

Abstract
Glial cells are critically important for maintenance of neuronal activity in the central nervous system (CNS), including the optic nerve (ON). However, the ON has several unique characteristics, such as an extremely high myelination level of retinal ganglion cell (RGC) axons throughout the length of the nerve (with virtually all fibers myelinated by 7 months of age in humans), lack of synapses and very narrow geometry. Moreover, the optic nerve head (ONH) - a region where the RGC axons exit the eye - represents an interesting area that is morphologically distinct in different species. In many cases of multiple sclerosis (demyelinating disease of the CNS) vision problems are the first manifestation of the disease, suggesting that RGCs and/or glia in the ON are more sensitive to pathological conditions than cells in other parts of the CNS. Here, we summarize current knowledge on glial organization and function in the ON, focusing on glial support of RGCs. We cover both well-established concepts on the important role of glial cells in ON health and new findings, including novel insights into mechanisms of remyelination, microglia/NG2 cell-cell interaction, astrocyte reactivity and the regulation of reactive astrogliosis by mitochondrial fragmentation in microglia.
Keywords: Optic nerve (ON), glia, oligodendrocytes, astrocytes, neuron glial 2 (NG2) cells, microglia
1. Introduction
The optic nerve (ON) is a specialized sensory nerve that is responsible for vision. It connects the neural retina to the brain: the axons of the ON are extensions of the retinal ganglion cells (RGC) that form the nerve fiber layer of the neural retina (Lee et al., 2020). These unmyelinated axons then make a sharp turn and exit the eye at the optic nerve head (ONH) region and continue as a series of myelinated bundles toward the brain. The fibrous sheaths that cover the ON are a direct continuation of all three meningeal layers of the brain (dura, arachnoid, and pia mater), so the ON is considered to be part of the brain rather than a true cranial nerve (Figure 1) (Rea, 2014). Consistent with being a tract of the brain from the anatomical point of view, its physiological reactions to various lesions and degenerative processes are somewhat similar to those observed in the white matter of the brain (Hogan and Zimmerman, 1962). Importantly, since the ON is directly connected to both the brain (at its chiasm end) and the retina (at its ON head end) (Figure 1A), it becomes vulnerable to diseases affecting both the ocular and central nervous systems (CNS) (Fischer and Leibinger, 2012; Yanoff and Sassani, 2015).
Figure 1: The structure of the optic nerve A.
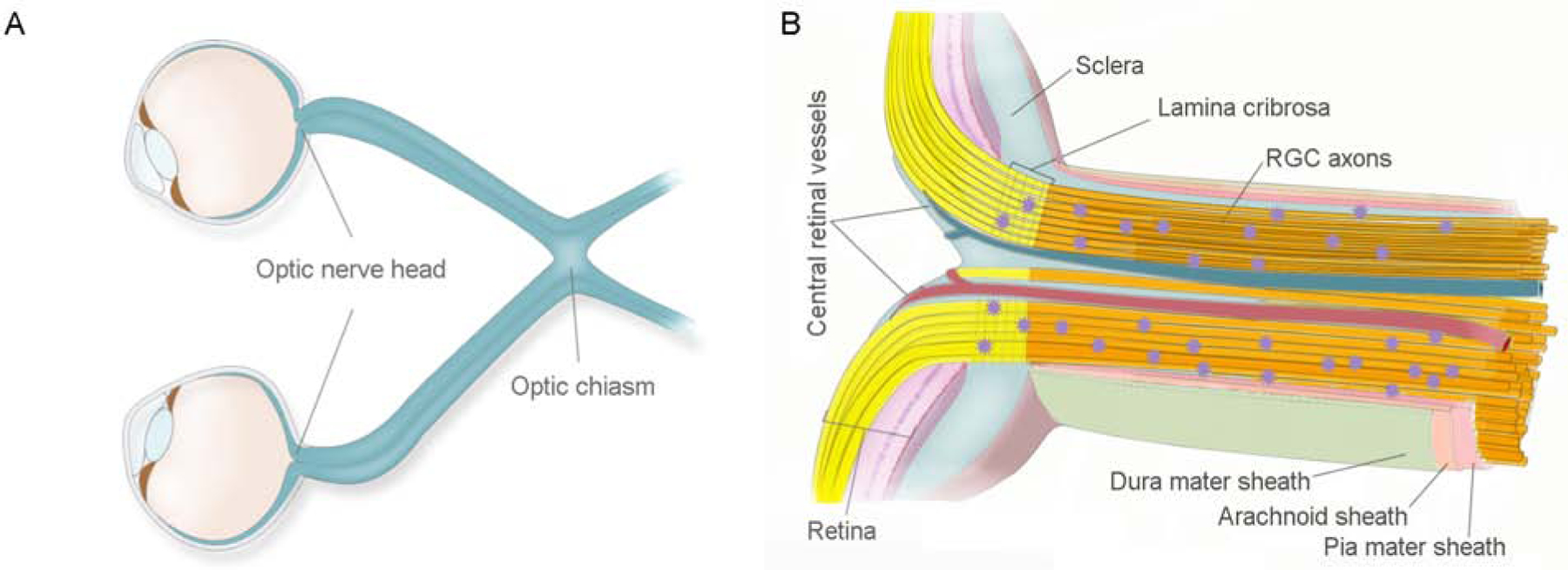
Optic nerve is a white matter tract of the brain that connects the eye (from the optic nerve head end) with the brain. The left and right optic nerves intersect at the chiasm, which is an X-shaped structure that is located in the forebrain, in front of the hypothalamus. B. The optic nerve is composed of myelinated axons of the retinal ganglion cells (RGCs) (yellow bundles) and interstitial glial cells (purple stars). Numerous septa of neuroglia, plus elastic and collagenous fibers divide the nerve into sharply defined bundles that are encapsulated by the three meningeal layers of the brain (dura, arachnoid, and pia mater). The main structural element of the optic nerve head is the dense collagenous lamina cribrosa that acts as a pressure barrier between the intraocular and retrobulbar spaces.
The ON is composed of myelinated nerve fibers, which are the axons of the RGC; interstitial glial cells (oligodendrocytes, neuron glial 2 (NG2) cells, astrocytes, and microglia); and the fibrovascular septa of the pia mater (Figure 1B). The estimated numbers of nerve fibers within an ON vary in different organisms (Bruesch and Arey, 1942). In humans, between 770,000 and 1.7 million fibers have been reported, with loss of fibers increasing as one ages, especially after the age of 60 (Jonas et al, 1992). Glial cells are critically important residents of the ON that work as a team to maintain its neuronal activity and structural stability (Figure 2). Oligodendrocytes form short interfascicular rows of adjacent cells along the length of the ON, which are separated from each other by solitary interfascicular astrocytes. This oligodendrocyte-astrocyte mesh is complemented with regularly spaced NG2 cells and microglia. Numerous septa of neuroglia, plus elastic and collagenous fibers divide the nerve into sharply defined bundles (Butt et al., 2004).
Figure 2: Schematic diagram of glial cells in the optic nerve.
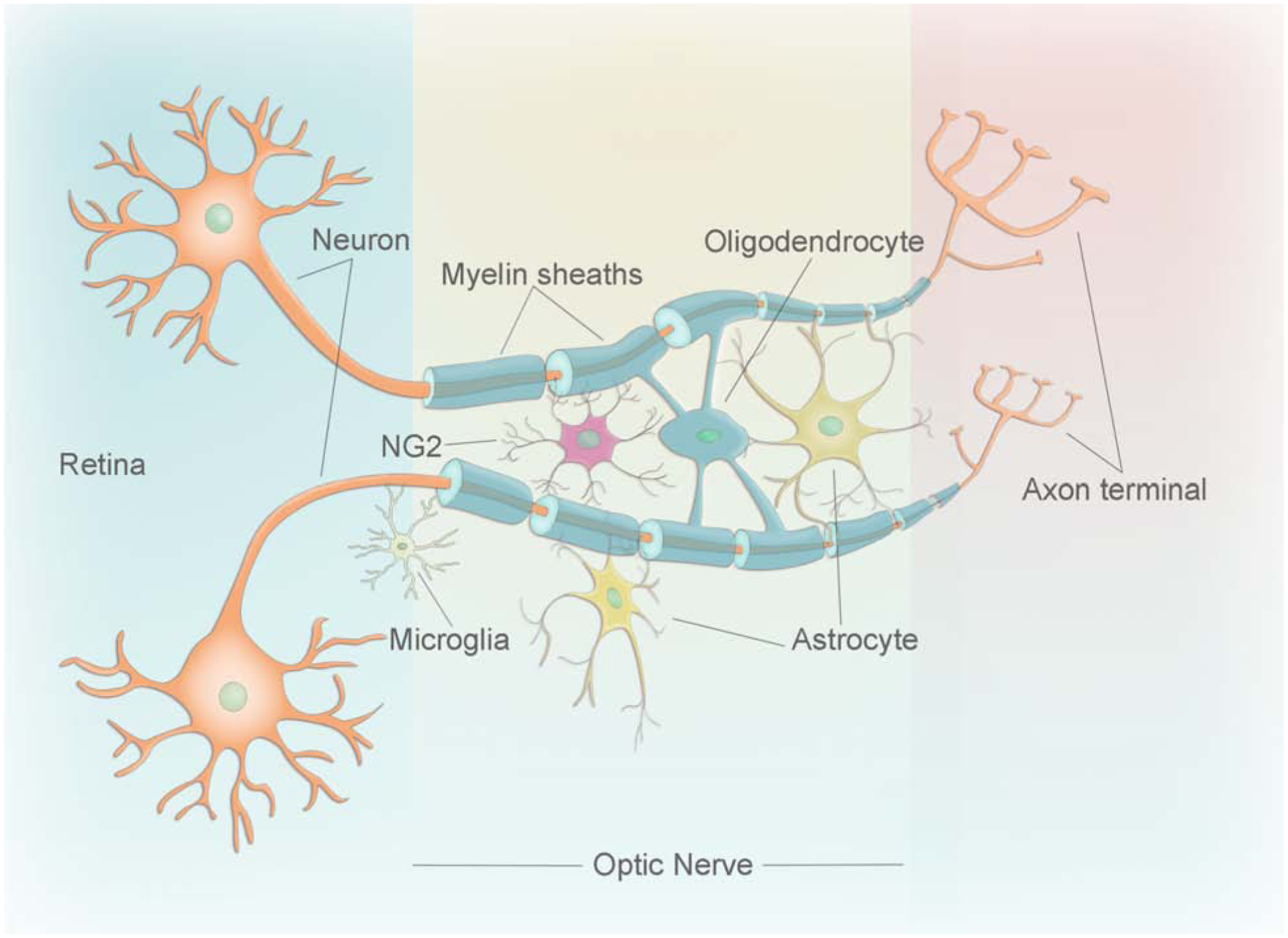
Cartoon showing the different types of glial cells (astrocytes, oligodendrocytes, NG2 cells, and microglia) in the optic nerve and their association with the neuron.
Glial cells play a wide range of physiological and pathological roles including response to any form of pathological insult, such as inflammation, injury, or ischemia (Vecino et al., 2016; Berry et al., 2002). Glial cell types and their roles are briefly summarized in Figure 3. Astrocytes were originally considered to be passive structural elements, but it is clear now that they are dynamic multifunctional cells that are important for potassium homeostasis, metabolism and axon-glial signaling, as well as the physiology of nodes of Ranvier (periodic gaps in the myelin sheaths that encapsulate the axon) and the blood-brain barrier (Khakh and Deneen, 2019). Recent studies have been focusing on deciphering the details of astrocyte reactivity - morphological and functional changes that occur in astrocytes in response to insults to the CNS (Liddelow et al., 2017; Liddelow and Barres, 2017; Li et al., 2019). Significant progress has been made, however, very little is known about fibrous white matter astrocytes, especially those in the ON that look completely different from cortical protoplasmic astrocytes (Sun and Jakobs, 2012; Sun et al., 2009).
Figure 3. Schematic representation of the simplified interactions between glial cells in the optic nerve.
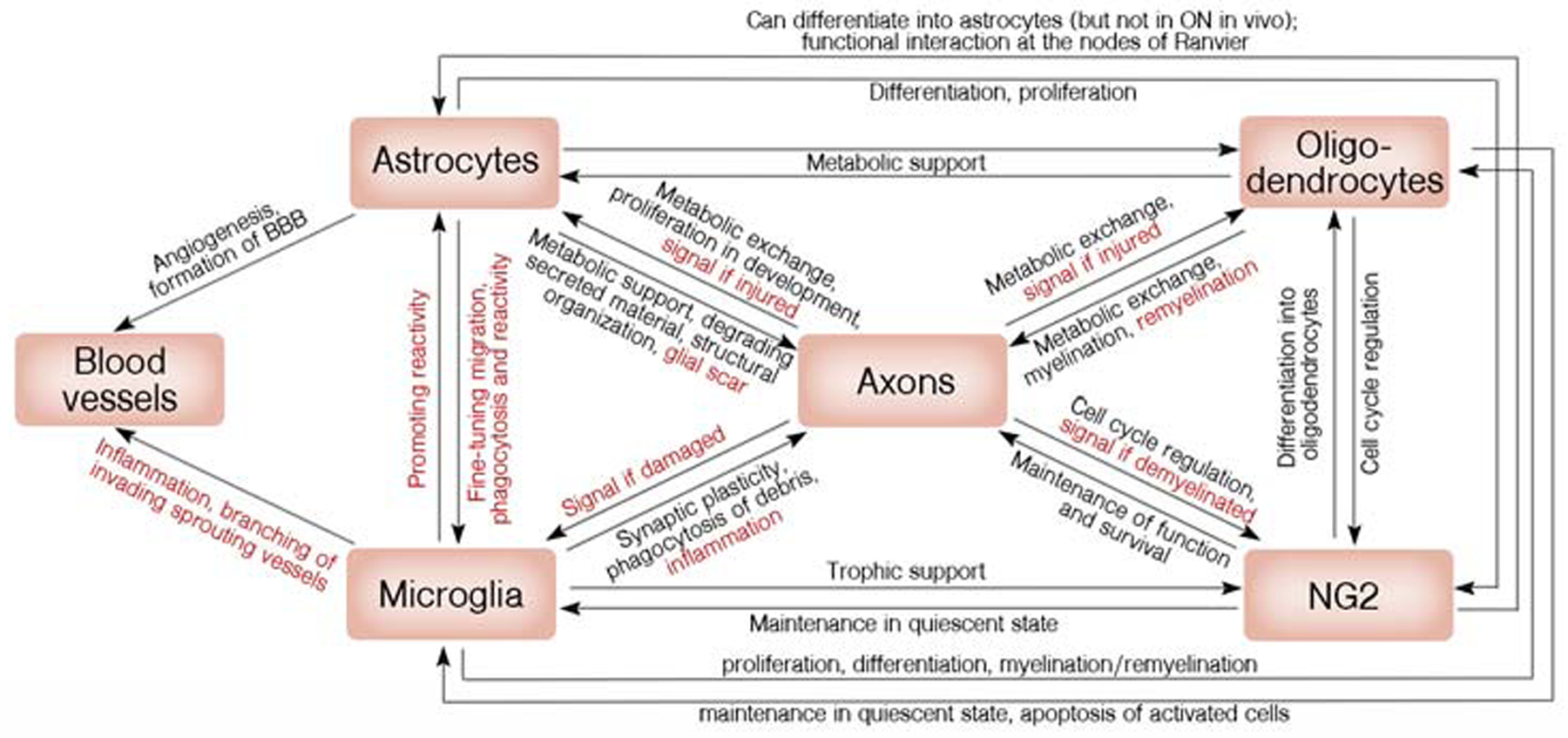
Glial cells work as a team to ensure both healthy homeostasis (black annotations) and response to insults (red) in the optic nerve.
Oligodendrocytes form the myelin sheaths around axons that facilitate rapid and metabolically efficient conduction of action potentials and are essential for axon integrity and nodal function. They are exceptionally sensitive to any insult to the CNS, which results in the loss of myelin followed by secondary axon degeneration (Butt et al., 2004). This sensitivity is especially striking in the ON, as in many cases of multiple sclerosis (MS, demyelinating disease of the central nervous system) vision problems are the first manifestation of the disease. In addition, they provide important metabolic support. Interestingly, recent studies demonstrate that oligodendrocyte-derived lactate is critical to fuel RGC axons in the ON, whereas the axons in the corpus callosum prefer glucose that is also derived from oligodendrocytes (Meyer et al., 2018; Brown et al., 2003; Fünfschilling et al., 2012).
Until recently, NG2 glia had been considered to be just adult oligodendrocyte progenitor cells, but it is now accepted that these cells are also involved in interactions with the nodes of Ranvier, axon-glial signaling, and glial scar formation following axon injury (Yang et al., 2013; Parolisi and Boda, 2018; Butt et al., 2003). Very recent studies also proposed a role for NG2 cells in inflammation through their interaction with microglia; however, this interaction has not yet been studied in the ON (Liu and Aguzzi, 2019; Zhang et al., 2019).
Finally, microglia monitor the integrity of the CNS via a range of ion channels and neurotransmitter receptors that enables rapid response to changes in neuronal functions (Liu et al., 2016). Activated microglia secrete cytokines and trophic factors, which can be either protective or destructive. Microglia play an important role in the pathogenesis of glaucoma and during acute injury, and recent studies have considered targeting microglia as a therapeutic approach to prevent ON degeneration (Wang et al., 2016; Hilla et al., 2017). Moreover, recent studies have demonstrated that mitochondrial fragmentation in microglia can regulate reactive astrogliosis (Joshi et al., 2019).
In addition to interaction with the axons, glial cells constantly interact with each other, forming a strong and highly efficient network supporting the functionality of the ON. Importantly, since the ON does not contain any synapses, but just axons that span the length of the nerve, it has become a popular model to study the interactions of glial cells with axons without having to consider their complex role in maintaining synapse functionality. However, this feature also demands caution when applying knowledge obtained while working with the rest of CNS to the ON. In this review, we summarize the current knowledge on glial organization and function in the ON under physiological and pathological conditions. We will focus on the regions of the ON that contain glial cells - the ONH and the retrobulbar part of the ON. Discussing both of these regions is important, since as discussed in detail below, the ONH is very different from the remaining portion of the nerve both in terms of cellular composition and myelination.
2. Evolution of optic nerve glia
It is likely that glial cells originated independently in two or more stem groups during evolution, supporting the importance of these cells for the survival of the species (reviewed extensively in Hartline, 2011). Interestingly, the development of the sophisticated glial network (in contrast to simple support cells) coincided with the increased complexity of the nervous system, and particularly with the emergence of complex intelligence (Freeman and Rowitch, 2013; Losada-Perez, 2018). The complexity of the glial network can be estimated by the increase of both the glia:neuron ratio (1:9 in invertebrates vs 1:1 in vertebrates) and the diversity of glial cell types, which can be fully appreciated when considering the structural organization of one particular white matter tract - the ON (Hartline, 2011; Losada-Perez, 2018).
Indeed, in Drosophila melanogaster, the total pool of macroglia is typically categorized into four major types (peripheral glia, cortex glia, surface glia and neuropil glia); however, only one type of glia (ensheathing peripheral glia) is found wrapping the Bolwig nerve (larval ON in Drosophila) (Losada-Perez, 2018). In vertebrates, evolution dictated the need for better conduction of the nerve impulse: neuronal axons span extraordinary lengths from the neuronal cell body to distant synapses. For example, axons that comprise the spinal tract of a blue whale, are estimated to be longer than 30 meters, while the longest human axons are estimated to be around 1 meter in length. Moreover, the development of the large brain has an expensive metabolic toll: while weighing only 2% of the average body, it consumes 20–25% of all oxygen since generating action potentials requires a tremendous amount of ATP. Lack of available energy could significantly limit the size of the brain as well as lead to susceptibility to damage during anoxia or ischemia (Attwell and Laughlin, 2001). One of the solutions to meet the essential requirement of a rapid and reliable transfer of action potentials from one part of the body to another with a significant conservation of energy was to develop a way to insulate the axons. As a result myelin, a specialized membranous sheath that consists of 70%–80% lipids and approximately 20% proteins, emerged during evolution (Miller, 2002; Dhaunchak et al., 2010; Jahn et al., 2009). The evolutionary requirement for myelination resulted in the emergence of two types of specialized glial cells - oligodendrocytes and their precursors (NG2 cells). There is an interesting hypothesis, that myelin first evolved to meet the energy demands of large vertebrates, and only later, after the end-Permian mass extinction, which killed off >95% of marine species and the majority of terrestrial species that populated the planet, acquired the function of facilitating rapid signal transduction (Stiefel et al., 2013). This hypothesis is based on the faunal changes that occurred after the extinction: animals became faster and predation became more widespread.
It is estimated that myelin significantly reduces the energy cost of action potentials: it insulates the axons to allow metabolically costly ion exchange only at gaps in the myelin shield that are called nodes of Ranvier, rather than throughout the entire length of the axons (Arancibia-Carcamo and Attwell, 2014; for comprehensive mathematical models see Namazi and Kulish, 2013; Crotty et al., 2006; Attwell, Laughlin, 2001; Pan et al., 2012). A unique feature of myelin is its extraordinary stability - myelin dissected from the Tyrolean Ice Man, who lived 5000 years ago, had preserved ultrastructure, as observed under the electron microscope (Hess et al., 1998; Simons and Nave, 2015). Myelination also adds an additional level of regulation of axonal function: different parts of the brain, and even different parts of the ON, have different levels of myelination. Thus, even though the length of human RGC axons is only about 5 cm, they are projected through a variety of regions in the skull and the brain, dictating differential needs in myelination and adding to the complexity of glial cell support. RGC axons are unmyelinated in the retina and almost completely myelinated throughout the length of the nerve (reaching 98.8% myelination in the mouse) (Young et al., 2013), Interestingly, in the ONH, the area where RGC axons exit the eye, the levels of myelination depend on the species (studied extensively by Fujita et al, 2000). For example, in humans, myelination of the RGC axons stops at the lamina cribrosa (the mesh-like structure formed by multiple layers of connective tissue that occupies the hole in the sclera through which the ON fibers exit the eye), while in rabbits and dogs it extends beyond the lamina cribrosa. A number of animals (such as rodents, turtles and some species of fish) do not develop a lamina cribrosa per se, but instead manifest a glial “pseudo-lamina” (Wolburg, 1981).
Evolutionary pressure in the CNS also dictated the need for better homeostatic maintenance of the synapses, leading to diversification of astrocytes. In fact, there is a growing body of evidence that astrocytes have been one of the most divergent cell types during evolution of the nervous system, resulting in much better control of synaptic development, function and plasticity in primates, and especially in humans (reviewed in Krencik et al, 2017, Verkhratsky and Nedergaard 2016). Moreover, while fruit flies could afford having a multifunctional single cell type that would carry out many immune functions, development of a sophisticated immune system in vertebrates required additional cell specialization. As a result, various types of immune cells have emerged, including microglia - macrophages that function specifically in the CNS, including the ON (Hartenstein and Giangrande, 2018).
As a result of these evolutionary requirements, the vertebrate ON contains four types of glial cells - astrocytes, oligodendrocytes, NG2 cells and microglia - that need to be arranged in a very specific, highly regulated pattern. As discussed below, the emergence of the four types of glial cells in vertebrates has provided a strong foundation on which to form a complex network of physical and functional interactions that maintain the functionality of the ON, and allow it to adequately respond to injury or disease.
3. Astrocytes in the optic nerve
Astrocytes represent a diverse population of glial cells in the CNS. Two main types of astrocytes have been described in the human ON. Type 1 astrocytes are characterized by the expression of Glial Fibrillary Acidic Protein (GFAP) and represent the majority of astrocytes in the unmyelinated part of the ON, while type 2 astrocytes are GFAP-negative and are abundant in the myelinated part of the ON (Kobayashi et al., 1997; Ye and Hernandez, 1995). Type 1 astrocytes can be further divided into two subtypes - type 1A (NCAM-negative) and 1B (NCAM-positive). Type 1B astrocytes are the major astrocyte population in the ONH, distributed throughout both the prelaminar and laminar regions (Schneider and Fuchshofer, 2016). In contrast to humans, in rodents the classification of astrocytes is mainly based on anatomical features instead of antigen expression. Rodent astrocytes can be divided into protoplasmic and fibrous subtypes. Protoplasmic astrocytes, mainly present in the gray matter are absent in the ON, where fibrous astrocytes are located (Oberheim et al., 2009). Fibrous astrocytes can be further divided into random, transverse and longitudinal types, depending on the orientation of their processes along the nerve. The unmyelinated region of the ONH is mostly populated with transverse astrocytes that are important for the structural support of the rodent ON (Sun et al., 2009).
Astrocytes are multifunctional cells. They are known to maintain extracellular homeostasis by degrading material that has been secreted by neurons (Lee et al., 2017). Astrocytes are also essential for functionality of the blood-brain and blood-retina barriers, and they provide nutrients to neurons by connecting them to the blood vessels (Bouzier-Sore and Pellerin, 2013; Ransom et al., 2003). In the case of neuronal injury, astrocytes migrate to the site of a lesion to form a glial scar. Previously it was thought that the formation of a glial scar blocks axonal regrowth, however a recent study demonstrated that astrocyte scar formation aids rather than prevents CNS axon regeneration (Anderson et al., 2016; Hernandez, 2000; Ridet et al., 1997). In addition, astrocytes are capable of restructuring the extracellular matrix and releasing growth factors, cytokines and other cellular mediators (Hernandez, 2000; Schwab et al., 2000) as well as maintaining the health of RGC mitochondria (Davis et al., 2014; Munemasa and Kitaoka, 2015). Below we discuss a few astrocyte functions that are especially important in the context of the ON.
3.1. Astrocytes in the development of the ON
Astrocytes play an important role in CNS development. In the developing ON, proliferation of astrocyte precursor cells and mature astrocytes is dependent on signals from RGCs, particularly via sonic hedgehog (Shh) (Dakubo et al., 2008). In addition to the proliferation and specification that happens early in development, astrocytes need to form a cribriform network with a fine lattice of processes that plays a structural role in organizing RGC axons into sharply defined bundles. In rats, this organization is achieved by postnatal day 6 (P6), coinciding with abundant expression of phosphorylated epidermal growth factor receptor (p-EGFR) (Liu and Neufeld, 2004). Strikingly, activation of EGFR in postnatal astrocytes in vitro stimulated organization of astrocytes into cribriform structures that looked very similar to those observed in the ON in vivo, indicating the critical role of this signaling pathway in the development of the ON. Moreover, inhibition of EGFR by a specific inhibitor resulted in disorganization of the RGC axons inside the ON. Thus, at P10, multiple vacuolated spaces were observed between the axonal bundles as well as in individual axons, indicating their possible degeneration. By P21 the vacuolated spaces disappeared, but the axons were not properly divided into bundles and the shape of axons looked irregular. Importantly, even though immunostaining revealed a strong GFAP signal indicating the presence of differentiated astrocytes, the fine network of astrocytic processes failed to be formed. These data indicate not only the importance of EGFR signaling, but also highlights the role of astrocytes in the development of the ON (Liu and Neufeld, 2004).
Astrocytes are also critical for angiogenesis in the CNS. It has been well established that astrocytes are capable of sensing oxygen levels in the tissues and secreting VEGF to promote vessel growth into hypoxic areas (Nakamura-Ishizu, 2012). The ON (especially at the ONH region) is well vascularized (reviewed in Mackenzie and Cioffi, 2008), and there is no reason to doubt that astrocytes were not involved in angiogenesis during the development of the ON. Although no studies have been reported on the role of astrocytes in angiogenesis of the ON in vivo, it has been shown that astrocytes isolated from healthy adult ONH are capable of stimulating angiogenesis in vitro (Rudzinski et al., 2008).
3.2. Astrocytes in inflammation
Inflammation is the major consequence of insults to the CNS, including acute trauma, infection, or chronic neurodegenerative diseases (see Sofroniew, 2015). Microglia (discussed later in the review) and astrocytes are the main regulators of inflammation in the CNS depending on the mode of injury (Zamanian et al., 2012; Anderson et al., 2016; Crotti and Ransohoff, 2016; Liddelow et al., 2017; Herz et al., 2017; Klein and Hunter, 2017). Resident microglia and infiltrating immune cells are known to be involved in promoting astrocyte reactivity, thus coordinated interactions between these cells are critical for managing and fine-tuning the response to inflammation (Liddelow et al., 2017). Astrocytes also use their processes (astrocytic endfeet) to closely surround endothelial cells and pericytes, thus mediating the formation of the neurovascular unit and acting as a bridge between neurons and blood vessels (Selvam et al., 2018). This assembly is crucial for the structure of the blood-brain barrier (BBB) that creates a unique “immune privileged” environment, which provides protection from the invasion of peripheral immune cells (Volterra and Meldolesi, 2005).
It is well established that during ischemia and traumatic injuries, the pathophysiological changes in the CNS and signs of neurodegeneration result from the loss of normal astrocyte function (Kajihara et al., 2001; Giffard and Swanson, 2005; Liu and Chopp, 2016). Specific types of injury can result in either acute or chronic changes in the reactive astrocyte transcriptome, leading to different morphology and function (reviewed in Liddelow and Barres, 2017). Upon stimulation, astrocytes can become reactive and release extracellular molecules, such as neurotrophic factors (for example BDNF, VEGF and bFGF) and inflammatory factors (such as IL-1β, TNF-α and NO). In broad terms, reactive astrocytes can be considered to have either a neuroprotective or neurotoxic effect (such as provoking inflammation or increasing damage). For example, neuroinflammation and ischemia led to the identification of two different types of reactive astrocytes, termed “A1” and “A2,” respectively. Such terminology was selected to match the “M1” and “M2” macrophage nomenclature: further, it has been shown that the phenotype of microglia in the CNS could range from pro-inflammatory “M1-like” to an immunosuppressive “M2-like” phenotype (Block et al., 2007; Boche et al., 2013; Liddelow and Barres, 2017). Similarly, A1 neuroinflammatory reactive astrocytes were shown to upregulate a number of genes known to be harmful to synapses, while A2 reactive astrocytes promoted neuronal survival and growth through upregulation of neurotrophic factors and facilitated synapse repair through upregulation of thrombospondins (Liddelow et al., 2017; Liddelow and Barres, 2017; Li et al., 2019). Interestingly, a new function emerged for the A1 reactive astrocytes: they were shown to secrete a yet-to-be-identified neurotoxin that induced apoptosis in neurons and oligodendrocytes without affecting other CNS cell types. Importantly, A1 reactive astrocytes were rapidly generated after CNS injury and were responsible for the death of damaged CNS neurons, while microglia were insufficient to induce death of neurons or oligodendrocytes by themselves (Liddelow and Barres, 2017). In contrast to A1 reactive astrocytes, A2 scar-forming reactive astrocytes promote survival of neurons and synapse repair.
Most data relating to A1 and A2 reactive astrocytes has been obtained using gray matter protoplasmic astrocytes; very little is known about fibrous white matter astrocytes, especially those in the ON that look completely different from cortical protoplasmic astrocytes (Sun and Jakobs, 2012; Sun et al., 2009). Moreover, the studies on A1 and A2 astrocytes have been performed in the context of synapse maintenance and repair - a context that is not applicable for the astrocytes in the ON. Therefore, caution needs to be taken in describing results from the brain and inferring that they apply to all other astrocytes. Further studies are needed to understand the effect of different populations of reactive astrocytes in the pathogenesis of different types of ON disorders. Moreover, it is important to note that the classification of astrocytes having a neuroprotective or neurotoxic effect might be simplistic and in different cases very complex feedback loops may exist (Boghdadi et al., 2020). It is likely that different types of astrocytes are not only produced in different ON degenerative disorders, but even at different stages of the same disease. More extensive studies are needed to address these questions.
In general, astrocyte reactivity is usually characterized by two features: cellular hypertrophy and overexpression of intermediate filament proteins (such as GFAP and vimentin) (Okada et al., 2006). Importantly, astrocyte reactivity should not be confused with proliferation since an increase in GFAP expression could be misinterpreted. For example, recently it has been clearly demonstrated that astrocyte proliferation is limited during progression of diseases which lead to neuroinflammation and neurodegeneration in the brain (Liddelow and Barres, 2017; Li et al., 2019). The higher density of GFAP+ cells in that case results from the overexpression of this protein and astrocyte hypertrophy, rather than from astrocyte proliferation (Serrano-Pozo et al., 2013). In contrast to the pathogenesis of neurodegenerative disorders in the brain, studies in a rat glaucoma model caused by early pressure-induced ONH injury revealed that both proliferation and IL-6-type cytokine expression in astrocytes were greatly increased. Dramatic astrocyte proliferation in ONH injured by elevated intraocular pressure (IOP) was shown to be regulated by activation of the Janus kinase (JAK) - signal transducer and activator of transcription (STAT) pathway (Lozano et al., 2019).
Some reactive astrocytes have been shown to exhibit NF-κB activation that contributes to RGC loss in various pathologies. Inactivation or inhibition of NF-κB in astrocytes promotes survival of retinal neurons after ischemic injury and reduces RGC loss in experimental optic neuritis (Brambilla et al., 2012). It has been demonstrated that the JAK-STAT3 pathway mediates the formation of reactive astrocytes with an overall protective function, akin to the A2 astrocytes described by Liddelow and Barres (Liddelow and Barres, 2017; Herrmann et al., 2008; Anderson et al., 2016; Ceyzeriat et al., 2016). STAT3 is expressed by all astrocytes in the ON, brain and spinal cord, and its expression markedly increases after injuries and during progression of neurodegenerative diseases, as well as in a rat model of transient IOP elevation (Justicia et al., 2000; Yamauchi et al., 2006; Zhang et al., 2013; Ben et al., 2015). Recent data suggest that knocking out STAT3 specifically in astrocytes results in attenuated astrocyte hypertrophy, reactive remodeling after the injury and increased loss of RGCs. Thus, STAT3-mediated astrocyte reactivity plays a protective role in experimental glaucoma and the ON crush model, supporting RGC survival (Sun et al., 2017). In addition, it has been postulated that ciliary neurotrophic factor (CNTF) activates astrocytes, thereby inducing fibroblast growth factor 2 (FGF2). Convincing evidence has been provided showing that during ON injury resulting from ocular inflammation, CNTF is produced by retinal astrocytes and binds to RGCs via receptor tyrosine kinases triggering the JAK-STAT pathway to switch RGCs to a regenerative stage (Fischer and Leibinger, 2012; Müller et al., 2007; Peterson et al., 2000) . Taken together, these findings suggest that drugs that inhibit NF-κB or promote STAT3 activation might hold therapeutic potential in neurodegenerative disorders.
3.3. Metabolic role of astrocytes
Neurons are the main energy-consuming cells in the brain, but glial cells provide the solution to the energy demand problem. Astrocytes play their role by facilitating the transfer of energy substrates from the blood to the neurons. To perform this task, astrocytes express different receptors such as glucose transporter 1 (GLUT1) (Allen and Messier, 2013), monocarboxylate transporter (MCT) (Pierre and Pellerin, 2005) and fatty acid translocase (FAT) (Husemann and Silverstein 2001; Morita et al., 2019).
Glucose is an indispensable fuel that is required for adenosine triphosphate (ATP) production, regulation of oxidative stress, synthesis of secreted neurotransmitters and neuromodulators as well as structural components of the cell (Dienel, 2019). Importantly, the network of glial cells and neurons provides metabolic and physical compartmentalization (extensively reviewed by Rich et al, 2019). It has been well-documented that not all cells in the brain take up glucose to completely metabolize it: the ratio between glucose consumption and oxygen uptake suggests an incomplete oxidation of glucose (Dienel, 2019). When the astrocyte-neuron lactate shuttle hypothesis emerged, the glycolytic capacity was attributed to astrocytes, with oxidative metabolism taking place in neurons (Dienel, 2017). Briefly, it has been demonstrated that glutamate from the synapse was taken up by astrocytes and converted to glutamine. Glutamate uptake also triggered glucose uptake by astrocytes, followed by its glycolytic conversion to lactate, which is then transported to neurons for oxidative metabolism (Dienel, 2017; Mason, 2017).
Although the interdependence between neurons and astrocytes has been widely studied in regard to glucose metabolism, the mechanisms by which astrocytes and neurons metabolize lipids to provide energy to the CNS and affect neurotransmission largely remain unknown. (Barber and Raben, 2019; Joyal et al., 2018). Lipids have been shown to be integral components of CNS structure and can affect cell function and neurotransmission (Puchkov and Haucke 2013). It has recently been shown that fatty acids, including essential fatty acids, can cross the BBB and be transported into neurons by fatty acid transporters (Tracey et al., 2018). This suggests fatty acids may play a role in neuronal function and neurotransmission. It is well documented that astrocytes are the energy storing cells of the CNS. Fatty acid oxidation primarily occurs in astrocytes, producing metabolites such as FADH2, acetyl CoA, NADH, and ketones that can be transported and utilized by neurons. Additionally, recent studies have shown that under stressful conditions, astrocytes play a crucial role in survival of neurons by ameliorating fatty acid toxicity (Morita et al., 2019; Mukherjee and Suresh 2019).
Under stressful conditions, levels of reactive oxygen species (ROS) are increased, leading to peroxidation of fatty acids in neurons. Peroxidized fatty acids are very toxic to neurons and must be removed. It has been shown that these toxic fatty acids bind to lipoproteins for release from the stressed neuron. Then, neighboring astrocytes come into action endocytosing lipoprotein-fatty acid particles and storing the fatty acids in lipid droplets. Collectively, these studies reveal the vital role of astrocytes as a neuronal defense mechanism against fatty acid toxicity. (Bailey et al., 2015; Ioannou et al., 2019; Liu et al., 2015). It has also been shown that astrocytes directly or indirectly transport several membrane lipids, including phosphatidic acid, to neurons (Perea et al., 2009; Zhu et al., 2016).
In the context of lipid metabolism in neurons and astrocytes, cholesterol is a molecule that has been well studied. Cholesterol is a key component of the synaptic membrane (Chen et al., 2013). In the ON, which does not have synapses, it is required for oligodendrocytes to make myelin. Cholesterol may be expelled from astrocytes by both lipid-free apolipoproteins and lipoproteins, but cholesterol efflux from neurons is tightly regulated by lipoproteins (Camargo et al., 2012; Chen et al., 2013). However, given that lipoproteins are specifically produced by astrocytes, cholesterol efflux from neurons is also regulated by astrocytes. Recently it has been found that the genes involved in cholesterol synthesis were downregulated in the ON astrocytes in experimental autoimmune encephalomyelitis (EAE) - a widely used animal model for MS (Itoh et al., 2018; Tassoni et al., 2019). The authors speculate that the reduction of cholesterol synthesis in ON astrocytes during the progression of EAE may result in a shortage of cholesterol available for transport to neurons and/or oligodendrocytes, which, in turn, may lead to limited reparative plasticity and remyelination (Itoh et al., 2018). Thus, lipid metabolism in astrocytes plays a central role in regulation of neuronal viability.
As RGC axons span from the retina into the brain, they experience a variety of stressors (light, varying IOP and poor oxygen supply) that differ depending on the particular location along the axon (Risner et al., 2018; Thonas et al., 2014). In addition, the functionality of the RGCs depends on the active transport of organelles and vesicles and the maintenance of membrane potential along the entire length of the axon, which, in turn, requires a steady supply of ATP from axonal mitochondria (Yu-Wai-Man et al., 2011; Hayakawa et al., 2016). Thus, proper distribution and function of mitochondria within RGCs is critical for visual transduction (Carelli et al., 2004; Maes et al., 2017; Yu et al., 2013). Maintaining a healthy complement of mitochondria depends on a balance between de novo biogenesis and the elimination of damaged mitochondria (Berridge et al., 2016; Ito and Di Polo, 2017). One of the mechanisms to remove damaged mitochondria is mitophagy (selective autophagy). This housekeeping process is essential for the survival, development and homeostasis of the cell and is usually maintained at a basal level (Boya et al., 2016; Yazdankhah et al., 2014; 2019). It has been demonstrated that defective mitophagy is associated with impairment of neuronal function and susceptibility to neurodegeneration (Rani and Mondal, 2019; Lou et al., 2019).
Interestingly, in the axons at the ONH, mitochondria are clustered near the sites of astrocyte membranes. Moreover, large numbers of mitochondria are packed into axonal avulsions and internalized by astrocytes after shedding, followed by lysosomal degradation inside the astrocytes. Interestingly, astrocytes processes engulfing axonal mitochondria were observed under normal, physiological conditions. This phenomenon of transcellular degradation, termed “transmitophagy” is not unique to the ON, but has also been found to be present elsewhere in the nervous system (Davis et al., 2014). There is still an enigma concerning whether the axonal mitochondria that have been transferred to astrocytes are degraded through autophagy or phagocytosis, but studying this process might shed light on the details of the quality control mechanisms involved in elimination of mitochondria from neurons (Davis and Marsh-Armstrong, 2014).
3.4. Role of astrocytes in the regulation of extracellular matrix: implications in glaucoma
Astrocytes are one of the major cell types involved in the production of extracellular matrix (ECM) in the ONH, and their activation is thought to be important in restructuring the lamina cribrosa region of the ONH (Schneider and Fuchshofer, 2016). This process may contribute to the progression of glaucoma - a progressive optic neuropathy, which is characterized by structural changes in the ONH (with increased IOP being one of the major risk factors) that result in RGC death (Burgoyne et al., 2005; Fenwick et al., 2019). The healthy lamina cribrosa contains connective tissue beams that consist of proteoglycans, laminin, fibronectin, collagen types I, -III, -IV, -V and -VI and elastin fibers. During the progression of glaucoma, the ECM composition of the lamina cribrosa is significantly altered (Downs and Girkin, 2017). It has been demonstrated that astrocytes residing in the ONH of patients with glaucoma express higher levels of laminin, fibulin and collagens type II, -IV and -XI and lower levels of proteoglycans (specifically aggrecan), compared to healthy controls (Lukas et al., 2008; Zhong et al., 2013).
In addition to synthesis, ECM deposition is also regulated by the process of degradation that is governed in turn by the expression of matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs) and serine proteinases (Hausman 2007). Importantly, astrocytes residing in the ONH have been demonstrated to express a number of MMPs as well as their inhibitors TIMP1 and TIMP2, suggesting a role in ECM remodeling (Agapova et al., 2001; 2003; Neumann et al., 2008). Disruption of the homeostatic balance of growth factors might in turn result in altered balance between ECM production and degradation in astrocytes. For example, TGF-β2 is commonly elevated in the ONH of patients with glaucoma, and it has been demonstrated that it can drastically affect ECM remodeling by astrocytes. Treatment of ONH astrocytes with TGF-β2 resulted in elevated expression of TIMPs and increased synthesis of elastin, collagens type I, -IV and -VI and fibronectin, leading to profound changes in the ECM composition and structure in the glaucomatous ONH (Fuchshofer et al., 2005; Neumann et al., 2008; Zode et al., 2011).
Normal-tension glaucoma (NTG) results in optic disc excavation and visual field defects that are not caused by eye pressure exceeding the normal range (Flammer et al., 2002; Trivli et al., 2019). The pathogenesis of NTG remains to be elucidated, but one study has demonstrated that the level of endothelin-1 (ET-1) was significantly increased in the peripheral blood circulation in patients with NTG relative to normal subjects (Sugiyama et al., 1995; Cellini et al., 1997). On the other hand, ET-1 is a mitogenic factor for ONH astrocytes and ET-1- mediated proliferation of ONH astrocytes plays an important role in promoting astrogliosis and glaucomatous optic neuropathy (Prasanna et al., 2002, 2011; He et al., 2007).
Thus, astrocytes are important for ECM remodeling in the ONH and this role is implicated in the progression of glaucoma since remodeling of the ECM can subsequently lead to axonal degeneration in RGCs (Munemasa and Kitaoka, 2012).
4. Oligodendrocytes in the optic nerve
As discussed above, oligodendrocytes evolved in vertebrates to provide energy efficiency and rapid conduction of nerve impulses within the CNS (Dezawa and Adachi-Usami, 2000; Miller, 2002). Generation of oligodendrocytes requires well-designed mechanisms of oligodendrocyte precursor cell (OPC) specification, proliferation, and migration, followed by their differentiation (van Tilborg et al., 2018). Oligodendrocyte differentiation is a multistep process that involves transition among several oligodendrocyte progenitor states. It is now known that as OPCs slowly change from bipolar to multipolar cells, they adjust gene expression to express stage dependent antigens (Ono et al., 2001, Hirahara et al., 2017; Baracskay et al., 2007). Most researchers divide oligodendrocyte development into four maturation stages (Barateiro and Fernandes, 2014): OPCs, pre-oligodendrocytes (or late OPCs), immature (or pre-myelinating) oligodendrocytes and mature (or myelinating) oligodendrocytes. See Figure 4 for markers that delineate each developmental stage.
Figure 4: Four different phases of oligodendrocyte (OL) maturation in the oligodendroglial lineage: OPC, pre-OLs, immature OLs and mature OLs.
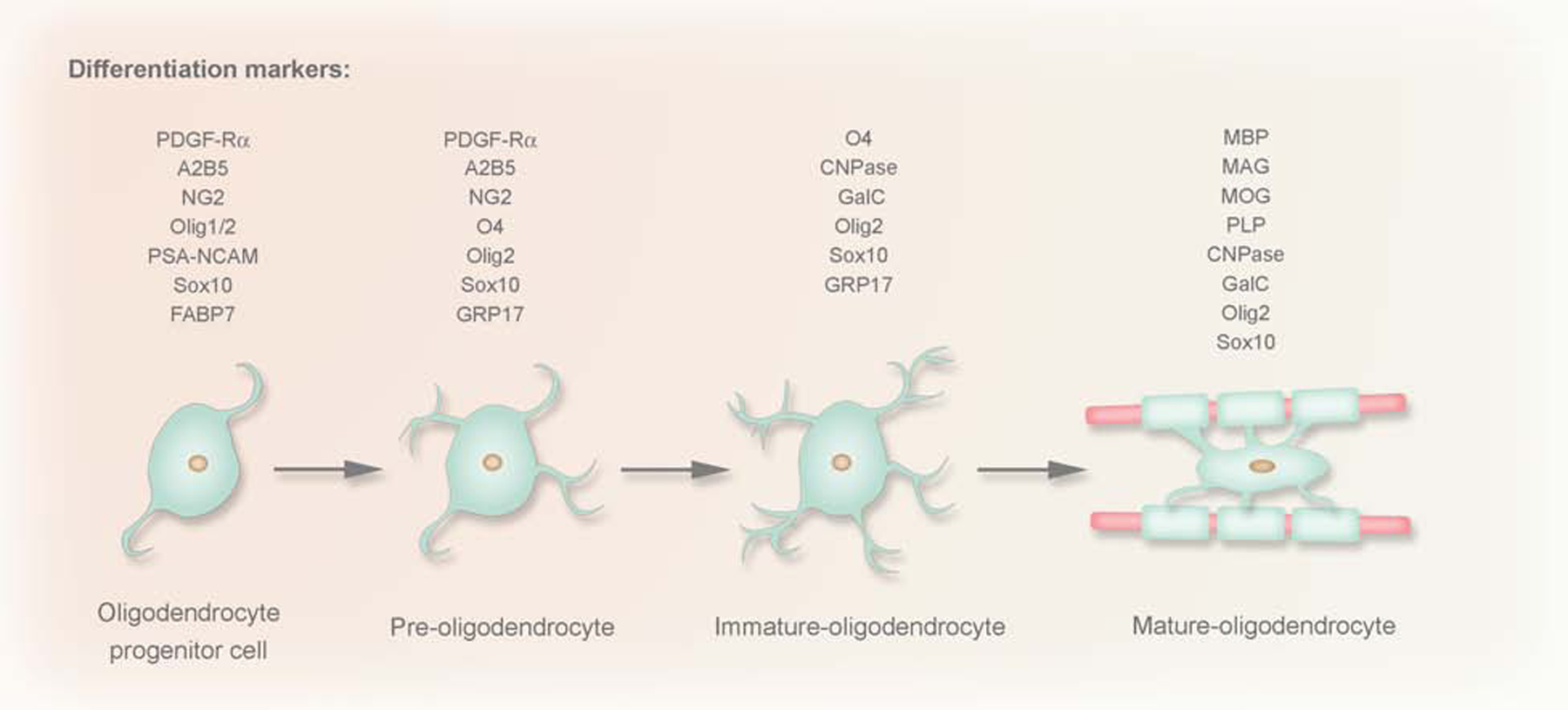
These phases were established based on the increasingly complex morphology and the expression pattern of well-defined markers. CNPase, 2′,3′-cyclic nucleotide 3′-phosphodiesterase; FABP, fatty-acid-binding proteins; GalC, galactocerebroside C; MAG, myelin associated glycoprotein; MBP, myelin basic protein; MOG, myelin OL glycoprotein;Olig1/2 two basic helix-loop-helix transcription factors; PDGF-Rα, platelet-derived growth factor; PLP, proteolipid protein; PSA-NCAM, polysialic acid-neural cell adhesion molecule.
OPCs do not originate in the ON. Recently, lineage tracing of Olig2-positive cells revealed that OPCs appear by E12.5 in the preoptic area of the brain and migrate to the ON through the chiasmal region (Ono et al., 2017). In rats, the chiasmal-to-retinal gradient is retained until at least P4, with the overall number of OPCs rapidly increasing (Ueda et al., 1999; Kajigaya et al., 2011). In the adult rat ON, the distribution of OPCs becomes uniform (with the exception of the ONH, where OPCs are not present, coinciding with the lack of myelination) and the population of NG2-positive OPCs reaches 7% of all glial cells (Butt et al., 2004; Serwanski et al., 2017).
4.1. NG2 cells
OPCs and pre-oligodendrocytes are proliferative cells, with a great migratory capacity. Since, these cells express a great amount of chondroitin sulfate proteoglycan (CSPG4), also called NG2, these cells are referred to as NG2 glia or NG2 cells (Barateiro and Fernandes, 2014). NG2 cells represent 5–10% of all glia in the developing and adult CNS and are uniformly distributed in both grey and white matter, including the ON (Ueda et al., 1999; Viganò and Dimou, 2016). Morphologically, NG2 cells have long thin processes; however the number and length of these processes vary depending on the location of the cells and age of the animal (Nishiyama et al., 2009). Originally NG2 cells were thought to be just progenitors of mature glial cells (extensively reviewed in Nishiyama et al., 2016; Valny et al., 2017). They are the main, if not the only, source of oligodendrocytes in the brain (Dimou et al., 2008; Kang et al., 2010) and can also give rise to astrocytes (Zhu et al., 2008; Zhao et al., 2009; Huang et al., 2018). The potential of NG2 cells to differentiate into different types of glial cells strictly depends on local needs and the presence of particular growth factors in the environment. For example, NG2 cells served as exclusive oligodendrocyte progenitors when transplanted into the healthy neonatal rat brain, whereas transplantation into a glia-depleted environment (where white matter of the spinal cord in the adult rat was depleted by X-irradiation and ethidium bromide injection) resulted in generation of both oligodendrocytes and astrocytes (Espinosa de los Monteros et al., 1993; Franklin et al., 1995). Transplanted cells preferentially migrate along white matter tracts, so the lack of differentiation of NG2 cells into astrocytes under normal physiological conditions indicates that the local environment of the white matter does not support differentiation of NG2 cells into astrocytes (Zhu et al., 2008). This hypothesis is further supported by the observation that NG2 cells can serve as bi-potential progenitors for both oligodendrocytes and a subpopulation of protoplasmic astrocytes in the gray matter, but not for fibrous astrocytes in the white matter (Zhu et al., 2008). Consistent with these observations, NG2 cells do not generate astrocytes in the healthy ON, an exclusively white matter tract, in vivo (Nishiyama et al., 2016). However, in vitro NG2 glia that were isolated from the ON could differentiate into both oligodendrocytes and astrocytes, with the preference for astrocytes in medium containing 10% fetal calf serum, and oligodendrocytes in hormone-supplemented, serum-free medium (Stallcup and Beasley, 1987). The difference in the in vivo and in vitro differentiation capabilities of the ON NG2 glia supports the hypothesis that NG2 cell differentiation is dictated by the local environment rather than the origin of the cells. Interestingly, the location of NG2 glia seems to affect not only their differentiation potential, but also their cell cycle progression: the rate of NG2 glial cell division in vivo is generally slower in gray matter than in white matter and two times slower in ON compared to the corpus callosum, the white matter of forebrain (Young et al., 2013).
No strict correlation between the density, distribution and proliferation of NG2 glia and the presence and density of myelinating fibers was found in adult human or rodent CNS (Parolisi and Boda, 2018). The lack of direct correlation might indicate that the role of NG2 glia is not limited to producing oligodendrocytes for myelination. Intriguingly, NG2 glia are known to establish physical and functional interactions with neurons (extensively reviewed in Yang et al., 2013; Parolisi and Boda, 2018). The abundance of physical contacts varies, depending on location: in the rat ON, 48% of nodes of Ranvier are contacted by NG2 glia, which is comparable to the percentage of contacts found in the spinal cord, whereas in the corpus callosum, the percentage of these contacts drops to 33% (Serwanski et al., 2017). The vast majority of the nodes that are contacted by NG2 glia are also contacted by astrocytes (Serwanski et al., 2017). Functionally, the interactions between NG2 glia and the nodes of Ranvier can be considered as bi-directional: neurons modulate proliferation and differentiation of NG2 glia, whereas NG2 glia, in turn, help maintain neuronal function and survival (reviewed in Yang et al., 2013; Parolisi and Boda, 2018).
NG2 glia have been demonstrated to respond to neuronal activity through expression of a variety of ion channels and neurotransmitter receptors. The expression of these “neuronal” proteins is not uniform and differs even within the population of NG2 cells residing in the same region of the CNS (Parolisi and Boda, 2018). In the ON, signals are relayed to NG2 glia through at least two mechanisms involving glutamate that is released by RGC axons at the nodes of Ranvier and/or ATP that is released by astrocytes (Hamilton et al., 2009; Hamilton et al., 2010). Glutamate acts through α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, whereas ATP in the ON is predominantly sensed through P2Y G-protein coupled receptors, resulting in stimulation of Ca2+ signaling in NG2 glia. Moreover, NG2 glia in the ON have been demonstrated to express synaptophysin, a component of the neuroendocrine secretory granule membrane, indicating that they might be capable of signaling back to the neurons and astrocytes (Hamilton et al., 2010). Such a complex system for communication among axons, NG2 glia and astrocytes is supported by the following: most nodes of Ranvier in the ON are contacted by both NG2 cells and astrocytes (Serwanski et al., 2017); the processes of NG2 glia and astrocytes often enwrap each other (Hamilton et al., 2010); the capability of NG2 glia to receive glutamate/ATP signals; and the expression of synaptophysin. This network is further strengthened by the functional interaction between oligodendrocytes and astrocytes (as discussed earlier in Section 4.3). Recently it has been demonstrated that NG2 cells also play an important role in inflammation, particularly in maintaining microglia homeostasis (discussed later in the review) (Liu and Aguzzi, 2020; Zhang et al., 2019), adding to the complexity of glial cell interactions.
4.2. Role of oligodendrocytes in myelination
It has been shown that both oligodendrogenesis and myelination continue throughout life. Nearly half of oligodendrocytes are generated after 4 months of age in rodents (Hughes et al., 2018). Dynamic regulation of myelination throughout the lifespan of the animal is linked to cognitive processes including social interaction, learning, and memory (Fields, 2008; Fields, 2015). Interestingly, adult-born oligodendrocytes generate more abundant and shorter internodes compared to oligodendrocytes formed during early development (Young et al., 2013). There is also a significant difference in the levels of axon myelination depending on the location. In the rodent ON, about 85% of axons are myelinated by P28, and by 6–8 months of age myelination reaches 98.8%, while in contrast, in the corpus callosum, >70% of the axons remain unmyelinated even in adult mice, but may become targets for myelination later (Young et al., 2013).
It is well-known that the velocity of action potential conduction is greatly dependent on axonal myelination thickness and the length of uninterrupted myelin internodes (Devaux et al., 2002; Chang et al, 2014). Given that myelination has such a critical effect on neuronal function, it needs to be tightly regulated both spatially and temporally (Mayoral et al., 2018; Almeida, 2018). A growing body of evidence indicates that dynamic neuronal signaling may affect myelination by regulating OPC proliferation and differentiation (Gibson et al., 2014; Hill et al., 2014; McKenzie et al., 2014), selection of axon targets (Hines et al., 2015), the number of myelinated internodes, and the thickness of myelin sheaths (Liu et al., 2012; Makinodan et al., 2012; Gibson et al., 2014; Mensch et al., 2015). Importantly, myelination does not necessarily start at the place where the first OPCs appear, suggesting the existence of additional fine-tuning signals (Williamson and Lyons, 2018). Moreover, this observation is consistent with the evidence that, in addition to being progenitor cells, OPCs can also play a role in maintenance of axonal function (discussed later in this review).
Upon reaching their final destination, OPCs differentiate into myelin-forming oligodendrocytes. To do that, they stop proliferating and start extending numerous radial processes, initially without wrapping nearby axons (Zuchero and Barres, 2013). Platelet-derived growth factor (PDGF) is the most important mitogen regulating the proliferation and differentiation of OPCs both in vivo and in vitro: overexpression of PDGF results in an increased proliferation of OPCs and excessive ectopic production of oligodendrocytes (Calver et al., 1998). To ensure that the number of pre-oligodendrocytes matches the number of axons, an excess of cells is generated during development, and a significant portion of these extra cells is later eliminated by apoptosis (Caprariello et al., 2015). Approximately 50% of pre-oligodendrocytes and newly formed oligodendrocytes in the ON undergo apoptosis within 2–3 days of being generated (Michalski and Kothary, 2015). In parallel with differentiation of OPCs into pre-oligodendrocytes, the number of PDGF receptors is promptly reduced, leading to competition among cells for reaching non-myelinated axons (Klingseisen and Lyons, 2018). Such competition for the limited amount of target-derived survival factors, such as PDGF-A, may play an important role in determining the final number of mature oligodendrocytes (Miller, 2002).
It has been shown that in the ON, changes in the activity of RGCs caused by manipulating the visual stimuli through monocular deprivation in mouse models, lead to altered action potential conduction and axonal myelination (Etxeberria et al., 2016). Moreover, elimination of dynamic neuronal signaling along the developing ON promoted oligodendrocyte differentiation, but disrupted myelination (Mayoral et al., 2018). In general, myelination is thought to be dependent on the biophysical properties of the target surface (mainly axon diameter) that are recognized by the oligodendrocyte to determine whether or not the axon should be myelinated (Lee et al., 2012; Bechler et al., 2015). However, it is still uncertain how oligodendrocytes recognize axonal diameter. During development, all axons in the ON with the minimum axon diameter size of 200–300 nm are myelinated (Simons and Nave, 2015), however, most parts of the brain remain unmyelinated despite having many axons of this size (Goebbels et al., 2017). Therefore, it seems that axons in these areas may express some type of inhibitory signal to prevent myelination (Almeida, 2018).
Negative or repulsive signals include some of the ephrin molecules expressed in oligodendrocyte lineage cells and axons. For instance, binding of axonal ephrin-A1/B2 to EphA/B receptors on oligodendrocyte lineage cells leads to a reduction in myelination both in vitro and in vivo (Linneberg et al., 2015; Harboe et al., 2018). Expression of ephrin-A1 in neurons reduces the level of myelination in the zebrafish spinal cord by interacting with the EphA4 receptor on oligodendrocyte lineage cells, while downregulation of ephrin-A1 expression rescues this phenotype (Harboe et al., 2018). Polysialylated neural cell-adhesion molecule (PSA-NCAM) is another negative regulator that reduces the number of myelinated axons in vivo. Consistent with this role, inhibiting the function of PSA-NCAM promotes myelination (Charles et al., 2000; Fewou et al., 2007). Notch1 signaling is known to negatively regulate OPC differentiation and myelination: Notch1 receptor haploinsufficiency resulted in more myelinated axons in the ON and in the molecular layer of the cerebellum (which normally remains non-myelinated), suggesting the importance of Notch signaling in spatiotemporal regulation of myelination (Givogri et al., 2002). RGCs express Jagged1, which is the transmembrane ligand for the Notch1 receptor expressed in oligodendrocytes (Wang et al., 1998). On the other hand, several axonal molecules are known to drive CNS myelination. For example, pan-neuronal overexpression of Neuregulin 1 type III increases the number of ON and cortex axons targeted for myelination, including small-diameter axons that usually remain unmyelinated (Brinkmann et al., 2008). Loss of Nectin-like 1, a neuronal member of the synaptic cell adhesion molecule family, transiently reduces the number of axons targeted for myelination in the spinal cord and ON (Park et al., 2008).
Interestingly, recent studies point to the involvement of lysosomal proteins in spatiotemporal regulation of myelination (Sun et al., 2018; Meireles et al., 2018). For example, it has been shown that transcription factor EB (TFEB), a master regulator for lysosome biogenesis and autophagy, is highly expressed in pre-oligodendrocytes and that the TFEB-PUMA-Bax-Bak pathway controls the timing and location of myelination (Sardiello et al., 2009; Settembre et al., 2011). The genetic deletion of TFEB in oligodendrocyte lineage cells led to retention of ectopic pre-oligodendrocytes during development (Sun et al., 2018). Consistently, another recent study also indicates that a lysosomal G protein (RagA), a repressor of TFEB activity, may be involved in regulation of myelination (Efeyan et al., 2013; Efeyan et al., 2014; Efeyan et al., 2015, Meireles et al., 2018). In rraga−/− oligodendrocytes, myelination was greatly downregulated; however, loss of TFEB function was sufficient to rescue myelination in RagA mutants. Taken together, the complex spatiotemporal regulation of myelination involves both extracellular (via cell surface receptors) and intracellular (via lysosomal proteins) signaling pathways.
4.3. Mechanisms of re-myelination
Destruction of myelin sheaths is a nonspecific process that may occur clinically as a result of nutritional deficiencies, viral infections, immunopathologic reactions, toxins, or genetically determined enzyme deficiencies and can be produced experimentally by a variety of techniques. Use of the term “demyelinating disease” is usually restricted to inflammatory disorders in which the demyelinating process occurs early, often out of proportion to the damage observed in neurons and supporting structures. If myelin loss happens on an otherwise intact axon, this process is called primary demyelination, whereas the loss of myelin together with the axon is called Wallerian degeneration (Franklin and Goldman 2015). Restoration of myelin sheaths is critical for the functionality of the CNS and is called remyelination.
The ON is damaged in a number of demyelinating diseases, with optic neuritis being the most prevalent one (Levin et al., 2013; Watanabe and Fukuda 2002). It is an inflammation of the ON that can be caused by various factors, including infections and immune-related illnesses, such as MS, as well as by some viral diseases such as mumps, measles, and influenza. Importantly, the severity of optic neuritis can serve as an important indicator of disease progression and treatment outcome for patients with MS, since remyelination of the ON can be easily documented based on the patient’s self-reported changes in vision, as well as by clinical and electrophysiological assessment (Cohen and Tesar 2017). Demyelination is the characteristic change that appears early in the disease, followed by glial replacement in the later stages (Lampron et al., 2015). Myelin debris is phagocytized by microglia and demyelinating lesions are filled by proliferating OPCs and astrocytes. Even when a remarkable return of vision occurs, considerable gliosis of the nerve is still evident (Bosco et al., 2016; Pajoohesh-Ganji and Miller, 2019).
Typically, if demyelination is caused by injury in an otherwise healthy CNS (including the ON), the remyelination is complete, but the thickness of restored myelin sheaths and the length of the internodes are decreased compared to those that were originally formed during development. Importantly, such thin myelin sheaths do not seem to have a detrimental effect on the functionality of axons which remain stable and may persist indefinitely even in animals with a long lifespan (Duncan et al., 2017), however this may significantly increase the energy consumption (Zambonin et al., 2011). Despite the overall high efficiency of remyelination, some conditions, such as MS or EAE, create an environment that is intrinsically hostile to the oligodendrocytes, making remyelination challenging due to an adaptive immune response (Franklin and Goldman, 2015).
It is widely accepted that remyelination is a multi-step process requiring the generation of new oligodendrocytes from NG2 cells (reviewed in Franklin and Goldman, 2015); however, a recent study has demonstrated that previously existing oligodendrocytes can also effect remyelination (Duncan et al., 2017). In the latter scenario, the surviving mature oligodendrocytes re-extend their processes to contact axons that have lost myelin sheaths, in addition to maintaining mature myelin internodes that they have formed previously (Duncan et al., 2017). The conventional remyelination pathway from NG2 cells is more complex. First, a demyelinating injury activates microglia and astrocytes to release factors that act on NG2 cells to promote their switch from quiescence to the regenerative state. These events hallmark the beginning of the first stage of remyelination - the recruitment phase (Franklin and Goldman, 2015). Importantly, this stage is a limiting stage in remyelination in the ON due to the geometrical constraints of the ON compared to other parts of the brain (Klistorner et al., 2010). Indeed, the typical lesions in the brain have oval or elliptical shapes, fully surrounded by normal white matter, making them accessible to NG2 cells migrating from any direction. In contrast, in the ON, the lesions often occupy the whole cross-section of the nerve, thus limiting the area accessible to migrating NG2 cells to the proximal and distal ends of the lesion (Klistorner et al., 2010). In addition, ON-specific constraints in NG2 access to the lesion site can lead to an incomplete remyelination of the lesion, as is sometimes observed during optic neuritis (Klistorner et al., 2010).
The second stage of remyelination is the differentiation phase, when the recruited NG2 cells undergo differentiation into mature oligodendrocytes. This phase, in turn, can be subdivided into three intermediate steps: 1) establishing physical contact with the axon that needs to be remyelinated, 2) expression of myelin genes, and 3) wrapping and compaction to form the new myelin sheath (Franklin and Goldman, 2015). Interestingly, the differentiation phase of remyelination can be negatively affected by the shape constraints of the ON. It has been demonstrated that there is a specific “window of time” for successful completion. If the lesion in the ON is especially long, the local environment of the lesion may no longer be supportive to remyelination by the time NG2 cells finally reach that area, resulting in incomplete remyelination (Klistorner et al., 2010). The differentiation stage of remyelination is an attractive target for the development of novel therapies to treat MS and optic neuritis. For example, a recent clinical trial has demonstrated some level of healing after treating optic neuritis with clemastine fumarate, a drug that is known to stimulate the differentiation of NG2 cells in vitro (Cohen and Tesar, 2017).
One more factor contributing to successful remyelination is the involvement of the immune response (Franklin and Goldman, 2015). Initial tissue damage (and thus demyelination) is mediated by an adaptive immune response that is hostile to the myelin repair. However, this initial tissue damage is known to result in acute inflammation that is caused by the innate immune response and is manifested by the production of proinflammatory cytokines and other factors that actually attract oligodendrocyte progenitors and microglia, thus facilitating remyelination. Taken together, the success of remyelination depends on a balance between the actions of innate and adaptive immunity at the lesion site. This consideration might also dictate the “deadline” that exists for the completion of remyelination. Thus, in MS patients, the timeframe for myelin repair is usually considered to be 6 months, coinciding with the timeline for acute inflammation in the ON (Klistorner et al., 2010).
4.4. Metabolic supportive function of oligodendrocytes
Over the past decade, an accrued body of evidence based on achievements in genetic mouse models and human pathologies implies a critical role for oligodendroglia in the long-term survival of neurons (Morrison et al., 2013; Nave, 2010; Simons and Nave, 2015). It is now evident that oligodendrocytes are actively involved in metabolic support of neurons, a function known to depend on the expression and activity of monocarboxylate transporters (MCTs) (Fünfschilling et al., 2012; Lee et al., 2012; Nave and Trapp, 2008). MCTs are a family of proton-linked plasma membrane channels that transport molecules with one carboxylate group, such as lactate, pyruvate, and ketone bodies (Pierre and Pellerin, 2005). MCTs are differentially expressed by different cell types in the CNS. MCT1 is expressed predominantly in oligodendroglia and is localized in the myelin sheath surrounding axons, while MCT2 is expressed mainly in neurons. MCT3 expression is restricted to retinal pigmented epithelial (RPE) cells (Fields et al., 2019), whereas MCT4 is expressed in astrocytes (Bergersen et al., 2002; Rafiki et al., 2003; Pierre and Pellerin, 2005). Oligodendrocyte-specific inhibition of MCT1 in organotypic spinal cord slice cultures results in axonal injury, which can be treated by adding l-lactate supplements to the culture medium (Lee et al., 2012). Moreover, oligodendrocyte-specific MCT1 depletion in vivo causes severe axonal injury and motor neuron death (Lee et al., 2012).
Studying cellular distribution and physiologic properties of MCTs resulted in the development of the lactate shuttle hypothesis that explains how energy is transferred from astrocytes into neurons (Pellerin et al., 1998). This hypothesis states that cells with high levels of glycolytic flux generate large amount of pyruvate that is converted to lactate and is transported into the neurons (dependent on oxidative metabolism) to produce ATP (Magistretti and Pellerin, 2000). It has been demonstrated that neurons preferentially metabolize lactate over glucose (Wyss et al., 2011; Boumezbeur et al., 2010; Glenn et al., 2015). Moreover, studying ATP generation in neurons from acutely isolated ONs revealed that even in the presence of glucose, the electrical activity of the neurons was highly dependent on lactate metabolism (Trevisiol et al., 2017). Recent studies suggest that oligodendrocytes also provide neuronal metabolic support through MCTs that is further regulated by neuronal activity through N-methyl-D-aspartate receptors (NMDARs) and glutamate receptors, which are expressed on the noncompacted cell surface of oligodendrocytes. An ex vivo study on ONs isolated from NMDA-mutant mice suggests that glutamate enhances Ca2+ influx in oligodendrocytes resulting in elevated glucose uptake through GLUT1 and an increase in the level of lactate through glycolysis (Saab et al., 2016). In addition, oligodendrocytes are connected with each other and with astrocytes through gap junctions, which may provide oligodendrocytes with essential nutrients. It has also been shown that oligodendrocyte-astrocyte connections are required for normal myelination (Rouach et al., 2008; Niu et al., 2016; Giaume et al., 2013; Hirrlinger and Nave, 2014).
4.5. Oligodendrocyte-astrocyte interactions
Perivascular end-feet of astrocytes are intimately connected with endothelial cells and are the main location of glucose uptake. Aside from the astrocyte-neuron lactate shuttle hypothesis (discussed in 4.4), which explains how glucose-derived metabolites are provided to the neurons, glucose and astrocyte-derived lactate can be transferred into oligodendrocytes through gap junctions (Nagy et al., 2004). Impairment of gap junctions that connect astrocytes and oligodendrocytes, mainly affects the early phase of myelination because of the increased lipid synthesis and metabolic demand during this phase (Rinholm et al., 2011). Later, when myelination is complete, oligodendrocyte-derived lactate supports the neuronal metabolic needs in response to NMDAR signaling (Fünfschilling et al., 2012; Saab et al., 2016). The major findings regarding mechanisms of energy supply to axons in white matter have been derived from studies on ON, where lactate is crucial to fuel axons (Brown et al., 2003; Fünfschilling et al., 2012). However, it is worth mentioning that oligodendrocytes seem to use different mechanisms for delivering energy supplies into axons in different parts of the CNS. For example, it has been recently shown that in contrast to ON, axons in the corpus callosum, do not prefer lactate, and glucose is supplied from oligodendrocytes to axons via gap junction-coupled glial networks (Meyer et al., 2018).
It is well known that astrocytes play an important role in the development of oligodendrocytes. Astrocytes produce and secrete growth factors such as PDGF (Kernt et al., 2010), FGF2 (Messersmith et al., 2000), and CNTF (Modi et al., 2013) that affect the proliferation and differentiation of OPCs. Astrocytes also respond to various forms of CNS insult, such as ischemia, trauma, inflammation and neurodegeneration, by changing their morphology, gene expression, and physiology to become reactive (Sofroniew and Vinters, 2010). Reactive astrocytes were thought to be detrimental to myelin regeneration, mainly due to scar formation around the lesion (do Carmo Cunha et al., 2007), causing inhibition of OPC migration and neurite outgrowth (Wang et al., 2011). However, it is now accepted that reactive astrocytes can be beneficial for myelin repair, because they create a permissive environment for OPC differentiation and secrete various trophic factors for maintenance of the cells surrounding a lesion. Furthermore, it has been shown that elimination of reactive astrocytes after certain CNS insults leads to lesion expansion, severe demyelination, and greater oligodendrocyte cell death (Sofroniew and Vinters, 2010; Haroon et al., 2011). For example, astrocyte-deficient GFAP knockout mice exhibit a greater abnormality in white matter than in gray matter (Liedtke et al., 1996).
A body of evidence indicates that the ON has particular sensitivity to demyelination that significantly distinguishes it from other parts of the CNS. For instance, in MS a vision problem caused by inflammation and demyelination of the ON is commonly the first symptom of disease (Kale, 2016). Considering this particular sensitivity to demyelination of the ON, an animal model with selective ON degeneration and glial cell abnormality would be very helpful in investigating the critical role of astrocyte-oligodendrocyte interaction in the pathogenesis of some clinically important ON disorders.
Here, we introduce a unique rat model in which two separate mutations act synergistically to produce severe ON degeneration with profound demyelination. The first mutation is in Cryba1 (in Nuc1 rats) which encodes βA3/A1-crystallin, a protein expressed in the lysosomal lumen of astrocytes that regulates autophagy (Valapala et al., 2013, 2014a). The second mutation is in Bckdk, which encodes branched chain ketoacid dehydrogenase kinase, an enzyme involved in the catabolism of the branched-chain amino acids - leucine, isoleucine and valine, - and that is essential for mitochondrial function (Oyarzabal et al., 2016). This spontaneous mutation, called frogleg, was initially observed because homozygous rats have severely splayed hind limbs (Zigler et al., 2016). Neither of the mutations in isolation has significant effect on ON development or function, but animals homozygous for both mutations (double mutant) exhibit very serious ON degeneration with profound demyelination (Figure 5). Interestingly, by the age when the double mutant rats exhibited demyelination of the ON, the other myelinated parts of the brain, such as the cortex and brainstem remain unaffected, suggesting selective vulnerability of ON to demyelination in this model. Importantly, astrocytes are the only cells in the ON where both of the mutated genes are expressed. Therefore, our ongoing studies using this animal model may help to unravel the molecular mechanisms whereby astrocyte-oligodendrocyte interaction may regulate the pathogenesis of ON disorders and may serve as a foundation for the development of novel treatments.
Figure 5: Rats homozygous for both Cryba1 and Bckdk mutations exhibit very serious optic nerve degeneration with profound demyelination.
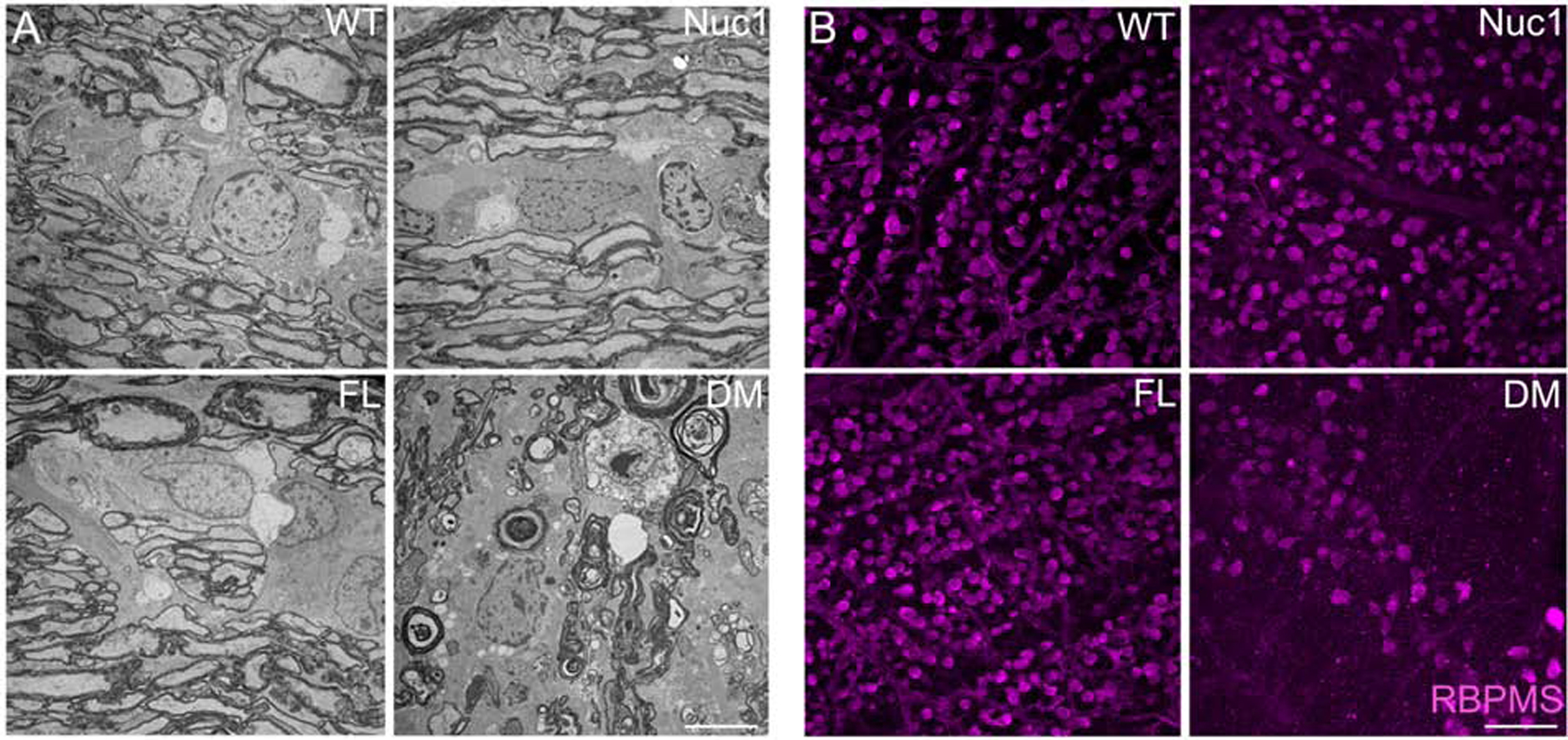
To understand the roles of Bckdk and Cryba1 in optic nerve survival, the optic nerves and retinas were dissected from wild type (WT), Nuc1 (mutation in Cryba1), FL (mutation in Bckdk, named frogleg (FL) because of a severe splaying of the hind limbs) and DM (double mutant, mutations in both Cryba1 and Bckdk) rats. (A) TEM revealed a severe optic nerve degeneration in the DM rats compared to other genotypes at 4 months of age. Bar: 5 μm. (B) Immunohistochemistry of retinal flat mounts against RBPMS (Anti-RNA binding protein with multiple splicing) showed degeneration of RGCs in 4 month old DM rats relative to other genotypes. Bar: 100 μm.
5. Microglia in the optic nerve
Microglia comprise around 10% of cells found within the CNS (von Bartheld, 2016) and they are the resident macrophage population in the CNS which acts as the main mediator of the immune response (Chen et al., 2019; Filiano et al., 2015; Karlstetter et al., 2015). Strikingly, the abundance of microglia was significantly different in different parts of the CNS and appeared to be inversely related to the weights of the analyzed parts, being 1,140 cells per mg of ON and just 280 cells per mg of the cerebral cortex. However, the authors speculate that this inverse correlation might result from a hampered microglia separation in dense suspension (De Hass et al., 2007).
Fate map studies demonstrate that microglia arise and develop within a very short time window during embryonic development. In the mouse, microglia originate from a pool of primitive macrophages present in the yolk sac at E8.5. Microglial precursors can be also detected at the base of the fourth ventricle at approximately E13. In humans, microglial-like cells can be detected at 13 weeks of gestation, while ramified microglia can be distinguished at 21 weeks (Ginhoux et al., 2013). In human ONs, microglial cells have been observed in 8, 10, 12, 14, 15 and 18 week post-conception embryos. During the initial weeks of gestation (8–10 weeks), most microglia were undifferentiated, and the cytoplasm contained numerous large vacuoles similar to those in amoeboid microglia (Christensen et al., 2006), which become well-differentiated in older embryos. Microglia were most prevalent within axonal bundles. Throughout development, the microglial population of the ON contained cellular debris and lipid droplets, indicating their intrinsic ability to phagocytose cellular waste products (Sturrock, 1984).
5.1. The role of microglia in the healthy ON.
Microglia are responsible for general maintenance of the CNS, including the ON, as they constantly engulf cellular debris, plaques, damaged neurons, and infectious agents, thereby preventing potentially fatal damage (Dissing-Olesen et al., 2007). In the healthy CNS, microglia are known to provide essential support to neurons. Their secretion of insulin-like growth factor 1 (IGF-1) provides support to neurons during development and they are involved in the induction of programmed cell death during development via release of neurotrophins (Frade and Barde, 1998). They also contribute to the regulation of neurogenesis through phagocytosis of apoptotic neuronal progenitor cells. In addition, they play an important role in vascular networking during development: in mice and zebrafish, the branching of invading sprouting vessels is regulated by microglia (Fantin et al., 2010; Kubota et al., 2009). Microglial cells are known to make direct contact with pre- and postsynaptic structures in the healthy CNS (Wake et al., 2009; Eyo et al., 2014; Li et al., 2012).
5.2. Microglia in acute and chronic stress
It is known that microglia and other innate immune cells provide the first line of defense against tissue injury or pathogens. However, unregulated activation of these innate immune cells along with the inflammatory molecules that they release cause chronic inflammation, leading to several diseases (Medzhitov, 2008). Microglia are extremely sensitive to any pathological change in the CNS, due to the presence of unique potassium channels on their surface that respond to miniscule changes in potassium levels (Nguyen et al., 2017). One of the main roles of microglia is sensing the tissue microenvironment in order to respond rapidly to neuronal injury in CNS, including the ON. They phagocytose potentially harmful neuronal debris to limit damage, secrete local pro-inflammatory mediators, and release signals to attract other potential immune effector cells (Hanisch and Kettenmann, 2007).
Acute injury and chronic diseases highlight the diverse functions of immune cells such as microglia. Cumulative evidence indicates the involvement of microglia in the degeneration of the ON and RGCs (Madeira et al., 2015). The ON acts as a battlefield for activated and infiltrating microglia and other retinal or peripheral myeloid cells following ON injury (Heuss et al., 2018). In acute injuries to the ON, as demonstrated in animal models of ON crush, the injury attracts retinal microglia to the dying RGCs and their axons (Heuss et al., 2018). Microglia regulate neuroglial remodeling during ON injury, resulting in marked upregulation of inflammatory marker genes, such as Il17f, Tnfsf11, Ccl4, Il1a, Ccr2, Il4, Il5, and Csf2, which are associated with ON damage, increased RGC loss, and retinal nerve fiber layer thinning (Paschalis et al., 2019). ON crush also causes increase in the TNF-α gene and protein expression in microglia, which is associated with conferring protection to RGCs (Mac Nair et al., 2014; Wei et al., 2019). However, in contrast to this, inflammatory cytokines released from microglia after injury are also associated with ON degeneration and RGC loss. It has been shown that IL-1 plays an important role in animal models of blast-mediated traumatic brain injury and blocking IL-1R1 (IL-1 receptor 1) with a specific antagonist resulted in preservation of RGC function and RGC layer thickness (Evans et al., 2020).
Crosstalk between microglia and other retinal cells is dictated by the cell surface molecules on microglia. The interaction between Cluster of Differentiation 200 (CD200) and its receptor (CD200R) is an important example that induces an inhibitory signal limiting tissue damaging activation of microglia. CD200 is broadly expressed in RGCs, photoreceptors, vascular endothelium and the RPE, while CD200R, is predominantly expressed in retinal microglia. Activation of CD200R signaling reduces ON injury in experimental autoimmune uveoretinitis animal models (Copland et al., 2007; Horie et al., 2013). Further, C-X3-C Motif Chemokine Ligand 1 (CX3CL1), a neuron-derived constitutively expressed neuro-immune regulator that binds to its receptor CX3CR1, is expressed in microglia and maintains them in a quiescent mode (Wolf et al., 2013). Transplantation of mesenchymal stem cells secreting CX3CR1 into the subretinal space reduced microglial activation and the expression of pro-inflammatory factors in light-induced retinal degeneration in rats (Huang et al., 2013). Also, loss of CX3CL1 signaling worsens dysfunction in axonal transport in an animal model of chronic glaucoma (Breen et al., 2016). In contrast, deletion of CX3CR1 in the rd10 retinal degeneration mouse model amplified microglial activation and infiltration into the photoreceptor layers along with increased photoreceptor loss, indicating that the CX3CL1-CX3CR1 signaling axis has an important role in microglial activity (Zabel et al., 2016).
Immune cells or retinal cells also modulate microglia function and enable their differential response depending on the type of stress. It was shown that in a rat model of ON crush injury, autoreactive T cells induced accumulation of macrophages/microglia and B cells at the site of injury, and the production of neurotrophic factors, which is the basis of the protective effect conferred by T cell-mediated autoimmunity on injured neurons (Barouch and Schwartz, 2002). During glutamate toxicity, antigen-specific autoimmune T cells alter the microglia phenotype, increasing the ability to remove excess glutamate. Moreover, this T-cell-mediated effect could be simulated by the Th1 cytokine interferon (IFN)-gamma and was significantly reduced by neutralizing anti-IFN-gamma antibodies (Shaked et al., 2005), indicating that T cells or their cytokines can regulate microglial phenotype and facilitate glutamate clearance and restoration of homeostasis.
The transition from acute to chronic immune response, as observed in various chronic or multifactorial diseases including ON disorders, depends on several factors such as genetic predisposition, environmental factors and lifestyle (Medzhitov, 2008; Allen at al., 2015; Newman et al., 2004; Fan et al., 2004). The differential effect of microglia function during acute and chronic stress is further evident from studies done on human tissues and in animal models. Microglia that repopulate the injury site are the usual suspects for causing redox imbalance and inflammation and thus further exacerbating the neurotoxicity. In a rat model of MS that frequently leads to optic neuritis, an increased infiltration of inflammatory microglia plays an important role in demyelination, loss of RGCs and axon degeneration in the ON (Fairless et al., 2012). The early infiltration of microglia to the ON, at the onset of optic neuritis can be explained by the fact that microglia are the major innate immune cells of the CNS and are responsible for triggering inflammation the ON. However, in chronic diseases like MS and glaucoma (discussed below), the constant activation of microglia due by various endogenous factors associated with such diseases, could be responsible for its hyperactivation leading to a transition from para inflammation to chronic inflammation (Medzhitov, 2008).
Overall, microglial activation clearly has important implications for ON health. Microglial activation during acute injury or at the early stages of a disease may lead to a more robust and protective response, whereas in a chronic disease state it can induce pathologic sequelae in the microglia (hyper-activated state), potentiating degenerative changes in the ON.
5.3. Role of microglia in glaucoma
Microglia are critical in the pathogenesis of glaucoma. In human glaucoma, microglia located at the ONH become enlarged/activated and accumulate in the parapapillary chorioretinal region, which is the non-myelinated region of the ON, where the RPE and Bruch’s membrane terminate, and where the RGC axons bypass the retina and the marginal choroidal capillaries (Neufeld, 1999). The increase in microglial density may be caused by migration of activated microglia from the disorganized prelaminar and laminar tissue of the ON. The altered activation state of microglia in the parapapillary chorioretinal region in glaucoma is thought to be responsible for some major histopathological changes associated with parapapillary chorioretinal atrophy in human glaucoma patients (Neufeld, 1999).
In animal models of glaucoma, particularly the DBA/2J mouse model of inherited glaucoma, which develops progressive RGC degeneration of variable severity with age, microglial activation serves as an indicator of future neurodegeneration severity (Bosco et al., 2015). Reduction in microglial activation in the DBA/2J mouse model improved ON integrity (Bosco et al., 2008). In the chondroitin sulfate - rat glaucoma model, activated microglia have been shown to trigger alterations in axoglial networks of the ON (Bordone et al., 2017). It is now accepted that even though the activated microglia are seen in the early stages of glaucoma probably as a protective measure, as they get hyperactivated with the disease progression, they cause neuronal damage (Wang et al., 2016).
Microglia secrete a plethora of pro-inflammatory mediators such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and complement 1q (C1q), which serve as key signaling modulators for protective and/or degenerative changes in the ON (Zeng et al., 2018). Over-activation of microglia results in the production of pro-inflammatory cytokines and an increase in the release of reactive oxygen species and nitric oxide (NO), causing damage to retinal neurons (Figure 6) (Karlstetter et al., 2010). Pro-inflammatory cytokines, such as TNF-α which is secreted by macrophages in the periphery and microglia in the ON and ONH, are involved in the process of glaucomatous neurodegeneration (Al-Gayyar et al., 2013; Xu et al., 2009). Moreover, in vitro and, particularly, in vivo studies in animal models of glaucoma revealed that treatment with a TNF-α inhibitor prevented RGC apoptosis and inflammation (Tezel and Wax, 2000; Roh et al., 2012). However, in contrast to these observations, it has been demonstrated in mouse models of ON injury, that TNF-α renders protection to RGCs and helps with tissue maintenance and regeneration (Veroni et al., 2010; Fontaine et al., 2002; Bartsch et al., 2010). Both TNF-α and IL-1β are released by microglia upon activation and inhibiting microglial activation reverses inflammation and RGC death in animal models of glaucoma and ischemic injury (Almasieh et al., 2012; Huang et al., 2011, Dang et al., 2014, Li et al., 2014). Increased expression of the anti-inflammatory cytokine TGF-β indicates that microglia initially combat the degenerative processes in the glaucomatous ONH. However, with the progression of the disease, microglia get hyperactivated and trigger a vicious inflammatory cycle causing neurodegeneration and degradation of the ECM of the ON (Rashid et al., 2019).
Figure 6: Microglia activation during disease progression.
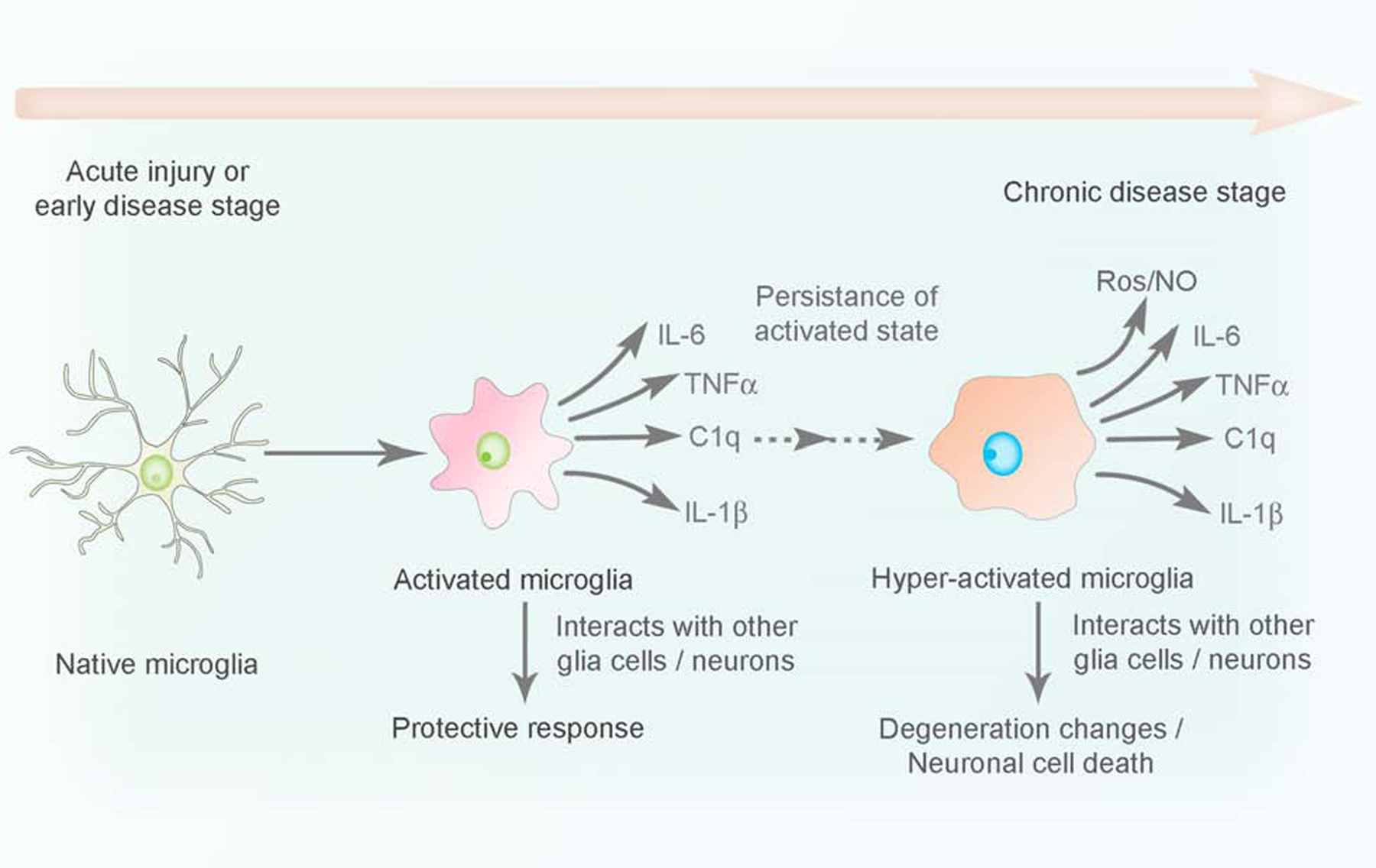
Naïve microglia undergo inflammatory transition to an activated state (releasing inflammatory cytokines, chemokines) to confer protection during acute infection or tissue injury in the CNS. However, persistent activation of microglia upon stress stimuli rendered by genetic predisposition or environmental factors, leads to a state of hyper activation associated with an uncontrolled release of pro-inflammatory mediators (chronic inflammation), which in turn causes neuronal damage in the CNS.
Furthermore, specific kinases like the mitogen-activated protein kinases (MAPKs) (p42/44 MAPK, SAPK/JNK, p38 MAPK), play an important role in the initiation of inflammation and development of retinal pathology in glaucoma, due to their action in the ONH, where the initial insult to RGC axons likely occurs. Each of the MAPKs shows a specific spatio-temporal expression and activation pattern in the ON following IOP elevation during glaucoma. p38 MAPK was expressed by a population of microglia which significantly increased in number after IOP elevation. However, this activation was seen in microglia only after 3 days of IOP elevation, and specifically in the ONH and ON, whereas in the retina it was solely activated in RGC (Mammone et al., 2018). Therefore, it can be assumed that inhibiting specific kinases could reduce the pro-inflammatory signaling in the ON in early stages of glaucoma, thereby conferring protection from ON degeneration as the disease progresses.
Targeting microglia as a therapeutic approach to prevent ON degeneration in animal models of glaucoma or acute injury has been considered. Neuroprotective strategies that inhibit activated microglia are thought to be novel treatment approaches for glaucomatous optic neuropathy and other retinal and ON diseases (Wang et al., 2016). However, experiments involving microglial depletion by using the colony stimulating factor 1 receptor (CSF1R) inhibitor PLX5622 (Liddelow et al., 2017), which specifically eliminates microglia in murine retinae and ONs with high efficiency, revealed that RGC degeneration and induction of regeneration-associated genes upon ON injury remained unaffected. However, astrocyte repopulation at the site of ON injury was significantly delayed upon microglia depletion (Hilla et al., 2017), thereby indicating that microglia may be involved in ON regeneration rather than neurodegeneration in acute injury conditions.
5.4. Crosstalk between microglia, astrocytes, NG2 cells and oligodendrocytes
As mentioned previously, glial cells work as a team to maintain the functionality of the CNS (Figure 3), and microglia are not an exception. A strong bidirectional communication exists between microglia and astrocytes (reviewed extensively by Jha et al., 2019). Being immune cells, microglia constantly sense changes in the CNS, provide rapid response to pathological stimuli, and secrete signals to induce astrocyte activation. In turn, astrocytes are capable of changing the activation state of microglia and fine-tuning their behavior, such as migration and phagocytosis (Vainchtein et al., 2018). This bidirectional astrocyte-microglia crosstalk is also known to modulate oligodendrocyte homeostasis and myelination (Domingues et al., 2016).
Recent studies suggest that astrocytes can serve as a part of the innate immune system, but this function is tightly controlled by their interactions with microglia (Refolo and Stefanova, 2019). For example, astrocytes express a number of innate immune pattern recognition receptors, such as Toll-like receptors (TLRs), NOD-like receptors (NLRs), mannose receptors, scavenger receptors and complement receptors. The response of astrocytes to stimulation of TLR2, TLR3 and TLR4 is likely to be dependent on the presence of microglia (Holm et al., 2012). If functional microglia are present, their activation through stimulation of CSF1 receptors triggers the production of cytokines, such as IL-1α, TNF-α and the C1q, which in turn leads to the induction of A1 reactive astrocytes both in vivo and in vitro. In animal models that lack Csf1r, Ilα, Tnfα and C1qα genes, microglia fail to trigger A1 reactive astrocytes (Liddelow et al., 2017). Moreover, in the absence of microglia, lipopolysaccharide (LPS) stimulation of astrocytes does not result in the production of adequate amounts of proinflammatory factors (such as TNF-α and NO) to trigger astrocyte reactivity, thus supporting the important role of microglia in reactive astrogliosis (Chen et al., 2015).
An interesting novel mechanism of astrocyte activation to the A1 state has been proposed recently (Joshi et al., 2019). In this study, activation of astrocytes was shown to be mediated by the release of fragmented and dysfunctional mitochondria from microglia. This concept is indeed very interesting. The authors speculate that during evolution, mitochondria were made immunologically privileged in order to retain the endosymbiosis that was extremely beneficial to the host eukaryotic cells. On the other hand, the host organism had to be extremely hostile to the phylogenetically related pathogenic bacteria. As a result, damage or fragmentation of mitochondria, followed by the release of the fragments outside of the cells, can result in the loss of immune-privileged status of mitochondria and misidentification of them as bacterial invaders that may trigger a severe innate immune response. The authors have demonstrated that activation of microglia results in an increase in Drp1-Fis1-mediated mitochondrial fission, which, in turn, leads to the release of fragmented and damaged mitochondria into the neuronal milieu and subsequent activation of astrocytes to A1 state. Importantly, inhibition of mitochondrial fragmentation using peptide P110 results in inhibition of reactive astrogliosis and protection of neurons from the innate immune response. Overall, the study suggests that inhibition of mitochondrial fragmentation in microglia might become a novel therapeutic strategy to prevent neurodegeneration (Joshi et al., 2019).
Upon stimulation by microglia, reactive astrocytes in turn start secreting neurotrophic factors, including factors that facilitate permeability of the BBB, thus promoting the recruitment of more immune cells and amplifying the initial response (Alvarez et al., 2013). Interestingly, at the late stages of inflammation, astrocytes are capable of alleviating neuroinflammation by reducing microglia activation. This can be done through secretion of orosomucoid-2 (ORM2) by astrocytes: ORM2 binds microglial C-C chemokine receptor type 5 (CCR5) to deactivate microglia (Jo et al., 2017). On the other hand, reactive astrocytes can secrete high levels of lipocalin-2 (LCN2) that counteracts ORM2 and results in microglia activation (Bi et al., 2013; Ghosh et al., 2017; Tripathi et al., 2017). A number of other factors, such as monocyte chemoattractant protein (MCP)-1/CCL2 and interferon gamma (IFN-γ) inducible protein (IP)-10/CXCL10 are also involved in microglia activation (Tanuma et al., 2006).
In addition to the regulation of the activation state of microglia, astrocytes are known to modulate microglial behavior (Jha et al., 2019). During inflammation, both astrocytes and microglia secrete plasminogen activator inhibitor type 1 (PAI-1), which has a dual role in regulating microglia function. First, it acts through the low-density lipoprotein receptor-related protein (LPR)-1/JAK/STAT1 axis to promote microglial migration. Second, it affects vitronectin and TLR2/6 to modulate microglial phagocytic activity. Additional regulation of microglial phagocytosis is mediated through the secretion of PTX3 (Jeon et al, 2012).
Astrocyte-microglia crosstalk is critical for myelination, demyelination and remyelination, both under physiological and pathological conditions. During development, resting astrocytes and microglia facilitate differentiation of oligodendrocytes from OPCs (Zuchero and Barres, 2015). When microglia are activated and astrocytes become reactive, they both secrete mediators to directly regulate demyelination/remyelination, that can be either beneficial or harmful, depending on the nature and severity of the injury (Nave and Werner, 2014). Astrocyte-derived cytokines and chemokines, including CCL2 and CXCL10 promote inflammation and further microglia recruitment that results in myelin degeneration and oligodendrocyte loss. On the other hand, expression of CXCL10 that results in microglia recruitment is beneficial for demyelination because phagocytosis of myelin debris is a critical step in this process. Astrocytes and microglia can also recruit OPCs and promote OPC differentiation through secretion of CXCL1, CXCL8 and CXCL10 (Skripuletz et al., 2013; Patel et al.; 2012, Tanuma et al., 2006). Oligodendrocytes, in turn, can affect microglia activation and survival (reviewed in Peferoen et al., 2013; Zeis et al., 2016). For example, oligodendrocytes are known to express CD200 that, as discussed earlier in this review, is known to suppress microglial activation. Moreover, oligodendrocytes can initiate apoptosis of the activated microglia by expressing Semaphorin 3A (SEMA3A) (Zeis et al., 2016).
In addition, recent studies have found a strong functional interaction between NG2 cells and microglia, suggesting an important role of NG2 cells in neuroinflammation (Liu and Aguzzi, 2019; Zhang et al., 2019). Currently it is not known whether this interaction exists in the ON, but in the adult brain (at least in cerebrocortex, hippocampus, striatum, and substantia nigra) NG2 glia have been demonstrated to secrete TGF-β2, which, in turn is known to suppress the activation and proliferation of microglia through CX3CR1, thus maintaining microglia in a quiescent state under normal physiological conditions. Ablation of NG2 glia in the brain resulted in overactivation of microglia in response to LPS stimulus, which could be attenuated by overexpressing CX3CR1. Interestingly, it has been shown that NG2 proliferation in the brain diminishes with age, and the authors speculate that this might contribute to the development of age-related neurodegenerative diseases (Zhang et al., 2019).
Taken together, interactions between glial cells are complex and context dependent. The diverse roles of these cells are fascinating: under healthy physiological conditions, they work together to ensure proper neuronal homeostasis, however, under pathological conditions, their interplay can be either detrimental or beneficial, depending on the circumstances.
6. Future Directions
At present, we are in a new era of glial cell biology — full of exciting possibilities for discoveries that are now feasible due to a variety of recently developed innovative tools and techniques, including isolation of highly enriched glial cells from both rodents and humans and glial cell multi-omics analyses (genomics, epigenomics, transcriptomics, metabolomics, etc.) at the single cell level. Such techniques have revealed additional details on glial heterogeneity and enabled us to identify a variety of specialized astrocytes, microglia and oligodendrocytes depending on the different anatomical regions of the CNS in health and disease.
The microglia-astroglia interactions have long been under-estimated and have just recently started to gain serious attention. Indeed, many of the most recent studies demonstrate that microglia are able to guide astrocytes towards different reactive states and functions (Liddelow et al., 2017; Shinozaki et al., 2017; Yun et al., 2018). What are the specific signaling pathways that regulate the induction of astrocyte reactivity? What are the astrocyte-derived toxins that lead to neuronal and oligodendrocyte cell death? Understanding the molecular mechanisms that govern the induction of reactive astrocytes might enable us to develop novel therapeutic strategies to promote repair in neurodegenerative diseases such as glaucoma or optic neuritis.
In the context of demyelinating diseases, the complex interactions between astrocytes and oligodendrocytes largely remain unknown. These studies are complicated by the lack of an animal model to study such interactions without the systemic health complications caused by a global glial cell abnormality in the CNS. Our unique spontaneous double mutant rat (with mutations in both Cryba1 and Bckdk), where the defect in astrocytes results in selective vulnerability of the ON for a profound demyelination without obviously affecting the rest of the CNS, represents an interesting novel model to study astrocyte-oligodendrocyte interactions. Studies using this novel animal model may enable us to unravel molecular mechanisms of astrocyte-mediated regulation of myelination (discussed in the section 4.3).
Taken together, significant progress has been made in recent years toward understanding the complexity of the glial cell network in the CNS, however, a lot of important questions still remain unanswered. The ON can serve as a unique and invaluable tissue of interest to study important aspects of glial cell biology due to its heterogeneous glial populations characterizing different areas of the ON, as well as the relative simplicity of the ON due to its lack of synapses.
9. Article Highlights:
The glial cells in the ON are critical to its functional integrity and homeostasis.
The glial cell network maintains functional interactions among glia and the axons.
The nuances of glial interactions in the ON may differ from other parts of the CNS.
Acknowledgements:
We thank Professor (Dr.) Morton F. Goldberg, Wilmer Eye Institute, The Johns Hopkins University School of Medicine for critical reading and discussions regarding this review article.
Funding Sources: This work was supported by Knights Templar Eye Foundation (MY), NIH EY019037 (DS), NIH EY019037-S (DS), NIH EY018636 (DS), IPA from National Eye Institute, NIH (DS), University of Pittsburgh start-up funds (DS), Jennifer Salvitti Davis, M.D. Chair in Ophthalmology (DS), and an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology, University of Pittsburgh.
Abbreviations:
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ATP
adenosine triphosphate
- BBB
blood-brain barrier
- Bckdk
branched chain ketoacid dehydrogenase kinase
- C1q
complement 1q
- CCR5
C-C chemokine receptor type 5
- CD200
cluster of differentiation 200
- CD200R
CD200 receptor
- CNS
central nervous system
- CNTF
ciliary neurotrophic factor
- CSF1R
colony stimulating factor 1 receptor
- CX3CL1
C-X3-C Motif Chemokine Ligand 1
- CX3CR1
CX3C chemokine receptor 1
- EAE
experimental autoimmune encephalomyelitis
- EGFR
epidermal growth factor receptor
- ECM
extracellular matrix
- ET-1
endothelin-1
- FAT
fatty acid translocase
- FGF2
fibroblast growth factor 2
- FL
frogleg
- GFAP
glial fibrillary acidic protein
- GLUT1
glucose transporter 1
- IFN-γ
interferon gamma
- IGF-1
insulin-like growth factor 1
- IL-1β
interleukin-1 beta
- IL-6
interleukin-6
- IOP
intraocular pressure
- LCN2
lipocalin-2
- LPR
lipoprotein receptor-related protein
- LPS
lipopolysaccharide
- MAG
myelin associated glycoprotein
- MAPK
mitogen-activated protein kinases
- MCP
monocyte chemoattractant protein
- MCT
monocarboxylate transporter
- MBP
myelin basic protein
- MMP
matrix metalloproteinases
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- NG2
neuron glial 2 cells
- NLR
NOD-like receptors
- NMDAR
N-methyl-D-aspartate receptors
- NTG
normal-tension glaucoma
- ON
optic nerve
- ONH
optic nerve head
- OPC
oligodendrocyte progenitor cells
- ORM2
orosomucoid-2
- PAI-1
plasminogen activator inhibitor type 1
- PDGF
platelet-derived growth factor
- PLP
proteolipid protein
- RGC
retinal ganglion cell
- RPE
retinal pigment epithelium
- SEMA3A
Semaphorin 3A
- TFEB
transcription factor EB
- TIMP
tissue inhibitors of metalloproteinases
- TLR
Toll-like receptors
- TNF-α
tumor necrosis factor-alpha
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. References:
- Agapova OA, Kaufman PL, Lucarelli MJ, Gabelt BAT, Hernandez MR, 2003. Differential expression of matrix metalloproteinases in monkey eyes with experimental glaucoma or optic nerve transection. Brain Res, 967, 132–143. [DOI] [PubMed] [Google Scholar]
- Agapova OA, Ricard CS, Salvador-Silva M, Hernandez MR, 2001. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human optic nerve head astrocytes. Glia. 33, 205–216. [DOI] [PubMed] [Google Scholar]
- Al-Gayyar MM, Elsherbiny NM, 2013. Contribution of TNF-α to the development of retinal neurodegenerative disorders. Eur. Cytokine. Netw 24, 27–36. [DOI] [PubMed] [Google Scholar]
- Alkozi HA, Navarro G, Franco R, Pintor J, 2019. Melatonin and the control of intraocular pressure. Prog Retin Eye Res. 25, 100798. [DOI] [PubMed] [Google Scholar]
- Allen A, Messier C, 2013. Plastic changes in the astrocyte GLUT1 glucose transporter and beta-tubulin microtubule protein following voluntary exercise in mice. Behav Brain Res 240: 95–102. [DOI] [PubMed] [Google Scholar]
- Allen KF, Gaier ED, Wiggs JL, , 2015. Genetics of Primary Inherited Disorders of the Optic Nerve: Clinical Applications. Cold Spring Harb Perspect Med 5(7): a017277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A, 2012. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res. 31(2), 152–81. [DOI] [PubMed] [Google Scholar]
- Almeida RG, 2018. The Rules of Attraction in Central Nervous System Myelination. Front. Cell Neurosci 12, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez JI, Katayama T, Prat A, 2013. Glial influence on the blood brain barrier. Glia. 61(12), 1939–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV, 2010. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 29(2), 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, and Sofroniew MV, 2016. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 532, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancibia-Carcamo IL, Attwell D, 2014. The node of Ranvier in CNS pathology. Acta Neuropathol. 128, 161–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D and Laughlin SB, 2001. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21(10): 1133–1145. [DOI] [PubMed] [Google Scholar]
- Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, et al. , 2015. Antioxidant role for lipid droplets in a stem cell niche of drosophila. Cell. 163, 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracskay KL, Kidd GJ, Miller RH, Trapp BD, 2007. NG2-positive cells generate A2B5-positive oligodendrocyte precursor cells. Glia. 55, 1001–1010. [DOI] [PubMed] [Google Scholar]
- Barateiro A, Fernandes A 2014. Temporal oligodendrocyte lineage progression: in vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta 1843, 1917–29. [DOI] [PubMed] [Google Scholar]
- Barber CN, Raben DM, 2019. Lipid Metabolism Crosstalk in the Brain: Glia and Neurons. Front Cell Neurosci. May 21;13:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch R, Schwartz M, 2002. Autoreactive T cells induce neurotrophin production by immune and neural cells in injured rat ON: implications for protective autoimmunity. FASEB. J 16, 1304–6. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC, 1992. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 70, 31–46. [DOI] [PubMed] [Google Scholar]
- Barres BA, Raff MC, 1994. Control of oligodendrocyte number in the developing rat ON. Neuron. 12, 935–942. [DOI] [PubMed] [Google Scholar]
- Bartsch JW, Wildeboer D, Koller G, Naus S, Rittger A, Moss ML, Minai Y, Jockusch H, 2010. Tumor necrosis factor-alpha (TNF-alpha) regulates shedding of TNF-alpha receptor 1 by the metalloprotease- disintegrin ADAM8: evidence for a protease-regulated feedback loop in neuroprotection. J. Neurosci 30, 12210–12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechler ME, Byrne L, Ffrench-Constant C, 2015. CNS myelin sheath lengths are an intrinsic property of oligodendrocytes. Curr. Biol 25, 2411–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben HL, Ceyzériat K, Carrillo-de Sauvage MA, Aubry F, Auregan G, Guillermier M, Ruiz M, Petit F, Houitte D, Faivre E, Vandesquille M, Aron-Badin R, Dhenain M, Déglon N, Hantraye P, Brouillet E, Bonvento G, Escartin C, 2015. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J Neurosci. 35(6), 2817–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergersen L, Rafiki A, Ottersen OP, 2002. Immunogold cytochemistry identifies specialized membrane domains for monocarboxylate transport in the central nervous system. Neurochem. Res 27, 89–96. [DOI] [PubMed] [Google Scholar]
- Berridge MV, Schneider RT, McConnell MJ, 2016. Mitochondrial Transfer from Astrocytes to Neurons following Ischemic Insult: Guilt by Association? Cell Metab. 24(3), 376–378. [DOI] [PubMed] [Google Scholar]
- Berry M, Hubbard P, Butt AM, 2002. Cytology and lineage of NG2-positive glia. J. Neurocytol 31, 457–467. [DOI] [PubMed] [Google Scholar]
- Bi F, Huang C, Tong J, Qiu G, Huang B, Wu Q, Li F, Xu Z, Bowser R, Xia XG, Zhou H, 2013. Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci U S A. 110(10), 4069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Zecca L, and Hong JS, 2007. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci 8, 57–69. [DOI] [PubMed] [Google Scholar]
- Boche D, Perry VH, and Nicoll JA, 2013. Review: activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol 39, 3–18. [DOI] [PubMed] [Google Scholar]
- Boghdadi AG, Teo L and Bourne JA, 2020. The Neuroprotective Role of Reactive Astrocytes after Central Nervous System Injury. J Neurotrauma 37(5): 681–691. [DOI] [PubMed] [Google Scholar]
- Bordone MP, González Fleitas MF, Pasquini LA, Bosco A, Sande PH, Rosenstein RE, Dorfman D, 2017. Involvement of microglia in early axoglial alterations of the ON induced by experimental glaucoma. J. Neurochem 142, 323–337. [DOI] [PubMed] [Google Scholar]
- Bosco A, Breen KT, Anderson SR, Steele MR, Calkins DJ, Vetter ML, 2016. Glial coverage in the optic nerve expands in proportion to optic axon loss in chronic mouse glaucoma. Exp Eye Res. 150, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, Inman DM, Steele MR, Wu G, Soto I, Marsh-Armstrong N, Hubbard WC, Calkins DJ, Horner PJ, Vetter ML, 2008. Reduced retina microglial activation and improved ON integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest. Ophthalmol. Vis. Sci 49, 1437–46. [DOI] [PubMed] [Google Scholar]
- Bosco A, Romero CO, Breen KT, Chagovetz AA, Steele MR, Ambati BK, Vetter ML, 2015. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Dis. Model Mech 8, 443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumezbeur F, Petersen KF, Cline GW, Mason GF, Behar KL, Shulman GI, Rothman DL, 2010. The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J. Neurosci 30, 13983–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Pellerin L, 2013. Unraveling the complex metabolic nature of astrocytes. Front. Cell Neurosci 7, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Esteban-Martínez L, Serrano-Puebla A, Gómez-Sintes R, Villarejo-Zori B, 2016. Autophagy in the eye: Development, degeneration, and aging. Prog Retin Eye Res. 55, 206–245. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Dvoriantchikova G, Barakat D, Ivanov D, Bethea JR, Shestopalov VI, 2012. Transgenic inhibition of astroglial NF-κB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J Neuroinflammation 9, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen KT, Anderson SR, Steele MR, Calkins DJ, Bosco A and Vetter ML, 2016. Loss of Fractalkine Signaling Exacerbates Axon Transport Dysfunction in a Chronic Model of Glaucoma. Front Neurosci 10: 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann BG, Agarwal A, Sereda MW, Garratt AN, Müller T, Wende H, Stassart RM, Nawaz S, Humml C, Velanac V, Radyushkin K, Goebbels S, Fischer TM, Franklin RJ, Lai C, Ehrenreich H, Birchmeier C, Schwab MH, Nave KA, 2008. Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron. 59, 581–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Tekkök SB, Ransom BR, 2003. Glycogen regulation and functional role in mouse white matter. J. Physiol 549, 501–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruesch SR, Arey LB, 1942. The Number of Myelinated and Unmyelinated Fibers in the ON of Vertebrates. J. Comp. Neurol 77, 169–191. [Google Scholar]
- Burgoyne CF, Downs JC, Bellezza AJ, Suh JK, Hart RT, 2005. The optic nerve head as a biomechanical structure: a new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog Retin Eye Res. 24(1), 39–73. [DOI] [PubMed] [Google Scholar]
- Butt AM, Hubbard P, Kiff J, Ibrahim M, Berry M, 2003. Synantocytes: new functions for novel NG2 expressing glia. J. Neurocytol 31, 551–565. [DOI] [PubMed] [Google Scholar]
- Butt AM, Pugh M, Hubbard P, James G, 2004. Functions of ON glia: axoglial signalling in physiology and pathology. Eye (Lond). 18, 1110–1121. [DOI] [PubMed] [Google Scholar]
- Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, Richardson WD, 1998. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 20, 869–82. [DOI] [PubMed] [Google Scholar]
- Camargo N, Brouwers JF, Loos M, Gutmann DH, Smit AB, and Verheijen MH, 2012. High-fat diet ameliorates neurological deficits caused by defective astrocyte lipid metabolism. FASEB J. 26, 4302–4315. [DOI] [PubMed] [Google Scholar]
- Caprariello AV, Batt CE, Zippe I, Romito-DiGiacomo R, Karl M, Miller RH, 2015. Apoptosis of Oligodendrocytes during Early Development Delays Myelination and Impairs Subsequent Responses to Demyelination. J Neurosci. 35(41): 14031–14041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carelli V, Ross-Cisneros FN, Sadun AA, 2004. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 23(1), 53–89. [DOI] [PubMed] [Google Scholar]
- Carrero I, Gonzalo MR, Martin B, Sanz-Anquela JM, Arévalo-Serrano J, Gonzalo-Ruiz A, 2012. Oligomers of b-amyloid protein (Ab1–42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1b, tumour necrosis factor-a, and a nuclear factor k-B mechanism in the rat brain. Exp. Neurol 236, 215–227. [DOI] [PubMed] [Google Scholar]
- Cellini M, Possati GL, Profazio V, Sbrocca M, Caramazza N, Caramazza R, 1997. Color Doppler imaging and plasma levels of endothelin-1 in low-tension glaucoma. Acta Ophthalmol Scand Suppl. 224, 11–3. [DOI] [PubMed] [Google Scholar]
- Ceyzeriat K, Abjean L, Carrillo-de Sauvage MA, Ben HL, Escartin C, 2016. The complex STATes of astrocyte reactivity: How are they controlled by the JAK-STAT3 pathway? Neuroscience. 330, 205–218. [DOI] [PubMed] [Google Scholar]
- Chang KJ, Zollinger DR, Susuki K, Sherman DL, Makara MA, Brophy PJ, Cooper EC, Bennett V, Mohler PJ, Rasband MN, 2014. Glial ankyrins facilitate paranodal axoglial junction assembly. Nat. Neurosci 17, 1673–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Hernandez MP, Stankoff B, Aigrot MS, Colin C, Rougon G, Zalc B, Lubetzki C, 2000. Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc. Natl. Acad. Sci. U. S. A 97, 7585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang X, Kusumo H, Costa LG, and Guizzetti M, 2013. Cholesterol efflux is differentially regulated in neurons and astrocytes: implications for brain cholesterol homeostasis. Biochim. Biophys. Acta Mol. Cell. Biol 1831, 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Luo C, Zhao J, Devarajan G, Xu H, 2018. Immune regulation in the aging retina. Prog Retin Eye Res 69, 59–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SH, Oyarzabal EA, Sung YF, Chu CH, Wang Q, Chen SL, Lu RB, Hong JS, 2015. Microglial regulation of immunological and neuroprotective functions of astroglia. Glia. 63(1), 118–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen RN, Ha BK, Sun F, Bresnahan JC, Beattie MS, 2006. Kainate induces rapid redistribution of the actin cytoskeleton in ameboid microglia. J. Neurosci. Res 84, 170–81. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Tesar PJ, 2017. Clemastine fumarate for promotion of ON remyelination. Lancet. 390, 2421–2422. [DOI] [PubMed] [Google Scholar]
- Colello RJ, Devey LR, Imperato E, Pott U, 1995. The chronology of oligodendrocyte differentiation in the rat ON: evidence for a signaling step initiating myelination in the CNS. J. Neurosci 15, 7665–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copland DA, Calder CJ, Raveney BJ, Nicholson LB, Phillips J, Cherwinski H, Jenmalm M, Sedgwick JD, Dick AD, 2007. Monoclonal antibody-mediated CD200 receptor signaling suppresses macrophage activation and tissue damage in experimental autoimmune uveoretinitis. Am J Pathol 171(2): 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti A, Ransohoff RM, 2016. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity. 44, 505–515. [DOI] [PubMed] [Google Scholar]
- Crotty P, Sangrey T, Levy WB, 2006. Metabolic energy cost of action potential velocity. J Neurophysiol 96(3): 1237–1246. [DOI] [PubMed] [Google Scholar]
- Dakubo GD, Beug ST, Mazerolle CJ, Thurig S, Wang Y, Wallace VA 2008. Control of glial precursor cell development in the mouse optic nerve by sonic hedgehog from retinal ganglion cells. Brain Res 1228: 27–42. [DOI] [PubMed] [Google Scholar]
- Dang Y, Xu Y, Wu W, Li W, Sun Y, Yang J, Zhu Y, Zhang C, 2014. Tetrandrine suppresses lipopolysaccharide-induced microglial activation by inhibiting NF-κB and ERK signaling pathways in BV2 cells. PLoS One. 9, e102522. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N, 2014. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A, 111(26), 9633–9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CH, Marsh-Armstrong N, 2014. Discovery and implications of transcellular mitophagy. Autophagy. 10(12), 2383–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haas AH, Boddeke HW, Brouwer N, Biber K, 2007. Optimized isolation enables ex vivo analysis of microglia from various central nervous system regions. Glia 55(13): 1374–1384. [DOI] [PubMed] [Google Scholar]
- Devaux J, Gola M, Jacquet G, Crest M, 2002. Effects of K channel blockers on developing rat myelinated CNS axons: identification of four types of K channels. J. Neurophysiol 87, 1376 – 1385. [DOI] [PubMed] [Google Scholar]
- Dezawa M, Adachi-Usami E, 2000. Role of Schwann cells in retinal ganglion cell axon regeneration. Prog Retin Eye Res.19(2):171–204. [DOI] [PubMed] [Google Scholar]
- Dhaunchak AS, Huang JK, De Faria Junior O, Roth AD, Pedraza L, Antel JP, Bar-Or A, Colman DR, 2010. A proteome map of axoglial specializations isolated and purified from human central nervous system. Glia. 58, 1949–1960. [DOI] [PubMed] [Google Scholar]
- Dienel GA, 2017. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J Neurosci Res. 95(11), 2103–2125. [DOI] [PubMed] [Google Scholar]
- Dienel GA, 2019. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev 99, 949–1045. [DOI] [PubMed] [Google Scholar]
- Dimou L, Simon C, Kirchhoff F, Takebayashi H, Götz M, 2008. Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J. Neurosci 28, 10434–10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissing-Olesen L, Ladeby R, Nielsen HH, Toft-Hansen H, Dalmau I, Finsen B, 2007. Axonal lesion-induced microglial proliferation and microglial cluster formation in the mouse. Neuroscience 149, 112–22. [DOI] [PubMed] [Google Scholar]
- do Carmo Cunha J, de Freitas Azevedo Levy B, de Luca BA, de Andrade MS, Gomide VC, Chad IG, 2007. Responses of reactive astrocytes containing S100 beta protein and fibroblast growth factor-2 in the border and in the adjacent preserved tissue after a contusion injury of the spinal cord in rats: implications for wound repair and neuroregeneration. Wound Repair Regen. 15, 134–46. [DOI] [PubMed] [Google Scholar]
- Domingues HS, Portugal CC, Socodato R, Relvas JB, 2016. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol 4, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JC, Girkin CA, 2017. Lamina cribrosa in glaucoma. Curr Opin Ophthalmol. 28(2), 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan ID, Marik RL, Broman AT, Heidari M, 2017. Thin myelin sheaths as the hallmark of remyelination persist over time and preserve axon function. Proc. Natl. Acad. Sci. U. S. A 114, E9685–E9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Comb WC, Sabatini DM, 2015. Nutrient-sensing mechanisms and pathways. Nature. 517, 302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Schweitzer LD, Bilate AM, Chang S, Kirak O, Lamming DW, Sabatini DM, 2014. RagA, but not RagB, is essential for embryonic development and adult mice. Dev. Cell 29, 321–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM, 2013. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 493, 679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa de los Monteros A, Zhang M, De Vellis J, 1993. O2A progenitor cells transplanted into the neonatal rat brain develop into oligodendrocytes but not astrocytes. Proc. Natl. Acad. Sci. U. S. A 90, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etxeberria A, Hokanson KC, Dao DQ, Mayoral SR, Mei F, Redmond SA, Ullian EM, Chan JR, 2016. Dynamic Modulation of Myelination in Response to Visual Stimuli Alters ON Conduction Velocity. J. Neurosci 36, 6937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans LP, Woll AW, Wu S, Todd BP, Hehr N, Hedberg-Buenz A, Anderson MG, Newell EA, Ferguson PJ, Mahajan VB, Harper MM, Bassuk AG, 2020. Modulation of Post-Traumatic Immune Response Using the IL-1 Receptor Antagonist Anakinra for Improved Visual Outcomes. J Neurotrauma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu LJ, 2014. Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J Neurosci. 34(32), 10528–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairless R, Williams SK, Hoffmann DB, Stojic A, Hochmeister S, Schmitz F, Storch MK, Diem R, 2012. Preclinical retinal neurodegeneration in a model of multiple sclerosis. J Neurosci 32(16): 5585–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan BJ, Leung YF, Wang N, Lam SC, Liu Y, Tam OS, Pang CP, 2004. Genetic and environmental risk factors for primary open-angle glaucoma. Chin Med J (Engl) 117(5): 706–710. [PubMed] [Google Scholar]
- Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, Peri F, Wilson SW, Ruhrberg C, 2010. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 116(5), 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick EK, Man RE, Aung T, Ramulu P, Lamoureux EL, 2019. Beyond intraocular pressure: Optimizing patient-reported outcomes in glaucoma. Prog Retin Eye Res. 100801. [DOI] [PubMed] [Google Scholar]
- Fewou SN, Ramakrishnan H, Büssow H, Gieselmann V, Eckhardt M, 2007. Down-regulation of polysialic acid is required for efficient myelin formation. J. Biol. Chem 282, 16700–16711. [DOI] [PubMed] [Google Scholar]
- Fields MA, Del Priore LV, Adelman RA, Rizzolo LJ, 2019. Interactions of the choroid, Bruch’s membrane, retinal pigment epithelium, and neurosensory retina collaborate to form the outer blood-retinal-barrier.. Prog Retin Eye Res. 100803. [DOI] [PubMed] [Google Scholar]
- Fields RD, 2008. White matter in learning, cognition and psychiatric disorders. Trends Neurosci. 31, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, 2015. A new mechanism of nervous system plasticity: activity-dependent myelination. Nat. Rev. Neurosci 16, 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiano AJ, Gadani SP, Kipnis J, 2015. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 1617, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Leibinger M, 2012. Promoting optic nerve regeneration. Prog Retin Eye Res. 31, 688–701 [DOI] [PubMed] [Google Scholar]
- Flammer J, Orgül S, Costa VP, Orzalesi N, Krieglstein GK, Serra LM, Renard JP, Stefánsson E, 2002. The impact of ocular blood flow in glaucoma. Prog Retin Eye Res. 21(4), 359–93. [DOI] [PubMed] [Google Scholar]
- Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U, 2002. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci 22, RC216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade JM, Barde YA, 1998. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron. 20(1), 35–41. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Bayley SA, Milner R, Ffrench-Constant C, Blakemore WF, 1995. Differentiation of the O-2A progenitor cell line CG-4 into oligodendrocytes and astrocytes following transplantation into glia-deficient areas of CNS white matter. Glia. 13, 39–44. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Goldman SA, 2015. Glia Disease and Repair-Remyelination. Cold Spring Harb. Perspect. Biol 7, a020594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR, Rowitch DH, 2013. Evolving concepts of gliogenesis: a look way back and ahead to the next 25 years. Neuron. 80, 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchshofer R, Birke M, Welge-Lussen U, Kook D, Lutjen-Drecoll E, 2005. Transforming growth factor-beta 2 modulated extracellular matrix component expression in cultured human optic nerve head astrocytes. Investig. Ophthalmol. Vis. Sci, 46, 568–578. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Imagawa T, Uehara M, 2000. Comparative study of the lamina cribrosa and the pial septa in the vertebrate optic nerve and their relationship to the myelinated axons. Tissue Cell. 32, 293–301. [DOI] [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, Nave KA, 2012. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 485, 517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Shang P, Yazdankhah M, Bhutto I, Hose S, Montezuma SR, Luo T, Chattopadhyay S, Qian J, Lutty GA, Ferrington DA, Zigler JS Jr., Sinha D, 2017. Activating the AKT2-nuclear factor-κB-lipocalin-2 axis elicits an inflammatory response in age-related macular degeneration. J. Pathol 241, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaume C, Leybaert L, Naus CC, Sáez JC, 2013. Connexin and pannexin hemichannels in brain glial cells: properties, pharmacology, and roles. Front. Pharmacol 4, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, Barres BA, Woo PJ, Vogel H, Monje M, 2014. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. 344, 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giffard RG, Swanson RA, 2005. Ischemia-induced programmed cell death in astrocytes. Glia 50, 299–306. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Lim S, Hoeffel G, Low D, Huber T, 2013. Origin and differentiation of microglia. Front. Cell Neurosci 7, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G, Kavanagh TJ, Costa LG, 2009. Mouse cerebellar astrocytes protect cerebellar granule neurons against toxicity of the polybrominated diphenylether (PBDE) mixture DE-71. Neurotoxicology. 30, 326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givogri MI, Costa RM, Schonmann V, Silva AJ, Campagnoni AT, Bongarzone ER, 2002. Central nervous system myelination in mice with deficient expression of Notch1 receptor. J. Neurosci. Res 67, 309–320. [DOI] [PubMed] [Google Scholar]
- Glenn TC, Martin NA, Horning MA, McArthur DL, Hovda DA, Vespa P, Brooks GA, 2015. Lactate: brain fuel in human traumatic brain injury: a comparison with normal healthy control subjects. J. Neurotrauma 32, 820–32.Trevisi [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Wieser GL, Pieper A, Spitzer S, Weege B, Yan K, Edgar JM, Yagensky O, Wichert SP, Agarwal A, Karram K, Renier N, Tessier-Lavigne M, Rossner MJ, Káradóttir RT, Nave KA, 2017. A neuronal PI (3,4,5) P3-dependent program of oligodendrocyte precursor recruitment and myelination. Nat. Neurosci 20, 10–15. [DOI] [PubMed] [Google Scholar]
- Hamilton N, Hubbard PS, Butt AM, 2009. Effects of glutamate receptor activation on NG2-glia in the rat ON. J. Anat 214, 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton N, Vayro S, Wigley R, Butt AM, 2010. Axons and astrocytes release ATP and glutamate to evoke calcium signals in NG2-glia. Glia. 58, 66–79. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H, 2007. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci 10, 1387–94. [DOI] [PubMed] [Google Scholar]
- Harboe M, Torvund-Jensen J, Kjaer-Sorensen K, Laursen SL, 2018. Ephrin-A1-EphA4 signaling negatively regulates myelination in the central nervous system. Glia. 66, 934–950. [DOI] [PubMed] [Google Scholar]
- Haroon F, Drögemüller K, Händel U, Brunn A, Reinhold D, Nishanth G, Mueller W, Trautwein C, Ernst M, Deckert M, Schlüter D, 2011. Gp130-dependent astrocytic survival is critical for the control of autoimmune central nervous system inflammation. J. Immunol 186, 6521–31. [DOI] [PubMed] [Google Scholar]
- Hartenstein V, Giangrande A, 2018. Connecting the nervous and the immune systems in evolution. Commun. Biol 1, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartline DK, 2011. The evolutionary origins of glia. Glia. 59, 1215–1236 [DOI] [PubMed] [Google Scholar]
- Hausman RE, 2007. Ocular extracellular matrices in development. Prog Retin Eye Res. 26(2),162–88. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH, 2016. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 535(7613), 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Prasanna G, Yorio T, 2007. Endothelin-1-mediated signaling in the expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in astrocytes. Invest Ophthalmol Vis Sci. 8, 3737–45. [DOI] [PubMed] [Google Scholar]
- Hernandez MR, 1992. Ultrastructural immunocytochemical analysis of elastin in the human lamina cribrosa. Changes in elastic fibers in primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci 33, 2891–2903. [PubMed] [Google Scholar]
- Hernandez MR, 2000. The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Prog Retin Eye Res. 19, 297–321. [DOI] [PubMed] [Google Scholar]
- Hernandez MR, Yang J, Ye H, 1994. Activation of elastin mRNA expression in human ON heads with primary open-angle glaucoma. J. Glaucoma 3, 214–225. [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, and Sofroniew MV, 2008. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci 28, 7231–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Filiano AJ, Smith A, Yogev N, and Kipnis J, 2017. Myeloid cells in the central nervous system. Immunity 46, 943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess MW, Kirschning E, Pfaller K, Debbage PL, Hohenberg H, Klima G, 1998. 5000-year-old myelin: uniquely intact in molecular configuration and fine structure. Curr. Biol 8, 512–3. [DOI] [PubMed] [Google Scholar]
- Heuss ND, Pierson MJ, Roehrich H, McPherson SW, Gram AL, Li L, Gregerson DS, 2018. Optic nerve as a source of activated retinal microglia post-injury. Acta Neuropathol Commun 6(1): 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuss ND, Pierson MJ, Roehrich H, McPherson SW, Gram AL, Li L, Gregerson DS, 2018. ON as a source of activated retinal microglia post-injury. Acta Neuropathol. Commun 6, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RA, Patel KD, Goncalves CM, Grutzendler J, Nishiyama A, 2014. Modulation of oligodendrocyte generation during a critical temporal window after NG2 cell division. Nat. Neurosci 17, 1518–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilla AM, Diekmann H, Fischer D, 2017. Microglia Are Irrelevant for Neuronal Degeneration and Axon Regeneration after Acute Injury. J. Neurosci 37, 6113–6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines JH, Ravanelli AM, Schwindt R, Scott EK, Appel B, 2015. Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci 18, 683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirahara Y, Wakabayashi T, Mori T, Koike T, Yao I, Tsuda M, Honke K, Gotoh H, Ono K, Yamada H, 2017. Sulfatide species with various fatty acid chains in oligodendrocytes at different developmental stages determined by imaging mass spectrometry. J. Neurochem 140, 435–450. [DOI] [PubMed] [Google Scholar]
- Hirrlinger J, Nave KA, 2014. Adapting brain metabolism to myelination and long-range signal transduction. Glia. 62, 1749–1761. [DOI] [PubMed] [Google Scholar]
- Hogan MJ, Zimmerman LE, 1962. Ophthalmic pathology. An atlas and textbook, second ed. W.B. Saunders Company, Philadelphia. [Google Scholar]
- Holländer H, Makarov F, Stefani FH, Stone J, 1995. Evidence of constriction of optic nerve axons at the lamina cribrosa in the normotensive eye in humans and other mammals. Ophthalmic Res. 27, 296–309. [DOI] [PubMed] [Google Scholar]
- Holm TH, Draeby D, Owens T, 2012. Microglia are required for astroglial Toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia. 60(4), 630–8. [DOI] [PubMed] [Google Scholar]
- Horie S, Robbie SJ, Liu J, Wu WK, Ali RR, Bainbridge JW, Nicholson LB, Mochizuki M, Dick AD, Copland DA, 2013. CD200R signaling inhibits pro-angiogenic gene expression by macrophages and suppresses choroidal neovascularization. Sci Rep 3: 3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW, 2007. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol 179(7): 1523–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SWM, 2007. [DOI] [PMC free article] [PubMed]
- Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y, 2013. A critical role of astrocyte-mediated nuclear factor-kB-dependent inflammation in Huntington’s disease. Hum. Mol. Genet 22, 1826–1842. [DOI] [PubMed] [Google Scholar]
- Huang L, Xu W, Xu G, 2013. Transplantation of CX3CL1-expressing mesenchymal stem cells provides neuroprotective and immunomodulatory effects in a rat model of retinal degeneration. Ocul Immunol Inflamm 21(4): 276–285. [DOI] [PubMed] [Google Scholar]
- Huang P, Xu Y, Wei R, Li H, Tang Y, Liu J, Zhang SS, Zhang C, 2011. Efficacy of tetrandrine on lowering intraocular pressure in animal model with ocular hypertension. J. Glaucoma 20,183–188. [DOI] [PubMed] [Google Scholar]
- Huang W, Bai X, Stopper L, Catalin B, Cartarozzi LP, Scheller A, Kirchhoff F, 2018. During Development NG2 Glial Cells of the Spinal Cord are Restricted to the Oligodendrocyte Lineage, but Generate Astrocytes upon Acute Injury. Neuroscience. 385, 154–165. [DOI] [PubMed] [Google Scholar]
- Hughes EG, Orthmann-Murphy JL, Langseth AJ, Bergles DE, 2018. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci 21, 696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husemann J, Silverstein SC, 2001. Expression of scavenger receptor class B, type I, by astrocytes and vascular smooth muscle cells in normal adult mouse and human brain and in Alzheimer’s disease brain. Am. J. Pathol 158, 825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, Pasolli HA, Xu CS, Pang S, Matthies D, Hess HF, Lippincott-Schwartz J, Liu Z, 2019. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell. 177(6):1522–1535. [DOI] [PubMed] [Google Scholar]
- Ito YA, Di Polo A, 2017. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion. 36, 186–192. [DOI] [PubMed] [Google Scholar]
- Jahn O, Tenzer S, Werner HB, 2009. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol. 40(1), 55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon H, Kim JH, Kim JH, Lee WH, Lee MS, Suk K, 2012. Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J Neuroinflammation 9, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha MK, Jo M, Kim JH, Suk K, 2019. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist. 25(3), 227–240. [DOI] [PubMed] [Google Scholar]
- Jo M, Kim JH, Song GJ, Seo M, Hwang EM, Suk K, 2017. Astrocytic orosomucoid-2 modulates microglial activation and neuroinflammation. J Neurosci. 37(11), 2878–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas JB, Schmidt AM, Müller-Bergh JA, Schlötzer-Schrehardt UM, Naumann GO, 1992. Human ON fiber count and optic disc size. Invest. Ophthalmol. Vis. Sci 33, 2012–8. [PubMed] [Google Scholar]
- Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GW 2nd, Mochly-Rosen D, 2019. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci. 22(10), 1635–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyal JS, Gantner ML, Smith LEH, 2018. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Prog Retin Eye Res. 64, 131–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justicia C, Gabriel C, Planas AM, 2000. Activation of the JAK/STAT pathway following transient focal cerebral ischemia: signaling through Jak1 and Stat3 in astrocytes. Glia. 30(3), 253–270. [DOI] [PubMed] [Google Scholar]
- Kajigaya H, Tanaka KF, Hayashi A, Suzuki A, Ishibashi T, Ikenaka K, Baba H, 2011. Increased numbers of oligodendrocyte lineage cells in the ONs of cerebroside sulfotransferase knockout mice. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci 87, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajihara H, Tsutsumi E, Kinoshita A, Nakano J, Takagi K, Takeo S, 2001. Activated astrocytes with glycogen accumulation in ischemic penumbra during the early stage of brain infarction: immunohistochemical and electron microscopic studies. Brain Res. 909, 92–101. [DOI] [PubMed] [Google Scholar]
- Kale N, 2016. Optic neuritis as an early sign of multiple sclerosis. Eye Brain. 8, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE, 2010. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 68, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T, 2015. Retinal microglia: just bystander or target for therapy? Prog Retin Eye Res. 45:30–57. [DOI] [PubMed] [Google Scholar]
- Karlstetter M, Ebert S, Langmann T, 2010. Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology. 215, 685–691. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ, 1996. Synaptic activity and the construction of cortical circuits. Science. 274(5290), 1133–1138. [DOI] [PubMed] [Google Scholar]
- Kernt M, Liegl RG, Rueping J, Neubauer AS, Haritoglou C, Lackerbauer CA, Eibl KH, Ulbig MW, Kampik A, 2010. Sorafenib protects human ON head astrocytes from light-induced overexpression of vascular endothelial growth factor, platelet-derived growth factor, and placenta growth factor. Growth Factors. 28, 211–20. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Deneen B, 2019. The Emerging Nature of Astrocyte Diversity. Annu Rev Neurosci 42: 187–207. [DOI] [PubMed] [Google Scholar]
- Klein RS, Hunter CA, 2017. Protective and pathological immunity during CNS infections. Immunity. 46, 891–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingseisen A, Lyons DA, 2018. Axonal Regulation of Central Nervous System Myelination: Structure and Function. Neuroscientist. 24, 7–21. [DOI] [PubMed] [Google Scholar]
- Klistorner A, Arvind H, Garrick R, Yiannikas C, Paine M, Graham SL 2010. Remyelination of ON lesions: spatial and temporal factors. Mult. Scler 16, 786–795. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Vidal I, Pena JD, Hernandez MR, 1997. Expression of neural cell adhesion molecule (NCAM) characterizes a subpopulation of type 1 astrocytes in human optic nerve head. Glia. 20, 262–273. [DOI] [PubMed] [Google Scholar]
- Krencik R, van Asperen JV, Ullian EM, 2017. Human astrocytes are distinct contributors to the complexity of synaptic function. Brain Res. Bull 129, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M, Saya H, Suda T, 2009. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med. 206(5), 1089–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampron A, Larochelle A, Laflamme N, Préfontaine P, Plante MM, Sánchez MG, Yong VW, Stys PK, Tremblay MÈ, Rivest S, 2015. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. 212(4), 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Han JC, Kee C, 2017. A novel hypothesis for the pathogenesis of glaucomatous disc hemorrhage. Prog Retin Eye Res. 60, 20–43. [DOI] [PubMed] [Google Scholar]
- Lee EJ, Han JC, Park DY, Kee C, 2020. A neuroglia-based interpretation of glaucomatous neuroretinal rim thinning in the optic nerve head. Prog Retin Eye Res. e100840. [DOI] [PubMed] [Google Scholar]
- Lee S, Leach MK, Redmond SA, Chong SY, Mellon SH, Tuck SJ, Feng ZQ, Corey JM, Chan JR, 2012. A culture system to study oligodendrocyte myelination processes using engineered nanofibers. Nat. Methods 9, 917–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin MH, Bennett JL, Verkman AS, 2013. Optic neuritis in neuromyelitis optica. Prog Retin Eye Res. 36, 159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Li J, Zheng J, Qin S, 2019. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 10(3), 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yang C, Lu J, Huang P, Barnstable CJ, Zhang C, Lu J, Zhang S, 2014. Tetrandrine protects mouse retinal ganglion cells from ischemic injury. Drug Des. Devel. Ther 8, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Du XF, Liu CS, Wen ZL, Du JL, 2012. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell. 23(6), 1189–1202. [DOI] [PubMed] [Google Scholar]
- Lian H, Yang L, Cole A, Sun L, Chiang AC-A, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu HC, Zheng H, 2015. NFkB activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron. 85, 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA, 2017. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity. 46(6), 957–967. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS, 1996. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron. 17, 607–15. [DOI] [PubMed] [Google Scholar]
- Linneberg C, Harboe M, Laursen LS, 2015. Axo-Glia interaction preceding CNS myelination is regulated by bidirectional eph-ephrin signaling. ASN Neuro. 7, 1759091415602859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Neufeld AH, 2004. Activation of epidermal growth factor receptors directs astrocytes to organize in a network surrounding axons in the developing rat optic nerve. Dev Biol 273(22): 297–307. [DOI] [PubMed] [Google Scholar]
- Liu H, Leak RK, Hu X, 2016. Neurotransmitter receptors on microglia. Stroke Vasc Neurol 1(22): 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Dietz K, DeLoyht JM, Pedre X, Kelkar D, Kaur J, Vialou V, Lobo MK, Dietz DM, Nestler EJ, Dupree J, Casaccia P, 2012. Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nat. Neurosci 15, 1621–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, et al. , 2015. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 160, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Aguzzi A, 2020. NG2 glia are required for maintaining microglia homeostatic state. Glia 68(2):345–355. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chopp M, 2016. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. 144, 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada-Perez M, 2018. Glia: from ‘just glue’ to essential players in complex nervous systems: a comparative view from flies to mammals. J. Neurogenet 32, 78–91. [DOI] [PubMed] [Google Scholar]
- Lou G, Palikaras K, Lautrup S, Scheibye-Knudsen M, Tavernarakis N, Fang EF, 2019. Mitophagy and Neuroprotection. Trends Mol Med. S1471–4914(19)30176–5. [DOI] [PubMed] [Google Scholar]
- Lozano DC, Choe TE, Cepurna WO, Morrison JC, Johnson EC, 2019. Early Optic Nerve Head Glial Proliferation and Jak-Stat Pathway Activation in Chronic Experimental Glaucoma. Invest Ophthalmol Vis Sci. 60(4):921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas TJ, Miao H, Chen L, Riordan SM, Li W, Crabb AM, Wise A, Du P, Lin SM, Hernandez MR, 2008. Susceptibility to glaucoma: differential comparison of the astrocyte transcriptome from glaucomatous African American and Caucasian American donors. Genome Biol. 9, R111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Nair CE, Fernandes KA, Schlamp CL, Libby RT, Nickells RW, 2014. Tumor necrosis factor alpha has an early protective effect on retinal ganglion cells after optic nerve crush. J Neuroinflammation 11: 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie PJ, Cioffi GA, 2008. Vascular anatomy of the optic nerve head. Can J Ophthalmol 43(3): 308–312. [DOI] [PubMed] [Google Scholar]
- Madeira MH, Boia R, Santos PF, Ambrósio AF, Santiago AR, 2015. Contribution of microglia-mediated neuroinflammation to retinal degenerative diseases. Mediators Inflamm. 2015, 673090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes ME, Schlamp CL, Nickells RW, 2017. BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog Retin Eye Res. 57, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L, 2000. The astrocyte-mediated coupling between synaptic activity and energy metabolism operates through volume transmission. Prog Brain Res. 125, 229–240. [DOI] [PubMed] [Google Scholar]
- Makinodan M, Rosen KM, Ito S, Corfas G, 2012. A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science. 337, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammone T, Chidlow G, Casson RJ, Wood JPM, 2018. Expression and activation of mitogen-activated protein kinases in the ON head in a rat model of ocular hypertension. Mol. Cell Neurosci 88, 270–291. [DOI] [PubMed] [Google Scholar]
- Mason S, 2017. Lactate Shuttles in Neuroenergetics-Homeostasis, Allostasis and Beyond. Front Neurosci 11:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayoral SR, Etxeberria A, Shen YA, Chan JR, 2018. Initiation of CNS Myelination in the ON Is Dependent on Axon Caliber. Cell Rep. 25, 544–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, Richardson WD, 2014. Motor skill learning requires active central myelination. Science. 346, 318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, 2008. Origin and physiological roles of inflammation. Nature 454(7203): 428–435. [DOI] [PubMed] [Google Scholar]
- Meireles AM, Shen K, Zoupi L, Iyer H, Bouchard EL, Williams A, Talbot WS, 2018. The Lysosomal Transcription Factor TFEB Represses Myelination Downstream of the Rag-Ragulator Complex. Dev. Cell 47, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensch S, Baraban M, Almeida R, Czopka T, Ausborn J, Manira A, Lyons DA, 2015. Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci 18, 628–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messersmith DJ, Murtie JC, Le TQ, Frost EE, Armstrong RC, 2000. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J. Neurosci. Res 62, 241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, Richter N, Fan Z, Siemonsmeier G, Pivneva T, Jordan P, Steinhäuser C, Semtner M, Nolte C, Kettenmann H, 2018. Oligodendrocytes in the Mouse Corpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep. 22, 2383–2394. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Shen S, Barres BA, 1997. Astrocytes induce oligodendrocyte processes to align with and adhere to axons. Mol. Cell. Neurosci 14, 385–397. [DOI] [PubMed] [Google Scholar]
- Michalski JP, Kothary R, 2015. Oligodendrocytes in a Nutshell. Front Cell Neurosci. 9: 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RH, 2002. Regulation of oligodendrocyte development in the vertebrate CNS. Prog. Neurobiol 67, 451–67. [DOI] [PubMed] [Google Scholar]
- Modi KK, Sendtner M, Pahan K, 2013. Up-regulation of ciliary neurotrophic factor in astrocytes by aspirin: implications for remyelination in multiple sclerosis. J. Biol. Chem 288, 18533–18545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Ikeshima-Kataoka H, Kreft M, Vardjan N, Zorec R, Noda M, 2019. Metabolic Plasticity of Astrocytes and Aging of the Brain. Int J Mol Sci. 20(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BM, Lee Y, Rothstein JD, 2013. Oligodendroglia: metabolic supporters of axons. Trends Cell. Biol 23, 644–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Suresh SN, 2019. Neuron-Astrocyte Liaison to Maintain Lipid Metabolism of Brain. Trends Endocrinol Metab. 30(9),573–575. [DOI] [PubMed] [Google Scholar]
- Müller A, Hauk TG, Fischer D, 2007. Astrocyte-derived CNTF switches mature RGCs to a regenerative state following inflammatory stimulation. Brain. 130, 3308–3320. [DOI] [PubMed] [Google Scholar]
- Munemasa Y, Kitaoka Y, 2012. Molecular mechanisms of retinal ganglion cell degeneration in glaucoma and future prospects for cell body and axonal protection. Front Cell Neurosci 6: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemasa Y, Kitaoka Y, 2015. Autophagy in axonal degeneration in glaucomatous optic neuropathy. Prog Retin Eye Res. 47, 1–18. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Dudek FE, Rash JE, 2004. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res. Rev 47,191–215. [DOI] [PubMed] [Google Scholar]
- Nakamura-Ishizu A, Kurihara T, Okuno Y, Ozawa Y, Kishi K, Goda N, Tsubota K, Okano H, Suda T, Kubota Y, 2012. The formation of an angiogenic astrocyte template is regulated by the neuroretina in a HIF-1-dependent manner. Dev Biol 363(1): 106–114. [DOI] [PubMed] [Google Scholar]
- Namazi H, Kulish VV, 2013). A mathematical based calculation of a myelinated segment in axons. Comput Biol Med 43(6): 693–698. [DOI] [PubMed] [Google Scholar]
- Nave KA, 2010. Myelination and support of axonal integrity by glia. Nature 468, 244–252. [DOI] [PubMed] [Google Scholar]
- Nave KA, Trapp BD, 2008. Axon-glial signaling and the glial support of axon function. Annu. Rev. Neurosci 31, 535–561. [DOI] [PubMed] [Google Scholar]
- Nave KA, Werner HB, 2014. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol 30, 503–33. [DOI] [PubMed] [Google Scholar]
- Neufeld AH, 1999. Microglia in the ON head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch. Ophthalmol 117, 1050–6. [DOI] [PubMed] [Google Scholar]
- Neumann C, Yu A, Welge-Lussen U, Lutjen-Drecoll E, Birke M, 2008. The effect of TGF-2 on elastin, Type VI collagen, and components of the proteolytic degradation system in human optic nerve astrocytes. Investig. Ophthalmol. Vis. Sci, 49, 1464–1472. [DOI] [PubMed] [Google Scholar]
- Newman NJ, Biousse V, 2004. Hereditary optic neuropathies. Eye (Lond) 18(11): 1144–1160. [DOI] [PubMed] [Google Scholar]
- Nguyen HM, Blomster LV, Christophersen P, Wulff H, 2017. Potassium channel expression and function in microglia: Plasticity and possible species variations. Channels (Austin) 11(4): 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Boshans L, Goncalves CM, Wegrzyn J, Patel KD, 2016. Lineage, fate, and fate potential of NG2-glia. Brain Res. 1638, 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X, 2009. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat. Rev. Neurosci 10, 9–22. [DOI] [PubMed] [Google Scholar]
- Niu J, Li T, Yi C, Huang N, Koulakoff A, Weng C, Li C, Zhao CJ, Giaume C, Xiao L, 2016. Connexin-based channels contribute to metabolic pathways in the oligodendroglial lineage. J. Cell Sci 129, 1902–14. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, Xu Q, Wyatt JD, Pilcher W, Ojemann JG, Ransom BR, Goldman SA, Nedergaard M, 2009. Uniquely hominid features of adult human astrocytes. J. Neurosci, 29, 3276–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H, 2006. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med, 12, 829–834. [DOI] [PubMed] [Google Scholar]
- Ono K, Kagawa T, Tsumori T, Yokota S, Yasui Y, 2001. Morphological changes and cellular dynamics of oligodendrocyte lineage cells in the developing vertebrate central nervous system. Dev Neurosci. 23(4–5), 346–55. [DOI] [PubMed] [Google Scholar]
- Ono K, Yoshii K, Tominaga H, Gotoh H, Nomura T, Takebayashi H, Ikenaka K, 2017. Oligodendrocyte precursor cells in the mouse ON originate in the preoptic area. Brain Struct. Funct 222, 2241–2248. [DOI] [PubMed] [Google Scholar]
- Oyarzabal A, Bravo-Alonso I, Sánchez-Aragó M, Rejas MT, Merinero B, García-Cazorla A, Artuch R, Ugarte M, Rodríguez-Pombo P, 2016. mitochondrial response to the BCKDK-deficiency: Some clues to understand the positive dietary response in this form of autism. Biochim. Biophys. Acta 1862, 592–600. [DOI] [PubMed] [Google Scholar]
- Pajoohesh-Ganji A, Miller RH, 2019. Targeted Oligodendrocyte Apoptosis in Optic Nerve Leads to Persistent Demyelination. Neurochem Res. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan BX, Ross-Cisneros FN, Carelli V, Rue KS, Salomao SR, Moraes-Filho MN, Moraes MN, Berezovsky A, Belfort R, Sadun AA, 2012. Mathematically modeling the involvement of axons in Leber’s hereditary optic neuropathy. Invest Ophthalmol Vis Sci 53(12): 7608–7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Liu B, Chen T, Li H, Hu X, Gao J, Zhu Y, Zhu Q, Qiang B, Yuan J, Peng X, Qiu M, 2008. Disruption of Nectin-like 1 cell adhesion molecule leads to delayed axonal myelination in the CNS. J. Neurosci 28, 12815–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolisi R, Boda E, 2018. NG2 Glia: Novel Roles beyond Re-/Myelination. Neuroglia 1, 151–175. [Google Scholar]
- Paschalis EI, Lei F, Zhou C, Chen XN, Kapoulea V, Hui PC, Dana R, Chodosh J, Vavvas D, Dohlman CH, 2019. Microglia Regulate Neuroglia Remodeling in Various Ocular and Retinal Injuries. J. Immunol 202, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JR, Williams JL, Muccigrosso MM, Liu L, Sun T, Rubin JB, Klein RS, 2012. Astrocyte TNFR2 is required for CXCL12-mediated regulation of oligodendrocyte progenitor proliferation and differentiation within the adult CNS. Acta Neuropathol. 124(6), 847–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peferoen L, Kipp M, van der Valk P, van Noort JM, Amor S, 2014. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 141(3): 302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, Stella N, Magistretti PJ, 1998. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci 20, 291–9. [DOI] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A, 2009. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431. [DOI] [PubMed] [Google Scholar]
- Peterson WM, Wang Q, Tzekova R, Wiegand SJ, 2000. Ciliary neurotrophic factor and stress stimuli activate the Jak-STAT pathway in retinal neurons and glia. J Neurosci. 20(11), 4081–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre K, Pellerin L, 2005. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J. Neurochem 94, 1–14. [DOI] [PubMed] [Google Scholar]
- Prasanna G, Krishnamoorthy R, Clark AF, Wordinger RJ, Yorio T 2002. Human optic nerve head astrocytes as a target for endothelin-1. Invest Ophthalmol Vis Sci. 8, 2704–13. [PubMed] [Google Scholar]
- Prasanna G, Krishnamoorthy R, Yorio T 2011. Endothelin, astrocytes and glaucoma. Exp Eye Res. 2,170–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchkov D, Haucke V, 2013. Greasing the synaptic vesicle cycle by membrane lipids. Trends Cell Biol. 23, 493–503. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD, 1993. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 262, 695–700. [DOI] [PubMed] [Google Scholar]
- Rafiki A, Boulland JL, Halestrap AP, Ottersen OP, Bergersen L, 2003. Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience. 122, 677–88. [DOI] [PubMed] [Google Scholar]
- Rani L, Mondal AC, 2019. Emerging concepts of mitochondrial dysfunction in Parkinson’s disease progression: Pathogenic and therapeutic implications. Mitochondrion. 50, 25–34. [DOI] [PubMed] [Google Scholar]
- Ransom B, Behar T, Nedergaard M, 2003. New roles for astrocytes (stars at last). Trends Neurosci. 26, 520–2. [DOI] [PubMed] [Google Scholar]
- Rashid K, Akhtar-Schaefer I, Langmann T, 2019. Microglia in Retinal Degeneration. Front Immunol 10: 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea P, 2014. Clinical Anatomy of the Cranial Nerves, Academic press, Cambridge, Massachusetts. [Google Scholar]
- Refolo V, Stefanova N, 2019. Neuroinflammation and Glial Phenotypic Changes in Alpha-Synucleinopathies. Front Cell Neurosci. 13, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich LR, Harris W, Brown AM, 2019. The Role of Brain Glycogen in Supporting Physiological Function. Front Neurosci. 13, 1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridet JL, Malhotra SK, Privat A, Gage FH, 1997. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 20, 570–577. [DOI] [PubMed] [Google Scholar]
- Rinholm JE, Hamilton NB, Kessaris N, Richardson WD, Bergersen LH, Attwell D, 2011. Regulation of oligodendrocyte development and myelination by glucose and lactate. J. Neurosci 31, 538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risner ML, Pasini S, Cooper ML, Lambert WS, Calkins DJ, 2018. Axogenic mechanism enhances retinal ganglion cell excitability during early progression in glaucoma. Proc Natl Acad Sci U S A. 115(10), E2393–E2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh M, Zhang Y, Murakami Y, Thanos A, Lee SC, Vavvas DG, Benowitz LI, Miller JW, 2012. Etanercept, a widely used inhibitor of tumor necrosis factor-α (TNF-α), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS One. 7, e40065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C, 2008. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 322, 1551–1555. [DOI] [PubMed] [Google Scholar]
- Rudzinski MN, Chen L, Hernandez MR, 2008. Antiangiogenic characteristics of astrocytes from optic nerve heads with primary open-angle glaucoma. Arch Ophthalmol 126(5): 679–685. [DOI] [PubMed] [Google Scholar]
- Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, Möbius W, Goetze B, Jahn HM, Huang W, Steffens H, Schomburg ED, Pérez-Samartín A, Pérez-Cerdá F, Bakhtiari D, Matute C, Löwel S, Griesinger C, Hirrlinger J, Kirchhoff F, Nave KA, 2016. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron. 91, 119–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A, 2009. A gene network regulating lysosomal biogenesis and function. Science. 325, 473–7. [DOI] [PubMed] [Google Scholar]
- Schneider M, Fuchshofer R, 2016. The role of astrocytes in optic nerve head fibrosis in glaucoma. Exp Eye Res. 142, 49–55. [DOI] [PubMed] [Google Scholar]
- Schwab JM, Postler E, Nguyen TD, Mittelbronn M, Meyermann R, Schluesener HJ, 2000. Connective tissue growth factor is expressed by a subset of reactive astrocytes in human cerebral infarction. Neuropathol. Appl. Neuro-biol 26, 434–440. [DOI] [PubMed] [Google Scholar]
- Selvam S, Kumar T, Fruttiger M, 2018. Retinal vasculature development in health and disease. Prog Retin Eye Res. 63, 1–19. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, Muzikansky A, Gómez-Isla T, Growdon JH, Betensky RA, Frosch MP, Hyman BT, 2013. Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J Neuropathol Exp Neurol. 72(6), 462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwanski DR, Jukkola P, Nishiyama A, 2017. Heterogeneity of astrocyte and NG2 cell insertion at the node of ranvier. J. Comp. Neurol 525, 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A, 2011. TFEB links autophagy to lysosomal biogenesis. Science. 332, 1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, Hart RP, Schwartz M, 2005. Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem 92(5): 997–1009. [DOI] [PubMed] [Google Scholar]
- Shinozaki Y, Shibata K, Yoshida K, Shigetomi E, Gachet C, Ikenaka K, Tanaka KF, Koizumi S, 2017. Transformation of Astrocytes to a Neuroprotective Phenotype by Microglia via P2Y1 Receptor Downregulation. Cell Rep. 19, 1151–1164. [DOI] [PubMed] [Google Scholar]
- Simons M, Nave KA 2015. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol 8, a020479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skripuletz T, Hackstette D, Bauer K, Gudi V, Pul R, Voss E, Berger K, Kipp M, Baumgärtner W, Stangel M, 2013. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain. 136(Pt 1), 147–67. [DOI] [PubMed] [Google Scholar]
- Smith SB, Wang J, Cui X, Mysona BA, Zhao J, Bollinger KE, 2018. Sigma 1 receptor: A novel therapeutic target in retinal disease. Prog Retin Eye Res. 67, 130–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV, 2010. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, 2015. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci, 6(5), 249–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallcup WB, Beasley L 1987. Bipotential glial precursor cells of the ON express the NG2 proteoglycan. J. Neurosci 7, 2737–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiefel KM, Torben-Nielsen B, Coggan JS, 2013. Proposed evolutionary changes in the role of myelin. Front Neurosci 7:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturrock RR, 1984. Microglia in the human embryonic ON. J. Anat 139, 81–91. [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Moriya S, Oku H, Azuma I, 1995. Association of endothelin-1 with normal tension glaucoma: clinical and fundamental studies. Surv Ophthalmol. 39(1), S49–56. [DOI] [PubMed] [Google Scholar]
- Sun D, Jakobs TC, 2012. Structural remodeling of astrocytes in the injured CNS. Neuroscientist 18(6): 567–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Moore S, Jakobs TC, 2017. Optic nerve astrocyte reactivity protects function in experimental glaucoma and other nerve injuries. J Exp Med. 214(5), 1411–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Lye-Barthel M, Masland RH, Jakobs TC, 2009. The morphology and spatial arrangement of astrocytes in the optic nerve head of the mouse. J. Comp. Neurol 516, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LO, Mulinyawe SB, Collins HY, Ibrahim A, Li Q, Simon DJ, Tessier-Lavigne M, Barres BA, 2018. Spatiotemporal Control of CNS Myelination by Oligodendrocyte Programmed Cell Death through the TFEB-PUMA Axis. Cell. 175, 1811–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanuma N, Sakuma H, Sasaki A, Matsumoto Y, 2006. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 112(2), 195–204. [DOI] [PubMed] [Google Scholar]
- Tezel G, Wax MB, 2000. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J. Neurosci 20, 8693–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos S, Böhm MR, Meyer zu Hörste M, Prokosch-Willing V, Hennig M, Bauer D, Heiligenhaus A, 2014. Role of crystallins in ocular neuroprotection and axonal regeneration. Prog Retin Eye Res. 42:145–61. [DOI] [PubMed] [Google Scholar]
- Tracey TJ, Steyn FJ, Wolvetang EJ, Ngo ST, 2018. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front Mol Neurosci. January 23, 11:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay MÈ, Lowery RL, Majewska AK, 2010. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8(11):e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisiol A, Saab AS, Winkler U, Marx G, Imamura H, Möbius W, Kusch K, Nave KA, Hirrlinger J, 2017. Monitoring ATP dynamics in electrically active white matter tracts. Elife. 6, e24241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi P, Rodriguez-Muela N, Klim JR, de Boer AS, Agrawal S, Sandoe J, Lopes CS, Ogliari KS, Williams LA, Shear M, Rubin LL, Eggan K, Zhou Q, 2017. Reactive Astrocytes Promote ALS-like Degeneration and Intracellular Protein Aggregation in Human Motor Neurons by Disrupting Autophagy through TGF-β1. Stem Cell Reports. 9(2), 667–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivli A, Koliarakis I, Terzidou C, .Goulielmos GN, Siganos CS, Spandidos DA, Dalianis G, Detorakis ET, 2019. Normal-tension glaucoma: Pathogenesis and genetics. Exp Ther Med. 17(1), 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter J, Karram K, Nishiyama A, 2010. NG2 cells: Properties, progeny and origin. Brain Res. Rev 63, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Levine JM, Miller RH, Trapp BD, 1999. Rat ON oligodendrocytes develop in the absence of viable retinal ganglion cell axons. J. Cell Biol 146, 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA, Nguyen PT, Nakao-Inoue H, Dorman LC, Akil O, Joshita S, Barres BA, Paz JT, Molofsky AB, Molofsky AV, 2018. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 359, 1269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valapala M, Hose S, Gongora C, Dong L, Wawrousek EF, Zigler JS Jr., Sinha D, 2013. Impaired endolysosomal function disrupts Notch signaling in ON astrocytes. Nat. Commun 4, 1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valapala M, Wilson C, Hose S, Bhutto IA, Grebe R, Dong A, Greenbaum S, Gu L, Sengupta S, Cano M, Hackett S, Xu G, Lutty GA, Dong L, Sergeev Y, Handa JT, Campochiaro P, Wawrousek E, Zigler JS Jr., Sinha D, 2014a. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/βA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy. 10, 480–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valny M, Honsa P, Kriska J, Anderova M, 2017. Multipotency and therapeutic potential of NG2 cells. Biochem. Pharmacol 141, 42–55. [DOI] [PubMed] [Google Scholar]
- van Tilborg E, de Theije CGM, van Hal M, Wagenaar N, de Vries LS, Benders MJ, Rowitch DH, Nijboer CH, 2018. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia. 66, 221–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC, 2016. Glia-neuron interactions in the mammalian retina. Prog Retin Eye Res. 51, 1–40. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Nedergaard M, 2016. The homeostatic astroglia emerges from evolutionary specialization of neural cells. Philos. Trans. R. Soc. Lond. B. Biol. Sci 371, 20150428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veroni C, Gabriele L, Canini I, Castiello L, Coccia E, Remoli ME, Columba-Cabezas S, Aricò E, Aloisi F, Agresti C, 2010. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell Neurosci 45, 234–244. [DOI] [PubMed] [Google Scholar]
- Viganò F, Dimou L, 2016. The heterogeneous nature of NG2-glia. Brain Res. 1638, 129–137. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J, 2005. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci, 6, 626–640. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS, Bahney J, Herculano-Houzel S, 2016. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J Comp Neurol 524(18): 3865–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J, 2009. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 29(13), 3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Chen SD, Zhang XL, Jonas JB, 2016. Retinal Microglia in Glaucoma. J. Glaucoma 25, 459–65. [DOI] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, Barres BA, 1998. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 21, 63–75. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cheng X, He Q, Zheng Y, Kim DH, Whittemore SR, Cao QL, 2011. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J. Neurosci 31, 6053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Fukuda Y, 2002. Survival and axonal regeneration of retinal ganglion cells in adult cats. Prog Retin Eye Res. 21(6), 529–53. [DOI] [PubMed] [Google Scholar]
- Wei X, Cho KS, Thee EF, Jager MJ, Chen DF, 2019. Neuroinflammation and microglia in glaucoma: time for a paradigm shift. J Neurosci Res 97(1): 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson JM, Lyons DA, 2018. Myelin Dynamics Throughout Life: An Ever-Changing Landscape? Front Cell Neurosci. 12: 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolburg H, 1981. Myelination and remyelination in the regenerating visual system of the goldfish. Exp Brain Res. 43(2), 199–206. [DOI] [PubMed] [Google Scholar]
- Wolf Y, Yona S, Kim KW, Jung S, 2013. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci 7: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B, 2011. In vivo evidence for lactate as a neuronal energy source. J. Neurosci 31, 7477–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Chen M, Forrester JV, 2009. Para-inflammation in the aging retina. Prog Retin Eye Res. 28(5), 348–68. [DOI] [PubMed] [Google Scholar]
- Yamauchi K, Osuka K, Takayasu M, Usuda N, Nakazawa A, Nakahara N, Yoshida M, Aoshima C, Hara M, Yoshida J, 2006. Activation of JAK/STAT signaling in neurons following spinal cord injury in mice. J Neurochem. 96(4), 1060–1070. [DOI] [PubMed] [Google Scholar]
- Yang QK, Xiong JX, Yao ZX, 2013. Neuron-NG2 cell synapses: novel functions for regulating NG2 cell proliferation and differentiation. Biomed. Res. Int 402843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanoff M, Sassani J, 2015. Ocular Pathology, seventh ed. Cambridge, Massachusetts. [Google Scholar]
- Yazdankhah M, Farioli-Vecchioli S, Tonchev AB, Stoykova A, Cecconi F, 2014. The autophagy regulators Ambra1 and Beclin 1 are required for adult neurogenesis in the brain subventricular zone. Cell Death Dis. 5:e1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdankhah M, Shang P, Ghosh S, Bhutto IA, Stepicheva N, Grebe R, Hose S, Weiss J, Luo T, Mishra S, Riazuddin SA, Ghosh A, Handa JT, Lutty GA, Zigler JS Jr, Sinha D, 2019. Modulating EGFR-MTORC1-autophagy as a potential therapy for persistent fetal vasculature (PFV) disease. Autophagy. 1, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Hernandez MR, 1995. Heterogeneity of astrocytes in human optic nerve head. [DOI] [PubMed] [Google Scholar]
- Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, Richardson WD, 2013. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77, 873–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ, Balaratnasingam C, Morgan WH, Yu PK, Su EN, 2013. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog Retin Eye Res. 36, 217–46. [DOI] [PubMed] [Google Scholar]
- Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, Kim S, Oh N, Kim NA, Lee S, Brahmachari S, Mao X, Lee JH, Kumar M, An D, Kang SU, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL, Lee S, Dawson TM, Ko HS, 2018. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 24(7), 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Griffiths PG, Chinnerya PF, 2011. Mitochondrial optic neuropathies - Disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 30(2–2): 81–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, Fariss RN, Wong WT, 2016. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 64(9): 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, and Barres BA, 2012. Genomic analysis of reactive astrogliosis. J. Neurosci 32, 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambonin JL, Zhao C, Ohno N, Campbell GR, Engeham S, Ziabreva I, Schwarz N, Lee SE, Frischer JM, Turnbull DM, Trapp BD, Lassmann H, Franklin RJ, Mahad DJ, 2011. Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain 134(Pt 7): 1901–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeis T, Enz L, Schaeren-Wiemers N, 2016. The immunomodulatory oligodendrocyte. Brain Res 1641(Pt A): 139–148. [DOI] [PubMed] [Google Scholar]
- Zeng HL, Shi JM, 2018. The role of microglia in the progression of glaucomatous neurodegeneration- a review. Int. J. Ophthalmol 11, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Li W, Wang W, Zhang SS, Huang P, Zhang C, 2013. Expression and activation of STAT3 in the astrocytes of optic nerve in a rat model of transient intraocular hypertension. PLoS One. 8(1), e55683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SZ, Wang QQ, Yang QQ, Gu HY, Yin YQ, Li YD, Hou JC, Chen R, Sun QQ, Sun YF et al. , 2019. NG2 glia regulate brain innate immunity via TGF-β2/TGFBR2 axis. BMC Med 17(1):204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JW, Raha-Chowdhury R, Fawcett JW, Watts C, 2009. Astrocytes and oligodendrocytes can be generated from NG2+ progenitors after acute brain injury: intracellular localization of oligodendrocyte transcription factor 2 is associated with their fate choice. Eur. J. Neurosci 29, 1853–1869. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Wang J, Luo X, 2013. Integrins in trabecular meshwork and optic nerve head: possible association with the pathogenesis of glaucoma. Biomed Res Int. 2013, 202905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, Nishiyama A, 2008. NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development. 135, 145–157. [DOI] [PubMed] [Google Scholar]
- Zhu YB, Gao W, Zhang Y, Jia F, Zhang HL, Liu YZ, et al. , 2016. Astrocyte-derived phosphatidic acid promotes dendritic branching. Sci. Rep 6:21096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigler JS Jr., Hodgkinson CA, Wright M, Klise A, Sundin O, Broman KW, Hejtmancik F, Huang H, Patek B, Sergeev Y, Hose S, Brayton C, Xaiodong J, Vasquez D, Maragakis N, Mori S, Goldman D, Hoke A, Sinha D, 2016. A Spontaneous Missense Mutation in Branched Chain Keto Acid Dehydrogenase Kinase in the Rat Affects Both the Central and Peripheral Nervous Systems. PLoS One. 11, e0160447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigler JS Jr., Sinha D, 2015. βA3/A1-crystallin: more than a lens protein. Prog Retin Eye Res. 44, 62–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zode GS, Sethi A, Brun-Zinkernagel AM, Chang IF, Clark AF, Wordinger RJ, 2011. Transforming growth factor-beta2 increases extracellular matrix proteins in optic nerve head cells via activation of the Smad signaling pathway. Mol. Vis, 17, 1745–1758. [PMC free article] [PubMed] [Google Scholar]
- Zuchero JB, Barres BA, 2013. Intrinsic and extrinsic control of oligodendrocyte development. Curr. Opin. Neurobiol 23, 914–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchero JB, Barres BA, 2015. Glia in mammalian development and disease. Development. 142(22), 3805–380. [DOI] [PMC free article] [PubMed] [Google Scholar]