

Abstract
The plant pathogen Agrobacterium tumefaciens causes crown gall disease and is a widely used tool for generating transgenic plants owing to its virulence. The pathogenic process involves a shift from an independent to a living form within a host plant. However, comprehensive analyses of metabolites, genes, and reactions contributing to this complex process are lacking. To gain new insights about the pathogenicity from the viewpoints of physiology and cellular metabolism, a genome‐scale metabolic model (GSMM) was reconstructed for A. tumefaciens. The model, referred to as iNX1344, contained 1,344 genes, 1,441 reactions, and 1,106 metabolites. It was validated by analyses of in silico cell growth on 39 unique carbon or nitrogen sources and the flux distribution of carbon metabolism. A. tumefaciens metabolic characteristics under three ecological niches were modelled. A high capacity to access and metabolize nutrients is more important for rhizosphere colonization than in the soil, and substantial metabolic changes were detected during the shift from the rhizosphere to tumour environments. Furthermore, by integrating transcriptome data for tumour conditions, significant alterations in central metabolic pathways and secondary metabolite metabolism were identified. Overall, the GSMM and constraint‐based analysis could decode the physiological and metabolic features of A. tumefaciens as well as interspecific interactions with hosts, thereby improving our understanding of host adaptation and infection mechanisms.
Keywords: Agrobacterium tumefaciens, crown gall, genome‐scale metabolic model, plant pathogen, systems biology
The genome‐scale metabolic model for Agrobacterium tumefaciens exhibited its physiological features, and metabolic response to its living condition change, and decoded its infection process at cellular metabolic level.
1. INTRODUCTION
Agrobacterium tumefaciens is a gram‐negative α‐proteobacterium of the Rhizobiaceae family. Taxa that belong to Rhizobiaceae can live independently in the soil or rhizosphere environment or coexist with plant nodules. Unlike Rhizobium in the same phylogenetic family, A. tumefaciens causes crown gall disease in plants. A. tumefaciens can transfer a part of the tumour‐inducing (Ti) plasmid into plant cells, with the subsequent integration of segments into the host genome. A. tumefaciens is a highly efficient tool in nature for generating transgenic dicotyledonous plants and, in laboratory conditions, some monocots (Nester, 2014). Within the past two decades, A. tumefaciens has become a model organism for studies of many biological processes, such as pathogen–host interactions (McCullen & Binns, 2006), nucleoprotein complex transport (Guo et al., 2019), the type IV secretion system (Guo et al., 2007), and quorum sensing (Lang & Faure, 2014). A. tumefaciens research has focused on the mechanism underlying transfer DNA (T‐DNA) transfer and virulence induction.
Genetic transformation by A. tumefaciens is related to its pathogenicity and involves a shift from free‐living to plant‐associated forms. Soilborne A. tumefaciens is widely found in natural habitats, such as the bulk soil and rhizosphere (Meyer et al., 2019), where it is subjected to nutrient limitation and external stress, including pollutants. On sensing phenols and other nutrients secreted from plant wounds, A. tumefaciens moves to wound sites and mediates crown gall formation to create a better environment for survival. The movement and adaptability of A. tumefaciens is spontaneous and dynamically controlled by metabolic regulation, therefore knowledge of metabolic differences between free‐living and saprophytic A. tumefaciens could improve our understanding of the pathogenesis of crown gall disease and provide a basis for optimizing transgenic plant development. The cellular metabolism of A. tumefaciens is not well‐characterized, and most studies have focused on specific metabolites that accumulate in tumours (Deeken et al., 2006; Lang et al., 2014; Simoh et al., 2009; Vigouroux & El Sahili, 2017). Gene expression data related to the exploitation of host resources have recently been published (Gonzalez‐Mula et al., 2018, 2019), but these transcriptomics analyses do not reflect comprehensive metabolic changes in A. tumefaciens.
With the development of systems biology, genome‐scale metabolic models (GSMMs) have become an effective and systemic platform for evaluating associations among metabolites, genes, and reactions in a living organism. By integrating various computational constraint‐based algorithms, GSMMs could identify requirements for cell growth, predict metabolism in pathogen–host systems, and analyse interspecific interactions (Xu et al., 2018). GSMMs for Rhizobiaceae microbes, like Sinorhizobium meliloti (Dicenzo et al., 2016; Zhao et al., 2012), Rhizobium etli (Resendis‐Antonio et al., 2007), Sinorhizobium fredii (Contador et al., 2020), and Bradyrhizobium diazoefficiens (Yang et al., 2017), have been used for studies of nitrogen fixation, niche adaptation, growth, fitness, and other biological process. For some leguminous bacteria, multiple GSMMs have been developed (Resendis‐Antonio et al., 2007, 2011).
A. tumefaciens C58 is a model strain within the species whose genome was sequenced in the early 2000s (Goodner et al., 2001; Wood et al., 2001). In this study, based on genome annotation and biochemical data mining, the first GSMM for A. tumefaciens C58 was reconstructed using the Matlab platform and was validated with respect to growth phenotypes by qualitative and quantitative analyses. Moreover, omics data integration was performed for a systematic analysis of the pathogenicity of A. tumefaciens and its interactions with plant hosts.
2. RESULTS AND DISCUSSION
2.1. Genome‐scale metabolic model for A. tumefaciens
The GSMM iNX1344 for A. tumefaciens contained 1,106 metabolites and 1,441 biochemical reactions in the cytosolic and extracellular compartments (Data S1). The 1,441 reactions were distributed in 77 metabolic pathways, which were classified into nine metabolic subsystems, except for exchange reactions, transfer RNA (tRNA) charging, and biomass synthesis. As shown in Figure 1, transport subsystems included the largest number of genes, with 380 genes for 150 metabolites. Among these, 120 reactions belonged to adenosine 5'‐triphosphate (ATP)‐binding cassette transporters. Abundant transporters in A. tumefaciens can facilitate the adaption of A. tumefaciens to different environments, such as barren bulk soil and complex tumour nodules. The subsystem of amino acids contained the largest number of reactions. A. tumefaciens could synthesize 20 kinds of amino acids de novo, and this result was validated by its growth on synthetic minimal medium. With respect to gene‐associated reactions, all the reactions in amino acid metabolism and energy metabolism were associated with genes, and the ratio of gene‐associated reactions in the nine metabolic subsystems was, on average, 97.1%. The high number of gene‐associated reactions suggests that the iNX1344 model sufficiently annotated the A. tumefaciens genome.
FIGURE 1.
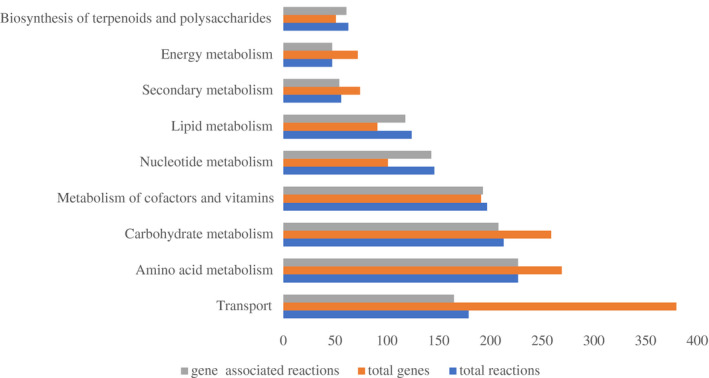
Genes, reactions, and gene‐associated reactions for each metabolic subsystem of the model iNX1344. Total genes and total reactions denote the number of genes or reactions in each metabolic subsystem. Gene‐associated reactions denote the number of reactions by annotating their encoding genes. The metabolic subsystems were based on KEGG pathway maps
Compared with GSMMs of closely related taxa within Rhizobiaceae, the model size for A. tumefaciens was similar to that of the iGD1575 model for S. meliloti (Dicenzo et al., 2016) and was much larger than that of the iOR450 model for R. etli (Resendis‐Antonio et al., 2011). The open reading frame (ORF) coverages of iNX1344 and iGD1757 were roughly 25% (Table 1). The ratios of gene‐associated reactions to total reactions, and the ratios of unique metabolites to total metabolites in iNX1344 were both 4% higher than those in iGD1757. For 251 genes in iNX1344, no homologous genes were found in the other two models; these genes participated in 309 metabolic reactions. Fewer than one‐third of the unique metabolites (305) in iNX1344 were shared by the two models, reflecting the metabolic differences between the plant pathogen Agrobacterium and plant symbionts (Sinorhizobium and Rhizobium). A total of 164 unique metabolites were found only in iNX1344, and they participated in 224 reactions. The overlapping nonhomologous genes and unique metabolites were involved in 105 reactions in 32 metabolic pathways (Table S1). Metabolic pathways with more than three reactions were listed here. They included 19 reactions involved in the biosynthesis of odd‐carbon acids and cardiolipin (Aktas et al., 2014). Additionally, 25 transport and exchange reactions were related to the response to stress and uptake of nutrients in tumours. Nine reactions in pyrimidine metabolism contributed to the uracil degradation III pathway. Seven reactions in biotin metabolism were involved in biotin biosynthesis from 8‐amino‐7‐oxononanoate (Feng et al., 2013). Four reactions in pentose and glucuronate interconversions participated in pectate degradation (Park et al., 2003).
TABLE 1.
Comparison among metabolic models for Agrobacterium tumefaciens (iNX1344), Sinorhizobium meliloti (iGD1575), and Rhizobium etli (iOR450)
| Model contents | iNX1344 | iGD1575 | iOR450 |
|---|---|---|---|
| Genes | 1,344 | 1,575 | 450 |
| Gene coverage (%) | 25.1 | 25.3 | 7.5 |
| Total reactions | 1,441 | 1,825 | 405 |
| Internal reactions | 1,096 | 1,324 | 380 |
| Transport reactions | 179 | 233 | 11 |
| Exchange reactions | 166 | 268 | 13 |
| Gene‐associated reactions | 1,237 | 1,479 | 402 |
| Metabolites | 1,106 | 1,307 | 377 |
| Unique metabolites | 942 | 1,061 | 364 |
| Compartments | c, e | c, e | c, e |
c, cytosolic compartment; e, extracellular compartment.
2.2. Essential gene analysis
A total of 195 metabolic genes were predicted to be essential for cell growth on synthetic minimal medium (Table S2). They participated in 287 biochemical reactions distributed in 49 metabolic pathways. For 189 of 195 genes, homologous genes were detected in the Database of Essential Genes (DEG) (Luo et al., 2014) using identity >40% and E‐value <10−5 as thresholds, and only six genes were not validated (Figure 2). The high proportion of homologous genes supported the accuracy of the iNX1344 model. Most of the essential genes participated in the metabolism of amino acids, lipids, cofactors, vitamins, and nucleotides, and relatively few essential genes were involved in carbohydrate metabolism in minimal medium (Xu et al., 2013). In addition, metabolic genes indispensable for tumour formation were simulated using the iNX1344 model. It is difficult to implement in vivo knockouts during nodulation, and in silico deletion is a complementary approach. A total of 189 metabolic genes were predicted to be essential for nodulation using the iNX1344 model. These genes were involved in two processes: biomass precursor synthesis and colonization of hosts. For 158 of the 189 genes, homologous genes were obtained in the DEG database, and these were mostly chromosomal housekeeping genes. Of the additional 31 genes, seven were validated using transposon mutants on tryptone yeast (TY) medium (Gonzalez‐Mula et al., 2019). The other 24 genes without experimental support were closely related to metabolic interactions with plants. For example, transporters (Atu4783‐Atu4785, Atu6025‐Atu6028, Atu4620‐Atu4621, Atu6138‐Atu6143, and Atu4471) import nutrient resources (e.g., opine, sugars, and iron) from plant tumours. Some essential genes could improve conditions for survival in plant tumours, including the degradation of phenolics, like ferulate, which impedes cell growth in tumours (Aznar et al., 2015), and the biosynthesis of exopolysaccharides, like 1,2‐glucan, which resist environmental osmolarities (Binns & Thomashow, 1988). Analyses of essential genes in the nodulation stage clarify the genomic basis of interactions with host plants.
FIGURE 2.

Distribution of essential genes of Agrobacterium tumefaciens in nine metabolic subsystems. The pie chart represents the metabolic subsystem distribution of 287 metabolic reactions associated with 195 essential genes for growth on synthetic minimal medium. The table shows the gene names and functions of six invalidated essential genes on synthetic minimal medium
2.3. The validation of model iNX1344
A total of 39 substrates were used to evaluate A. tumefaciens cell growth. The simulation results were in complete agreement with the corresponding experimental results (Table S3) and data from the literature (Table 2). A. tumefaciens was able to grow on 30 organic carbon sources, including sugars, carboxylic acids, amino acids, alcohol, alkaloids, and some unassigned carbohydrates. The discrepancy between the in silico and in vivo growth occurred when arginine and ornithine were utilized as sole carbon sources, which may be caused by no expression of the genes (Atu3949, Atu4758, Atu6016) encoding ornithine cyclodeaminase. Capric acid and phenylacetic acid as the sole carbon source could not support growth owing to a deficiency of key enzymes in A. tumefaciens. The diversity of carbon sources was related to the extensive transport systems. A total of 746 proteins encoded by the A. tumefaciens C58 genome were annotated with homologue sequences in the Transporter Classification Database (TCDB), and more than half of the proteins were in the iNX1344 model. A large number of transporters sustained efficient and steady access to nutritional sources. The utilization of some specific carbon sources, in addition to common sugars, organic acids, and amino acids, reflected the host plant interaction. For example, A. tumefaciens C58 could convert cellobiose to glucose via β‐glucosidase (Atu4485), which might involve the biological degradation of cellulose (Goodner et al., 2001). γ‐Aminobutyric acid (GABA), which accumulates at the highest concentrations in plant tumours (Gonzalez‐Mula et al., 2019), was converted to succinate semialdehyde via GABA aminotransferase (Atu3300) in A. tumefaciens C58, providing a carbon source to the tricarboxylic acid cycle (TCA) cycle. Nopaline as an energy source was converted to arginine via nopaline dehydrogenase (Atu6015, Atu6017, Atu6019, and Atu6021).
TABLE 2.
Validation of growth phenotypes under various sole carbon and nitrogen sources for Agrobacterium tumefaciens
| Substrate | In silico growth | In vivo growth | Reference | |
|---|---|---|---|---|
| Saccharides | Glucose | + | + | This study |
| Sucrose | + | + | This study | |
| Arabinose | + | + | This study | |
| Palatinose | + | + | De Costa et al. (2003) | |
| Maltose | + | + | De Costa et al. (2003) | |
| Ribose | + | + | This study | |
| Cellobiose | + | + | Marasco et al. (1995) | |
| Mannose | + | + | Behura et al. (2015) | |
| Carboxylic acid | Succinate | + | + | This study |
| Malic acid | + | + | This study | |
| Gluconate | + | + | This study | |
| Pyruvate | + | + | This study | |
| γ‐Aminobutyric acid | + | + | Gonzalez‐Mula et al. (2019) | |
| Hydroxybutyric acid | + | + | Parke (2000) | |
| Ferulate | + | + | Baude et al. (2016) | |
| Coumarate | + | + | Baude et al. (2016) | |
| Alginate | + | + | Hirayama et al. (2016) | |
| D‐galacturonate | + | + | Vetting et al. (2016) | |
| Cyanuric acid | + | + | Galíndez‐Nájera et al. (2009) | |
| Glucose−1‐phosphate | + | + | This study | |
| Hydroxybenzoate | + | + | Parke (2000) | |
| Capric acid | − | − | This study | |
| Galactarate | + | + | Groninger‐Poe et al. (2014) | |
| Phenylacetic acid | ‐ | − | This study | |
| Amino acids | Homoserine | + | + | Vanderlinde et al. (2014) |
| Arginine | + | − | This study | |
| Glumate | + | + | This study | |
| Proline | + | + | This study | |
| Ornithine | + | − | This study | |
| Alkaloids | Glycine betaine | + | + | Boncompagni et al. (1999) |
| Choline | + | + | Boncompagni et al. (1999) | |
| Nopaline | + | + | Parke (2000) | |
| Alcohols | Glycerol | + | + | Ugalde et al. (2003) |
| Mannitol | + | + | This study | |
| Inorganic salts | Nitrate | + | + | This study |
| Nitrite | + | + | This study | |
| NH3 | + | + | This study | |
| Other | N‐acetyl‐glucosamine | + | + | Alippi et al. (2010) |
| Methylamine | + | + | Chen et al. (2010) | |
+, growth; −, no growth.
The utilization of 11 nitrogen sources by A. tumefaciens C58 was predicted qualitatively by the iNX1344 model and flux balance analysis (FBA). Eight organic nitrogen compounds (glutamate, arginine, proline, ornithine, nopaline, glycine betaine, choline, and N‐acetyl‐glucosamine) and three inorganic salts (NH3, nitrate, or nitrite) could support A. tumefaciens C58 growth as the sole nitrogen source. The existence of genes related to denitrification suggests that nitrate may act as an electron transfer acceptor for environmental adaptability (Rossbach et al., 1987). Another sole nitrogen source, methylamine, is used in different pathways in methylotrophic bacteria. In the nonmethylotroph A. tumefaciens C58, methylamine was metabolized to formaldehyde and ammonium via methylglutamate synthase (Atu4227, Atu4228, and Atu4229) and methylglutamate dehydrogenase (Atu4224, Atu4223, and Atu4221).
In addition, to assess iNX1344 quantitatively, the specific growth rate was set as an objective function in the iNX1344 model under the constraints of 4.5 mmol⋅g−1 dry weight⋅hr−1 glucose uptake, based on previously reported estimates (Fuhrer et al., 2005). The distribution of carbon flux was compared with published data for 22 central carbon metabolic reactions. In general, in silico fluxes were roughly equal to those reported in the literature (Figure 3). Three routes from glucose to pyruvate were annotated in A. tumefaciens C58: the glycolysis pathway, pentose phosphate (PP) pathway, and Entner–Doudoroff (ED) pathway. Among the three pathways, substantial carbon flux through the ED pathway, less carbon flux through the PP pathway, and no flux through the glycolysis pathway were observed. The slight discrepancy in small reactions could be explained by the complex model structure of iNX1344. The flux from citrate to α‐ketoglutarate in the model iNX1344 was previously reported (Fuhrer et al., 2005) as only 7.2% carbon flux, which was diverted to the glyoxylate cycle. A 3.3‐fold greater flux to oxaloacetate in the model iNX1344 was detected from pyruvate carboxylase compared to the literature data previously reported. Oxaloacetate was transferred to α‐ketoglutarate and aspartate by aminotransferase in the iNX1344 model.
FIGURE 3.
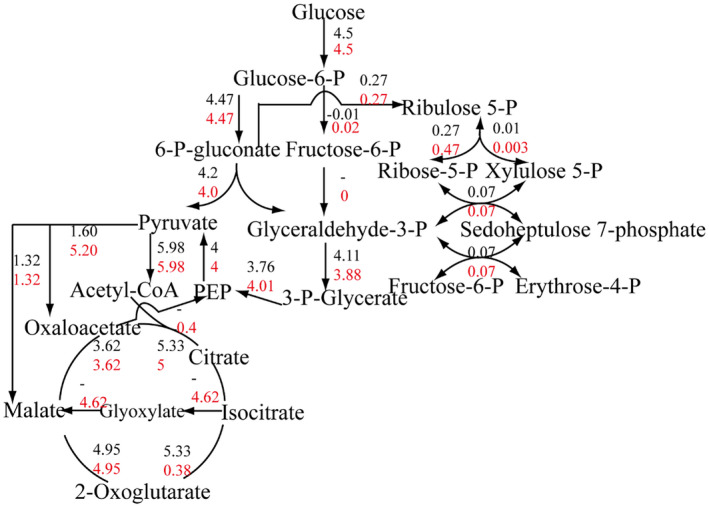
Comparison of central metabolic flux distributions between the model predictions and the published data. Values in black are the flux from the literature; values in red are the flux from the model iNX1344. “−” represents no associated value in the literature. Estimated fluxes in the iNX1344 and the literature are expressed in mmol·g −1 dry weight·hr−1
2.4. In silico evaluation of A. tumefaciens metabolic characteristics under an environment shift
The metabolic shifts experienced by A. tumefaciens during the transition between bulk soil, the rhizosphere, and the nodule were modelled using in silico representations of the nutritional composition of each environment. In the bulk soil and rhizosphere environments, the model was optimized for the production of biomass, whereas in the nodule environment the model was optimized for the process of nodulation. In total, 404, 407, and 430 reactions carried flux in the soil, rhizosphere, and nodule niches, respectively (Table S4). Among these active reactions, 251 reactions shared by all three environments, excluding 25 transport and exchange reactions, were assigned to the following metabolic subsystems: lipids (24.3%), amino acids (17.5%), nucleotide (14.3%), carbohydrates (13.5%), cofactors and vitamins (12.7%), and cell wall biosynthesis (7.6%). Furthermore, reactions carrying flux only in certain environments were investigated (Table S4). In bulk soil, 28 unique metabolic reactions were related to lysine biosynthesis, adenine and uridine‐related metabolism, cystathionine metabolism, and lauroyl‐CoA elongation. Two reactions catalysed by 3‐phospho‐d‐glycerate dehydrogenase and pyridine nucleotide transhydrogenase regulate intracellular redox levels. In the rhizosphere, 25 unique metabolic reactions were involved in cysteine and methionine metabolism (such as serine catabolism and H2S‐related reactions), arginine and proline metabolism (such as the oxidation‐reduction of 1‐pyrroline‐4‐hydroxy‐2‐carboxylate), and carbohydrate metabolism (such as the degradation of melibiose and stachyose). A greater number of reactions were active in tumours alone than in the other two environments. In addition to the 54 transport and exchange reactions, 75 metabolic reactions covered the metabolism of cofactors and vitamins (28), carbohydrates (24), secondary metabolites (9), amino acids (6), nucleotides (5), and lipids (3). Specifically, the two main cofactor metabolic pathways observed were the biotin metabolic pathway for biotin biosynthesis, and the porphyrin and chlorophyll metabolic pathway for heme biosynthesis. Carbohydrate metabolism specific to tumours included the conversion of inositol to acetyl‐CoA and glyceraldehyde 3‐phosphate in the inositol phosphate pathway, d‐gluconate metabolism in the PP pathway, polysaccharide synthesis in the starch and sucrose metabolism pathway, and some metabolic reactions of galactarate, galacturonate, glucarate, glycerol, and formaldehyde. In particular, secondary metabolic pathways were uniquely active in tumours, such as those involved in the metabolism of opines, vanillin, ferulate, and quinate. The low proportion of unique amino acid, nucleotide, and lipid metabolism pathways might reflect abundant nutrients in the tumour environment. In addition to common intermediate metabolites of central metabolic pathways and amino acids, some secondary metabolites (opines, ferulate, spermidine, and quinate), iron, inositol, and hypoxanthine were also predicted to be exchangeable from the tumour environment.
To investigate metabolic shifts from one lifestyle to another, the cellular metabolism of A. tumefaciens was compared between the soil and rhizosphere environments. As shown in Figure 4, the reaction flux in both conditions was correlated (p < .01, Spearman's correlation coefficient = .78). Similarly, fitness in the two environments was highly correlated (p < .01, Spearman's correlation coefficient = .93). The metabolic network was globally similar in bulk soil and the rhizosphere. A total of 169 reactions were considerably different (Table S5). In particular, 78 reactions were active in only one environment with no flux in the other. The flux difference between the two conditions was >50% for 80 reactions and another 11 reversible reactions changed direction. Of 169 reactions, 59 were supported by fitness data. The flux values for 62 reactions in the rhizosphere were outside of the flux variability analysis (FVA) range in soil, whereas rhizosphere values for the additional 48 reactions were within the FVA range in soil. By setting the rhizosphere values to the constraints of the 48 reactions, cell growth was individually simulated in soil. Flux through at least one other reaction shifted to outside the rhizosphere FVA range to sustain growth rates in bulk soil. Among the 169 reactions occurring in the rhizobium/soil niches, 61 reactions were activated or showed more than 50% increased flux in the rhizosphere. These mainly included 22 transport and exchange reactions, 16 reactions in carbohydrate metabolism, and 16 reactions in amino acid metabolism. Some sugars and amino acids, such as sucrose, raffinose, stachyose, rhamnose, serine, glutamate, lysine, and aspartate, were exported from the rhizosphere. With respect to carbohydrate metabolism, glyoxylate cycle and galactose metabolism were activated in the rhizosphere. With respect to amino acid metabolism, the conversion from serine or cysteine into pyruvate, and the flux into glucuronate metabolism from proline were found in the rhizosphere.
FIGURE 4.
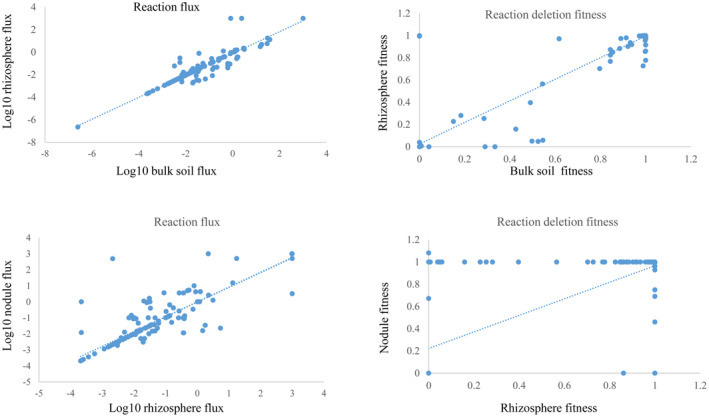
Correlations of the reaction flux and fitness effect in the model iNX1344 between bulk soil, the rhizosphere, and the nodule. Four scatter plots showing the flux correlations or the fitness correlations under the nodule/rhizobium and rhizobium/soil conditions are shown. Among these, flux correlations of the reactions were converted to a base‐10 logarithm of the absolute value, excluding the zero‐flux reactions. Blue dotted lines are the trend lines
The metabolic shift from the rhizosphere to nodule environments was also modelled. As shown in Figure 4, the correlation coefficients for the relationships between both reaction flux and reaction deletion fitness in the rhizosphere and nodule were approximately .50 (p < .01, Spearman). The moderate correlation indicated that substantial metabolic changes occurred in the rhizosphere to nodule transition. There were 407 and 430 reactions with flux in the rhizosphere and nodule environments, respectively. Among 441 reactions with significant changes, 289 were active in only one environment, 137 showed at least 50% changes in flux between the two environments, while 17 switched directions (Table S6). Of 441 reactions, 110 were supported by fitness data. The flux values for 230 reactions in the nodule were outside of the FVA range for the reaction in the rhizosphere. The remaining 101 reactions were validated by a constraint‐based analysis following the same procedure used for the soil to rhizosphere transition.
Generally, nearly 5‐fold reactions in various metabolic pathways, including the TCA cycle; glycine, serine, and threonine metabolism; biotin metabolism; and fatty acid biosynthesis, showed increased flux (>50%) in the nodule compared to that in the rhizosphere, suggesting that cellular metabolism in the nodule was more intense. In carbohydrate subsystems, stronger central carbon metabolism generates more ATP. When A. tumefaciens utilized fructose as the main carbon source, the glycolysis pathway became active. Greater carbon flow via the PP pathway and glyoxylate cycle was observed in the rhizosphere, accompanied by the uptake of glucose (Shih et al., 2018). Additionally, ascorbate and aldarate metabolism activation was detected in the nodule environment for the degradation of glucarate, galactarate, and galacturonate. Galactose metabolism observed in the rhizosphere participated in the utilization of stachyose and melibiose, which confers a competitive advantage in this environment (Meyer et al., 2018). Among amino acid subsystems, arginine and proline metabolism showed a change in metabolic importance in tumour nodules (Haudecoeur et al., 2009). The urea cycle and arginine degradation (from arginine to GABA) were active, and the accumulation of proline might have a protective effect under stress. Among cofactor subsystems, strengthened porphyrin and chlorophyll metabolism in nodules would affect the colonization of A. tumefaciens by regulating heme and Fe–S concentrations (Hibbing & Fuqua, 2011). Several subsystems were uniquely changed in nodules, including some secondary metabolic processes (vanillin, 3‐oxoadipate, ferulate, γ‐hydroxybutyrate, and opine) and energy subsystems (formate dehydrogenase in methane metabolism, glutathione synthesis in glutathione metabolism, nitrite oxidoreductase, and carbonate dehydratase in nitrogen metabolism).
2.5. Transcriptome‐based modelling of the metabolic capacity of A. tumefaciens in tumours
To understand A. tumefaciens behaviour during plant infection, A. tumefaciens gene expression was studied in Arabidopsis thaliana tumours or with enrichment for three compounds (opines, γ‐aminobutyric acid, and γ‐hydroxybutyric acid) in tumours. RNA‐Seq data reveal transcript abundances of metabolic genes; however, cell metabolism is not a static set of reactions and pathways but adapts to different environments to allow for the uptake of metabolites or the production of required compounds (Rodenburg et al., 2019). An integrative analysis of transcriptome data and the iNX1344 model showed that differentially expressed genes were enriched in various biological processes, including central carbon metabolism, alternative carbon metabolism, various amino acid metabolism, porphyrin and chlorophyll metabolism, and lipopolysaccharide biosynthesis.
We focused on changes in carbon and nitrogen metabolism (Figure 5). ATP was an up‐regulated metabolite, reflecting abundant energy production and conversion in A. tumefaciens. To prosper in plant tumours, A. tumefaciens needs to cope with drought and oxidative and hypoxic stress, and the absorption of diverse nutrient resources from plant tumours is dependent on ATP‐binding cassette transporters. In central carbon metabolism, glycolysis was identified as a down‐regulated pathway. 2‐Dehydro‐3‐deoxy‐6P‐gluconate was identified as an up‐regulated reporter metabolite, located at the node linking the ED pathway and glycolysis pathway. The ED pathway, as the main route for sugar degradation and entry to the TCA cycle, was more important than the glycolysis pathway in the nodule environment (Fuhrer et al., 2005). 5‐phospho‐α‐D‐ribose 1‐diphosphate (PRPP), a downstream metabolite of the PP pathway, and xylose, a precursor of xylulose 5‐phosphate, were both down‐regulated, indicating a reduced carbon flux into the PP pathway. In addition, carbohydrates, including rhamnose, glycerol‐3‐phosphate, ethanol, glycerol, succinyl‐CoA, and propanoyl‐CoA, were identified as reporter metabolites, reflecting the diversification of carbon metabolism in plant tumours.
FIGURE 5.
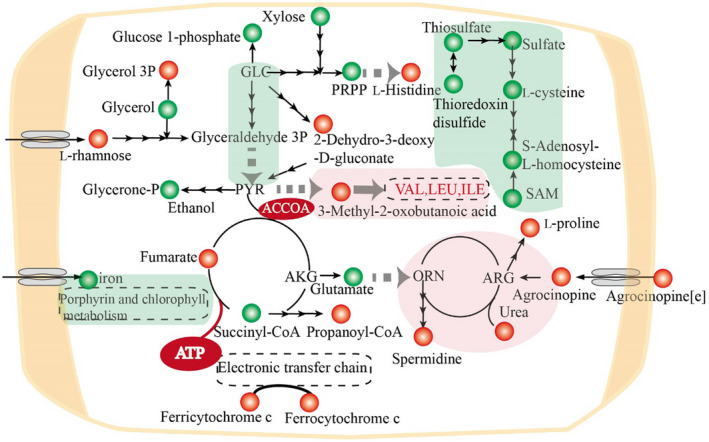
Significantly changed metabolites and pathways of Agrobacterium tumefaciens in plant tumours. Red circles and green circles represent the up‐regulated and down‐regulated reporter metabolites. Red backgrounds and green backgrounds represent the up‐regulated and down‐regulated reporter pathways. Metabolic pathways are shown in dotted rounded rectangles. A small black arrow represents one reaction, and a large grey arrow represents more than three steps connecting two metabolites or between one metabolite and one pathway. GLC, l‐glucose; PYR, pyruvate, AKG, α‐ketoglutaric acid; ORN, l‐ornithine; ARG, l‐arginine; SAM, S‐adenosyl‐l‐methionine; VAL, valine; LEU, leucine; ILE, isoleucine; [e], extracellular compartment
Seven kinds of amino acids were identified as reporter metabolites, including up‐regulated (histidine, valine, leucine, and isoleucine) and down‐regulated (glutamate and cysteine) amino acids. In the up‐regulated urea cycle, arginine was metabolized into urea and ornithine by arginases, which was increased in host‐colonizing A. tumefaciens. Proline and spermidine were significantly up‐regulated metabolites in A. tumefaciens and were enriched in plant tumours. The addition of exogenous spermidine enhances biofilm formation (Wang et al., 2016). The increased spermidine in A. tumefaciens could facilitate attachment to the plant root. The accumulation of proline as an osmoprotectant might be related to stress resistance in plant tumours. Both cytosolic and extracellular agrocinopine, as reporter metabolites, were highly degraded in the nodule environment. Agrocinopine metabolized into arabinose‐2‐phosphate could stimulate quorum‐sensing and T‐DNA transfer in A. tumefaciens (El Sahili et al., 2015). Five compounds involved in cysteine and methionine metabolism and thioredoxin disulphide were identified as down‐regulated reporter metabolites, and sulphur metabolism was identified as a reporter metabolic pathway. The down‐regulation of sulphur‐containing amino acids would affect the cytosolic redox state. For example, decreased thioredoxin might be related to the maintenance of intracellular homeostasis in oxidative stress (Vattanaviboon et al., 2007).
The interaction of A. tumefaciens with host plants involves multiple processes of motility, biological adaptation, and T‐DNA transfer to the host. After A. tumefaciens senses the secretions and moves to wound sites via chemotaxic systems, ensuring the survival becomes the basis of its interaction with host plants. Essential gene analysis predicted 189 metabolic genes to be indispensable for both biomass precursor synthesis and colonization of hosts. Considering that A. tumefaciens is the known plant pathogen of crown gall disease, these essential genes also provide potential antimicrobial targets (Koduru et al., 2020). To adapt to its host plants, A. tumefaciens attempts to settle in and reconstruct otherwise inhabitable niches. Previous studies have focused on the use of opines for this process (Lang et al., 2014, 2017; Marty et al., 2016), which was also identified as one of the reporter metabolites in the model iNX1344. In addition to opines, our study expands the nutrient exploitation and intracellular metabolic response to these substances for host adaptation. A total of 47 active exchange reactions in the host tumour were identified, which was more than those identified in the soil (29) and in the rhizosphere (33). Overall, 25 nutrient types were found to be exchanged in both the soil and rhizosphere environments. However, the overlap of active exchange reactions in the host tumour included only 12 reactions with the soil and 16 reactions with the rhizosphere. Active exchange reactions in the host tumour cover the saccharides (4), alkaloids (2), amino acids (13), carboxylic acids (16), alcohols (3), hypoxanthine, inorganic salts, and metal ions (8). The diversity of active substance exchange with the host sustains the adaptive advantage of A. tumefaciens. With respect to the molecular mechanism of infection, A. tumefaciens transfers its T‐DNA and related vir genes into host plants; 3‐oxo‐octonanoylhomoserine lactone, opines, and γ‐aminobutyric acid act as direct or indirect quorum‐sensing signals to regulate the horizontal transfer of the Ti plasmid (Lang & Faure, 2014). Using the model iNX1344, a significant metabolic response to the signals was analysed under transcriptome constraints. For example, the urea cycle of A. tumefaciens was identified to be significantly up‐regulated, which indicated that adding certain intermediates or increasing the expression levels of some genes in the urea cycle could be potential targets for the infection process. Thus, the model iNX1344 provides a useful tool to understand the interspecific interactions between A. tumefaciens and tumours from the levels of genes, metabolites, and pathways.
In conclusion, a well‐established GSMM for A. tumefaciens, including 1,344 genes, 1,441 reactions, and 1,106 metabolites, agreed with the corresponding growth phenotypes. Using this model, A. tumefaciens was modelled under the bulk soil, rhizosphere, and nodule environments and the metabolic differences were compared between the three lifestyles. Furthermore, the significant metabolic response of A. tumefaciens in the tumour was predicted by integrating transcriptomic data. The model iNX1344 is expected to serve as a useful research tool for exploring key functions in interspecific interactions by integrating GSMMs of different plant tissues. The prediction capacity of a microbial GSMM is important to simulate both the physiology and metabolism, which are in turn determined by the background of target microorganisms (Yilmaz & Walhout, 2017). Gene function in most studied microorganisms tends to be comparatively comprehensively annotated. More types of constraints used in modelling could improve the prediction accuracy. Some common constraints include metabolite exchange and gene on/off switching at certain nodes. Some advanced constraints should be integrated into GSMMs, including omics data, thermodynamics, and enzyme kinetic parameters (O'Brien et al., 2015; Xu et al., 2018). Moreover, model predictions should be related to the biomass equation. Unlike universal biomass components used in automated construction, the biomass equation of the model iNX1344 covers all compounds that comprise species‐ or even strain‐specific biomass. Although the total contents of proteins, nucleic acids, and lipids of the model iNX1344 referred to the related species Zymomonas mobilis (Widiastuti et al., 2011), the model iNX1344 could reflect the cellular metabolism of A. tumefaciens, especially for gene essentiality and growth phenotypes (Cankorur‐Cetinkaya et al., 2017). The biomass of microbial cells also varies with experimental and genetic conditions. Hence, context‐dependent biomass composition could improve predictive capabilities in the further development of GSMMs.
3. EXPERIMENTAL PROCEDURES
3.1. Model reconstruction
A GSMM of A. tumefaciens C58 was generated by three main steps: draft model construction, gap filling, and simulating cell growth. The draft model of A. tumefaciens C58 was obtained by combining automated and manual construction methods. The chromosome and plasmid sequences were annotated using the Rapid Annotation using Subsystem Technology (RAST) server, and the RAST annotations were used for automated construction using Model SEED. The genome sequences of S. meliloti, R. etli, and Escherichia coli were compared with those of A. tumefaciens by a local sequence similarity search (Altschul et al., 1990) (BLASTp). The BLASTp results with both an E‐value of ≤10−5 and identity ≥40% (Tian & Skolnick, 2003) were considered homologues. The gene–protein–reaction relationships were established by integrating the results of the two procedures. Cytoplasmic and extracellular compartments were considered for the draft model of A. tumefaciens C58. Transport reactions were measured by comparing A. tumefaciens C58 protein sequences with the transporter classification database (TCDB) (Saier et al., 2006). The draft model had a number of metabolic gaps that were identified using gapAnalysis. Some gaps were filled by adding reactions and genes from physiological and metabolic studies of A. tumefaciens C58, some gaps were filled using BLASTp with relevant protein sequences in UniProtKB/SwissProt (Boutet et al., 2007). Gap filling was an iterative process until the nonexistence of gaps that blocked biomass precursor formation. The mass‐balanced and charge‐balanced reactions were converted into a stoichiometric matrix S (m × n), where m and n were the numbers of metabolites and reactions, respectively. The in silico cell growth was debugged and compared with experimental data and previously published data. Simulations were performed using the COBRA Toolbox (Heirendt et al., 2019) by the GUROBI mathematical optimization solver (Gurobi Optimization, 2014) on the MATLAB interface.
3.2. Biomass composition
Similar to the biomass of S. meliloti (Dicenzo et al., 2016), the macromolecular composition of A. tumefaciens has not been reported. A. tumefaciens biomass contained proteins (60.5%), DNA (2.7%), RNA (19.5%), lipids (8.5%), glycogen (1.5%), peptidoglycan (3%), lipopolysaccharides (3%), and cofactors (1.3%), determined with reference to near‐relative microorganisms (Dicenzo et al., 2016; Widiastuti et al., 2011). The compositions of nucleotides and amino acids were calculated from genome sequence information for A. tumefaciens C58. Contents of free fatty acids and lipids (Geske et al., 2013), glycogen (Zevenhuizen, 1971), and lipopolysaccharides (Manasse & Corpe, 1967) were obtained from the literature. Details are provided in Table S7.
3.3. Objective function
An appropriate objective function could accurately reflect metabolic processes using GSMMs (Schuetz et al., 2007). Biomass formation is the most common objective function when simulating cell growth under synthetic medium and analogous bulk soil or rhizosphere environments. The pathogenic lifestyle involves survival inside a host and interactions with the host. A suitable objective function was proposed to mimic the nodulation stage, including eight compounds (β‐1,2‐glucan, cellulose, 5‐dehydro‐4‐deoxy‐d‐glucarate, nopaline, arabinose‐phosphate, γ‐hydroxybutyrate, catalase, and 3,4‐dihydroxybenzoate). The objective function was developed as follows: The biosynthesis of β‐1,2‐glucan and cellulose in A. tumefaciens is important for early attachment to plant cells (Cangelosi et al., 1989; Tomlinson & Fuqua, 2009). 5‐Dehydro‐4‐deoxy‐d‐glucarate is a degradation product of pectin; the pectin network in targeted cell walls is collapsed so that bacterial T‐DNA or proteins easily enter the host plant (Goodner et al., 2001). The T‐DNA region causes plants to develop galls, which excrete opines that are used as nutrients used by A. tumefaciens. γ‐Hydroxybutyrate is a metabolic intermediate of γ‐butyrolactone, which was assimilated to partially balance the costs of replication and T‐DNA transfer (Carlier et al., 2004). A. tumefaciens also needs to respond to the stress induced by plant defence mechanisms, including the accumulation of H2O2 (Torres et al., 2006) and trans‐ferulate phenolics (Vellosillo et al., 2010). Generally, the metabolism of the eight compounds in the objective function is representative of the typical infection process.
3.4. Cell growth test
A. tumefaciens C58 growth was evaluated using Agrobacterium (AB) minimal medium, which contained 50 ml of 20 × AB salts per litre (MgSO4.7H2O 6 g, KCl 3 g, CaCl2 0.2 g, FeSO4.7H2O 50 mg) and 50 ml of 20 × AB buffer per litre (K2HPO4 60 g, KH2PO4 20 g). The pH of AB buffer was adjusted to 7.0 using KOH or H3PO4. Carbon sources included sucrose, glucose, ribose, arabinose, pyruvate, succinate, malate, potassium gluconate, capric acid, phenylacetic acid, mannitol, arginine, glutamate, proline, and ornithine. Nitrogen sources included nitrate, nitrite, NH4Cl, arginine, glutamate, proline, and ornithine. All carbohydrates or nitrogenous compounds were set to an equal carbon or nitrogen concentration. The molar masses of the individual carbon and nitrogen sources were the same as those of 5 g of sucrose and 1 g of NH4Cl, respectively. A. tumefaciens C58 from an agar slant was inoculated in 20 ml MG/L medium (10 g mannitol, 2.32 g sodium glutamate, 0.5 g KH2PO4, 0.2 g NaCl, 0.2 g MgSO4.7H2O, 2 mg biotin, 500 ml LB, per L, pH 7) and the inoculum was incubated at 28 °C for 12 hr on a shaker at 200 rpm. After the incubation period, 2 ml of the culture was washed twice using AB buffer and inoculated into 25 ml of synthetic minimal medium with the sole carbon and nitrogen source in 250‐ml flasks. The flask culture was grown for 36 hr and then cell growth was evaluated using optical density (OD600).
3.5. Model simulation and analysis
Under a pseudosteady‐state, an FBA (Orth et al., 2010) seeks to optimize an objective function or to identify the best flux distribution subjected to an optimized objective function. FBA was used to evaluate cell growth by solving a linear programming problem constrained by changing the exchange reaction rates based on the literature or experimental data. A flux variability analysis (Gudmundsson & Thiele, 2010) could be used to calculate the range of flux for each reaction while still maintaining optimal flux through the objective function.
Essential genes and reactions were analysed using the singleGeneDeletion and singleRxnDeletion functions. If the deletion of a gene and reaction inhibits growth, they are defined as essential. The constraint data, used for simulating metabolic characteristics with an environment shift, included the uptake rates of nutrients under the three environmental conditions. The lower bounds of nutrients are provided in Table S8. Fitness was defined as the flux through the objective function in the deletion mutants relative to that in the wild type.
Transcriptome data (GSE106257 and GSE121889) for A. tumefaciens C58 related to plant tumours was download from Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) and analysed using the piano library in the R language (Väremo et al., 2013). Furthermore, if an identified reporter pathway or reporter metabolite with a value of p < .05 was found in more than two sets in the transcriptome analysis, it was identified as a significantly changed metabolite and pathway.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (21808196, 31870118), the China Postdoctoral Science Foundation (2018M632389), and the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (18KJB180030).
Xu N, Yang Q, Yang X, Wang M, Guo M. Reconstruction and analysis of a genome‐scale metabolic model for Agrobacterium tumefaciens . Mol Plant Pathol. 2021;22:348–360. 10.1111/mpp.13032
DATA AVAILABILITY STATEMENT
The authors declare that the data supporting the findings of this study are available within the article and its Supporting Information. Scripts for simulation in this study are available from the corresponding author upon reasonable request.
REFERENCES
- Aktas, M. , Danne, L. , Möller, P. & Narberhaus, F. (2014) Membrane lipids in Agrobacterium tumefaciens: biosynthetic pathways and importance for pathogenesis. Frontiers in Plant Science, 5, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alippi, A.M. , Lopez, A.C. , & Balatti, P.A. (2010) First report of Agrobacterium rubi and A. rhizogenes causing crown and root gall and hairy root on blueberry in Argentina. Plant Disease, 94, 1064. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. & Lipman, D.J. (1990) Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Aznar, A. , Chen, N.W. , Thomine, S. & Dellagi, A. (2015) Immunity to plant pathogens and iron homeostasis. Plant Science, 240, 90–97. [DOI] [PubMed] [Google Scholar]
- Baude, J. , Vial, L. , Villard, C. , Campillo, T. , Lavire, C. , Nesme, X. , & Hommais, F. (2016) Coordinated regulation of species‐specific hydroxycinnamic acid degradation and siderophore biosynthesis pathways in Agrobacterium fabrum . Applied and Environmental Microbiology, 82, 3515–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behura, R. , Kumar, S. , Saha, B. , Panda, M.K. , Dey, M. , Sadhukhan, A. , Mishra, S. , Alam, S. , Sahoo, D.P. , Sugla, T. & Sahoo, L. (2015) Cowpea [Vigna unguiculata (L.) Walp]. Methods in Molecular Biology, 1223, 255–264. [DOI] [PubMed] [Google Scholar]
- Binns, A.N. & Thomashow, M.F. (1988) Cell biology of Agrobacterium infection and transformation of plants. Annual Review of Microbiology, 42, 575–606. [Google Scholar]
- Boncompagni, E. , Østerås, M. , Poggi, M.‐C. , & le Rudulier, D. (1999) Occurrence of choline and glycine betaine uptake and metabolism in the family Rhizobiaceae and their roles in osmoprotection. Applied and Environmental Microbiology, 65, 2072–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet, E. , Lieberherr, D. , Tognolli, M. , Schneider, M. & Bairoch, A. (2007) UniProtKB/Swiss‐Prot. Methods in Molecular Biology, 406, 89–112. [DOI] [PubMed] [Google Scholar]
- Cangelosi, G.A. , Martinetti, G. , Leigh, J.A. , Lee, C.C. , Thienes, C. & Nester, E.W. (1989) Role for [corrected] Agrobacterium tumefaciens ChvA protein in export of β‐1,2‐glucan. Journal of Bacteriology, 171, 1609–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cankorur‐Cetinkaya, A. , Dikicioglu, D. & Oliver, S.G. (2017) Metabolic modeling to identify engineering targets for Komagataella phaffii: The effect of biomass composition on gene target identification. Biotechnology and Bioengineering, 114, 2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier, A. , Chevrot, R. , Dessaux, Y. & Faure, D. (2004) The assimilation of γ‐butyrolactone in Agrobacterium tumefaciens C58 interferes with the accumulation of the N‐acyl‐homoserine lactone signal. Molecular Plant‐Microbe Interactions, 17, 951–957. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , McAleer, K.L. , & Murrell, J.C. (2010) Monomethylamine as a nitrogen source for a nonmethylotrophic bacterium, Agrobacterium tumefaciens . Applied and Environmental Microbiology, 76, 4102–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contador, C.A. , Lo, S.K. & Chan, S.H.J. (2020) Metabolic analyses of nitrogen fixation in the soybean microsymbiont Sinorhizobium fredii using constraint‐based modeling. mSystems, 5, e00516–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Costa, D.M. , Suzuki, K. , & Yoshida, K. (2003) Structural and functional analysis of a putative gene cluster for palatinose transport on the linear chromosome of Agrobacterium tumefaciens MAFF301001. Journal of Bacteriology, 185, 2369–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeken, R. , Engelmann, J.C. , Efetova, M. , Czirjak, T. , Muller, T. , Kaiser, W.M. et al. (2006) An integrated view of gene expression and solute profiles of Arabidopsis tumors: A genome‐wide approach. The Plant Cell, 18, 3617–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicenzo, G.C. , Checcucci, A. , Bazzicalupo, M. , Mengoni, A. , Viti, C. , Dziewit, L. et al. (2016) Metabolic modelling reveals the specialization of secondary replicons for niche adaptation in Sinorhizobium meliloti . Nature Communications, 7, 12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Sahili, A. , Li, S.Z. , Lang, J. , Virus, C. , Planamente, S. , Ahmar, M. et al. (2015) A pyranose‐2‐phosphate motif is responsible for both antibiotic import and quorum‐sensing regulation in Agrobacterium tumefaciens . PLoS Pathogens, 11, e1005071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Y. , Zhang, H. & Cronan, J.E. (2013) Profligate biotin synthesis in α‐proteobacteria—A developing or degenerating regulatory system? Molecular Microbiology, 88, 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer, T. , Fischer, E. & Sauer, U. (2005) Experimental identification and quantification of glucose metabolism in seven bacterial species. Journal of Bacteriology, 187, 1581–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galíndez‐Nájera, S.P. , Llamas‐Martínez, M.A. , Ruiz‐Ordaz, N. , Juárez‐Ramírez, C. , Mondragón‐Parada, M.E. , Ahuatzi‐Chacón, D. et al. (2009) Cyanuric acid biodegradation by a mixed bacterial culture of Agrobacterium tumefaciens and Acinetobacter sp. in a packed bed biofilm reactor. Journal of Industrial Microbiology & Biotechnology, 36, 275–284. [DOI] [PubMed] [Google Scholar]
- Geske, T. , Vom Dorp, K. , Dörmann, P. & Hölzl, G. (2013) Accumulation of glycolipids and other non‐phosphorous lipids in Agrobacterium tumefaciens grown under phosphate deprivation. Glycobiology, 23, 69–80. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Mula, A. , Lachat, J. , Mathias, L. , Naquin, D. , Lamouche, F. , Mergaert, P. et al. (2019) The biotroph Agrobacterium tumefaciens thrives in tumors by exploiting a wide spectrum of plant host metabolites. New Phytologist, 222, 455–467. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Mula, A. , Lang, J. , Grandclement, C. , Naquin, D. , Ahmar, M. , Soulere, L. et al. (2018) Lifestyle of the biotroph Agrobacterium tumefaciens in the ecological niche constructed on its host plant. New Phytologist, 219, 350–362. [DOI] [PubMed] [Google Scholar]
- Goodner, B. , Hinkle, G. , Gattung, S. , Miller, N. , Blanchard, M. , Qurollo, B. et al. (2001) Genome sequence of the plant pathogen and biotechnology agent Agrobacterium tumefaciens C58. Science, 294, 2323–2328. [DOI] [PubMed] [Google Scholar]
- Groninger‐Poe, F.P. , Bouvier, J.T. , Vetting, M.W. , Kalyanaraman, C. , Kumar, R. , Almo, S.C. , et al. (2014) Evolution of enzymatic activities in the enolase superfamily: galactarate dehydratase III from Agrobacterium tumefaciens C58. Biochemistry, 53, 4192–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson, S. & Thiele, I. (2010) Computationally efficient flux variability analysis. BMC Bioinformatics, 11, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. , Jin, S. , Sun, D. , Hew, C.L. & Pan, S.Q. (2007) Recruitment of conjugative DNA transfer substrate to Agrobacterium type IV secretion apparatus. Proceedings of the National Academy of Sciences of the United States of America, 104, 20019–20024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. , Ye, J. , Gao, D. , Xu, N. & Yang, J. (2019) Agrobacterium‐mediated horizontal gene transfer: Mechanism, biotechnological application, potential risk and forestalling strategy. Biotechnology Advances, 37, 259–270. [DOI] [PubMed] [Google Scholar]
- Gurobi Optimization, I. (2014) Gurobi optimizer reference manual. http://www.gurobi.com [Accessed 22 December 2020]. [Google Scholar]
- Haudecoeur, E. , Planamente, S. , Cirou, A. , Tannières, M. , Shelp, B.J. , Moréra, S. et al. (2009) Proline antagonizes GABA‐induced quenching of quorum‐sensing in Agrobacterium tumefaciens . Proceedings of the National Academy of Sciences of the United States of America, 106, 14587–14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heirendt, L. , Arreckx, S. & Pfau, T. (2019) Creation and analysis of biochemical constraint‐based models using the COBRA Toolbox vol 3.0. Nature Protocols, 14, 639–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbing, M.E. & Fuqua, C. (2011) Antiparallel and interlinked control of cellular iron levels by the Irr and RirA regulators of Agrobacterium tumefaciens . Journal of Bacteriology, 193, 3461–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama, M. , Hashimoto, W. , Murata, K. , & Kawai, S. (2016) Comparative characterization of three bacterial exo‐type alginate lyases. International Journal of Biological Macromolecules, 86, 519–524. [DOI] [PubMed] [Google Scholar]
- Koduru, L. , Kim, H.Y. , Lakshmanan, M. , Mohanty, B. , Lee, Y.Q. , Lee, C.H. et al. (2020) Genome‐scale metabolic reconstruction and in silico analysis of the rice leaf blight pathogen, Xanthomonas oryzae . Molecular Plant Pathology, 21, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, J. & Faure, D. (2014) Functions and regulation of quorum‐sensing in Agrobacterium tumefaciens . Frontiers in Plant Science, 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, J. , Vigouroux, A. , El Sahili, A. , Kwasiborski, A. , Aumont‐Nicaise, M. , Dessaux, Y. et al. (2017) Fitness costs restrict niche expansion by generalist niche‐constructing pathogens. The ISME Journal, 11, 374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, J. , Vigouroux, A. , Planamente, S. , El Sahili, A. , Blin, P. , Aumont‐Nicaise, M. et al. (2014) Agrobacterium uses a unique ligand‐binding mode for trapping opines and acquiring a competitive advantage in the niche construction on plant host. PLoS Pathogens, 10, e1004444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, H. , Lin, Y. , Gao, F. , Zhang, C.T. & Zhang, R. (2014) DEG 10, an update of the database of essential genes that includes both protein‐coding genes and noncoding genomic elements. Nucleic Acids Research, 42, D574–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manasse, R.J. & Corpe, W.A. (1967) Chemical composition of cell envelopes from Agrobacterium tumefaciens . Canadian Journal of Microbiology, 13, 1591–1603. [DOI] [PubMed] [Google Scholar]
- Marasco, R. , Lago, C.T. , & De Felice, M. (1995) Utilization of cellobiose and other β‐D‐glucosides in Agrobacterium tumefaciens . Research in Microbiology, 146, 485–492. [DOI] [PubMed] [Google Scholar]
- Marty, L. , Vigouroux, A. , Aumont‐Nicaise, M. , Dessaux, Y. , Faure, D. & Moréra, S. (2016) Structural basis for high specificity of amadori compound and mannopine opine binding in bacterial pathogens. Journal of Biological Chemistry, 291, 22638–22649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullen, C.A. & Binns, A.N. (2006) Agrobacterium tumefaciens and plant cell interactions and activities required for interkingdom macromolecular transfer. Annual Review of Cell and Developmental Biology, 22, 101–127. [DOI] [PubMed] [Google Scholar]
- Meyer, T. , Thiour‐Mauprivez, C. , Wisniewski‐Dye, F. , Kerzaon, I. , Comte, G. , Vial, L. et al. (2019) Ecological conditions and molecular determinants involved in Agrobacterium lifestyle in tumors. Frontiers in Plant Science, 10, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, T. , Vigouroux, A. , Aumont‐Nicaise, M. , Comte, G. , Vial, L. , Lavire, C. et al. (2018) The plant defense signal galactinol is specifically used as a nutrient by the bacterial pathogen Agrobacterium fabrum . Journal of Biological Chemistry, 293, 7930–7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nester, E.W. (2014) Agrobacterium: Nature's genetic engineer. Frontiers in Plant Science, 5, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien, E.J. , Monk, J.M. & Palsson, B.O. (2015) Using genome‐scale models to predict biological capabilities. Cell, 161, 971–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth, J.D. , Thiele, I. & Palsson, B. (2010) What is flux balance analysis? Nature Biotechnology, 28, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S.Y. , Lee, S.J. , Oh, T.K. , Oh, J.W. , Koo, B.T. , Yum, D.Y. et al. (2003) AhlD, an N‐acylhomoserine lactonase in Arthrobacter sp., and predicted homologues in other bacteria. Microbiology, 149, 1541–1550. [DOI] [PubMed] [Google Scholar]
- Parke, D. (2000) Positive selection for mutations affecting bioconversion of aromatic compounds in Agrobacterium tumefaciens: analysis of spontaneous mutations in the protocatechuate 3,4‐dioxygenase gene. Journal of Bacteriology, 182, 6145–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resendis‐Antonio, O. , Hernández, M. , Salazar, E. , Contreras, S. , Batallar, G.M. , Mora, Y. et al. (2011) Systems biology of bacterial nitrogen fixation: High‐throughput technology and its integrative description with constraint‐based modeling. BMC Systems Biology, 5, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resendis‐Antonio, O. , Reed, J.L. , Encarnación, S. , Collado‐Vides, J. & Palsson, B. (2007) Metabolic reconstruction and modeling of nitrogen fixation in Rhizobium etli . PLoS Computational Biology, 3, 1887–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenburg, S.Y.A. , Seidl, M.F. , Judelson, H.S. , Vu, A.L. , Govers, F. & de Ridder, D. (2019) Metabolic model of the Phytophthora infestans–tomato interaction reveals metabolic switches during host colonization. mBio, 10, 00454–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossbach, S. , Schell, J. & de Bruijn, F.J. (1987) The ntrC gene of Agrobacterium tumefaciens C58 controls glutamine synthetase (GSII) activity, growth on nitrate and chromosomal but not Ti‐encoded arginine catabolism pathways. Molecular and General Genetics, 209, 419–426. [DOI] [PubMed] [Google Scholar]
- Saier, M.H. Jr. , Tran, C.V. & Barabote, R.D. (2006) TCDB: the transporter classification database for membrane transport protein analyses and information. Nucleic Acids Research, 34, D181–D186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz, R. , Kuepfer, L. & Sauer, U. (2007) Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli . Molecular Systems Biology, 3, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih, P.Y. , Chou, S.J. , Müller, C. , Halkier, B.A. , Deeken, R. & Lai, E.M. (2018) Differential roles of glucosinolates and camalexin at different stages of Agrobacterium‐mediated transformation. Molecular Plant Pathology, 19, 1956–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoh, S. , Quintana, N. , Kim, H.K. , Choi, Y.H. & Verpoorte, R. (2009) Metabolic changes in Agrobacterium tumefaciens‐infected Brassica rapa . Journal of Plant Physiology, 166, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Tian, W. & Skolnick, J. (2003) How well is enzyme function conserved as a function of pairwise sequence identity? Journal of Molecular Biology, 333, 863–882. [DOI] [PubMed] [Google Scholar]
- Tomlinson, A.D. & Fuqua, C. (2009) Mechanisms and regulation of polar surface attachment in Agrobacterium tumefaciens . Current Opinion in Microbiology, 12, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres, M.A. , Jones, J.D. & Dangl, J.L. (2006) Reactive oxygen species signaling in response to pathogens. Plant Physiology, 141, 373–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugalde, J.E. , Parodi, A.J. , & Ugalde, R.A. (2003) De novo synthesis of bacterial glycogen: Agrobacterium tumefaciens glycogen synthase is involved in glucan initiation and elongation. Proceedings of the National Academy of Sciences of the United States of America, 100, 10659–10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väremo, L. , Nielsen, J. & Nookaew, I. (2013) Enriching the gene set analysis of genome‐wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Research, 41, 4378–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlinde, E.M. , Hynes, M.F. , & Yost, C.K. (2014) Homoserine catabolism by Rhizobium leguminosarum bv. viciae 3841 requires a plasmid‐borne gene cluster that also affects competitiveness for nodulation. Environmental Microbiology, 16, 205–217. [DOI] [PubMed] [Google Scholar]
- Vattanaviboon, P. , Tanboon, W. & Mongkolsuk, S. (2007) Physiological and expression analyses of Agrobacterium tumefaciens trxA, encoding thioredoxin. Journal of Bacteriology, 189, 6477–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellosillo, T. , Vicente, J. , Kulasekaran, S. , Hamberg, M. & Castresana, C. (2010) Emerging complexity in reactive oxygen species production and signaling during the response of plants to pathogens. Plant Physiology, 154, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting, M.W. , Bouvier, J.T. , Gerlt, J.A. , & Almo, S.C. (2016) Purification, crystallization and structural elucidation of D‐galactaro‐1,4‐lactone cycloisomerase from Agrobacterium tumefaciens involved in pectin degradation. Acta Crystallographica Section F Structural Biology Communications, 72, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigouroux, A. & El Sahili, A. (2017) Structural basis for high specificity of octopine binding in the plant pathogen Agrobacterium tumefaciens . Scientific Reports, 7, 18033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Kim, S.H. , Natarajan, R. & Heindl, J.E. (2016) Spermidine inversely influences surface interactions and planktonic growth in Agrobacterium tumefaciens . Journal of Bacteriology, 198, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widiastuti, H. , Kim, J.Y. , Selvarasu, S. , Karimi, I.A. , Kim, H. , Seo, J.S. et al. (2011) Genome‐scale modeling and in silico analysis of ethanologenic bacteria Zymomonas mobilis . Biotechnology and Bioengineering, 108, 655–665. [DOI] [PubMed] [Google Scholar]
- Wood, D.W. , Setubal, J.C. , Kaul, R. , Monks, D.E. , Kitajima, J.P. , Okura, V.K. et al. (2001) The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science, 294, 2317–2323. [DOI] [PubMed] [Google Scholar]
- Xu, N. , Liu, L. , Zou, W. , Liu, J. , Hua, Q. & Chen, J. (2013) Reconstruction and analysis of the genome‐scale metabolic network of Candida glabrata . Molecular BioSystems, 9, 205–216. [DOI] [PubMed] [Google Scholar]
- Xu, N. , Ye, C. & Liu, L. (2018) Genome‐scale biological models for industrial microbial systems. Applied Microbiology and Biotechnology, 102, 3439–3451. [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Hu, X.P. & Ma, B.G. (2017) Construction and simulation of the Bradyrhizobium diazoefficiens USDA110 metabolic network: A comparison between free‐living and symbiotic states. Molecular BioSystems, 13, 607–620. [DOI] [PubMed] [Google Scholar]
- Yilmaz, L.S. & Walhout, A.J. (2017) Metabolic network modeling with model organisms. Current Opinion in Chemical Biology, 36, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zevenhuizen, L.P.T.M. (1971) Chemical composition of exopolysaccharides of Rhizobium and Agrobacterium . Journal of General Microbiology, 68, 239–243. [Google Scholar]
- Zhao, H. , Li, M. , Fang, K. , Chen, W. & Wang, J. (2012) In silico insights into the symbiotic nitrogen fixation in Sinorhizobium meliloti via metabolic reconstruction. PLoS One, 7, e31287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and its Supporting Information. Scripts for simulation in this study are available from the corresponding author upon reasonable request.