

Summary
Stalling during translation triggers ribosome quality control (RQC) to maintain proteostasis. Recently, stalling has also been linked to the activation of integrated-stress response (ISR) by Gcn2. How the two processes are coordinated is unclear. Here we show that activation of RQC by Hel2 suppresses that of Gcn2. We further show that Hel2 and Gcn2 are activated by a similar set of agents that cause ribosome stalling, with maximal activation of Hel2 observed at a lower frequency of stalling. Interestingly, inactivation of one pathway was found to result in the overactivation of the other, suggesting that both are activated by the same signal of ribosome collisions. Notably, the processes do not appear to be in direct competition with each other; ISR prefers a vacant A site, whereas RQC displays no preference. Collectively, our findings provide important details about how multiple pathways that recognize stalled ribosomes coordinate to mount the appropriate response.
Keywords: Integrated-stress response, Ribosome-quality control, Gcn2, Hel2, alkylation, RNA damage, ribosome rescue
Graphical Abstract
eTOC blurb
Ribosome stalling activates ribosome-quality control (RQC) and integrated stress response, but how the two process are coordinated is unclear. Yan and Zaher show that while both processes are activated in response to ribosome collisions, RQC is more sensitive to changes to translation dynamics and responds quickly to lower collision frequency.
Introduction
A defining feature of all organisms is their ability to sense and respond to a constantly changing environment. T8hey excel at recognizing fluctuations in nutrient availability and respond by reprogramming gene expression to meet metabolic needs (Hinnebusch, 2005; Pakos-Zebrucka et al., 2016). They are also able to react to endogenous and exogenous stresses. Among the different cellular responses, the Integrated Stress Response (ISR), by which eukaryotic cells alter their translational and transcriptional output in response to a number of homeostatic imbalances and disease conditions, is perhaps one of the most important adaptive pathways (Hetz et al., 2013; Kroemer et al., 2010; Masson, 2019). To this end, dysregulation of ISR signaling has been linked to various pathological disorders that include diabetes, cancer and neurodegeneration (Augusto et al., 2019; Bi et al., 2005; Delépine et al., 2000; Eyries et al., 2014; Lin et al., 2013; Ma et al., 2013; Mouton-Liger et al., 2012; Ye et al., 2010).
Unique to ISR is its activation in response to diverse stress conditions (Pakos-Zebrucka et al., 2016). These stimuli all lead to the phosphorylation of eukaryotic initiation factor 2α (eIF2α). Mammals have four distinct eIF2α kinases: GCN2, PERK, PKR and HRI; which are activated in response to amino-acid deprivation, ER stress, double-stranded RNA (typically associated with viral infections) and heme availability, respectively. Phosphorylation of eIF2α by these kinases shifts the factor from a substrate to a competitive inhibitor for the guanine exchange factor eIF2B, inhibiting recycling of the GDP-bound eIF2 to a GTP-bound one (Gomez et al., 2002; Krishnamoorthy et al., 2001; Sudhakar et al., 2000). This results in depletion of initiatortRNA•eIF2•GTP ternary complex and leads to a global reduction of protein synthesis, but allows translation of a set of mRNAs whose protein products are required for cell survival and recovery, including ATF4 in mammals and Gcn4 in yeast. Both undergo an atypical-initiation mechanism (Dever et al., 1992; Harding et al., 2000; Hinnebusch, 1993, 2005; Vattem and Wek, 2004), and are the main effectors of ISR as they upregulate biosynthetic pathways as well as autophagy (B’Chir et al., 2013; Hinnebusch, 2005; Kroemer et al., 2010; Natarajan et al., 2001; Valasek et al., 2002).
Saccharomyces cerevisiae has only one eIF2α kinase, Gcn2, and the organism has proven to be an invaluable model for studying ISR (Masson, 2019), especially in response to amino-acid starvation (Sonenberg and Hinnebusch, 2009). Initial studies focused on clues afforded by its domain architecture and possible analogies to the stringent response in bacteria, which is activated by Rel-A mediated recognition of deacylated tRNAs bound to the ribosome (Atkinson et al., 2011). In addition to its kinase domain, Gcn2 harbors ribosome- and tRNA-binding domains (Ramirez et al., 1991; Wek et al., 1995; Zhu and Wek, 1998; Zhu et al., 1996). The latter is homologous to His-tRNA synthetase (HisRS-like), and allows the factor to preferentially bind deacylated tRNA over aminoacylated ones (Dong et al., 2000; Lageix et al., 2015). These observations led to the hypothesis that Gcn2 is activated in response to the accumulation of deacylated tRNA. Several biochemical and genetics studies supported this model; for instance mutations that abrogate binding to the ribosome or deacylated tRNAs inhibit activation of ISR (Dong et al., 2000; Lageix et al., 2015; Qiu et al., 2001, 2002; Ramirez et al., 1992). Furthermore in yeast, for full activation of ISR in response to amino-acid depletion, Gcn2 requires two additional factors: Gcn1 and Gcn20 (Garcia-Barrio, 2000; Marton et al., 1997; Sattlegger and Hinnebusch, 2005). Each of these factors possesses a domain similar to elongation factor eEF3 (Marton et al., 1997; Visweswaraiah et al., 2012), which removes deacylated tRNA from the E site of the ribosome. Although this model of Gcn2 activation is appealing, several new studies have called it into question and suggested a more ribosome-centric mode of activation. For example, in mice lacking a tRNA isoacceptor and a ribosome recycling factor, which leads to ribosome stalling, eIF2α is phosphorylated in a GCN2-dependent manner without accumulation of uncharged tRNAs (Han et al., 2018; Ishimura et al., 2016). In agreement with this model, two recent studies have shown the factor to be robustly activated by the P stalk of the ribosome relative to deacylated tRNAs (Harding et al., 2019; Inglis et al., 2019). These observations suggest a direct role for ribosome slowing or stalling in activating Gcn2.
In eukaryotes, ribosome stalling also activates the mRNA-surveillance pathway of no-go decay (NGD) and the associated ribosome-quality-control pathway (RQC). Briefly, upon stalling on defective mRNA, the trailing ribosomes collide into the stalled ribosome (Yan and Zaher, 2019). The E3 ligase Hel2 (Znf598 in mammals) recognizes collided ribosomes and adds ubiquitin to a number of ribosomal proteins (Ikeuchi et al., 2019; Juszkiewicz et al., 2018; Simms et al., 2017). The ubiquitination appears to play an important role in recruiting downstream factors involved in ribosome rescue, mRNA degradation and nascent-peptide ubiquitination (Garzia et al., 2017; Juszkiewicz and Hegde, 2017; Matsuo et al., 2017; Sundaramoorthy et al., 2017). How ISR activation is coordinated with that of RQC during stalling is unknown.
Here we show that, in yeast, Gcn2 is activated under conditions that promote ribosome stalling, including alkylation and oxidation stresses known to modify mRNAs and activate RQC. We further show that activation of RQC, through Hel2-mediated ubiquitination of ribosomal proteins, antagonizes activation of ISR, and vice versa. Notably, Hel2 was found to suppress the premature activation of Gcn2 under nonstress conditions. Finally, we provide evidence that suggests that, similar to RQC, Gcn2 is activated by ribosome collisions, but prefers the A site of the stalling ribosome to be empty. Altogether, our findings suggest that cells carefully monitor ribosome speed to sense changes in their environment and mount an appropriate response.
Results
Hel2 antagonizes the activation of Gcn2
We recently showed that alkylation and oxidation stresses modify mRNAs and trigger ribosome stalling (Yan et al., 2019). The resulting ribosome collisions were in turn found to activate K63-linked ubiquitination of ribosomal proteins by the E3 ligase Hel2. Interestingly, our group and others have identified several conserved targets for Hel2 on colliding ribosomes (Matsuo et al., 2017; Yan et al., 2019), suggesting that ribosome collisions are likely to serve important roles for multiple signals. As a result, we were motivated to investigate whether ribosomal-protein ubiquitination is utilized by organisms to alter gene expression in response to stress. We carried out RNA-sequencing analysis of wild-type and hel2Δ yeast cells in the absence and presence of MMS (Figure 1, Figure S1 and Table S1). We note that deletion of HEL2 completely inhibits MMS-mediated ribosomal-protein ubiquitination (Yan et al., 2019) and hence any changes to RNA-expression levels in this strain is likely to be the result of the absence of ribosome modification.
Figure 1∣. Transcriptional profiling of wild-type and hel2Δ cells in the absence and presence of MMS.

A) Volcano plots of −log10p values against log2 relative-fold-changes in the RNA levels of all genes in the hel2Δ strain to the wild-type one. Genes that are significantly upregulated are shown in red and those that are significantly downregulated are shown in blue. B) Similar to A, but for the relative change between wild-type cells treated with MMS and mock-treated ones. C) Volcano plot used to follow the relative change in mRNA levels for hel2Δ cells in the presence of MMS versus wild-type cells in the presence of MMS, but only for genes that were upregulated in the presence of MMS in the wild-type cells, i.e. genes shown in red in B. D) Similar to C, but for genes that were downregulated in the presence of MMS in the wild-type cells, i.e. those shown in blue dots in B. E) Bar graphs of −log10p values of the gene-ontology (GO) analysis conducted on the 100 most upregulated genes in the hel2Δ cells relative to the wild-type ones in the presence of MMS. F) Heat-map of the relative mRNA levels of Gcn4-regulated genes in MMS-treated cells to mock-treated ones for each biological replicate. The genes are hierarchically organized based on their fold difference.
We initially focused on differences between wild-type cells and those lacking HEL2 under normal conditions. Our analysis showed the deletion of HEL2 to result in modest changes to mRNA levels. A closer examination of the data, however, revealed that these cells display a signature typically observed in response to environmental and physiological stresses. In particular, we observed an overexpression of genes associated with DNA-damage response as well as heat-shock ones (Figure S1, Table S1). We further noted an upregulation of genes typically associated with amino-acid deprivation conditions, including those involved in the arginine-biosynthetic pathway (Figure 1A). These observations were consistent with recent reports showing that deletion of HEL2 leads to the activation of ISR (Meydan and Guydosh, 2020). It, thus, seems that the absence of Hel2 triggers an unwarranted stress response and that ribosome-protein ubiquitination could suppress its activation.
We were next interested in exploring how this signature is altered in response to alkylation stress. As expected (Jelinsky and Samson, 1999), the addition of MMS was found to have profound effects on gene expression. We observed induction of genes involved in multiple processes; including those involved in DNA-damage, heat-shock and integrated-stress response (Figure 1B). In the DNA-damage and heat shock responses, DDR2, HSP12 and RNR3 were induced by almost a hundredfold. In ISR, genes in the arginine-biosynthetic pathway (ARG1, ARG4 and ARG7) stood out and were induced by more than tenfold. Consistent with MMS-induced activation of ISR, expression of ribosomal-protein genes was significantly reduced. Collectively, our analysis of MMS-stressed cells revealed that multiple stress responses are robustly activated in response to alkylation stress, consistent with earlier observations in budding (Natarajan et al., 2001) as well as fission yeast (Berlanga et al., 2010).
Since Hel2 appears to suppress the activation of a stress response in the absence of any stressors, we wondered if the factor could also play a role in altering the response under alkylation stress. RNA-sequencing analysis of genes induced in response to MMS revealed that most of these genes are further induced, by as much as 30-fold, in the absence of Hel2 (Figure 1C). In contrast, most of the genes that were repressed in the presence of MMS were further repressed in the absence of the factor (Figure 1D). These observations suggest that Hel2 antagonizes the pathways that are activated in response to alkylation stress. To gain an unbiased insight into the identity of these pathways, we performed a gene-ontology (GO) analysis of the top 100 genes that were further induced in Hel2Δ cells relative to the wild-type ones. As expected, GO analysis identified amino-acid synthetic pathways as the top ten processes that are modulated by Hel2 (Figure 1E). Since many of the genes involved in these processes are targets of Gcn4 (the main ISR effector), this suggests that Hel2 suppresses its activation. Indeed, analysis of the expression levels of the Gcn4 regulon (Rawal et al., 2018) showed that deletion of HEL2 results in significant over-induction of these targets (Figure 1F). Collectively, our RNA-sequencing analysis suggests that Hel2 suppresses the premature activation of Gcn2 and attenuates it under alkylation stress.
Deletion of Hel2 increases eIF2a phosphorylation by Gcn2
During ISR, yeast eIF2α is phosphorylated by Gcn2, which suppresses global translation initiation while simultaneously promoting the translation of Gcn4 (Hinnebusch, 2005), which our RNA-sequencing analysis suggested to be induced. To add further support for these observations, we investigated the phosphorylation status of eIF2α in response to MMS addition and presence/absence of Hel2. As expected, the addition of the drug was found to result in the accumulation of phospho-eIF2α in a time-dependent manner (Figure 2A, 2B), and in agreement with our model, deletion of HEL2 accelerated its accumulation (Figure 2A, 2B). Notably, in the absence of Hel2, the levels of phospho-eIF2α increased by more than fivefold in the absence of MMS (Figure 2B). These observations were confirmed by directly immunoblotting for Gcn4, which showed the transcription factor to accumulate to much higher levels in the absence of HEL2 (Figure 2A). qPCR analysis of the ARG1 transcript, a Gcn4 target, confirmed that ISR is prematurely activated in the absence of Hel2. More specifically, we observe an almost twofold increase in ARG1 transcript levels in the hel2Δ cells relative to wild-type ones (Figure 2C), corroborating our RNA-sequencing data (Figure 1A). Additionally, while the addition of MMS was found to increase the levels of ARG1 transcript by as much as 25-fold, deletion of HEL2 increased its levels more rapidly and almost to a hundredfold after only 15 minutes of incubation with the drug (Figure 2D). To confirm that the increased phosphorylation of eIF2α observed in the absence of Hel2 is mediated by Gcn2, we deleted the kinase from wild-type and hel2D cells. As expected, we could not detect any phospho-eIF2α in hel2Δ; gcn2Δ in the absence or presence of MMS (Figure 2E). Altogether, our western-blot and qPCR analyses confirmed our transcriptomic analysis that Hel2 suppresses the activation of ISR.
Figure 2∣. Ribosome stalling activates eIF2α phosphorylation in a process that is antagonized by Hel2.
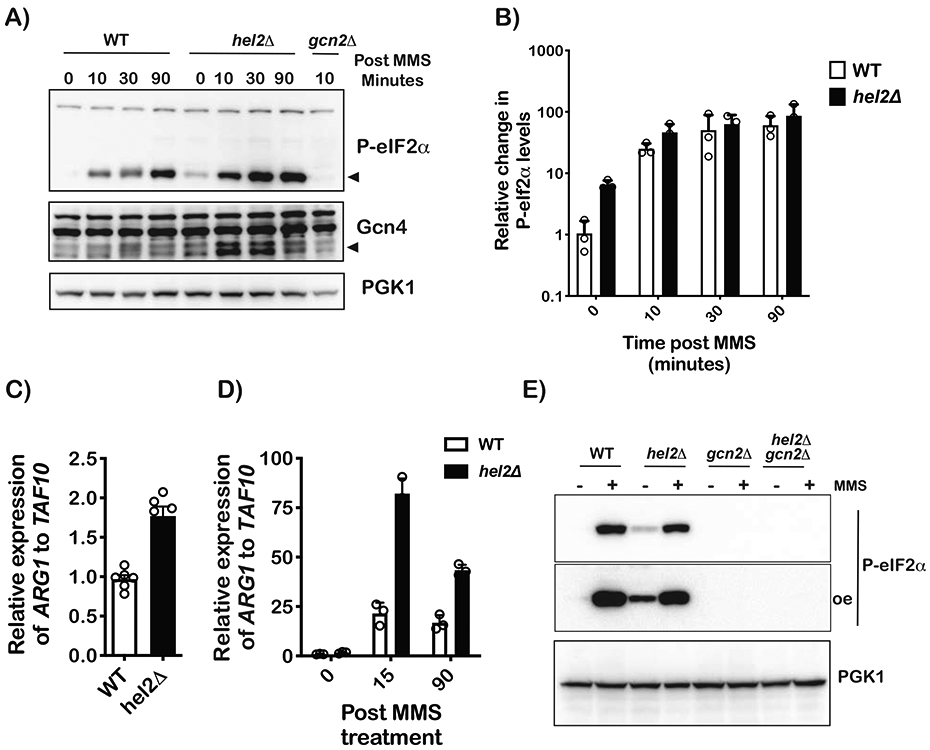
A) Western-blot analysis of total proteins isolated from wild-type and hel2Δ cells treated with 0.1% MMS for the indicated times. B) Bar graph of western-blot analysis used to follow phospho-eIF2 α levels in WT and hel2Δ cells upon MMS treatment for the indicated times. The signal for phospho-eIF2α was normalized to that of PGK1. p-value for time point 0 is 0.0018 as determined by a multiple-t test in GraphPad Prism. C) Bar graph of qPCR analysis used to measure the relative levels of the ARG1 transcript in the indicated cells with no treatment. D) Similar to C, but for cells treated with MMS for the depicted times. E) Western-blot analysis of proteins isolated from the indicated cells that were grown in the absence of presence of 0.1% MMS for 30 minutes. In all cases, plotted is the mean values determined from at least three biological repeats with the error bars representing the standard deviation around it.
Gcn2 and its coactivators suppress RQC activation
Our analysis so far focused on the suppressive effect of Hel2 on Gcn2 activation and our findings suggest that two pathways may compete with each other. Consistent with this proposal, we document overactivation of Gcn2 in the absence of Hel2 (Figures 1 and 2). Logically, this model would also predict an overactivation of RQC in the absence of Gcn2 and its coactivators. As expected, in the absence of Gcn1 and Gcn2 we observe no accumulation of phospho-eIF2α upon addition of MMS. In the absence of Gcn20, phosphorylation of eIF2α was reduced but not completely inhibited (Figure 3A). This is in agreement with previous reports showing Gcn1 and Gcn2 are essential for eIF2α phosphorylation, but Gcn20 is not (Hinnebusch, 2005). More relevant, however and corroborating our prediction, was the observation that MMS-induced ubiquitination was increased in the absence of Gcn1 and Gcn2, suggesting that the factors compete with Hel2 for collided ribosomes (Figures 3A and 3B).
Figure 3∣. RQC and ISR do not appear to directly compete with each other.
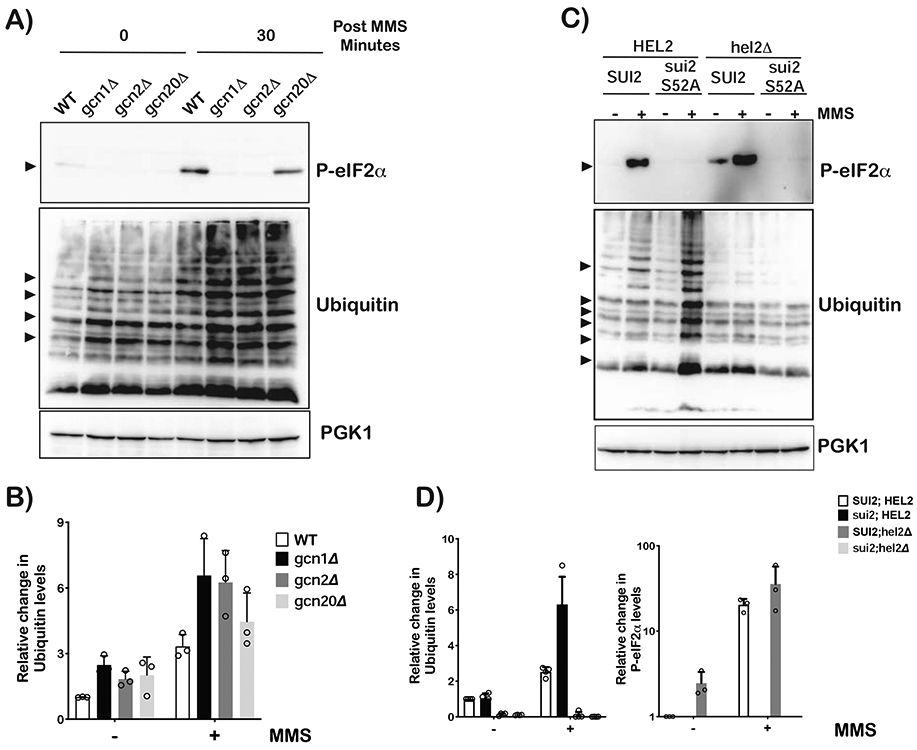
A) Western-blot analysis used to follow eIF2α phosphorylation and ubiquitination as a function of MMS in wild-type, gcn1Δ, gcn2Δ and gcn2Δ cells. B) Bar-graph analysis of three independent immuno blots similar to the one shown in (A), used to follow the relative levels of indicated ubiquitin bands in the denoted samples. C) Western-blot analysis of total protein isolated from wild-type, sui2-S52A (eIF2α cannot be phosphorylated), hel2Δ and sui2-S52A;hel2Δ cells that had been grown in the absence or presence of 0.1% MMS for 30 minutes. D) Relative levels of ubiquitin bands and phospho-eIF2α in the indicated cells, respectively. Analysis was carried out on three independent immuno blots similar to the one shown in (C). For all bar graphs, plotted is the mean values determined from three biological repeats with the error bars representing the standard deviation around it.
An inability to phosphorylate eIF2α results in increased Hel2-mediated ubiquitination
The finding that deletion of Gcn1 and Gcn2 was accompanied by increased ribosomal-protein ubiquitination (Figure 2B) can be rationalized by at least two non-mutually exclusive models. In the first one, Gcn1 and/or Gcn2 compete with Hel2 for the same interface on the collided ribosomes. In the second model, deletion of GCN1 or GCN2 suppresses the effect of MMS on translation initiation- allowing loading of ribosomes even when many of the mRNAs are damaged- and hence increases the occurrence of collisions. In this latter model, bypassing the requirement of Gcn1/Gcn2 deletion to inhibit MMS-induced eIF2α phosphorylation would result in increased Hel2-mediated ubiquitination. To accomplish this, we generated SUI2- (the gene encoding eIF2α) mutant strains where serine 52 was altered to alanine in wild-type and hel2Δ backgrounds. As expected, addition of MMS to sui2-S52A cells resulted in no phosphorylation of eIF2α, irrespective of the background (Figure 3C). Most important, however, was the observation that ubiquitination significantly increased in sui2 cells upon the addition of MMS by almost threefold relative to SUI2 cells (Figure 3D). This increase in ubiquitination is Hel2 mediated, as no MMS-induced ubiquitination was observed in hel2Δ strains (Figure 3C). Since Gcn1 and Gcn2 are present in sui2 cells, the increase in ubiquitination from MMS addition is likely to be the result of increased initiation on damaged mRNA rather than direct competition between Gcn1/Gcn2 with Hel2.
Ribosome collisions activate Gcn2
MMS is an alkylating agent known to modify nucleic acids, including RNA (Lawley, 1974). Consequently, this leads to ribosome stalling and eventual collisions, which we previously showed to activate Hel2 (Yan et al., 2019). Ribosome stalling has been recently found to be important for activating GCN2 in mammals (Ishimura et al., 2016). If the observed MMS-induced activation of Gcn2 is the result of ribosome stalling, one would predict that addition of compounds that promote readthrough to suppress the accumulation of P-eIF2α. The aminoglycoside paromomycin is such a compound and has been documented to lead to readthrough of rare codons by allowing for the misincorporation of near-cognate aa-tRNAs (Kramer et al., 2010). Addition of the drug to yeast cells on its own resulted in no observable change to the phosphorylation status of eIF2α. However, its addition to cells in the presence of MMS slightly delayed the accumulation of P-eIF2α (Figure S2). We note that this slight effect of paromomycin is expected, given that the drug cannot suppress the impact of many of the MMS-induced modified nucleotides on tRNA selection.
Although our data so far suggest that MMS activates Gcn2 through stalling, whether this activation is mediated by collisions is currently unknown. To address this, we titrated MMS from low to very-high concentrations (0-3%) and assessed P-eIF2α accumulation, reasoning that at high concentrations mRNAs are damaged at multiple positions. As this would stall multiple ribosomes on a given transcript, the probability of ribosomes colliding into each other is reduced (Figure 4A). Strikingly, we observe maximal phosphorylation of eIF2α to occur at intermediate concentration of 0.33%-1% (Figures 4B and 4C), suggesting that Gcn2 is activated in response to ribosome collision. As expected, and since Hel2 is known to be activated in response to ribosome collisions, ubiquitination of uS3 (as assessed by immunoblotting for FLAG-tagged uS3) was also found to saturate at intermediate concentrations of MMS (0.12%) (Figures 4B and 4C). Similarly, using an anti-ubiquitin antibody we observe accumulation of smaller ubiquitinated-protein products at these intermediate concentrations of MMS (Figure 4B). No ubiquitination of these proteins was observed in hel2Δ cells (Figure 4D), suggesting that anti-ubiquitin blots can be used to follow Hel2 activity circumventing the need to tag individual ribosomal proteins in all of our cells.
Figure 4∣. Intermediate concentrations of MMS and tigecycline are required for maximal activation of Gcn2.

A) Schematic showing how intermediate, but not high, concentrations of inhibitors lead to ribosome collision. B) Western-blot analysis used to follow phosphorylation of eIF2α and uS3 ubiquitination as a function of MMS concentration. In all cases cells were treated with the indicated concentration of MMS for 30 minutes. C) Relative levels of phospho-eIF2α, Ub-uS3 and indicated Ubiquitin bands in (B) collected from cells treated with the depicted concentration of MMS for 30 minutes. Quantification was conducted by densitometry analysis of at least three independent immuno-blots similar to the ones shown in (B); band intensities were normalized to Pgk1 signal. D) Western-blot analysis of total proteins isolated from wild-type and hel2Δ cells treated with the indicated MMS concentration for 30 minutes. E) Bar graph showing the relative levels of phospho-eIF2α and Ubiquitin bands in WT and hel2Δ cells treated with the indicated concentration of MMS for 30 minutes. Analysis was conducted as per (C). F) and G), similar to D) and E), respectively, but for cells treated with tigecycline for 30 minutes. H) Western-blot analysis of total proteins isolated from wild-type and hel2Δ cells treated with the indicated compounds for the indicated amount of time. I) Bar graph analysis of three biological repeats of the immuno-blots shown in (H) used to assess the relative levels of phospo-eIF2α and Ubiquitin bands. J) Fluorescence image of an ethidium-bromide-stained acid-PAGE gel used to resolve tRNAs isolated from wild-type and hel2Δ cells treated with the indicated compounds for 30 minutes. Note a sample, which was isolated from wild-type cells in the absence of any compounds, was deacylated with mild-base treatment and resolved on the left most lane. In all cases, plotted is the mean values determined from at least three biological repeats with the error bars representing the standard deviation around it.
Interestingly, for eIF2α phosphorylation, the levels of phospho-eIF2α saturate at higher concentrations of MMS compared to that of Hel2-mediated ubiquitination; 0.3% and 0.04%, respectively (Figures 4B, C, D and E), suggesting that higher frequency of ribosome collisions are needed to activate Gcn2. In agreement with this idea, in hel2Δ cells, for which collided ribosomes cannot be rescued through RQC, the levels of phosphorylated factor saturate at half the concentration of MMS (0.12%) (Figure 4E). As a result, it is highly likely that Gcn2 is activated by collided ribosomes and that under low frequency of collisions Hel2 resolves these ribosomes before they can initiate ISR.
Drugs that stall translation but stabilize A-site tRNA binding do not activate Gcn2
We previously showed that induction of ribosome collisions through the addition of cycloheximide at an intermediate concentration, around its Kd for the ribosome, results in Hel2-mediated ubiquitination of ribosomal proteins (Simms et al., 2017). Similarly, in mammals, the addition of emetine at intermediate concentrations activates ZNF598-mediated ubiquitination (Juszkiewicz et al., 2018). As a result, we hypothesized that if collisions are also responsible for Gcn2 activation that cycloheximide addition to intermediate concentrations should result in the accumulation of phospho-eIF2α. We were, therefore, surprised to observe no significant phosphorylation of eIF2α at any concentrations tested (0.015-100 mg/mL) independent of the presence of Hel2 (Figure S3A). Notably, we observed robust Hel2-dependent ribosome ubiquitination at intermediate concentrations of the inhibitor (Figure S3B). This was in disagreement with our model and suggested that activation of Gcn2 by stalled ribosomes is distinct from that of Hel2. In support of these ideas, we observe a similar pattern in the presence of the unrelated translation inhibitor anisomycin. The drug has no observable effect on eIF2α phosphorylation but mediates Hel2-dependent ubiquitination (Figure S3C).
It was not immediately clear how disparate classes of compounds robustly activate RQC, yet only one class is capable of activating Gcn2. We reasoned that these differences stem from the mode by which these compounds stall the ribosome. MMS modifies mRNAs and is hypothesized to prevent aa-tRNAs from recognizing the A-site codon (Thomas et al., 2020; You et al., 2017). Cycloheximide prevents translocation, locking the ribosome in a pre-translocated state with the A-site tRNA adopting a hybrid state. Anisomycin inhibits peptidyl transfer and stabilizes the A-site tRNA in a pre-accommodated state (Wu et al., 2019). Since MMS is the only compound of the three that prevents the tRNA from occupying the A site of the ribosome, it is feasible that activation of Gcn2 requires an empty A site. In bacteria, the tetracycline class of antibiotics inhibits binding of the A-site tRNA (Brodersen et al., 2000), but most of these do not work in eukaryotes. Recently, tigecycline has been used effectively in lysates to carry out profiling of ribosomes with an empty A site (Wu et al., 2019). The compound, however, does not work efficiently in live yeast cells, presumably due to its permeability. Nevertheless, we can show that addition of the compound to very high concentrations (2 mg/mL) inhibits cellular growth, suggesting that we can overcome some of the permeability issues (Figure S3D). This in turn allowed us to address whether the compound can activate Gcn2 at intermediate concentrations. Addition of tigecycline was found to robustly activate phosphorylation of eIF2α at concentrations of 2.5-5 mg/mL, and to lower levels at 10 mg/mL (Figure 4F). Deletion of HEL2 was found to decrease the concentration of drug required for robust activation of Gcn2 (Figure 4F). In particular, we measured a maximal increase of ~sevenfold for Phospho-eIF2α levels at tigecycline concentration of 5 mg/mL for wild-type cells compared to that of ~tenfold at tigecycline concentration of 2.5 mg/mL for hel2Δ ones (Figure 4G). The observation that tigecycline and MMS robustly induce both RQC and ISR activation, but cycloheximide and anisomycin only activate RQC, suggests that ISR activation by Gcn2 prefers the stalled ribosomes to have an empty A site. In agreement with this model, addition of 4-NQO, which oxidizes RNA and inhibits tRNA selection (Simms et al., 2014; Thomas et al., 2019) activates both processes. Similarly, the addition of 3-Amino-1,2,4-triazole (3-AT)- which inhibits histidine synthesis, resulting in depletion of His-tRNAHis- also activates both processes. We note that the addition of hydroxyurea (HU), which has been shown to activate ISR through an unknown mechanism (Hughes et al., 2000), activates Hel2-mediated ubiquitination (Figures 4H and 4I). Collectively, these observations suggest that activation of ISR and RQC converges on collided ribosomes; with ISR being sensitive to the status of the A site, whereas RQC is not.
In addition to ribosome-mediated activation of Gcn2, the factor is activated in response to accumulation of deacylated tRNAs. As a result, it is likely that many of the conditions used in our previous experiments can result in tRNA deacylation. We isolated total RNA from wild-type and hel2Δ cells in the absence of any compound and in the presence of MMS, 4-NQO and HU, and resolved the tRNAs on acid-PAGE gels (Varshney et al., 1991). We deacylated tRNAs through mild-base treatment to serve as a control. As predicted, none of the conditions resulted in general changes to the global levels of deacylated tRNAs as assessed by their mobility-shift relative to the deacylated control (Figure 4J). In contrast, nitrogen starvation, which activated ISR (Figure S3E) resulted in a slight increase of deacylated-tRNA levels (Figure S3F). While our data are consistent with a model whereby alkylation and oxidation stresses activate Gcn2 through ribosome stalling, given that the concentration of total tRNAs is estimated to be > 1000 fold in excess of that of Gcn2, we cannot rule out a role for tRNA deacylation in the process.
Stalling by a catalytically inactive release factor activates RQC but not ISR
Our analysis with translation inhibitors, and alkylation and oxidation agents suggests that Gcn2 activation is sensitive to the status of the A site of the ribosome. To add further support for this model that the A site needs to be empty for Gcn2 activation, we sought to employ a distinct strategy that does not rely on addition of compounds. More specifically, we took advantage of a dominant-negative variant of release factor eRF1, for which the universal GGQ motif in the active site that is required to engage the peptidyl-transferase center of the ribosome (Brown et al., 2015; Frolova et al., 1999) is mutated to GAQ. We and others have shown that this mutation has no effect on binding, but almost completely inhibits peptide release (Petropoulos et al., 2014; Shaw and Green, 2007). As a result, expression of this mutant factor will cause ribosome stalling and collisions at stop codons with the lead ribosome having its A site occupied with the release factor (Figure 5A). The wild-type eRF1 and GAQ mutant were introduced on a plasmid into wild-type cells under galactose induction. As expected, only the GAQ mutant promoted ubiquitination (Figure 5B), in agreement with the idea that it induces ribosome collisions. We note that, ubiquitination was observed in the absence of galactose most likely due to leaky induction as these cells were cultured in Raffinose (absence of glucose). Indeed, even in the absence of galactose, the growth rate of these cells was slower than that of ones transformed with wild-type eRF1. Furthermore, induction of the GAQ variant was much less than that of the GGQ wild-type factor (Figure 5B) due to its toxic effect. More important than its effect on ubiquitination was the observation that induction of the GAQ mutant resulted in little to no change in the levels of phsopho-eIF2α (Figure 5B), especially when compared to that observed in the presence of MMS (Figure S4). These observations suggest that ribosome collisions induced by an eRF1-bound-lead ribosome do not trigger ISR. To directly confirm that expression of GAQ leads to ribosome collisions, we conducted sucrose-gradient fractionation of polysomes isolated from cells expressing eRF1GGQ and eRF1GAQ. Polysome profiles of the GAQ cells indicated that they accumulated significant amount of higher-order-polysome structures (Figure 5C, top). These structures were found to be resistant to RNase I treatment (Figure 5C, bottom). Under normal conditions, during which ribosomes are sparsely distributed across mRNAs, addition of RNase I collapses polysomes into monosomes. Under conditions where ribosomes collide, RNase I is incapable of accessing mRNA between them and polysomes collapse to higher order structures such as disomes instead (Guydosh and Green, 2014). These observations suggest that eRF1GAQ induces significant ribosome collisions, but yet fails to activate Gcn2.
Figure 5∣. Collided ribosomes with an eRF1-bound-lead ribosome do not activate ISR but activate RQC.

A) Schematic showing how a catalytically dead mutant (GAQ) of eRF1 can be used to cause ribosome collisions at stop codons with the factor bound to the A site of the lead ribosome. B) Western-blot analysis of total proteins isolated from cells expressing wild type or catalytically dead mutant of eRF1 in the absence and presence of induction by galactose. C) Polysome profiles of lysates isolated from cells expressing the indicated-release-factor variants. Top shows profiles of untreated lysates; bottom shows those of RNase I-treated lysates.
Inhibition of collided-ribosomes clearance activates Gcn2 and ribosomal-protein ubiquitination
During RQC, following Hel2-mediated ubiquitination of ribosomal proteins, stalled ribosomes are rescued through the action of Slh1 (Ikeuchi et al., 2019). Hence, in the absence of this factor, collided ribosomes are likely to linger a while longer. Consequently, in our model of ribosome collisions being responsible for Gcn2 activation, one would predict that deletion of SLH1 would result in greater phosphorylation of eIF2α. Furthermore, one should also observe greater MMS-induced ubiquitination. In complete agreement with this prediction, in the absence of Slh1, we observe significant accumulation of phospho-eIF2α in the absence of any stressors (Figure 6A). We measured a ~fourfold increase in the levels of P-eIF2 phospho-eIF2α levels in slhlΔ cells relative to wild-type ones (Figure 6B). In agreement with these observations, qPCR analysis revealed that MMS induction of the Gcn4 target ARG1 is twofold higher in the slh1Δ strain relative to the wild-type one; upon the addition of MMS, ARG1 expression is increased by less than tenfold in the wild-type strain, whereas in slh1Δ its induction is increased by > twentyfold (Figure 6C). In addition to its impact on Gcn2 activation, deletion of SLH1, resulted in a significant increase in MMS-induced protein ubiquitination (Figures 6A and B). These findings greatly suggest that in the absence of Slh1, collided ribosomes are not cleared efficiently and as a result activate both Gcn2 and Hel2.
Figure 6∣. Ribosome collisions activate Gcn2.
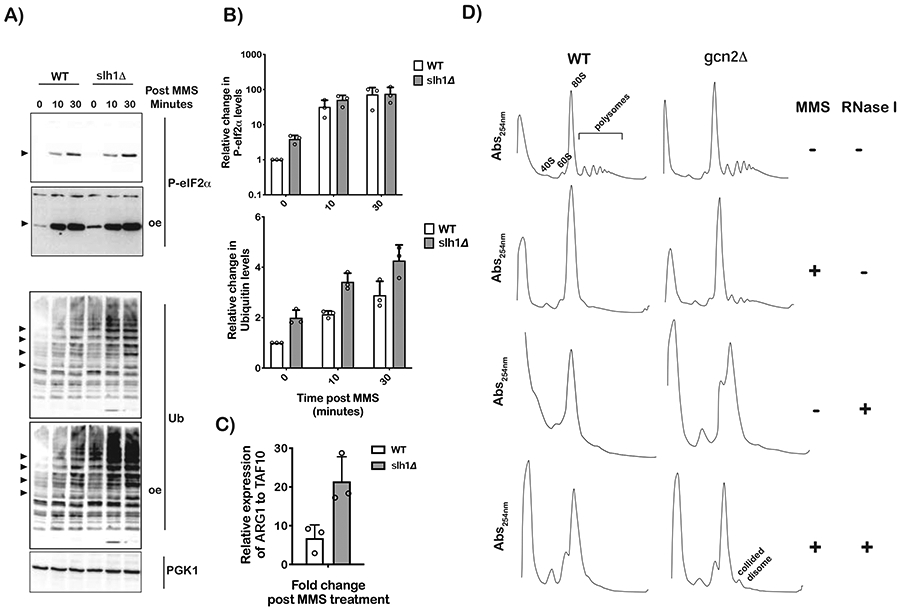
A) Western-blot analysis of total proteins isolated from the depicted cells treated with 0.1% MMS for the indicated times (oe indicates overexpression). B) Bar-graph analysis of three independent immuno blots similar to the one shown in (A), used to follow the relative levels of phospho-eIF2α and ubiquitin bands in the indicated samples. C) qPCR analysis of the AGR1 transcript in wild-type and slh1Δ cells after a 10-minute incubation period with 0.1% MMS. D) Ribosome profiles of lysates isolated from the indicated strains that were incubated in the absence or presence of 0.1% MMS for 30 minutes. The lysates were either mock treated or RNase I treated, as shown, before resolving them over a 10-50% sucrose gradient. The disome peak that appears to be resistant to RNase I is labeled as such. For all bar graphs, plotted is the mean values determined from three biological repeats with the error bars representing the standard deviation around it.
Alkylation stress causes ribosome collisions.
The observation that MMS and 4-NQO activate RQC, suggests that these compounds lead to ribosome collisions, but we could not demonstrate that this actually occurs in vivo (Yan et al., 2019). Our observation that these compounds also induce ISR (Figure 4) gave us a rationale as to why collisions were not detected under these conditions. In particular, since these compounds cause eIF2α phosphorylation, their addition also results in global inhibition of translation and, as a result, loading and eventual collision of ribosomes on mRNAs is severely limited. These ideas were confirmed by utilizing a GCN2-deletion strain. We used an RNaseprotection assay of mRNAs in polysomes and in complete agreement with our model, upon addition of MMS, only in the gcn2Δ strain were we able to observe significant accumulation of disomes (Figure 6D). These findings suggest that indeed alkylation stress results in ribosome collisions.
Gcn1 associates with collided ribosomes
Previous studies have shown that Gcn1, Gcn2 and Gcn20 co-sediment with polysomes during sucrose-gradient fractionation (Marton et al., 1997; Sattlegger, 2000; Sattlegger and Hinnebusch, 2005). This together with the observation that Gcn1 and Gcn20 have a domain homologous to elongation factor 3 (Visweswaraiah et al., 2012), suggest that the factors directly bind to ribosomes. But whether the factors recognize specific conformations of the ribosomes that accumulate under stress is currently unknown. The observation that conditions that increase levels of collided ribosomes activate Gcn2 suggests that at least one of the factors interact with collided ribosomes. To test this prediction, we set out to conduct sucrose-gradient fractionation of RNAse I-treated lysates and investigate the association of Gcn1 with different ribosome fractions as function of MMS addition. We note that addition of cycloheximide, which is used routinely during profiling, results in significant levels of collided ribosomes even in the absence of any stressors (Figure S5). As a result, we opted not to use any antibiotics in our profiling analysis. The absence of inhibitor explains why we observe significantly lower levels of polysomes due to increased ribosome runoff during our lysate preparation. This, however, should not affect our analysis of stalled ribosomes especially in the absence of Hel2, during which collided ribosomes cannot be rapidly resolved.
As expected, the addition of MMS to wild-type cells resulted in an almost complete loss of polysomes resulting from phospho-eIF2α-induced inhibition of translation (Figure S6). In contrast, the profile of the sui2-mutant cells exhibited polysomes in the presence of MMS (Figure S6), due to the inability of these cells to inhibit initiation in the presence of alkylation stress. Interestingly, further deletion of HEL2 from these cells resulted in increased levels of polysomes in the presence of MMS; this is in agreement with the fact that these cells are incapable of resolving collided ribosomes. As a result, we used these cells to assess the association between Gcn1 and ribosomes (Figure 7A). Similar to what others have reported (Marton et al., 1997; Sattlegger, 2000; Sattlegger and Hinnebusch, 2005), Gcn1 was observed to only marginally associate with polysomes. Remarkably, in the presence of MMS, Gcn1 was found to associate with the heavy fraction of the polysomes, suggesting that it prefers stalled ribosomes (Figure 7A). We then conducted polysome profiling on RNase I-treated lysates, which collapsed most of the polysomes into a monosome. Even under these conditions, Gcn1 was observed to remain associated with the heavy fractions, suggesting that factor associates with collided ribosomes (Figure 7B). Quantification of the signal corresponding to Gcn1 to assess its distribution across the sucrose gradient confirmed that the protein indeed migrates with the heavy polysome fractions (Figure 7C). This is in contrast to proteins that are known not to stably associate with ribosomes such as PGK1 and eRF1 (Figure 7C and Figure S7). Notably, similar analysis of RNaseI-treated lysates revealed that in the presence of MMS, the factor remains associated with the resulting collided ribosomes (Figure 7D). In particular, in the absence of MMS, ~7% of the protein was found to remain associated with the collided ribosomes (fractions 5-7) relative to 20% that was observed to associate with polysomes when lysates were not treated with RNase I (Figures 7C and 7D). In contrast, for MMS-treated cells the relative association of the factor with polysomes only slightly decreased from 30% to 27% as a result of RNase I treatment. The observation that under conditions that activate Gcn2, more of its coactivator associates with collided ribosomes is in agreement with our model that collisions are the primary signal for ISR.
Figure 7∣. Gcn1 associates with collided ribosomes.

A) Ribosome profiles of lysates isolated from sui2-S52A;hel2Δ cells incubated in the absence and presence of 0.1% MMS for 30 minutes. Shown is the absorbance at 254 nm of lysates fractionated over a 10-35% sucrose gradient. Below the profiles is western-blot analysis of fractions collected from sucrose-gradient fractions for the indicated proteins. B) Similar to A), but lysates were treated with RNase I prior to fractionation over a 10-35% sucrose gradient. C) and D) Densitometry analysis of immuno blots similar to the ones shown in (A) and (B), respectively, which was used to analyze the relative distribution of the indicated proteins across sucrose gradients. Analysis was conducted on three biological repeats, with the mean values plotted and the error bars representing the standard deviation around them.
Discussion
During protein synthesis, the ribosome receives multiple cues to ensure that the correct protein is made at the right place, right time and at the right concentration. These cues are the result of signals triggered by varying cellular needs and environmental conditions such as proliferation and stress. The integrated-stress response (ISR), which responds to stresses through the activation of initiation factor eIF2α kinases, is one such important pathway in eukaryotes. Phosphorylation of eIF2α represses global translation, but also derepresses translation of key pro-survival mRNAs. In yeast, ISR is activated by the eIF2α kinase Gcn2 in response to amino-acid deprivation. The most widely accepted model evokes an active role for deacylated tRNA in the process (Dong et al., 2000). In this model, under amino-acid-deprivation conditions, deacylated tRNAs accumulate and are recognized by Gcn2 and its coactivators Gcn1 and Gcn20. Interestingly, Gcn1 and Gcn20 have been shown to bind the ribosome, and they are thought to deliver the deacylated tRNA from the A site of the ribosome to Gcn2, activating its kinase activity (Sattlegger, 2000). In addition to amino-acid deprivation, recent data have shown Gcn2 to respond to other stressors that do not change the levels of deacylated tRNAs, suggesting an alternative mechanism for its activation (Harding et al., 2019; Inglis et al., 2019; Ishimura et al., 2016). In this alternative model, ribosome stalling plays a more critical role in activating Gcn2. In agreement with this model, here we showed that alkylation and oxidation agents that modify mRNAs and stall the ribosome robustly activate Gcn2 as assessed by accumulation of phospho-eIF2α (Figures 1, 2, 3 and 4). Interestingly, ribosome stalling also activates the quality-control pathways NGD and RQC. Both of these processes depend critically on an E3 ligase Hel2. Notably, the very same alkylating and oxidizing agents that activate Gcn2 also activate Hel2 (Yan et al., 2019), suggesting that ISR and RQC must be coordinated somehow. Indeed, here we provide compelling evidence that suggests that the two processes antagonize each other (Figures 1, 2, 3, 4 and 6).
In addition to this apparent competition between the two processes, ISR and RQC appear to be exquisitely sensitive to the concentration of stressor. Optimum activation was observed to take place at intermediate concentrations of damaging agents (Figure 4B), with RQC activation occurring at lower concentrations. It thus appears that Hel2 is more sensitive to changes to the translation apparatus and responds faster than Gcn2. The sensitivity of Hel2 to the concentration of translation inhibitors has been noted by several groups (Ikeuchi et al., 2019; Juszkiewicz et al., 2018; Simms et al., 2017), and has been used to formulate a model for its activation through ribosome collisions (Simms et al., 2017). Structural studies provided a molecular rational for this activation, for which collided ribosomes form an interface that can be recognized by the ligase (Ikeuchi et al., 2019; Juszkiewicz et al., 2018). Interestingly, however, promoting ribosome collisions through the addition of translation inhibitors at intermediate concentrations, known to activate RQC, was found not to activate ISR (Figure S3A-C). A simple model that could explain these observations is that Gcn2 is not activated by ribosome collisions, and instead the factor responds to ribosome stalling through a different mechanism, although this model cannot explain why stalling by translation inhibitors is incapable of triggering ISR. In an alternative model, Gcn2 is sensitive to the status of the stalled ribosome during collisions. Given that chemical agents and translation inhibitors stall the ribosome through different mechanisms, this model could explain the observed disparities between the effect of the two classes of molecules on ISR activation. Specifically, the ISR activators MMS, 4-NQO and 3-AT leave the A site of the ribosome empty; whereas inhibitors such as cycloheximide and anisomycin keep the A site occupied by a tRNA (Wu et al., 2019). As a result, we hypothesized that Gcn2 prefers the A site to be empty for its activation. To test this hypothesis, we looked for inhibitors that prevent aminoacyl-tRNA from binding the A site. For instance, the tetracycline class of antibiotics is known to accomplish this in bacteria (Brodersen et al., 2000), but most have no effect on translation in eukaryotes. Fortuitously, the antibiotic tigecycline was recently shown to be effective in preventing A-site occupancy in yeast lysate (Wu et al., 2019), suggesting that the drug could be used to inhibit aa-tRNA from binding the A site in vivo. Indeed, the addition of drugs to very high concentrations (> 2 mg/mL) was found to inhibit yeast growth (Figure S3D). Most important and in contrast to cycloheximide and anisomycin, tigecycline was found to activate Gcn2 (Figure 4F). Furthermore, robust activation was observed at intermediate concentrations of drug, around those that inhibit growth modestly. Therefore, similar to Hel2, Gcn2 appears to be activated by ribosome collisions, but its activation is sensitive to the status of the A site of the ribosome. In agreement with this model, overexpression of a catalytically dead mutant of eRF1, which causes ribosome collisions on stop codon with the A site of the lead ribosomes occupied by the factor, does not trigger ISR (Figure 5).
To add further support for this model of collision-mediated activation of Gcn2, we sought to confirm that stressors that activate the factor also lead to the formation of collided ribosomes. To this end, we used an RNase I-protection assay, for which collided ribosomes are not accessible to the enzyme and remain intact in their higher-order form. As expected, addition of MMS to yeast cells that cannot phosphorylate eIF2α (gcn2Δ and sui2-S52A) results in the formation of stable higher order collided polysomes, as assessed by sucrose-gradient fractionation of RNase I-treated yeast lysate (Figures 6 and 7, and Figure S6). More notable was the observation that Gcn1 (a Gcn2 coactivator and is required for ISR) remains associated with the polysomes even after the RNase I treatment (Figure 7). Moreover, addition of MMS was observed to result in further association between the factor and polysomes, suggesting that Gcn1 is likely to prefer collided ribosomes over spread-out polysomes.
Our data suggest that both Hel2 and Gcn2 are activated by the very same signal of ribosome collisions. We find that inhibition of RQC, through deletion of HEL2, results in the overactivation of ISR, and vice versa (Figures 1, 2, 3 and 6). There are at least three models that can explain this connection between the two processes. In the first one, Gcn2 coactivators compete with Hel2 for the same interface on the ribosome. In the second one, ubiquitination of ribosomal proteins by Hel2 inhibits the activation of Gcn2. In the third and simplest model, inhibition of ISR or RQC pathways results in increased ribosome collisions, through derepression of translation initiation or inhibition of collided-ribosome clearance, respectively. This increased frequency of collision would then lead to over-activation of whichever of the two pathways that remains intact. We favor this last model, since inhibition of eIF2α phosphorylation by using the S52A mutant of the factor was found to result in a significant increase in Hel2-mediated ubiquitination in the presence of MMS (Figure 3). Nevertheless, we cannot rule out that multiple models may contribute to this apparent competition between the two pathways.
During revision of this study, Wu et al. (Wu et al., 2020) reported that collided ribosomes activate the stress-activated protein kinase (SAPK) pathway through the MAPKKK ZAKa in mammals. In particular, the authors found the factor to preferentially associate with collided ribosomes and that this association was responsible for its activation of the SAPK (p38/JNK) and GCN2, but whether the factor displays a preference of the status of the A site of the stalled ribosome was not immediately clear. Notably, Wu and colleagues used the translation inhibitors anisomycin and emetine at intermediate concentrations to promote ribosome collisions and showed that both of these inhibitors activate Gcn2. As emetine is known not to be able to inhibit protein synthesis in yeast (Battaner and Vazquez, 1971), we could only test anisomycin. Interestingly and in contrast to what was observed in mammalian cells, we could not detect significant accumulation of phospho-eIF2α in the presence of anisomycin (Figure S3). This distinction between mammals and yeast might be explained by differences in the mode of Gcn2 activation. For instance, it is unclear whether yeast has a ZAK homologue that acts upstream of Gcn1, Gcn2 and Gcn20 during ISR activation. Alternatively, and given how conserved ISR activation is, it is likely that this observed difference is the result of disparate mode of interactions between the drug and the ribosome. Anisomycin inhibits peptidyl-transfer, and as a result the A-site tRNA adopts a so-called A/T state (Valle et al., 2002). This tRNA has been shown to be unstable in this state and readily dissociates from the ribosome (Dorner et al., 2006), which may give ZAKα or GCN1 enough time to bind collided ribosomes.
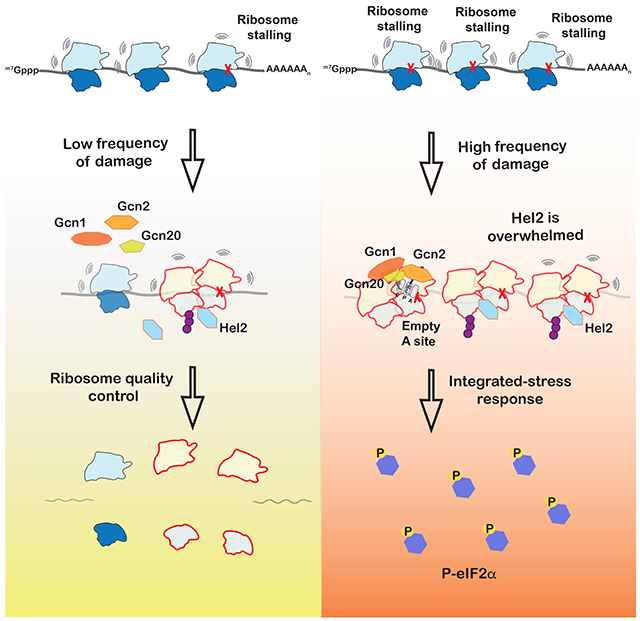
The model of ribosome-collision-mediated ISR activation is appealing because it easily rationalizes how Gcn2 and its coactivators (Gcn1 and Gcn20) are activated by ribosomes only under stress. Under normal conditions, ribosome collisions occur infrequently as a result of aberrant mRNAs, and it makes sense that Hel2 is activated to trigger NGD and RQC. Under stress conditions, ribosome collisions occur frequently, Hel2 is likely to get overwhelmed and Gcn2 gets activated. The finding that Hel2-mediated ubiquitination is more sensitive to drug concentration is consistent with these ideas (Figure 2). Multiple factors could contribute to the ability of Hel2 to recognize collided ribosomes and activate RQC before the activation of Gcn2. For instance, the factor’s relatively higher affinity, concentration or faster kinetics to those of Gcn2 and its coactivators could ensure RQC is activated prior to ISR activation. Future experiments aimed at dissecting the thermodynamic and kinetics contributions for both processes are likely to shed important insights into how RQC and ISR are coordinated.
Limitations of Study
Our findings reported here provide new insights about the interplay between the activation of ISR by Gcn2 that of RQC by Hel2, and suggest that Hel2 is more apt at recognizing stalled ribosomes. However, important molecular details about this apparent competition are lacking and are needed to expand our understanding of these two important processes. In particular, it would be interesting to investigate the thermodynamic and kinetic parameters that govern the activation of both processes. Furthermore, complementary high-resolution structural studies of stalled ribosomes in complex with Gcn1, Gcn20 and Gcn2 as well as with Hel2 are more than likely to identify the molecular rationale for how these factors recognize collided ribosomes.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests and information for reagents and resources will be fulfilled by Dr. Hani Zaher (hzaher@wustl.edu).
Materials Availability
All stable reagents from this study are available from the Lead Contact.
Data and Code Availability
The RNAseq raw data reported in this paper can be accessed through GEO: GSE150790. All data generated in this study are available from the Lead Contact. Raw gel images are available at http://dx.doi.org/10.17632/n5svwx4rt9.1 through Mendeley Data.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strain Construction
Yeast strains used in this study are listed below. Yeast cells were grown at 30°C in YPD media. Deletion, mutant and chromosomally tagged yeast strains were constructed in the BY4741 background (MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0) using common PCR-knockout techniques. Oligonucleotide sequences are listed in Table S2.
Media Preparation
All solid and liquid media have been prepared according to standard lab protocols. Unless otherwise stated, MMS, 4-NQO, cycloheximide, HU and 3-AT were added to final concentrations of 0.1%, 5 μg/mL, 100 μg/mL, 100 mM and 100 mM respectively. Cells were incubated with drugs for 30 min, unless stated otherwise.
METHOD DETAILS
RNA seq
The RNA-seq data sets were prepared from total RNA isolated using the hot-phenol extraction method (Simms et al., 2017) from mid-log phase yeast cells in the absence and presence of 0.1% MMS incubated for 90-min at 30°C. For each condition, 30 μg of total RNA from three biological replicates were processed and mRNA libraries were generated and processed by the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine. Briefly, polyA enriched libraries were prepared from total RNA using Oligo-dT beads. Samples were prepared according to library kit manufacturer’s protocol, indexed, pooled, and sequenced on an Illumina NovaSeq. Basecalls and demultiplexing were performed with Illumina’s RTA version 1.9 and bcl2fastq2 software with a maximum of one mismatch in the indexing read. It was then sequenced with 2 X 150-bp paired-end reads. RNA-seq reads were then aligned to the Ensembl release 76 (GCA_000146045.2) primary assembly with STAR version 2.5.1a (Dobin et al., 2013). Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 1.4.6-p5 (Liao et al., 2014). Isoform expression of known Ensembl transcripts were estimated with Salmon version 0.8.23. Sequencing performance was assessed for the total number of aligned reads, total number of uniquely aligned reads, and features detected. The ribosomal fraction, known junction saturation, and read distribution over known gene models were quantified with RSeQC version 2.6.2 (Wang et al., 2012). Genes with more than 20 reads were normalized over all read counts across all samples. Differentially expressed genes were identified by Perseus 1.6.12 (Tyanova et al., 2016) using pairwise comparisons between genotypes/conditions. Significant changes in transcript abundance were calculated by ANOVA contrasts (p ≤ 0.05), excluding transcripts with an FDR > 0.05 to account for multiple comparisons.
Polysome profiling with RNase I
Yeast cultures from mid-log phase yeast cells were harvested by centrifuging at room temperature. Cells were then resuspended in polysome-lysis buffer (20 mM Tris pH 7.5, 140 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 200 μg/mL heparin, 1% Triton), washed once and lysed with glass beads using a FastPrep-24 (MP Biomedical). Supernatants from cleared lysates corresponding to 500 ug of total RNA were treated with 100 U of RNase I (Thermo Fisher, cat#AM2294) at 30°C for 30 minutes. The treated lysates were then layered over a 10–50% sucrose gradient and centrifuged at 35,000 rpm for 160 min in an SW41Ti (Beckman) swinging-bucket rotor. Gradients were then fractionated using a Brandel tubepiercing system combined with continuous absorbance reading at 254 nm. Proteins were isolated through a TCA precipitation method and resuspended in HU buffer (8 M Urea, 5% SDS, 200 mM Tris pH 6.8, 100 mM DTT, 1 mM ethylenediaminetetraacetic acid (EDTA), bromophenol blue).
Overexpression of eRF1 and its GAQ mutant
Sequences encoding wild-type and GAQ eRF1 were PCR amplified and cloned into pYES2 plasmid (ThermoFisher) by Gibson assembly (NEB). Plasmids were transformed into yeast using the lithium acetate method (Gietz and Woods, 2002). Individual colonies were inoculated into -Ura complete synthetic media with raffinose as the sole carbon source. Overnight cultures were diluted to an Abs600nm of 0.05 and grown to an Abs of 0.4 before cells were divided into two cultures and galactose was added to a final concentration of 2% to only one of the cultures. Cells were grown for an additional 3 hours before collecting them for western-blot and ribosome-profile analyses.
Quantitative RT-PCR
Total RNA from yeast cells was isolated following the hot phenol method (Köhrer and Domdey, 1991) . cDNA was generated with M-MuLV reverse transcriptase (NEB, cat#M0253L) from 1-10 μg of total RNA that was treated with DNase I (Thermo Scientific, cat# EN0601) using a random hexamer (Thermo Scientific, cat# SO142) for priming. Quantitative RT-PCR was conducted using iTaq Universal SYBR Green Supermix (BIO-RAD, cat#1725121) with ~ 50 ng of cDNA. The relative fold change was obtained by following the ΔΔCt method. The relative transcript abundance for each gene from three biological repeats was determined by normalizing to the expression level of the TAF10 gene.
Western blotting
Total lysates from mid-log-phase cells were harvested and lysed in 1 mL of ice-cold lysis buffer (300 mM NaOH, 1% β-mercaptoethanol). Proteins were precipitated through the addition of TCA to 7.5% and resuspended in HU buffer (8 M Urea, 5% SDS, 200 mM Tris pH 6.8, 100 mM DTT, 1 mM ethylenediaminetetraacetic acid (EDTA), bromophenol blue) using a volume that was normalized to the amount of cells harvested. Proteins were separated by SDS–PAGE and analyzed by immunoblotting. The following antibodies were used: rabbit anti phospho- eIF2α (Ser51) (Cell Signaling Technology, cat# 9721S), rabbit anti-Gcn4 (from Thomas Dever lab, 1:2000 v/v dilution), rabbit anti Gcn1 (from Alan Hinnebusch lab, 1:2000 v/v dilution), mouse anti-ubiquitin HRP (Santa Cruz, Cat#: sc8017; 1:2,000 v/v dilution), mouse anti-FLAG (Sigma; Cat#: F1804; 1:5,000 v/v dilution), rabbit anti-uS4 (Rps9) (Abcam, Cat#:ab117861; 1:5,000 v/v dilution), and rabbit anti-eRF1(Eyler et al., 2013) (1:2000 dilution). Secondary antibodies of goat anti mouse IgG HRP (Thermo Scientific, Cat#: 31430) and goat anti rabbit IgG HRP (Thermo Scientific, Cat#: 31460) were used at (1:10,000 v/v dilution). Densitometry analysis was conducted using Image Lab (Bio-Rad Laboratories). Unless otherwise stated, relative signal of the protein of interest was obtained by normalizing to that of PGK1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical and graphs analysis for RT-PCR (from 3 biological repeats) and western blot (from 3 biological repeats) results were processed by GraphPad Prism 8.4.3 (Graphpad Software, La Jolla, CA). For RNA seq analysis, differentially expressed genes from 3 biological repeats were identified by Perseus 1.6.12 (Tyanova et al., 2016) using pairwise comparisons between genotypes/conditions. Significant changes in transcript abundance were calculated by ANOVA contrasts (p ≤ 0.05), excluding transcripts with an FDR > 0.05 to account for multiple comparisons.
Supplementary Material
Document S1: Figures S1-S7, Table S2, Data S1.
Table S1: RNAseq analysis of WT and hel2Δ cells in the absence and presence of MMS, related to Figure 1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti phosphor-eIF2a (Ser51) | Cell Signaling Technology | 9721S |
| rabbit anti-GCN4 | Thomas Dever lab | N/A |
| rabbit anti GCN1 | Alan Hinnebusch lab | N/A |
| mouse anti-ubiquitin HRP | Santa Cruz | sc8017 |
| mouse anti-FLAG | Sigma | F1804 |
| rabbit anti-uS4 (Rps9) | Abcam | ab117861 |
| rabbit anti-eRF1 | Rachel Green lab | N/A |
| goat anti mouse IgG HRP | Thermo Scientific | 31430 |
| goat anti rabbit IgG HRP | Thermo Scientific | 31460 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phusion High Fidelity DNA polymerase | NEB | Cat#: M0530 |
| Ambion RNase I | Thermo Scientific | AM2294 |
| iTaq Universal SYBR Green Supermix | BIO-RAD | cat#1725121 |
| M-MuLV reverse transcriptase | NEB | cat#M0253L |
| Random hexamer | Thermo Scientific | SO142 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Scientific | Cat#: 34095 |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo Scientific | Cat#: 30480 |
| N-Ethylmaleimide | Millipore Sigma | Cat#: E3876 |
| Xbal | NEB | Cat#: R0145 |
| BamHI | NEB | Cat#: R0136 |
| XhoI | NEB | Cat#: R0146 |
| T4 DNA Ligase | NEB | Cat#: M0202 |
| DNA Polymerase I, Large (Klenow) Fragment | NEB | Cat#: M0210 |
| DNase I | Thermo Scientific | Cat#: 89836 |
| cycloheximide ultra pure | VWR | Cat#: 94271 |
| 4-nitroquinoline 1-oxide | Millipore Sigma | N8141-1G |
| Methyl methanesulfonate | Millipore Sigma | 129925-25G |
| Hydroxyurea | Millipore Sigma | H8627-5G |
| Phenylmethylsulfonyl fluoride | GoldBio | P-470-10 |
| anisomycin | Millipore Sigma | A5862 |
| tigecycline | Fisher Scientific | T3589-100mg |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO:GSE150790 |
| Experimental Models: Organisms/Strains | ||
| BY4741 (matA) | Dharmacon lab | N/A |
| DOM34Δ | Doma and Parker, 2006 | N/A |
| SUI2 (iosgenic strain for sui2-S52A) | This paper | N/A |
| sui2-S52A | This paper | N/A |
| SUI2, HEL2Δ | This paper | N/A |
| sui2-S52A; HEL2 Δ | This paper | N/A |
| HEL2Δ | Yan et al., 2019 | N/A |
| GCN1Δ | Dharmacon | N/A |
| GCN2Δ | This paper | N/A |
| GCN4Δ | Dharmacon lab | N/A |
| Hel2Δ; GCN2Δ | This paper | N/A |
| Rps3-FLAG | Simms et al., 2017 | N/A |
| GCN20Δ | Dharmacon lab | N/A |
| SLH1Δ | This paper | N/A |
| Oligonucleotides | ||
| DNA oligos for cloning: See Table S2 | N/A | N/A |
| Recombinant DNA | ||
| pYES2-eRF1 | This paper | N/A |
| pYES2-eRF1(GAQ) | This paper | N/A |
| Software and Algorithms | ||
| Ensembl release 76 | N/A | GCA_000146045.2 |
| STAR version 2.5.1a | Dobin et al., 2013 | 2.5.1a |
| Subread:featureCount | Liao et al., 2014 | version 1.4.6-p5 |
| Salmon | N/A | version 0.8.23 |
| RSeQC | Wang et al., 2012 | version 2.6.2 |
| Perseus | Tyanova et al., 2016 | 1.6.12 |
| Image Lab | Bio-Rad | 6.1 |
| GraphPad Prism | GraphPad | 8.4.3 |
Highlights.
Hel2 attenuates Gcn2-mediated activation of integrated stress response and vice versa
Gcn2 is activated in response to conditions that promote ribosome collisions
Stalling-induced integrated stress response prefers ribosomes with empty A sites
Hel2-mediated ubiquitination is more sensitive to stalling relative to Gcn2 activation
Acknowledgments
We would like to thank Alan Hinnebusch and Thomas Dever for their gift of Gcn1 and Gcn4 antibodies. We thank Carrie Simms for careful reading of the manuscript. We thank Fionn McLoughlin for assisting the analysis the RNA seq data. We are also grateful to members of the Zaher laboratory for useful discussions on earlier versions of this manuscript. This work was supported by a grant from the National Institutes of Health to HSZ (R01GM112641).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Atkinson GC, Tenson T, and Hauryliuk V (2011). The RelA/SpoT Homolog (RSH) Superfamily: Distribution and Functional Evolution of ppGpp Synthetases and Hydrolases across the Tree of Life. PLoS One 6, e23479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augusto L, Amin PH, Wek RC, and Sullivan WJ (2019). Regulation of arginine transport by GCN2 eIF2 kinase is important for replication of the intracellular parasite Toxoplasma gondii. PLOS Pathog. 15, e1007746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, and Bruhat A (2013). The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaner E, and Vazquez D (1971). Inhibitors of protein synthesis by ribosomes of the 80-S type. BBA Sect. Nucleic Acids Protein Synth. 254, 316–30. [DOI] [PubMed] [Google Scholar]
- Berlanga JJ, Rivero D, Martin R, Herrero S, Moreno S, and de Haro C (2010). Role of mitogen-activated protein kinase Sty1 in regulation of eukaryotic initiation factor 2α kinases in response to environmental stress in Schizosaccharomyces pombe. Eukaryot. Cell 9, 194–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al. (2005). ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J. 24, 3470–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen DE, Clemons WM, Carter AP, Morgan-Warren RJ, Wimberly BT, and Ramakrishnan V (2000). The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 103, 1143–1154. [DOI] [PubMed] [Google Scholar]
- Brown A, Shao S, Murray J, Hegde RS, and Ramakrishnan V (2015). Structural basis for stop codon recognition in eukaryotes. Nature 524, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delépine M, Nicolino M, Barrett T, Golamaully M, Mark Lathrop G, and Julier C (2000). EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet 25, 406–409. [DOI] [PubMed] [Google Scholar]
- Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, and Hinnebusch AG (1992). Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68, 585–596. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Qiu H, Garcia-Barrio M, Anderson J, and Hinnebusch AG (2000). Uncharged tRNA Activates GCN2 by Displacing the Protein Kinase Moiety from a Bipartite tRNA-Binding Domain. Mol. Cell 6, 269–279. [DOI] [PubMed] [Google Scholar]
- Dorner S, Brunelle JL, Sharma D, and Green R (2006). The hybrid state of tRNA binding is an authentic translation elongation intermediate. Nat. Struct. Mol. Biol 13, 234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler DE, Wehner KA, and Green R (2013). Eukaryotic Release Factor 3 Is Required for Multiple Turnovers of Peptide Release Catalysis by Eukaryotic Release Factor 1. J. Biol. Chem 288, 29530–29538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyries M, Montani D, Girerd B, Perret C, Leroy A, Lonjou C, Chelghoum N, Coulet F, Bonnet D, Dorfmuller P, et al. (2014). EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat. Genet 46, 65–69. [DOI] [PubMed] [Google Scholar]
- Frolova LY, Tsivkovskii RY, Sivolobova GF, Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI, and Kisselev LL (1999). Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA 5, 1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Barrio M (2000). Association of GCN1-GCN20 regulatory complex with the N-terminus of eIF2alpha kinase GCN2 is required for GCN2 activation. EMBO J. 19, 1887–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzia A, Jafarnejad SM, Meyer C, Chapat C, Gogakos T, Morozov P, Amiri M, Shapiro M, Molina H, Tuschl T, et al. (2017). The E3 ubiquitin ligase and RNA-binding protein ZNF598 orchestrates ribosome quality control of premature polyadenylated mRNAs. Nat. Commun 8, 16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, and Woods RA (2002). Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350, 87–96 [DOI] [PubMed] [Google Scholar]
- Gomez E, Mohammad SS, and Pavitt GD (2002). Characterization of the minimal catalytic domain within eIF2B: the guanine-nucleotide exchange factor for translation initiation. Embo J. 21, 5292–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guydosh NR, and Green R (2014). Dom34 Rescues Ribosomes in 3′ Untranslated Regions. Cell 156, 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Guy MP, Kon Y, and Phizicky EM (2018). Lack of 2’-O-methylation in the tRNA anticodon loop of two phylogenetically distant yeast species activates the general amino acid control pathway. PLoS Genet. 14, e1007288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, and Ron D (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding HP, Ordonez A, Allen F, Parts L, Inglis AJ, Williams RL, and Ron D (2019). The ribosomal P-stalk couples amino acid starvation to GCN2 2 activation in mammalian cells. Elife. 8, e50149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Chevet E, and Harding HP (2013). Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov 12, 703–719. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG (1993). Gene-specific translational control of the yeast GCN4 gene by phosphorylation of eukaryotic initiation factor 2. Mol. Microbiol 10, 215–223. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG (2005). Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol 59, 407–450. [DOI] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, et al. (2000). Functional Discovery via a Compendium of Expression Profiles. Cell 102, 109–126. [DOI] [PubMed] [Google Scholar]
- Ikeuchi K, Tesina P, Matsuo Y, Sugiyama T, Cheng J, Saeki Y, Tanaka K, Becker T, Beckmann R, and Inada T (2019). Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 38, e100276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis AJ, Masson GR, Shao S, Perisic O, McLaughlin SH, Hegde RS, and Williams RL (2019). Activation of GCN2 by the ribosomal P-stalk. Proc. Natl. Acad. Sci. USA 116, 4946–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura R, Nagy G, Dotu I, Chuang JH, and Ackerman SL (2016). Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. Elife 5, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinsky SA, and Samson LD (1999). Global response of Saccharomyces cerevisiae to an alkylating agent. Proc. Natl. Acad. Sci. USA 96, 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszkiewicz S, and Hegde RS (2017). Initiation of Quality Control during Poly(A) Translation Requires Site-Specific Ribosome Ubiquitination. Mol. Cell 65, 743–750.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, and Hegde RS (2018). ZNF598 Is a Quality Control Sensor of Collided Ribosomes. Mol. Cell 72, 469–481.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrer K, and Domdey H (1991). [27] Preparation of high molecular weight RNA. In Methods in Enzymology, pp. 398–405. [DOI] [PubMed] [Google Scholar]
- Kramer EB, Vallabhaneni H, Mayer LM, and Farabaugh PJ (2010). A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA 16, 1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, and Hinnebusch AG (2001). Tight Binding of the Phosphorylated α Subunit of Initiation Factor 2 (eIF2α) to the Regulatory Subunits of Guanine Nucleotide Exchange Factor eIF2B Is Required for Inhibition of Translation Initiation. Mol. Cell. Biol 21, 5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Marino G, and Levine B (2010). Autophagy and the Integrated Stress Response. Mol. Cell 40, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lageix S, Zhang J, Rothenburg S, and Hinnebusch AG (2015). Interaction between the tRNA-Binding and C-Terminal Domains of Yeast Gcn2 Regulates Kinase Activity In Vivo. PLOS Genet. 11, e1004991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley PD (1974). Some chemical aspects of dose-response relationships in alkylation mutagenesis. Mutat. Res. Mol. Mech. Mutagen 23, 283–295. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin W, Lin Y, Li J, Fenstermaker AG, Way SW, Clayton B, Jamison S, Harding HP, Ron D, and Popko B (2013). Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J. Neurosci 33, 5980–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, and Klann E (2013). Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci 16, 1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton MJ, Vazquez de Aldana CR, Qiu H, Chakraburtty K, and Hinnebusch AG (1997). Evidence that GCN1 and GCN20, translational regulators of GCN4, function on elongating ribosomes in activation of eIF2alpha kinase GCN2. Mol. Cell. Biol 17, 4474–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson GR (2019). Towards a model of GCN2 activation. Biochem. Soc. Trans 47, 1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Ikeuchi K, Saeki Y, Iwasaki S, Schmidt C, Udagawa T, Sato F, Tsuchiya H, Becker T, Tanaka K, et al. (2017). Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat. Commun 8, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meydan S, and Guydosh NR (2020). Disome and Trisome Profiling Reveal Genome-wide Targets of Ribosome Quality Control. Mol. Cell 79, 588–602.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton-Liger F, Paquet C, Dumurgier J, Lapalus P, Gray F, Laplanche JL, and Hugon J (2012). Increased cerebrospinal fluid levels of double-stranded rna-dependant protein kinase in alzheimer’s disease. Biol. Psychiatry 71, 829–835. [DOI] [PubMed] [Google Scholar]
- Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, and Marton MJ (2001). Transcriptional Profiling Shows that Gcn4p Is a Master Regulator of Gene Expression during Amino Acid Starvation in Yeast. Mol. Cell. Biol 21, 4347–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO Rep. 17, 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos AD, McDonald ME, Green R, and Zaher HS (2014). Distinct Roles for Release Factor 1 and Release Factor 2 in Translational Quality Control. J. Biol. Chem 289, 17589–17596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H (2002). Mutations that bypass tRNA binding activate the intrinsically defective kinase domain in GCN2. Genes Dev. 16, 1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Dong J, Hu C, Francklyn CS, and Hinnebusch AG (2001). The tRNA-binding moiety in GCN2 contains a dimerization domain that interacts with the kinase domain and is required for tRNA binding and kinase activation. EMBO J. 20, 1425–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez M, Wek RC, and Hinnebusch AG (1991). Ribosome association of GCN2 protein kinase, a translational activator of the GCN4 gene of Saccharomyces cerevisiae. Mol. Cell. Biol 11, 3027–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez M, Wek RC, Vazquez de Aldana CR, Jackson BM, Freeman B, and Hinnebusch AG (1992). Mutations activating the yeast eIF-2 alpha kinase GCN2: isolation of alleles altering the domain related to histidyl-tRNA synthetases. Mol. Cell. Biol 12, 5801–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawal Y, Chereji RV, Valabhoju V, Qiu H, Ocampo J, Clark DJ, and Hinnebusch AG (2018). Gcn4 Binding in Coding Regions Can Activate Internal and Canonical 5’ Promoters in Yeast. Mol. Cell 70, 297–311.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattlegger E (2000). Separate domains in GCN1 for binding protein kinase GCN2 and ribosomes are required for GCN2 activation in amino acid-starved cells. EMBO J. 19, 6622–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattlegger E, and Hinnebusch AG (2005). Polyribosome binding by GCN1 is required for full activation of eukaryotic translation initiation factor 2α kinase GCN2 during amino acid starvation. J. Biol. Chem 280, 16514–16521. [DOI] [PubMed] [Google Scholar]
- Shaw JJ, and Green R (2007). Two distinct components of release factor function uncovered by nucleophile partitioning analysis. Mol Cell 28, 458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Eyler DE, and Green R (2010). Dom34:Hbs1 Promotes Subunit Dissociation and Peptidyl-tRNA Drop-Off to Initiate No-Go Decay. Science 330, 369–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms CL, Hudson BH, Mosior JW, Rangwala AS, and Zaher HS (2014). An Active Role for the Ribosome in Determining the Fate of Oxidized mRNA. Cell Rep. 9, 1256–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms CL, Yan LL, and Zaher HS (2017). Ribosome Collision Is Critical for Quality Control during No-Go Decay. Mol. Cell 68, 361–373.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, and Hinnebusch AG (2009). Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhakar A, Ramachandran A, Ghosh S, Hasnain SE, Kaufman RJ, and Ramaiah KVA (2000). Phosphorylation of serine 51 in initiation factor 2α (eIF2α) promotes complex formation between eIF2α(P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry 39, 12929–12938. [DOI] [PubMed] [Google Scholar]
- Sundaramoorthy E, Leonard M, Mak R, Liao J, Fulzele A, and Bennett EJ (2017). ZNF598 and RACK1 Regulate Mammalian Ribosome-Associated Quality Control Function by Mediating Regulatory 40S Ribosomal Ubiquitylation. Mol. Cell 65, 751–760.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EN, Simms CL, Keedy HE, and Zaher HS (2019). Insights into the base-pairing preferences of 8-oxoguanosine on the ribosome. Nucleic Acids Res. 47, 9857–9870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EN, Kim KQ, McHugh EP, Marcinkiewicz T, and Zaher HS (2020). Alkylative damage of mrna leads to ribosome stalling and rescue by trans translation in bacteria. Elife 9, e61984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Valasek L, Nielsen KH, and Hinnebusch AG (2002). Direct eIF2-eIF3 contact in the multifactor complex is important for translation initiation in vivo. EMBO J. 21, 5886–5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle M, Sengupta J, Swami NK, Grassucci RA, Burkhardt N, Nierhaus KH, Agrawal RK, and Frank J (2002). Cryo-EM reveals an active role for aminoacyl-tRNA in the accommodation process. EMBO J. 21, 3557–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney U, Lee CP, and RajBhandary UL (1991). Direct analysis of aminoacylation levels of tRNAs in vivo: Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J. Biol. Chem 266, 24712–24718. [PubMed] [Google Scholar]
- Vattem KM, and Wek RC (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 101, 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visweswaraiah J, Lee SJ, Hinnebusch AG, and Sattlegger E (2012). Overexpression of Eukaryotic Translation Elongation Factor 3 Impairs Gcn2 Protein Activation. J. Biol. Chem 287, 37757–37768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wek SA, Zhu S, and Wek RC (1995). The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol. Cell. Biol 15, 4497–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CCC, Zinshteyn B, Wehner KA, and Green R (2019). High-Resolution Ribosome Profiling Defines Discrete Ribosome Elongation States and Translational Regulation during Cellular Stress. Mol. Cell 73, 959–970.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CCC, Peterson A, Zinshteyn B, Regot S, and Green R (2020). Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell. 182, 404–416.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LL, and Zaher HS (2019). How do cells cope with RNA damage and its consequences? J. Biol. Chem 294, 15158–15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LL, Simms CL, McLoughlin F, Vierstra RD, and Zaher HS (2019). Oxidation and alkylation stresses activate ribosome-quality control. Nat. Commun 10, 5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, and Koumenis C (2010). The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 29, 2082–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You C, Dai X, and Wang Y (2017). Position-dependent effects of regioisomeric methylated adenine and guanine ribonucleosides on translation. Nucleic Acids Res. 45, 9059–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, and Wek RC (1998). Ribosome-binding domain of eukaryotic initiation factor-2 kinase GCN2 facilitates translation control. J. Biol. Chem 273, 1808–1814. [DOI] [PubMed] [Google Scholar]
- Zhu S, Sobolev AY, and Wek RC (1996). Histidyl-tRNA synthetase-related sequences in GCN2 protein kinase regulate in vitro phosphorylation of eIF-2. J. Biol. Chem 271, 24989–24994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1: Figures S1-S7, Table S2, Data S1.
Table S1: RNAseq analysis of WT and hel2Δ cells in the absence and presence of MMS, related to Figure 1.
Data Availability Statement
The RNAseq raw data reported in this paper can be accessed through GEO: GSE150790. All data generated in this study are available from the Lead Contact. Raw gel images are available at http://dx.doi.org/10.17632/n5svwx4rt9.1 through Mendeley Data.