

Abstract
Background:
Histamine (HA), a wake-promoting monoamine implicated in stress-related arousal states, is synthesized in histidine decarboxylase (HDC)-expressing hypothalamic neurons of the tuberomammillary nucleus (TMN). HDC-containing varicosities diffusely innervate striatal and mesolimbic networks, including the nucleus accumbens (NAc). The nucleus accumbens (NAc) integrates diverse monoaminergic inputs to coordinate motivated behavior. While the NAc expresses various HA receptor subtypes, mechanisms by which HA modulates NAc circuit dynamics are undefined.
Methods:
Utilizing male D1tdTomato transgenic reporter mice, whole-cell patch clamp electrophysiology, and input-specific optogenetics, we employed a targeted pharmacological approach to interrogate synaptic mechanisms recruited by HA signaling at glutamatergic synapses in the NAc. We incorporated an immobilization stress protocol to assess whether acute stress engages these mechanisms at glutamatergic synapses onto D1 receptor-expressing [D1(+)] medium spiny neurons (MSNs) in the NAc core.
Results:
HA negatively regulates excitatory gain onto D1(+)-MSNs via presynaptic H3 receptor-dependent long-term depression (LTD) that requires Gβγ-directed Akt-GSK3β signaling. Furthermore, HA asymmetrically regulates glutamatergic transmission from the prefrontal cortex (PFC) and mediodorsal thalamus (MDT), with inputs from the PFC undergoing robust HA-induced LTD. Finally, we report that acute immobilization stress attenuates this LTD by recruiting endogenous H3R signaling in the NAc at glutamatergic synapses onto D1(+)-MSNs.
Conclusion:
Stress-evoked HA signaling in the NAc recruits H3 heteroreceptor signaling to shift thalamocortical input onto D1(+)-MSNs in the NAc. Our findings provide novel insight into an understudied neuromodulatory system within the NAc and implicate HA in stress-associated physiological states.
Keywords: Nucleus accumbens, glutamatergic transmission, histamine, histamine H3 receptor, synaptic plasticity, stress
Introduction
The nucleus accumbens (NAc) coordinates goal-directed behavior by integrating information encoded by distinct neuromodulatory systems. While monoaminergic influences on mesolimbic network activity are well-characterized, less is known how non-canonical monoamines, such as histamine (HA), contribute to NAc circuit function. HA, synthesized primarily in L-histidine decarboxylase (HDC)-expressing hypothalamic neurons of the tuberomammillary nucleus (TMN), promotes wakefulness, sleep-wake transitions, and attention, with a purported regulatory role in appetitive and motivational behavior(1–3). Although HA-containing varicosities moderately innervate the NAc, multiple HA receptor subtypes, including H1, H2 and H3, are abundantly expressed in the NAc, indicating that HA may broadly influence circuit dynamics in this region(4,5).
NAc output is gated by the strength of glutamatergic synapses onto D1 and D2 dopamine (DA) receptor-expressing GABAergic medium spiny neurons (MSNs)(6,7). Experience-dependent adaptations at corticolimbic synapses onto D1(+) and D1(−)-MSNs drive distinct reward-related motivational outcomes(8,9). An unexplored mechanism that may scale excitatory gain in the NAc is the HA-containing ascending arousal system. HA has been shown to regulate limbic and paralimbic glutamatergic synapses via Gi/o-coupled G protein-coupled receptor (GPCR), H3 receptor (H3R)(10–12). While the effects of HA on NAc glutamatergic transmission remain unknown, intra-NAc HA infusion elicits H3R-dependent biphasic effects on locomotor activity(13). Furthermore, manipulating endogenous H3R activity in vivo modulates NAc-dependent motivational responding to drugs of abuse(14). These findings suggest that H3R signaling may result in complex cell type- and synapse-specific adaptations capable of shifting NAc output.
Here, we interrogated mechanisms by which HA signaling modulates NAc circuit function. We find that HA differentially modulates glutamatergic synapses onto D1(+) and D1(−)-MSNs via presynaptically-localized H3R. H3R activity is sufficient to induce long-term depression (HA-LTD) of glutamatergic transmission by mobilizing the Gβγ complex to recruit the Akt-GSK3β effector pathway. Afferent-specific optogenetics revealed that HA biases thalamocortical balance onto D1(+) MSNs, with corticoaccumbens synapses displaying greater sensitivity to inhibitory H3R activity. Finally, we provide evidence that acute immobilization stress recruits endogenous H3R function at glutamatergic synapses onto D1(+)-MSNs in the NAc core.
Materials and Methods
Animal Use
Animals were bred and housed at Vanderbilt University Medical Center in accordance with IACUC. Male mice 8–12-weeks-of-age were housed in groups of 3–5/cage on a 12-hr light-dark cycle with ad lib access to standard food and water. Whole-cell patch-clamp electrophysiology were obtained in acute brain slice preparations from C57BL/6J wild-type (WT) mice (no MSN subtype-specificity) or bacterial artificial chromosome (BAC) transgenic mice carrying the tdTomato fluorophore under control of the Drd1a (D1 receptor) promoter (Jackson labs), as described previously and in Supplemental Methods(15,16). Optogenetic viral injections with AAV-CaMKII-ChR2-eYFP (Addgene) into the PFC and MDT were performed as previously described(17,18). For a subset of experiments, mice underwent 30-min acute immobilization stress (AIS) in a cylindrical holding tube followed by a 20-min recovery period.
Electrophysiology
See Supplemental Methods for a detailed description of electrophysiological methods.
Microinfusions
Bilateral guide cannulas (26 gauge, 3-mm length, 2-mm center-to-center distance, C235GS-5–2.0/SPC- Plastics One, 22 Roanoke VA) were ventral to the NAcC (AP: 1.45, ML: ±1.00, DV: 3.00). H3R antagonist, JNJ 5207852, was infused at a dose of 0 (saline) or 3.89-μg/μL at rate of 0.4-μL over 60-sec and was allowed to absorb for 90-sec before cannula was gently removed. Animals were then permitted 5-min prior to entering the immobilization apparatus. See Supplemental Methods for more detail.
Pharmacology
All pharmacological agents were purchased from Tocris Biosciences.
Statistics and Data Analysis
Electrophysiological experiments were analyzed using Clampfit 10.4 and GraphPad Prism v7.0. Errors bars depicted in figures represent SEM. For all analyses, α was set as 0.05, with P values < α indicating a statistically significant difference. Statistical analyses and results are presented in the Figure Legends. For a more detailed descriptions of analyses, see Supplemental Materials and Methods.
Results
HA recruits a presynaptic gain control mechanism that is differentially expressed at glutamatergic synapses onto D1(+) and D1(−) MSNs in the NAc
To interrogate if HA modulates glutamatergic transmission in the NAc, electrically-evoked EPSCs were recorded from non-specific MSNs in the NAc core (NAcC) and shell (NAcSh) (Fig. S1A,D). Following a 10-min EPSC baseline, HA (10 μM) was bath-applied for 15-min, resulting in a significant depression in EPSC amplitude in approximately half of MSNs tested [HA-positive-(+)] in NAcC and shell NAcSh subterritories (Fig. S1,B–F). The differential actions of HA on dorsal striatal MSNs prompted us to examine whether MSN subtype dictates which synapses in the NAc are regulated by HA(11,19). To test this hypothesis, we prepared acute brain slices from D1tdTomato BAC transgenic mice in which tdTomato-positive D1 dopamine (DA) receptor-expressing [D1(+)] MSNs are differentiable from D2 dopamine receptor-expressing [D1(−)] MSNs(15,16,20). EPSCs were recorded at −70-mV from D1(+) and D1(−)-MSNs in the NAcC (Fig. 1A). Following a 10-min EPSC baseline, HA (10 μM) was bath-applied for 15-min, resulting in a significant depression in EPSC amplitude at synapses onto D1(+)-MSNs that persisted following drug wash-out (Fig. 1B,D,F). However, at D1(−)-MSN synapses, HA failed to elicit a significant reduction in EPSC amplitude (Fig. 1C–F). These data suggest that HA differentially regulates glutamatergic transmission onto D1(+) and D1(−)-MSN synapses in the NAc, with synapses onto D1(+)-MSNs undergoing a HA-induced depression in synaptic strength (Fig. 1F).
Figure 1. HA recruits a gain control mechanism that differentially modulates glutamatergic transmission onto D1(+) and D1(−) MSNs in the NAc core.
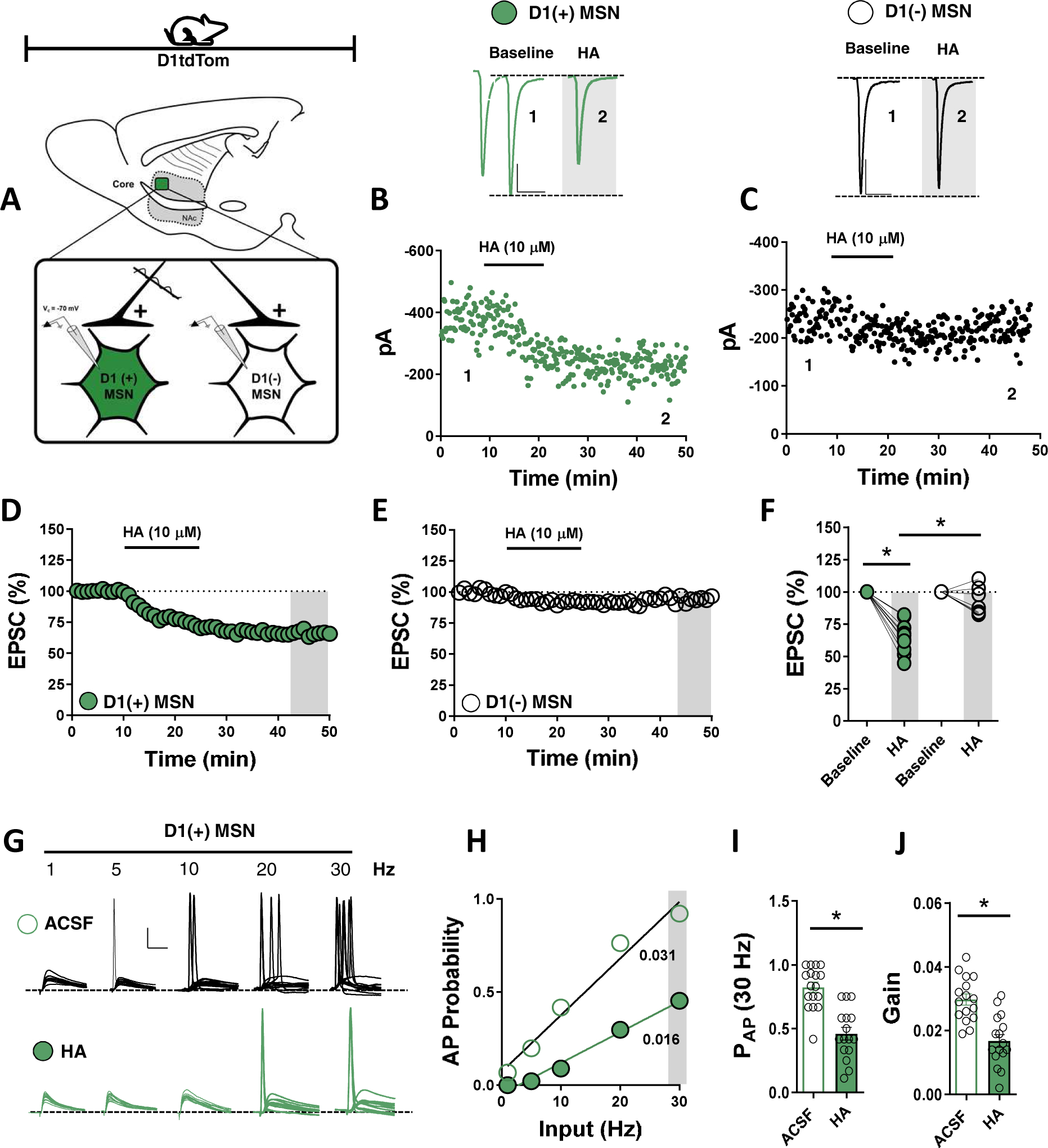
(A) Schematic of sagittal mouse brain slice depicting electrophysiological recording location in the dorsomedial NAc core. (B) Representative experiment and traces (above) of EPSCs obtained at baseline and in the presence of HA (10 μM) from tdTomato-expressing [D1(+)] MSNs. (C) Representative experiment and traces (above) of EPSCs obtained at baseline and in the presence of HA (10 μM) from tdTomato-negative [D1(−), putative D2] MSNs. Scale bars: 100 pA/50-ms. (D) Time-course summary of normalized EPSCs in D1(+) MSNs depicting the HA-induced depression in EPSC amplitude that persists post-drug wash out [64.55±4.03%, n=10, p<0.0001]. (E) Time-course summary of normalized EPSCs in D1(−) MSNs depicting a modest HA-induced depression in EPSC amplitude that returns to baseline [92.94±4.47%, n=7, p=0.126]. (F) Average EPSC amplitude in D1(+) (blue circles) and D1(−) MSNs (open circles) obtained at t(gray)= 45–50-min [2-way RM-ANOVA, effect of MSN subtype, F(3,27)=35.95, Sidak’s post-hoc analysis, p<0.0001]. (G) Representative traces from D1(+) MSNs showing EPSPs and APs obtained following 1, 5, 10, 20 and 30 Hz stimulation of glutamatergic synapses in ACSF and HA-superfused slices Scale bars: 20 mV/20-ms. (H) Input-output gain curve with AP probability (y-axis) plotted as a function of input frequency (x-axis) in ACSF and HA. (I) Average AP probability obtained at 30 Hz in ACSF (open bar) and HA (closed green bar). (J) Gain of EPSCs in ACSF (open bar) and HA (closed green bar) [PAP 30 Hz ACSF: 0.921±0.039, n=16; PAP 30 Hz HA: 0.453±0.055, n=16, p<0.001; Gain ACSF: 0.029±0.001, n=16; Gain HA: 0.017±0.002, n=16, p<0.001]. Error bars indicate SEM with (*) signifying p < 0.05.
To examine whether HA scales excitatory gain onto D1(+)-MSNs, we obtained an input-output function in which action potential (AP) probability was assessed at a plateau potential of −70 mV following increasing stimulus frequencies (1, 5, 10, 20 and 30 Hz). We first confirmed that synaptically-evoked AP firing in D1(+)-MSNs was due to AMPAR-mediated excitatory postsynaptic potentials (EPSPs). Bath-application of AMPAR antagonist, NBQX (50 μM), completely blocked synaptically-evoked AP firing, indicating that increasing AP output in D1(+)-MSNs is due to spatiotemporal summation of AMPAR-mediated EPSPs (data not shown). D1(+) MSNs in ACSF alone demonstrated a linear increase in AP fidelity following sequential increases in input frequency (Fig. 1G,H). In contrast, HA superfusion significantly reduced synaptically-evoked AP firing that coincided with a negative shift in gain (Fig. 1G,I). Together, these findings suggest that HA function on glutamatergic synapses in the NAcC contributes to input-output inequities that alter D1(+)-MSN output.
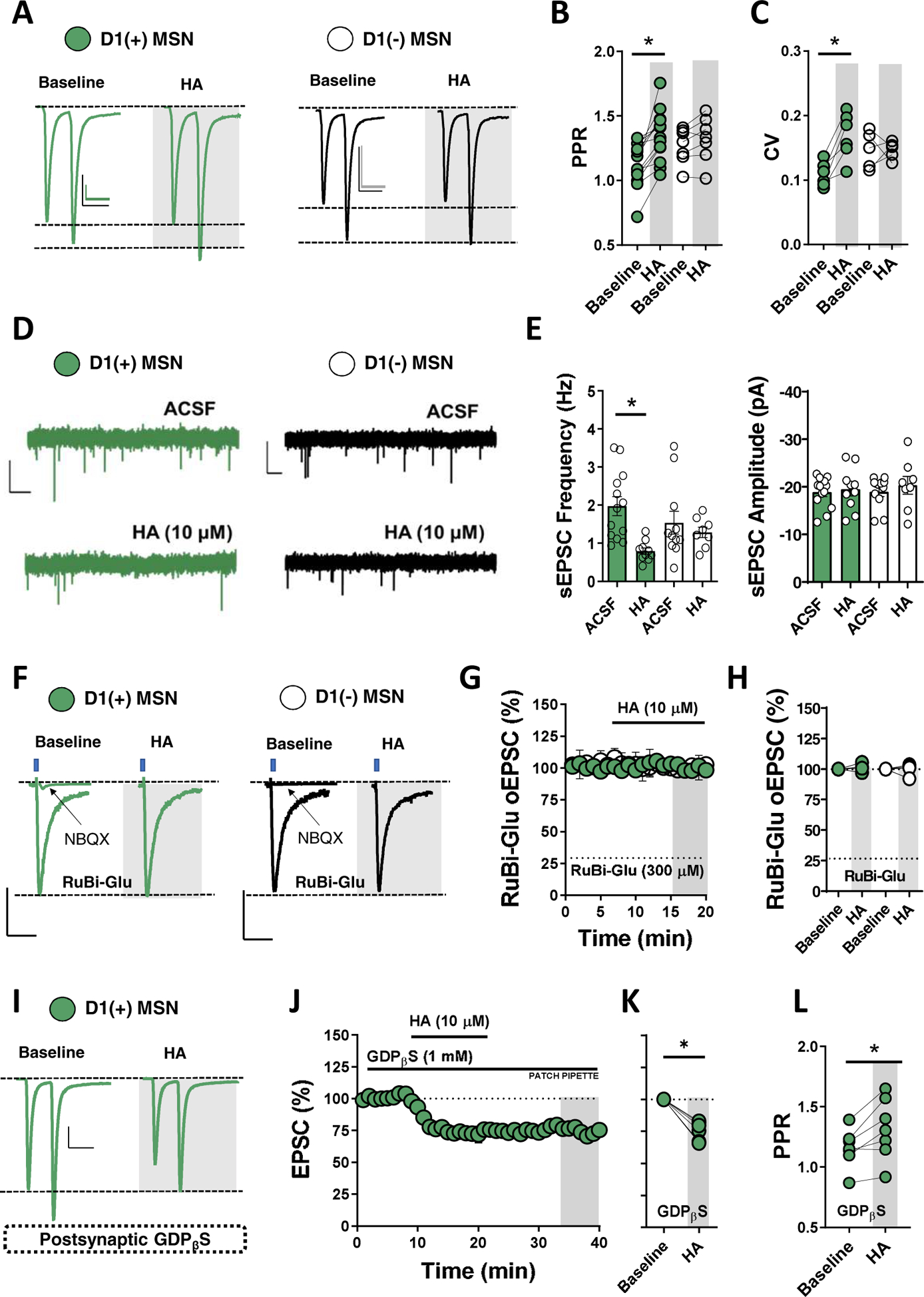
To determine if HA modulates glutamatergic synapses via pre- or postsynaptic mechanisms, we examined the effects of HA on paired-pulse ratio (PPR) and coefficient of variance (CV). HA significantly increased PPR and CV at glutamatergic synapses onto D1(+)-MSNs but not D1(−)-MSNs, indicating a presynaptic locus of action that is restricted to D1(+) MSNs (Fig. 2A–C). HA also decreased spontaneous EPSC (sEPSC) frequency but not amplitude at D1(+)-MSNs without altering sEPSCs at D1(−)-MSNs (Fig. 2D,E). To examine the synaptic localization of HA function more closely, we performed glutamate uncaging experiments with ruthenium-bipyridine-trimethylphosphine (RuBi) glutamate (RuBi-Glu, 300 μM), a caged-glutamate compound released by 473-nm blue light(21). Optical stimulation of RuBi-Glu-containing ACSF reliably elicited NBQX-sensitive AMPA receptor (AMPAR)-mediated optical EPSCs (RuBi-Glu oEPSCs) in D1(+) and D1(−)-MSNs (Fig. 2F). HA had no effect on RuBi-Glu oEPSC amplitude at D1(+) or D1(−)-MSNs (Fig. 2F–H). If the effects of HA are exclusively presynaptic, disabling postsynaptic GPCR signaling should have negligible effect on the HA-induced depression in glutamatergic transmission. Consistent with this hypothesis, incorporating GDP analog, GDPβS (1 mM), into the intracellular solution of the patch pipette failed to prevent the HA-induced depression in EPSC amplitude and increase in PPR (Fig. 2I–L). Collectively, these data indicate that HA decreases NAcC glutamatergic synaptic efficacy through a presynaptic mechanism preferentially expressed at D1(+)-MSN synapses.
Figure 2. HA decreases glutamatergic synaptic efficacy onto D1(+) MSNs via a presynaptic locus of action.
(A) Representation traces of 50-ms ISI paired-pulse EPSCs obtained in D1(+) (blue circles) and D1(−) MSNs (open circles) at baseline and following HA (10 μM) bath-application with light-blue shaded region indicating ΔPPR. Scale bars [D1(+)]: 50-pA, 100-pA/50-ms. Scale bars [D1(−)]: 50-pA/50-ms. (B) Average PPR obtained at baseline and post-HA at t(gray)= 45–50-min in D1(+) and D1(−) MSNs [D1(+) PPR baseline: 1.12±0.05, D1(+) PPR HA: 1.33±0.07, n=11, p=0.007; D1(−) PPR baseline: 1.24±0.06, D1(−) PPR HA: 1.31±0.06, n=8, p=0.187]. (C) Average coefficient of variance (CV) of EPSCs obtained during 10-min baseline and post-HA at t(gray)= 45–50-min in D1(+) and D1(−) MSNs [D1(+) CV baseline: 0.11±0.01, n=5, D1(+) CV HA: 0.17±0.02, n=5, p=0.012;; D1(−) CV baseline: 0.15±0.007, CV HA: 0.146±0.006, n=5, p=0.483]. (D) Representative traces of sEPSCs in D1(+) (blue traces) and D1(−) MSNs (black traces) in ACSF alone and in HA-containing ACSF. Scale bars: 20 pA/1-s. (E) Average sEPSC frequency (Hz) and amplitude (pA) in D1(+) (blue bars) and D1(−) (open bars) in ACSF alone and in the presence of HA [sEPSC frequency = D1(+) baseline: 1.97±0.304 Hz, n=13 D1(+) HA: 0.80±0.09 Hz, n=10, p<0.001; D1(−) baseline: 1.54±0.316 Hz, n=11, D1(−) HA: 1.29±0.15 Hz, n=8, p=0.483; sEPSC amplitude = D1(+) baseline: −18.8±1.09 pA, n=13, D1(+) HA: −19.49±1.48 pA, n=10, p=0.3524; D1(−) baseline: −18.99±1.04 pA, n=11, D1(−) HA: −20.34±1.99 pA, n=8, p=0.2483. (F) Representative traces of optically-evoked RuBi-Glu EPSCs (RuBi-Glu oEPSCs) in D1(+) and D1(−) MSNs at baseline and in the presence of HA. Superimposed traces show that AMPAR-antagonist, NBQX, abolishes RuBi-Glu oEPSCs in both MSNs. Scale bars: 20-pA/50-ms. (G) Time-course summary of RuBi-Glu oEPSCs in D1(+) (blue circles) and D1(−) MSNs (open circles) showing that HA has no effect on RuBi-Glu oEPSC amplitude. (H) Average RuBi-Glu oEPSC amplitude obtained at t(gray) = 15–20-min in D1(+) and D1(−) MSNs [D1(+): 101.26±1.96, n=4, p=0.5113; D1(−) 99.65±2.99%, n=4, p=0.899]. (I) Representative traces of eEPSCs obtained from D1(+) MSNs postsynaptically-loaded with GDPβS at baseline and in the presence of HA. Scale bars: 100-pA/50-ms (J) Time-course summary of EPSCs in D1(+) MSNs showing the inhibitory actions of HA remain intact in GDPβS-loaded cells. (K) Quantification of average EPSC amplitude pre- and post-HA in GDPβS-loaded cells at t(gray) = 45–50-min [HA GDPβS: 75.39±13.04%, n=6, p<0.001]. (L) Average PPR pre- and post-HA in GDPβS-loaded cells [PPR = pre-HA: 1.20±.043, post-HA: 1.38±0.08, n=8, p=0.0163]. Error bars indicate SEM with (*) signifying p < 0.05.
H3 heteroreceptors trigger long-term depression of glutamatergic transmission onto D1(+) MSNs and are tonically active at D1(−) MSN synapses
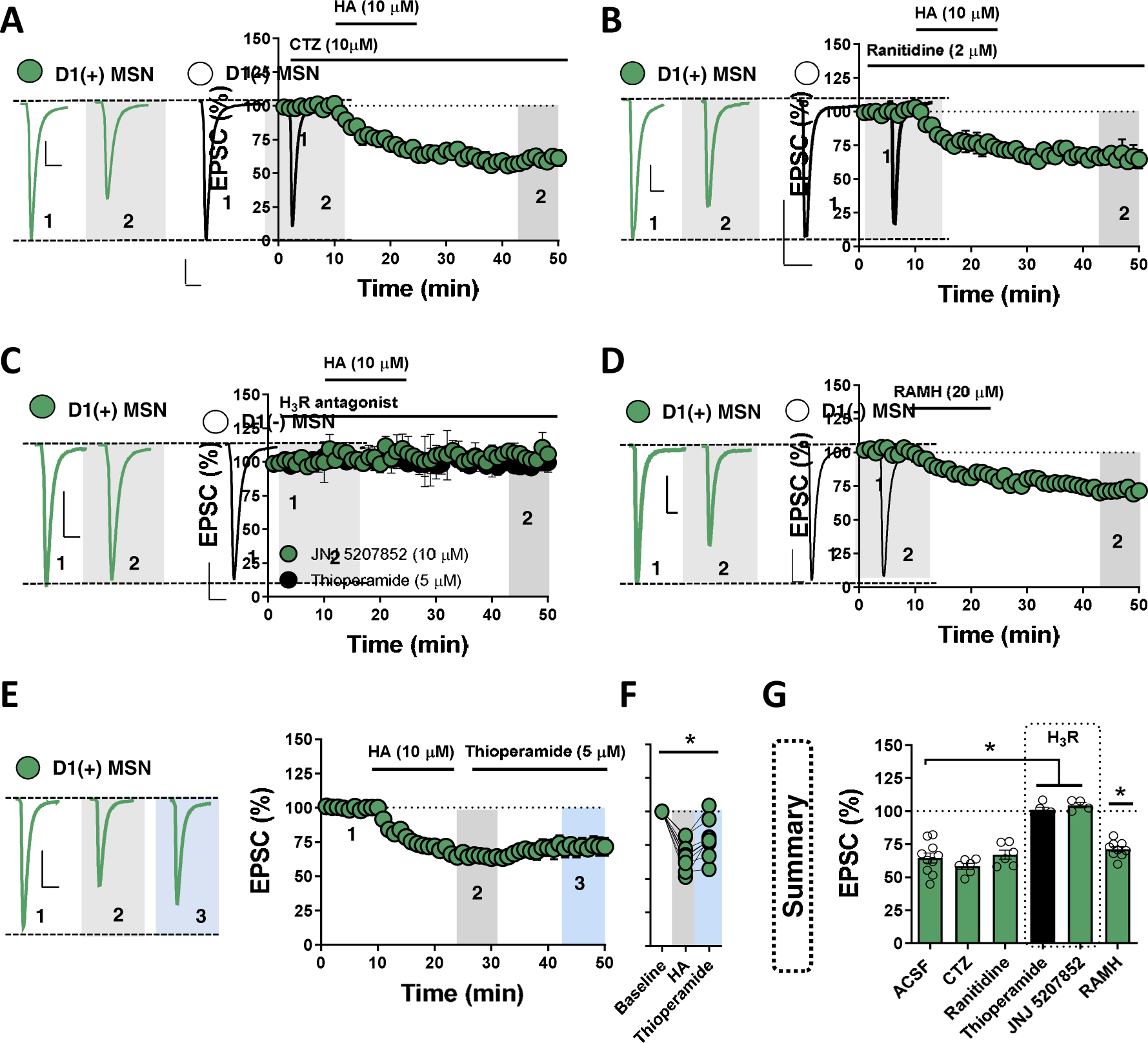
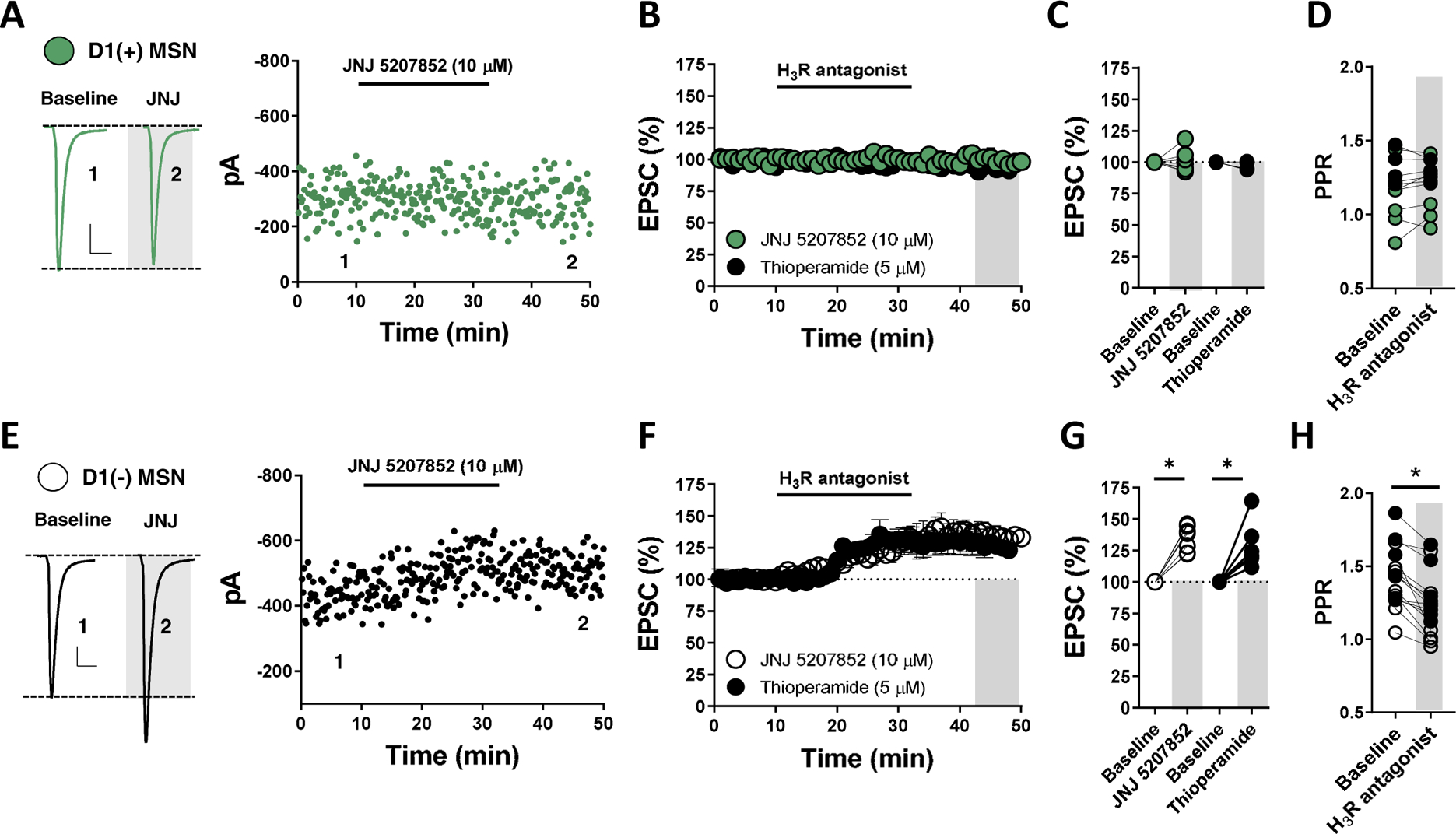
HA modulates striatal circuit function through various HA receptor subtypes, including Gαq− and Gαs-coupled H1 and H2 receptors, respectively, and Gαi-coupled H3 receptors(11,22,23). The HA receptor mediating synaptic depression at NAcC D1(+)-MSN synapses has not been determined. To assess the contribution of H1 receptors (H1R) to the actions of HA, we superfused selective H1R antagonist, cetirizine (CTZ, 1 μM), into the ACSF bath prior to HA. Prior application of CTZ had no effect on the HA-induced decrease in EPSC amplitude at D1(+)-MSN synapses (Fig. 3A, G). Similarly, bath-application of a selective H2 receptor (H2R) antagonist, ranitidine (20 μM), failed to block the effects of HA at D1(+)-MSN synapses, indicating that HA reduces glutamate release probability independently of H1 and H2 receptors (Fig. 3b,G). To determine if HA functions through H3 heteroreceptors, H3R antagonists thioperamide (5 μM) and a chemically-distinct non-imidazole H3R antagonist, JNJ5207852 (10 μM), were superfused into the ACSF bath for 30-min prior to HA. Both H3R antagonists abolished the HA-induced depression in EPSC amplitude at glutamatergic synapses onto D1(+)-MSNs (Fig. 3C,G). Consistent with an H3R-mediated effect, selective H3R agonist, R-(−)-α-methylhistamine (RAMH, 1 μM), recapitulated the HA-induced depression in EPSC amplitude at D1(+)-MSN synapses (Fig. 3D,G). To determine whether HA induces long-term depression (LTD) of glutamatergic transmission onto D1(+)-MSNs, thioperamide was incorporated into the superfusate immediately following HA. The HA-induced depression in EPSC amplitude persisted in the presence of thioperamide, indicating that HA triggers H3R-dependent LTD of glutamatergic transmission (HA-LTD) onto D1(+)-MSNs in the NAc (Fig. 3E,F). While H3R blockade alone had no effect on basal EPSC amplitude at synapses onto D1(+)-MSNs, we observed an increase in EPSC amplitude at D1(−)-MSN synapses following bath-application of either H3R antagonist (JNJ5207852 or thioperamide) (Fig. 4A–F). The H3R antagonist-induced increase in EPSC amplitude at D1(−) MSN synapses was accompanied by a significant reduction in PPR, indicating that tonic presynaptic H3R signaling occurs at glutamatergic synapses onto D1(−) MSN synapses (Fig. 4C,F).
Figure 3. HA acts via H3 heteroreceptors to elicit long-term depression of glutamatergic transmission onto D1(+) MSNs.
(A) Representative traces (left) and normalized time-course summary of EPSCs obtained in D1(+) MSNs (blue circles) showing the effects of HA in the presence of H1R antagonist, cetirizine (CTZ) [D1(+) HA: 58.12±2.60%, n=6, p=0.247]. (B) Representative traces (left) and time-course summary of EPSCs obtained in D1(+) MSNs showing the effects of HA in the presence of H2R antagonist, ranitidine [D1(+) HA: 67.11±3.76%, n=6, p=0.656]. (C) Representative traces (left) and time-course summary of EPSCs obtained in D1(+) MSNs showing that imidazole or non-imidazole H3R antagonists, thioperamide (black) and JNJ 5207852 (green), respectively, completely block the effects of HA [D1(+) HA in thioperamide: 100.98±2.24%, n=5, p=0.005; HA in JNJ: 104.46±3.02%, n=4, p<0.001]. (D) Representative traces (left) and time-course summary of EPSCs obtained in D1(+) MSNs showing the effects of selective H3R agonist, R-(−)-α-methylhistamine (RAMH) [D1(+) RAMH: 71.06±2.16%, n=9, p<0.001]. (E) Representative traces (left) and time-course summary of EPSCs obtained in D1(+) MSNs showing the effects of HA chased with H3R antagonist, thioperamide [: D1(+) HA: 66±3.70, D1(+) thioperamide chase: 79.68±4.86%, n=9, p=0.002]. (F) Average EPSC amplitude in D1(+) MSNs obtained at baseline, t(grey) = 25–30-min, and t(blue) = 45–50-min. (G) Summary table of average EPSC amplitude at D1(+) MSNs following each pharmacological treatment. Scale bars: 100 pA/20-ms. Error bars indicate SEM with (*) signifying p < 0.05.
Figure 4. H3R activity tonically regulates glutamatergic synapses onto D1(−) MSNs but not D1(+) MSNs.
(A) Representative traces (left) and experiments of EPSCs in D1(+) MSNs depicting the effects of H3R antagonist, JNJ 5207852. (B) Time-course summary of EPSCs during bath-application of imidazole or non-imidazole H3R antagonists, thioperamide (black) and JNJ 5207852 (green), respectively [D1(+) JNJ: 100.02±3.33%, n=8, p=0.497; D1(+) in thioperamide: 95.69±1.49%, n= 4, p=0.065]. (C) Quantification of average EPSC amplitude in D1(+) MSNs at baseline in the presence of thioperamide or JNJ 5207852 at t(grey) = 45–50-min. (D) PPR in D1(+) MSNs at baseline and in the presence of thioperamide (black) or JNJ 5207852 (green) [D1(+) PPR baseline: 1.92±0.07, D1(+) PPR H3R antagonist: 1.22±0.05, n=11, p=0.309]. (E) Representative traces (left) and experiments of EPSCs in D1(−) MSNs depicting the effects of H3R antagonist, JNJ 5207852. (F) Time-course summary of EPSCs during bath-application of H3R antagonists, thioperamide (black) and JNJ 5207852 (open) in D1(−) MSNs [D1(−) JNJ: 134.72±4.06%, n=7, p<0.001; D1(−) in thioperamide: 128.10±6.57%, n=7, p=0.005]. (G) Quantification of average EPSC amplitude in D1(−) MSNs at baseline in the presence of thioperamide or JNJ 5207852 at t(grey) = 45–50-min. (H) PPR in D1(+) MSNs at baseline and in the presence of thioperamide (black) or JNJ 5207852 (open) [D1(−) PPR baseline: 1.42±0.05, D1(−) PPR H3R antagonist: 1.24±0.06, n=15, p<0.001]. Scale bars: 50-pA/20-ms. Error bars indicate SEM with (*) signifying p < 0.05.
Gβγ-directed recruitment of the Akt-GSK3β axis mediates HA-LTD at glutamatergic synapses onto D1(+) MSNs
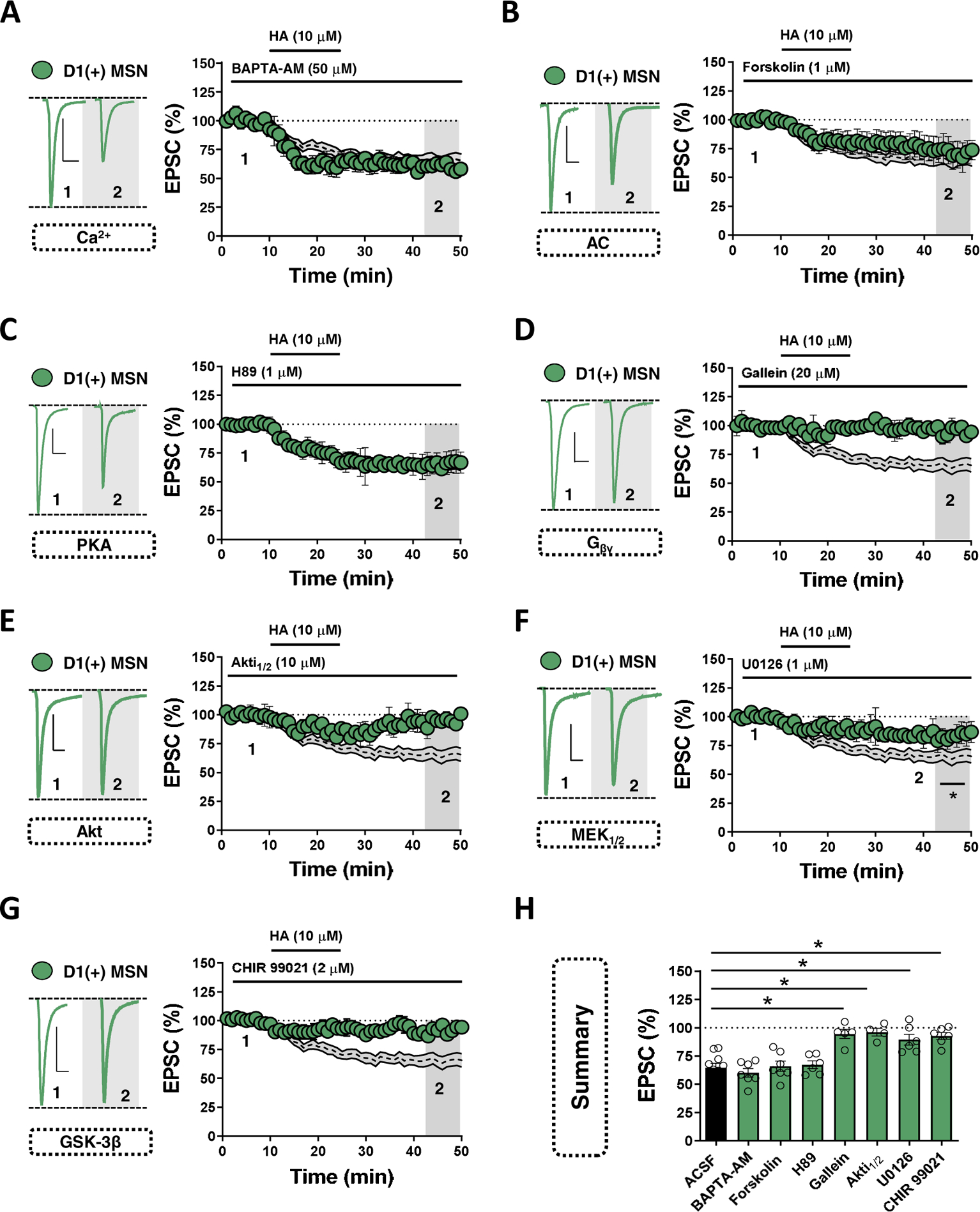
H3Rs recruit diverse intracellular effectors to reduce neurotransmitter release probability(2,19,24). While striatal H3R have been shown to modulate synaptosomal glutamate release, in part, by inhibiting voltage-gated calcium channels (VGCCs), recent reports implicate mitogen-activated protein kinase (MAPK)- and Akt/glycogen synthase kinase-3β (GSK3β)-dependent signaling pathways(25). To interrogate the synaptic mechanism of HA at synapses onto D1(+) MSNs in the NAc, we first assessed whether HA-LTD requires a shift in Ca2+ dynamics. In slices incubated in cell-permeant Ca2+-chelator, BAPTA-AM (50 μM), HA induced a depression in EPSC amplitude that was indistinct from control conditions (Fig. 5A,H). We next tested if HA recruits a Gαi signaling pathway dependent on adenylyl cyclase (AC) and protein kinase A (PKA) activity. Bath-application of AC activator, forskolin (10 μM), had no effect on HA-LTD (Fig. 5B,H). If HA-LTD proceeds independently of AC/PKA signaling, then inhibiting downstream PKA function should also fail to occlude the expression of HA-LTD. Indeed, HA-LTD remained intact in the presence of PKA inhibitor, H89 (1 μM) (Fig. 5C,H). These data suggest HA-LTD of glutamatergic transmission onto D1(+)-MSNs in the NAcC is AC/PKA-independent.
Figure 5. Gβγ-dependent recruitment of the Akt-GSK3β axis mediates HA-LTD at glutamatergic synapses onto D1(+) MSNs.
(A) Representative traces (left) and normalized time-course summary of EPSCs obtained in D1(+) MSNs (green circles) showing the effects of HA in slices incubated in Ca2+ chelator, BAPTA-AM [HA in BAPTA-AM: 60.02±3.74%, n=7, 1-way ANOVA, F(7,44)=13.78, Sidak’s post-hoc analysis, ACSF vs. BAPTA-AM, p=0.935)]. (B) Representative traces (left) and normalized time-course summary of EPSCs obtained in D1(+) MSNs showing the effects of HA in the presence of AC activator, forskolin [D1(+) HA in forskolin: 65.80±4.81%, n=7, 1-way ANOVA, ACSF vs. FSK, p=0.999]. (C) Representative traces (left) and time-course summary of EPSCs obtained in D1(+) MSNs showing the effects of HA in the presence of PKA inhibitor, H89 [: D1(+) HA in H89: 67.12±3.76%, n=6, 1-way ANOVA, ACSF vs. H89, p=0.997]. (D) Representative traces (left) and time-course summary of EPSCs obtained in D1(+) MSNs showing the effects of HA in the presence of Gβγ inhibitor, gallein [D1(+) HA in gallein: 94.38±4.51%, 1-way ANOVA, ACSF vs. gallein, n=6, p<0.001]. (E) Representative traces (left) and time-course summary of EPSCs obtained in D1(+) MSNs showing the effects of HA in the presence of Akt-1/2 inhibitor, Akti1/2 [D1(+) HA in Akti1/2 : 91.38±3.01%, 1-way ANOVA, ACSF vs. Akti1/2, n=4, p<0.001]. (F) Representative traces (left) and time-course summary of EPSCs obtained in D1(+) MSNs showing the effects of HA in the presence of MAPK (MEK1/2) inhibitor, U0126 [D1(+) HA in U0126: 87.02±5.99%, 1-way ANOVA, ACSF vs. U0126, n=6, p=0.002]. (G) Representative traces (left) and time-course summary of EPSCs obtained in D1(+) MSNs showing the effects of HA in the presence of GSK-3 inhibitor, CHIR 99021 [: D1(+) HA in CHIR 99021: 92.45±3.45%, 1-way ANOVA, ACSF vs. CHIR, n=6, p<0.001]. (H) Quantification of average HA-induced depression during each pharmacological manipulation at t(grey) = 45–50-min. Scale bars: 100-pA/20-ms. Error bars indicate SEM with (*) signifying p < 0.05.
Having ruled out canonical effectors targeted by Gαi signaling, we next asked if H3R activation mobilizes the Gβγ complex to elicit HA-LTD at D1(+)-MSN synapses. To test this hypothesis, we incubated slices in biased Gβγ complex inhibitor, gallein (20 μM). Pre-incubation in gallein completely blocked HA-LTD, indicating that HA-LTD is mediated by an intracellular pathway targeted by the Gβγ complex (Fig. 5D,H). A downstream target of Gβγ signaling associated with long-term changes in synaptic strength is the phosphoinositide 3-kinase (PI3K) pathway-Akt pathway. Intracellular recruitment of the PI3K-Akt signaling pathway underlies GPCR-induced synaptic plasticity in various regions(26–28). Superfusion of HA in the presence of Akt-1/2 inhibitor, Akti1/2 (10 μM), induced a transient depression in EPSC amplitude that returned to baseline following drug wash-out, indicating that Akt signaling contributes to the expression of HA-LTD at D1(+)-MSN synapses in the NAcC (Fig. 5E,H).
Several intracellular targets of Akt signaling can induce heterosynaptic forms of LTD, most notably MAPKs (e.g., MEK1/2) and glycogen synthase kinase-3β (GSK-3β)(29). To determine if MAPK signaling contributes to HA-LTD, HA was bath-applied in the presence of MEK1/2 inhibitor, U0126 (1 μM). HA-LTD at D1(+)-MSNs was only partially disrupted following MEK1/2 blockade, indicating that MAPK signaling likely serves as a parallel effector in the mechanism engaged by H3R (Fig. 5F,H). Given the apparent contingency of Gβγ-directed Akt-MAPK function on HA-LTD, we postulated that HA requires a proximal shift in the activity of GSK-3β, as phospho-regulation of GSK-3β can mediate the expression of PI3K/Akt-dependent forms of LTD(19,27). Thus, we incubated slices in GSK-3 inhibitor, CHIR 99021 (2 μM). Bath-application of HA in the presence of CHIR 99021 completely blocked HA-LTD at D1(+)-MSN synapses (Fig. 5G,H). Together, these findings elucidate a novel form of plasticity in the NAc wherein H3R activation engages Akt signaling to induce GSK-3β-dependent LTD of glutamatergic transmission onto D1(+)-MSNs.
Thalamocortical drive onto D1(+) MSNs in the NAc is differentially regulated by HA signaling
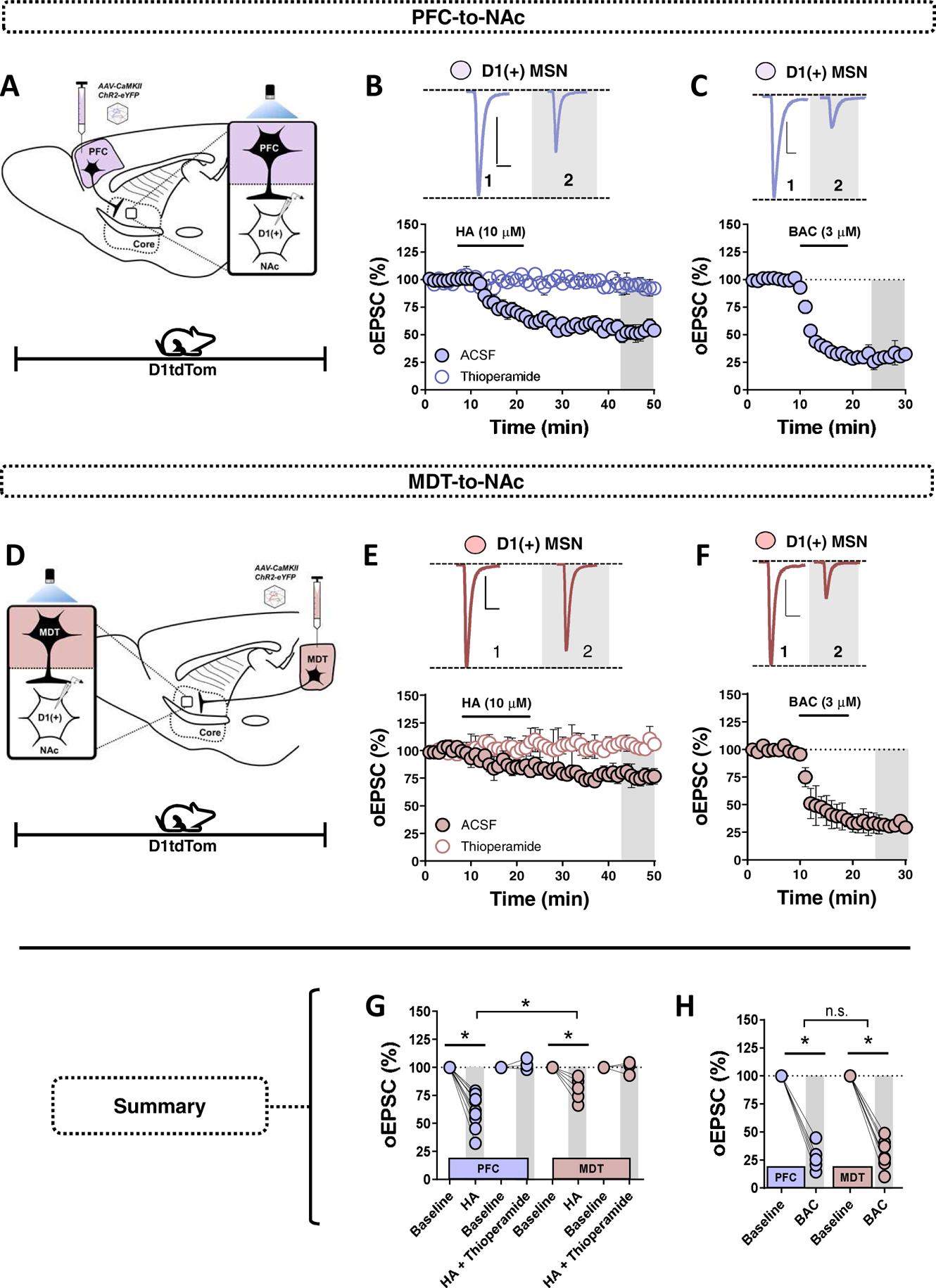
We next hypothesized that HA acutely tunes MSN responsiveness to specific glutamatergic afferents, an effect recently observed with the opioid, cannabinoid, and metabotropic glutamate receptor systems in the NAc and striatum(18,30,31). To test this hypothesis, we employed optogenetics to determine if specific glutamatergic inputs onto D1(+)-MSNs are differentially sensitive to HA. We examined inputs from the prefrontal cortex (PFC) and mediodorsal thalamus (MDT), as glutamatergic inputs from these regions exert divergent effects on NAc-dependent motivational states, with the PFC supporting reward-related behavioral outcomes and the MDT contributing to negative emotional valence(32,33). The viral construct, AAV-CaMKII-ChR2-eYFP, was stereotaxically injected into the medial PFC and periventricular MDT of D1tdTomato mice, as described previously(17,18) (Fig. 6A,D).
Figure 6. Thalamocortical drive onto D1(+) MSNs in the NAc is differentially regulated by HA signaling.
(A) Schematic of (1) stereotaxic delivery of ChR2-eYP-harboring viral vectors into the medial PFC of D1tdTomato mice; (2) patch-clamp recordings in NAc core D1(+) MSNs of mice expressing ChR2 in the PFC (purple). (B) Representative traces (above) and time-course summary of optically-evoked EPSCs (oEPSCs) from the PFC at baseline and in the presence of HA (closed circles) and HA in thioperamide (open circles) in D1(+) MSNs [PFC-to-D1(+) HA: 60.66±4.79%, n=10, p<0.001]. Scale bars (PFC): 50-pA/20-ms. (C) Representative traces and time-course summary of oEPSCs from the PFC at baseline and in the presence of BAC (3 μM). Scale bars: 50-pA/20-ms (D) Schematic of (1) stereotaxic delivery of ChR2-eYP-harboring viral vectors into the MDT of D1tdTomato mice; (2) patch-clamp recordings in NAc core D1(+) MSNs of mice expressing ChR2 in the MDT (red). (E) Representative traces (above) and time-course summary of optically-evoked EPSCs (oEPSCs) from the MDT at baseline and in the presence of HA (closed red circles) and HA+ thioperamide (open circles) in D1(+) MSNs [MDT-to-D1(+) HA: 80.25±5.15%, n=6, p<0.001; 1-way RM-ANOVA, input effect: F3,28 = 39.4, p=0.0017, Sidak’s post-hoc analysis]. (F) Representative traces (above) and time-course summary MDT oEPSCs obtained in D1(+) MSNs showing the effects of BAC on oEPSC amplitude. Scale bars (MDT): 100-pA/20-ms. (G) Average oEPSC amplitude at PFC- and MDT-to-NAc D1(+) MSN synapses obtained at t(grey) = 45–50-min post-HA and post-HA + thioperamide. (H) Average BAC-induced oEPSC amplitude at PFC- and MDT-to-NAc D1(+) MSN synapses obtained at t(grey) = 25–30-min [PFC-to-D1(+) BAC: 27.49±4.6%, n=6, p<0.001; MDT-to-D1(+) BAC: 30.10±5.28%, n=7, p<0.001; 1-way ANOVA, input effect: F3,22 = 158, p=0.994]. Error bars indicate SEM with (*) signifying p < 0.05.
To determine if PFC-to-NAc inputs are regulated by HA, oEPSCs were obtained in D1(+)-MSNs from D1tdTomato mice expressing ChR2 in the medial PFC (Fig. 6A). HA (10 μM) was superfused into the ACSF bath for 15-min, resulting in a robust depression in oEPSC amplitude (Fig. 6B,G). In contrast to PFC-to-NAc synapses, HA resulted in a significantly smaller depression in oEPSC amplitude at MDT-to-D1(+)-MSN synapses (Fig. 6E,G). These data suggest that HA modulates thalamocortical gain in the NAc such that MSN responsiveness is biased toward glutamatergic afferents originating from the MDT. To ensure that the differential effects of HA were afferent-specific, we examined presynaptic GABAB receptor (GABABR) function at both synaptic inputs, as GABABR is highly expressed in the NAc and significantly regulates presynaptic glutamate release(15). Bath-application of GABABR agonist, baclofen (BAC, 3 μM), resulted in a robust depression in EPSC amplitude at PFC- and MDT-to-D1(+)- MSN synapses that was indistinct between inputs (Fig. 6C,F,H). Together, these data support the hypothesis that HA differentially regulates PFC- and MDT inputs to the NAcC.
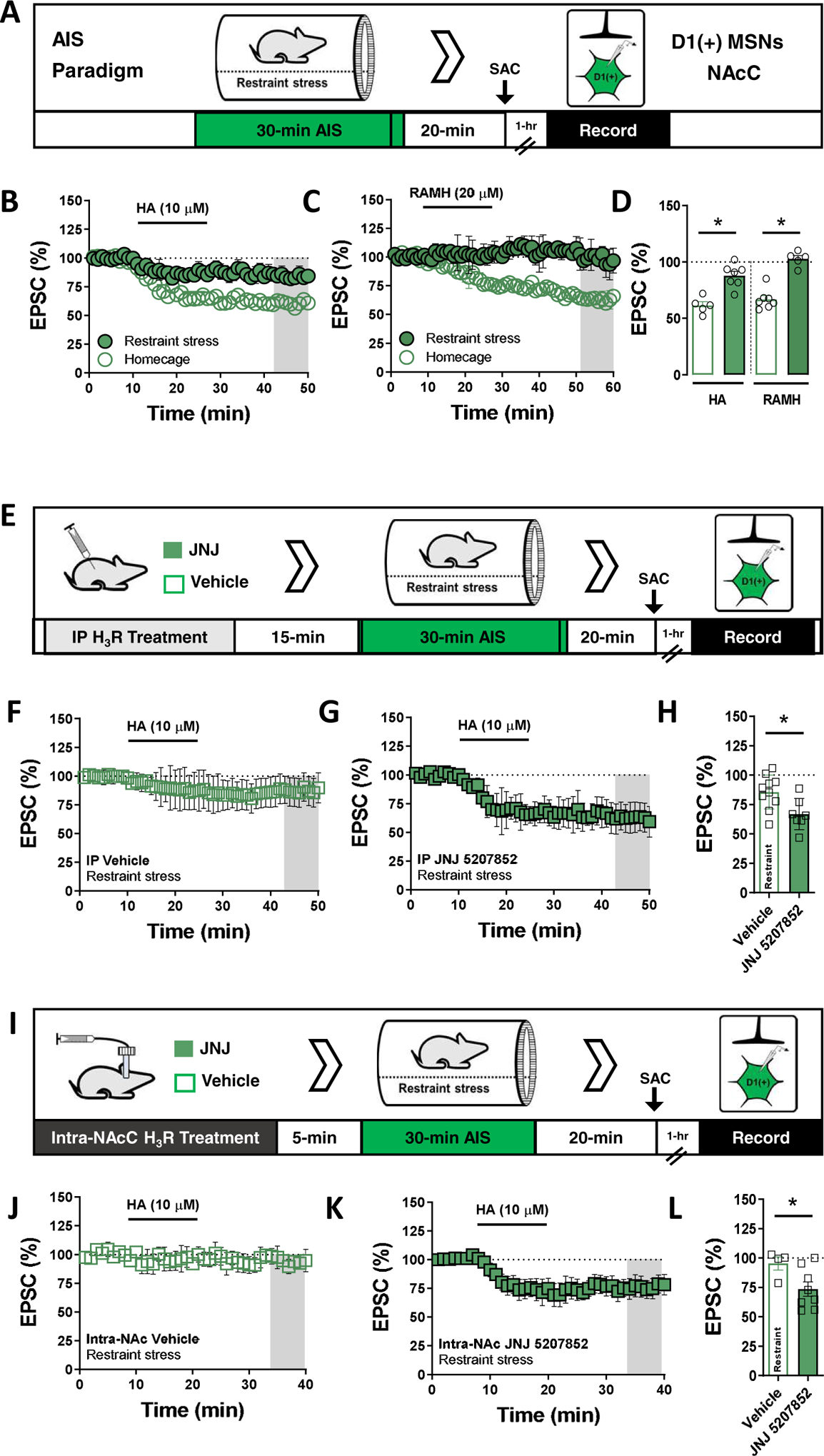
Acute stress recruits endogenous H3R signaling at glutamatergic synapses onto D1(+) MSNs in the NAcC
HA-containing TMN neuron output increases during behavioral states requiring heightened awareness of salient environment stimuli, including acute stress, fear learning, and systemic metabolic strain(34–36). Thus, we asked whether acute stress recruits endogenous HA signaling at glutamatergic synapses in the NAcC. We employed an acute immobilization stress (AIS) paradigm in which mice were restrained for 30-min followed by a 20-min recovery period, after which brain slices were prepared for ex vivo electrophysiology (Fig. 7A). We hypothesized that AIS engages TMN-to-NAc volume transmission, thereby altering the expression of HA-LTD at glutamatergic synapses onto D1(+)-MSNs. Similar to experiments performed in naïve mice, bath-application of HA in control mice elicited a robust depression in EPSC amplitude at D1(+)-MSN synapses. However, in mice that underwent AIS, HA elicited a slight depression in EPSC amplitude that was significantly attenuated relative to control mice (Fig. 7B,C). In contrast, bath-application of HA had no significant effect at glutamatergic synapses onto D1(−)-MSNs in both AIS-exposed and control mice (Fig. S2A–C).
Figure 7. Acute immobilization stress recruits endogenous H3R signaling at glutamatergic synapses onto D1(+) MSNs in the NAc core.
(A) Schematic depicting 30-min acute immobilization stress (AIS) paradigm and recording strategy in D1(+) MSNs of control (open circles) and AIS-exposed D1tdTomato mice (green circles), (B) Time-course summary of electrically-evoked EPSCs in control homecage mice and AIS-exposed mice depicting the effects of HA at synapses onto D1(+) MSNs in the NAcC [D1(+) HA control: 61.48±3.64%, n=5, N(animals)=4; D1(+) HA AIS: 87.88±4.13%, n=7, N(animals)=5, p<0.001]. (C) Time-course summary of EPSCs in control mice (left) and AIS-exposed mice (right) depicting the effects of H3R agonist, RAMH, at synapses onto D1(+) MSNs [D1(+) RAMH control: 66.78±3.61%, n=7, N (animals)=4; D1(+) RAMH AIS: 93.08±7.13%, n=7, N(animals)=5, p=0.004]. (D) Average HA and RAMH-induced EPSC amplitude obtained at t(gray) = 45–50 min in control and AIS-exposed mice. (E) Schematic depicting prophylactic treatment with water-soluble non-imidazole H3R antagonist, JNJ 5207852, or vehicle (saline) prior to AIS exposure. (F) Time-course summary of EPSCs in D1(+) MSNs of vehicle-treated (open squares) AIS-exposed mice. (G) Time-course summary of EPSCs in D1(+) MSNs of JNJ 5207852-treated (green squares) AIS-exposed mice [D1(+) HA-VEH: 85.61±4.82%, n=10, N(animals)=5; D1(+) HA-JNJ: 66.85±5.38%, n=7, N(animals)=6, p=0.008]. (H) Average HA-induced EPSC amplitude obtained at t(gray) = 45–50 min in vehicle vs. JNJ 5207852-treated AIS-exposed mice. (I) Schematic depicting intra-NAc microinfusion of 3.86 μg/μL JNJ 5207852 or vehicle prior to AIS exposure. (J) Time-course summary of EPSCs in D1(+) MSNs of intra-NAc vehicle-infused (open squares) AIS-exposed mice. (K) Time-course summary of EPSCs in D1(+) MSNs of JNJ 5207852-infused (green squares) AIS-exposed mice [Fig. 8j-l: HA-VEH: 95.35±5.62%, n=4, N(animals)=2; HA-JNJ: 73.65±6.14%, n=8, N(animals)=4, p=0.0481]. (L) Average HA-induced EPSC amplitude obtained at t(gray) = 35–40 min in intra-NAc vehicle vs. JNJ 5207852-infused AIS-exposed mice. Error bars indicate SEM with (*) signifying p < 0.05.
To determine if the reduction in HA-LTD was due to an experience-dependent shift in H3R function, we repeated these experiments with H3R agonist, RAMH. In control mice, RAMH significantly decreased EPSC amplitude at D1(+)-MSN synapses that persisted throughout the recording period. However, bath-application of RAMH in AIS mice had no significant effect on EPSC amplitude, supporting the hypothesis that AIS modulates presynaptic H3R function at glutamatergic synapses onto D1(+)-MSNs (Fig. 7D,E). One explanation for these findings is that stress-evoked H3R activity occludes subsequent HA-LTD assessed ex vivo. If this hypothesis is correct, in vivo blockade of H3R should protect HA-LTD at D1(+)-MSN synapses in the NAc.
Thus, 15-min prior to AIS, mice received an intraperitoneal (IP) injection of vehicle (saline) or brain-penetrant H3R antagonist, JNJ5207852 (10 mg/kg) (Fig. 7F). JNJ520785 was chosen over thioperamide due to its solubility profile and minimal effects on hepatic cytochrome P450 activity(14,37). Similar to prior experiments performed in AIS-exposed mice, HA-LTD in vehicle-treated mice AIS mice was significantly attenuated. Prior administration of JNJ5207852 in AIS mice significantly increased HA-LTD relative to vehicle-treated mice (Fig. 7G,H). Together, these data suggest that AIS occludes HA-LTD at glutamatergic synapses onto D1(+)-MSNs by recruiting endogenous HA signaling via H3R.
Our findings suggest that AIS alters presynaptic H3R function at glutamatergic synapses onto D1(+) MSNs. However, stress may evoke NAc- and HA-independent synaptic adaptations that attenuate the expression of HA-LTD at these synapses. To assess whether AIS recruits HA-induced H3R signaling specifically at synapses in the NAc, we microinfused JNJ5207852 directly into the NAcC of D1tdTomato mice 5-min prior to AIS exposure. Following AIS and the 20-min recovery period, cannulae were extruded and brain slices were prepared for electrophysiological assessment of HA-LTD at D1(+)-MSNs in the NAcC. Bath-application of HA in vehicle-infused mice had minimal effect on EPSC amplitude, congruent with the initial effects of AIS observed at this synapse. In JNJ5207852-infused mice, HA resulted in a decrease in glutamatergic transmission at D1(+)-MSNs that was significantly greater than vehicle-infused control mice. Taken together, these data suggest that AIS-evoked HA signaling elicits NAc-specific H3R activity at glutamatergic synapses onto D1(+)-MSNs.
Discussion
We report that HA recruits a presynaptic gain control mechanism at glutamatergic synapses in the NAc. At D1(+)-MSN synapses in the NAcC, HA induces H3R-dependent LTD by mobilizing the Gβγ complex to engage Akt-GSK3β signaling. Furthermore, PFC-to-NAc synapses exhibit greater sensitivity to HA than MDT-to-NAc synapses, shifting thalamocortical balance onto D1(+)-MSNs. Finally, HA signaling in the NAc is recruited by AIS in an H3R-dependent manner, suggesting that acute stress engages endogenous HA signaling at glutamatergic synapses in the NAc. These findings offer novel insight into an understudied neuromodulatory system regulating NAc circuit function.
Our initial findings in WT mice indicate that HA decreases glutamatergic transmission uniformly across NAc core and shell subterritories. However, a subset of synapses failed to undergo HA-induced changes in synaptic strength, pointing to differentiable populations of HA-responsive [HA-(+)] MSN synapses. Consistent with this hypothesis, HA decreased synaptic efficacy onto D1(+)-MSNs with minimal effect at synapses onto D1(−)-MSNs. The effect at D1(+)-MSN synapses was abolished by two chemically-distinct H3R antagonists and recapitulated with a selective H3R agonist, indicating that H3R mediates the effect of HA on excitatory transmission in the NAc. Although we do not fully explain the cell-type specific actions of HA, it is unlikely that HA-containing varicosities preferentially synapse onto D1(+)-MSNs, as striatal HA signaling occurs predominately via volume transmission(11,38). Alternatively, we found that glutamatergic synapses onto D1(−)-MSNs, but not D1(+)-MSNs, exhibit tonic presynaptic H3R activity. While we do not test synapses-specific differences in tonic HA release, H3Rs exhibit constitutive receptor activity that may occlude additive effects of HA observed at D1(−)-MSN synapses(39).
An intriguing question that remains is whether glutamatergic afferents in the NAc express varying levels or isoforms of H3R that couple to distinct intracellular effectors. Our input-specific optogenetic analysis encourages this notion, as HA strongly inhibits PFC-to-NAc inputs onto D1(+)-MSNs with only modest efficacy at MDT synapses. Thalamocortical rearrangements in the NAc exert divergent effects on NAc-directed motivational behavior(33,40,41). Given that no overt differences have been described between the connectivity of these regions onto D1(+) and D1(−)-MSNs, these findings suggest that HA may direct NAc responsiveness to inputs from the MDT over the PFC(14,42). By improving the signal-to-noise element of MDT-to-NAc transmission and preferentially acting at D1(+)-MSN synapses, the physiological actions of HA at these synapses may explain the effects of central H3R function on drug reward behavior, though future studies are needed to test this hypothesis(43,44).
We report that HA recruits a presynaptic locus of action at D1(+)-MSN synapses. Although presynaptic H3R function has been shown to also negatively regulate glutamate release in the dorsal striatum, recent reports indicate that H3R is also expressed on striatal D1 and D2-MSNs(25,45,46). It is tempting to speculate that HA engages a parallel postsynaptic effector pathway. However, disabling postsynaptic GPCR function with GDPβS and optically-uncaging RuBi-Glu AMPAR currents failed to unmask a postsynaptic effect of HA on glutamatergic transmission. Because MSN output is reliant on concerted glutamatergic input to reach AP threshold, decreasing glutamatergic drive onto MSNs should reduce functional NAc output. Our findings support this notion, as HA reduces the gain and synaptically-evoked AP fidelity by imposing a filter on glutamatergic inputs onto D1(+)-MSNs. Altogether, our data suggests that HA redistributes NAc circuit activity by presynaptically decreasing glutamatergic synaptic efficacy onto D1(+)-MSNs.
Although heterosynaptic HA function has been demonstrated elsewhere in the CNS, few studies define the temporal dynamics of H3R on synaptic transmission or the mechanism(s) engaged by H3R in native tissue preparations. We offer insight into the mechanism by which HA depresses glutamatergic synapses onto D1(+)-MSNs, showing that HA triggers H3R-dependent LTD that requires Gβγ-directed Akt-GSK3β signaling. While the depression in glutamatergic transmission and increase in PPR and CV persisted following drug wash-out, interpreting this finding is complicated by reports that HA may be retained in slices post-application(10). Thus, we assessed whether HA induces LTD by chasing HA with thioperamide. Thioperamide resulted in a modest uptrend toward baseline that remained depressed throughout the recording period, indicating that HA indeed triggers LTD of excitatory transmission onto D1(+)-MSNs.
Our findings point to a critical role for the Gβγ effector arm in the induction of H3R-dependent LTD. While Gβγ signaling can activate multiple intracellular signaling pathways, recent in vivo data suggest H3R activation engages striatal Akt-MAPK-GSK3β signaling without affecting AC/cAMP/PKA function, consistent with our findings(19,47). Moreover, GSK-3β is associated with H3R function and contributes to NAc-dependent motivational states(48,49). Our data proposes that H3R activation mobilizes the Gβγ complex, which activates the Akt pathway, leading to the phosphorylation of MAPKs (MEK1/2) and GSK-3β. Altering presynaptic GSK-3β activity may shift the phosphorylation state of exocytotic release machinery, leading to the expression of HA-LTD(27). Multiple forms of presynaptic LTD in the NAc core proceed through molecular alterations in SNARE-associated release machinery, such as RIM1α(50). Although our pharmacological analysis points to a presynaptic Akt-GSK3 effector system in mediating the effects of HA, we do not provide direct biochemical evidence of this interaction in the NAcC. Moreover, our data does not rule out the possibility that multiple signaling mechanisms are engaged in parallel by presynaptic H3R activity, as inhibiting MAPKs blunted but did not completely block HA-LTD. Additional studies and tools will be needed to determine exactly how HA-induced H3R signaling elicits a prolonged decrease in glutamate release probability onto D1(+)-MSNs in the NAc.
HA transmission gates sleep-wake transitions, arousal, and attentional control(3,42,51). Accordingly, in vivo TMN activity is enhanced during bouts of acute stress, such as forced swim, foot shock, and AIS(34,35). Thus, we employed AIS as a means to recruit the TMN axis and assess whether endogenous HA signaling modulates glutamatergic transmission in the NAcC. We find that HA-LTD at D1(+)-MSN synapses is substantially reduced following AIS in a NAc-specific H3R-dependent manner, as pre-treatment with an H3R antagonist IP or microinfused into the NAcC protects this plasticity. Although HAergic afferents from the TMN are the most probable source of endogenous HA signaling in the NAc, HA derived from mast cells cannot be excluded, as several studies indicate that HA degranulation is triggered by acute stress(52,53). Nevertheless, while mast cell degranulation contributes to overall CNS HA content, direct synaptic effects of mast cell-derived HA remain to be determined.
While the involvement of HA in stress-associated behavioral states is beyond the scope of this study, our findings encourage investigative interest in the role of NAc HA in stress-related pathologies, including depression, anxiety, and addiction. Early pre-clinical studies suggest that pharmacologically targeting H3R with non-imidazole-based compounds modulates the rewarding properties of drugs of abuse, including psychostimulants and alcohol(43,44). Given the established role of the NAc in goal-directed behavior, it is enticing to speculate broadly that HA and/or H3R signaling gates stress-induced decisional economic strategies communicated by NAc-projecting top-down control centers, such as the PFC and MDT. These studies highlight a novel neuromodulatory network in the NAc with potential therapeutic avenues for the management of stress-related motivational states.
Supplementary Material
KEY RESOURCES TABLE
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Chemical Compound, Drug | Histamine dihydrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 5818 | |
| Chemical Compound, Drug | Thioperamide | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 3035905 | |
| Chemical Compound, Drug | JNJ 5207852 dihydrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 90488903 | |
| Chemical Compound, Drug | Cetirizine dihydrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 55182 | |
| Chemical Compound, Drug | Ranitidine hydrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 657344 | |
| Chemical Compound, Drug | (R)-(−)-α-methylhistamine dihydrobromide | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 45037031 | |
| Chemical Compound, Drug | RuBi-Glutamate | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 90488860 | |
| Chemical Compound, Drug | (RS)-baclofen | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 2284 | |
| Chemical Compound, Drug | Akti-1/2 | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 10196499 | |
| Chemical Compound, Drug | CHIR 99021 | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 9956119 | |
| Chemical Compound, Drug | Gallein | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 73685 | |
| Chemical Compound, Drug | Forskolin | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 47936 | |
| Chemical Compound, Drug | NBQX disodium salt | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 3272523 | |
| Chemical Compound, Drug | Picrotoxin | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 6473767 | |
| Chemical Compound, Drug | H89 dihydrochloride | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 5702541 | |
| Chemical Compound, Drug | BAPTA-AM | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 2293 | |
| Chemical Compound, Drug | U0126 | Tocris/Bio-Techne, Minneapolis, MN | PubChem ID: 3006531 | |
| Organism, Strain | Mouse:B6.Cg-Tg(Drd1a-tdTomato)6Calak/J | The Jackson Laboratory, Sacramento, CA | Stock No: 016204 | |
| Organism, Strain | Mouse: C57BL/6J | The Jackson Laboratory, Sacramento, CA | Stock No: 000664 | |
| Bacterial or viral strain | Virus: pAAV5-CaMKIIα-hChR2(H134R)-EYFP | Addgene, Watertown, MA | Plasmid No: 26969 | |
| Software, Algorithm | EthoVision XT | Noldus Information Technology, Leesburg, VA | www.noldus.com | |
| Software, Algorithm | pClamp 10 | Molecular Devices, Sunnyvale, CA | www.moleculardevices.com | |
| Software, Algorithm | GraphPad Prism | GraphPad Software, Inc., La Jolla, CA | www.graphpad.com/scientific-software/prism/ |
Acknowledgements
We thank members of the Grueter lab for their helpful comments. This study was supported by National Institute on Drug Abuse (NIDA) grant R01DA040630 (to B.A.G).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures
All authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Bunney BS, Aghajanian GK (1975): Evidence for drug actions on both pre- and postsynaptic catecholamine receptors in the CNS. Psychopharmacol Bull 11: 8–10. [PubMed] [Google Scholar]
- 2.Passani MB, Blandina P (2011): Histamine receptors in the CNS as targets for therapeutic intervention. Trends Pharmacol Sci 32: 242–249. [DOI] [PubMed] [Google Scholar]
- 3.Yu X, Ye Z, Houston CM, Zecharia AY, Ma Y, Zhang Z, et al. (2015): Wakefulness Is Governed by GABA and Histamine Cotransmission. Neuron 87: 164–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takagi H, Morishima Y, Matsuyama T, Hayashi H, Watanabe T, Wada H (1986): Histaminergic axons in the neostriatum and cerebral cortex of the rat: a correlated light and electron microscopic immunocytochemical study using histidine decarboxylase as a marker. Brain Res 364: 114–123. [DOI] [PubMed] [Google Scholar]
- 5.Shoblock J, O’Donnell P (2000): Histamine Modulation of Nucleus Accumbens Neurons. Annals of the New York Academy of Sciences 909: 270–2. [DOI] [PubMed] [Google Scholar]
- 6.Turner BD, Kashima DT, Manz KM, Grueter CA, Grueter BA (2018): Synaptic Plasticity in the Nucleus Accumbens: Lessons Learned from Experience. ACS Chem Neurosci 9: 2114–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baimel C, McGarry LM, Carter AG (2019): The Projection Targets of Medium Spiny Neurons Govern Cocaine-Evoked Synaptic Plasticity in the Nucleus Accumbens. Cell Rep 28: 2256–2263.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A (2012): Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76: 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, Lüscher C (2014): Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509: 459–464. [DOI] [PubMed] [Google Scholar]
- 10.Brown RE, Reymann KG (1996): Histamine H3 receptor-mediated depression of synaptic transmission in the dentate gyrus of the rat in vitro. J Physiol (Lond) 496 ( Pt 1): 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellender TJ, Huerta-Ocampo I, Deisseroth K, Capogna M, Bolam JP (2011): Differential modulation of excitatory and inhibitory striatal synaptic transmission by histamine. J Neurosci 31: 15340–15351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takei H, Yamamoto K, Bae Y-C, Shirakawa T, Kobayashi M (2017): Histamine H3 Heteroreceptors Suppress Glutamatergic and GABAergic Synaptic Transmission in the Rat Insular Cortex. Front Neural Circuits 11: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bristow LJ, Bennett GW (1988): Biphasic effects of intra-accumbens histamine administration on spontaneous motor activity in the rat; a role for central histamine receptors. Br J Pharmacol 95: 1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brabant C, Alleva L, Quertemont E, Tirelli E (2010): Involvement of the brain histaminergic system in addiction and addiction-related behaviors: a comprehensive review with emphasis on the potential therapeutic use of histaminergic compounds in drug dependence. Prog Neurobiol 92: 421–441. [DOI] [PubMed] [Google Scholar]
- 15.Manz KM, Baxley AG, Zurawski Z, Hamm HE, Grueter BA (2019): Heterosynaptic GABAB receptor function within feedforward microcircuits gates glutamatergic transmission in the nucleus accumbens core. J Neurosci. 10.1523/JNEUROSCI.1395-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Báldi R, Ghosh D, Grueter BA, Patel S (2016): Electrophysiological Measurement of Cannabinoid-Mediated Synaptic Modulation in Acute Mouse Brain Slices. Curr Protoc Neurosci 75: 6.29.1–6.29.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joffe ME, Grueter BA (2016): Cocaine Experience Enhances Thalamo-Accumbens N-Methyl-D-Aspartate Receptor Function. Biol Psychiatry 80: 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner BD, Rook JM, Lindsley CW, Conn PJ, Grueter BA (2018): mGlu1 and mGlu5 modulate distinct excitatory inputs to the nucleus accumbens shell. Neuropsychopharmacology 43: 2075–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rapanelli M, Frick LR, Horn KD, Schwarcz RC, Pogorelov V, Nairn AC, Pittenger C (2016): The Histamine H3 Receptor Differentially Modulates Mitogen-activated Protein Kinase (MAPK) and Akt Signaling in Striatonigral and Striatopallidal Neurons. J Biol Chem 291: 21042–21052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ade KK, Wan Y, Chen M, Gloss B, Calakos N (2011): An Improved BAC Transgenic Fluorescent Reporter Line for Sensitive and Specific Identification of Striatonigral Medium Spiny Neurons. Front Syst Neurosci 5: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fino E, Araya R, Peterka DS, Salierno M, Etchenique R, Yuste R (2009): RuBi-Glutamate: Two-Photon and Visible-Light Photoactivation of Neurons and Dendritic spines. Front Neural Circuits 3: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aquino-Miranda G, Escamilla-Sánchez J, González-Pantoja R, Bueno-Nava A, Arias-Montaño J-A (2016): Histamine H3 receptor activation inhibits dopamine synthesis but not release or uptake in rat nucleus accumbens. Neuropharmacology 106: 91–101. [DOI] [PubMed] [Google Scholar]
- 23.Zhuang Q-X, Xu H-T, Lu X-J, Li B, Yung W-H, Wang J-J, Zhu J-N (2018): Histamine Excites Striatal Dopamine D1 and D2 Receptor-Expressing Neurons via Postsynaptic H1 and H2 Receptors. Mol Neurobiol 55: 8059–8070. [DOI] [PubMed] [Google Scholar]
- 24.Nuutinen S, Vanhanen J, Mäki T, Panula P (2012): Histamine H3 Receptor: A Novel Therapeutic Target in Alcohol Dependence? Front Syst Neurosci 6 10.3389/fnsys.2012.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rapanelli M, Frick L, Jindachomthong K, Xu J, Ohtsu H, Nairn AC, Pittenger C (2018): Striatal Signaling Regulated by the H3R Histamine Receptor in a Mouse Model of tic Pathophysiology. Neuroscience 392: 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lüscher C, Huber KM (2010): Group 1 mGluR-Dependent Synaptic Long-Term Depression: Mechanisms and Implications for Circuitry and Disease. Neuron 65: 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradley CA, Peineau S, Taghibiglou C, Nicolas CS, Whitcomb DJ, Bortolotto ZA, et al. (2012): A pivotal role of GSK-3 in synaptic plasticity. Front Mol Neurosci 5 10.3389/fnmol.2012.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joffe ME, Santiago CI, Stansley BJ, Maksymetz J, Gogliotti RG, Engers JL, et al. (2019): Mechanisms underlying prelimbic prefrontal cortex mGlu3/mGlu5-dependent plasticity and reversal learning deficits following acute stress. Neuropharmacology 144: 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horwood JM, Dufour F, Laroche S, Davis S (2006): Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur J Neurosci 23: 3375–3384. [DOI] [PubMed] [Google Scholar]
- 30.Tejeda HA, Wu J, Kornspun AR, Pignatelli M, Kashtelyan V, Krashes MJ, et al. (2017): Pathway- and Cell-Specific Kappa-Opioid Receptor Modulation of Excitation-Inhibition Balance Differentially Gates D1 and D2 Accumbens Neuron Activity. Neuron 93: 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deroche MA, Lassalle O, Manzoni OJ (2019): Cell-type and endocannabinoid specific synapse connectivity in the adult nucleus accumbens core. bioRxiv 613497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascoli V, Turiault M, Lüscher C (2011): Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature 481: 71–75. [DOI] [PubMed] [Google Scholar]
- 33.Zhu Y, Wienecke CFR, Nachtrab G, Chen X (2016): A thalamic input to the nucleus accumbens mediates opiate dependence. Nature 530: 219–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor KM, Snyder SH (1971): Brain histamine: rapid apparent turnover altered by restraint and cold stress. Science 172: 1037–1039. [DOI] [PubMed] [Google Scholar]
- 35.Dismukes K, Snyder SH (1974): Histamine turnover in rat brain. Brain Res 78: 467–481. [DOI] [PubMed] [Google Scholar]
- 36.Miklós IH, Kovács KJ (2003): Functional heterogeneity of the responses of histaminergic neuron subpopulations to various stress challenges. Eur J Neurosci 18: 3069–3079. [DOI] [PubMed] [Google Scholar]
- 37.Barbier AJ, Berridge C, Dugovic C, Laposky AD, Wilson SJ, Boggs J, et al. (2004): Acute wake-promoting actions of JNJ-5207852, a novel, diamine-based H3 antagonist. Br J Pharmacol 143: 649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giannoni P, Passani M-B, Nosi D, Chazot PL, Shenton FC, Medhurst AD, et al. (2009): Heterogeneity of histaminergic neurons in the tuberomammillary nucleus of the rat. Eur J Neurosci 29: 2363–2374. [DOI] [PubMed] [Google Scholar]
- 39.Morisset S, Rouleau A, Ligneau X, Gbahou F, Tardivel-Lacombe J, Stark H, et al. (2000): High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature 408: 860–864. [DOI] [PubMed] [Google Scholar]
- 40.Lee BR, Ma Y-Y, Huang YH, Wang X, Otaka M, Ishikawa M, et al. (2013): Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat Neurosci 16: 1644–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sweis BM, Larson EB, Redish AD, Thomas MJ (2018): Altering gain of the infralimbic-to-accumbens shell circuit alters economically dissociable decision-making algorithms. Proc Natl Acad Sci USA 115: E6347–E6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blandina P, Munari L, Provensi G, Passani MB (2012): Histamine neurons in the tuberomamillary nucleus: a whole center or distinct subpopulations? Front Syst Neurosci 6 10.3389/fnsys.2012.00033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox GB, Esbenshade TA, Pan JB, Radek RJ, Krueger KM, Yao BB, et al. (2005): Pharmacological properties of ABT-239 [4-(2-{2-[(2R)-2-Methylpyrrolidinyl]ethyl}-benzofuran-5-yl)benzonitrile]: II. Neurophysiological characterization and broad preclinical efficacy in cognition and schizophrenia of a potent and selective histamine H3 receptor antagonist. J Pharmacol Exp Ther 313: 176–190. [DOI] [PubMed] [Google Scholar]
- 44.Galici R, Rezvani AH, Aluisio L, Lord B, Levin ED, Fraser I, et al. (2011): JNJ-39220675, a novel selective histamine H3 receptor antagonist, reduces the abuse-related effects of alcohol in rats. Psychopharmacology (Berl) 214: 829–841. [DOI] [PubMed] [Google Scholar]
- 45.Moreno E, Hoffmann H, Gonzalez-Sepúlveda M, Navarro G, Casadó V, Cortés A, et al. (2011): Dopamine D1-histamine H3 receptor heteromers provide a selective link to MAPK signaling in GABAergic neurons of the direct striatal pathway. J Biol Chem 286: 5846–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreno E, Moreno-Delgado D, Navarro G, Hoffmann HM, Fuentes S, Rosell-Vilar S, et al. (2014): Cocaine disrupts histamine H3 receptor modulation of dopamine D1 receptor signaling: σ1-D1-H3 receptor complexes as key targets for reducing cocaine’s effects. J Neurosci 34: 3545–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jernigan KK, Cselenyi CS, Thorne CA, Hanson AJ, Tahinci E, Hajicek N, et al. (2010): Gbetagamma activates GSK3 to promote LRP6-mediated beta-catenin transcriptional activity. Sci Signal 3: ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scala F, Nenov MN, Crofton EJ, Singh AK, Folorunso O, Zhang Y, et al. (2018): Environmental Enrichment and Social Isolation Mediate Neuroplasticity of Medium Spiny Neurons through the GSK3 Pathway. Cell Rep 23: 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aceto G, Colussi C, Leone L, Fusco S, Rinaudo M, Scala F, et al. (2020): Chronic mild stress alters synaptic plasticity in the nucleus accumbens through GSK3β-dependent modulation of Kv4.2 channels. Proc Natl Acad Sci USA 117: 8143–8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grueter BA, Brasnjo G, Malenka RC (2010): Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci 13: 1519–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Venner A, Mochizuki T, De Luca R, Anaclet C, Scammell TE, Saper CB, et al. (2019): Reassessing the role of histaminergic tuberomammillary neurons in arousal control. J Neurosci. 10.1523/JNEUROSCI.1032-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Theoharides TC, Spanos C, Pang X, Alferes L, Ligris K, Letourneau R, et al. (1995): Stress-induced intracranial mast cell degranulation: a corticotropin-releasing hormone-mediated effect. Endocrinology 136: 5745–5750. [DOI] [PubMed] [Google Scholar]
- 53.Baldwin AL (2006): Mast cell activation by stress. Methods Mol Biol 315: 349–360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.