

Abstract
Directed evolution, which applies the principles of Darwinian evolution to a laboratory setting, is a powerful strategy for generating biomolecules with diverse and tailored properties. This technique can be implemented in a highly efficient manner using continuous evolution, which enables the steps of directed evolution to proceed seamlessly over many successive generations with minimal researcher intervention. Phage-assisted continuous evolution (PACE) enables continuous directed evolution in bacteria by mapping the steps of Darwinian evolution onto the bacteriophage life cycle and allows directed evolution to occur on much faster timescales compared to conventional methods. This protocol provides detailed instruction on evolving proteins using PACE and phage-assisted non-continuous evolution (PANCE), and includes information on the preparation of selection phage and host cells, assembly of a continuous flow apparatus, and performing and analyzing evolution experiments. This protocol can be performed in as little as two weeks to complete more than 100 rounds of evolution (complete cycles of mutation, selection, and replication) in a single PACE experiment.
Keywords: PACE, PANCE, continuous evolution, protein evolution, directed evolution
Introduction
A single round of conventional directed evolution of gene-encoded biomolecules requires four discrete steps: mutagenesis, gene expression, screening or selection, and replication1,2. Typically, performing these four steps to complete a single round of evolution requires several days and extensive researcher engagement. Because it often takes several rounds or more to evolve desirable properties, the ability to carry out laboratory evolution in an automated or self-sustaining manner can greatly increase the efficiency of obtaining optimal protein variants. The first demonstration of so-called “continuous” evolution was reported by Spiegelman and coworkers over five decades ago3. In this seminal work, the Qβ phage RNA genome was minimized and optimized through serial amplification with its cognate RNA-dependent RNA-replicase. Thirty years later, Joyce and coworkers established the feasibility of the continuous evolution of catalytic function4. This groundbreaking work was nevertheless limited to in vitro RNA evolution and not easily adaptable to proteins or in vivo settings.
Phage-assisted continuous evolution (PACE) was reported in 2011 as a versatile continuous method for protein evolution in bacterial cells5. By mapping the discrete steps of laboratory evolution onto the life cycle of M13 filamentous bacteriophage and using mutagenesis plasmids to diversify in situ, PACE requires minimal researcher intervention during evolution and allows for the evolution of new or improved activity in significantly shorter timespans compared to traditional methods. Here, we describe a protocol for PACE with instructions for preparation of cell lines, phage, and an apparatus for continuous flow. We also include instructions to adapt these components for phage-assisted non-continuous evolution (PANCE) and describe approaches for the analysis of evolution experiments.
Development of the protocol
During PACE, propagation of an evolving population of filamentous bacteriophage (“selection phage,” SP; Box 1) is dependent on the activity of a protein of interest (POI; Box 1) encoded in the phage genome (Fig. 1a)2,5. The SP exist in a “lagoon” of fixed volume that is continuously diluted with a culture of host E. coli cells. The SP population can only persist in the lagoon if their rate of propagation from infecting and replicating in host cells exceeds their rate of dilution. The SP are engineered to lack gene III (gIII), which encodes the minor coat protein pIII. Because pIII is absolutely essential for both host cell infection and phage particle detachment, it can be used to link POI activity and phage propagation. In PACE, gIII is provided on an accessory plasmid (AP; Box 1)6 in the host cells and its expression is triggered by the desired activity from the POI encoded in its place on the SP genome (Fig. 1b). Therefore, only SP encoding an active POI can induce pIII production and propagate faster than their rate of dilution5.
Box 1. Abbreviations and definitions.
POI – “protein of interest.” The protein whose activity is selected for in evolution.
SP – “selection phage.” M13 bacteriophage with gIII removed from the genome. The POI is encoded on the SP genome in its place under the control of the gIII promoter (PgIII)
AP – “accessory plasmid.” A plasmid in the host cells that encodes the phage survival genes, usually comprised of gIII transcribed from a promoter that is triggered by POI activity.
CP – “complementary plasmid.” A plasmid in the host cells that expresses any non-evolving proteins or other elements that are important for supporting the selection. Often, this plasmid encodes a polymerase that is activated by POI activity.
MP – “mutagenesis plasmid.” A plasmid in the host cells that expresses mutagenic genes upon induction with arabinose. The most commonly used mutagenesis plasmid, MP6, expresses the genes dnaQ926, dam, seqA, emrR, ugi, and cda1 from an arabinose inducible promoter
DP – “drift plasmid.” A plasmid in the host cells that expresses mutagenic genes upon induction with arabinose and also allows for genetic drift through the expression of gIII upon induction with anhydrotetracycline and phage infection. The most common drift plasmid, DP6, expresses the genes dnaQ926, dam, seqA, emrR, ugi, and cda1 from an arabinose inducible promoter and gIII from a hybrid phage shock/Tet promoter
S2060 – The E. coli cell line engineered for use in PACE (see Materials). The parent strain is DH10B. Modifications were made to enable infection by M13 phage, reduce biofilming, enable the use of n-hybrid systems, and enable colorimetric or luminescent detection of phage infection. The strain has the genotype F’ proA+B+ Δ(lacIZY) zzf∷Tn10 lacIQ1 PN25-tetR luxCDE Ppsp(AR2) lacZ luxR Plux groESL / endA1 recA1 galE15 galK16 nupG rpsL ΔlacIZYA araD139 Δ(ara,leu)7697 mcrA Δ(mrr-hsdRMS-mcrBC) proBA∷pir116 araE201 ΔrpoZ Δflu ΔcsgABCDEFG ΔpgaC λ–
S2208 – S2060 cells transformed with pJC175e, a plasmid that expresses gIII under the control of the phage shock promoter, allowing for activity-independent phage growth.
PACE host cells – S2060 cells transformed with a combination of the AP(s)/CP(s) needed for a given selection
Figure 1. Overview of phage-assisted continuous evolution (PACE).

(a) In a fixed-volume vessel, the “lagoon”, selection phage 3 encoding an evolving protein of interest (POI) trigger gIII expression from an accessory plasmid 6 in host E. coli cells, resulting in the production of the essential phage protein, pIII. The lagoon is continuously diluted with a culture of fresh host cells. All replicating DNA within the lagoon is mutagenized via an engineered mutagenesis plasmid 7 to provide genetic diversity. Only the SP containing genes encoding functional POI variants are capable of replicating faster than the rate of dilution, allowing them to persist in the lagoon. (b) Gene variants on the SP that encode active POIs trigger the expression of gIII from the AP, typically through the activation or recruitment of an RNA polymerase (RNAP). The MP expresses mutagenic genes under the control of the arabinose promoter; induction of mutagenesis occurs upon addition of arabinose to the growth media. (c) Established PACE selection strategies for various POI (blue) activities, including RNA polymerase activity (upper left), protein:DNA binding (upper middle), protein solubility (upper right), proteolysis of specific amino acid sequences (middle left), protein:protein binding( middle right), base editing (lower left), and incorporation of non-canonical amino acids by orthogonal aminoacyl-tRNA synthetases (lower right), and base editing (lower right). (d) Negative selections can be performed in PACE by linking the expression of a dominant-negative version of gIII, gIII-neg, to undesired activity, such that phage propagation requires SP encoding POI variants with the desired activity and lacking the undesired activity.
In order to simultaneously evolve and select for function, the host cells also contain a mutagenesis plasmid (MP; Box 1) 5,7,8. The MP subjects all DNA in the host cell to random mutagenesis, including the SP and host cell genomes, as well as any plasmids the cell contains (i.e. the AP and MP) (Fig. 1b). Critically, because the host cells replicate more slowly than the rate of lagoon turnover, any cells that accumulate mutations in their plasmids or genome are washed out of the system and replaced with fresh host cells. Thus, the only persisting genetic variation occurs in the SP, which replicate at a much faster rate of ~15 min per life cycle9.
PACE was initially validated by evolving T7 RNA polymerase (T7 RNAP) mutants that transcribe from the T3 promoter (PT3) (Fig. 1c)5. After 8 days of continuous evolution in PACE (corresponding to ~200 generations of T7 RNAP mutation, selection, and replication), promiscuous RNAPs that could initiate transcription from both the native (PT7) and target (PT3) promoters emerged5. While this initial example demonstrated the power of protein continuous evolution, the study also highlighted limitations in the early PACE technology that were addressed through subsequent developments.
For example, applications in cellular or therapeutic contexts would likely require PACE-evolved proteins to be highly selective in order to avoid detrimental off-target activity. Therefore, our laboratory developed negative selection PACE by linking undesired activity to the expression of a dominant-negative version of pIII, pIII-neg (Fig. 1d)10, which contains a deletion of 70 amino acids in its C domain that results in the production of phage particles unable to detach from the host cell membrane even in the presence of wild-type pIII11. Negative selection can be implemented simultaneously with a positive selection during PACE such that desired activity leads to gIII expression and undesired activity leads to gIII-neg expression10.
The ability to simultaneously mutagenize and select for activity is a critical component for continuous evolution. Mutagenesis must occur at levels sufficient to access active sequences during selection. The initial mutagenesis plasmid, MP1, increased mutagenesis rates ~100-fold over the basal E. coli mutation rate by expressing a dominant-negative variant of the E. coli DNA polymerase III proofreading domain (dnaQ926) and the error prone DNA polymerase V (polV) (Table 1)5. However, the resulting mutational rate of ~10−8 substitutions per base pair per generation was low compared to in vitro mutagenesis techniques, impeding access to variants containing many mutations1. To address this limitation, our laboratory developed inducible mutagenesis plasmids (MP2-MP6) with a wide range of cellular mutagenesis rates (Table 1)8. These MPs increase mutagenesis rates by expressing additional proteins that increase DNA methylation and deamination (dam and cda1), inhibit cellular repair pathways (ugi), and retain mutagenic bases in the cell (emrR)12. Uninduced background mutagenesis was decreased by strategically orienting the genes to remove potential unwanted promoters and by adding seqA to modulate DNA supercoiling8. In E. coli, full induction of the most mutagenetic MP (MP6) results in a ~300,000-fold increase in the mutation rate of chromosomal DNA (Table 1), which corresponds to ~2.3 substitutions per kb per generation in phage8. The greatly increased rate of mutagenesis provided by MP6 allowed evolution of T7 RNAP variants active on the T3 promoter in less than 10 h, compared to ~200 h when using MP1.
Table 1.
Inducible bacterial mutagenesis plasmids
| Plasmid | Genes | Mutagenesis rate (per bp per generation) |
Uninduced background mutagenesis (per bp per generation) |
|---|---|---|---|
| MP1 | dnaQ926, polV | 6.4 × 10−8 | 8.2 × 10−10 |
| MP2 | dnaQ926 | 9.9 × 10−8 | 2.8 × 10−10 |
| MP3 | dnaQ926, dam | 2.7 × 10−7 | 1.7 × 10−8 |
| MP4 | dam, seqA, dnaQ926 | 4.4 × 10−7 | 2.7 × 10−10 |
| MP5 | dam, seqA, dnaQ926, ugi, cda1 | 2.0 × 10−6 | 4.8 × 10−9 |
| MP6 | dam, seqA, dnaQ926, ugi, cda1, emrR | 6.2 × 10−6 | 2.4 × 10−9 |
In directed evolution, selection stringency impacts both the feasibility of evolution and the activity of surviving proteins. Selection stringency in PACE has been modulated using several strategies, which are discussed further below: (1) changing the rate of lagoon dilution, (2) using evolutionary stepping-stones to guide the evolving SP through a series of progressively more challenging targets, (3) applying drift by using a drift plasmid (DP), which mutagenizes DNA while providing pIII in a selection-independent manner to generate diversity with little selection pressure, and (4) adjusting the selection circuit on the AP that relates desired activity to pIII production5,7,8,10,13-18.
Increasing the lagoon dilution rate in PACE increases the required rate of replication for SP persistence. Thus, the SP must trigger gIII production at a higher rate, which in turn correlates to higher POI activity. Because selection in PACE is kinetically controlled, POIs that act slowly can be more difficult to evolve, since flow rates below 0.5 V/h can lead to biofilm formation in pump tubing. In these cases, phage-assisted non-continuous evolution (PANCE) can present a lower stringency alternative18-20. PANCE uses the same selection principles as PACE but is performed through serial dilution instead of under continuous flow (Supplementary Fig. 1a). In addition to lower stringency, PANCE also enables multiplexing of phage-based evolution as it can be performed easily in parallel (i.e. in deep-well plates)18,21. However, PANCE is slower and can be more time-intensive than PACE.
Stepping-stone approaches are particularly useful when evolving POIs with no or minimal starting activity7,14,17-19. In this strategy, an intermediate target encoded on a stepping-stone AP in PACE is used to bridge the wild-type activity and desired target activity. When POI variants with activity on the stepping-stone target can be accessed during PACE, but POI variants with activity on the final target are difficult to access, stepping-stone strategies can increase the likelihood of successful evolution. For example, a hybrid T7/T3 promoter was used to facilitate evolution of T7 RNAP to transcribe from the T3 promoter, as wild-type T7 RNAP had no detected activity on the T3 promoter5. Similarly, protease target stepping-stones14, protein binding target stepping-stones7, small-molecule substrate concentration stepping-stones17,18, and DNA sequence stepping-stones19,21 have also been used to guide evolution to successful endpoints during PACE.
Another option when evolving proteins that have slow kinetics or minimal starting activity is to implement evolutionary drift by using a drift plasmid (DP)7,15,21. DPs express the same mutagenic genes as MPs, but in addition express gIII from a hybrid phage shock/Tet promoter8. Induction of gIII in the presence of mutagenesis allows for diversification of SP in the absence of selection pressure to produce a starting library of mutants in the initial stages of a PACE experiment. Following this drift phase, gIII production from the drift plasmid can be reduced to increase selection pressure.
Finally, modulating stringency by tuning the selection circuit on the AP can be especially effective5,15,21,22. AP tuning tailors copy number, promoter strength, ribosome-binding site strength, and other parameters that define the relationship between target activity and gIII expression, and is usually required for optimal PACE outcomes. Optimal selection tuning usually requires insight into the basic mechanism of the POI, and will sometimes require adjustment beyond simple modifications on the AP, such as changes to the form or expression level of the POI on the SP.
Applications of the method
PACE is, in principle, capable of evolving any protein whose activity can be linked to expression of an essential phage gene in bacteria. In most cases, this essential phage gene is gIII; however, gVI has also been used21,23. Phage-based continuous evolution has been used to evolve polymerases5,10,22,24-26, DNA-binding proteins including TALE arrays12, Cas919,21, and transcription factors23, protein-protein interactions including antibody-like proteins16 and insecticidal proteins7, proteases14,27, aminoacyl-tRNA synthetases17,20, dehydrogenases18, and cytidine and deoxyadenosine deaminases used for base editing15,16,28 (Fig 1c).
PACE was initially applied to evolve RNA polymerases (RNAPs) using APs that placed gIII expression directly under the control of a specific promoter5 . Using this circuit in both positive and negative selection forms evolved highly active T7 RNAP variants with ~10,000-fold altered selectivity for PT3 over PT710. PACE has also been used to evolve T7 RNAP variants tolerant to C-terminal fusions24. Similar approaches using phage-derived transcription factors have been reported for phagemid-based evolution (PACEmid). In these studies, gVI was used as the selection marker because high gIII background expression from the synthetic promoters employed rendered host cells uninfectable23. PACEmid also allows for the generation of large combinatorial libraries of transcription factors and is compatible with continuous and non-continuous evolution23,29.
Evolving proteins other than polymerases requires more elaborate systems to couple protein activity to gene expression. A modification of the RNAP selection is a circuit in which POI DNA binding recruits a RNA polymerase to a promoter upstream of gIII using a 1-hybrid approach12. In these cases, the POI is linked to the ω subunit of E. coli RNAP (rpoZ) such that successful DNA binding recruits endogenous RNAP to enable gIII transcription from a nearby weak promoter. This strategy was used to evolve TALE array proteins12 and Cas9 variants19,21 with increased DNA specificity or altered DNA recognition requirements.
Analogously, a 2-hybrid system can be used to evolve protein-protein interactions in PACE by linking the POI (prey) to rpoZ and the bait to a DNA-binding protein such as the 434 cI phage repressor7. Our laboratory has used such a selection to evolve the insecticidal toxin Cry1Ac to bind a non-native receptor found in an insect pest resistant to the wild-type toxin7 and antibody-like proteins with improved soluble expression16.
In addition to recruitment, PACE selections can use polymerase activation by the POI in order to trigger gIII expression and phage propagation. For example, our laboratory created a selection for protease activity by first inactivating T7 RNAP through tethering to its natural inhibitor T7 lysozyme, then placing a substrate for proteolysis in the linker region between the two proteins, thus rendering liberation of T7 RNAP and transcription of gIII from PT7 dependent on protease cleavage of the target substrate in the linker. This selection was used to evolve drug-resistant HCV proteases27, as well as a TEV protease variant that recognizes and cleaves human IL-2314. T7 RNAP can also be inactivated by splitting into two halves. Split T7 RNAP complementation has been used as a folding reporter selection to evolve proteins with improved soluble expression16, as well as to select protein-protein interaction-dependent RNA polymerases24,25.
Modifications to the sequence of T7 RNAP can result in inactivation that is then corrected by POI activity. For example, aminoacyl-tRNA synthetases were evolved in PACE using amber suppression of premature stop codons in the T7 RNAP gene, as well as in gIII itself17. This selection was used to improve the selectivity of a promiscuous aminoacyl-tRNA synthetase towards incorporation of p-iodophenylalanine. T7 RNAP activation through sequence modification can also be used to evolve DNA modifying enzymes. For example, T7 RNAP inactivated through C-terminal fusion to a degron was used to evolve cytidine deaminases in base editors that could introduce a stop codon in the linker region through a successful C to T transition15. A similar approach was used to evolve deoxyadenosine deaminases in base editors that could fix a stop codon placed within the coding region of T7 RNAP28. These selections yielded new deaminase variants with increased activity and altered target sequence compatibility.
PACE can also be used to evolve proteins that act on small molecules, such as enzymes involved in metabolite biosynthesis. Our laboratory recently evolved methanol dehydrogenase (Mdh) variants with increased activity18. Mdh activity was linked to gIII production through use of a formaldehyde-responsive promoter. This work was also a key example of using PANCE as a strategy for evolving POIs with poor starting activity.
Comparisons with other methods
While many methods exist for discrete in vitro evolution, only a few examples of continuous directed evolution systems have been reported. These systems typically employ either viral vectors or orthogonal error-prone replication systems2. PACE and other phage/phagemid-based systems are the only bacterial continuous evolution methods available. While continuous evolution using continuous flow enables incredibly fast evolution times (often reaching dozens of generations per 24 hours) with minimal researcher intervention, it limits the scope of evolvable POIs towards those that can express and function in bacterial cells.
Chang Liu and coworkers created a system for continuous evolution in yeast (OrthoRep) which uses a linear plasmid system replicated using a dedicated DNA polymerase (DNAP1) orthogonal to endogenous yeast polymerases30-32. DNAP1 was engineered to increase its mutagenesis rate to exceed genomic error thresholds. As an initial example, OrthoRep was used to evolve drug resistant variants of malarial dihydrofolate reductases (DHFR)31. Like PACE, this system selects for organismal survival and should accommodate any selection that can link the activity of interest with yeast propagation. Though theoretically slower than PACE, OrthoRep enables continuous evolution in a eukaryotic system33 and should be compatible with POIs that require more complex post-translational modification or that can only be expressed functionally in eukaryotic cells.
Continuous evolution systems have also been developed for mammalian cells by relying upon viral life cycles to mutagenize and transfer genetic information. To date, two viruses, adenovirus type 5 and the RNA virus Sindbis, have been used for mammalian continuous evolution. Matthew Shoulders and coworkers used an engineered adenovirus type 5 with both its dedicated DNA polymerase (AdPol) and essential protease (AdProt) deleted from its genome6. Instead, an engineered error-prone AdPol variant (~10−6 mutations per bp per generation) and AdProt are provided by the mammalian cell genome. The evolving POI encoded on the viral genome is selected for its ability to trigger expression of AdProt and, therefore, viral propagation. This system was used to evolve doxycycline resistant tetracycline transactivator proteins. Bryan Roth and coworkers engineered the Sindbis virus to remove the capsid proteins E1, E2, and E3 from the viral genome and instead encode a POI34. These proteins are then provided in a POI activity-dependent manner on a vector in the mammalian cell host. Diversification is achieved by the naturally error-prone Sindbis viral RNA-dependent RNA polymerase, which is capable of mutagenizing the viral RNA genome at ~10−4 mutations per bp per generation. This system, known as “viral evolution of genetically actuating sequences” (VEGAS), has been used successfully to evolve transcription factors, GPCRs, and nanobodies.
Limitations of PACE
While the versatility of PACE has been demonstrated through its application to many classes of proteins, evolution of a given POI using PACE is only possible if its desired activity can be linked to gene transcription in bacteria. The selection circuit resulting from this coupling must have good dynamic range: we suggest activation of at least 10-fold. Furthermore, as low levels of pIII expression will render a host cell uninfectable9, the selection circuit must possess low background gIII expression levels or alternatively use a different phage gene such as gVI in place of gIII21,23.
Since the POI must be made faithfully in the E. coli cytosol, poor folding or the requirement for post-translational modifications such as disulfide bond formation or complex glycosylation will preclude evolution using PACE. Another limitation on the POI is imposed by the packaging limit of the SP (~11 kb max, corresponding to a ~5 kb limit on the POI/POIs)9: Thus, very large proteins or protein clusters may be unsuitable for PACE. To date, the largest single protein that has been evolved in PACE is ω-dSpCas9 (~4.4 kb)19,21.
PACE selections involving small-molecule substrates can be challenging to implement. The small molecule must be either synthesized by the host cell or readily cell permeable and available at a sufficiently high concentration to sustain the selection. In the case of exogenous addition, a large quantity of small molecule may be required as a single PACE experiment often turns over >10 L of media; substrate cost therefore can limit evolution of enzymes that catalyze transformations of precious small molecules. Additionally, free diffusion of a given enzymatic product between host cells can significantly complicate selections by triggering or repressing gene circuits in trans. In some cases, local concentrations of product molecules (such as formaldehyde in the methanol dehydrogenase example described above18) can activate gene circuits before their diffusion and dilution takes place, or an exogenous chemical sink can be used to limit in trans circuit modulation18.
Finally, selection in PACE takes place very rapidly, as a complete phage life cycle typically requires only ~15-30 min9. If the activity of interest proceeds more slowly than phage replication, selection in PACE could prove too stringent, even when lagoon flow rates are minimized and selection circuits are optimized.
Experimental Design.
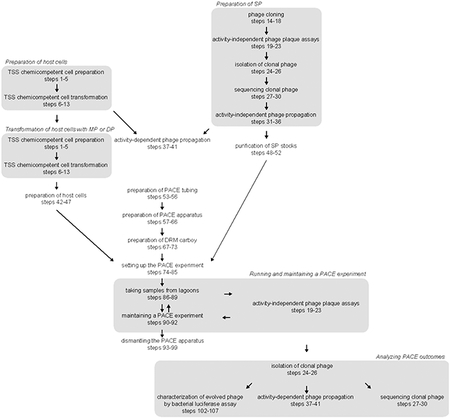
Preparation of PACE host cells and phage (Steps 1-52)
Like other members of the Inovirus genus, M13 filamentous bacteriophage form long, pilus-like particles of various coat proteins encasing a circular, single-stranded genome9,35. Eleven phage proteins (gI-gXI) are encoded on the wild-type genome; the engineered SP used in PACE encode all of these except for gIII. The SP genome can be transformed as circular dsDNA into host cells that provide gIII in trans to produce fully packaged and infectious phage particles. Accordingly, both cloning and expansion of SP stocks are typically performed using strain S2208 (Box 1), which contains a plasmid (pJC175e) that supplies gIII under the control of the phage shock promoter 5. Typically, after transformation or transduction into S2208 followed by a period of overnight growth to allow propagation, SP particles can be easily isolated by collecting the culture supernatant and passing it through a 0.22-μm filter to remove any residual host cells. The concentration of phage particles (titer) in SP stocks can be determined by performing a plaque assay using S2208 cells (Steps 19-23). Additionally, as the SP in each plaque are clonal, picking individual plaques (Steps 24-26) allows for the isolation of clonal SP populations for sequencing (Steps 27-30). SP stocks can be amplified if needed using S2208 cells (Steps 31-36).
Our laboratory engineered the PACE host cell strain S2060 (Box 1) from strain DH10β through several rounds of engineering5,10,12. Modifications include deletion of flu, pgaC and csgABCDEFG to substantially reduce biofilming and deletion of rpoZ to enable the use of n-hybrid systems utilizing fusions to the RNAP ω subunit. Furthermore, S2060 cells contain an F plasmid, which allows phage infection via the expressed F pilus, modified to incorporate lacI, tetR, and luxCDE, as well as lacZ under the control of the phage shock promoter. Phage infection triggers transcription from the phage shock promoter, leading to lacZ production that enables colorimetric visualization of phage infection or plaque activity by addition of Bluo-gal or related substrates to the growth media.
Prior to starting a PACE experiment, we recommend benchmarking the starting activity of the SP using S2060s transformed with the AP and complementary plasmid (CP), if using. Overnight propagation experiments in the absence of mutagenesis (Steps 37-41) can be performed with these cells to estimate the ability of the starting SP to survive under conditions of continuous flow. We have discovered empirically that SP will persist in a lagoon at a flow rate of 0.5 volumes/h if their overnight propagation on the host cells equals or exceeds ~103-fold. Typically, 102- to 103-fold overnight propagation is sufficient activity to begin a PACE experiment with the expectation that any SP that acquire a fitness-increasing mutation will be able to persist in the system. Selections with lower than 102-fold overnight propagation levels may still be used to initiate a PACE experiment by enabling an initial period of evolutionary drift using a DP. On the other hand, overnight propagation magnitudes that equal or exceed 105-fold imply significant starting activity of the POI and suggest that selection stringency should be increased before PACE is attempted.
Mutagenesis plasmids (MPs)8 are always transformed immediately before a PACE experiment, as increased background mutagenesis due to the leakiness of the arabinose promoter can lead to slow accumulation of mutations in host cells over time. To help prevent leaky expression of MP components, strains containing the MP or DP are always grown using glucose-containing media to further repress the arabinose promoter and are never stored for more than a week. Similarly, after their initial cloning, the SP used in PACE are re-isolated prior to being used to infect the lagoons in a process we term purification (Steps 48-52). SP purification decreases the likelihood of random recombination with host cell-encoded gIII to produce recombinant phage in the SP stock that can rapidly poison a PACE experiment.
Setting up the PACE apparatus (Steps 53-73)
The lagoons in PACE are continually diluted with host cell culture at a flow rate that is faster than the rate of host cell division, but slower than the replication rate of propagating phage. Continuous dilution is implemented by using two peristaltic pumps to cycle the chemostat (host cell culture) and the lagoons, respectively. The chemostat is diluted with fresh media to maintain constant cell density, thus ensuring that F pilus expression in the host cells is preserved, while the lagoons are diluted with fresh host cells from the chemostat for SP infection and propagation. Induction of the mutagenesis plasmid present in the host cells occurs solely in the lagoons such that the phage experience a high rate of mutagenesis while the chemostat cells remain un-mutagenized.
This protocol lists all necessary materials and instructions for setting up a PACE experiment using one chemostat and two lagoons (Fig. 2, Supplementary Fig. 2). We typically perform PACE experiments in 2-4 replicate lagoons, where one chemostat provides host cells to multiple identical lagoons evolving at the same time. While this protocol can be scaled to multiple chemostats, because of the resource-intensive nature of PACE, running many evolutions in parallel can be challenging. If a high degree of parallelization or throughput is required, PANCE may offer an attractive alternative (Steps 108-118).
Figure 2. Assembly of a PACE apparatus.

(a) List of parts required for assembly of a PACE apparatus. (b) Schematic for preparation of PACE tubing segments using parts listed in (a). (c) Construction of PACE tubing using parts shown in (b). (d) An example of a media carboy suitable for supporting the host-cell chemostat during PACE. (e) Assembly of the full PACE apparatus from parts depicted in (a-d).
Beginning PACE (Steps 74-85)
The initial stage of M13 phage infection involves binding of pIII to the F pilus9. Because expression of the F pilus decreases when temperatures are lower than 37 °C and when cells enter the late exponential stage of growth, maintaining the host cells at 37 °C and in mid-exponential growth in the chemostat is critical for the success of a PACE experiment. Mutagenesis and drift, if used, should be induced in the lagoon host cells at least an hour prior to infection with phage. This delay allows for adequate time for the host cells to begin expressing the mutagenic factors from the MP. Immediately after SP addition, the flow in and out of the lagoons is temporarily halted in order to allow infection to occur in the absence of dilution. Flow is typically resumed ~10 minutes post-infection.
Monitoring PACE (Steps 86-99)
The infection of lagoons with SP marks the beginning of the PACE experiment. After this point, the researcher should monitor the titer of phage in each lagoon to inform adjustments to flow rate or drift induction (Fig. 3a). During the course of the PACE experiment, samples of the lagoons can be drawn. We typically remove ~1 mL of culture from the lagoon every 8-16 h to determine the lagoon phage titer or to sequence the evolving gene pool.
Figure 3. Monitoring the PACE experiment.

(a) Simulated lagoon phage titers (pfu/mL) from a theoretical PACE experiment. In general, titers range from 105 to 108 pfu/mL. Flow rates can be increased when titers increase or remain stable to exert greater selection pressure. (b) Simulated lagoon phage titers (pfu/mL) from a theoretical PACE experiment incorporating drift through use of a drift plasmid (DP). [aTc] = anhydrotetracycline concentration. (c) Representative examples of plaque assays using wild-type M13 phage (top), ΔgIII selection phage5 containing a ~2-kb gene encoding the protein of interest (middle), and ΔgIII SP with a ~4-kb insert (bottom) using S2208 cells. From the top left quadrant, movement clockwise along the plate represents a 100-fold dilution of phage relative to the previous quadrant.
Phage titer can be estimated by qPCR (Steps 119-126); however, the most precise way to measure titer is through plaque assays (Steps 19-23) (Fig. 3b). A drop in lagoon phage titer (reduction of 10-fold or greater) is typically associated with POIs with little or no activity. A lagoon whose titer decreases to ≤103 pfu (plaque-forming units)/mL is considered to have “washed out”: as library size is proportionate to SP population size, such a small phage population is unlikely to find a fitness-increasing mutation to recover. Conversely, an increase in lagoon titer (expansion of 10-fold or greater) often indicates the acquisition of a fitness-increasing mutation.
Thus, lagoon flow rates should be increased if titers either remain stable (suggesting a lack of selection pressure) or increase (Fig. 3a). We suggest increasing flow rates in increments of 0.5 to 1 volume/h. After such an increase, lagoon titers will usually drop. A recovery of titer after such a decrease can signify an increase in fitness from the acquisition of beneficial mutations. In the course of a typical PACE, flow rate is increased every 24-48 h with rates ranging between 0.5 and 3 volumes/h. As replication time of M13 phage is proportionate to genome size, SP carrying smaller POIs replicate faster and can persist at higher rates of dilution compared to SP encoding larger POIs.
If using drift, the researcher should expect lagoon phage titers to remain consistently high (108 pfu/mL or greater) during DP induction (Fig. 3c). Decreases in titer are typically observed after drift is discontinued; however, if a solution that improves fitness has been found, titers should increase subsequently. Once the lagoons reach a flow rate of 3 volumes/h or higher while maintaining a high titer of phage, it is unlikely that further improvements in POI fitness will be achieved without changing selection stringency in ways other than increasing flow rate. At this point, we recommend ending the PACE experiment.
Characterization of evolved phage (Steps 100-107)
Clonal phage from lagoon samples can be isolated using plaque assays. These assays can be performed in either a POI activity-independent (with S2208 cells, Steps 19-23) or POI activity-dependent manner (with host cells containing the APs used for evolution, Steps 37-41). The latter option can be advantageous as it selects for SP encoding POIs with robust activity on the selection circuit encoded by the AP.
The POI can be directly amplified by PCR from plaques and the resulting PCR product can be used for Sanger DNA sequencing (Steps 27-30). We typically sequence at least 8 plaques from the last PACE sample collected per lagoon, although we will often sequence plaques from earlier samples to arrive at a clearer picture of when specific mutations arose, if they arose simultaneously with other mutations or independently, and if the solutions found are similar across different replicates (lagoons) of the same evolution.
While there are many ways to validate the activity of evolved POIs, the simplest way to compare between multiple phage clones is to perform luciferase assays (Steps 102-107). In most APs cloned in our laboratory, pIII is translationally coupled to bacterial luciferase, allowing POI activity to be estimated by infecting host cells with an excess of phage and using luminescence as an approximate readout for gIII expression (Supplementary Fig. 1b). An alternative approach is to subclone promising hits into an expression plasmid for use with this luciferase assay.
PANCE (Steps 108-118)
PANCE uses the same selection strategies as PACE but with iterative dilution of lagoons instead of continuous dilution (Supplementary Fig. 1a)18-20. PANCE may be a good alternative to PACE for POIs that have low or slow activity, or when a higher throughput is desired. The lower stringency nature of PANCE comes from the increased time allowed for phage propagation: whereas the lagoons in PACE are turned over every ~20 minutes to 2 h, PANCE allows phage to incubate with the same host cells for 6-18 h. As a result, evolution in PANCE is much slower than in PACE.
Parallel evolution is more easily achieved using PANCE than PACE. While PACE becomes prohibitive in space, equipment, and cost once several chemostat/lagoon pairs are running simultaneously, PANCE can be easily performed in 96 well deep-well plates18. PANCE in multi-well plates enables the parallel evolution towards many different targets or many replicates of the same evolution. Increasing the number of simultaneous evolutions can make determination of phage titers tedious due to the large number of plaque assays required. For this reason, we often use a qPCR method for phage titer determination when faced with a large number of samples (Steps 119-126).
In place of lagoon flow rate adjustments, stringency in PANCE can be modulated by changing the dilution factor of the phage or the incubation time. PANCE is also compatible with drift; however, we do not recommend performing drift for greater than 6-8 h as it may lead to an increase in gIII recombination.
Materials
Biological materials
E. coli S2060 (Addgene 105064), derived from DH10β, genotype F’ proA+B+ Δ(lacIZY) zzf∷Tn10 lacIQ1 PN25-tetR luxCDE Ppsp(AR2) lacZ luxR Plux groESL / endA1 recA1 galE15 galK16 nupG rpsL ΔlacIZYA araD139 Δ(ara,leu)7697 mcrA Δ(mrr-hsdRMS-mcrBC) proBA∷pir116 araE201 ΔrpoZ Δflu ΔcsgABCDEFG ΔpgaC λ–
E. coli S2208, defined as strain S2060 transformed with plasmid pJC175e
Plasmids and phage
Mutagenesis plasmid MP6 (Addgene 69669)
Drift plasmid DP6 (Addgene deposition in progress)
pJC175e (Addgene 79219)
- Selection phage5, derived from bacteriophage M13 (V00604.2, ATCC 15669-B1) with the following modifications, see Supplementary Note 1:
- Removal of gIII
- Substitution of gIII RBS with synthetic RBS36
- Insertion of gene of interest immediately following synthetic RBS
- Insertion of the following sequence, containing an artificial promoter and RBS for production of downstream gVI: tctagaaggagattttcaacatgggctagcacagccctaggtattatgctagcgtggtgtatttggaataaggagtcaaaa
Primers
AB1396 (ACAGAGAGAATAACATAAAAACAGGGAAGC) (Integrated DNA Technologies)
AB1792 (TAATGGAAACTTCCTCATGAAAAAGTCTTTAG) (Integrated DNA Technologies)
M13fwd (CACCGTTCATCTGTCCTCTTT) (Integrated DNA Technologies)
M13rev (CGACCTGCTCCATGTTACTTAG) (Integrated DNA Technologies)
Reagents
2xYT broth (United States Biological T9200)
LB broth (United States Biological L1505)
- Davis Rich Media:
- Harvard Custom Media A (United States Biological CS050H-001)
- Harvard Custom Media C (United States Biological CS050H-003)
- TWEEN-20 (Sigma-Aldrich P1379) or TWEEN-80 (Sigma-Aldrich P4780)
Agar (United States Biological A0930)
SOC media (New England Biolabs B9020S)
Carbenicillin or ampicillin (Gold Biotechnology C-103)
Chloramphenicol (Gold Biotechnology C-105)
Tetracycline (Gold Biotechnology T-101)
Streptomycin (Gold Biotechnology S-150)
L-arabinose (Gold Biotechnology A-300)
Glucose (Sigma-Aldrich G7021)
Anhydrotetracycline (Sigma-Aldrich 27919)
Bluo-gal (Gold Biotechnology B-673-10)
- Trace metals:
- (NH4)6Mo7O24 × 4H2O (Sigma-Aldrich 431346)
- H3BO3 (Sigma-Aldrich B6768)
- CoCl2 (Sigma-Aldrich 232696)
- CuSO4 (Sigma-Aldrich C1297)
- MnCl2 (Sigma-Aldrich 328146)
- ZnSO4 × 7H2O(Sigma-Aldrich Z0251)
Milli-Q filtered water
Ethanol (Koptec V1016)
CAUTION: ethanol is highly flammable. Keep away from open flames and work in a well-ventilated area.
N,N-Dimethylformamide (Sigma-aldrich D4551)
CAUTION: N,N-Dimethylformamide is considered toxic. Wear the appropriate personal protective equipment when handling.
DNAseI with DNAseI buffer (New England Biolabs M0303)
Phusion HF (ThermoFisher F530)
dNTPs (New England Biolabs N0447S)
Sybr Green (Invitrogen S7563)
Agarose (Lonza, 50004)
QIAprep Spin Miniprep Kit (Qiagen 27106)
TempliPhi Amplification kit (GE Healthcare 25640050)
Equipment
General
Incubator (Fisher Scientific isotemp incubator, similar: 15-103-0515)
Biological shaker (New Brunswick innova44, M1282)
Benchtop centrifuge (Eppendorf centrifuge 5920R)
Water bath, 42 °C (Thermo microprocessor controlled 280 series, 3166686)
PCR machine (Biorad C1000 Touch thermal cycler)
qPCR machine (Biorad CFX96 Real-Time system)
Plate reader (Tecan infinite M1000 pro)
Culture tubes (Corning 352059)
96 deep well plate (Eppendorf 951033502)
50mL Bioreactor (Corning 431720)
Petri dish (Corning 351029)
Petri X-plate (VWR 25384-308)
Eppendorf tubes (Eppendorf 022431021)
PCR strip tubes (Corning PCR-0208-C)
Clear 96-well plate seal (BioRad MSB1001)
White-well PCR plates (BioRad HSP9655)
Clear PCR strip tube cap (BioRad TCS0803)
White-well PCR strip tubes (BioRad TLS0851)
Library strips (VWR 83009-680)
Bottle-top vacuum filter system (0.22 μm; Corning 431097)
Syringe filter, PES, 30 mm (0.22 μm; Santa Cruz Biotechnology sc-395291)
Syringe filter, PVDF, 13 mm (0.22 μm; Santa Cruz Biotechnology sc-358811)
PACE-specific
Warm room (temperature-controlled to 37 °C)
Masterflex L/S Digital Drive, 100 RPM, 115/230 VAC (Cole-Parmer EW-07522-30)
Masterflex L/S 8-channel multichannel pump head for microbore tubing (Cole-Parmer EW-07534-08)
Six Channel Programmable Syringe Pump (New Era NE-1600)
Cimarec i Poly 15 and Multipoint Stirrers, 15 point (Thermo Scientific 50093538)
Microbore 2-stop, silicone (platinum cured) 0.89 mm ID (Cole-Parmer EW-06421-26); referred to in this protocol as “0.89 mm ID 2-stop line”
Microbore 2-stop, silicone (platinum cured) 1.42 mm ID (Cole-Parmer EW-06421-34); referred to in this protocol as “1.42 mm ID 2-stop line”
Microbore 2-stop, silicone (platinum cured) 2.06 mm ID (Cole-Parmer EW-06421-42); referred to in this protocol as “2.06 mm ID 2-stop line”
Cole-Parmer clear tubing, 1/16 in ID x 1/8 in OD (Cole-Parmer EW-06422-02); referred to in this protocol as “1/16 in ID PVC tubing”
Male Luer with lock ring x 1/16 in ID hose barb, PVDF (Cole-Parmer WU-45513-00); referred to in this protocol as “1/16 in ID male Luer adapter”
Male Luer with lock ring x 3/32 in ID hose barb, PVDF (Cole-Parmer WU-45513-02); referred to in this protocol as “3/32 in ID male Luer adapter”
Male Luer with lock ring x 1/8 in ID hose barb, PVDF (Cole-Parmer WU-45513-04); referred to in this protocol as “1/8 in ID male Luer adapter”
Male Luer with lock ring x ¼ in ID hose barb, Nylon (Cole-Parmer EW-45505-19); referred to in this protocol as “1/4 in ID male Luer adapter”
Male Luer Cap, PVDF (Cole-Parmer EW-45513-56)
Female Luer x 1/16 in ID hose barb adapter, PVDF (Cole-Parmer WU-45512-00); referred to in this protocol as “1/16 in ID female Luer adapter”
Female Luer x 3/32 in ID hose barb adapter, PVDF (Cole-Parmer WU-45512-14); referred to in this protocol as “3/32 in ID female Luer adapter”
Female Luer x 1/8 in ID hose barb adapter, PVDF (Cole-Parmer WU-45512-04); referred to in this protocol as “1/8 in ID female Luer adapter”
Female Luer x ¼ in ID hose barb adapter, Nylon (EW-45502-20); referred to in this protocol as “1/4 in ID female Luer adapter”
Female Luer Cap, PVDF (Cole-Parmer EW-45512-28)
Barbed Y-Connectors, Polypropylene, 1/16 in ID (Cole-Parmer EW-40727-41); referred to in this protocol as “Y-connector”
Sterile Luer-Lock syringes, 50 mL capacity (VWR 80076-428)
Blunt fill needle, 18 G x 1 ½ in (VWR BD305180); referred to in this protocol as “18 G x 1.5 in needle”
Air-Tite veterinary needle, 16 G x 4 in (VWR 89219-278); referred to in this protocol as “16 G x 4 in needle”
Air-Tite veterinary needle, 22 G x 4 in (VWR 89219-270); referred to in this protocol as “22 G x 4 in needle”
Media bottle, 100 mL w/out Cap (VWR 89012-114)
Mono-Mold Standard Stir Bar, 1 1/2 in length x 3/8 in (Cole-Parmer UX-04770-50); referred to in this protocol as “1.5 in stir bar”
Corning High-Temperature PBT Cap, Red, Open (VWR 11310-688)
PTFE-Faced Silicone Septa (Bellco Glass 5637-00030)
Glass vials, 22 mL volume with PTFE/silicone septa (Cole-Parmer EW-08922-48)
½ in x 1/8 in disposable stir bars (VWR 58947-140); referred to in this protocol as “1/2 in stir bar”
Nalgene heavy-duty polypropylene carboy, 10 L with cap (Thermo Fisher 2226-0020)
Nalgene polypropylene 3-port filling/venting closure, cap size 83B, fits tubing 6.4 mm (1/4 in) ID (VWR 16225-229)
Reagent setup
0.1 M CaCl2 solution
Dissolve CaCl2 to a final concentration of 0.1 M in water and sterilize using a 0.22 μm syringe filter. Store at room temperature (~25 °C) indefinitely.
Trace metals solution
Mix together equal volumes of the following six solutions. Store at room temperature indefinitely.
(NH4)6Mo7O24 × 4H2O solution: Dissolve (NH4)6Mo7O24 × 4H2O to a final concentration of 3.75 mg/mL in water. Store at room temperature indefinitely.
H3BO3 solution: Dissolve H3BO3 to a final concentration of 25 mg/mL in water. Store at room temperature indefinitely.
CoCl2 solution: Dissolve CoCl2 to a final concentration of 7.14 mg/mL in water. Store at room temperature indefinitely.
CuSO4 solution: Dissolve CuSO4 to a final concentration of 2.6 mg/mL in water. Store at room temperature indefinitely.
MnCl2 solution: Dissolve MnCl2 to a final concentration of 15.8 mg/mL in water. Store at room temperature indefinitely.
ZnSO4 × 7H2O solution: Dissolve ZnSO4 × 7H2O to a final concentration of 2.88 mg/mL in water. Store at room temperature indefinitely.
Davis Rich Media (DRM)
To 900 mL water, add Harvard Custom Media A (18.1 g) and TWEEN-20 or TWEEN-80 (1 mL), then autoclave to sterilize at 121.0 °C. Allow to cool completely. Separately, add Harvard Custom Media C (5.9 g), 5 μL of 0.1 M CaCl2 solution, and 6 μL of Trace Metals solution to 100 mL water and sterilize using a Corning bottle-top vacuum filter system. Add the Harvard Custom Media C solution to the cooled Harvard Custom Media A solution to make complete DRM. Store in the dark at 4 °C for up to a year.
LB liquid media
Add LB media powder to a final concentration of 25 g/L in water. Mix to dissolve, then autoclave to sterilize at 121.0 °C. Store at room temperature indefinitely.
2xYT liquid media
Add 2xYT media powder to a final concentration of 31 g/L in water. Mix to dissolve, then autoclave to sterilize at 121.0 °C. Store at room temperature indefinitely.
2xYT agar
Add 2xYT media powder and agar to a final concentration of 31 g/L and 1.5% (w/v), respectively, in water. Autoclave to sterilize at 121.0 °C. Store at room temperature in the dark for up to six months.
2xYT top agar
Melt 2xYT agar using a microwave until completely liquid. Dilute hot 2xYT agar with room-temperature 2xYT liquid media to a final agar concentration of 0.6% (w/v) and mix thoroughly. Incubate in a water bath at 55 °C to keep top agar liquid until use. Can be kept in the 55 °C water bath for up to 1 day.
Carbenicillin/ampicillin stock solution
Dissolve carbenicillin or ampicillin to a final concentration of 50 mg/mL in water. Store at −20 °C for up to 6 months.
Chloramphenicol stock solution
Dissolve chloramphenicol to a final concentration of 25 mg/mL in ethanol. Store at −20 °C for up to 6 months.
Tetracycline stock solution
Dissolve tetracycline to a final concentration of 10 mg/mL in 50% (v/v) ethanol in water. Store at −20 °C for up to 6 months.
Streptomycin stock solution
Dissolve streptomycin to a final concentration of 50 mg/mL in water. Store at −20 °C for up to 6 months.
L-arabinose stock solution
Dissolve L-arabinose to a final concentration of 1 M in water and sterilize using a Corning bottle-top vacuum filter system. Store at room temperature for up to 3 months.
Glucose stock solution
Dissolve glucose to a final concentration of 1 M in water and sterilize using a Corning bottle-top vacuum filter system. Store at room temperature for up to 3 months.
Anhydrotetracycline stock solution
Dissolve anhydrotetracycline to a final concentration of 1 mg/mL in ethanol. Store at −20 °C for up to 6 months.
Bluo-gal stock solution
Dissolve Bluo-gal to a final concentration of 2% (w/v) in DMF. Store in the dark at −20 °C for up to 6 months.
2X TSS
Add MgCl2, PEG 3350, and DMSO to LB media at final concentrations of 20 mM, 10% w/v, and 5% v/v, respectively, and sterilize using a Corning bottle-top vacuum filter system. Store at 4 °C indefinitely.
5X KCM solution
Add KCl, CaCl2, and MgCl2 to H2O at final concentrations of 100 mM, 30 mM, and 50 mM, respectively. Store at room temperature indefinitely.
Procedure
TSS chemicompetent cell preparation • Timing 6 h
CRITICAL: This is a general protocol for any E. coli strain used in this protocol. Protocol can be scaled to any necessary volume.
Dilute a saturated culture of cells 1,000-fold into 50 mL of 2xYT liquid media supplemented with the appropriate antibiotics and grow at 37 °C in a biological shaker to OD600 ~0.4–0.6 (typically 4-6 h).
Pellet cells by centrifugation at 4,000 g for 10 min at 4 °C
Discard supernatant and resuspend the pellet by gentle stirring in 2 mL of ice-cold LB media
Add 2 mL of ice-cold 2X TSS, stir to mix the cell suspension completely
-
Aliquot into 100 μL portions in microcentrifuge tubes and freeze either on dry ice or in liquid N2. Store at −80 °C until use.
■ PAUSE POINT Prepared chemicompetent cells can be stored for up to 6 months at −80 °C without freeze-thaw cycles
TSS chemicompetent cell transformation • Timing 2 h
CRITICAL: This is a general protocol for any E. coli strain used in this protocol. Protocol can be scaled to any necessary volume.
-
6.
Thaw a 100 μL aliquot of competent cells on ice
-
7.
Prepare a mixture of plasmids (1-2 μL each (< 100 ng); up to three plasmids per transformation) in a final volume of 100 μL 1X KCM solution. Chill on ice.
-
8.
Add the thawed competent cells to the pre-chilled plasmid mixture on ice. Stir gently with a pipette tip to mix.
-
9.
Incubate on ice for 10 min
-
10.
Heat shock at 42 °C in a water bath for 75 s.
-
11.
Transfer cells back onto ice, then immediately add 500 μL of SOC media.
-
12.
Recover cells at 37 °C in a biological shaker for 1-1.5 h CRITICAL STEP: Note that cells transformed with plasmids containing ampicillin or carbenicillin resistance do not require this recovery step and can be plated directly after heat shock.
-
13.
Plate on Petri dishes containing 2xYT/agar with the appropriate antibiotics and incubate at 37 °C for 16-18 h.
■ PAUSE POINT Transformed cell colonies plated on 2xYT/agar media can be stored at 4 °C for up to 1 week
? TROUBLESHOOTING
Phage cloning • Timing 2 d
-
14.
Transform assembled phage genomes into S2208 cells by repeating Steps 1-10 using S2208 cells and substituting the plasmid used in Step 7 with the entire yield of assembled phage genome
-
15.
Immediately after the heat shock step (Step 10), dilute the cell mixture into 10 mL of antibiotic-free 2xYT liquid media in a culture tube
-
16.
Grow at 37 °C for 16-18 h in a biological shaker.
-
17.
Pellet 1 mL of the resultant culture by centrifugation at 8000 g for 2 min. Collect and filter the supernatant using a 3 mL syringe fitted with a 13 mm 0.22 μm PVDF syringe filter to remove residual cells.
■ PAUSE POINT Filtered phage can be stored at 4 °C or −20 °C indefinitely; however, phage titers can gradually decrease over time.
-
18.
Collected phage may be clonally isolated through plaque assays (Steps 19-23) and sequenced (Steps 27–30).
? TROUBLESHOOTING
Activity-independent phage plaque assays • Timing 2 d
-
19.
Dilute a saturated culture of S2208 cells in 2xYT liquid media supplemented with 50 μg/mL carbenicillin or ampicillin and grow to an OD600 of 0.6–0.9 at 37 °C in a biological shaker.
■ PAUSE POINT When cells reach the appropriate density, they may be kept at room temperature until use for up to 6 h.
-
20.
Dilute phage serially in three 100-fold increments to yield four total samples (undiluted, 102-, 104-, and 106-fold diluted)
-
21.
Transfer 10 μL of each dilution into one tube of a library strip (or other disposable tube that can hold up to 1.5 mL of liquid).
-
22.
To each phage dilution, add 150 μL of S2208 cells, followed by 1 mL of warm (~55 °C) 2xYT top agar. (Optional: add Bluo-gal to 2xYT top agar at a final concentration of 0.04% (w/v) to produce more visually distinct plaques.)
-
23.
Mix by pipetting up and down once, then plate each phage dilution onto one quadrant of a Petri X-plate containing 2 mL of solidified 2xYT agar in each quadrant. Incubate plates overnight (16-20 h) at 37 °C. The titer of the phage stock solution can be determined by the following formula: Titer (in pfu/mL) = (# plaques in quadrant)*(dilution factor of quadrant)*100
? TROUBLESHOOTING
Isolation of clonal phage • Timing 2 d
-
24.
Pick a single plaque from a plaque assay plate (Steps 19-23) (this corresponds to a single clonal phage) by gently touching a P10 or P20 pipette tip to the surface of the top agar. Place the pipette tip into 2-3 mL of DRM and grow at 37 °C in a biological shaker for 16-20 h.
-
25.
Pellet the cells by centrifuging at 8000 g for 2 min, then collect and filter the supernatant using a 3 mL syringe fitted with a 13 mm 0.22 μm PVDF syringe filter to remove residual cells.
■ PAUSE POINT Filtered phage can be stored at 4 °C or −20 °C indefinitely; however, phage titers can gradually decrease over time. Isolated clonal phage should be purified before use in PANCE or PACE (Steps 48-52).
-
26.
Determine the phage titer using an activity-independent phage plaque assay (Steps 19-23).
Sequencing clonal phage • Timing 1 d
-
27.
Pick a single plaque from a plaque assay plate (Steps 19-23) (this corresponds to a single clonal phage) by gently touching a P10 or P20 pipette tip to the surface of the top agar.
-
28.
Amplify the phage DNA, using either option A to amplify the POI insert on the phage by PCR or option B to amplify the entire phage genome by rolling circle amplification (RCA).
- Amplify the POI insert on the phage by PCR
- Place the tip from Step 27 into a PCR reaction mixture containing the following:
Component Amount Final concentration Nuclease-free water 20 μL 5x Phusion HF Buffer 5 μL 1x dNTPs 0.5 μL 0.2 mM each dNTP Phusion HF polymerase 0.5 μL 1 U AB1396 0.125 μL 1 μM AB1792 0.125 μL 1 μM Total 26.25 μL - Let the tip sit in the reaction for at least 30 sec before removing. Do not discard tip after removing; the remaining material will be used to inoculate a culture to amplify the phage clone in Step 29.
- Run the PCR reaction using an extension time that is appropriate for the size of the gene encoding the POI (approximately 15 s per kb for Phusion HF polymerase).
Cycle number Denature Anneal Extend Final 1 98 °C, 2 min 2-41 98 °C, 10s 60 °C, 20 s 72 °C, 15 s per kb 42 72 °C, 2 min 12 °C, 30 s - PCR product can be used directly for Sanger sequencing
- Amplify the entire phage genome by rolling circle amplification (RCA)
- Using the TempliPhi Amplification Kit, place the tip from Step 27 into the provided sample buffer and follow the provided directions for amplification. Do not discard tip after removing from the sample buffer; the remaining material will be used to inoculate a culture to amplify the phage clone in Step 29.
- Submit amplified DNA directly for Sanger sequencing.
? TROUBLESHOOTING-
29.Place the pipette tip saved from Step 28 in a culture tube containing 3 mL of DRM and incubate in a biological shaker at 37 °C for 16-20 h.
-
30.Pellet the cells by centrifuging at 8000 g for 2 min, then collect and filter the supernatant using a 3 mL syringe fitted with a 13 mm 0.22 μm PVDF syringe filter to remove residual cells.■ PAUSE POINT Filtered phage can be stored at 4 °C or −20 °C indefinitely; however, phage titers can gradually decrease over time. Isolated clonal phage should be purified before use in PANCE or PACE
SP stock amplification (activity-independent phage propagation) • Timing 2 d
-
31.
Dilute a saturated culture of S2208 cells 1,000-fold into DRM media supplemented with 50 μg/mL carbenicillin or ampicillin and grow at 37 °C in a biological shaker to OD600 ~0.4-0.6.
-
32.
Infect cells with the desired phage to be amplified at a starting titer of ~104 pfu/mL
-
33.
Grow at 37 °C in a biological shaker for 16-20 h
-
34.
Pellet the cells by centrifuging at 8000 g for 2 min and filter the supernatant using a 3 mL syringe fitted with a 13 mm 0.22 μm PVDF syringe filter to remove residual cells.
-
35.
Store the filtered supernatant at 4 °C.
■ PAUSE POINT Filtered phage can be stored at 4 °C or −20 °C indefinitely; however, phage titers can gradually decrease over time. Isolated clonal phage should be purified before use in PANCE or PACE
-
36.
Determine phage titer using an activity-independent phage plaque assay (step 19-23).
Activity-dependent phage propagation • Timing 2 d
-
37.
Transform S2060 cells with the AP(s)/CP(s) of interest (steps 1-13).
-
38.
Dilute a saturated culture of the cells 1,000-fold into DRM media supplemented with the appropriate antibiotics and grow at 37 °C in a biological shaker to OD600 of 0.4–0.6.
-
39.
Infect cells with phage at a starting titer of ~104 pfu/mL, then grow at 37 °C in a biological shaker for another 16-20 h.
-
40.
Pellet the cells by centrifuging at 8000 g for 2 min and filter the supernatant using a 3 mL syringe fitted with a 13 mm 0.22 μm PVDF syringe filter to remove residual cells. Store the filtered supernatant at 4 °C.
■ PAUSE POINT Filtered phage can be stored at 4 °C or −20 °C indefinitely; however, phage titers can gradually decrease over time.
-
41.
Determine phage titer using an activity-independent phage plaque assay (steps 19-23). To calculate fold propagation, divide the final phage titer by the starting titer.
? TROUBLESHOOTING
Preparation of host cells • Timing 2 d
-
42.
Transform either MP6 or DP6 into S2060 cells containing desired AP(s)/CP(s) (Steps 1-13) to obtain PACE host cells (Box 1).
-
43.
Plate on Petri dishes containing 2xYT agar supplemented with 100 mM glucose and the appropriate antibiotics.
▲ CRITICAL STEP Cells containing MP6 or DP6 should always be plated on and cultured in media containing glucose (i.e. DRM) to suppress mutagenesis of host strains. Cells containing MP6 or DP6 should be transformed immediately before a PACE experiment and should never be stored longer than 1 week at 4 °C.
■ PAUSE POINT Ideally, cells transformed with MP6 or DP6 should be used immediately, but can be stored up to 1 week at 4 °C.
-
44.
The day before starting a PACE experiment, pick a single colony of PACE host cells into 1 mL of DRM containing the appropriate antibiotics. Then, either in individual culture tubes or in the wells of a 96 deep well plate, create 8 1:10 serial dilutions of this resuspended colony. Incubate in a biological shaker at 37 °C for 16-20 h.
-
45.
The next morning, select a serially diluted culture of PACE host cells that is in mid-log phase (OD600 of 0.2-0.8) and use this culture to inoculate 30-40 mL of DRM containing the appropriate antibiotics in either a 100 mL baffle flask or in a Corning Mini Bioreactor Centrifuge Tube.
-
46.
Grow this larger culture in a biological shaker at 37 °C until it reaches mid-log phase (OD600 ~0.2-0.8)
-
47.
Use this culture to start chemostats (step 74)
▲ CRITICAL STEP To ensure maximum phage infectibility, host cells should never surpass OD600 = 0.8.
Purification of SP stocks • Timing 2 d
-
48.
Plaque sequence-verified, clonal SPs using an activity-independent plaque assay (step 19-23)
-
49.
Using a P1000 pipette tip, punch out the section of agar containing a single plaque and resuspend the entire cross-section in 2 mL of DRM without antibiotics
-
50.
Incubate in a biological shaker for 6-8 h, or until the culture reaches mid-log phase (OD600 of 0.2-0.8)
-
51.
Isolate phage by pelleting the cells by centrifuging at 8000 g for 2 min, then collecting and filtering the supernatant using a 3 mL syringe fitted with a 13 mm 0.22 μm PVDF syringe filter to remove residual cells.
■ PAUSE POINT Filtered phage can be stored at 4 °C or −20 °C indefinitely; however, phage titers can gradually decrease over time.
-
52.
Determine the phage titer using an activity-independent phage plaque assay (step 19-23).
▲ CRITICAL STEP This step prepares a SP stock that can be used to infect a PACE experiment with reduced likelihood of producing wild-type recombinants (SP whose genomes have re-acquired gIII through recombination with the gIII sequence present on either APs or pJC175e in S2208 cells), which will quickly poison a PACE experiment.
Preparation of PACE tubing • Timing 1-4 h
-
53.For each chemostat and two lagoons, fit the specified 2-stop silicone lines with the corresponding male Luer adapter as described below (Fig. 2 a, b):
- 17: Fit one 1.42 mm ID 2-stop line (1) with two 3/32 in ID Male Luer adapters (6b) – for media to chemostat
- 18: Fit two 0.89 mm ID 2-stop lines (2) with two 1/16 in ID Male Luer adapters (6a) each – for chemostat to lagoons
- 19: Fit three 2.06 mm ID 2-stop lines (3) with two 1/8 in ID Male Luer adapters (6c) each – for chemostat and lagoons to waste
-
54.Assemble the connecting 1/16 in ID PVC tubing (Fig. 2 a, b). Note: line lengths will vary depending on the positioning of your apparatus.
- 20: Fit each of four 30 in segments of 1/16 in ID PVC tubing (4a) with one 1/16 in ID Male Luer adapter (6a) on one end and one 1/16 in Female Luer adapter (7a) on the other end.
- 21: Fit each of eight 12 in segments of 1/16 in ID PVC tubing (4b) with one 1/16 in ID Male Luer adapter (6a) on one end and one 1/16 in Female Luer adapter (7a) on the other end.
- 22: Connect one side of a Y-connector (8) to two 5 in segments of 1/16 in ID PVC tubing (4c). Fit two 1/16 in ID Female Luer adapters (7a) to the other end of the tubing. Then, connect the other side of the Y-connector to a 12 in segment of 1/16” ID PVC (4b) tubing. Fit a 1/16 in ID Male Luer adapter (6a) on the other end.
-
55.Attach assembled 2-stop silicone lines 17-19 to the assembled 1/16 in ID PVC tubing segments 20-22 by tightly connecting the Male and Female Luer adapters as shown in Fig. 2c and Supplementary Fig. 2a to make final PACE tubing parts C-E. Specifically:
- C: Connect 17 to 20 and 21.
- D: Connect 2 × 18 to 4 × 21. Join the two lines with 22.
-
E: Connect 19 to 20 and 21. Repeat to make three total of this part.Inducer tubing: The remaining 2 × 21 are used to connect the lagoons with the syringes for inducer addition.
-
56.
Wrap any open ends of the tubing with aluminum foil and autoclave to sterilize at 121.0 °C.
■ PAUSE POINT Assembled and sterilized PACE tubing can be stored indefinitely provided the interior of the tubing is not exposed.
Preparation of PACE apparatus • Timing 1-4 h
-
57.
Prepare the lagoons. For each lagoon: Add one 1/2 in stir bar (16) to each 22 mL glass vial fitted with PFTE/silicone septum (9). Make the cap (A) by placing the following four needles in the septum in the arrangement shown in Fig. 2b: one 18G x 1.5 in needle (12), two 22G x 4 in needles (13), and one 16G x 4 in needle (14). Wrap the inlets of each needle with aluminum foil and autoclave to sterilize at 121.0 °C.
-
58.
Prepare the chemostats. For each chemostat: Add one 1.5 in stir bar (15) to a 100 mL media bottle and lid with PTFE septum (10). Make the cap (B) by placing the following four needles in the septum in the arrangement shown in Figure 2b: two 18G x 1.5 in needles (12) and two 16G x 4 in needles (14). Wrap the inlets of each needle with aluminum foil and autoclave to sterilize at 121.0 °C.
-
59.
Attach a sterile 0.22 μm 13 mm PVDF syringe filter to one 18G x 1.5 (12) in needle in each sterilized lagoon and chemostat to allow for aeration.
-
60.
Secure the lagoons to prevent them from toppling over (i.e. by placing inside a test tube rack) and place both the lagoons and the chemostat on the multipoint stirrer.
-
61.
Attach the 2-stop segment of PACE tubing (C-E) to the Masterflex L/S 8-channel multichannel pump heads as shown in Fig. 2e. For simplicity, set both pumps to rotate in the same direction. To one Masterflex L/S Digital Drive pump with a Masterflex L/S 8-channel multichannel pump head (pump 1), attach the 2-stop segments of 1xC and 1xE to the pump head with the short segment of PVC tubing on C and the long segment on E facing the direction of outgoing flow. To a second Masterflex L/S Digital Drive pump with a Masterflex L/S 8-channel multichannel pump head (pump 2), attach the two 2-stop segments of part D to the pump head with the non-Y-joint segments of PVC tubing facing the direction of outgoing flow. Similarly, attach the 2-stop segment of 2xE to the pump head with the longer segments of PVC tubing facing the direction of outgoing flow.
-
62.
Connect the shorter (outgoing) segment of C attached to pump 1 to the remaining 18G x 1.5 in needle (12) on the chemostat (B) (media to chemostat), as shown in Fig. 2e and Supplementary Fig. 2b. Leave the other (incoming) segment of C unconnected until PACE is started.
-
63.
Connect the shorter (incoming) segment of part E attached to pump 1 to one 16G x 4 in needle (14) on the chemostat (B) (chemostat to waste) as shown in Fig. 2e and Supplementary Fig. 2b. Leave the other (incoming) segment of E unconnected until PACE is started.
-
64.
Connect the shorter (incoming) segment of each part E attached to pump 2 to the 16G x 4 in needle (14) in each lagoon (A) (lagoon to waste) as shown in Fig. 2e and Supplementary Fig. 2b. Leave the other (outgoing) segment of E unconnected until PACE is started.
-
65.
Connect the Y-joint side segment (incoming) of part D attached to pump 2 to the other 16G x 4 in needle (14) on the chemostat (B) (chemostat to lagoons) as shown in Fig. 2e and Supplementary Fig. 2b.
-
66.
Connect the outgoing end of each part D attached to pump 2 to one 22G x 4 in needle (13) in each of the lagoons (A) (chemostat to lagoons) as shown in Fig. 2e and Supplementary Fig. 2b.
▲ CRITICAL STEP Make sure to avoid contamination while removing foil covers and connecting apparatus parts.
▲ CRITICAL STEP The PACE apparatus should be set up either in a warm room set at 37 °C or in a biological incubator set at 37 °C large enough to contain the multipoint stirrer, chemostat, and lagoons.
■ PAUSE POINT PACE apparatus can be left partially assembled provided that that the chemostat, lagoons, and tubing are kept sterile until the experiment begins.
Preparation of DRM carboy • Timing 2 h
-
67.
To 9.75 L of water in a 10 L Nalgene heavy-duty polypropylene carboy with a Nalgene polypropylene 3-port filling/venting closure cap, add 181 g of Harvard Custom Media A and 10 mL of TWEEN-20.
-
68.
Attach three 5 in segments of 1/4 in ID Masterflex BPT tubing (5a) to the three ports on the top side of the cap. Insert one Female (7b) and two Male (8d) 1/4 in ID Luer adapters into the tubing (Fig. 2d). Cap one Male Luer adapter and place foil over the other ends for autoclaving.
-
69.
Attach one 30 in segment of 1/4 in ID Masterflex BPT tubing (5b) to the underside of the cap so that it is attached to the other side of the 5 in segment containing the Female Luer adapter (Fig. 2d).
-
70.
Autoclave to sterilize at 121.0 °C.
■ PAUSE POINT Sterilized carboys containing Harvard Custom Media A can be stored at room temperature for up to 3 months.
-
71.
Separately, add Harvard Custom Media C (59 g), 50 μL of 0.1 M CaCl2 solution, 120 μL of Trace Metals solution, and any appropriate antibiotics to 250 mL water. Mix to dissolve all solids, then sterilize using a Corning bottle-top vacuum filter system.
-
72.
Add the filtered Harvard Custom Media C solution to the cooled Harvard Custom Media A solution in the carboy and mix to combine.
-
73.
Attach one 30 mm 0.22 μm syringe filter to the remaining Male Luer adapter.
Setting up the PACE experiment • Timing 1 d
-
74.
Transfer 30 mL of the OD600 ~0.2-0.8 culture of host cells prepared in step 46 to the chemostat.
-
75.
Connect the media to the chemostat: Connect the longer (incoming) segment of part C attached to pump 1 to the Female Luer adapter on the media carboy (Figure 2e, Supplementary Fig. 1b.
-
76.
Connect the chemostat to waste: Connect the outgoing segment of part E attached to pump 1 to the waste (Figure 2e, Supplementary Fig. 1b).
-
77.
Begin media flow into the chemostat: Hold the rapid dispense button on pump 1 until media begins flowing into the chemostat. Adjust the height of the waste needle (part 14 attached to part E) in the chemostat to maintain an approximate volume of 40 mL. The waste needle should never be lower than the needle for lagoon outflow (part 14 attached to part D). Set the flow rate of pump 1 to 40 mL/h (1 volume/h) and allow the chemostat to turn over at a flow rate of 1 volume/h for at least 1 h to ensure that the chemostat OD600 is maintained at 0.4-0.6
? TROUBLESHOOTING
-
78.
Connect the lagoons to waste: Connect the outgoing end of each part E attached to pump 2 to the waste (Figure 2e, Supplementary Fig. 1b).
-
79.
Fill the lagoons: Hold the rapid dispense button on pump 2 until the chemostat cell culture begin to flow into the lagoons. Adjust the height of the waste needle (16G x 4 in needle attached to part E) in the lagoons to maintain an approximate volume of 15mL. Set the flow rate of pump 2 to 15 mL/h (1 volume/h) and allow lagoons to turn over at a flow rate of 1 volume/h for at least 1 h to ensure that the steady-state chemostat volume is unchanged.
? TROUBLESHOOTING
-
80.
Prepare induction syringes (Fig. 2e, Supplementary Fig. 2b): Add 50 mL of 250 mM arabinose to each 50 mL syringe (Note: both syringes must be filled with the same volume of solution). If inducing drift from DP6, add aTc to the arabinose solution to the desired final concentration. The rate of aTc addition typically ranges between 0-50 ng/mL/h (fig. 3c). Attach the female Luer adapters of the inducer lines (21) to the syringes. Place syringes in the syringe pump. Connect the male Luer adapters of the inducer lines (21) to the other 22G x 4 in needle in each lagoon (A)
-
81.
Clear the air from the inducer lines by turning on the syringe pump and setting a high flow rate (e.g. 10 mL/h). Allow the syringe pump to pump until the inducer lines are filled with inducer solution and approximately 0.6 mL of inducer solution has been added to each lagoon.
-
82.
Continue induction of mutagenesis and drift, if using, by setting the syringe pump to a pump rate of 0.6 mL/h. Allow the arabinose (or arabinose and aTc) solution to pump for at least 1 h before infecting the lagoons with phage.
-
83.
Infect lagoons: Fill two 1 mL syringes with 1 mL of 107 pfu/mL purified SP in DRM (see steps 48-52 for phage purification). Stop all pumps. Disconnect PACE tubing part E from the lagoon 16G x 4 in waste needles. Inject 1 mL of the 107 pfu/mL SP directly into the lagoon (A) through the 16G x 4 in waste needle. Reconnect the male Luer adapter from PACE tubing part E to the lagoon 16G x 4 in waste needles. Wait 10 min before collecting the first sample from the lagoon or turning on the pumps.
-
84.
Collect a sample from each lagoon (steps 86-89). This sample corresponds to the 0 h timepoint.
-
85.
Turn on all pumps to begin the PACE experiment. We typically start at a lagoon flow rate of 0.5 volumes/h (pump 2: 7.5 mL/h; syringe pump: 0.3 mL/h) and increase flow rates as the experiment continues (see steps 90-92).
Taking samples from lagoons • Timing 10 min
-
86.
Disconnect the lagoon waste lines (E) from the lagoon 16G x 4 in waste needles.
-
87.
Fit a 3 mL syringe to the 16G x 4 in waste needle and draw up approximately 1 mL from each lagoon.
-
88.
Reconnect the male Luer adapter from PACE tubing part E to the lagoon 16G x 4 in waste needles.
-
89.
Transfer lagoon samples to Eppendorf tubes and isolate the phage (step 25).
Maintaining a PACE experiment • Timing ~7 d
-
90.
Lagoon titers can be determined by performing activity-independent plaque assays (step 19-23) with collected lagoon samples (step 86-89).
-
91.
Increase the flow rate when titers either remain constant or increase over the span of ~24 hours (Fig. 3a).
-
92.
The experiment can be discontinued when titers remain constant (or fall below detection) for over 12 hours at a flow rate exceeding 3 volumes/h.
? TROUBLESHOOTING
Dismantling the PACE apparatus • Timing 3 h
-
93.
Disconnect Part C from the media carboy and Part D from the chemostat and place them directly into a 1 L bottle containing a solution of 10% bleach in water.
-
94.
Continue to run the peristaltic pumps to flush the system with about 500 mL of the bleach solution.
-
95.
Swap the bleach solution for a 1 L bottle containing water and continue to run the pumps to flush about 500 mL of water through the system.
-
96.
Stop the pumps
-
97.
Detach all PACE tubing from pump heads and disconnect the chemostats and lagoons. Dispose of needles securely in a sharps container. Soak tubing and chemostat components in a solution of 10% bleach in water for 20 min.
▲ CRITICAL STEP We have found that autoclaving or sterilization with ethanol is insufficient to destroy phage particles. Therefore, to avoid contaminating between PACE runs, we strongly recommend disinfecting all reused parts thoroughly with 10% bleach in water solution.
-
98.
Rinse tubing and chemostat components several times with water to remove all traces of bleach.
-
99.
Air-dry the PACE tubing. PACE tubing and chemostats may be reused but should be sterilized again first (steps 56-58).
Isolating and sequencing single clones from lagoon samples • Timing 1 d
-
100.
Clonal phage from lagoon samples can be isolated using an activity-independent plaque assay (step 19-23) or, if the POI encoded on the phage is sufficiently active, using an activity-dependent plaque assay with host cells containing the appropriate AP(s) and CP(s), if applicable (step 37-41).
-
101.
Sequence clonal phage as described in steps 27-30.
? TROUBLESHOOTING
Characterization of evolved phage by bacterial luciferase assay (optional) • Timing 3-4 d
-
102.
Transform S2060 host cells with the relevant AP(s)/CP(s) (steps 1-13). The AP(s) should contain bacterial luciferase (luxAB) translationally coupled to gIII (Supplementary Fig. 1b).
-
103.
Pick a single colony of the host cells into DRM supplemented with the appropriate antibiotics and incubate in a biological shaker at 37 °C for 16-20 h.
-
104.
Make a 1:100 dilution of the resultant saturated culture of host cells into DRM supplemented with the appropriate antibiotics and incubate in a biological shaker at 37 °C until the diluted culture reaches an OD600 of 0.4-0.6.
-
105.
Infect the host cells with clonal phage isolated from PACE samples at a 10:1 phage:host cell ratio.
-
106.
Incubate cells in a biological shaker at 37 °C for 1-2 h.
-
107.Read luminescence using a plate reader using the following settings:
- Set temperature to 37 °C
- Shake (orbital) for 10s with 1 mm amplitude and 582 rpm
- Read Absorbance at 600 nm (number flashes = 10, settle time = 0 ms)
- Read luminescence (attenuation = none, settle time = 0 ms, integration time = 500 ms)
Phage-assisted non-continuous evolution (PANCE) (optional) • Timing 1d per passage
-
108.
To generate host cells for PANCE, transform either MP6 or DP6 into S2060 cells containing desired AP(s)/CP(s) as described in steps 1-13.
-
109.
Plate on Petri dishes containing 2xYT agar supplemented with 100 mM glucose and the appropriate antibiotics.
▲ CRITICAL STEP Cells containing MP6 or DP6 should always be plated on and cultured in media containing glucose (i.e. DRM) to suppress mutagenesis of host strains. Cells containing MP6 or DP6 should be transformed immediately before a PANCE experiment and should never be stored longer than 1 week at 4 °C.
-
110.
Pick a single colony of the host cells into DRM supplemented with the appropriate antibiotics and incubate in a biological shaker at 37 °C for 16-20 h.
-
111.
Make a 1:100 dilution of the resultant saturated culture of host cells into DRM supplemented with the appropriate antibiotics and incubate in a biological shaker at 37 °C until the diluted culture reaches an OD600 of 0.4-0.6 (Supplementary Fig. 1a).
-
112.
Induce mutagenesis and drift (if using DP6) by adding L-arabinose and aTc to a final concentration of 10 mM and 50 ng/μL, respectively. The aTc concentration can be adjusted between 0 and 50 ng/μL to vary the amount of drift.
-
113.
Infect the culture with the desired SP at a final titer of 106-107 pfu/mL. Infection at higher starting titers lowers the stringency of selection by reducing the fold propagation required for the SP to persist in PANCE
-
114.
Incubate in a biological shaker for 6-18 h at 37 °C. Longer incubation times lower the stringency of selection by providing more time for phage propagation.
-
115.
Isolate phage by pelleting the cells by centrifuging at 8000 g for 2 min, then collecting and filtering the supernatant using a 3 mL syringe fitted with a 13 mm 0.22 μm PVDF syringe filter to remove residual cells.
-
116.
Repeat steps 111-115 by using the phage pool to infect the next passage of induced host cells (Supplementary Fig. 1a), adjusting starting titer (in other words, the dilution factor of the phage pool from the previous passage), drift induction, and incubation time as desired to increase selection stringency.
-
117.
The titers of the PANCE phage pools after each passage can determined either by an activity-independent plaque assay (steps 19-23) or by qPCR (steps 119-126)
-
118.
If the phage pools resulting from PANCE exhibit high activity in phage propagation assays using host cells transformed with the appropriate AP(s)/CP(s) (steps 1-13) or in luciferase assays (steps 102-107), they can be used to start a PACE experiment. Alternatively, clonal phage can be isolated from advanced PANCE pools and analyzed by Sanger sequencing (steps 27-30).
? TROUBLESHOOTING
qPCR titer determination (optional) • Timing 3-4 h
-
119.
Transfer 25-50 μL of phage stocks of unknown titer to PCR tubes
-
120.
Create a titration curve using phage of known titer by making eight 1:10 serial dilutions starting from a phage stock of ~109 or 1010 pfu/mL. Transfer 25-50 μl of each dilution into a PCR tube
-
121.
Heat at 80 °C for 30 min to disrupt polyphage
-
122.
To digest polyphage genomes, dilute 5 μl of heat-treated phage into 45 μl of 1x DNaseI buffer containing 1 unit (0.5 μL) of DNaseI
-
123.
Heat at 37 °C for 20 min, then 95 °C for 20 min
-
124.
Set up the qPCR reactions. Each reaction contains the following:
| Component | Amount | Final concentration |
|---|---|---|
| Nuclease-free water | 20 μL | |
| 5x Phusion HF Buffer | 5 μL | 1x |
| dNTPs | 0.5 μL | 0.2 mM each dNTP |
| Phusion HF Polymerase | 0.5 μL | 1 U |
| 100x Sybr Green | 0.25 μL | 1x |
| M13fwd primer | 0.125 μL | 1 μM |
| M13rev primer | 0.125 μL | 1 μM |
| Treated phage | 1.5 μL | |
| Total | 28 μL |
Run the qPCR with the following program: 98 °C for 2 min, 40 cycles of: [98 °C for 10 s, 60 °C for 20 s, and 72 °C for 15 s], and 12 °C for 30 s
| Cycle number | Denature | Anneal | Extend | Final |
|---|---|---|---|---|
| 1 | 98 °C, 2 min | |||
| 2-41 | 98 °C, 10s | 60 °C, 20 s | 72 °C, 15 s | |
| 42 | 12 °C, 30 s |
-
125.
Calculate phage titers using the titration curve derived from the phage stock of known concentration
Troubleshooting
Timing
Preparation of cell lines
Steps 1-5, TSS chemicompetent cell preparation: 6 h
Steps 6-13, TSS chemicompetent cell transformation: 2 h
Preparation and analysis of phage
Steps 14-18, phage cloning: 2d
Steps 19-23, activity-independent phage plaque assays: 2 d
Steps 24-26 Isolation of clonal phage: 2 d
Steps 27-30, sequencing clonal phage: 1d
Steps 31-36, SP stock amplification (activity-independent phage propagation): 2 d
Steps 37-41, activity-dependent phage propagation: 2 d
Preparing and running a PACE experiment
Steps 42-47, preparation of host cells, 2 d
Steps 48-52, Purification of SP stocks: 2 d
Steps 53-56, preparation of PACE tubing: 1-4 h
Steps 57-66, preparation of PACE apparatus: 1-4 h
Steps 67-73, preparation of DRM carboy: 2 h
Steps 74-85 setting up the PACE experiment: 1 d
Steps 86-89, taking samples from lagoons: 10 min
Steps 90-92, maintaining a PACE experiment: ~7 d
Steps 93-99, dismantling the PACE apparatus: 3 h
Analysis of PACE experiments
Steps 100-101, isolating and sequencing single clones from lagoon samples: 1 d
Steps 102-107, characterization of evolved phage by bacterial luciferase assay (optional): 3-4 d
Phage-assisted non-continuous evolution
Steps 108-118, Phage-assisted non-continuous evolution (PANCE) (optional): 1 d per passage
Steps 119-126, qPCR titer determination (optional): 3-4 h
Anticipated Results
PACE experiments are typically performed for up to 7 days, although with care, they can run for several weeks. During PACE, lagoon titers typically range between 105-108 pfu/mL. Titers will vary depending on fitness, the selection circuit, and POI size. During the experiment, titers should drop and recover as selection pressure is increased and the evolving phage pool discovers fitness-increasing mutations in response to changing selection conditions. Drops in phage titer are often observed within 12-24 h of the initial phage infection, flow rate increases, or cessation of drift induction. If the SP discover fitness-increasing mutations, their titers should recover within an additional 12-24 h. Phage titers may also remain stable throughout a PACE experiment even if flow rate is increased. This observation may indicate that the selection stringency is too low, given the properties of the current evolved genes, to support the additional evolution of improved activity.
PACE experiments may fail in a variety of ways. For example, lagoon phage titers may drop below a limit (~102-103 pfu/mL) beyond which recovery is either extremely unlikely or impossible. This “phage wash out” can occur when the selection is too stringent or if the host cells are uninfectable. Another possible failure mode is recombination of gIII into the SP genome, which leads to phage that can short-circuit the selection and therefore rapidly take over the lagoon population. Recombination is frequently accompanied by the loss of the now-dispensable POI insert, which can be detected by a decrease in the size of the corresponding PCR amplification product (step 28). Strategies for addressing these failures are listed in the troubleshooting (Table 2).
Table 2.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 13 | No colonies | Poor quality plasmid prep | Re-prep plasmid; verify by Sanger Sequencing |
| Wrong selection antibiotic | Use correct antibiotic | ||
| Cells have low competency | Keep cells chilled on ice throughout procedure. Never vigorously pipette or vortex competent cells. |
||
| 18 | No phage | Cells used for propagation lacks F pilus | Use F+ E. coli |
| Did not use gIII producing cell line to propagate | Use S2208s or S2060s containing an AP | ||
| Cells were grown to improper densities | Grow cells to OD600 ~0.4–0.6 | ||
| Fault in phage backbone | Ensure that cloning method retains all essential phage genes besides gIII | ||
| Incorrect phage insert | Inefficient cloning | Choose different cloning method or increase scale of assembly | |
| Insert is large in size (> 3 kbp) | Clone in an activity-dependent manner (transform into cells containing an AP) | ||
| 23 | No plaques | Cells were grown to improper densities | Grow cells to OD600 ~0.4–0.6 |
| Plaques are too small or difficult to see | Add Bluo-gal to top agar at 0.04% w/v or decrease agar concentration of top agar. | ||
| 28 | No PCR product | Incorrect primers | Ensure that primers AB1396 and AB1792 anneal to your phage backbone. If not, use different primers |
| Too little phage in PCR reaction | Add more template | ||
| PCR protocol not optimized | Ensure that annealing temperature, extension time, and number of cycles are correct | ||
| 41 | No propagation | POI in phage has minimal activity on selection circuit encoded by AP | Decrease selection stringency, choose different circuit |
| Cells were infected at improper densities | Grow cells to OD600 ~0.4–0.6 | ||
| 77, 79 | Host cells do not maintain correct OD600 | Flow rate is too low or high | Adjust waste needle height or peristaltic pump rate |
| Incorrect antibiotics in PACE media | Ensure that the correct antibiotics have been added | ||
| 92 | SP exhibit gIII recombination | Phage stocks contaminated with wild-type recombinants | Purify phage (steps 48-52) |
| Drift induced for too long | Limit drift to <= 48 h | ||
| Phage washout | Stringency too high | Run PACE at lower stringency – drift with DP6, lower stringency of selection circuit on AP, use lower flow rates | |
| No change in phage titers over multiple flow rate increases | Stringency too low to select for improved POI activity | Increase stringency of selection circuit on AP | |
| Lagoons contaminated with wild-type recombinants (verify by Sanger Sequencing lagoon samples) | Purify phage (steps 48-52) | ||
| Phage titer decreasing in the absence of stringency change | Ability of host cells in chemostat to be infected diminishes over long periods of continuous culture | Replace chemostat cells with fresh host cells picked from a bacterial colony (steps 44-47, re-transform MP/DP if plated cells are more than one week old). | |
| 101, 118 | No mutations observed in POI | MP/DP not induced | Ensure correct concentration of arabinose has been added to lagoons to induce mutagenesis from MP or DP |
| MP/DP non-functional | Verify MP/DP by Sanger Sequencing. Alternatively, perform phenotypic verification: transform MP/DP into cells and plate on 2xYT/agar supplemented with glucose or arabinose. Cells plated on arabinose should exhibit notable growth inhibition due to mutagenesis induction compared to cells plated on glucose. If the MP/DP is non-functional, re-prep the plasmid. Ensure that the media used to grow cells for plasmid prep is supplemented with the correct antibiotics and glucose to repress mutagenesis. | ||
| Stringency too low to select for improved POI activity | Run PACE with higher stringency –remove drift, increase dilution rate, increase the stringency of the AP selection circuit | ||
| PACE is optimizing SP backbone | Changes in SP backbone to optimize SP propagation, POI expression levels, etc., often occur before mutations begin to accumulate in the POI. Their effects on SP fitness can be confirmed using propagation assays (steps 37-41). If the SP isolated from PACE exhibit drastically improved propagation on cells containing the AP, continue to evolve this population in PACE using higher selection stringency. |
After completion of a PACE or PANCE experiment, mutations that may have accumulated in the POI can be identified by sequencing clonal SP isolated from lagoon samples at later timepoints (PACE) or passages (PANCE). We recommend sequencing the POI encoded by eight or more clonal phage to allow for identification of consensus mutations. Mutations can accumulate in both the SP backbone and the POI gene insert. The number of mutations observed will vary from selection to selection and can be influenced by factors such as selection stringency, POI size, the duration of the PACE/PANCE experiment, and the nature of the POI itself. Additionally, significant increases in fitness can occur simply because of mutations in the SP backbone that optimize replication efficiency. Observation of a low number of consensus mutations does not preclude a successful evolution, as POI activity can be increased by just a single mutation. However, a lack of any shared mutations altogether may indicate that the evolution lacked sufficient stringency to enrich for POI variants with improved fitness, or that a large fraction of sequence space contains variants with sufficient fitness to survive the present selection conditions.
The outcome of an evolution experiment can be quickly assessed using either bacterial luciferase (steps 102-107) or activity-dependent phage propagation assays (steps 37-41); activity in these assays should increase for both the evolved SP pool and isolated SP clones relative to SP encoding the wild-type or starting POI. Ultimately, individual POI variants isolated from a PACE or PANCE experiment should be assessed using appropriate and orthogonal activity assays.
Supplementary Material
Supplementary Figure 1. Phage-assisted non-continuous evolution (PANCE) and higher throughput characterization of evolved phage. (a) Schematic representation of PANCE. (b) Example of transcriptional coupling of gIII to luxAB in APs used in luciferase assays.
Supplementary Figure 2. Photographs of PACE tubing and apparatus. (a) PACE tubing as depicted in Fig. 2c. (b) PACE apparatus as depicted in in Fig. 2e. Parts are labeled identically to Fig. 2.
Acknowledgements
We thank Kevin Esvelt, Jacob Carlson, Bryan Dickinson, Ahmed Badran, Basil Hubbard, Michael Packer, David Bryson, Johnny Hu, Tim Roth, Ben Thuronyi, Michelle Richter, and Kevin Zhao for their contributions to the development of PACE, and Travis Blum for helpful discussion. S.M.M. was supported by an NSF Graduate Research Fellowship. T.W. was supported by a Ruth L. Kirchstein National Research Service Awards Postdoctoral Fellowship (F32GM119228). We are grateful for support from US NIH R01 EB027793, U01 AI142756, RM1 HG009490, R35 GM118062, HHMI, and the Bill and Melinda Gates Foundation.
Footnotes
Competing interests
The authors declare competing financial interests: S.M.M., T.W., and D.R.L. have filed patent applications on PACE technologies and PACE-evolved proteins.
Data availability
The 2060 cell line and plasmids pJC175e, MPs, DP6, and various CPs and APs are available through Addgene. Specific phage are available upon request.
REFERENCES
- 1.Packer MS & Liu DR Methods for the directed evolution of proteins. Nat Rev Genet 16, 379–394, doi: 10.1038/nrg3927 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Badran AH & Liu DR In vivo continuous directed evolution. Curr Opin Chem Biol 24, 1–10, doi: 10.1016/j.cbpa.2014.09.040 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mills DR, Peterson RL & Spiegelman S An extracellular Darwinian experiment with a self-duplicating nucleic acid molecule. Proc Natl Acad Sci U S A 58, 217–224, doi: 10.1073/pnas.58.1.217 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wright MC & Joyce GF Continuous in vitro evolution of catalytic function. Science 276, 614–617, doi: 10.1126/science.276.5312.614 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Esvelt KM, Carlson JC & Liu DR A system for the continuous directed evolution of biomolecules. Nature 472, 499–503, doi: 10.1038/nature09929 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berman CM et al. An Adaptable Platform for Directed Evolution in Human Cells. J Am Chem Soc 140, 18093–18103, doi: 10.1021/jacs.8b10937 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Badran AH et al. Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance. Nature 533, 58–63, doi: 10.1038/nature17938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badran AH & Liu DR Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat Commun 6, 8425, doi: 10.1038/ncomms9425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbas CF Phage display : a laboratory manual. (Cold Spring Harbor Laboratory Press, 2001). [Google Scholar]
- 10.Carlson JC, Badran AH, Guggiana-Nilo DA & Liu DR Negative selection and stringency modulation in phage-assisted continuous evolution. Nat Chem Biol 10, 216–222, doi: 10.1038/nchembio.1453 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett NJ & Rakonjac J Unlocking of the filamentous bacteriophage virion during infection is mediated by the C domain of pIII. J Mol Biol 356, 266–273, doi: 10.1016/j.jmb.2005.11.069 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Hubbard BP et al. Continuous directed evolution of DNA-binding proteins to improve TALEN specificity. Nat Methods 12, 939–942, doi: 10.1038/nmeth.3515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leconte AM et al. A population-based experimental model for protein evolution: effects of mutation rate and selection stringency on evolutionary outcomes. Biochemistry 52, 1490–1499, doi: 10.1021/bi3016185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Packer MS, Rees HA & Liu DR Phage-assisted continuous evolution of proteases with altered substrate specificity. Nat Commun 8, 956, doi: 10.1038/s41467-017-01055-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thuronyi BW et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat Biotechnol 37, 1070–1079, doi: 10.1038/s41587-019-0193-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Badran AH, Huang TP & Liu DR Continuous directed evolution of proteins with improved soluble expression. Nat Chem Biol 14, 972–980, doi: 10.1038/s41589-018-0121-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bryson DI et al. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat Chem Biol 13, 1253–1260, doi: 10.1038/nchembio.2474 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roth TB, Woolston BM, Stephanopoulos G & Liu DR Phage-Assisted Evolution of Bacillus methanolicus Methanol Dehydrogenase 2. ACS Synth Biol 8, 796–806, doi: 10.1021/acssynbio.8b00481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63, doi: 10.1038/nature26155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki T et al. Crystal structures reveal an elusive functional domain of pyrrolysyl-tRNA synthetase. Nat Chem Biol 13, 1261–1266, doi: 10.1038/nchembio.2497 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller SM et al. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat Biotechnol, doi: 10.1038/s41587-020-0412-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dickinson BC, Leconte AM, Allen B, Esvelt KM & Liu DR Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc Natl Acad Sci U S A 110, 9007–9012, doi: 10.1073/pnas.1220670110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodel AK, Jaramillo A & Isalan M Engineering orthogonal dual transcription factors for multi-input synthetic promoters. Nat Commun 7, 13858, doi: 10.1038/ncomms13858 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pu J, Zinkus-Boltz J & Dickinson BC Evolution of a split RNA polymerase as a versatile biosensor platform. Nat Chem Biol 13, 432–438, doi: 10.1038/nchembio.2299 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zinkus-Boltz J, DeValk C & Dickinson BC A Phage-Assisted Continuous Selection Approach for Deep Mutational Scanning of Protein-Protein Interactions. ACS Chem Biol 14, 2757–2767, doi: 10.1021/acschembio.9b00669 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Pu J, Disare M & Dickinson BC Evolution of C-Terminal Modification Tolerance in Full-Length and Split T7 RNA Polymerase Biosensors. Chembiochem 20, 1547–1553, doi: 10.1002/cbic.201800707 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dickinson BC, Packer MS, Badran AH & Liu DR A system for the continuous directed evolution of proteases rapidly reveals drug-resistance mutations. Nat Commun 5, 5352, doi: 10.1038/ncomms6352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richter MF Z. K. T.; Eton E; Lapinaite A; Newby GA; Thuronyi BW; Wilson C; Koblan LW; Zeng J; Bauer DE; Doudna JA; Liu DR . Phage-Assisted Evolution of an Adenine Base Editor with Enhanced Cas Domain Compatibility and Activity. Nat Biotechnol In press (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brodel AK, Jaramillo A & Isalan M Intracellular directed evolution of proteins from combinatorial libraries based on conditional phage replication. Nat Protoc 12, 1830–1843, doi: 10.1038/nprot.2017.084 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Javanpour AA & Liu CC Genetic Compatibility and Extensibility of Orthogonal Replication. ACS Synth Biol 8, 1249–1256, doi: 10.1021/acssynbio.9b00122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ravikumar A, Arzumanyan GA, Obadi MKA, Javanpour AA & Liu CC Scalable, Continuous Evolution of Genes at Mutation Rates above Genomic Error Thresholds. Cell 175, 1946–1957 e1913, doi: 10.1016/j.cell.2018.10.021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong Z, Ravikumar A & Liu CC Tunable Expression Systems for Orthogonal DNA Replication. ACS Synth Biol 7, 2930–2934, doi: 10.1021/acssynbio.8b00400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhong Z et al. Automated Continuous Evolution of Proteins in Vivo. ACS Synth Biol, doi: 10.1021/acssynbio.0c00135 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.English JG et al. VEGAS as a Platform for Facile Directed Evolution in Mammalian Cells. Cell 178, 1030, doi: 10.1016/j.cell.2019.07.036 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barbas CF 3rd. Recent advances in phage display. Curr Opin Biotechnol 4, 526–530, doi: 10.1016/0958-1669(93)90072-5 (1993). [DOI] [PubMed] [Google Scholar]
- 36.Ringquist S et al. Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol Microbiol 6, 1219–1229, doi: 10.1111/j.1365-2958.1992.tb01561.x (1992). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Phage-assisted non-continuous evolution (PANCE) and higher throughput characterization of evolved phage. (a) Schematic representation of PANCE. (b) Example of transcriptional coupling of gIII to luxAB in APs used in luciferase assays.
Supplementary Figure 2. Photographs of PACE tubing and apparatus. (a) PACE tubing as depicted in Fig. 2c. (b) PACE apparatus as depicted in in Fig. 2e. Parts are labeled identically to Fig. 2.
Data Availability Statement
The 2060 cell line and plasmids pJC175e, MPs, DP6, and various CPs and APs are available through Addgene. Specific phage are available upon request.