

SUMMARY
We examined the role of repeat expansions in the pathogenesis of frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) by analyzing whole-genome sequence data from 2,442 FTD/ALS patients, 2,599 Lewy body dementia (LBD) patients, and 3,158 neurologically healthy subjects. Pathogenic expansions (range: 40 to 64 CAG repeats) in the huntingtin (HTT) gene were found in three (0.12%) patients diagnosed with pure FTD/ALS syndromes, but were not present in the LBD or healthy cohorts. We replicated our findings in an independent collection of 3,674 FTD/ALS patients. Postmortem evaluations of two patients revealed the classical TDP-43 pathology of FTD/ALS, as well as huntingtin-positive, ubiquitin-positive aggregates in the frontal cortex. The neostriatal atrophy that pathologically defines Huntington’s disease was absent in both cases. Our findings reveal an etiological relationship between HTT repeat expansions and FTD/ALS syndromes, and indicate that genetic screening of FTD/ALS patients for HTT repeat expansions should be considered.
Keywords: amyotrophic lateral sclerosis, frontotemporal dementia, Lewy body dementia, Huntington’s disease, whole-genome sequencing, huntingtin, repeat expansions
Graphical Abstract
eTOC
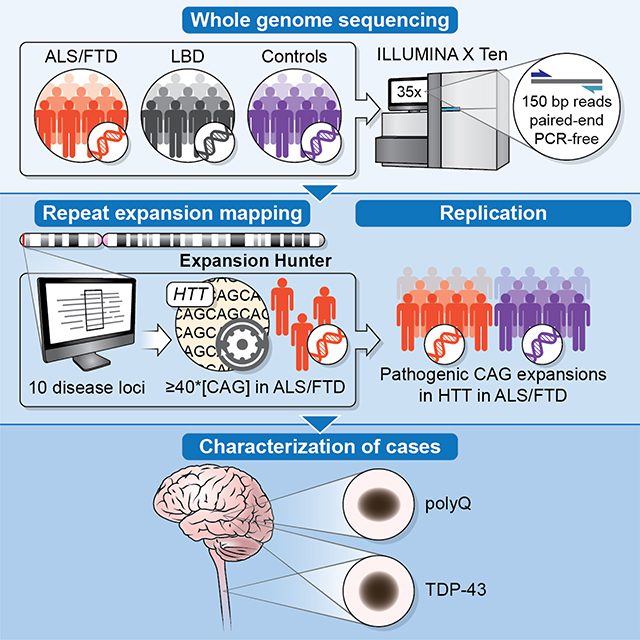
Using large-scale whole-genome sequencing, Dewan et al identifies pathogenic HTT repeat expansions in patients diagnosed with FTD/ALS neurodegenerative disorders. Autopsies confirm the TDP-43 pathology expected in FTD/ALS and show polyglutamine inclusions within the frontal cortices, but no striatal degeneration. These data broaden the phenotype resulting from HTT repeat expansions.
INTRODUCTION
Frontotemporal dementia (FTD, OMIM #600274) and amyotrophic lateral sclerosis (ALS, OMIM #105400) are progressive neurological disorders that are characterized clinically by cognitive deficits, language abnormalities, and muscle weakness (Neary et al., 2005; Rowland and Shneider, 2001). These aggressive illnesses typically occur between the ages of 40 and 70, leading to death within three to eight years of symptom onset (Chiò et al., 2013; Neary et al., 2005). Approximately 15,000 individuals die of FTD or ALS in the United States annually (Arthur et al., 2016), and there are no treatments that halt the degenerative process. Clinical, genetic, and neuropathologic data demonstrate that FTD and ALS are closely related conditions that exist along a spectrum of neurological disease (Lillo and Hodges, 2009).
Though progress has been made, much remains unclear about the genetic etiology of the FTD/ALS spectrum. Approximately 40% of FTD cases are familial, and causative mutations have been identified, most notably in MAPT, GRN, C9orf72, and VCP (Ferrari et al., 2019). In ALS, 10% of patients report a family history of the disease. The genetic etiology is known for two-thirds of these familial cases, whereas the underlying gene is recognized in 10% of sporadic cases (Chia et al., 2018; Renton et al., 2014). The intronic repeat expansion of the C9orf72 gene is the most common cause of FTD and ALS (Majounie et al., 2012). Other repeat expansions have been implicated in neurological diseases. These include polyglutamine repeats observed in Huntington’s disease (McDonald et al., 1993) and spinobulbar muscular atrophy (La Spada et al., 1991), and more complex expansions in the RFC1 gene that were recently associated with autosomal recessive cerebellar ataxia (Cortese et al., 2019). Together, these data suggest that repeat expansions play a critical role in the pathogenesis of neurodegenerative diseases. This type of mutation may be amenable to antisense oligonucleotide therapy, adding further incentive to their identification (Tabrizi et al., 2019).
The discovery of new genetic causes of FTD and ALS provides insights into the cellular mechanisms of neurodegeneration (Renton et al., 2014). From a clinical perspective, the molecular characterization of the genetic causes of disease in a patient helps to establish an accurate diagnosis and the genetic counseling of the patients and their families. It is also a necessary first step towards future precision medicine. To explore the genetic architecture of neurodegenerative disorders, we performed whole-genome sequencing in patients diagnosed with FTD/ALS, LBD, and neurologically healthy individuals and systematically assessed the role of previously identified, disease-causing repeat expansions.
RESULTS
Repeat analysis identifies FTD/ALS patients harboring HTT CAG repeat expansions
After quality control, whole-genome sequence data from 2,442 patients diagnosed with FTD/ALS, 2,599 LBD patients, and 3,158 neurologically healthy individuals were available for analysis. We used the ExpansionHunter – Targeted tool to assess ten repeat expansion motifs that have been previously associated with neurological disease and have been experimentally validated for this algorithm (Dolzhenko et al., 2019; Dolzhenko et al., 2017). The examined genes included AR, ATN1, ATXN1, ATXN3, C9orf72, DMPK, FMR1, FXN, HTT, and PHOX2B (Table S1). Consistent with the previous publications (Dolzhenko et al., 2019; Dolzhenko et al., 2017), we found excellent concordance between the identification of the known C9orf72 repeat expansion using the ExpansionHunter – Targeted algorithm and a repeat-primed PCR assay for C9orf72 (Majounie et al., 2012) among our samples (n = 7 (0.3%) discordance out of 2,147 samples with information for both assays, kappa = 0.974).
We identified three FTD/ALS patients who carried full-penetrance pathogenic repeat expansions (≥ 40) in the HTT gene, representing 0.12% of the patients diagnosed with FTD/ALS disorders. The sizes of the detected HTT expansions were 41, 40, and 40 repeats, which are deemed to be fully-penetrant (Table 1). In contrast, none of the LBD cases or healthy control subjects carried pathogenic HTT expansions. The lengths of the repeat expansions in these three FTD/ALS patients were confirmed by repeat-primed PCR and by cloning and Sanger sequencing (Figure 1A and Figure S1). We did not observe a higher rate of intermediate (27–35) or low-penetrance (36–39) HTT repeat expansions among patients diagnosed with FTD/ALS or LBD compared to control subjects (Figure 1B). Aside from C9orf72 as a common cause of FTD and ALS, none of the other repeat expansions evaluated by the ExpansionHunter - Targeted algorithm displayed a similar pattern of being present in cases and absent in control subjects (see Table S1). For this reason, we focused our efforts on the HTT repeat expansion in FTD/ALS patients.
Table 1.
Pathogenic HTT repeat expansions within the discovery and replication cohorts
| Discovery cohort | Replication cohorta | |||
|---|---|---|---|---|
| Number carriers/number screened | Rate | Number carriers/number screened | Rate | |
| FTD/ALSb | 3/2442 | 0.1% | 5/3674 | 0.1% |
| LBD | 0/2,599 | 0 | - | - |
| Controls | 0/3,158 | 0 | 10/31,583 | 0.03% |
The replication control-cohort included 210 neurologically-healthy controls, 13,670 population controls from Gardiner et al., 2019 (Gardiner et al., 2019), and 17,703 neurologically-healthy individuals from the UK 100K Genomes Project. The replication case-cohort included 1,236 samples analyzed by repeat-primed PCR for HTT repeats and 2,648 samples analyzed by nextgeneration sequencing. All samples that whole-genome sequencing predicted to possess 40 or more CAG repeats were verified by repeat-primed PCR and by cloning and Sanger sequencing.
Within the FTD/ALS discovery cohort, 3/1,377 (0.2%) FTD patients and 0/1,065 ALS patients carried a pathogenic HTT repeat expansion. Within the FTD/ALS replication cohort, 2/1,009 (0.2%) FTD patients and 3/2,665 (0.1%) ALS patients carried a pathogenic HTT repeat expansion.
Figure 1. Repeat sizing analysis identifies FTD/ALS patients harboring HTT CAG repeat expansions.

(A) An ideogram of chromosome 4 showing the HTT gene location at 4p16.3, the gene transcript (exon 1 in red), and representative repeat-primed PCR chromatograms depicting wild-type and HTT CAG repeat expansions. (B) The distributions of HTT CAG repeat expansions in the FTD/ALS (n = 2,442), LBD (n = 2,599), and control (n = 3,158) discovery cohorts. Insets are magnified views showing the number of cases carrying CAG repeat expansions ε 36 repeats. (C) Ages at symptom onset among FTD/ALS patients versus the size of their HTT repeat expansions. The curve represents the estimated onset age and corresponding standard deviation based on the number of CAG repeats (Langbehn et al., 2010). (D) The allelic structure of patients carrying HTT repeat expansions. The pathogenic repeat sequence is represented by [CAG]n, where n corresponds to the number of repeats. The trailing CAG-CAA glutamine sequence, the CCG-CCA proline sequence, and the [CCT]n codons are also shown (Ciosi et al., 2019). See also Figures S1, S2, S3, and S6.
To replicate our findings, we assessed the HTT CAG repeat length in an independent cohort of 3,674 patients diagnosed with FTD/ALS spectrum disorders and 210 healthy control participants. We detected an additional five patients diagnosed with FTD/ALS that carried pathogenic HTT repeat expansions, representing 0.14% of this replication cohort (sizes 64, 40, 44, 40, 41 for Patients #4–8 in Table 1). We compared this to published data on the occurrence of HTT repeat expansions in the general population, which was only 0.03% (10 individuals had ≥ 40 repeats out of 31,463)(Gardiner et al., 2019; Peplow, 2016).
Overall, the carrier rate among patients diagnosed with FTD/ALS spectrum disorders in the discovery and replication cohorts was 4.4 times higher than that observed among healthy individuals (Fisher’s exact test p-value = 2.68×10−3, odds ratio = 4.55, 95% confidence interval = 1.56–12.80, Table 1). None of the patients found to carry the HTT full-penetrance expansions had additional disease-causing mutations in fifty other genes implicated in FTD/ALS and other neurodegenerative diseases (see Methods for gene list). We performed a genome-wide association study (GWAS) on the FTD/ALS discovery cohort, and single-variant analysis similarly failed to identify loci that would account for our findings (Figure S2).
FTD/ALS patients with pathogenic HTT repeats have multiple haplotypes and exhibit somatic instability
The FTD/ALS patients carrying HTT full-penetrance repeat expansions harbored several different haplotypes that have previously been associated with this locus (Figure S3). The presence of multiple haplotypes indicated diverse ancestral sources among our samples, making it unlikely that another genetic variant outside of the expansion was causing disease in these patients. Furthermore, we did not detect interruptions within the HTT repeat expansion in any of the patients. We only encountered the loss of the CAA-CAG trailing sequence in a single individual (patient #8, Figure 1D).
Additionally, we examined somatic instability across multiple brain regions obtained from the postmortem tissues of two patients diagnosed with FTD/ALS (Patient #5, Patient #8), and from a Huntington’s disease patient carrying 41 CAG repeats (Figure 2). We utilized repeat-primed PCR GeneMapper chromatograms to quantify CAG repeat length distributions, as recently published (Mouro Pinto et al., 2020) (Figure S4). The motor cortex and globus pallidus/putamen regions displayed the most instability in Patient #5 and Patient #8, whereas the spinal cord was among the most stable. A similar pattern was observed in the Huntington’s disease patient. The range of expansion indices (range = 0.042–0.985) observed in the FTD/ALS patients was also consistent with reports from Huntington’s disease patients carrying lower pathogenic-range HTT repeat expansions (Mouro Pinto et al., 2020).
Figure 2. Quantitative analysis of somatic HTT mosaicism across brain regions in FTD/ALS and Huntington’s disease patients.

(A-C) Measurements of somatic mosaicism across brain regions obtained at autopsy for Patient #5, Patient #8, and a Huntington’s disease patient. (D) Heat map of the expansion indices aggregated for the three patients, with tissues displayed in order of mean expansion index. Grey boxes indicate regions that were not assessed. M Ctx, Motor Ctx = motor cortex; GP/Put = globus pallidus/putamen; Corp C, Corpus C = corpus callosum; P Ctx, Pariet Ctx = parietal cortex; F Ctx, Frontal Ctx = superior frontal cortex; Cd/Acb/Put = caudate/accumbens/putamen; Temp Pole = temporal pole; Cing Gyrus = cingulate gyrus; Hipp head = head of the hippocampus; Mb/SN = midbrain/substantia nigra; Hipp = hippocampal formation; L Pons = lower pons; Subthal N = subthalamic nucleus; U Pons = upper pons; SC Cerv = cervical spinal cord; Dent Gyrus = dentate gyrus; SC Lumb = lumbar spinal cord; SC Thor = thoracic spinal cord. See also Figure S4.
Clinicopathologic analysis reveals huntingtin-positive aggregates predominantly in the frontal cortex
The clinical details of the eight patients carrying the HTT full-penetrance, pathogenic repeat expansions are summarized in Table 2 and described in the supplemental information. None of the patients reported choreoathetosis. Four patients reported a family history of neurological disease. Patient #4 (Table 2) was an outlier among the cases we identified as carrying HTT pathogenic repeat expansions. She presented at 17 years of age with cognitive decline, supranuclear vertical gaze palsy, parkinsonism, postural instability, gait disturbance, spasticity, and hyperreflexia in the lower limbs. Treatment with levodopa did not yield a clinical improvement. Genetic screening of HTT performed at the time of presentation was incorrectly reported as normal, and she was initially diagnosed with FTD-PSP despite her young age. A repeat genetic panel conducted several years later to investigate possible causes of young-onset dementia correctly identified an expanded HTT repeat allele. Her diagnosis was updated to young-onset Huntington’s disease (Westphal variant). Her father had presented with a similar syndrome of cognitive decline, gait disorder, and dysarthria in his late twenties, and, post hoc after his daughter’s genetic diagnosis, was diagnosed with Huntington’s disease.
Table 2.
Clinical details of the eight patients carrying a full-penetrance pathogenic HTT repeat expansion
| Patient | Cohort | CAG repeats (A1/A2) | Clinical diagnosis | Age at onset (y) | Sex | Family history | Presenting symptoms |
|---|---|---|---|---|---|---|---|
| 1 | Discovery | 41/21 | PSP-FTD | 68 | M | No | - |
| 2 | Discovery | 40/16 | bvFTD | 56 | F | Yes | Behavioral changes |
| 3 | Discovery | 40/17 | nfvPPA | 57 | F | No | Language disturbance Academic decline, |
| 4 | Replication | 64/17 | PSP-FTD | 17 | F | Yes | dysarthria, bradykinesia, and gait disturbance |
| 5 | Replication | 40/15 | ALS | 56 | F | - | - |
| 6 | Replication | 44/28 | bvFTD | 44 | M | Yes | Personality changes and apathy |
| 7 | Replication | 40/19 | ALS | 76 | M | Yes | Lower limb weakness |
| 8 | Replication | 41/17 | ALS | 61 | M | No | Right foot weakness |
Clinical diagnoses include progressive supranuclear palsy - frontotemporal dementia type (PSP-FTD), behavioral variant frontotemporal dementia (bvFTD), nonfluent variant primary progressive aphasia subtype of FTD (nfvPPA), and amyotrophic lateral sclerosis (ALS). Family history refers to a family history of neurological disease. A1, allele 1; A2, allele 2. DNA for Patient #5 and Patient #8 was extracted from frozen cerebellar tissue, and DNA for the other six patients was extracted from blood. See also Methods S1.
We examined postmortem brains obtained from two of our patients harboring HTT full-penetrance CAG repeats. The first patient was a woman carrying 40 HTT CAG repeats, who developed ALS symptoms at the age of 56 and died eleven years later of respiratory failure following a typical course of motor neuron disease (Table 2, Patient #5). Postmortem examination showed mild atrophy of the precentral gyrus and thinning of the spinal cord anterior roots. Microscopic examination revealed classical neuropathological features of ALS: there was a loss of the motor neurons of the spinal cord and hypoglossal nuclei (Figure 3A–B). Staining with anti-TDP-43 antibodies showed rare neurons with nuclear to cytoplasmic TDP-43 translocation, and occasional neuropil skeins confined to the prefrontal cortex (Brodmann area 9, BA9; Figure 3C). Neither aberrant TDP-43 translocation nor ubiquitinated inclusions were detected in the dentate gyrus.
Figure 3. Neuropathologic changes observed in an ALS patient carrying a full-penetrance pathogenic HTT repeat expansion (Patient #5).

(A) A representative cervical cord section showing pallor of the lateral (*) and anterior corticospinal tracts (**) with ventral horn atrophy. (B) The loss of motor neurons of the anterior horns was severe. (C) Nucleocytoplasmic TDP-43 translocation (arrows) in the prefrontal cortex (BA9 area). (D) Frequent p62 (red arrow) and huntingtin (black arrow) dystrophic neurites (Insert), intranuclear huntingtin (black arrow), and p62 (red arrow) inclusions were noted within the prefrontal cortex. (E) The neostriatum was apparently normal, for example, at the level of the nucleus accumbens, and neither neuronal loss nor reactive gliosis was detectable. (F) & (G) Occasional huntingtin aggregates were seen within the neuropil of the nucleus accumbens. (H) The tail of the caudate nucleus was not atrophic, and the neuronal density was normal and without reactive gliosis. (I) & (J) Rare huntingtin aggregates involve the neuropil of the tail of the caudate nucleus (arrows). Scale bars: A: 1 mm, and C-D: 50 microns. See also Figure S5.
Interestingly, dual staining of the prefrontal cortex and striatum using anti-huntingtin/p62 antibodies showed nuclear and cytoplasmic aggregates of huntingtin and p62 with the highest density observed in the infragranular layers of the prefrontal cortex (Figure 3D). Ubiquitinated nuclear inclusions were found in the tail of the caudate nucleus (Figure 3H) and the frontal cortex (not shown). However, there was no atrophy, neuronal loss or active gliosis in the striatum (Figures 3E–F, 3H–I).
The second brain evaluated postmortem was that of a man carrying 41 CAG repeats in HTT, who presented with right foot weakness at age 61. He was diagnosed with ALS based on disease progression and electromyography, and he died from respiratory failure nine years after symptom onset following a typical course of motor neuron disease (Patient #8, Table 2). Postmortem examination showed mild atrophy of the precentral gyrus and thinning of the anterior spinal roots. There was otherwise no cerebral cortical or striatal atrophy (Figure 4A), or evidence of neuronal loss or gliosis in the striatum on microscopic examination (Figure 4B). Staining for ubiquitin (Figure 4C) and polyglutamine showed scattered nuclear and cytoplasmic and neuropil aggregates within the striatum (Figure 4D) and motor cortex (Figure 4E). Polyglutamine aggregates were not detected in the spinal cord. There was a marked loss of spinal motor neurons (Figure 4F) and degeneration of the corticospinal tracts. Staining with anti-phospho-TDP-43 antibodies showed ALS-type TDP-43 cytoplasmic inclusions within some residual motor neurons (Figure 4F, inset).
Figure 4. Neuropathologic changes observed in an ALS patient carrying a full-penetrance pathogenic HTT repeat expansion (Patient #8).

(A) A coronal section of the fresh brain showing that the caudate, putamen, and globus pallidus was intact with no evidence of atrophy. (B) Luxol fast blue/hematoxylin and eosin staining of the caudate nucleus showed no neuronal loss or gliosis. (C) Ubiquitin immunostaining of the caudate nucleus showed extranuclear aggregates (arrow) and rare intranuclear inclusions (arrowhead). (D-E) Immunohistochemistry for polyglutamine expansions showed occasional extranuclear inclusions within the caudate nucleus (D) and the peri-Rolandic cortex (E, arrows). (F) There was severe motor neuron loss within the anterior horn of the spinal cord (Luxol fast blue/hematoxylin and eosin). (Inset) A remaining motor neuron with a TDP-43 cytoplasmic inclusion. Scale bars: B: 50 microns, C-D: 20 microns, and F: 100 microns.
To confirm the specificity of our pathological findings, we examined the prefrontal cortex (BA9) obtained from postmortems of individuals without Huntington’s disease using the anti-huntingtin (2B4) immunostain previously used. These specimens consisted of four neurologically healthy individuals, three patients diagnosed with FTD/ALS, two patients with Alzheimer’s disease, and one patient with diffuse LBD. All ten cases showed weak cytoplasmic immunostaining within neurons (Figure S5A–B) and did not show aggregation as seen in the two ALS cases with CAG expansions (Figures 3–4) or in Huntington’s disease patients (Figure S5C). Microscopic examination of the spinal cords of nine patients diagnosed with typical Huntington’s disease did not show spinal motor neuron loss.
DISCUSSION
Our data indicate that pathogenic CAG repeat expansions in HTT can give rise to FTD/ALS syndromes that are clinically distinct from the classical Huntington’s disease syndrome. A careful review of the clinical features of the patients carrying pathogenic HTT expansions confirmed the diagnosis of FTD or ALS in seven out of the eight patients. None of the patients manifested choreoathetoid movements during their illness or reported a family history of Huntington’s disease. Furthermore, the postmortem findings of two of our patients with HTT full-penetrance repeat expansions displayed the classical neuropathologic features of ALS, including loss of motor neurons of the anterior horns and hypoglossal nuclei, and the presence of TDP-43-positive inclusions, thereby ruling out mimic syndromes as an explanation of our findings. However, the pathogenic repeat expansions’ effects were corroborated by the occurrence of polyglutamine/huntingtin co-pathology within the frontal lobes. Finally, pathogenic HTT repeat expansions were specific to FTD/ALS cases, as they were not found in a similar-sized cohort of patients diagnosed with LBD.
It is possible that the patients carrying a HTT repeat expansion were simply misdiagnosed cases of atypical Huntington’s disease or suffered from two different neurodegenerative diseases by chance, and they would have developed the classic Huntington’s disease symptoms had they lived long enough. We believe that these are unlikely scenarios for several reasons. First, we identified multiple FTD/ALS patients in our discovery cohort following the same clinical pattern and found a similar occurrence rate in our replication cohort. In contrast, full-penetrance pathogenic HTT expansions were not present in our LBD or control whole-genome sequence data. Second, the apparently healthy striatum in both patients who underwent postmortem evaluation diminishes the likelihood of subclinical Huntington’s disease as an explanation for their symptoms. The choreoathetoid movements observed in Huntington’s disease originate from the striatum, and the lack of detectable neuronal loss on general surveys or reactive gliosis in this region of our FTD/ALS patients implies that the motor neuron disease was not masking these symptoms. Third, two of our eight patients lived at least nine years after symptom onset and did not manifest motor signs of Huntington’s disease during this extended survival period. Fourth, the prevalence rates of FTD (22 per 100,000 population) (Onyike and Diehl-Schmid, 2013), ALS (6 per 100,000) (Chiò et al., 2013), and Huntington’s disease (3 per 100,000) (Pringsheim et al., 2012) indicate that, by chance, there should only be three cases of disease co-occurrence in the entire United States population of 327 million. Instead, we identified eight patients among a moderately-sized cohort of FTD/ALS cases (n = 6,116 patients). Finally, the age at onset among our patients overlapped with the predicted age at onset of Huntington’s disease based on their CAG repeat length (Figure 1C).
Regardless of the semantic debate as to whether it is correct to designate HTT repeat expansions as a genetic cause of FTD/ALS spectrum disorders, our findings have direct implications for the clinical care of patients presenting with these neurological conditions and the neuropathologic staging of disease. Although there have been previous reports of the coexistence of FTD/ALS and Huntington’s disease (Chhetri et al., 2014; Kanai et al., 2008; Nielsen et al., 2010; Papageorgiou et al., 2006; Phukan et al., 2010; Rubio et al., 1996; Sadeghian et al., 2011; Tada et al., 2012), pathogenic HTT mutations have not been described in cases of pure FTD and ALS. Even though these expansions account for less than 1% of FTD and ALS cases, clinical practice should be adapted to include regular screening of these patient populations for this mutation, particularly in light of the antisense oligonucleotide treatments targeting the HTT locus that are undergoing clinical trials (Tabrizi et al., 2019). The uncertainty and challenge in obtaining an early accurate diagnosis for patient #4 provide further support for the clinical utility of genetic testing for HTT in FTD/ALS syndromes.
Pathogenic HTT repeats tend to expand in length. This somatic instability increases with age and longer repeat expansion lengths, and varies from tissue to tissue. In particular, specific brain regions, such as the striatum and the cerebral cortex, have consistently demonstrated greater somatic instability, while the spinal cord is relatively stable (Mouro Pinto et al., 2020; Kennedy et al., 2003; Swami et al., 2009; Telenius et al., 1994; De Rooij et al., 1995). Our somatic instability analysis revealed similar tissue-specific profiles among our two patients. Interestingly, the motor cortex is a vulnerable area of pathology for FTD/ALS and displayed the maximum level of somatic instability within our samples, a finding that is consistent with Huntington’s disease patients. Given the higher levels of somatic instability observed in the motor cortex, the clinical presentation and pathological findings may arise from a tissue-specific instability (Mouro Pinto et al., 2020). Under this paradigm, the patients presenting with an FTD/ALS phenotype may have a distinct pattern of HTT CAG repeats across the different brain regions that leads to specific downstream pathology and clinical presentations.
From a neuropathologic perspective, we have identified a pathologic subtype that is distinct from the classical features observed in the brains of Huntington’s disease patients (Vonsattel et al., 1985). This novel pattern is characterized by abundant huntingtin-positive, ubiquitin-positive inclusions in the frontal cortex and the absence of detectable neostriatal degeneration, with scarce TDP-43 positive co-pathology. The neuropathologic staging of Huntington’s disease, as defined by Vonsattel and colleagues in 1985 (Vonsattel et al., 1985), rests on the progressive striatal degeneration. Based on our work, an addendum of the neuropathologic consensus criteria for diagnosing Huntington’s disease should be considered to capture this subtype’s precise frequency among the disease population.
Our study has several limitations. Whereas our cohorts focused on individuals of European ancestry, future studies should determine the importance of HTT expansions among non-European FTD/ALS spectrum disorders. Moreover, our research has limited power due to the rarity of these disorders. Based on the carrier rate observed in our study, we would require approximately 150,000 samples to achieve a genome-wide significance level of 5×10−8 based on a Bonferroni correction and 80% power. Such a conservative threshold is an implicit obstacle to identifying rare genetic causes of any rare disease. We hope that future studies and meta-analyses can meet these standards, and indeed we have made our data publicly available with that goal in mind. In the meantime, researchers in the rare disease space must rely on objective collateral evidence to confirm the validity of their findings. In our example, the extra data pointing to HTT repeat expansions as a rare cause of FTD/ALS spectrum disorders include the similar mutation frequency across cohorts, the lack of similar findings in other neurological diseases, and the prior knowledge that similar-sized HTT repeat expansions cause neurodegeneration. Most convincingly, our autopsy data show a distinct pattern of huntingtin pathology in the frontal cortices in multiple patients that was not present in healthy individuals or patients with other neurological disorders.
There is an increasing consensus that molecularly-defined genetic causes of disease can present with heterogeneous neuropsychiatric syndromes. The polyglutamine expansion diseases spinocerebellar ataxia type 2 (SCA2) and 3 (SCA3) typically cause ataxia but can also cause levodopa-responsive parkinsonism (Simon-Sanchez et al., 2005). This consideration is particularly valid for frontal lobe diseases that can present with protean syndromes. For example, patients with MAPT mutations can present with behavioral variant FTD, nonfluent variant primary progressive aphasia, progressive supranuclear palsy, or corticobasal syndrome (van Swieten et al., 2000), and the pathogenic repeat expansion in the C9orf72 gene has united two clinically disparate neurologic illnesses, FTD and ALS, into a single disease entity (Majounie et al., 2012). It may be worthwhile to screen patients presenting with psychiatric symptoms later in life or with other dementias to elucidate the entire phenotypic spectrum associated with pathogenic HTT repeat expansions.
We have made the individual-level genome sequence data for our patients and control subjects publicly available on the dbGaP web portal as a resource for other researchers. Our research highlights the power of performing whole-genome sequencing in large cohorts of patients with complex neurodegenerative syndromes. We prioritized the screening of autopsy samples, as it allowed us to evaluate the neuropathologic changes associated with genetic variation quickly. As this technology’s cost decreases, the size of cohorts that can undergo whole-genome sequencing will increase, enhancing our ability to detect rare, clinically actionable genetic mutations underlying neurologic diseases. The emergence of high-throughput, long-range sequencing will further strengthen our ability to identify novel repeat expansions driving neuropsychiatric disorders (Sedlazeck et al., 2018).
Our work leads to an increase in diagnostic accuracy and a refinement of the phenotype characteristics associated with pathogenic HTT repeat expansions. Although our discovery accounts for a small subset of FTD/ALS patients, clinicians should be aware of this unusual presentation associated with pathogenic HTT repeat expansions. They should consider instituting testing for their FTD and ALS patients, especially as it paves the way for disease-modifying therapies in this small patient subset.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bryan Traynor (traynorb@mail.nih.gov).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The individual-level sequence data for the discovery genomes are available on dbGaP (accession number: phs001963.v1.p1 NIA DementiaSeq). The programming code used in this paper is available at https://github.com/dewanr2/Somatic_Instability.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study cohorts
Discovery cohort
The study workflow is depicted in Figure S6. The discovery cohort included (i) 1,377 patients diagnosed with FTD spectrum disorders including the known subtypes of behavioral variant FTD, primary progressive aphasia, and progressive supranuclear palsy (PSP), (ii) 1,065 patients diagnosed with ALS, (iii) 2,599 individuals diagnosed with Lewy body dementia (LBD), and (iv) 3,158 neurologically healthy participants. DNA was extracted from blood or frozen cerebellar tissue obtained at autopsy using standard methods [FTD-ALS cohort: 2,199 (90.0%) extracted from blood and 243 (10.0%) from brain tissue; LBD: 805 (31.0%) blood and 1,794 (69.0%) brains; Controls: 2,866 (90.8%) blood and 292 (9.2%) brains]. Patients with FTD were diagnosed according to the Neary criteria (Faber, 1999) or the Movement Disorders Society criteria for PSP (Höglinger et al., 2017). Patients with ALS were diagnosed according to the El Escorial criteria (Brooks, 1994), and the LBD cases were diagnosed with pathologically definite or clinically probable disease according to consensus criteria (Emre et al., 2007; McKeith et al., 2005). The LBD cases were included in this study as diseased control subjects. All participants included in the aged, healthy control cohort were free of neurological disease based on history and neurological examination (mean age = 77.0 years of age at collection, interquartile range = 69.0–86.0). All study participants were of European ancestry. Table S2 lists the demographic characteristics of the cohorts.
Replication cohort
For replication, we used DNA obtained from 1,009 patients diagnosed with FTD, 2,665 patients diagnosed with ALS, and 210 neurologically healthy individuals. DNA for 2,837 (77.2%) of the FTD/ALS cases were extracted from blood. Of the remainder, 657 (17.9%) were extracted from frozen CNS tissue (cerebellum, cortex, pons, spinal cord), 140 (3.8%) were extracted from frozen organ tissue (adrenal gland, diaphragm, kidney, liver, muscle, thyroid), 31 (0.8%) from saliva, and 9 (0.3%) from iPS cell lines. DNA for 177 (84.3%) of the healthy subjects was extracted from blood. Of the remainder, 20 (9.5%) were extracted from frozen CNS tissue (cerebellum, cortex, spinal cord, pons), 3 (1.4%) were extracted from frozen liver tissue, 4 (1.9%) from saliva, and 6 (2.9%) from iPS cell lines. The replication control cohort also included existing data for 13,670 population controls from Gardiner et al., 2019 (Gardiner et al., 2019), and 17,703 neurologically-healthy individuals from the UK 100K Genomes Project (Peplow, 2016). These HTT repeat expansion data were measured in DNA extracted from blood. The institutional review boards of participating institutions approved the study, and informed consent was obtained from all subjects or their surrogate decision-makers, according to the Declaration of Helsinki.
METHOD DETAILS
Data generation and pre-processing
Whole-genome sequencing
Genomic DNA was extracted from whole blood or cerebellar brain tissue using a Maxwell RSC Instrument (Promega Corp., Madison, WI, USA). Fluorometric quantitation of the genomic DNA samples was performed using the PicoGreen dsDNA assay (Thermo Fisher). PCR-free, paired-end, non-indexed libraries were constructed using the TruSeq PCR-free chemistry according to the manufacturer’s instructions (Illumina Corp., San Diego, CA, USA). Sequencing was performed on an HiSeq X Ten sequencer (version 2.5 chemistry, Illumina) using 150 base pair (bp), paired-end cycles, applying a single sample to each lane.
Sequence alignment and variant calling
Raw genome data in FASTQ file format were transferred to Google Cloud Storage. Paired-end sequences were processed by following the pipeline standard developed by the Centers for Common Disease Genomics (CCDG; https://www.genome.gov/27563570/). This standard allows for whole-genome sequence data processed by different groups to generate ‘functionally equivalent’ results (DePristo et al., 2011; Li and Durbin, 2009; Poplin et al., 2018; Regier et al., 2018). The GRCh38DH reference genome was used for alignment, as specified in the CCDG standard. For whole-genome sequence alignments and processing, the Broad Institute’s implementation of the functional equivalence standardized pipeline was used. This pipeline, which incorporates the GATK (2016) Best Practices, was implemented in the workflow description language (WDL) for deployment and execution on the Google Cloud Platform (Regier et al., 2018). Single-nucleotide variants (SNV) and insertion-deletions (InDels) variants were called from the processed whole-genome sequence data following the GATK Best Practices using another Broad Institute workflow for joint discovery and Variant Quality Score Recalibration (VQSR). The Broad workflows for whole-genome sequence sample processing and joint discovery are publically available (https://github.com/gatk-workflows/broad-prod-wgs-germline-snps-indels). The average sequencing read-depth after filtering by alignment quality was 35x, and mean coverage per genome was 36.3 (95% confidence interval: 29.3–43.3). All of the whole-genome sequence data (discovery and replication cohorts) were processed using the same pipeline.
Known gene analysis
Whole-genome sequence data was analyzed for pathogenic mutations in fifty genes associated with neurodegenerative diseases, including ALS2, APP, ATP13A2, ATXN2, BSN, C9orf72, CHCHD10, CHMP2B, CSF1R, DCTN1, DNAJC6, FBXO7, FIG4, FUS, GBA, GCH1, GRN, HNRNPA1, KIF5A, LRRK2, MAPT, MATR3, OPTN, PANK2, PARK2, PARK7, PFN1, PINK1, PLA2G6, POLG, PSEN1, PSEN2, RAB39B, SETX, SNCA, SOD1, SPG11, SPTLC1, SQSTM1, SYNJ1, TAF1, TARDBP, TBK1, TIA1, TUBA4A, UBQLN2, VAPB, VCP, VPS13C, and VPS35. The analysis was performed by subsetting gene regions from post-QC variant files using PLINK 2.0 and annotating variants in ANNOVAR (v. 2018–04/16).
Single-variant association analysis
We performed a GWAS in FTD/ALS (n = 2,451 cases and 4,029 controls) using generalized linear model association testing implemented in PLINK (version 2.0) with a minor allele frequency threshold of >5% based on the allele frequency estimates in the FTD case cohort. We used the step function in the R MASS package (version 7.3–53) to determine the minimum number of principal components (generated from common single nucleotide variants) required to correct for population substructure. Based on this analysis, we incorporated sex, age, and five principal components (PC1, PC2, PC3, PC5, PC8) as covariates in our model. Quantile-quantile plots revealed minimal residual population substructure, as estimated by the sample size-adjusted genome-wide inflation factor λ1000 of 1.005. The Bonferroni threshold for genome-wide significance was 5.0 × 10−8.
Repeat expansion analysis
ExpansionHunter - Targeted software (version 3.0.1) was used to estimate repeat lengths of ten known, disease-causing expansions in samples that had undergone whole-genome sequencing (Dolzhenko et al., 2019). This algorithm has been validated using experimentally-confirmed samples carrying pathogenic expansions, including HTT (Dolzhenko et al., 2019). Fully-penetrant pathogenic alleles in the huntingtin (HTT) gene were defined as those containing 40 or more CAG repeats according to the American College of Medical Genetics diagnostic criteria (Bean and Bayrak-Toydemir, 2014). The number of repeats was validated using a repeat-primed PCR assay (Jama et al., 2013) and by cloning and Sanger sequencing for each sample reported by ExpansionHunter - Targeted algorithm to carry 40 or more HTT CAG repeats.
Repeat-primed PCR assay
The CAG trinucleotide repeat length in HTT was quantified using a previously validated repeat-primed PCR method (see Table S3) (Jama et al., 2013). The repeat-primed PCR assay to measure HTT CAG repeat length was performed using the Eppendorf Mastercycler pro thermal cycler (Fisher Scientific, Houston, TX, USA) in a final volume of 22μl containing 1X FailSafe Premix J (Epicenter, Madison, WI, USA), 1.0U Platinum Taq DNA polymerase (Invitrogen Corp., Carlsbad, CA, USA), 0.5μM Primer Mix containing 0.5μmol/l each of forward primer 5′-ATGAAGGCCTTCGAGTCCCTCAAGTCC-3′, with a 5’ 6FAM fluorescent tag, and reverse primer 5′-CGGTGGCGGCTGTTGCTGCTGCTGCTGCTG-3′ (Eurofins Genomics LLC, Louisville, KY, USA), and 3μl of 25–50ng/μl genomic DNA. Cycling conditions were identical to those in Jama et al (Jama et al., 2013). The reverse primer 5’-end in this protocol contains a 15 bp sequence specific to the region between the HTT CAG and CCG trinucleotide repeats. This primer binding region fully encapsulates the trailing CAA-CAG sequence to prevent against any changes in primer specificity due to loss of interruption (i.e., conversion to CAG-CAG).
The fragment length analysis was performed on an ABI 3730xl genetic analyzer (Applied Biosystems Inc., Foster City, CA, USA) using a mixture of 2μl of repeat-primed PCR products, 0.5μl GeneScan 500 LIZ size standard (Thermo Fisher Scientific, Cincinnati, OH, USA), and 7.5μl HiDi formamide (Applied Biosystems Inc.). The mixture was heated at 95°C for 3 minutes and then immediately cooled on ice for at least 5 minutes before loading for capillary electrophoresis. Data were analyzed using GeneMapper software (version 4, Applied Biosystems Inc.). Repeat expansions were quantifiable with a 3-bp periodicity. GeneMapper chromatograms were used to quantify somatic instability by calculating an expansion index for each sample (Mouro Pinto et al., 2020). All samples analyzed from Patient #5, Patient #8, and a Huntington’s disease patient (for comparison) had the same modal allele length (CAG 40, 41, and 41 respectively).
Cloning and Sanger sequencing
The HTT region was PCR amplified using the forward (HD1F: 5′ CCGCTCAGGTTCTGCTTTTA 3′) and reverse (HD1FR 5′ GGCTGAGGCAGCAGCGGCTG 3′) primers with the following PCR components: 10ng genomic DNA, 2μl 10x PCR Buffer, 0.25μl Taq Polymerase, 0.4μl 10mM dNTP mix, 4μl Q Solution (Qiagen), 0.8μl 5uM forward primer, 0.8μl 5uM reverse primer, and water to a final reaction volume of 20μl. Thermocycling conditions are listed in Table S3. Amplified PCR products were assessed on 2% E-Gel EX Agarose Gels (Thermo Fisher) before cloning into the pCR4-TOPO TA vector supplied in the TOPO TA Cloning® Kits for Sequencing (Thermo Fisher). Cloning and the bacterial transformation were performed according to the manufacturer’s protocol. Colonies were picked, grown overnight, and plasmid was extracted using the QIAprep Spin Miniprep Kit (Qiagen). Plasmid with TA-cloned inserts were Sanger sequenced using the universal M13 forward primer.
Brain immunohistochemistry
Formalin-fixed, paraffin-embedded sections were prepared from postmortem brain and spinal cord regions and stained with Luxol fast blue counterstained with hematoxylin and eosin, and immunostained using polyclonal antibodies against huntingtin (clone 2B4), p62, and polyglutamine (clone 1c2) (see Table S4 for primary antibody conditions). The 2B4 antibody targets the N’-end of the huntingtin protein and stains soluble huntingtin and insoluble aggregates. The huntingtin/p62 dual stains were developed using a 3,3’-diaminobenzidine (DAB) and alkaline phosphatase dual staining system on a Leica Bond autostainer with a hematoxylin counterstain. Primary antibodies and staining methods are listed in Table S4.
QUANTIFICATION AND STATISTICAL ANALYSIS
Genomes were excluded from analysis for the following reasons: (1) high contamination rate (> 5% based on VerifyBamID freemix metric) (Jun et al., 2012), (2) excessive heterozygosity rate (exceeding +/− 0.15 F-statistic), (3) low call rate (≤ 95%), (4) discordance between reported sex and genotypic sex, and (5) duplicate samples (determined by pi-hat statistic). Samples of non-European ancestry based on comparison of principal components with the HapMap 3 Genome Reference Panel were flagged, but not excluded (Altshuler et al., 2010; Chang et al., 2015).
Trinucleotide repeat frequencies were compared between cohorts using Fisher’s exact test with a Bonferroni-corrected significance threshold of 0.005 (0.05 divided by ten repeat expansions tested). Concordance between identifying the C9orf72 repeat expansion using the ExpansionHunter – Targeted algorithm and a validated repeat-primed PCR assay for C9orf72 (Majounie et al., 2021) was assessed using the R package irr (version 0.84.1). Genotype data defining the common haplotypes in the HTT locus (Chao et al., 2017) were extracted from the whole-genome sequence data using PLINK (version 2.0). The ExpansionHunter - Targeted output for the CAG repeat length was merged with the genotype information, and phasing was performed using Eagle (version 2.4).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Huntingtin/ p62 double stain | Millipore/ Abcam | MAB5492/ ab207305 |
| Rabbit Anti-Ubiquitin | Dako | Z0458 |
| Anti-polyglutamine-Expansion Diseases Marker Antibody, clone 5TF1–1C2 | Millipore Sigma | MAB1574 |
| Anti-phospho TDP-43 (pS409/410) | Cosmo Bio Co., Ltd | CAC-TIP-PTD-M01 |
| Anti-TDP-43 | Proteintech | 10782–2-AP |
| Biological Samples | ||
| Human cerebral brain tissue and/or whole blood | Comprehensive list of study sites listed in Acknowledgements of this paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1X FailSafe Premix J | Lucigen | FSP995J |
| Q Solution | Qiagen | 201203 |
| Critical Commercial Assays | ||
| Maxwell RSC Tissue DNA Kit | Promega | AS1610 |
| PicoGreen dsDNA assay | Thermo Fisher | P7589 |
| TruSeq PCR-free Library Prep Kit | Illumina | 20015963 |
| HiSeq X Ten Reagent Kit | Illumina | FC-501–2501 |
| 2% E-Gel EX Agarose Gels | Thermo Fisher | G402002 |
| TOPO TA Cloning® Kit | Thermo Fisher | K4575J10 |
| QIAprep Spin Miniprep Kit | Qiagen | 27106 |
| Deposited Data | ||
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Individual-level sequence data for the discovery genomes | This paper | dbGaP (phs001963.v1.p1) |
| ExpansionHunter – Targeted summary data for the discovery genomes | This paper | Table S4 |
| Oligonucleotides | ||
| HTT RP-PCR primer sequence, forward:6FAM-ATGAAGGCCTTCGAGTCCCTCAAGTC | Jama et al., 2013 | N/A |
| HTT RP-PCR primer sequence, reverse:ATGAAGGCCTTCGAGTCCCTCAAGTC | Jama et al., 2013 | N/A |
| HTT PCR primer sequence, prior to cloning, forward:CCGCTCAGGTTCTGCTTTTA | Dabrowska et al., 2018 | N/A |
| HTT PCR primer sequence, prior to cloning, reverse:GGCTGAGGCAGCAGCGGCTG | Dabrowska et al., 2018 | N/A |
| Software and Algorithms | ||
| ANNOVAR v. 2018–04/16 | Wang et al., 2010 | https://doc-openbio.readthedocs.io/projects/annovar/en/latest/ |
| ExpansionHunter – Targeted v. 3.0.1 | Dolzhenko et al., 2019 | https://github.com/Illumina/ExpansionHunter |
| Eagle v. 2.4 | Loh et al., 2016 | https://data.broadinstitute.org/alkesgroup/Eagle/ |
| GATK | Broad Institute | https://software.broadinstitute.org/gatk |
| Pipeline-Standardization | CCDG | https://github.com/CCDG/Pipeline-Standardization/blob/master/PipelineStandard.md |
| PLINK v. 2.0 | Chang et al., 2015 | https://www.cog-genomics.org/plink/2.0/ |
| prod-wgs-germline-snps-indels | Broad Institute | https://github.com/gatk-workflows/broad-prod-wgs-germline-snps-indels |
| R | R Core Team | https://www.r-project.org |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
HIGHLIGHTS.
Pathogenic expansions in the HTT gene are a rare cause of FTD/ALS spectrum diseases
Autopsies showed both the expected TDP-43 pathology of FTD/ALS and polyQ inclusions
HTT repeat expansions were not seen in healthy subjects or Lewy body dementia cases
Clinicians should screen FTD/ALS patients for HTT repeat expansions
ACKNOWLEDGEMENTS
We thank contributors who collected samples used in this initiative, and the patients and families, whose help and participation made this work possible. This research was supported by the Intramural Research Program of the National Institutes of Health (National Institute on Aging, National Institute of Neurological Disorders and Stroke; project numbers: 1ZIAAG000935 [PI Bryan J. Traynor], 1ZIANS003154 [PI Sonja W. Scholz], 1ZIANS0030033 and 1ZIANS003034 [David S. Goldstein]). Drs. Sidransky, Lopez, and Tayebi were supported by the Intramural Research Program of the National Human Genome Research Institute. This research was supported by Italian Ministry of Health (Ricerca Corrente). Dr. Hickman is a Columbia University Irving Medical Center ADRC Research Education Component trainee (P30 AG066462-01 PI: Small, Scott), and is supported by grants from the Hereditary Disease Foundation and Huntington Disease Society of America. Dr Rowe is supported by the Wellcome Trust (103838) and National Institute for Health Research Cambridge Biomedical Research Centre. Dr. Landers was supported by the National Institutes of Health/National Institute of Neurological Disorders (R01NS073873). The American Genome Center is supported by an NHLBI grant: IAA-A-HL-007.001. The sequencing activities at NYGC were supported by the ALS Association (Grant # 19-SI-459) and The Tow Foundation. This study used the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, Maryland, USA (http://biowulf.nih.gov).
Footnotes
DECLARATION OF INTEREST
S.P-B, A.B.S., J.A.H., H.R.M, and B.J.T. hold US, EU, and Canadian patents on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of C9orf72. S.W.S. serves on the Scientific Advisory Council of the Lewy Body Dementia Association and is an editorial board member for the Journal of Parkinson’s Disease. B.J.T. is an editorial board member for JAMA Neurology, JNNP, and Neurobiology of Aging. V.S. is on the journal editorial boards of Amyotrophic Lateral Sclerosis, European Neurology, American Journal of Neurodegenerative Diseases, and Frontiers in Neurology. He has also received compensation for consulting services and speaking activities from AveXis, Cytokinetics, Italfarmaco, and Zambon. J.B.R is editor for Brain, and has received compensation for consulting services from Asceneuron, Biogen, UCB, Astex, and SV Health. J.E.L. is a member of the scientific advisory board for Cerevel Therapeutics, and a consultant and may provide expert testimony for Perkins Coie LLP.
CONSORTIA
The members of The American Genome Center (TAGC) are: Adelani Adeleye, Camille Alba, Dagmar Bacikova, Clifton L. Dalgard, Daniel N. Hupalo, Elisa McGrath Martinez, Harvey B. Pollard, Gauthaman Sukumar, Anthony R. Soltis, Meila Tuck, Coralie Viollet, Xijun Zhang, Matthew D. Wilkerson. The members of the FALS Sequencing Consortium are: Bradley N. Smith, Nicola Ticozzi, Claudia Fallini, Athina Soragia Gkazi, Simon D. Topp, Jason Kost, Emma L. Scotter, Kevin P. Kenna, Pamela Keagle, Jack W. Miller, Cinzia Tiloca, Caroline Vance, Eric W. Danielson, Claire Troakes, Claudia Colombrita, Safa Al-Sarraj, Elizabeth A. Lewis, Andrew King, Daniela Calini, Viviana Pensato, Barbara Castellotti, Jacqueline de Belleroche, Frank Baas, Anneloor L.M.A. ten Asbroek, Peter C. Sapp, Diane McKenna-Yasek, Russell L. McLaughlin, Meraida Polak, Seneshaw Asress, Jesús Esteban-Pérez, José Luis Muñoz-Blanco, Zorica Stevic, Sandra D’Alfonso, Letizia Mazzini, Giacomo P. Comi, Roberto Del Bo, Mauro Ceroni, Stella Gagliardi, Giorgia Querin, Cinzia Bertolin, Wouter van Rheenen, Frank P. Diekstra, Rosa Rademakers, Marka van Blitterswijk, Kevin B. Boylan, Giuseppe Lauria, Stefano Duga, Stefania Corti, Cristina Cereda, Lucia Corrado, Gianni Sorarù, Kelly L. Williams, Garth A. Nicholson, Ian P. Blair, Claire Leblond-Manry, Guy A. Rouleau, Orla Hardiman, Karen E. Morrison, Jan H. Veldink, Leonard H. van den Berg, Ammar Al-Chalabi, Hardev Pall, Pamela J. Shaw, Martin R. Turner, Kevin Talbot, Franco Taroni, Alberto García-Redondo, Zheyang Wu, Jonathan D. Glass, Cinzia Gellera, Antonia Ratti, Robert H. Brown, Jr., Vincenzo Silani, Christopher E. Shaw and John E. Landers. The members of The Genomics England Research Consortium are: John C. Ambrose, Prabhu Arumugam, Emma L. Baple, Marta Bleda, Freya Boardman-Pretty, Jeanne M. Boissiere, Christopher R. Boustred, H. Brittain, Mark J. Caulfield, Georgia C. Chan, Clare E.H. Craig, Louise C. Daugherty, Anna de Burca, Andrew Devereau, Greg Elgar, Rebecca E. Foulger, Tom Fowler, Pedro Furió-Tarí, Joanne M. Hackett, Dina Halai, Angela Hamblin, Shirley Henderson, James E. Holman, Tim J.P. Hubbard, Kristina Ibáñez, Rob Jackson, Louise J. Jones, Dalia Kasperaviciute, Melis Kayikci, Lea Lahnstein, Kay Lawson, Sarah E.A. Leigh, Ivonne U.S. Leong, Javier F. Lopez, Fiona Maleady-Crowe, Joanne Mason, Ellen M. McDonagh, Loukas Moutsianas, Michael Mueller, Nirupa Murugaesu, Anna C. Need, Chris A. Odhams, Christine Patch, Daniel Perez-Gil, Dimitris Polychronopoulos, John Pullinger, Tahrima Rahim, Augusto Rendon, Pablo Riesgo-Ferreiro, Tim Rogers, Mina Ryten, Kevin Savage, Kushmita Sawant, Richard H. Scott, Afshan Siddiq, Alexander Sieghart, Damian Smedley, Katherine R. Smith, Alona Sosinsky, William Spooner, Helen E. Stevens, Alexander Stuckey, Razvan Sultana, Ellen R.A. Thomas, Simon R. Thompson, Carolyn Tregidgo, Arianna Tucci, Emma Walsh, Sarah A. Watters, Matthew J. Welland, Eleanor Williams, Katarzyna Witkowska, Suzanne M. Wood, and Magdalena Zarowiecki. The members of The International ALS Genomics Consortium (iALSgc) are: Yevgeniya Abramzon, Sampath Arepalli, Pavan Auluck, Robert H. Baloh, Robert Bowser, Christopher B. Brady, Alexis Brice, James Broach, William Camu, Ruth Chia, Adriano Chiò, John Cooper-Knock, Philippe Corcia, Jinhui Ding, Carsten Drepper, Vivian E. Drory, Travis L. Dunckley, Faraz Faghri, Jennifer Farren, Eva Feldman, Mary Kay Floeter, Pietro Fratta, Glenn Gerhard, J. Raphael Gibbs, Summer B. Gibson, Jonathan D. Glass, Stephen A. Goutman, John A. Hardy, Matthew B. Harms, Terry D. Heiman-Patterson, Dena G. Hernandez, Ben Hoover, Lilja Jansson, Freya Kamel, Janine Kirby, Neil W. Kowall, Hannu Laaksovirta, John E. Landers, Francesco Landi, Isabelle Le Ber, Serge Lumbroso, Daniel JL. MacGowan, Nicholas J. Maragakis, Gabriele Mora, Kevin Mouzat, Liisa Myllykangas, Mike A. Nalls, Richard W. Orrell, Lyle W. Ostrow, Roger Pamphlett, Stuart Pickering-Brown, Erik Pioro, Stefan M. Pulst, John M. Ravits, Alan E. Renton, Wim Robberecht, Ian Robey, Ekaterina Rogaeva, Jeffrey D. Rothstein, Sonja W. Scholz, Michael Sendtner, Pamela J. Shaw, Katie C. Sidle, Zachary Simmons, Andrew B. Singleton, David J. Stone, Pentti J. Tienari, Bryan J. Traynor, John Q. Trojanowski, Juan C. Troncoso, Miko Valori, Philip Van Damme, Vivianna M. Van Deerlin, Ludo Van Den Bosch and Lorne Zinman. The members of the International FTD Genetics Consortium (IFGC) are: Diego Albani, Luisa Benussi, Roberta Ghidoni, Giuliano Binetti, Barbara Borroni, Alessandro Padovani, Amalia Bruni, Jordi Clarimon, Oriol Dols-Icardo, Ignacio Illán-Gala, Alberto Lleó, Adrian Danek, Daniela Galimberti, Elio Scarpini, Maria Serpente, Caroline Graff, Huei-Hsin Chiang, Behzad Khoshnood, Linn Öijerstedt, Maria L. Waldö, Christer F. Nilsson, Per M. Johansson, Stuart Pickering-Brown, Christopher M. Morris, Benedetta Nacmias, Sandro Sorbi, Jorgen E. Nielsen, Lynne E. Hjermind, Valeria Novelli, Annibale A. Puca, Pau Pastor, Ignacio Alvarez, Monica Diez-Fairen, Miquel Aguilar, Robert Perneczky, Janine Diehl-Schimd, Ekaterina Rogaeva, Mina Rossi, Paola Caroppo, Fabrizio Tagliavini, Agustin Ruiz, Mercè Boada, Isabel Hernández, Sonia Moreno-Grau and Johannes C Schlachetzki. The members of The International LBD Genomics Consortium (iLBDgc) are: Dag Aarsland, Yevgeniya Abramzon, Sarah Ahmed, Camille Alba, Marilyn S. Albert, Safa Al-Sarraj, Johannes Attems, Dagmar Bacikova, Matthew J. Barrett, Thomas G. Beach, Lynn M. Bekris, David A. Bennett, Lilah M. Besser, Eileen H. Bigio, Sandra E. Black, Bradley F. Boeve, Ryan C. Bohannan, Francesca Brett, Alexis Brice, Maura Brunetti, Chad A. Caraway, Jose-Alberto Palma, Andrea Calvo, Antonio Canosa, Ruth Chia, Adriano Chiò, Jordi Clarimon, Clifton L. Dalgard, Dennis Dickson, Monica Diez-Fairen, Jinhui Ding, Charles Duyckaerts, Kelley Faber, Tanis Ferman, Raffaele Ferrari, Luigi Ferrucci, Margaret E. Flanagan, Gianluca Floris, Tatiana M. Foroud, Juan Fortea, Ziv Gan-Or, Steve Gentleman, Bernardino Ghetti, Jesse Raphael Gibbs, Alison Goate, David Goldstein, Isabel González-Aramburu, Neill R. Graff-Radford, John A. Hardy, Angela K. Hodges, Heng-Chen Hu, Daniel Hupalo, Jon Infante, Alex Iranzo, Scott M. Kaiser, Horacio Kaufmann, Julia Keith, Ronald C. Kim, Gregory Klein, Rejko Krüger, Walter Kukull, Amanda Kuzma, Carmen Lage, Suzanne Lesage, Alberto Lleó, James B. Leverenz, Giancarlo Logroscino, Grisel Lopez, Seth Love, Qinwen Mao, Maria Jose Marti, Elisa Martinez-McGrath, Mario Masellis, Eliezer Masliah, Patrick May, Ian McKeith, Marek-Marsel Mesulam, Edwin S. Monuki, Christopher M. Morris, Huw R. Morris, Kathy L. Newell, Lucy Norcliffe-Kaufmann, Laura Palmer, Pau Pastor, Matthew Perkins, Stuart Pickering-Brown, Olga Pletnikova, Laura Molina-Porcel, Alan E. Renton, Susan M. Resnick, Regina H. Reynolds, Eloy Rodríguez-Rodríguez, Ekaterina Rogaeva, Jonathan D. Rohrer, Owen A. Ross, Mina Ryten, Marya S. Sabir, Pascual Sanchez-Juan, Clemens R. Scherzer, Sonja W. Scholz, Geidy E. Serrano, Vikram Shakkottai, Ellen Sidransky, Andrew B. Singleton, Toshiko Tanaka, Nahid Tayebi, Alan J. Thomas, Bension S. Tilley, Bryan J. Traynor, Claire Troakes, Juan C. Troncoso, Coralie Viollet, Ronald L. Walton, Randy Woltjer, Zbigniew K. Wszolek, Georgia Xiromerisiou and Chiara Zecca. The members of The NYGC ALS Consortium are as follows: Hemali Phatnani, Justin Kwan, Dhruv Sareen, James R. Broach, Zachary Simmons, Ximena Arcila-Londono, Edward B. Lee, Vivianna M. Van Deerlin, Neil A. Shneider, Ernest Fraenkel, Lyle W. Ostrow, Frank Baas, Noah Zaitlen, James D. Berry, Andrea Malaspina, Pietro Fratta, Gregory A. Cox, Leslie M. Thompson, Steve Finkbeiner, Efthimios Dardiotis, Timothy M. Miller, Siddharthan Chandran, Suvankar Pal, Eran Hornstein, Daniel J. MacGowan, Terry Heiman-Patterson, Molly G. Hammell, Nikolaos. A. Patsopoulos, Oleg Butovsky, Joshua Dubnau, Avindra Nath, Robert Bowser, Matt Harms, Eleonora Aronica, Mary Poss, Jennifer Phillips-Cremins, John Crary, Nazem Atassi, Dale J. Lange, Darius J. Adams, Leonidas Stefanis, Marc Gotkine, Robert H. Baloh, Suma Babu, Towfique Raj, Sabrina Paganoni, Ophir Shalem, Colin Smith, Bin Zhang, University of Maryland Brain and Tissue Bank and NIH NeuroBioBank, Brent Harris, Iris Broce, Vivian Drory, John Ravits, Corey McMillan, Vilas Menon, Lani Wu and Steven Altschuler. The members of the PROSPECT Consortium are as follows: Khaled Amar, Neil Archibald, Oliver Bandmann, Erica Capps, Alistair Church, Jan Coebergh, Alyssa Costantini, Peter Critchley, Boyd CP Ghosh, Michele T.M. Hu, Edwin Jabbari, Christopher Kobylecki, P. Nigel Leigh, Carl Mann, Luke A Massey, Huw R Morris, Uma Nath, Nicola Pavese, Dominic Paviour, James B. Rowe, Jagdish Sharma and Jenny Vaughan.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process
REFERENCES
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, de Bakker PI, Deloukas P, Gabriel SB, et al. (2010). Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur KC, Calvo A, Price TR, Geiger JT, Chiò A, and Traynor BJ (2016). Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun 7, 12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean L, and Bayrak-Toydemir P (2014). American College of Medical Genetics and Genomics Standards and Guidelines for Clinical Genetics Laboratories, 2014 edition: technical standards and guidelines for Huntington disease. Genet. Med 16, e2–e2. [DOI] [PubMed] [Google Scholar]
- Brooks BR (1994). El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J. Neurol. Sci 124 Suppl, 96–107. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, and Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MJ, Gillis T, Atwal RS, Mysore JS, Arjomand J, Harold D, Holmans P, Jones L, Orth M, Myers RH, et al. (2017). Haplotype-based stratification of Huntington’s disease. Eur. J. Hum. Genet 25, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhetri SK, Dayanandan R, Bindman D, Craufurd D, and Majeed T (2014). Amyotrophic lateral sclerosis and Huntington’s disease: neurodegenerative link or coincidence? Amyotroph. Lateral Scler. Frontotemporal Degener 15, 145–147. [DOI] [PubMed] [Google Scholar]
- Chia R, Chiò A, and Traynor BJ (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, and White LA (2013). Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 41, 118–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciosi M, Maxwell A, Cumming SA, Hensman Moss DJ, Alshammari AM, Flower MD, Durr A, Leavitt BR, Roos RAC, Holmans P, et al. (2019). A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine 48, 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, Humphrey J, Jaunmuktane Z, Sivakumar P, Polke J, et al. (2019). Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet 51, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowska M, Juzwa W, Krzyzosiak WJ, and Olejniczak M (2018). Precise Excision of the CAG Tract from the Huntingtin Gene by Cas9 Nickases. Front. Neurosci 12, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rooij KE, De Koning Gans PA, Roos RA, Van Ommen GJ and Den Dunnen JT (1995). Somatic expansion of the (CAG)n repeat in Huntington disease brains. Hum. Genet, 95, 270–274. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhenko E, Deshpande V, Schlesinger F, Krusche P, Petrovski R, Chen S, Emig-Agius D, Gross A, Narzisi G, Bowman B, et al. (2019). ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhenko E, van Vugt JJFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, Ajay SS, Rajan V, Lajoie BR, Johnson NH, et al. (2017). Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 27, 1895–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, Mizuno Y, Broe GA, Cummings J, Dickson DW, Gauthier S, et al. (2007). Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord 22, 1689–1707. [DOI] [PubMed] [Google Scholar]
- Faber R (1999). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 53, 1159. [DOI] [PubMed] [Google Scholar]
- Ferrari R, Manzoni C, and Hardy J (2019). Genetics and molecular mechanisms of frontotemporal lobar degeneration: an update and future avenues. Neurobiol. Aging 78, 98–110. [DOI] [PubMed] [Google Scholar]
- Gardiner SL, Boogaard MW, Trompet S, de Mutsert R, Rosendaal FR, Gussekloo J, Jukema JW, Roos RAC, and Aziz NA (2019). Prevalence of Carriers of Intermediate and Pathological Polyglutamine Disease-Associated Alleles Among Large Population-Based Cohorts. JAMA Neurol. 76, 650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, Mollenhauer B, Müller U, Nilsson C, Whitwell JL, et al. (2017). Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord 32, 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jama M, Millson A, Miller CE, and Lyon E (2013). Triplet repeat primed PCR simplifies testing for Huntington disease. J. Mol. Diagn 15, 255–262. [DOI] [PubMed] [Google Scholar]
- Jun G, Flickinger M, Hetrick KN, Romm JM, Doheny KF, Abecasis GR, Boehnke M, and Kang HM (2012). Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet 91, 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai K, Kuwabara S, Sawai S, Nakata M, Misawa S, Isose S, Hirano S, Kawaguchi N, Katayama K, and Hattori T (2008). Genetically confirmed Huntington’s disease masquerading as motor neuron disease. Mov. Disord 23, 748–751. [DOI] [PubMed] [Google Scholar]
- Kennedy L, Evans E, Chen CM, Craven L, Detloff PJ, Ennis M and Shelbourne PF (2003). Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet 12, 3359–3367. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, and Fischbeck KH (1991). Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79. [DOI] [PubMed] [Google Scholar]
- Langbehn DR, Hayden MR, and Paulsen JS (2010). CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am. J. Med. Genet. B Neuropsychiatr. Genet 153b, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Zhang J, Su AI, Walker JR, Wiltshire T, Kang K, Dragileva E, Gillis T, Lopez ET, Boily MJ, et al. (2010). A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst. Biol 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillo P, and Hodges JR (2009). Frontotemporal dementia and motor neurone disease: overlapping clinic-pathological disorders. J. Clin. Neurosci 16, 1131–1135. [DOI] [PubMed] [Google Scholar]
- Loh PR, Palamara PF, and Price AL (2016). Fast and accurate long-range phasing in a UK Biobank cohort. Nat. Genet 48, 811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, et al. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983. [DOI] [PubMed] [Google Scholar]
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 11, 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, et al. (2005). Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65, 1863–1872. [DOI] [PubMed] [Google Scholar]
- Mouro Pinto R, Arning L, Giordano JV, Razghandi P, Andrew MA, Gillis T, Correia K, Mysore JS, Grote Urtubey DM, Parwez CR, Von Hein SM, et al. (2020). Patterns of CAG repeat instability in the central nervous system and periphery in Huntington’s disease and in spinocerebellar ataxia type 1. Hum. Mol. Genet 29, 2551–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D, Snowden J, and Mann D (2005). Frontotemporal dementia. Lancet Neurol. 4, 771–780. [DOI] [PubMed] [Google Scholar]
- Nielsen TR, Bruhn P, Nielsen JE, and Hjermind LE (2010). Behavioral variant of frontotemporal dementia mimicking Huntington’s disease. Int. Psychogeriatr 22, 674–677. [DOI] [PubMed] [Google Scholar]
- Onyike CU, and Diehl-Schmid J (2013). The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry 25, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgiou SG, Antelli A, Bonakis A, Vassos E, Zalonis I, Kalfakis N, and Panas M (2006). Association of genetically proven Huntington’s disease and sporadic amyotrophic lateral sclerosis in a 72-year-old woman. J. Neurol 253, 1649–1650. [DOI] [PubMed] [Google Scholar]
- Peplow M (2016) The 100,000 Genomes Project. Br. Med. J 353, i1757. [DOI] [PubMed] [Google Scholar]
- Phukan J, Ali E, Pender NP, Molloy F, Hennessy M, Walsh RJ, and Hardiman O (2010). Huntington’s disease presenting as amyotrophic lateral sclerosis. Amyotroph. Lateral Scler 11, 405–407. [DOI] [PubMed] [Google Scholar]
- Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, et al. (2018). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 10.1101/201178. [DOI] [Google Scholar]
- Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, and Jette N (2012). The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov. Disord 27, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Regier AA, Farjoun Y, Larson DE, Krasheninina O, Kang HM, Howrigan DP, Chen BJ, Kher M, Banks E, Ames DC, et al. (2018). Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat. Commun 9, 4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Chiò A, and Traynor BJ (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci 17, 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LP, and Shneider NA (2001). Amyotrophic lateral sclerosis. N. Engl. J. Med 344, 1688–1700. [DOI] [PubMed] [Google Scholar]
- Rubio A, Steinberg K, Figlewicz DA, MacDonald ME, Greenamyre T, Hamill R, Shoulson I, and Powers JM (1996). Coexistence of Huntington’s disease and familial amyotrophic lateral sclerosis: case presentation. Acta Neuropathol. 92, 421–427. [DOI] [PubMed] [Google Scholar]
- Sadeghian H, O’Suilleabhain PE, Battiste J, Elliott JL, and Trivedi JR (2011). Huntington Chorea Presenting With Motor Neuron Disease. Arch. Neurol 68, 650–652. [DOI] [PubMed] [Google Scholar]
- Sayagues JM, Tabernero MD, Maillo A, Trelles O, Espinosa AB, Sarasquete ME, Merino M, Rasillo A, Vera JF, Santos-Briz A, et al. (2006). Microarray-based analysis of spinal versus intracranial meningiomas: different clinical, biological, and genetic characteristics associated with distinct patterns of gene expression. J. Neuropath. Exp. Neurol 65, 445–454. [DOI] [PubMed] [Google Scholar]
- Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, and Schatz MC (2018). Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Hanson M, Singleton A, Hernandez D, McInerney A, Nussbaum R, Werner J, Gallardo M, Weiser R, Gwinn-Hardy K, et al. (2005). Analysis of SCA-2 and SCA-3 repeats in Parkinsonism: evidence of SCA-2 expansion in a family with autosomal dominant Parkinson’s disease. Neurosci. Lett 382, 191–194. [DOI] [PubMed] [Google Scholar]
- Swami M Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH, and Wheeler VC (2009). Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Genet 18, 3039–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, Blair NF, Craufurd D, Priller J, Rickards H, et al. (2019). Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med 380, 2307–2316. [DOI] [PubMed] [Google Scholar]
- Tada M, Coon EA, Osmand AP, Kirby PA, Martin W, Wieler M, Shiga A, Shirasaki H, Makifuchi T, Yamada M, et al. (2012). Coexistence of Huntington’s disease and amyotrophic lateral sclerosis: a clinicopathologic study. Acta Neuropathol. 124, 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J, Adam S, Greenberg C, Ives EJ, Clarke LA, et al. (1994). Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat. Genet 6, 409–414. [DOI] [PubMed] [Google Scholar]
- van Swieten J, Rosso S, and Heutink P (2000). MAPT-Related Disorders In GeneReviews®, Adam M, Ardinger HH, Pagon RA, et al. , ed. (Seattle (WA): University of Washington, Seattle; ). [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, and Richardson EP Jr. (1985). Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol 44, 559–577. [DOI] [PubMed] [Google Scholar]
- Wang K, Mingyao L, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The individual-level sequence data for the discovery genomes are available on dbGaP (accession number: phs001963.v1.p1 NIA DementiaSeq). The programming code used in this paper is available at https://github.com/dewanr2/Somatic_Instability.