
Advancements in mass spectrometry instrumentation and fragmentation have pushed the boundaries of top-down proteomics to effectively analyze large proteoforms (>2 MDa) and complex mixtures of proteoforms.
A number of exciting applications of top-down proteomics have been demonstrated for biomedical research for better understanding and diagnosing of diseases, particularly for cardiovascular research.
Acknowledgement
Y.G. would also like to acknowledge NIH R01 HL096971 R01 GM117058, GM125085, and HL109810. K.A.B. and J.A.M would like to acknowledge support from the University of Wisconsin Cardiovascular Research Center Training Program in Translational Cardiovascular Science, T32 HL007936-19 and the University of Wisconsin Vascular Surgery Research Training Program, T32HL110853. J.A.M. would like to acknowledge support from the University of Wisconsin Cardiovascular Research Center Training Program in Translational Cardiovascular Science, T32 HL007936-20.
Footnotes
Financial & competing interest disclosure
The University of Wisconsin–Madison has filed a provisional patent application P180335US01, US serial number 62/682027 (June, 7 2018) on the basis of this work. Y.G. and K.A.B. are named as inventors on the provisional patent application. The University of Wisconsin—Madison has filed a provisional patent application serial No. 62/949,869 (December 18, 2019) on the basis of this work. Y.G. and D.S.R. are named as the inventors on the provisional patent application. The other authors declare no competing interests.
References
- 1.Smith LM, Kelleher NL. Proteoforms as the next proteomics currency. Science, 359(6380), 1106–1107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uhlén M, Fagerberg L, Hallström BM et al. Proteomics. Tissue-based map of the human proteome. Science, 347(6220), 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Smith LM, Kelleher NL, The Consortium for Top Down P et al. Proteoform: a single term describing protein complexity. Nature Methods, 10, 186 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aebersold R, Agar JN, Amster IJ et al. How many human proteoforms are there? Nat Chem Biol, 14(3), 206–214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; *An outline of mass spectrometry-based proteomics towards an understanding of the complexity of the human proteome.
- 5.Gregorich ZR, Ge Y. Top-down proteomics in health and disease: Challenges and opportunities. Proteomics, 14(10), 1195–1210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This review provides an introduction to top-down proteomics and its potential for characterizing proteoforms in health and disease.
- 6.Fert-Bober J, Murray CI, Parker SJ, Van Eyk JE. Precision Profiling of the Cardiovascular Post-Translationally Modified Proteome: Where There Is a Will, There Is a Way. Circulation research, 122(9), 1221–1237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; *An overview of studies linking various protein post-translational modifications to cardiovascular disease and how mass spectrometry-based proteomics has played a role in their discovery.
- 7.Cai WX, Tucholski TM, Gregorich ZR, Ge Y. Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert Review of Proteomics, 13(8), 717–730 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This review covers the technological advancements and applications of top-down proteomics up to 2016.
- 8.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature, 422(6928), 198–207 (2003). [DOI] [PubMed] [Google Scholar]; *An overview of mass spectrometry-based proteomics and its impact broadly on biology and medicine with a greater focus on the bottom-up approach.
- 9.Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng, 11, 49–79 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Fonslow BR, Shan B, Baek M-C, Yates JR 3rd. Protein analysis by shotgun/bottom-up proteomics. Chemical reviews, 113(4), 2343–2394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry, 5(11), 976–989 (1994). [DOI] [PubMed] [Google Scholar]
- 12.Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nature Biotechnology, 19(3), 242–247 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Toby TK, Fornelli L, Kelleher NL. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annual review of analytical chemistry, 9(1), 499–519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaffer LV, Millikin RJ, Miller RM et al. Identification and Quantification of Proteoforms by Mass Spectrometry. PROTEOMICS, 19(10), 1800361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen B, Brown KA, Lin Z, Ge Y. Top-Down Proteomics: Ready for Prime Time? Analytical Chemistry, 90(1), 110–127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Z, Jin Y, Chen B et al. Comprehensive Characterization of the Recombinant Catalytic Subunit of cAMP-Dependent Protein Kinase by Top-Down Mass Spectrometry. J Am Soc Mass Spectrom, 30(12), 2561–2570 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin Z, Guo F, Gregorich ZR et al. Comprehensive Characterization of Swine Cardiac Troponin T Proteoforms by Top-Down Mass Spectrometry. J Am Soc Mass Spectrom, 29(6), 1284–1294 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Ge Y. Comprehensive Analysis of Protein Modifications by Top-Down Mass Spectrometry. Circulation-Cardiovascular Genetics, 4(6), 711–+ (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riley NM, Westphall MS, Coon JJ. Activated Ion-Electron Transfer Dissociation Enables Comprehensive Top-Down Protein Fragmentation. Journal of Proteome Research, 16(7), 2653–2659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw JB, Li W, Holden DD et al. Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. Journal of the American Chemical Society, 135(34), 12646–12651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Speers AE, Wu CC. Proteomics of Integral Membrane ProteinsTheory and Application. Chemical Reviews, 107(8), 3687–3714 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Loo RR, Dales N, Andrews PC. Surfactant effects on protein structure examined by electrospray ionization mass spectrometry. Protein Sci, 3(11), 1975–1983 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donnelly DP, Rawlins CM, DeHart CJ et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat Methods, 16(7), 587–594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; *In this perspective, the Consortium for Top-Down Proteomics members descrobed robust protocols for mass analysis of intact proteins from mixtures of varying complexity (a decision tree) to guider researchers who are new comers to top-down proteomics.
- 24.Loo RRO, Dales N, Andrews PC. Surfactant effects on protein structure examined by electrospray ionization mass spectrometry. Protein Science, 3(11), 1975–1983 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Speers AE, Wu CC. Proteomics of integral membrane proteins--theory and application. Chem Rev, 107(8), 3687–3714 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Kachuk C, Doucette AA. The benefits (and misfortunes) of SDS in top-down proteomics. Journal of Proteomics, 175, 75–86 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Doucette AA, Vieira DB, Orton DJ, Wall MJ. Resolubilization of Precipitated Intact Membrane Proteins with Cold Formic Acid for Analysis by Mass Spectrometry. Journal of Proteome Research, 13(12), 6001–6012 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Yu YQ, Gilar M, Lee PJ, Bouvier ES, Gebler JC. Enzyme-friendly, mass spectrometry-compatible surfactant for in-solution enzymatic digestion of proteins. Anal Chem, 75(21), 6023–6028 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Saveliev SV, Woodroofe CC, Sabat G et al. Mass spectrometry compatible surfactant for optimized in-gel protein digestion. Anal Chem, 85(2), 907–914 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Chang Y-H, Gregorich ZR, Chen AJ et al. New Mass-Spectrometry-Compatible Degradable Surfactant for Tissue Proteomics. Journal of Proteome Research, 14(3), 1587–1599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown KA, Chen B, Guardado-Alvarez TM et al. A photocleavable surfactant for top-down proteomics. Nature Methods, 16, 417–420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This paper described the development of a photo-cleavable surfactant, 4-hexylphenylazosulfonate (Azo), which can be rapidly degraded by ultraviolet irradiation, for top-down proteomics. As demonstarted, Azo is MS-compatibel and could solubilize proteins effectively. The use of Azo enables the solubilization of membrane proteins for comprehensive characterization of post-translational modifications.
- 32.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol, 4(7), 566–577 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Lee S-H, Hadipour-Lakmehsari S, Murthy HR et al. REEP5 depletion causes sarco-endoplasmic reticulum vacuolization and cardiac functional defects. Nature Communications, 11(1), 965 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dautant A, Meier T, Hahn A, Tribouillard-Tanvier D, di Rago J-P, Kucharczyk R. ATP Synthase Diseases of Mitochondrial Genetic Origin. Frontiers in physiology, 9, 329–329 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He J, Ford HC, Carroll J et al. Assembly of the membrane domain of ATP synthase in human mitochondria. Proceedings of the National Academy of Sciences, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown KA, Tucholski T, Eken C et al. High‐Throughput Proteomics Enabled by a Photocleavable Surfactant. Angewandte Chemie, 132(22), 8484–8488 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y, Piehowski PD, Zhao R et al. Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nature Communications, 9(1), 882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou M, Uwugiaren N, Williams SM et al. Sensitive Top-Down Proteomics Analysis of a Low Number of Mammalian Cells Using a Nanodroplet Sample Processing Platform. Analytical Chemistry, 92(10), 7087–7095 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Lermyte F, Tsybin YO, O’Connor PB, Loo JA. Top or Middle? Up or Down? Toward a Standard Lexicon for Protein Top-Down and Allied Mass Spectrometry Approaches. Journal of the American Society for Mass Spectrometry, 30(7), 1149–1157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leney AC, Heck AJ. Native Mass Spectrometry: What is in the Name? J Am Soc Mass Spectrom, 28(1), 5–13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou M, Lantz C, Brown KA et al. Higher-order structural characterisation of native proteins and complexes by top-down mass spectrometry. Chemical Science, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blackwell AE, Dodds ED, Bandarian V, Wysocki VH. Revealing the Quaternary Structure of a Heterogeneous Noncovalent Protein Complex through Surface-Induced Dissociation. Analytical Chemistry, 83(8), 2862–2865 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crittenden CM, Novelli ET, Mehaffey MR et al. Structural Evaluation of Protein/Metal Complexes via Native Electrospray Ultraviolet Photodissociation Mass Spectrometry. Journal of the American Society for Mass Spectrometry, 31(5), 1140–1150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wongkongkathep P, Han JY, Choi TS, Yin S, Kim HI, Loo JA. Native top-down mass spectrometry and ion mobility MS for characterizing the cobalt and manganese metal binding of α-synuclein protein. Journal of the American Society for Mass Spectrometry, 29(9), 1870–1880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yen H-Y, Hoi KK, Liko I et al. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature, 559(7714), 423–427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolla JR, Agasid MT, Mehmood S, Robinson CV. Membrane Protein-Lipid Interactions Probed Using Mass Spectrometry. Annu Rev Biochem, 88, 85–111 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Urner LH, Liko I, Yen HY et al. Modular detergents tailor the purification and structural analysis of membrane proteins including G-protein coupled receptors. Nat Commun, 11(1), 564 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reading E, Liko I, Allison TM, Benesch JL, Laganowsky A, Robinson CV. The role of the detergent micelle in preserving the structure of membrane proteins in the gas phase. Angew Chem Int Ed Engl, 54(15), 4577–4581 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Laganowsky A, Reading E, Hopper JT, Robinson CV. Mass spectrometry of intact membrane protein complexes. Nat Protoc, 8(4), 639–651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barrera NP, Robinson CV. Advances in the mass spectrometry of membrane proteins: from individual proteins to intact complexes. Annu Rev Biochem, 80, 247–271 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Chorev DS, Baker LA, Wu D et al. Protein assemblies ejected directly from native membranes yield complexes for mass spectrometry. Science, 362(6416), 829 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anderson NL, Anderson NG. The Human Plasma Proteome. History, Character, and Diagnostic Prospects, 1(11), 845–867 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Thingholm TE, Jorgensen TJ, Jensen ON, Larsen MR. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat Protoc, 1(4), 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Villén J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nature Protocols, 3(10), 1630–1638 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol, 21(6), 660–666 (2003). [DOI] [PubMed] [Google Scholar]
- 56.Hwang L, Ayaz-Guner S, Gregorich ZR et al. Specific Enrichment of Phosphoproteins Using Functionalized Multivalent Nanoparticles. Journal of the American Chemical Society, 137(7), 2432–2435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen B, Hwang L, Ochowicz W et al. Coupling functionalized cobalt ferrite nanoparticle enrichment with online LC/MS/MS for top-down phosphoproteomics. Chem Sci, 8(6), 4306–4311 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roberts DS, Chen B, Tiambeng TN, Wu Z, Ge Y, Jin S. Reproducible large-scale synthesis of surface silanized nanoparticles as an enabling nanoproteomics platform: Enrichment of the human heart phosphoproteome. Nano Research, 12(6), 1473–1481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ntai I, Fornelli L, DeHart CJ et al. Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proceedings of the National Academy of Sciences, 115(16), 4140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ge Y, Rybakova IN, Xu Q, Moss RL. Top-down high-resolution mass spectrometry of cardiac myosin binding protein C revealed that truncation alters protein phosphorylation state. Proceedings of the National Academy of Sciences, 106(31), 12658 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, Guy MJ, Norman HS et al. Top-down quantitative proteomics identified phosphorylation of cardiac troponin I as a candidate biomarker for chronic heart failure. J Proteome Res, 10(9), 4054–4065 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Janes KA. Fragile epitopes—Antibody’s guess is as good as yours. Science Signaling, 13(616), eaaz8130 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baker M Reproducibility crisis: Blame it on the antibodies. In: Nature. (England, 2015) 274–276. [DOI] [PubMed] [Google Scholar]
- 64.Bradbury A, Plückthun A. Reproducibility: Standardize antibodies used in research. Nature, 518(7537), 27–29 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Zhang J, Guy MJ, Norman HS et al. Top-Down Quantitative Proteomics Identified Phosphorylation of Cardiac Troponin I as a Candidate Biomarker for Chronic Heart Failure. Journal of Proteome Research, 10(9), 4054–4065 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.La Vecchia L, Mezzena G, Zanolla L et al. Cardiac troponin I as diagnostic and prognostic marker in severe heart failure. The Journal of Heart and Lung Transplantation, 19(7), 644–652 (2000). [DOI] [PubMed] [Google Scholar]
- 67.Dong X, Sumandea CA, Chen Y-C et al. Augmented Phosphorylation of Cardiac Troponin I in Hypertensive Heart Failure. Journal of Biological Chemistry, 287(2), 848–857 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tiambeng TN, Roberts DS, Brown KA et al. Nanoproteomics enables proteoform-resolved analysis of low-abundance proteins in human serum. Nat Commun, 11(1), 3903 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This article describes the development of functionalized nanoparticles for the enrichment of low abundant cardiac troponin I (the gold standard biomarker for myocardial infarction) from serum for proteoform characterization using top-down proteomics.
- 69.Cai W, Tucholski T, Chen B et al. Top-Down Proteomics of Large Proteins up to 223 kDa Enabled by Serial Size Exclusion Chromatography Strategy. Anal Chem, 89(10), 5467–5475 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen Y, Tolić N, Piehowski PD et al. High-resolution ultrahigh-pressure long column reversed-phase liquid chromatography for top-down proteomics. J Chromatogr A, 1498, 99–110 (2017). [DOI] [PubMed] [Google Scholar]
- 71.Simone P, Pierri G, Foglia P et al. Separation of intact proteins on γ-ray-induced polymethacrylate monolithic columns: A highly permeable stationary phase with high peak capacity for capillary high-performance liquid chromatography with high-resolution mass spectrometry. J Sep Sci, 39(2), 264–271 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Liang Y, Jin Y, Wu Z et al. Bridged Hybrid Monolithic Column Coupled to High-Resolution Mass Spectrometry for Top-Down Proteomics. Analytical chemistry, 91(3), 1743–1747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gomes FP, Yates JR 3rd. Recent trends of capillary electrophoresis-mass spectrometry in proteomics research. Mass Spectrom Rev, 38(6), 445–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lubeckyj RA, McCool EN, Shen X, Kou Q, Liu X, Sun L. Single-Shot Top-Down Proteomics with Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry for Identification of Nearly 600 Escherichia coli Proteoforms. Anal Chem, 89(22), 12059–12067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carroll J, Fearnley IM, Walker JE. Definition of the mitochondrial proteome by measurement of molecular masses of membrane proteins. Proceedings of the National Academy of Sciences, 103(44), 16170 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gargano AFG, Roca LS, Fellers RT, Bocxe M, Domínguez-Vega E, Somsen GW. Capillary HILIC-MS: A New Tool for Sensitive Top-Down Proteomics. Anal Chem, 90(11), 6601–6609 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.VanAernum ZL, Busch F, Jones BJ et al. Rapid online buffer exchange for screening of proteins, protein complexes and cell lysates by native mass spectrometry. Nature Protocols, 15(3), 1132–1157 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muneeruddin K, Nazzaro M, Kaltashov IA. Characterization of intact protein conjugates and biopharmaceuticals using ion-exchange chromatography with online detection by native electrospray ionization mass spectrometry and top-down tandem mass spectrometry. Anal Chem, 87(19), 10138–10145 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Muneeruddin K, Bobst CE, Frenkel R et al. Characterization of a PEGylated protein therapeutic by ion exchange chromatography with on-line detection by native ESI MS and MS/MS. Analyst, 142(2), 336–344 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Chen B, Peng Y, Valeja SG, Xiu L, Alpert AJ, Ge Y. Online Hydrophobic Interaction Chromatography-Mass Spectrometry for Top-Down Proteomics. Anal Chem, 88(3), 1885–1891 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen B, Lin Z, Alpert AJ et al. Online Hydrophobic Interaction Chromatography–Mass Spectrometry for the Analysis of Intact Monoclonal Antibodies. Analytical Chemistry, 90(12), 7135–7138 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Regnier FE, Kim J. Proteins and Proteoforms: New Separation Challenges. Analytical chemistry, 90(1), 361–373 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Compton PD, Zamdborg L, Thomas PM, Kelleher NL. On the scalability and requirements of whole protein mass spectrometry. Anal Chem, 83(17), 6868–6874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tran JC, Zamdborg L, Ahlf DR et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature, 480(7376), 254–258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takemori A, Butcher DS, Harman VM et al. PEPPI-MS: Polyacrylamide-Gel-Based Prefractionation for Analysis of Intact Proteoforms and Protein Complexes by Mass Spectrometry. Journal of Proteome Research, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McCool EN, Lubeckyj RA, Shen X et al. Deep Top-Down Proteomics Using Capillary Zone Electrophoresis-Tandem Mass Spectrometry: Identification of 5700 Proteoforms from the Escherichia coli Proteome. Anal Chem, 90(9), 5529–5533 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu D, Wang Z, Cupp-Sutton KA, Liu X, Wu S. Deep Intact Proteoform Characterization in Human Cell Lysate Using High-pH and Low-pH Reversed-Phase Liquid Chromatography. J Am Soc Mass Spectrom, 30(12), 2502–2513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shen X, Kou Q, Guo R et al. Native Proteomics in Discovery Mode Using Size-Exclusion Chromatography-Capillary Zone Electrophoresis-Tandem Mass Spectrometry. Anal Chem, 90(17), 10095–10099 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Skinner OS, Haverland NA, Fornelli L et al. Top-down characterization of endogenous protein complexes with native proteomics. Nature Chemical Biology, 14(1), 36–41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gargano AFG, Shaw JB, Zhou M et al. Increasing the Separation Capacity of Intact Histone Proteoforms Chromatography Coupling Online Weak Cation Exchange-HILIC to Reversed Phase LC UVPD-HRMS. J Proteome Res, 17(11), 3791–3800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baghdady YZ, Schug KA. Online Comprehensive High pH Reversed Phase × Low pH Reversed Phase Approach for Two-Dimensional Separations of Intact Proteins in Top-Down Proteomics. Anal Chem, 91(17), 11085–11091 (2019). [DOI] [PubMed] [Google Scholar]
- 92.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science, 246(4926), 64 (1989). [DOI] [PubMed] [Google Scholar]
- 93.Susa AC, Xia Z, Williams ER. Small Emitter Tips for Native Mass Spectrometry of Proteins and Protein Complexes from Nonvolatile Buffers That Mimic the Intracellular Environment. Analytical Chemistry, 89(5), 3116–3122 (2017). [DOI] [PubMed] [Google Scholar]
- 94.Kocurek KI, Stones L, Bunch J, May RC, Cooper HJ. Top-Down LESA Mass Spectrometry Protein Analysis of Gram-Positive and Gram-Negative Bacteria. Journal of the American Society for Mass Spectrometry, 28(10), 2066–2077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mikhailov VA, Griffiths RL, Cooper HJ. Liquid extraction surface analysis for native mass spectrometry: Protein complexes and ligand binding. International Journal of Mass Spectrometry, 420, 43–50 (2017). [Google Scholar]
- 96.Valeja SG, Kaiser NK, Xian F, Hendrickson CL, Rouse JC, Marshall AG. Unit mass baseline resolution for an intact 148 kDa therapeutic monoclonal antibody by Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem, 83(22), 8391–8395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mamyrin BA. Time-of-flight mass spectrometry (concepts, achievements, and prospects). International Journal of Mass Spectrometry, 206(3), 251–266 (2001). [Google Scholar]
- 98.Fornelli L, Durbin KR, Fellers RT et al. Advancing Top-down Analysis of the Human Proteome Using a Benchtop Quadrupole-Orbitrap Mass Spectrometer. J Proteome Res, 16(2), 609–618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hebert AS, Richards AL, Bailey DJ et al. The one hour yeast proteome. Mol Cell Proteomics, 13(1), 339–347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Durbin KR, Fornelli L, Fellers RT, Doubleday PF, Narita M, Kelleher NL. Quantitation and Identification of Thousands of Human Proteoforms below 30 kDa. Journal of proteome research, 15(3), 976–982 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Makarov A, Denisov E. Dynamics of ions of intact proteins in the Orbitrap mass analyzer. J Am Soc Mass Spectrom, 20(8), 1486–1495 (2009). [DOI] [PubMed] [Google Scholar]
- 102.Shaw JB, Brodbelt JS. Extending the isotopically resolved mass range of Orbitrap mass spectrometers. Anal Chem, 85(17), 8313–8318 (2013). [DOI] [PubMed] [Google Scholar]
- 103.Fort KL, van de Waterbeemd M, Boll D et al. Expanding the structural analysis capabilities on an Orbitrap-based mass spectrometer for large macromolecular complexes. Analyst, 143(1), 100–105 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJR. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nature Methods, 9(11), 1084–1086 (2012). [DOI] [PubMed] [Google Scholar]
- 105.Kafader JO, Melani RD, Schachner LF et al. Native vs Denatured: An in Depth Investigation of Charge State and Isotope Distributions. Journal of the American Society for Mass Spectrometry, 31(3), 574–581 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huguet R, Mullen C, Srzentić K et al. Proton Transfer Charge Reduction Enables High-Throughput Top-Down Analysis of Large Proteoforms. Analytical Chemistry, 91(24), 15732–15739 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kafader JO, Durbin KR, Melani RD et al. Individual Ion Mass Spectrometry Enhances the Sensitivity and Sequence Coverage of Top-Down Mass Spectrometry. Journal of Proteome Research, 19(3), 1346–1350 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kafader JO, Melani RD, Durbin KR et al. Multiplexed mass spectrometry of individual ions improves measurement of proteoforms and their complexes. Nature Methods, 17(4), 391–394 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wörner TP, Snijder J, Bennett A, Agbandje-McKenna M, Makarov AA, Heck AJR. Resolving heterogeneous macromolecular assemblies by Orbitrap-based single-particle charge detection mass spectrometry. Nature Methods, 17(4), 395–398 (2020). [DOI] [PubMed] [Google Scholar]
- 110.Harper CC, Elliott AG, Oltrogge LM, Savage DF, Williams ER. Multiplexed Charge Detection Mass Spectrometry for High-Throughput Single Ion Analysis of Large Molecules. Analytical Chemistry, 91(11), 7458–7465 (2019). [DOI] [PubMed] [Google Scholar]
- 111.Kafader JO, Beu SC, Early BP et al. STORI Plots Enable Accurate Tracking of Individual Ion Signals. Journal of The American Society for Mass Spectrometry, 30(11), 2200–2203 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Macias LA, Santos IC, Brodbelt JS. Ion Activation Methods for Peptides and Proteins. Analytical Chemistry, 92(1), 227–251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Catherman AD, Durbin KR, Ahlf DR et al. Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence. Molecular & cellular proteomics : MCP, 12(12), 3465–3473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zubarev RA, Kelleher NL, McLafferty FW. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. Journal of the American Chemical Society, 120(13), 3265–3266 (1998). [Google Scholar]
- 115.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America, 101(26), 9528 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zenaidee M, Lantz C, Perkins T et al. Internal Fragments Generated by Electron Ionization Dissociation Enhances Protein Top-down Mass Spectrometry. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cleland TP, DeHart CJ, Fellers RT et al. High-Throughput Analysis of Intact Human Proteins Using UVPD and HCD on an Orbitrap Mass Spectrometer. J Proteome Res, 16(5), 2072–2079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brunner AM, Lössl P, Liu F et al. Benchmarking Multiple Fragmentation Methods on an Orbitrap Fusion for Top-down Phospho-Proteoform Characterization. Analytical Chemistry, 87(8), 4152–4158 (2015). [DOI] [PubMed] [Google Scholar]
- 119.Gomes FP, Diedrich JK, Saviola AJ, Memili E, Moura AA, Yates JR 3rd. EThcD and 213 nm UVPD for Top-Down Analysis of Bovine Seminal Plasma Proteoforms on Electrophoretic and Chromatographic Time Frames. Anal Chem, 92(4), 2979–2987 (2020). [DOI] [PubMed] [Google Scholar]
- 120.Riley NM, Westphall MS, Coon JJ. Sequencing Larger Intact Proteins (30–70 kDa) with Activated Ion Electron Transfer Dissociation. Journal of the American Society for Mass Spectrometry, 29(1), 140–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Riley NM, Sikora JW, Seckler HS et al. The Value of Activated Ion Electron Transfer Dissociation for High-Throughput Top-Down Characterization of Intact Proteins. Analytical Chemistry, 90(14), 8553–8560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; *The first demonstration of activated ion-electron transfer dissociation (AI-ETD) to LC-MS/MS for the characterization of intact proteins enabling improved proteoform sequence coverage and confidence scoring compared to conventional fragmentation approaches.
- 122.Greisch J-F, Tamara S, Scheltema RA et al. Expanding the mass range for UVPD-based native top-down mass spectrometry. Chemical Science, 10(30), 7163–7171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gault J, Liko I, Landreh M et al. Combining native and ‘omics’ mass spectrometry to identify endogenous ligands bound to membrane proteins. Nat Methods, 17(5), 505–508 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This work introduced ‘nativeomics’, a top-down technique wherein both proteoform information and the identification of ligands (lipids, drugs, etc.) bound to proteins can be determined using native mass spectrometry.
- 124.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. Journal of the American Society for Mass Spectrometry, 11(4), 320–332 (2000). [DOI] [PubMed] [Google Scholar]
- 125.Senko MW, Beu SC, McLaffertycor FW. Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. Journal of the American Society for Mass Spectrometry, 6(4), 229–233 (1995). [DOI] [PubMed] [Google Scholar]
- 126.Liu X, Inbar Y, Dorrestein PC et al. Deconvolution and database search of complex tandem mass spectra of intact proteins: a combinatorial approach. Molecular & cellular proteomics : MCP, 9(12), 2772–2782 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Marty MT, Baldwin AJ, Marklund EG, Hochberg GKA, Benesch JLP, Robinson CV. Bayesian Deconvolution of Mass and Ion Mobility Spectra: From Binary Interactions to Polydisperse Ensembles. Analytical Chemistry, 87(8), 4370–4376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jeong K, Kim J, Gaikwad M et al. FLASHDeconv: Ultrafast, High-Quality Feature Deconvolution for Top-Down Proteomics. Cell Syst, 10(2), 213–218.e216 (2020). [DOI] [PubMed] [Google Scholar]
- 129.Kou Q, Xun L, Liu X. TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics, 32(22), 3495–3497 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yuan ZF, Liu C, Wang HP et al. pParse: a method for accurate determination of monoisotopic peaks in high-resolution mass spectra. Proteomics, 12(2), 226–235 (2012). [DOI] [PubMed] [Google Scholar]
- 131.McIlwain SJ, Wu Z, Wetzel M et al. Enhancing Top-Down Proteomics Data Analysis by Combining Deconvolution Results through a Machine Learning Strategy. Journal of the American Society for Mass Spectrometry, 31(5), 1104–1113 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.LeDuc RD, Taylor GK, Kim YB et al. ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res, 32(Web Server issue), W340–345 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu X, Hengel S, Wu S, Tolić N, Pasa-Tolić L, Pevzner PA. Identification of Ultramodified Proteins Using Top-Down Tandem Mass Spectra. Journal of Proteome Research, 12(12), 5830–5838 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sun R-X, Luo L, Wu L et al. pTop 1.0: A High-Accuracy and High-Efficiency Search Engine for Intact Protein Identification. Analytical Chemistry, 88(6), 3082–3090 (2016). [DOI] [PubMed] [Google Scholar]
- 135.Karabacak NM, Li L, Tiwari A et al. Sensitive and Specific Identification of Wild Type and Variant Proteins from 8 to 669 kDa Using Top-down Mass Spectrometry. Molecular & Cellular Proteomics, 8(4), 846 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Frank AM, Bandeira N, Shen Z et al. Clustering millions of tandem mass spectra. J Proteome Res, 7(1), 113–122 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Park J, Piehowski PD, Wilkins C et al. Informed-Proteomics: open-source software package for top-down proteomics. Nature Methods, 14, 909 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cesnik AJ, Shortreed MR, Schaffer LV et al. Proteoform Suite: Software for Constructing, Quantifying, and Visualizing Proteoform Families. J Proteome Res, 17(1), 568–578 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wu Z, Roberts DS, Melby JA et al. MASH Explorer: A Universal Software Environment for Top-Down Proteomics. Journal of Proteome Research, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cai W, Tucholski TM, Gregorich ZR, Ge Y. Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert review of proteomics, 13(8), 717–730 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen Y-C, Ayaz-Guner S, Peng Y et al. Effective top-down LC/MS+ method for assessing actin isoforms as a potential cardiac disease marker. Analytical chemistry, 87(16), 8399–8406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Peng Y, Gregorich ZR, Valeja SG et al. Top-down Proteomics Reveals Concerted Reductions in Myofilament and Z-disc Protein Phosphorylation after Acute Myocardial Infarction. Mol Cell Proteomics, 13(10), 2752–2764 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.de Tombe PP, Solaro RJ. Integration of cardiac myofilament activity and regulation with pathways signaling hypertrophy and failure. Ann Biomed Eng, 28(8), 991–1001 (2000). [DOI] [PubMed] [Google Scholar]
- 144.Katrukha AG, Bereznikova AV, Esakova TV et al. Troponin I is released in bloodstream of patients with acute myocardial infarction not in free form but as complex. Clinical Chemistry, 43(8), 1379 (1997). [PubMed] [Google Scholar]
- 145.Soetkamp D, Raedschelders K, Mastali M, Sobhani K, Bairey Merz CN, Van Eyk J. The continuing evolution of cardiac troponin I biomarker analysis: from protein to proteoform. Expert Review of Proteomics, 14(11), 973–986 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Labugger R, Organ L, Collier C, Atar D, Van Eyk Jennifer E. Extensive Troponin I and T Modification Detected in Serum From Patients With Acute Myocardial Infarction. Circulation, 102(11), 1221–1226 (2000). [DOI] [PubMed] [Google Scholar]
- 147.Maron BJ. Clinical Course and Management of Hypertrophic Cardiomyopathy. N Engl J Med, 379(7), 655–668 (2018). [DOI] [PubMed] [Google Scholar]
- 148.Yotti R, Seidman CE, Seidman JG. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu Rev Genomics Hum Genet, 20, 129–153 (2019). [DOI] [PubMed] [Google Scholar]
- 149.Tucholski T, Cai W, Gregorich ZR et al. Distinct hypertrophic cardiomyopathy genotypes result in convergent sarcomeric proteoform profiles revealed by top-down proteomics. Proceedings of the National Academy of Sciences, 117(40), 24691 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; *This paper reported the use of top-down proteomics to analyze surgical heart tissue samples from hypertrophic cardiomyopathy (HCM) patients with severe outflow track obstruction, which revealed a common pattern of altered sarcomeric proteoforms across HCM tissues regardless of genotypes, opening the door for the development of interventions that target the HCM phenotype rather than the individual sarcomeric gene mutation.
- 150.Chen IY, Matsa E, Wu JC. Induced pluripotent stem cells: at the heart of cardiovascular precision medicine. Nature reviews. Cardiology, 13(6), 333–349 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cai W, Zhang J, de Lange WJ et al. An Unbiased Proteomics Method to Assess the Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes. Circ Res, 125(11), 936–953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Thompson LV. Age-related muscle dysfunction. Exp Gerontol, 44(1–2), 106–111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wei L, Gregorich ZR, Lin Z et al. Novel Sarcopenia-related Alterations in Sarcomeric Protein Post-translational Modifications (PTMs) in Skeletal Muscles Identified by Top-down Proteomics. Mol Cell Proteomics, 17(1), 134–145 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Melby JA, Jin Y, Lin Z et al. Top-Down Proteomics Reveals Myofilament Proteoform Heterogeneity among Various Rat Skeletal Muscle Tissues. Journal of Proteome Research, 19(1), 446–454 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Azad NS, Rasool N, Annunziata CM, Minasian L, Whiteley G, Kohn EC. Proteomics in Clinical Trials and Practice. Molecular & Cellular Proteomics, 5(10), 1819 (2006). [DOI] [PubMed] [Google Scholar]
- 156.Petricoin EF, Ardekani AM, Hitt BA et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet, 359(9306), 572–577 (2002). [DOI] [PubMed] [Google Scholar]
- 157.Toby TK, Fornelli L, Srzentić K et al. A comprehensive pipeline for translational top-down proteomics from a single blood draw. Nature Protocols, 14(1), 119–152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.He L, Rockwood AL, Agarwal AM et al. Diagnosis of Hemoglobinopathy and β-Thalassemia by 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometry and Tandem Mass Spectrometry of Hemoglobin from Blood. Clin Chem, 65(8), 986–994 (2019). [DOI] [PubMed] [Google Scholar]
- 159.Shaw JB, Liu W, Vasil′ ev YV et al. Direct Determination of Antibody Chain Pairing by Top-down and Middle-down Mass Spectrometry Using Electron Capture Dissociation and Ultraviolet Photodissociation. Analytical Chemistry, 92(1), 766–773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jin Y, Lin Z, Xu Q et al. Comprehensive characterization of monoclonal antibody by Fourier transform ion cyclotron resonance mass spectrometry. mAbs, 11(1), 106–115 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Larson EJ, Zhu Y, Wu Z et al. Rapid Analysis of Reduced Antibody Drug Conjugate by Online LC-MS/MS with Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Analytical Chemistry, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sawyer WS, Srikumar N, Carver J et al. High-throughput antibody screening from complex matrices using intact protein electrospray mass spectrometry. Proceedings of the National Academy of Sciences, 117(18), 9851 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li W, Srikumar N, Forrest WF et al. Characterizing and Quantitating Therapeutic Tethered Multimeric Antibody Degradation Using Affinity Capture Mass Spectrometry. Analytical Chemistry, 92(10), 6839–6843 (2020). [DOI] [PubMed] [Google Scholar]
- 164.Kellie JF, Tran JC, Jian W et al. Intact Protein Mass Spectrometry for Therapeutic Protein Quantitation, Pharmacokinetics, and Biotransformation in Preclinical and Clinical Studies: An Industry Perspective. Journal of the American Society for Mass Spectrometry, (2020). [DOI] [PubMed] [Google Scholar]