

Abstract
Immunotherapy of immunologically cold solid tumors may require multiple agents to engage immune effector cells, expand effector populations and activities, and enable immune responses in the tumor microenvironment (TME). To target these distinct phenomena, we strategically chose five clinical-stage immuno-oncology agents, namely, (a) a tumor antigen-targeting adenovirus-based vaccine (Ad-CEA) and an IL15 superagonist (N-803) to activate tumor specific T cells, (b) OX40 and GITR agonists to expand and enhance the activated effector populations, and (c) an IDO inhibitor (IDOi) to enable effector-cell activity in the TME. Flow cytometry, TCR sequencing, and RNAseq analyses showed that in the CEA-transgenic murine colon carcinoma (MC38-CEA) tumor model, Ad-CEA+N-803 combination therapy resulted in immune-mediated antitumor effects and promoted the expression of costimulatory molecules on immune subsets, OX40 and GITR, and the inhibitory molecule IDO. Treatment with Ad-CEA+N-803+OX40+GITR+IDOi, termed the pentatherapy regimen, resulted in the greatest inhibition of tumor growth and protection from tumor rechallenge without toxicity. Monotherapy with any of the agents had little to no antitumor activity, whereas combining two, three, or four agents had minimal antitumor effects. Immune analyses demonstrated that the pentatherapy combination induced CD4+ and CD8+ T-cell activity in the periphery and tumor, and antitumor activity associated with decreased regulatory T-cell (Treg) immunosuppression in the TME. The pentatherapy combination also inhibited tumor growth and metastatic formation in 4T1 and LL2-CEA murine tumor models. This study provides the rationale for the combination of multi-modal immunotherapy agents to engage, enhance, and enable adaptive antitumor immunity.
Keywords: Combination immunotherapy, cancer vaccine, IL15, costimulatory agonists, IDO, Ad-CEA, N-803, OX40, GITR, epacadostat
INTRODUCTION
An effective anti-cancer immune response requires the successful initiation and propagation of the cancer-immunity cycle (1). In cancer patients, one or more steps in the cancer-immunity cycle may be aberrant, leading to suboptimal generation of effector cells or the release of tumor cell-derived factors in the tumor microenvironment (TME) that result in the suppression of effector cell activity. Tumors with high immune infiltration, also referred to as “hot” tumors, respond well to immune checkpoint inhibitors (ICIs)(2). However, non-T cell-inflamed “cold” tumors, such as some colorectal cancers, fail ICI treatment. Hence, optimal therapy of established tumors, especially of cold tumors, may require multiple immuno-oncology (IO) agents that would engage the immune response and generate tumor specific effector cells, expand and enhance the number and breadth of the immune effector cells, and enable the antitumor activity of these immune cells in the TME.
Therapeutic cancer vaccines can activate tumor-specific T cells and engage the antitumor response (3,4). Human carcinoembryonic antigen (CEA), which is overexpressed in various forms of colorectal cancer, represents a potential target for active immunotherapy (5,6). In this study, CEA-transgenic mice were implanted with CEA-transgenic MC38 (MC38-CEA) murine colon carcinoma cells. CEA-transgenic mice express human CEA as a self-antigen in fetal tissues and various parts of the gut, mimicking the CEA-expression pattern in humans (7). CEA-transgenic mice are typically unresponsive immunologically to CEA and, hence, serve as a model to study whether immunotherapy can overcome host immune tolerance to prevent growth of CEA-expressing tumors without targeting CEA-expressing normal tissues (8). Additionally, PD-L1 blockade results in minimal induction of protective immunity in the MC38-CEA model (9), making this a model for therapy after ICI failure.
T-cell activation requires inflammatory stimulus; hence, cytokines such as IL15 have the potential to augment cancer vaccine effects (3,4). IL15 signaling results in the proliferation, survival, and effector function of T cells and NK cells (10). N-803, which is composed of an IL15 mutant (IL-15N72D) complexed to the sushi domain of IL15Rα (IL-15RαSu) and the Fc region of human IgG, has been shown to exhibit significant CD8+ T cell- and NK cell-driven antitumor activity in diverse murine tumor models (11–14).
Induction of tumor necrosis factor receptors (TNFRs), such as OX40, 4–1BB, CD40, and glucocorticoid-induced TNFR-related protein (GITR), can enhance cancer vaccine activity by providing costimulatory signals required for T-cell activation (4,15,16). Both OX40- and GITR-mediated costimulation play an important role in enhancement of T-cell effector functions and memory generation, as well as inhibition of regulatory T-cell (Treg) activity.
The tumor evolves with the changing immune landscape in order to evade the emerging effector immune cells (17). For instance, IFNγ, which is critical in antitumor responses, induces the expression of immunosuppressive molecules such as programmed death-ligand 1 (PD-L1), a checkpoint molecule, and indoleamine-2,3-deoxygenase (IDO), an enzyme that catabolizes tryptophan to kynurenine and other metabolites (17,18). The metabolic stress exerted by the IDO pathway results in effector T-cell anergy and apoptosis, while at the same time promoting Treg generation and activity (19,20). Mechanisms to block the IDO pathway could potentially recondition the immunosuppressive TME to enable effector cell antitumor activity.
In this study, we show for the first time that a CEA-targeting adenovirus-based vaccine (Ad-CEA)+N-803 combination therapy resulted in tumor growth suppression associated with activation of CEA-specific T cells, increased TCR clonality, decreased T-cell exhaustion, and decreased Treg populations in a MC38-CEA tumor model. We identified OX40 and GITR as potential costimulatory targets to enhance T-cell activity and the IDO pathway as a potential inhibitory mechanism upon Ad-CEA+N-803 combination treatment. A rationally designed Ad-CEA+N-803+OX40+GITR+IDOi (termed “pentatherapy”) combination regimen showed that the pentatherapy regimen outperformed single, duplet, triplet, and quadruplet permutations of the agents in inhibiting tumor growth and stimulating CEA-specific CD4+ and CD8+ T-cell responses in the MC38-CEA tumor model. The pentatherapy regimen was also effective in controlling tumor metastasis in murine 4T1 breast and LL2-CEA lung carcinoma models. The pentatherapy regimen promoted CD8+ T cells in the tumor and was dependent on their activity for the therapeutic response. The combination also reprogrammed the metabolic state of tumors by decreasing kynurenine, thereby decreasing Treg function in the TME. The pentatherapy regimen was well-tolerated in mice, and the antitumor effects of the combination were not associated with any adverse effects. Overall, we demonstrated that this strategic combination of multiple IO agents, that could engage, expand, and enable the immune response, was essential to address diverse immune-tumor interactions in order to mount optimal antitumor therapy.
Materials and Methods
Cell lines
MC38-CEA, LL2-CEA, and MC38 cells were generated in our laboratory, and cultured as previously described (5,21). 4T1 and LLC cells were purchased from American Type Culture Collection in 2019. All cell lines were passaged for less than 6 months, free of Mycoplasma and cultured at 37°C, 5% CO2.
Animals and tumor models
C57BL/6 CEA-Tg were provided by Dr. John Shively (City of Hope National Medical Center, Duarte, CA). C57BL/6 CEA-transgenic (Tg) and Balb/c mice were bred and maintained at the National Institutes of Health (Bethesda, MD). Ad-CEA and N-803 were provided through a Cooperative Research and Development Agreement (CRADAs) with the National Cancer Institute and ImmunityBio, NantOmics. The epacadostat was provided by InCyte through Cooperative Research and Development Agreements (CRADAs) with the National Cancer Institute. OX40 (OX86) and GITR (DTA-1) were purchased from BioXcell (Lebanon, NH).
For the Ad-CEA+N-803 combination, 8–16-week-old female C57BL/6 CEA-Tg mice were implanted on the right flank with MC38-CEA tumor cells (3×105 cells/mouse subcutaneous (s.c.)). MC38-CEA-bearing mice were treated with Ad-CEA (107 viral particles, s.c., ImmunityBio, Culver City, CA) on days 7, 14, and 21 and N-803 (1 μg, s.c.) on days 10 and 17. For the depletion experiment, anti-CD8 (2.43, 100 μg (BioXcell, Lebanon, NH) and anti-NK1.1 antibody (PK136, BioXCell, Lebanon, NH) were injected (i.p.) on days 3, 4, 5, 12, 19, and 26 post-tumor inoculation. For the kinetic studies, naïve female C57BL/6 CEA-Tg were injected concurrently with Ad-CEA and N-803.
For the pentatherapy combination, 8–16-week-old C57BL/6 CEA-Tg mice were implanted with MC38-CEA (3×105 cells/mouse, s.c.) or LL2-CEA (5×105 cells/mouse, intravenous (i.v.)). The tumor-bearing mice were injected with GITR agonist (500 μg, i.p.) on day 5 post-tumor inoculation; Ad-CEA (107 viral particles, s.c.) on days 7, 14, and 21; OX40 agonist (100 μg, i.p.) on days 7, 14, and 21; N-803 (1 μg, s.c.) on days 14 and 21; and IDO inhibitor (IDOi), which was incorporated in the food at 2.5mg/day to target 100 mg/kg, was given starting day 7 ad lib. IDO chow was purchased from Research Diets (New Brunswick, NJ). IDO, tryptophan, and kynurenine were measured by HPLC in mouse serum 21 days following start of treatment (28 days following tumor transplant).
For the delayed treatment schedule, MC38-CEA tumor-bearing mice were administered with GITR agonist on day 12 post-tumor inoculation; Ad-CEA on days 14, 21, and 28; OX40 agonist on days 14, 21, and 28; N-803 on days 21 and 28; and IDOi starting day 12 using the same concentration and route of administration stated above. 4T1-bearing Balb/c mice were treated similarly but instead of Ad-CEA, Ad-Twist (107 viral particles, s.c.) was administered. 8–16 week-old Balb/c mice were inoculated with 4T1 cells (5×104 cells/mouse, s.c.) on the mammary fat pad. Ad-Twist (ImmunityBio) is an adenovirus-based vaccine encoding the transcription factor Twist, and has been previously described (45). Mice free of tumors for 15 days were re-challenged with CEA-expressing or CEA-negative MC38 or LL2 cells (3×105 cells/mouse, s.c.).
For PD-L1 treatment, MC38-CEA, LL2-CEA, and 4T1 tumor-bearing mice were injected with blocking anti-PD-L1 (10F.9G2, 200 mg, intraperitoneal (i.p.); BioXcell, Lebanon, NH) on days 14 and 21 after tumor implantation.
For all animal studies, tumor growth was monitored two times per week. Animal studies were terminated as indicated in the figure legends or when the tumors reached the ethical limit (2 cm length or width). Lungs from LL2-CEA animals were collected and perfused with India ink (Amazon.com) and metastatic nodules were manually counted. 4T1 metastases were quantified as previously described (22). Clonogenic metastatic cell colonies were enumerated by culturing single-cell suspension of the lungs in a 6-thioguanine selection medium for at least 12 days. All animal studies were approved and conducted in accordance under an IACUC-approved animal studies protocol (LTIB-38) with the approval of NIH/NCI, an Institutional Animal Care and Use Committee (IACUC). To monitor for irAEs, an independent, board-certified pathologist analyzed hematoxylin and eosin (H&E) slides of brain, duodenum, heart, kidney, liver, and lung samples collected from untreated and pentatherapy regimen-treated animals in a blinded manner.
Serum analyses
On day 28 following tumor transplant, blood samples were taked by mandibular bleed and processed for serum. Sera were assessed for IFNγ using the Perkin Elmer (Waltham, MA) IFNγ ELISA kit as per manufacturer’s direction. Serum liver enzymes, including blood urea nitrogen (BUN), creatinine (CRE), aspartate aminotransferase (AST), alanine aminotransferease (ALT), alkaline phosphatase (ALK), and trypanothione reductase (TPR) were analyzed by the Pathology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health (NIH) (Bethesda, MD).
TCR and RNA analyses
Genomic DNA was extracted from tumors using the DNeasy Blood and Tissue Kit (Qiagen, Germantown, MD). TCRβ sequencing was performed by Adaptive Biotechnologies (Seattle, WA), and analyzed using the immunoSEQ analyzer software by Adaptive Biotechnologies (Adaptive Biotechnologies).
Total RNA was extracted from the indicated tumors on day 28 post tumor transplant using the RNeasy Mini kit (Qiagen, Germantown, MD). nCounter PanCancer Immune Profiling Panel (NanoString Technologies; Seattle, WA) analysis, run by the Genomics Laboratory, Frederick National Laboratory for Cancer Research (Frederick, MD), was performed on the RNA. Data files were analyzed using the nSolver analysis software v.4.0.70 (NanoString; Seattle, WA) with the mRNA counts normalized to housekeeping genes and with the untreated sample as the categorical reference value. Nanostring data are available in the GEO database under accession number GSE162799. Common genes associated with immune activation, suppression, and exhaustion were depicted.
Functional analyses
Splenocytes were collected from animal cohorts 33 days post-tumor inoculation, and CEA-specific CD4+ lymphoproliferation and CD8+ T-cell responses were evaluated as previously described (23). A Treg suppression assay was performed as previously described (21) with some modification: Tregs were isolated from tumors of control and treated animals via magnetic bead (Miltenyi Biotech, San Diego, CA) and CD4+ T-cell proliferation was measured.
High-performance liquid chromatography HPLC
IDO, tryptophan, and kynurenine were measured by HPLC in mouse serum 21 days following start of treatment (28 days following tumor transplant)(Incyte, Willmington, DE; brief protocol in Supplementary File S1).
Multiplex immunofluorescence staining and multispectral imaging
Tumor samples from control and pentatherapy regimen-treated animals were collected on day 33 post-tumor inoculation, mounted, and stained for multispectral imaging as previously described (24).
Flow cytometry
The following antibodies from BD Biosciences (Franklin Lakes, NJ) were used for staining: CD8-FITC (53.67), CD8-APC (KT15), Foxp3-PE (FJK-16s), CD3-AF700 (17A2), CD44-PerCP-Cy5.5 (IM7), CD4-FITC (RM4– 5), CD62L-BV421 (MEL-14), CD25-APC (PC61), Ki67-PE-Cy7 (SolA15), CTLA-4-PE (UC10–4F10–11), PD-1-BV421 (RMP1–30), granzyme B-PE (NGZB), OX40-BV711 (OX86), GITR-BV510 (DTA-1). MHC class I-restricted (H2-Db) PE-labeled CEA-tetramer (sequence: EAQNTTYL) was purchased from MBL International Corporation (Woburn, MA). Live/Dead fixable aqua stain and transcription factor staining buffer set were purchased from Thermo Fisher (Waltham, MA). Flow cytometry was performed on BD LSRFortessa or BD FACSVerse (BD Biosciences) and analyzed using FlowJo v.9.7.6 or v.10.5.3 (TreeStar). Cell viability was examined using trypan blue staining prior to data acquisition. Live cells were gated via forward and side scatter. Isotype control staining was <5% for all samples analyzed.
Statistical analyses
Student’s t-test was used to compare two groups. One-way or two-way ANOVA was used to compare more than two groups. Tukey’s or Sidak’s post hoc test was used to correct both one- and two-way ANOVA. P values less than 0.05 were considered significant. Error bars represent mean±SEM. Analyses were performed using GraphPad Prism 7.0 software.
Results
Antitumor activity of Ad-CEA and N-803 in a MC38-CEA tumor model
Ad-CEA, an adenovirus-based vaccine targeting the human CEA antigen, has been demonstrated to elicit tumor-associated antigen (TAA)-specific CD8+ T-cell responses (6). We investigated the synergistic activity of Ad-CEA with N-803, an IL15 superagonist shown to promote stimulation and proliferation of NK cells and memory CD8+ T cells (14,25,26). We first utilized MC38-CEA, a murine colon carcinoma that is unresponsive/minimally responsive to checkpoint therapy with anti-PD-L1 (Fig. 1A, inset for tumor growth). MC38-CEA-bearing C57BL/6 CEA-Tg mice were treated with Ad-CEA vaccine alone on days 7, 14, and 21, with N-803 alone on days 10 and 17 post-tumor inoculation, or with a combination of both IO agents (Fig. 1A). The Ad-CEA+N-803 combination significantly inhibited tumor progression compared to monotherapy treatments. The median survival of the animals that received the combination therapy was 31 days and was significantly higher (P<0.05) than the 22.5–25-day median survival observed in the no treatment and monotherapy groups. CD8+ T-cell depletion negatively impacted (P<0.05) the prolonged survival caused by the combination therapy (Fig. 1A). Our preliminary data demonstrated that treatment with N-803 prior to or concurrent with Ad-CEA priming did not result in increased survival (Supplementary Fig. S1).
Figure 1. The combination of Ad-CEA and N-803 inhibits MC38-CEA tumor growth and promotes CD8+ T-cell responses.
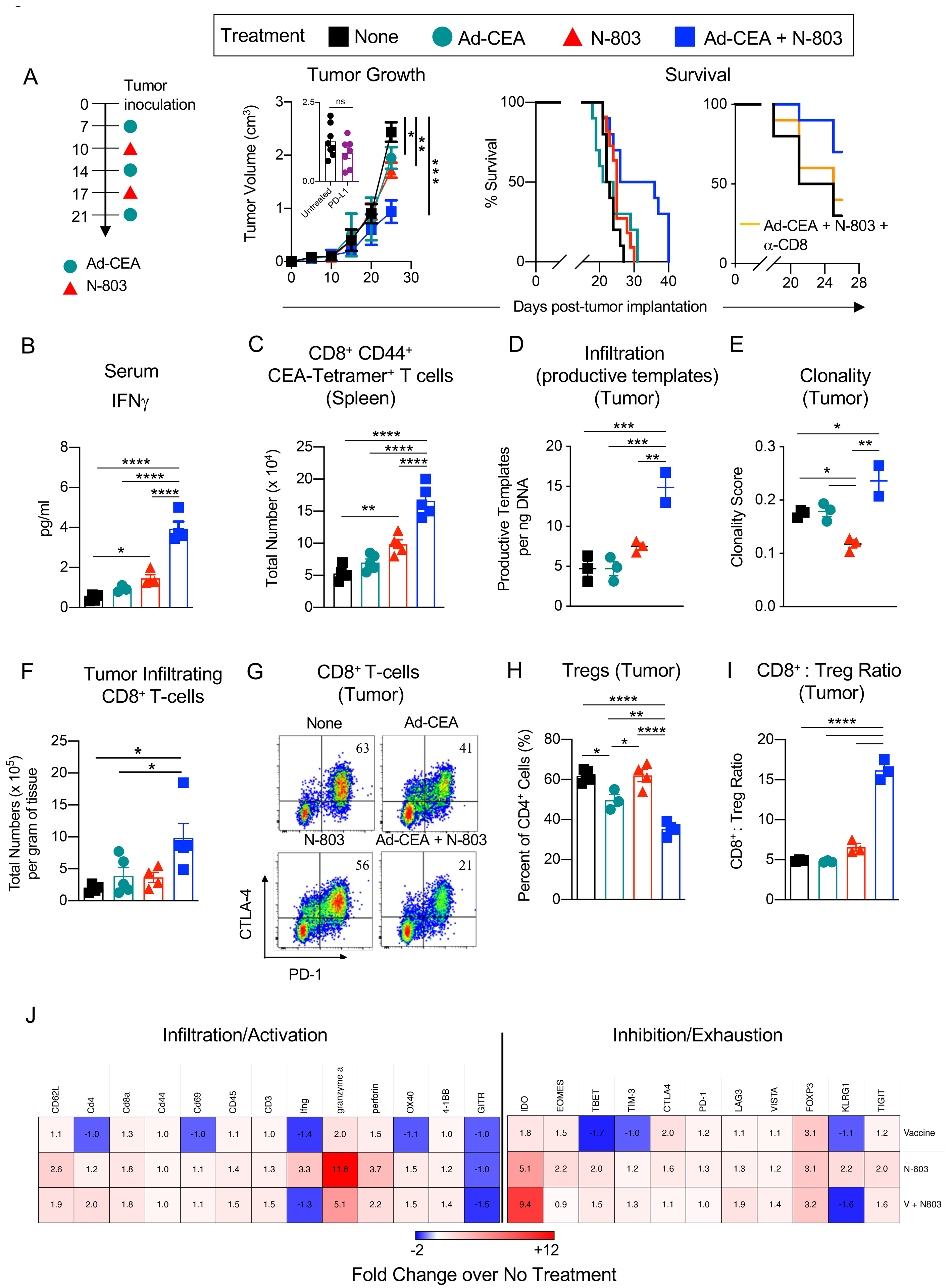
Female C57BL/6-CEA-Tg mice (10–16 weeks old) were inoculated with MC38-CEA tumors. A. Treatment schema: injected tumor-bearing mice (n=10/group) with Ad-CEA (Vaccine, V) on days 7, 14, and 21 post-tumor implantation, with the IL15Rα superagonist N-803 on days 10 and 17, or each agent alone. Tumor growth and animal survival were monitored. Subsets of mice (n=7–8/group) were also treated with blocking anti-PD-L1 on days 14 and 21 post-tumor implantation, and tumor volumes were reported on day 25 post-tumor implantation (inset). The effect of CD8+ T-cell depletion on the survival of animals treated with the combination therapy was also monitored. On day 25 post-tumor inoculation, sera, spleens, and tumors were collected (n=3–5/group). B. Serum IFNγ quantified by ELISA. C. CEA-specific CD8+CD44+ splenocytes were identified via CEA tetramer staining. D-E. Genomic DNA was isolated from the tumors and were analyzed for (D) productive TCRβ rearrangement and (E) TCRβ clonality. F. Frequency of live tumor-infiltrating CD8+ T cells was determined by flow cytometry, and total CD8+ T cells per gram of tumor tissue was calculated. G. PD-1 and CTLA-4 expression on tumor-infiltrating CD8+ T cells was measured by flow cytometry. H. Tumor-infiltrating Tregs (CD4+CD25+FoxP3+) were quantified via flow cytometry. I. CD8+ T cell-to-Treg ratio in the tumor was determined. J. Immune-related transcriptome of the tumor samples was analyzed using the nCounter PanCancer Immune Profiling Panel. Heatmap showing select genes with data presented as fold change values on scale of −2 (blue) to +12 (red). One-way or two-way ANOVA with Tukey’s post hoc test for group analyses, t-test for comparison of two groups, and Kaplan-Meier survival analysis. *P<0.05; **P<0.01; ***P<0.005, ****P<0.001. Error bars represent mean±SEM. These studies were repeated 3–4 times with similar results.
Next, immune activity that resulted from the Ad-CEA, N-803, and combination treatment on day 25 post-tumor inoculation was analyzed. Serum analysis showed that the proinflammatory cytokine IFNγ (Fig. 1B) was significantly elevated (P<0.01) in the Ad-CEA+N-803 group compared to the groups that received only one IO agent (Fig. 1B). Monotherapy with N-803 increased IFNγ compared to control (P<0.05). Tetramer staining revealed that activated CEA-specific CD8+ splenocytes were expanded in animals that received combination therapy (P<0.001; Fig. 1C). Ad-CEA alone did not promote expansion of CEA-specific T cells. Compared to control, N-803 increased CEA-specific CD8+ T cells but was still much lower than that induced by the combination therapy. This boost in CEA-specific T cells may be the result of general non-specific activation of T cells by N-803 (12).
Intratumoral T cells were also assessed. TCRβ sequencing of tumor genomic DNA showed that the amount of productive TCR templates in the tumor, which approximates the number of tumor-infiltrating T cells, was improved by the Ad-CEA+N-803 combination (Fig. 1D). Sequencing also revealed that the combination therapy resulted in higher T-cell clonality (Fig. 1E), which has been demonstrated to be a potential surrogate marker for immune responses (27,28). N-803 monotherapy, on the other hand, decreased TCR clonality and increased TCR diversity in the tumor compared to Ad-CEA alone and combination therapy.
Flow cytometric analysis of tumor-infiltrating lymphocytes (TILs) showed significant CD8+ T-cell infiltration after Ad-CEA+N-803 combination therapy but not with the other monotherapies (Fig. 1F). The CD8+ TILs in the combination therapy group were also less exhausted compared to control, as demonstrated by the three-fold decrease in CTLA-4+PD-1+ TILs (Fig. 1G). The CD4+CD25+FoxP3+ Treg population, on the other hand, was minimized by the combination therapy and by Ad-CEA treatment (Fig. 1H). The combination therapy resulted in an improvement of the CD8+ T-cell to Treg ratio (Fig. 1I). RNA analysis of bulk tumors demonstrated that compared to control, the Ad-CEA+N-803 combination, N-803, and, to a lower extent, Ad-CEA vaccine generally promoted the expression of genes associated with immune cell activation and infiltration (Fig. 1J). However, gene expression of some markers associated with immune inhibition and exhaustion, such as IDO, was also upregulated. Overall, the data suggest that tumor growth suppression by Ad-CEA+N-803 combination therapy is associated with robust CD8+ T-cell clonal expansion, activity, and tumor infiltration, accompanied by a decrease in Treg.
Therapy with Ad-CEA and N-803 drives OX40 and GITR expression in T cells
OX40 and 4–1BB expression increased with Ad-CEA + N-803 (Fig. 1J). GITR expression, on the other hand, appeared to decrease with the combination. However, previous studies have shown that GITR expression is upregulated 24–72 hours after TCR engagement (29,30). Because the RNA analysis was performed 96 hours after the last vaccination, we re-evaluated GITR expression over time on splenic T cells from naïve C57BL/6-CEA mice treated with Ad-CEA+N-803. An increase in the frequency, degree, and total number of CD4+ and CD8+ T cells that express OX40 and GITR was observed within 24 hours after the treatment (Fig. 2A–D). Likewise, 4–1BB expression on CD4+ and CD8+ splenocytes was also elevated within 24 hours after Ad-CEA+N-803 treatment (Supplementary Fig. S2). These data suggest that OX40, GITR, and 4–1BB were potentially valuable targets that could be activated to improve Ad-CEA+N-803 antitumor activity.
Figure 2. Combination of Ad-CEA+N-803 temporally upregulates OX40 and GITR expression in CD4+ and CD8+ splenocytes.
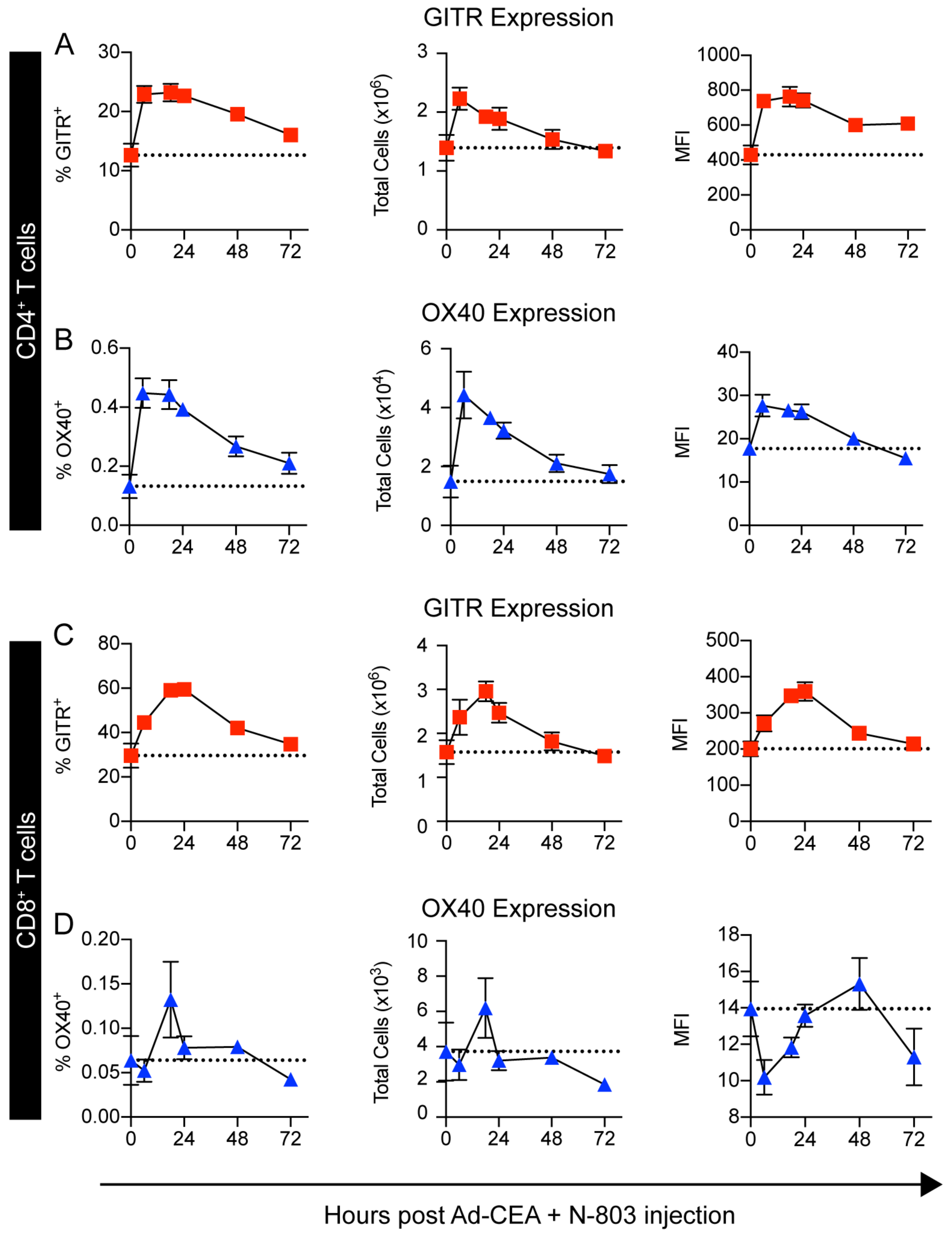
Naive female C57BL/6-CEA-Tg mice were injected concurrently with Ad-CEA (subcutaneous, s.c.) and N-803 (s.c.). Spleens were collected 0, 8, 18, 24, 48, and 72 hours after the injection. Frequency, total cell number, and mean fluorescence intensity (MFI) of GITR and OX40 expression on (A-B) CD4+ and (C-D) CD8+ splenocytes were analyzed via flow cytometry. Error bars represent mean±SEM. These studies were repeated 3–4 times with similar results.
Pentatherapy results in superior tumor growth suppression compared to monotherapies, duplets, triplets, and quadruplets of the IO agents
Based on the previous data, we hypothesized that OX40 and GITR agonists would further expand and enhance the T-cell activation, and that it was necessary to counter the IDO increase observed in tumors upon Ad-CEA+N-803 treatment (Fig. 1J). Thus, Ad-CEA+N-803 was combined with antibody agonists for OX40 and GITR to enhance antitumor immune activity, and epacadostat, an IDO inhibitor (IDOi), to enable immune effector activity in an otherwise immunosuppressive TME. In CEA-Tg mice bearing CEA-expressing tumors, we investigated the antitumor activity of the Ad-CEA+N-803+OX40+GITR+epacadostat (IDOi) pentatherapy combination and compared it to the efficacy of the different permutations of these IO agents (with Ad-CEA and N-803 always administered together; Fig. 3A). The tumor growth curves (Fig. 3B–P) show that monotherapy, doublet, triplet, and quadruplet iterations of the IO agents had little to moderate effect on the tumor growth of the MC38-CEA tumors in vivo. The fraction of animals with tumors smaller than 300 mm3 did not surpass 22% in these groups. On the other hand, the pentatherapy regimen (Fig. 3Q) resulted in significant tumor growth inhibition (P<0.001) in the MC38-CEA model, with 67% of the animals having <300 mm3 in tumor volume. Each of the components of the pentatherapy combination contributed to the antitumor effect; tumor growth inhibition was abrogated upon exclusion of IDOi (P<0.05; Fig. 3M), GITR (P<0.05; Fig. 3N), OX40 (P<0.0001; Fig. 3O), or Ad-CEA+N-803 (P<0.05; Fig. 3P).
Figure 3. Strategic combination of Ad-CEA, N-803, OX40, GITR, and IDOi (pentatherapy regimen) results in superior tumor growth suppression.
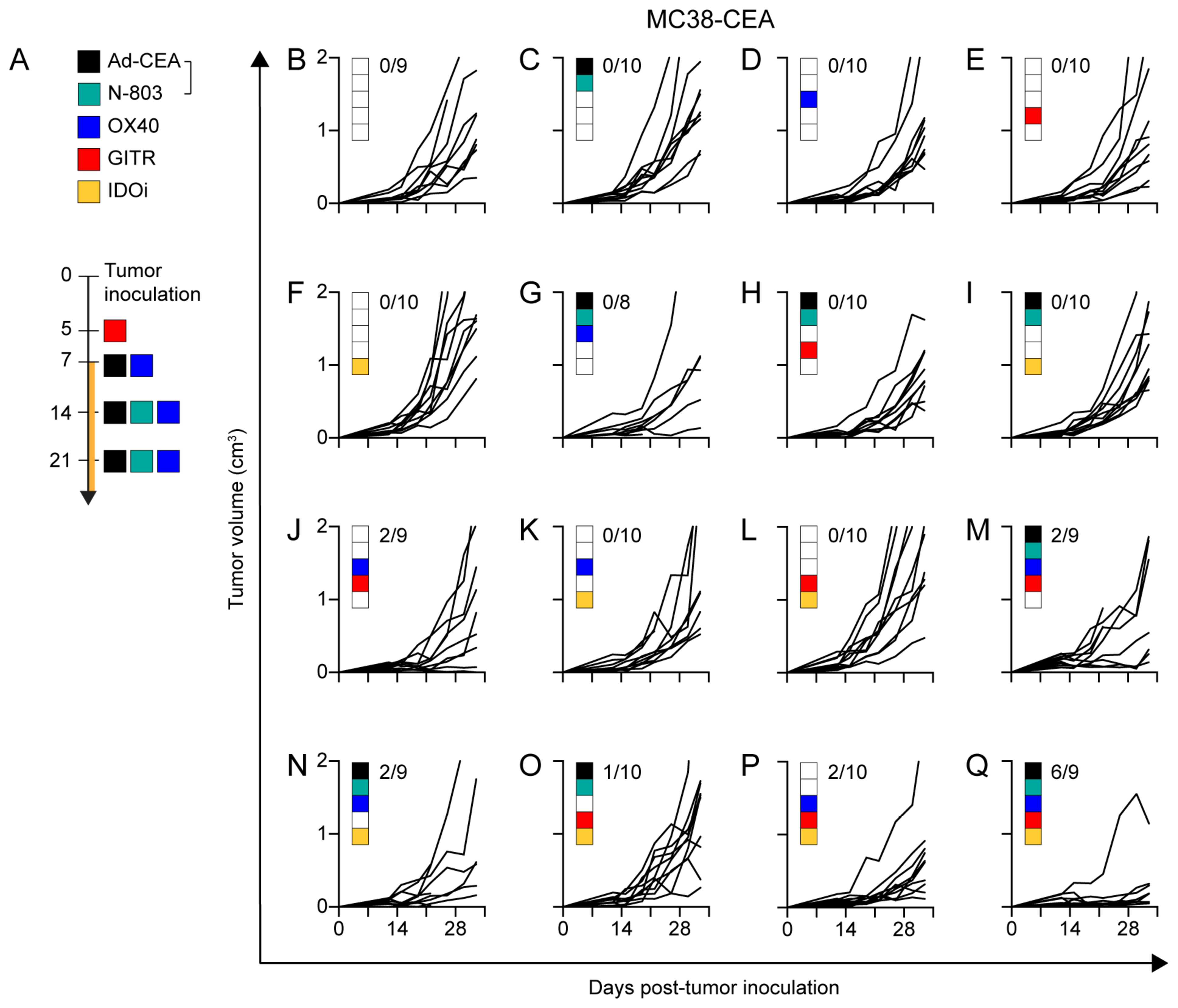
A. The pentatherapy combination treatment schedule wherein tumor-bearing mice were administered with: (1) the vaccine Ad-CEA on days 7, 14, and 21 post-tumor inoculation, subcutaneous (s.c.); (2) the superagonist N-803 on days 14, and 21, s.c.; (3) the costimulatory agonist OX40 on days 7, 14, and 21, intraperitoneally (i.p.); (4) the costimulatory agonist GITR on day 5, i.p.; and (5) the IDO inhibitor (IDOi) epacadostat feed starting at day 7. B-Q. Female C57BL/6-CEA-Tg mice (8–16 weeks old; n=10/group) were inoculated with 3×105 MC38-CEA cells on the flank, s.c. Sixteen different IO agent combinations were employed by following the treatment schedule described in (A). Tumor volume was monitored. Inset values represent fraction of mice with <300mm3 tumor volume at the endpoint for each group. Two-way or one-way ANOVA with Tukey’s post hoc test. Error bars represent mean±SEM. These studies were repeated 3–4 times with similar results.
CEA-specific CD8+ and CD4+ T-cell activities were investigated by collecting splenocytes and stimulating them with a H2Kb-CEA peptide or CEA protein, respectively. ELISAs demonstrated that the pentatherapy regimen resulted in elevated production of CEA peptide-induced IFNγ compared to monotherapy treatments (Fig. 4A). GITR+IDOi, OX40+GITR+IDOi, and Ad-CEA+N-803+OX40+GITR combinations also stimulated IFNγ. The pentatherapy regimen generated superior CD4+ T-cell proliferation compared to untreated, Ad-CEA+N-803, and OX40+GITR treatments, and the degree of proliferation was dependent on the concentration of CEA protein in the culture (Fig. 4B).
Figure 4. The pentatherapy regimen results in the activation of CEA-specific CD4+ and CD8+ T cells in the MC38-CEA tumor model.
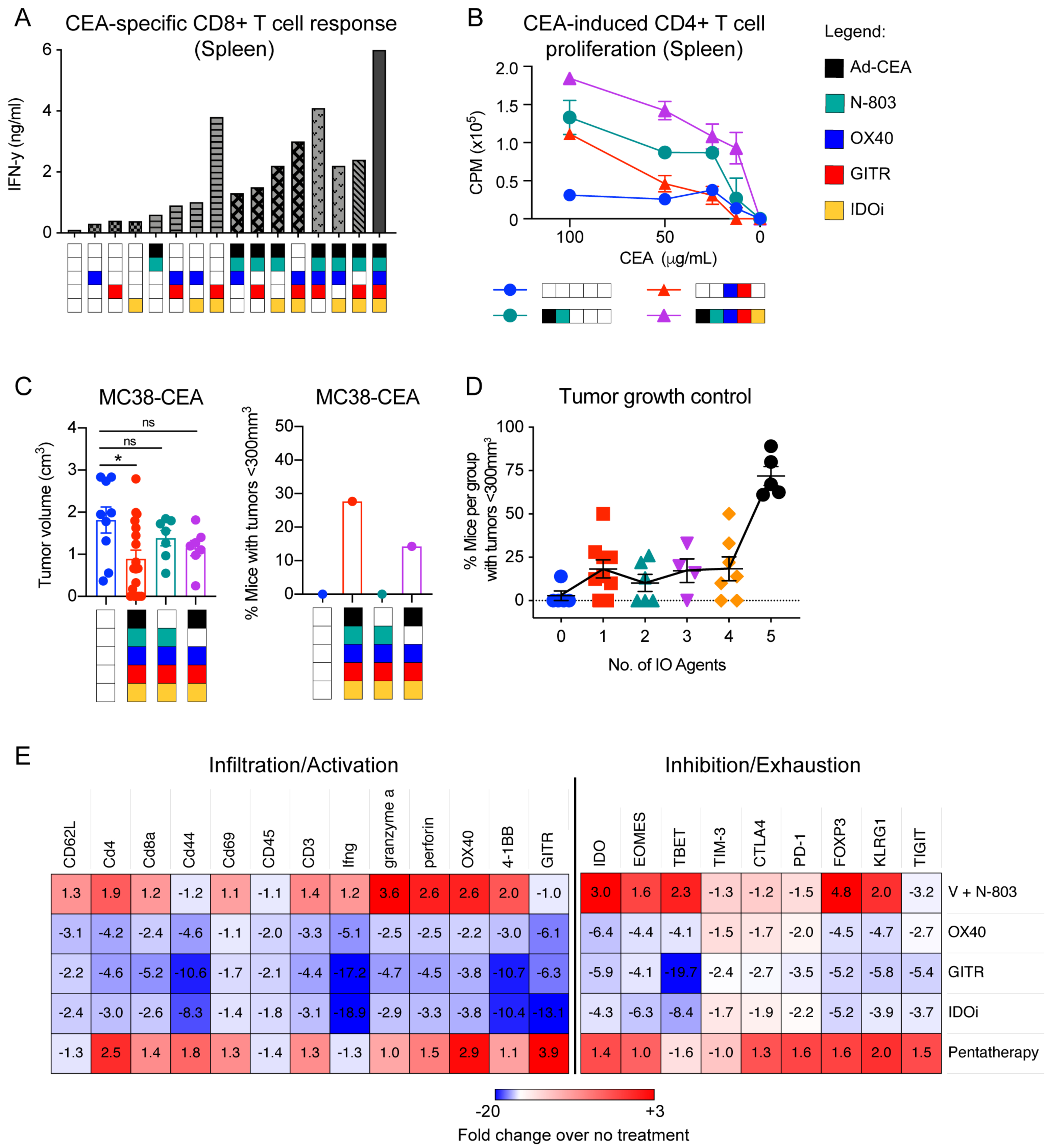
A. Splenocytes were collected on day 33 post-tumor inoculation and were analyzed via ELISA for IFNγ production after in vitro stimulation with an H2Kb-restricted CEA peptide. B. Proliferation of splenocytes from untreated, Ad-CEA+N-803, OX40+GITR, and the pentatherapy combination groups (n=5/group) was evaluated after ex vivo stimulation with I-Ab CEA peptide using a 3H-thymidine incorporation assay. C. Female C57BL/6-CEA-Tg mice (8–16 weeks old; n=10/group) were inoculated with 3×105 MC38-CEA cells on the flank. The tumor-bearing animals were treated with the pentatherapy regimen, N-803+OX40+GITR+IDOi (no Ad-CEA) or Ad-CEA+OX40+GITR+IDOi (no N-803). Tumor growth was monitored, and tumor volume and tumor growth control on day 28 are reported. D. For each treatment combination tested, the percentage of mice with tumor volume <300 mm3 was calculated and plotted against the number of IO agents received. Meta-analysis of four independent experiments is shown. E. Immune-related transcriptomes of tumor samples were analyzed using NanoString’s PanCancer Immune Profiling Panel. Heatmap showing select genes with data presented as fold-change values on scale of −20 (blue) to +3 (red). n.s., not significant. Error bars represent mean±SEM. These studies were repeated 3–4 times with similar results.
We next sought whether each of the IO agents was essential in the superior antitumor response associated with the pentatherapy regimen. MC38-CEA tumor-bearing animals were treated with the pentatherapy regimen, N-803+OX40+GITR+IDOi, or Ad-CEA+OX40+GITR+IDOi. Tumor volume and tumor growth suppression on day 28 post-tumor inoculation demonstrated that that removal of either Ad-CEA or N-803 abrogated tumor inhibition (Fig. 4C). A meta-analysis of the pentatherapy combination was performed, wherein for each group, the percent of mice that had tumors <300 mm3 was plotted against the number of agents administered to that group (Fig. 4D). Increasing the number of IO agents did not necessarily result in improved tumor growth control until agents that engaged, expanded, and enhanced the immune response were given.
RNA analysis of bulk tumors was performed on single modality treatments (Ad-CEA (V)+N-803, OX40, GITR, and IDOi) and the pentatherapy regimen. In concurrence with Fig. 1J, Ad-CEA+N-803 generally promoted expression of genes associated with immune cell activation and infiltration with concomitant increases in expression of immune inhibition and exhaustion markers (Fig. 4E). It appeared that the elevation of activation and inhibition markers observed in the pentatherapy regimen group was mainly driven by the Ad-CEA and N-803 components, as the expression pattern in the two groups was similar and OX40, GITR, and IDOi appeared to deregulate the expression these genes.
Next, the efficacy of the pentatherapy regimen was compared in small (53.3±4.4 mm3) and larger (163±23.7 mm3) MC38-CEA tumors (Fig. 5A). In order to obtain larger tumors, the pentatherapy treatment was initiated at day 14 rather than day 7. Compared to the untreated control, the pentatherapy regimen resulted in robust tumor growth suppression of both small (P<0.0001) and larger (P<0.0001) tumors. However, animals with small tumors were cured at the rate of 90%, whereas the animals with larger tumors were cured only at a 35% rate. This demonstrated that although the pentatherapy regimen resulted in a therapeutic advantage in both small and larger tumors, a more curative benefit could be achieved when the treatment was given to newly established tumors. Nevertheless, mice that were cured from small and large MC38-CEA tumors were all protected from tumor recurrence after MC38-CEA re-challenge (Fig. 5B). After 14 days, mice were challenged with MC38 (CEA-negative), and 80% (4/5 mice) were again protected from tumor.
Figure 5. The pentatherapy regimen has therapeutic benefits against larger MC38-CEA tumors and against additional cold tumor models LLC-CEA and 4T1.
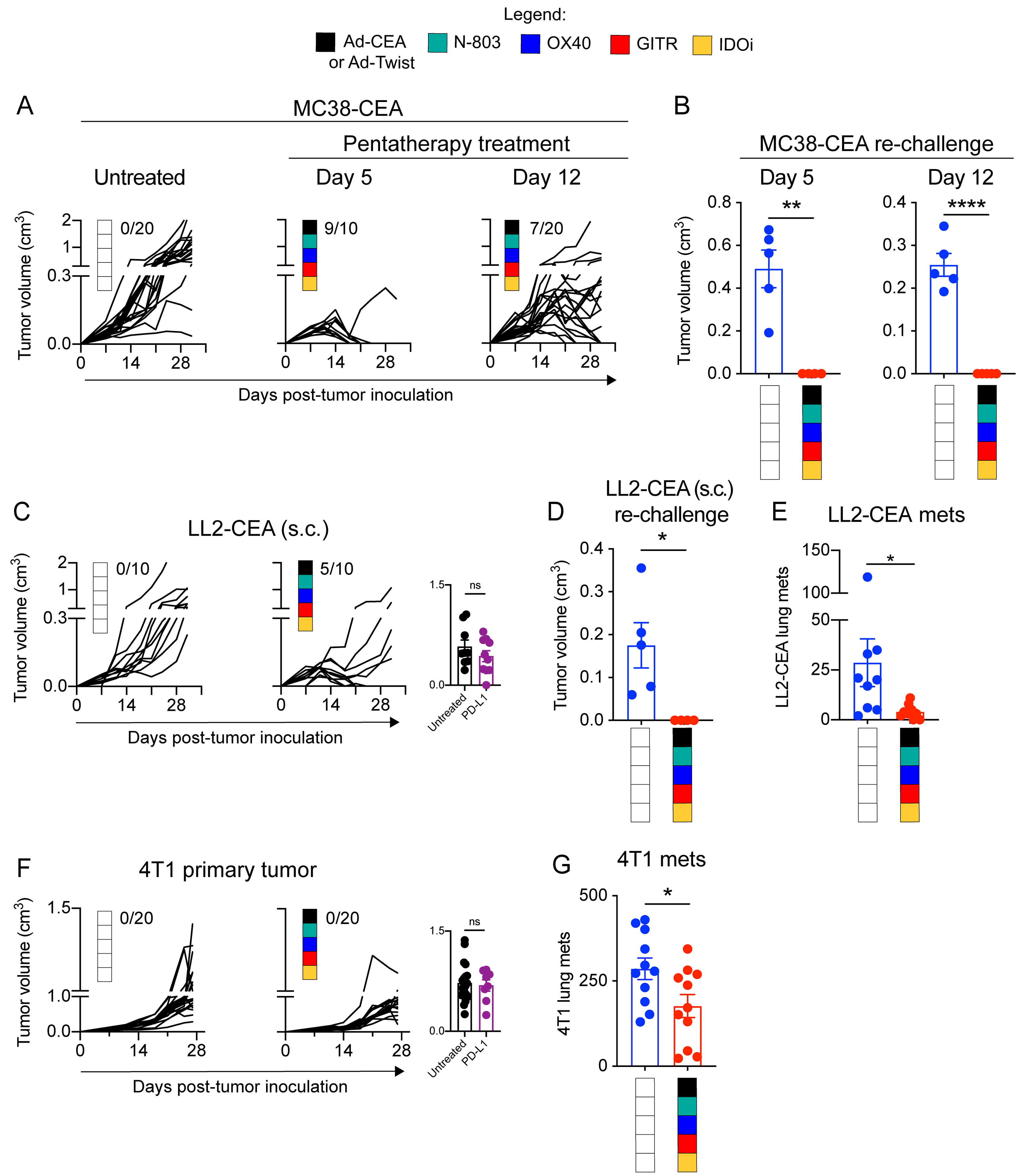
A. Female C57BL/6-CEA-Tg mice (8–16 weeks old; n=10–20/group) were inoculated with 3×105 MC38-CEA cells on the flank. The MC38-CEA tumor-bearing mice were treated with the pentatherapy regimen as described in Fig. 3A or given a delayed pentatherapy regimen treatment where Ad-CEA was given on days 14, 21 and 28 post-tumor inoculation, subcutaneous (s.c.); N-803 on days 21, and 28, s.c.; OX40 on days 14, 21 and 28, intraperitoneal (i.p.); (4) GITR on day 12, i.p.; and (5) the IDO inhibitor (IDOi) epacadostat feed starting at day 12. Tumor volume was monitored. Inset values represent fraction of mice that were cured in each group. B. Cured mice from (A) were re-challenged with MC38-CEA tumor cells (s.c.; 1×106 cells/mouse for the group that was treated using the original schedule, and 3×105 cells/mouse for the group that received delayed treatment), and tumor volume was reported on day 16 after tumor re-challenge. C. Female C57BL/6-CEA-Tg mice (8–16 weeks old; n = 10–20/group) were inoculated with 3×105 LL2-CEA cells on the flank (s.c.) and subsequently treated with the pentatherapy regimen as described in Fig. 3A. Tumor volume was monitored. Inset values represent fraction of mice that were cured in each group. D. Cured mice from (C) were re-challenged with 3×105 LL2-CEA tumor cells (s.c.), and tumor volume was reported on day 16 after tumor re-challenge. E. Female C57BL/6-CEA-Tg mice (8–16 weeks old, n=9–10/group) were injected with 5×105 LL2-CEA cells, intravenous (i.v.), and the animals were left untreated or administered with the pentatherapy combination as described in Fig. 3A. On day 28, lungs were collected, and metastatic tumor nodules were manually counted. F. Female Balb/c mice (8–16 weeks old) were inoculated with 5×104 4T1 cells on the mammary fat pad, s.c. 4T1-bearing mice (n=15/group) were either left untreated or administered with the pentatherapy combination, using Ad-Twist instead of Ad-CEA, as described in Fig. 3A. Primary tumor growth was monitored. Inset values represent fraction of mice that were cured in each group. G. On day 28 post-tumor inoculation, lung tissues were harvested from the 4T1-bearing animals. Clonogenic metastatic cell colonies were enumerated by culturing single-cell suspension of the lungs in a 6-thioguanine selection medium for at least 12 days. Two-way ANOVA with Tukey’s or Sidak’s post hoc test was used for grouped analyses; t-test was used to compare two groups. *P<0.05; **P<0.01; ****P<0.001. Error bars represent mean±SEM. These studies were repeated 3–4 times with similar results.
The efficacy of the pentatherapy combination was also interrogated in additional tumor models. Like the MC38-CEA tumor model, the CEA-transgenic Lewis lung carcinoma (LL2-CEA) subcutaneous model was also recalcitrant to PD-L1 treatment but was responsive to pentatherapy treatment (P<0.005; Fig. 5C). The pentatherapy treatment cured 50% of the LL2-CEA tumor-bearing mice, and these animals were protected from tumor recurrence after re-challenge (Fig. 5D). After 14 days, mice were challenged with LL2 (CEA-negative), and 75% (1 of 4 mice) were again protected from tumor. Likewise, in the LL2-CEA metastatic model, animals that received the pentatherapy treatment developed fewer tumor nodules in the lungs compared to control (P<0.05; Fig. 5E). Using the 4T1 orthotopic model of triple-negative breast cancer, another cold tumor unresponsive to PD-L1 blockade, we observed that the pentatherapy regimen using the Ad-Twist vaccine significantly inhibited primary tumor growth (P<0.001; Fig. 5F) and formation of lung metastases (P<0.01; Fig. 5G). The data demonstrated the potential applicability of the pentatherapy regimen in a wide range of tumor types.
The pentatherapy therapy did not result in toxicity in the MC38-CEA tumor model
One of the major concerns in cancer immunotherapy is immune-related adverse events (irAEs). To monitor for irAEs, an independent, board-certified pathologist analyzed H&E slides of brain, duodenum, heart, kidney, liver, and lung samples collected from untreated and pentatherapy regimen-treated animals in a blinded manner (Supplementary Fig. S3A). Overall, the tissues exhibited normal architecture and cellularity, with no perivascular inflammatory cells, such as lymphocytes, present. The pentatherapy regimen-treated animals were also within the normal range for other parameters measured, such as animal body weight, complete blood count, and liver enzymes (Supplementary Table. S1).
Pentatherapy promotes effector T-cell populations and inhibits Treg suppression
Tumor infiltration of effector T cells is critical in anti-cancer responses (31). Ad-CEA, N-803, OX40, GITR, and IDOi each invigorated effector T-cell responses. Hence, we predicted that the pentatherapy regimen would have a favorable effect on CD8+ and CD4+ T cells in the TME. Flow cytomety showed that the pentatherapy regimen promoted the expansion of splenic (P<0.01) and tumor-infiltrating CD8+ T cells (P<0.05; Fig. 6A). The increase in CD8+ T cells in the TME was confirmed via immunofluorescence (P<0.05; Fig. 6B). The Ki67 staining suggested that the pentatherapy regimen resulted in significant CD8+ T proliferation in the spleen (P<0.05) but not in the tumor (Fig. 6C). This indicated that the accumulation of intratumoral CD8+T cells was due to infiltration into the TME and not via local expansion. Pentatherapy combination did not induce greater frequencies of effector memory (CD44hi) and central memory (CD44+CD62L+) CD8+ T cells in the spleen or tumor compared to control (Fig. 6D). Nevertheless, a higher frequency of CD8+ T cells in the tumors were cytolytic based on granzyme B (P<0.01; Fig. 6E). The pentatherapy regimen resulted in higher frequency of CEA-specific T cells in the spleen and a higher number of CEA-specific T cells per tumor volume (P<0.05; Fig. 6F). In total, the pentatherapy combination improved the antitumor activity and tumor infiltration of the CD8+ T cells. When CD8+ T cells were depleted, the pentatherapy regimen failed to control tumor growth (P<0.01; Fig. 6G). Depletion of NK cells did not inhibit the therapeutic efficacy of the pentatherapy combination.
Figure 6. Antitumor response by Ad-CEA, N-803, OX40, GITR, and IDOi combination (pentatherapy) is correlated and dependent on CD8+ T-cell activation.
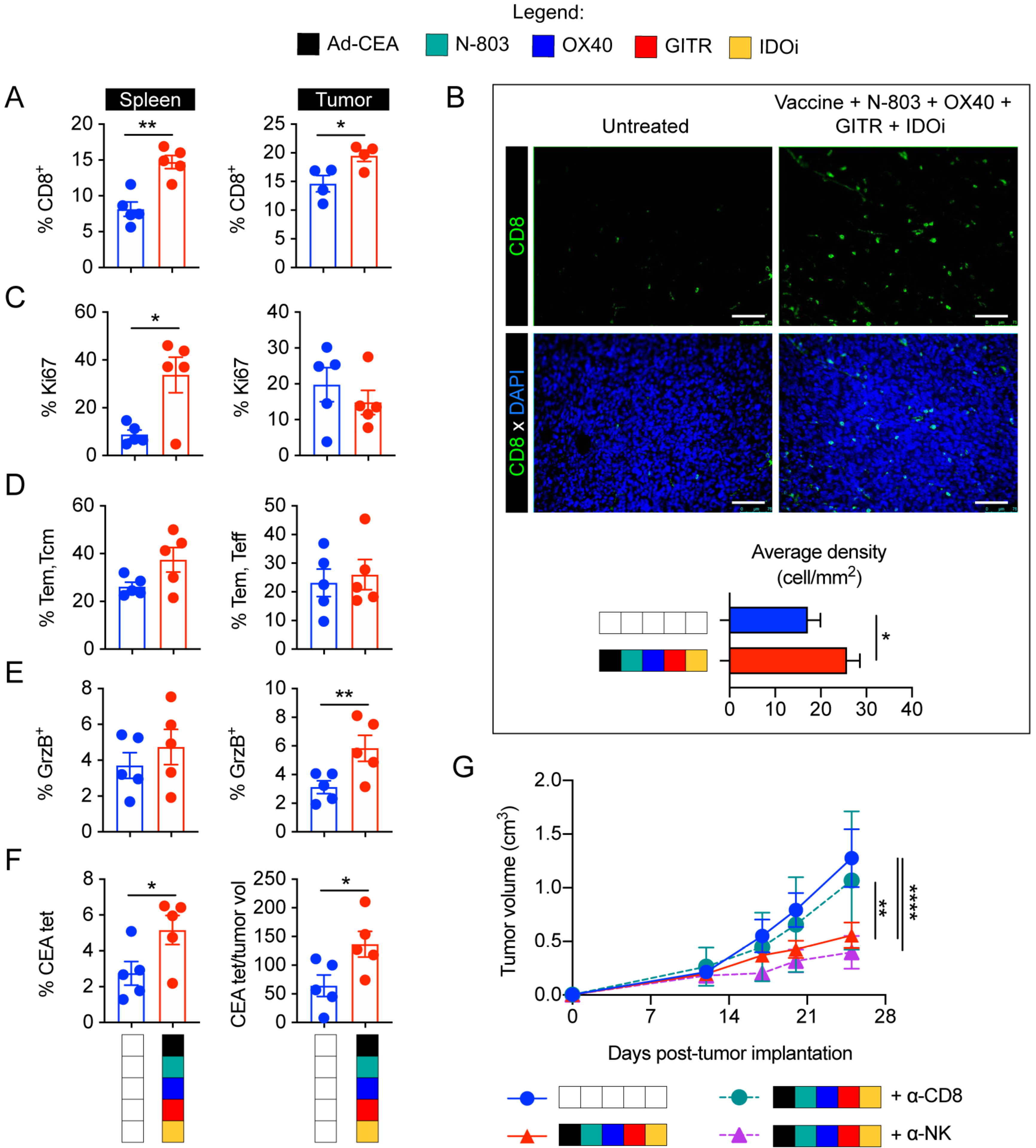
Female C57BL/6-CEA-Tg mice (8–16 weeks old; n=10/group) were inoculated with 3×105 MC38-CEA cells on the flank (subcutaneous, s.c.). Tumor-bearing mice were either left untreated or administered with the pentatherapy combination using the treatment schedule described in Fig. 3A. A. Spleens and tumors were collected on day 28 post-tumor inoculation and assessed via cytometry for CD8+ T cells (gated on live, CD3+ cells). B. Tumor infiltration of CD8+ T cells was assessed via immunofluorescence. C-F. Using flow cytometry, CD8+ T cells were further examined for (C) Ki67+ cells, (D) CD44+ and CD44+CD62L+ cells, and (E) granzyme B+ cells, as well as (F) CEA-tetramer staining. Student t-test. G. Pentatherapy-treated mice were injected with CD8- or NK-depleting antibodies on days 3, 4, 5, 12, 19, and 26 post-tumor inoculation. Tumor growth was monitored. Two-way ANOVA with Tukey’s post hoc test; *P<0.05; **P<0.01; ****P<0.001. Error bars represent mean±SEM. These studies were repeated 3–4 times with similar results.
Flow cytometry and immunofluorescence demonstrated that CD4+ T cells were enriched in spleens and tumors of pentatherapy-treated animals (Fig. 7A–B). The pentatherapy regimen promoted effector CD4+FoxP3− T cells over the CD4+FoxP3+ Tregs as demonstrated by the ratio of the two cell types. Ki67 staining implied that the pentatherapy combination did not significantly induce CD4+ T-cell proliferation (Fig. 7C). The pentatherapy regimen also increased the frequencies of tumoral effector memory (CD44hi) and central memory (CD44+CD62L+) CD4+ T cells (P<0.05), which are essential in facilitating and enhancing CD8+ T-cell antitumor responses (32).
Figure 7. Ad-CEA, N-803, OX40, GITR, and IDOi combination promotes effector CD4+ T cell populations and inhibits Treg suppression.
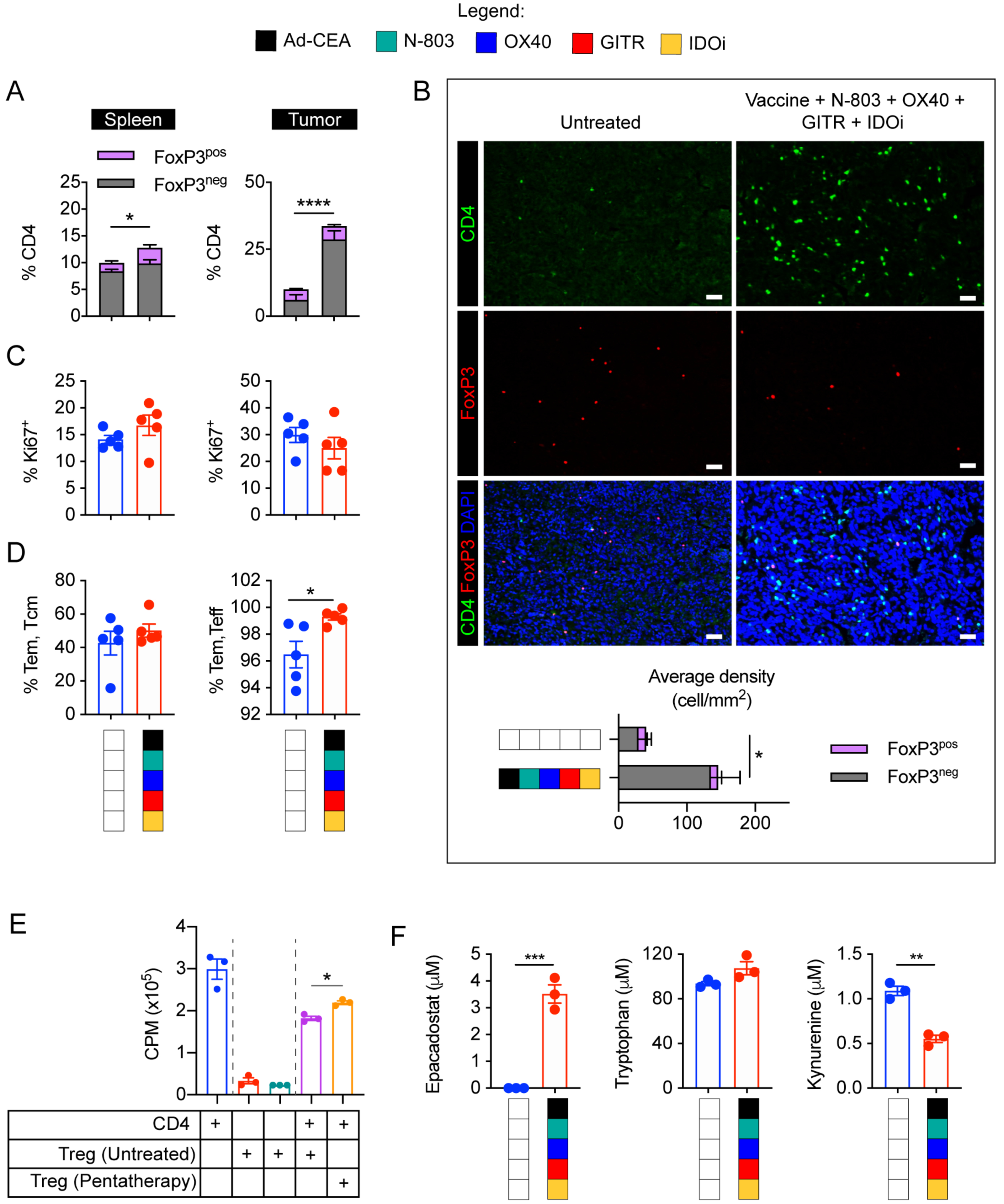
A. Spleens and tumors from Fig. 6A were evaluated for CD4+ T cells and CD4+FoxP3+ Tregs via flow cytometry (gated on live, CD3+). B. Tumor sections were evaluated for CD4 and FoxP3-expressing cells via immunofluorescence. C-D. CD4+ T cells were further analyzed by flow cytometry to determine (C) Ki67+ and (D) CD44+ and CD44+CD62L+ populations. E. Tregs isolated from untreated and pentatherapy-treated 4T1 primary tumors harvested 28 days post-tumor implantation were cocultured with CD4 cells purified from spleens of naïve mice. CD4+ T-cell proliferation was determined via 3H-thymidine incorporation assay. F. Epacadostat, tryptophan, and kynurenine in the serum of the untreated and pentatherapy-treated MC38-CEA tumor-bearing animals were determined via HPLC. One-way ANOVA or Student t-test. *P<0.05; **P<0.01; ***P<0.005, ****P<0.001. Error bars represent mean±SEM. These studies were repeated 3–4 times with similar results.
To further elucidate the effect of the pentatherapy combination on Treg activity, we isolated Tregs from tumors of untreated and pentatherapy-treated animals and tested their ability to inhibit CD4+ T-cell proliferation ex vivo (Fig. 7E). Pentatherapy combination dampened the suppressive activity of Tregs as demonstrated by the inability to hinder CD4+ T-cell proliferation (P<0.05). We also detected the presence of epacadostat in the serum of pentatherapy-treated mice, and it was associated with a decrease in kynurenine (Fig. 7F).
Discussion
Effective cancer immunotherapy entails the initiation and establishment of the cancer-immunity cycle (1). For immunologically “hot” tumors, treatment with ICI blocks the negative signals that hamper the activity of pre-existing immune effector cells, potentially resulting in favorable antitumor responses (2). Immunotherapy today is focused on patients who have failed ICI therapy or harbor ‘cold’ tumor types that are refractory to ICI, such as colon tumors. MC38-CEA, a murine colon carcinoma, LL2-CEA, a lung carcinoma, and 4T1, a breast carcinoma, were shown to be unresponsive/minimally responsive to anti-PD-L1 monotherapy (9), and tumor burden and/or mean overall survival were unimproved. Hence, for tumors that fail ICI, a combination of multiple agents targeting other immune-tumor interactions may be required.
Therapeutic cancer vaccines engage the immune response and elicit antigen-specific T cells (5,6,33) but require additional cancer treatment modalities to improve clinical outcomes (3). In this study, the Ad-CEA cancer vaccine, in combination with the IL15 superagonist N-803, was demonstrated to suppress tumor growth of MC38-CEA tumors and prolong overall survival and associated with increased tumoral CD8+ T cells, as well as improved CD8+ T cell to Treg ratio.
This study demonstrated that Ad-CEA+N-803 treatment shifted the tumor’s immune profile to a more activated and less exhausted phenotype. The costimulatory molecules OX40, GITR, and 4–1BB were upregulated, whereas PD-1 and CTLA-4 expression were downregulated. However, other markers for immune evasion, such as IDO, increased with Ad-CEA+N-803 treatment. Therefore, Ad-CEA+N-803+OX40+GITR+IDOi pentatherapy combination was designed to embody a multimodal immunotherapeutic approach aimed at targeting diverse immune-cancer interactions. N-803 and molecules targeting OX40, GITR, 4–1BB and IDO are all in clinical development (16,34).
The pentatherapy regimen resulted in tumor growth suppression and control of both small and larger MC38-CEA tumors, while avoiding irAEs. This treatment combination also had antitumor effects on other cold tumors, such as LL2-CEA and 4T1. Each of the agents contributed to the superior activity of the pentatherapy regimen, as the removal of any one of the IO agents resulted in significant loss of tumor inhibition. The relationship between the number of agents used and tumor growth control was non-linear and non-intuitive, indicating that in combination therapy, more is not necessarily better; rather, the ability to target diverse immune-tumor interactions with redundancies was essential. The pentatherapy regimen resulted in the expansion of CD8+ T cells, including CEA-specific CD8+ T cells, in the periphery followed by subsequent migration of these cells into the TME. The CD8+ TILs have improved cytolytic capacity, and their depletion removed the therapeutic advantage conferred by the pentatherapy regimen. The pentatherapy combination also promoted the proliferation and development of memory CD4+ T cells, while inhibiting Tregs, and resulted in 90% and 50% cures in the MC38-CEA and LL2-CEA tumor models, respectively. The cured animals were protected from tumor recurrence upon tumor re-challenge, as well as challenge with CEA-negative tumors, likely due to antigen cascade (35).
The timing and sequence of IO agent administration may be of utmost importance in the combination’s efficacy and safety and should be aligned with the temporal progression of anti-cancer responses (36–38). To re-condition the TME for the incoming effector immune cells, GITR was administered prior to primary vaccination. GITR is constitutively highly expressed on Tregs (39,40), and single administration of the GITR-targeting antibody, DTA-1, eliminates Tregs through T-cell lineage changes and cell depletion (41,42). GITR stimulation also breaks self-tolerance to TAAs (43) and could facilitate the activity of the Ad-CEA vaccine by helping to break the immune tolerance for CEA in the MC38-CEA model. The Ad-CEA vaccine was administered two days after the GITR agonist to prime and activate TAA-specific effector T cells (6). Antigen-experienced CD8+ T cells upregulate CD122 (IL2/IL15Rβ) (44), making them more receptive to N-803 stimulation (14). Hence, N-803 was applied after the primary vaccination with Ad-CEA. Our preliminary data demonstrated that treatment with N-803 prior to or concurrent with Ad-CEA priming did not result in increased survival. However, an OX40 agonist was co-administered with Ad-CEA, as the upregulation of the OX40 on the effector immune cells peaked within 24 hours of vaccination. Our group previously showed that vaccination and OX40 engagement coordinate together to induce greater CD4+ and CD8+ TAA-specific responses (45). Like the DTA-1 antibody, the OX86 agonist for mouse OX40 depletes Tregs in the TME (46). Hence, upon activation with Ad-CEA, N-803 and OX40 support T cells by enhancing effector functions, while counteracting immunosuppressive Tregs.
Although responses with monotherapy IDOi were underwhelming in clinical trials (47), this study showed that when used as a component of the pentatherapy combination, IDOi helped overcome barriers in the TME and was indispensable for the therapeutic effect of the pentatherapy regimen. IDOi was introduced in the animal chow at the same time as the first Ad-CEA treatment to prevent IDO and IDO-related metabolites that consequentially occur upon immune activation in the TME (48). Kynurenine and other downstream IDO metabolic products activate the aryl hydrocarbon receptor, which results in the differentiation of CD4+ T cells into Tregs and conversion of dendritic cells (DCs) and macrophages into immunosuppressive phenotypes (20). Kynurenine is a useful marker for therapeutic response to IDOi therapy (20), and we observed that pentatherapy treatment reduced serum kynurenine in tumor-bearing mice. The Tregs that OX40 and GITR antibodies failed to eradicate may be rendered less immunosuppressive as a consequence of IDOi activity (20). The uniqueness of IDOi in terms of its mechanism of action made it an integral component of the pentatherapy combination, even when the other IO agents had activities related to alleviating immunosuppression. This emphasizes the importance of using IO agents with overlapping roles but distinct mechanism of action.
Single- and double-agent immunotherapy rarely result in durable anti-tumor activity; hence, multimodal combination therapy with multiple IO agents is now becoming the cornerstone of cancer immunotherapy (49,50). An adaptive-design clinical trial offers a framework for expedient testing of combinatorial immunotherapy (51). The ongoing Quick Efficacy Seeking Trial (QuEST1; NCT03493945) aims to investigate the safety and efficacy of combination antitumor vaccine, TGFβ TRAP/anti-PD-L1, N-803, and epacadostat in metastatic castration-resistant prostate cancer. Novel clinical trial designs such as QuEST1 would allow for the rapid and safe examination of strategic immunotherapy combinations in the clinic.
Supplementary Material
Acknowledgements:
The authors thank the Center for Cancer Research Pathology Laboratory, NCI, NIH, and the Genomics Laboratory, Frederick National Laboratory for Cancer Research, for their assistance. The authors also thank Debra Weingarten for her editorial assistance in the preparation of this manuscript.
Financial Support: This work was funded by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute (NCI), National Institutes of Health, and via an NCI Cooperative Research and Development Agreement (CRADA) with Incyte, Corp., and an NCI CRADA with NantOmics/ImmunityBio.
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
Ethics approval and consent to participate: All animal experimental studies were performed under the approval of the NIH Intramural Animal Care and Use Committee. All mice were housed and maintained in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines.
References
- 1.Chen Daniel S, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39(1):1–10 doi 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Maleki Vareki S High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. Journal for ImmunoTherapy of Cancer 2018;6(1):157 doi 10.1186/s40425-018-0479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gatti-Mays ME, Redman JM, Collins JM, Bilusic M. Cancer vaccines: Enhanced immunogenic modulation through therapeutic combinations. Hum Vaccin Immunother 2017;13(11):2561–74 doi 10.1080/21645515.2017.1364322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. npj Vaccines 2019;4(1):7 doi 10.1038/s41541-019-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greiner JW, Zeytin H, Anver MR, Schlom J. Vaccine-based Therapy Directed against Carcinoembryonic Antigen Demonstrates Antitumor Activity on Spontaneous Intestinal Tumors in the Absence of Autoimmunity. Cancer Research 2002;62(23):6944. [PubMed] [Google Scholar]
- 6.Gabitzsch ES, Tsang KY, Palena C, David JM, Fantini M, Kwilas A, et al. The generation and analyses of a novel combination of recombinant adenovirus vaccines targeting three tumor antigens as an immunotherapeutic. Oncotarget 2015;6(31):31344–59 doi 10.18632/oncotarget.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eades-Perner AM, van der Putten H, Hirth A, Thompson J, Neumaier M, von Kleist S, et al. Mice transgenic for the human carcinoembryonic antigen gene maintain its spatiotemporal expression pattern. Cancer Res 1994;54(15):4169–76. [PubMed] [Google Scholar]
- 8.Kass E, Schlom J, Thompson J, Guadagni F, Graziano P, Greiner JW. Induction of Protective Host Immunity to Carcinoembryonic Antigen (CEA), a Self-Antigen in CEA Transgenic Mice, by Immunizing with a Recombinant Vaccinia-CEA Virus. Cancer Research 1999;59(3):676. [PubMed] [Google Scholar]
- 9.Knudson KM, Hicks KC, Alter S, Schlom J, Gameiro SR. Mechanisms involved in IL-15 superagonist enhancement of anti-PD-L1 therapy. J Immunother Cancer 2019;7(1):82 doi 10.1186/s40425-019-0551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nature Reviews Immunology 2006;6(8):595–601 doi 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 11.Rosario M, Liu B, Kong L, Collins LI, Schneider SE, Chen X, et al. The IL-15-Based ALT-803 Complex Enhances FcgammaRIIIa-Triggered NK Cell Responses and In Vivo Clearance of B Cell Lymphomas. Clin Cancer Res 2016;22(3):596–608 doi 10.1158/1078-0432.CCR-15-1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, Jones M, Liu B, Zhu X, Johnson CB, Edwards AC, et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor alphaSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res 2013;73(10):3075–86 doi 10.1158/0008-5472.CAN-12-2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhode PR, Egan JO, Xu W, Hong H, Webb GM, Chen X, et al. Comparison of the Superagonist Complex, ALT-803, to IL15 as Cancer Immunotherapeutics in Animal Models. Cancer immunology research 2016;4(1):49–60 doi 10.1158/2326-6066.CIR-15-0093-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim PS, Kwilas AR, Xu W, Alter S, Jeng EK, Wong HC, et al. IL-15 superagonist/IL-15RalphaSushi-Fc fusion complex (IL-15SA/IL-15RalphaSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget 2016;7(13):16130–45 doi 10.18632/oncotarget.7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M, et al. Agonists of Co-stimulation in Cancer Immunotherapy Directed Against CD137, OX40, GITR, CD27, CD28, and ICOS. Seminars in Oncology 2015;42(4):640–55 doi 10.1053/j.seminoncol.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol 2018;11(1):39 doi 10.1186/s13045-018-0582-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168(4):707–23 doi 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res 2012;72(21):5435–40 doi 10.1158/0008-5472.Can-12-0569. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010;115(17):3520–30 doi 10.1182/blood-2009-09-246124. [DOI] [PubMed] [Google Scholar]
- 20.Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol 2016;37(3):193–207 doi 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farsaci B, Sabzevari H, Higgins JP, Di Bari MG, Takai S, Schlom J, et al. Effect of a small molecule BCL-2 inhibitor on immune function and use with a recombinant vaccine. International Journal of Cancer 2010;127(7):1603–13 doi 10.1002/ijc.25177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol 2001;Chapter 20:Unit 20.2 doi 10.1002/0471142735.im2002s39. [DOI] [PubMed] [Google Scholar]
- 23.Hodge JW, Grosenbach DW, Aarts WM, Poole DJ, Schlom J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin Cancer Res 2003;9(5):1837–49. [PubMed] [Google Scholar]
- 24.Abdul Sater H, Marté JL, Donahue RN, Walter-Rodriguez B, Heery CR, Steinberg SM, et al. Neoadjuvant PROSTVAC prior to radical prostatectomy enhances T-cell infiltration into the tumor immune microenvironment in men with prostate cancer. Journal for ImmunoTherapy of Cancer 2020;8(1):e000655 doi 10.1136/jitc-2020-000655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han KP, Zhu X, Liu B, Jeng E, Kong L, Yovandich JL, et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 2011;56(3):804–10 doi 10.1016/j.cyto.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mathios D, Park CK, Marcus WD, Alter S, Rhode PR, Jeng EK, et al. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. Int J Cancer 2016;138(1):187–94 doi 10.1002/ijc.29686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schrama D, Ritter C, Becker JC. T cell receptor repertoire usage in cancer as a surrogate marker for immune responses. Seminars in Immunopathology 2017;39(3):255–68 doi 10.1007/s00281-016-0614-9. [DOI] [PubMed] [Google Scholar]
- 28.Hosoi A, Takeda K, Nagaoka K, Iino T, Matsushita H, Ueha S, et al. Increased diversity with reduced “diversity evenness” of tumor infiltrating T-cells for the successful cancer immunotherapy. Scientific Reports 2018;8(1):1058 doi 10.1038/s41598-018-19548-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gurney AL, Marsters SA, Huang A, Pitti RM, Mark M, Baldwin DT, et al. Identification of a new member of the tumor necrosis factor family and its receptor, a human ortholog of mouse GITR. Current Biology 1999;9(4):215–8 doi 10.1016/S0960-9822(99)80093-1. [DOI] [PubMed] [Google Scholar]
- 30.Ronchetti S, Zollo O, Bruscoli S, Agostini M, Bianchini R, Nocentini G, et al. Frontline: GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. European Journal of Immunology 2004;34(3):613–22 doi 10.1002/eji.200324804. [DOI] [PubMed] [Google Scholar]
- 31.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480(7378):480–9 doi 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang ML, Lukens JR, Bullock TNJ. Cognate Memory CD4+ T Cells Generated with Dendritic Cell Priming Influence the Expansion, Trafficking, and Differentiation of Secondary CD8+ T Cells and Enhance Tumor Control. The Journal of Immunology 2007;179(9):5829–38 doi 10.4049/jimmunol.179.9.5829. [DOI] [PubMed] [Google Scholar]
- 33.Balint JP, Gabitzsch ES, Rice A, Latchman Y, Xu Y, Messerschmidt GL, et al. Extended evaluation of a phase 1/2 trial on dosing, safety, immunogenicity, and overall survival after immunizations with an advanced-generation Ad5 [E1-, E2b-]-CEA(6D) vaccine in late-stage colorectal cancer. Cancer Immunol Immunother 2015;64(8):977–87 doi 10.1007/s00262-015-1706-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margolin K, Morishima C, Velcheti V, Miller JS, Lee SM, Silk AW, et al. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin Cancer Res 2018;24(22):5552–61 doi 10.1158/1078-0432.Ccr-18-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kudo-Saito C, Schlom J, Hodge JW. Induction of an antigen cascade by diversified subcutaneous/intratumoral vaccination is associated with antitumor responses. Clin Cancer Res 2005;11(6):2416–26 doi 10.1158/1078-0432.CCR-04-1380. [DOI] [PubMed] [Google Scholar]
- 36.Rothschilds AM, Wittrup KD. What, Why, Where, and When: Bringing Timing to Immuno-Oncology. Trends in Immunology 2019;40(1):12–21 doi 10.1016/j.it.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 37.Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, et al. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clin Cancer Res 2017;23(20):6165–77 doi 10.1158/1078-0432.CCR-16-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shrimali RK, Ahmad S, Verma V, Zeng P, Ananth S, Gaur P, et al. Concurrent PD-1 Blockade Negates the Effects of OX40 Agonist Antibody in Combination Immunotherapy through Inducing T-cell Apoptosis. Cancer immunology research 2017;5(9):755–66 doi 10.1158/2326-6066.CIR-17-0292. [DOI] [PubMed] [Google Scholar]
- 39.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nature Immunology 2002;3(2):135–42 doi 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 40.McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, et al. CD4+CD25+ Immunoregulatory T Cells: Gene Expression Analysis Reveals a Functional Role for the Glucocorticoid-Induced TNF Receptor. Immunity 2002;16(2):311–23 doi 10.1016/S1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 41.Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, et al. GITR pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer immunology research 2013;1(5):320–31 doi 10.1158/2326-6066.Cir-13-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med 2013;210(9):1685–93 doi 10.1084/jem.20130573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramirez-Montagut T, Chow A, Hirschhorn-Cymerman D, Terwey TH, Kochman AA, Lu S, et al. Glucocorticoid-Induced TNF Receptor Family Related Gene Activation Overcomes Tolerance/Ignorance to Melanoma Differentiation Antigens and Enhances Antitumor Immunity. The Journal of Immunology 2006;176(11):6434 doi 10.4049/jimmunol.176.11.6434. [DOI] [PubMed] [Google Scholar]
- 44.Ku CC, Murakami M, Sakamoto A, Kappler J, Marrack P. Control of Homeostasis of CD8+ Memory T Cells by Opposing Cytokines. Science 2000;288(5466):675 doi 10.1126/science.288.5466.675. [DOI] [PubMed] [Google Scholar]
- 45.Malamas AS, Hammond SA, Schlom J, Hodge JW. Combination therapy with an OX40L fusion protein and a vaccine targeting the transcription factor twist inhibits metastasis in a murine model of breast cancer. Oncotarget 2017;8(53):90825–41 doi 10.18632/oncotarget.19967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol Cell Biol 2014;92(6):475–80 doi 10.1038/icb.2014.26. [DOI] [PubMed] [Google Scholar]
- 47.Eynde BJVd Baren Nv, Baurain J-F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annual Review of Cancer Biology 2020;4(1):241–56 doi 10.1146/annurev-cancerbio-030419-033635. [DOI] [Google Scholar]
- 48.Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nature immunology 2013;14(10):1014–22 doi 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee KL, Benz SC, Hicks KC, Nguyen A, Gameiro SR, Palena C, et al. Efficient Tumor Clearance and Diversified Immunity through Neoepitope Vaccines and Combinatorial Immunotherapy. Cancer immunology research 2019;7(8):1359–70 doi 10.1158/2326-6066.Cir-18-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hicks KC, Knudson KM, Lee KL, Hamilton DH, Hodge JW, Figg WD, et al. Cooperative Immune-Mediated Mechanisms of the HDAC Inhibitor Entinostat, an IL15 Superagonist, and a Cancer Vaccine Effectively Synergize as a Novel Cancer Therapy. Clin Cancer Res 2020;26(3):704–16 doi 10.1158/1078-0432.Ccr-19-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Redman JM, Steinberg SM, Gulley JL. Quick efficacy seeking trial (QuEST1): a novel combination immunotherapy study designed for rapid clinical signal assessment metastatic castration-resistant prostate cancer. Journal for ImmunoTherapy of Cancer 2018;6(1):91 doi 10.1186/s40425-018-0409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.