

Abstract
Abundant intraperitoneal (IP) accumulation of extracellular mucus in patients with appendiceal mucinous carcinoma peritonei (MCP) causes compressive organ dysfunction and prevents delivery of chemotherapeutic drugs to cancer cells. We hypothesized that reducing extracellular mucus would decrease tumor-related symptoms and improve chemotherapeutic effect in patient-derived models of MCP.
Mucolysis was achieved using a combination of bromelain (BRO) and N-acetylcysteine (NAC). Ex vivo experiments of mucolysis and chemotherapeutic drug delivery/effect were conducted with MCP and non-MCP tissue explants. In vivo experiments were performed in mouse and rat patient-derived xenograft (PDX) models of early and late (advanced) MCP.
MCP tumor explants were less chemosensitive than non-MCP explants. Chronic IP administration of BRO + NAC in a mouse PDX model of early MCP and a rat PDX model of late (advanced) MCP converted solid mucinous tumors into mucinous ascites (mucolysis) that could be drained via a percutaneous catheter (rat model only), significantly reduced solid mucinous tumor growth and improved the efficacy of chemotherapeutic drugs.
Combination of BRO + NAC efficiently lyses extracellular mucus in clinically relevant models of MCP. Conversion of solid mucinous tumors into mucinous ascites decreases tumor bulk and allows for minimally invasive drainage of liquified tumors. Lysis of extracellular mucus removes the protective mucinous coating surrounding cancer cells and improves chemotherapeutic drug delivery/efficacacy in cancer cells. Our data provide a preclinical rationale for the clinical evaluation of BRO + NAC as a therapeutic strategy for MCP.
INTRODUCTION
Mucinous appendix cancers are a unique histologic subtype in which more than 50% of the tumor volume is composed of extracellular mucus. They have a high propensity for peritoneal dissemination, clinically referred to as mucinous carcinoma peritonei (MCP).(1–6) For low-grade MCP (LG-MCP), large volumes of extracellular mucus accumulate in the abdominal cavity and cause compressive organ dysfunction. For high-grade MCP (HG-MCP), extracellular mucus is less abundant (although still > 50% of the tumor mass) and provides a protective environment in which cancer cells thrive.(1, 5, 7–10) Peritoneal and liver metastases from mucinous colorectal cancers are known to be less responsive to palliative systemic chemotherapy than non-mucinous cancers, and mucinous rectal cancers are less likely to respond to neoadjuvant chemoradiation therapy than non-mucinous rectal cancers.(1, 11, 12) These data suggest that extracellular mucus presents a clinically relevant barrier to effective therapy in mucinous cancers. We hypothesized that extracellular mucolysis would decrease compressive effects from bulky mucinous tumor burden (especially for LG-MCP) and improve the efficacy of standard chemotherapeutic drugs by removing the protective mucus barrier surrounding neoplastic cells (especially for HG-MCP).(10, 13)
The viscoelastic extracellular mucus gel in MCP is predominantly comprised of a complex network of mucin 2 (MUC2) protein fibers, that make up 80% of the dry weight of mucus.(14–16) A variety of mucolytic drugs have been investigated in preclinical and clinical studies to treat benign mucin-hypersecreting diseases (e.g. cystic fibrosis, chronic obstructive pulmonary disease).(17–19) Recent preclinical in vitro and in vivo studies demonstrated promising mucus/MUC2 inhibitory and anti-proliferative effects of bromelain (BRO) and N-acetylcysteine (NAC) against MUC2-secreting colon cancer cells.(20, 21) Pillai and colleagues demonstrated the ability of BRO + NAC to lyse small fragments of mucus (derived from patients with appendiceal MCP) ex vivo and after implantation and treatment in nude rats for 48h.(22, 23) BRO is a natural aqueous extract of the pineapple fruit that contains a mixture of thiol proteases and non-protease components capable of hydrolyzing esters, amides and glycosidic linkages. NAC, naturally found in the Allium plant species (e.g. onions), is a thiol (sulfhydryl)-containing amino acid capable of reducing disulfide bonds. Since MCP is primarily composed of MUC2 protein, a large polymeric glycoprotein composed of peptide bonds, glycosidic linkages and disulfide bonds between individual MUC2 monomers, the combination of BRO and NAC is a promising candidate to dissolve extracellular mucus.
In this study, we evaluated the cytoprotective role of extracellular mucus and mucolytic effects of BRO + NAC ex vivo using patient-derived mucinous tumor explants. We also evaluated the safety and efficacy of chronic intraperitoneal (IP) delivery of BRO + NAC in clinically relevant patient-derived xenograft (PDX) models of early and late (advanced) MCP. Finally, we assessed the efficacy of standard chemotherapeutic drugs following mucolytic therapy. Our data demonstrate significant mucolysis and enhanced cytotoxicity following BRO + NAC treatment in clinically relevant models of MCP and provides a preclinical rationale for early phase clinical trials.
MATERIALS AND METHODS
Reagents
DMEM (Dulbecco’s Modified Eagle’s Medium) was obtained from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was obtained from Hyclone Laboratories (Logan, UT). Cell-culture plates were purchased from Costar (Cambridge, MA). BRO was obtained from Sigma (St. Louis, MO). NAC, oxaliplatin (OXAL), and mitomycin C (MMC) were obtained from Cayman Chemical (Ann Arbor, MI). CellTiter 96 Aqueous Assay was obtained from Promega Corporation (Madison, WI). Anti-rabbit and anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Tissue Path Disposable Base Molds, Tissue-Tek O.C.T compound Superfrost Plus microscope slides were obtained from Fisher Scientific (Pittsburgh, PA). MUC2 antibody (Catalog number sc-15334) for immunofluorescence assay (IF) and western blot were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and Abcam (Cambridge, MA), respectively. Reverse transcriptase-polymerase chain reaction (RT-PCR) kits, including primers and probe for MUC2, MUC5AC, MUC5B, MUC6 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), were obtained from Applied Biosystems (ABI, Foster City, CA). Anti-Cisplatin DNA Adducts Antibody, (clone ICR4) for immunofluorescence assay was obtained from Sigma. Anti-rabbit Alexa 647 and Alexa 488 were obtained from Cell Signaling Technology (Danvers, MA). SYTOX Orange for nucleic acid labeling was obtained from Life Technologies (Grand Island, NY).
Cell culture
LS174T cells were obtained from American Type Culture Collection (Manassas, VA). These cells were derived from a human mucinous colorectal cancer and demonstrate “characteristics of goblet-cells” by secreting relatively high levels of MUC2 protein. LS174T cells were grown in cell-culture plates in DMEM (supplemented with 4.5 g/L glucose, 10% fetal bovine serum, 2 mM L-glutamine, 20 mM HEPES, 100 IU/ml penicillin and 100 μg/ml streptomycin) at 37°C and 5% CO2.
Tumor explant culture
MCP (extracellular mucus/MUC2-rich) and non-MCP (extracellular mucus/MUC2-poor) tumor specimens were obtained during surgery, brought to the laboratory within one hour and processed for culture and experimental procedures. Demographic and clinicopathologic data, as well as details regarding in vitro experimental useage of collected human tumor samples are provided in Supplementary Table S1. Human tissue was collected under an Institutional Review Board (IRB)-approved protocol. The explant culture system was used according to a previously described method. (24, 25) Using a 4-mm biopsy puncher, cubes of tumor tissue were prepared and placed in antibiotic gentamicin containing DMEM and 10% FBS. The tumor explants were cultured at 37°C in a humidified atmosphere containing 5% CO2.
MTS assay
LS174T cells were seeded at a density of 10,000 cells per well in 96-well plate overnight. Cells were exposed to different concentrations of mucus and overlaid with OXAL for 24h. After treatment, cell viability was determined by MTS assay according to the manufactures instructions (Promega). Cells were treated with a combined solution of a tetrazolium compound MTS (3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium, inner salt) and an electron coupling reagent phenazine methosulfate for additional 2h at 37°C. The absorbance of the formazan product at 490 nm was measured.
Immunofluorescence assay
MCP and non-MCP explants (untreated or treated) were placed in Tissue path disposable Base Molds and snap-frozen in Tissue -Tek O.C.T compound. Using a cryostat microtome, 5 μM frozen section of the specimen was mounted on superfrost Plus microscope slides. The slides were incubated in 4% paraformaldehyde for 15 minutes, washed and blocked for 60 minutes at room temperature. The slides were then stained for 3h at room temperature with the MUC2 antibody. The slides were washed three times with 1X PBS and incubated with antirabbit Alexa 488 and SYTOX Orange for nucleic acid staining for 30 minutes at room temperature. The slides were washed three times with PBS. Confocal images were randomly taken of tissue sections using a Leica confocal microscope. Images of each slide were then analyzed using Image J software to quantify the average intensity of MUC2 protein.
Oxaliplatin-induced DNA adducts
Mucus tumor tissues were harvested from rat PDX models of late (advanced) MCP after 5 weeks of BRO + NAC treatment, treated ex vivo with OXAL, placed in Tissue path disposable Base Molds and snap-frozen in Tissue -Tek O.C.T compound. Using a cryostat microtome, 4 μM frozen section of the specimen was mounted on superfrost Plus microscope slides. Visualization and quantification of OXAL-induced intrastrand crosslinked in nuclear DNA of tumor samples were performed using IF staining.(26) Nuclear DNA was counterstained with Sytox orange. DNA adduct was visualized by Anti-Cisplatin DNA Adducts Antibody, clone ICR4 (rat anti-Guanine-Guanine; 1:1000 in 1%PBS/BSA; 3h; room temperature) and Alexa flour −488 secondary antibody (Goat anti-[rat Ig]; 30min; 37°C).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Apoptosis of MCP tissue was analyzed using terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay according to the manufacturer’s instruction. Sections were analyzed using an Olympus IX81 microscope. The percentage of cell death was determined by counting the number of TUNEL positive cells in three independent fields of different slides using Image J software.
Transwell assay
LS174T cells were seeded in the lower (acceptor) compartment at a density of 0.05 × 106 in 24-well plates overnight. Mucus (diluted or undiluted) was layered on the insert and different concentrations of OXAL (25μM, 50μM, and 100μM) were added to the upper (donor) compartment for 24h. After treatment, cells were collected from the lower compartment to measure the intracellular platinum levels by atomic absorption spectrophotometry (AAS). Mucus used for this experiment was taken from a single patient (Patient IDs #1 in Supplemetary Table 1). The mucus samples was taken fresh from the operating room to the laboratory within 1h and utilized for the transwell experiments.
Intracellular platinum concentration assay
Concentrations of intracellular platinum were quantitated by AAS (PerkinElmer, Waltham, MA) with a dynamic range of 50-5000 ng/mL. Briefly, 50 μl of cell pellets were used for the assay, and 50 μl of control human plasma/ultrafiltrates (50:50) were added to the samples, vortexed and, mixed 1:1 (v/v) with diluent buffer (0.5% ammonium dihydrogen phosphate and 0.03% magnesium nitrate 6-hydrate in water). An aliquot of 10 μl of this mixture was injected in the AAS, and concentrations were back-calculated relative to a calibration curve prepared in human plasma/ultrafiltrates (50:50). Ultrafiltrates were generated by centrifuging 0.5 mL of control plasma in Amicon Centrifree YM-30 (MPS, Millipore Billerica, MA) for 20 min at 5°C at 1500 x g.
Mucolysis with BRO plus NAC
One gram of MCP-derived mucus specimen was exposed to 10 ml of PBS containing BRO plus NAC at varying pH in 50 ml conical tubes. Control tubes were treated with only 10 ml of 1X PBS. The tubes were then incubated at 37°C for 0-24h. At the end of the incubation, the remaining mucus was retrieved and weighed after by passing the conical tube contents through a grade 703 blotting paper (VWR Radnor, PA).
Reverse transcription (RT) and real-time polymerase chain reaction (qPCR) analysis
Total RNA was isolated from human MCP tissue using RNeasy Mini Kit and quantified using Nanodrop ND-1000 spectrophotometer (Wilmington, DE). Each sample was reverse transcribed into cDNA in a Peltier Thermal Cycler (PTC-220 DNA Engine Dyad, MJ Research; Waltham, MA) using random hexamers and the GeneAmp RNA PCR Core Kit (ABI). Real-time PCR was then carried out in an ABI Prism SDS 7000 Cycler System (ABI), using commercially available primers and probes obtained from ABI, specific for MUC2, MUC5AC, MUC5B, MUC6 and GAPDH cDNA, for 40 cycles at 95°C for 15 seconds. Relative amounts of mucin mRNA were determined after normalization of mucin transcripts to that of GAPDH, using software supplied by the manufacturer (ABI).
Western blot analysis
Ten micrograms of soluble mucus proteins were run on a 1% agarose gel followed by blotting onto nitrocellulose membranes. After blocking the membranes with 5% fat-free dry milk powder for 60 min at room temperature, blots were incubated with the MUC2 antibody (1:1000) overnight at 4°C. The blots were then washed and incubated with HRP-conjugated anti-mouse secondary antibodies for 60 min at room temperature. After washing, blots were developed with enhanced Super Signal West Dura Extended Duration Substrate and visualized using a Bio-Rad imager with chemiluminescence capability.
Histopathology
Normal mice and rats were treated with BRO + NAC for 2 weeks. After treatment, animals were sacrificed and formalin-fixed spleen, liver, stomach, pancreas and kidney were submitted to the University of Pittsburgh Medical Center Hillman Cancer Center and Tissue and Research Pathology/Pitt Biospecimen Core shared resource for routine histology. Slides were stained with hematoxylin and eosin (H&E). The histology was evaluated by Lora Rigatti, VMC, DACVP, Senior Veterinary pathologist, University of Pittsburgh.
In vivo mouse PDX model
The development and utilization of our PDX model has been published.(24, 27) Fresh PMP tumor was processed and implanted in the peritoneal cavity of nude mice. The resulting model has been successfully passaged to subsequent generations in nude mice with 100% reliability and retains the clinical and pathological characteristics of the original human tumor. Mucinous tumor growth becomes clinically at 2 weeks with the progressive increase in abdominal girth and body weights over the following weeks. Animals were randomized at day 7, following tumor implantation, to different IP treatment groups (3-10 animals per group) and weekly measurements of gross body weight (grams) and abdominal girth (millimeters) were recorded. Following the completion of experiments, animals were sacrificed and abdominal tumor weight was measured.
In vivo rat PDX model
Fresh PMP tumor was processed and implanted in the peritoneal cavity of nude rats. Mucinous tumor growth becomes clinically at 2 weeks with the progressive increase in abdominal girth and body weights over the following weeks. Animals were randomized at day 14, following tumor implantation, to different treatment groups (3-10 animals per group) and weekly measurements of gross body weight (grams) and abdominal girth (millimeters) were recorded. At the fifth week, rats were anesthetized via a nose cone with 1-2% isoflurane and O2, positioned on an animal bed, and IP mucinous ascites was aspirated using a temporary catheter (14F [3.35 mm x 4.65 mm) x 12” silicone catheter (Norfolkaccess, Skokie, IL). Following ascites drainage, animals were sacrificed and solid mucinous tumor was harvested and weighed.
MRI Methods
Rats were anesthetized via a nose cone with 1-2% isoflurane and O2. positioned on an animal bed with the abdomen secured to reduce motion artifacts, and then placed in the scanner. Respiration rate was monitored, and body temperature was maintained using a warm air heating system, (SA Instruments, New York, NY, USA). MRI was performed on a 7T/30-cm AVIII spectrometer (Bruker Biospin, Billerica, MA) equipped with a 12 cm gradient set and using an 86 mm quadrature RF volume coil and Paravision 6.0.1. A T2-weighted RARE sequence was used to visualize the abdominal mucinous tumor, with the following parameters: repetition time (TR)/echo time (TE) = 4400/24 ms, anisotropic field of view (FOV) of 55-60 mm, acquisition matrix = 256 × 256, 41 slices with a slice thickness of 2 mm, 4 averages, and a RARE factor = 8.
STATISTICAL ANALYSIS
Statiscal analysis was carried out using GraphPad 7 software (GraphPad Software, Inc., San Diego, CA, USA). Two group comparisons were assessed using the Student’s t test. Comparisons among more than two groups were done using ANOVA with post hoc testing.
RESULTS
Extracellular mucus is cytoprotective for cancer cells and imparts chemoresistance.
Tumor tissues from 5 MCP and 3 non-MCP patients were exposed to OXAL (100 μM) for 24h ex vivo. Less apoptotic cells were identified in MCP explants by TUNEL assay compared to non-MCP explants (Figures 1A, B). Corresponding MUC2 staining for MCP and non-MCP explant tissue is shown in Supplemntary Figure S1. Similar results were seen when explants were treated with MMC (data not shown). We then performed MTS assay to demonstrate the cytoprotective properties of extracellular mucus. Cells were coated with varying dilutions of mucus harvested from MCP patients, varying concentrations of OXAL were added above the mucus layer, and viability of LS174T cells below the mucus layer was quantified by MTS assay at 24h. Minimal cytotoxicity was encountered when undiluted mucus coated the cells, while serial dilution of mucus coating the cells correlated with increasing cell death (Figure 1C). We then performed transwell experiments to demonstrate that mucus posed a barrier to OXAL delivery to cells. The upper chamber was separated from lower chamber by varying dilutions of extracellular mucus harvested from MCP patients, OXAL (100μM) was added to upper chamber, and intracellular platinum levels were quantified in LS174T cells in the lower chamber at 24h by AAS.
Figure 1. Extracellular mucus is cytoprotective for cancer cells and imparts chemoresistance.
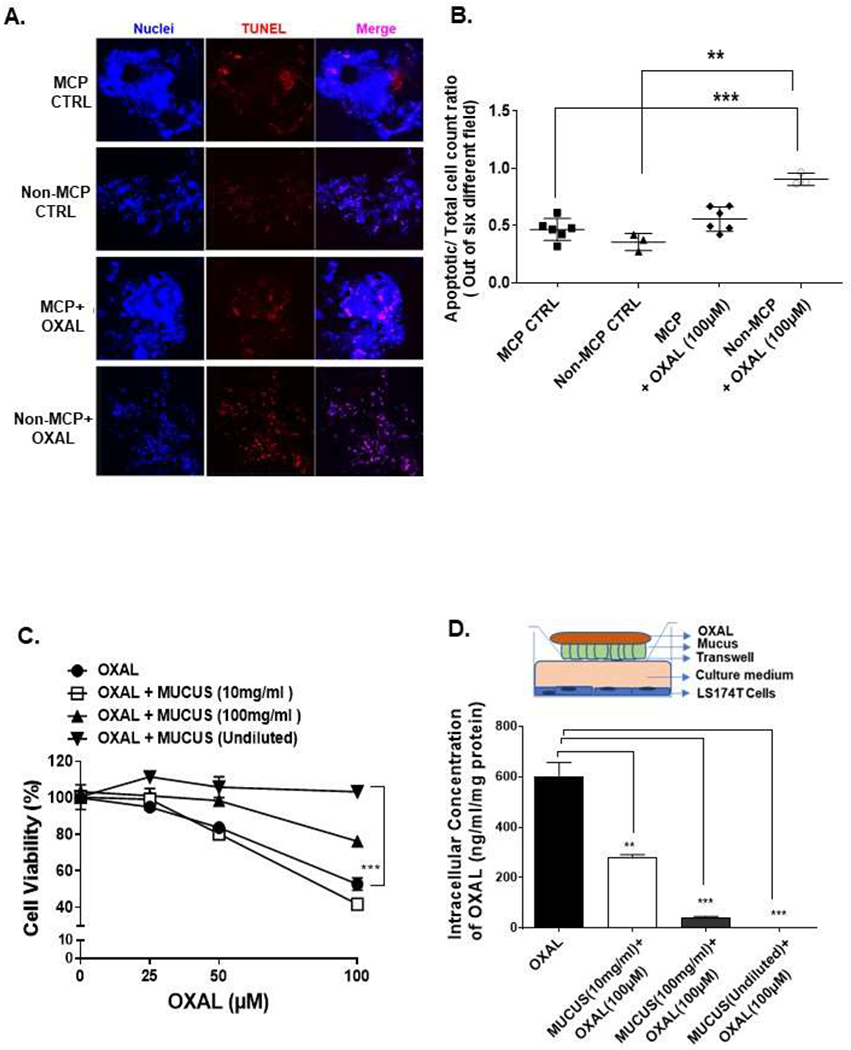
TUNEL assay was performed on MCP and non-MCP tumor tissues, with or without OXAL (100μM) treatment, for 24h ex vivo (A) and TUNEL-positive cell were quantified; images were randomly taken of 6 different fields and analyzed using Image J software to quantify apoptosis (B). LS174T cells were coated with undiluted or diluted mucus (diluted to 10 mg/ml or 100 mg/ml in PBS), treated with OXAL (0-100μM) for 24h, following which cell viability was analyzed by MTS assay (C). In a tranwell assay, delivery of OXAL (upper chamber) to LS174T cells (lower chamber) through undiluted or diluted mucus separating the chambers was determined by quantifying intracellular platinum using AAS; the insert is a pictorial representation of the transwell setup (D). Error bars represent standard deviation (SD) from triplicate experiments (**P< .01, ***P< .001).
Intracellular platinum levels correlated with serial dilution of mucus coating the cells, suggesting that undiluted mucus posed a barrier to OXAL delivery (Figure 1D).
Extracellular mucus can be lysed with a combination of BRO and NAC.
MCP tumor tissue fragments (1g) were exposed ex vivo to BRO (300 μg/ml) + NAC (250 mM) dissolved in 10 ml PBS solution (adjusted to pH 6.8 to 8.5) for 24h at 37°C (Figure 2A). Residual mucus gel was separated from the solution by filtration. There was a significant reduction in mucus gel weight following mucolytic therapy, especially at higher pH levels (Figure 2B). We were unable to detect MUC2 protein in the separated solution, especially at higher pH levels, consistent with complete destruction of MUC2 protein following mucolysis (Figure 2C). We measured mRNA levels for secretory mucins (MUC2, MUC5AC, MUC5B and MUC6) in pre-treated MCP tumor tissue and confirmed that MUC2 mRNA was the predominant mucin expressed in MCP as expected (Supplementary Figure S2). Other secretory mucins (MUC5AC, MUC5B, MUC6) are known to have a lower expression and minor role in the pathophysiology of MCP and were not further evaluated. There was no significant change in viscosity of the separated solution by syringe pump viscometer assay, suggesting that mucus and MUC2 protein were being completely lysed and disintegrated (Supplementary Figure S3). We found that complete lysis of 1g of mucus could be achieved at 2h when submerged at 37°C in 10 ml of PBS containing BRO (300 μg/ml) + NAC (250 mM) adjusted to pH 8.0 (Figure 2D).
Figure 2. Extracellular mucus can be lysed with a combination of BRO and NAC.
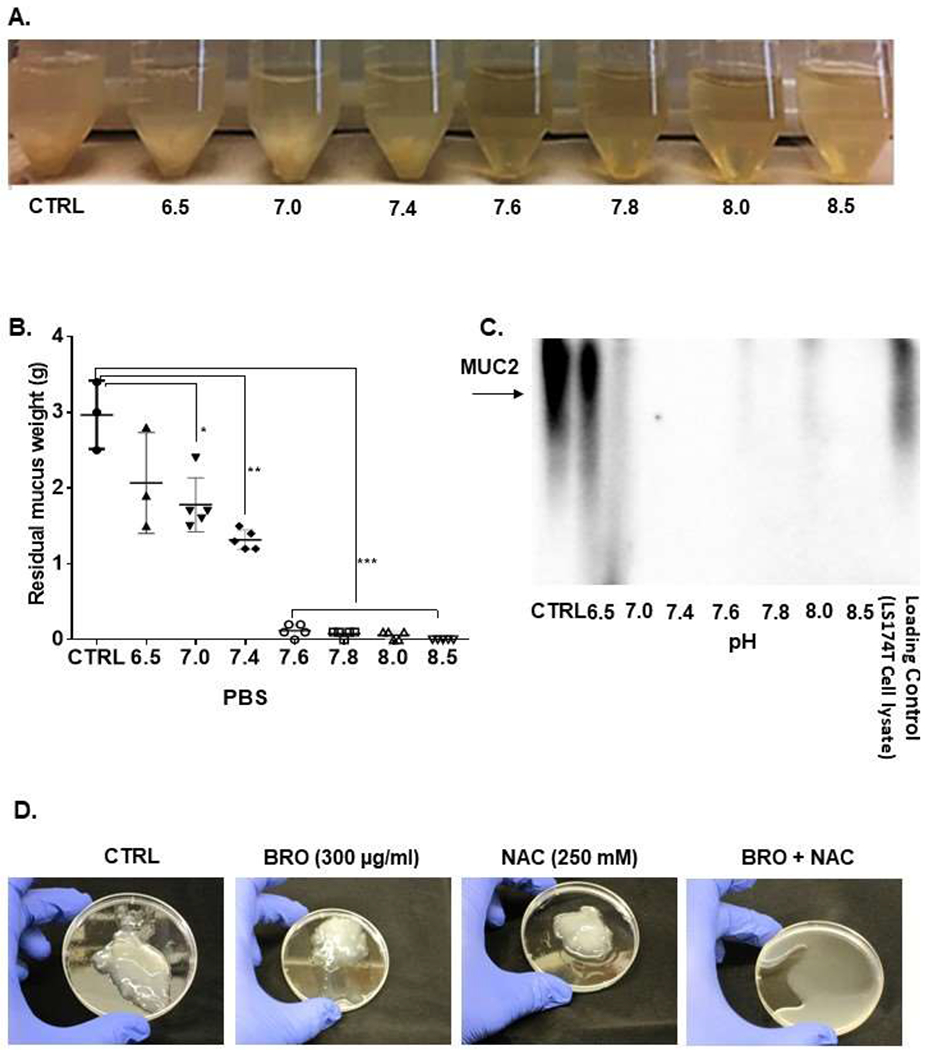
Extracellular mucus (1g) from MCP patients were treated with BRO (300 μg/ml) + NAC (250 mM) in 10 ml PBS solution or PBS alone (CTRL) at 37°C for 24h at varying pH (pH 6.5-8.5) (A). At 24h, the mucus solution from (A) was filtered and the residual mucus gel was weighed (B). Protein was extracted from the filtered solution and western blot assay for MUC2 protein was performed (C). Complete mucolysis of 1g of mucus was achieved when treated with BRO (300 μg/ml) + NAC (250mM) in 10 ml PBS (pH 8.0) at 37°C for 2h, while minimal mucolysis was achieved by BRO alone or NAC alone under the same condtions (D). Error bars represent standard deviation (SD) from triplicate experiments (**P< .01, ***P< .001).
Mucolytic therapy enhances drug delivery to cancer cells and improves chemosensitivity.
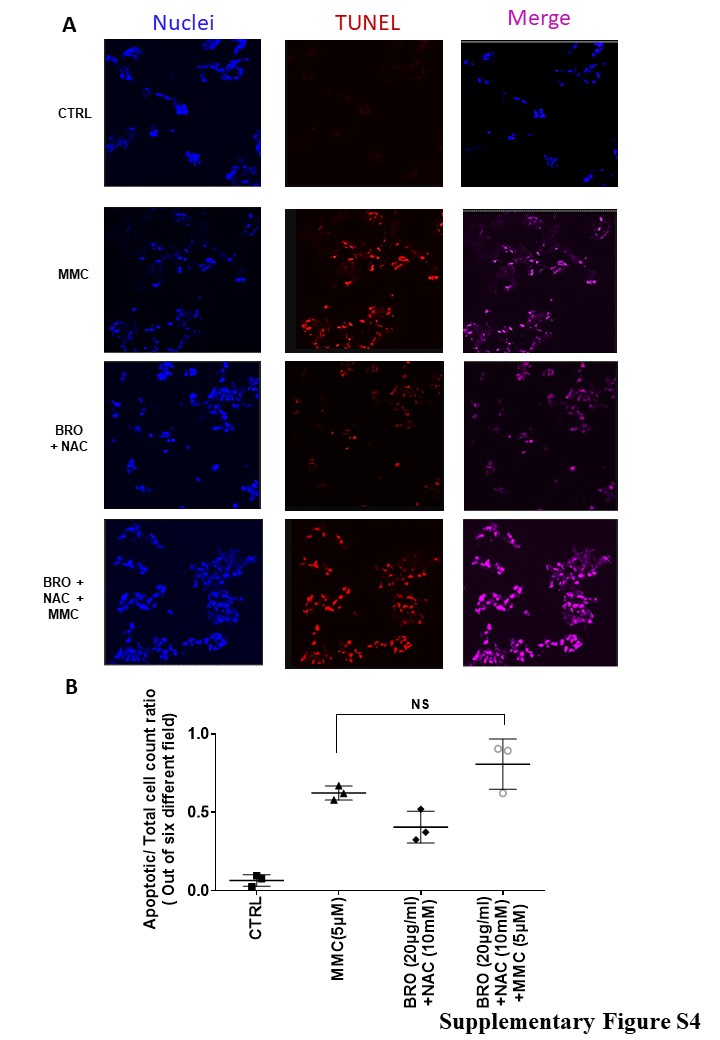
In transwell experiments, we compared intracellular platinum levels in LS174T cells (lower chamber) after OXAL (100 μM) treatment (upper chamber) for 24h, separated by either untreated mucus or pre-treated mucus (mucus pre-treated with BRO [300 μg/ml] + NAC [250 mM] in PBS soultion, pH adjusted to 8.0, for 2h). A higher concentration of intracellular platinum was detected by AAS and more cell death was identified by phase microscopy in the lower chamber LS174T cells when pre-treated mucus separated the chambers (Figures 3A, B). We confirmed these findings in MCP explants ex vivo. Significantly higher tumor cell apoptosis was seen at 24h when tumor tissue was exposed to a combination of BRO (20 μg/ml) + NAC (10 mM) + MMC (5 μM) compared to BRO + NAC alone or MMC alone (Figures 3C, D). Similar experiments were carried out using non MCP explants. We found that the degree to which apoptosis was induced at 24h by the combination of MMC + BRO + NAC over MMC alone was much more prominent in MCP tumors than in non-MCP tumors, consistent with the hypothesis that extracellular mucus provides a protective coating around cancer cells and that mucolysis overcomes this barrier effect (Supplementary Figure S4).
Figure 3. Mucolytic therapy enhances drug delivery to cancer cells and improves chemosensitivity.
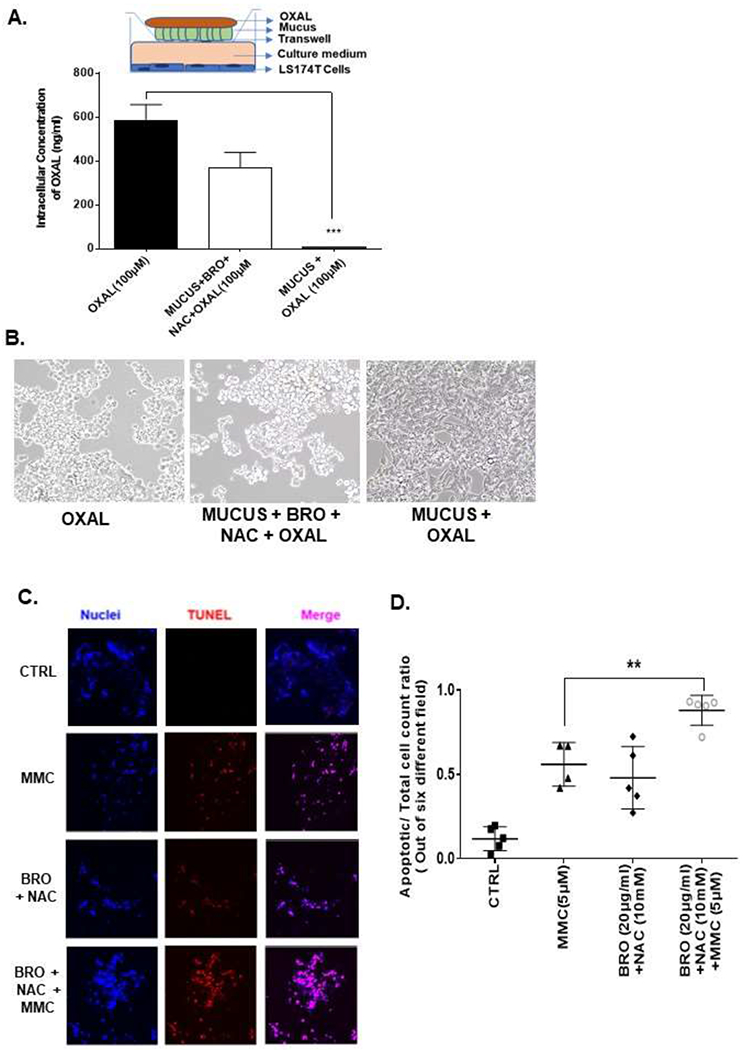
In a tranwell assay undiluted mucus, or mucus lysed with BRO (300 μg/ml) + NAC (250mM) in 10 ml PBS solution at 37°C for 2h, was coated onto the insert, OXAL (100μM) was added to the upper chamber for 24h, following which intracellular platinum in LS174T cells in the lower chamber was measured by AAS; insert is a pictorial representation of the transwell setup (A). Apoptotic cells following treatment in (A) were analyzed by phase-contrast microscopy (B). MCP tumor explants were treated with MMC (5 μM), BRO (20 μg/ml) + NAC (10 mM) or combination of MMC + BRO + NAC for 24h and apoptosis was measured by TUNEL assay (C). Confocal images were randomly taken and analyzed using Image J software to quantify the average intensity of TUNEL positivity (D). Error bars represent standard deviation (SD) from triplicate experiments (***P< .001).
Mucolytic therapy decreases mucinous tumor growth in a mouse PDX model of early MCP.
The early MCP mouse model mimics a clinical picture of low peritoneal disease burden without significant abdominal distention and tumor-related compressive symptoms. Toxicity studies in non tumor bearing nude mice demonstrated that IP injection of 200 μl of BRO (6 mg/kg) + NAC (0.8 g/kg) solution (in PBS, pH 8.0) or PBS alone (control) every other day for 2 weeks (3 mice per group) was well tolerated, without clinical or pathologic evidence of toxicity (Figures 4A, B). For the early MCP model, PDXs were implanted IP in nude mice and IP mucolytic therapy (BR + NAC) or PBS (control) was injected every other day for 3 weeks, starting on day 7 following tumor implantation (10 animals per group). The BRO + NAC treated animals demonstrated significant reduction in weekly body weight and abdominal girth measurements compared to control mice, and also had less abdominal tumors when they were sacrificed (day 28 after tumor implantation) (Figures 4C–E). The same treatment regimen was attempted in a mouse PDX model for late (advanced) MCP, starting treatment on day 14 following tumor implantation (high peritoneal disease burden with significant abdominal distention and tumor-related compressive symptoms). However the same treatment volume/frequency was not tolerated by the animals due to significant abdominal distension from larger tumor burden at this stage (Figure 4F).
Figure 4. Mucolytic therapy decreases mucinous tumor growth in a mouse PDX model of early MCP.
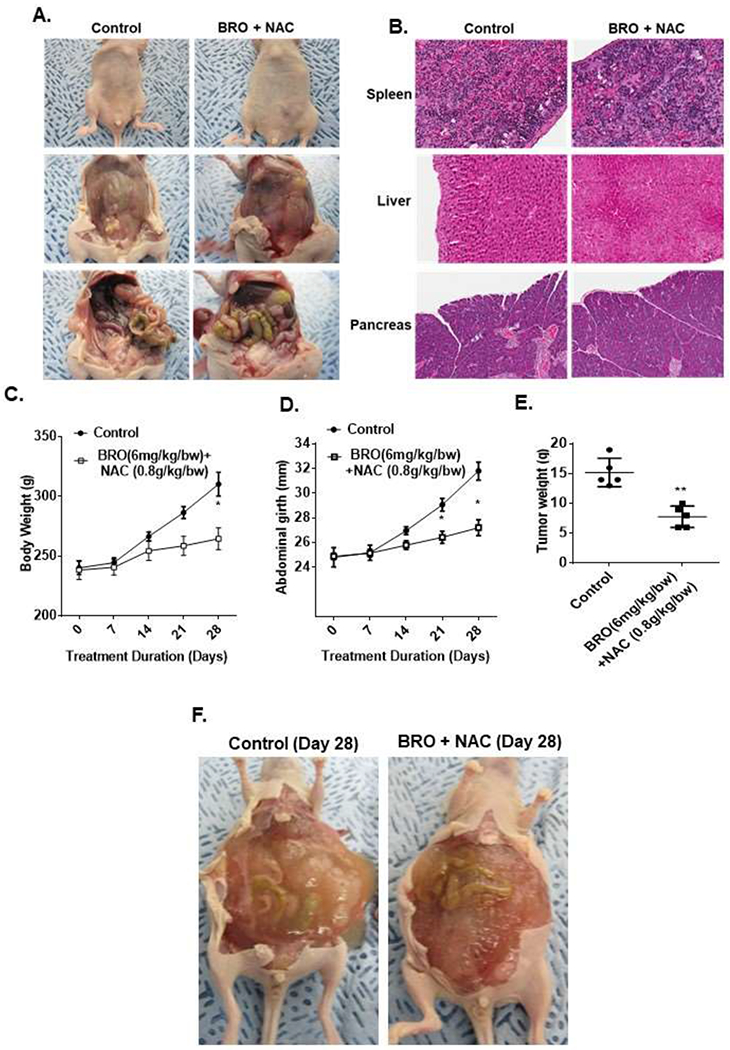
Non-tumor bearing nude mice were administered 200 μl of BRO (6 mg/kg bw) + NAC (0.8 g/kg bw) solution (in PBS, pH 8.0) or PBS (control) IP every other day for 2 weeks; gross evaluation (A) and pathological microscopic evaluation of abdominal organs (with H&E staining) (B) for toxicity is shown. Early MCP model of IP mucolytic (versus control) therapy, at same dose/frequency as in toxicity studies, demonstrating serial changes in gross body weight (C) and abdominal girth (D); and abdominal tumor content weight following sacrifice at day 28 (E). Late (advanced) MCP model of IP mucolytic (versus control) therapy showing gross pictures of advanced tumor burden unresponsive to mucolytic therapy at day 28 (F). Error bars represent standard deviation (SD) from triplicate experiments (*P< .05, **P< .01).
Mucolytic therapy decreases mucinous tumor growth in a rat PDX model of late (advanced) MCP.
The late (advanced) MCP rat model mimics a clinical picture of high peritoneal disease burden with significant abdominal distention and tumor-related compressive symptoms. Toxicity studies in non tumor bearing nude rats demonstrated that IP injection of 5 ml of BRO (6 mg/kg) + NAC (0.8 g/kg) solution (in PBS, pH 8.0) or PBS alone (control) for 2 weeks (3 rats per group) was well tolerated, without clinical or pathologic evidence of toxicity (Figures 5A,B). For the late (advanced) MCP model, PDXs were implanted IP in nude rats and IP mucolytic therapy (BR + NAC) or PBS (control) was injected every other day for 3 weeks, starting on day 14 following tumor implantation (10 animals per group). Following 3 weeks of treatment, the animals were anesthetized and mucinous ascites was drained completely using a surgically placed silicone catheter. Comparing pre-and post-drainage MRIs, the BRO + NAC treated animals had significantly less imageable tumor/mucinous ascites than control animals (Figure 5C). A significantly larger volume of mucinous ascites could be drained from BRO + NAC treated animals compared to control animals (Figures 5D, E) At the time of sacrifice (week 5), the mucolytic combination treated animals had significantly less solid mucinous tumor burden, lower gross body weight, and lower ratio of solid mucinous tumor weight to mucinous ascites volume, compared to control animals (Figures 5F–H). We randomly harvested 0.5 cm3 sections of mucinous tumors from control and treated animals at the time of sacrifice, immersed them in 2 ml PBS containing OXAL (100 μM) for 24h, and performed IF assay on cryosections to quantify DNA adducts. We found significantly higher DNA adducts in tissues harvested from BRO + NAC treated animals than control animals (Figures 5I, J). These data suggest that BRO + NAC reduced the protective mucus coating around neoplastic cells and allowed higher OXAL delivery to the cells.
Figure 5. Mucolytic therapy decreases mucinous tumor growth in a rat PDX model of late (advanced) MCP.
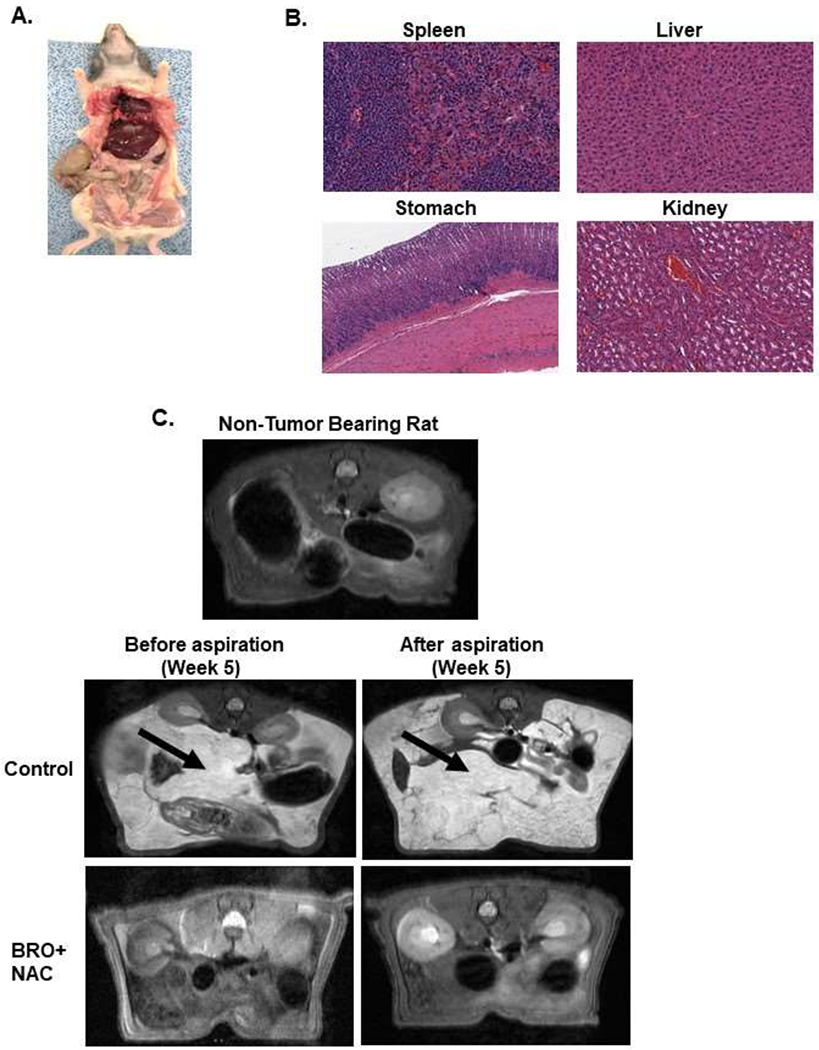
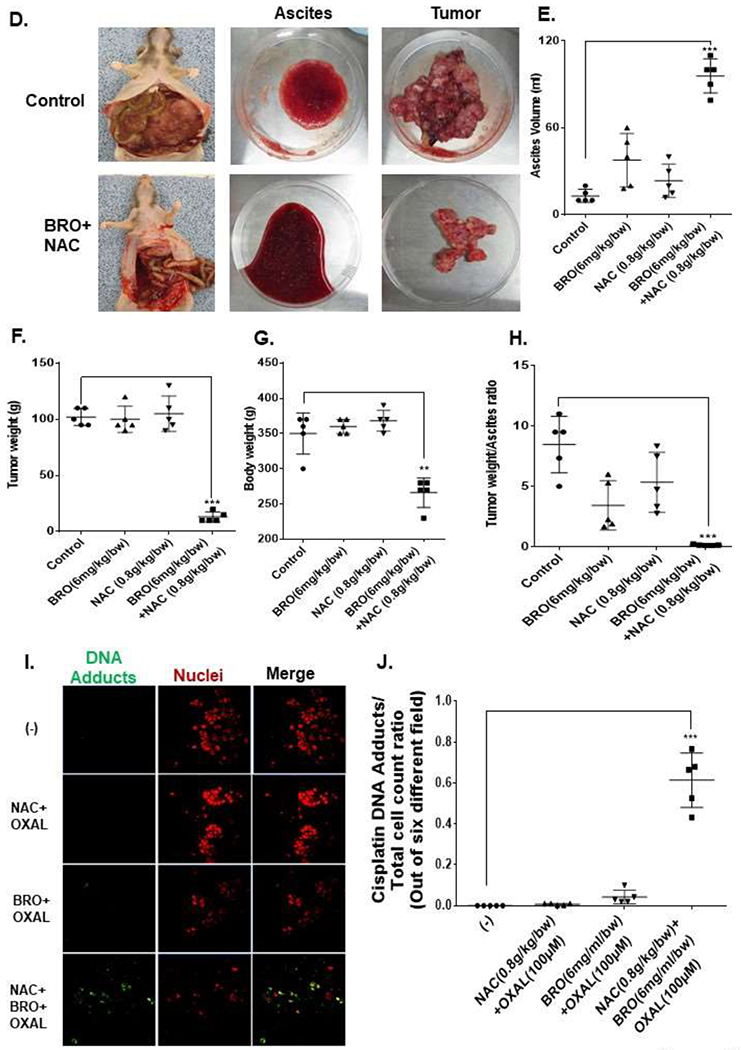
Non-tumor bearing nude rats were administered 5 ml of BRO (6 mg/kg bw) + NAC (0.8 g/kg bw) solution (in PBS, pH 8.0) or PBS (control) IP every other day for 2 weeks; gross evaluation (A) and pathological microscopic evaluation of abdominal organs (with H&E staining) (B) for toxicity is shown. Late (advanced) MCP model of IP mucolytic (versus control) therapy at same dose/frequency as in toxicity studies; pre-and post ascites drainage MRIs at week 5 (MRI of non-tumor bearing rat is also shown) (C); gross pictures of mucinous ascites drained and residual abdominal mucinous tumor at sacrifice (week 5) (D); volume of muinous ascites (E), residual mucinous tumor weight (F), gross body weight (G), and ratio of ascites volume to tumor weight (H). Residual mucinous tumor fragments following IP mucolytic (or control) therapy, treated with OXAL (100 μM) for 24h ex vivo, and subjected to IF assay to quantify DNA adducts (I, J). Error bars represent standard deviation (SD) from triplicate experiments (**P< .01, ***P< .001).
DISCUSSION
Patients suffering from appendiceal MCP require novel and innovative therapeutic strategies because current surgical therapies for this malignant process are morbid and lack long-term efficacy. Standard surgical therapy, cytroreductive surgery to remove macroscopic disease and hyperthermic intraperitoneal chemotherapy to treat residual microscopic disease is associated with major postoperative complications in 25-35% of patients and surgery-related mortality in 2-10% of patients, making this one of the most challenging and potentially morbid surgical procedures.(6, 28) Systemic chemotherapy is ineffective for low-grade MCP. Response rates for high-grade MCP remain low (partial response rates of 10-40%) and even if the neoplastic epithelium responds to chemotherapy, the mucus component of the disease (> 50% of tumor volume by definition) does not.(29) We postulated that dissolving extracellular mucus would potentially (a) decrease disease symptoms related to mass-effect from mucinous ascites and bulky mucinous tumor nodules; (b) improve the efficacy of chemotherapeutic drugs by removing the protective mucus barrier within which cancer cells thrive; and (c) reduce mucinous tumor growth. In addition, effective mucolytic therapy may reduce the need for and/or frequency of morbid surgical procedure and/or improve resectability of patients with extensive MCP.
In our experiments, MCP explant tissues exposed ex vivo to chemotherapeutic drugs were less chemosensitive than non-MCP explant tissues. This is consistent with published clinical data in which mucinous colorectal liver and peritoneal metastases are less responsive to palliative systemic chemotherapy than their non-mucinous counterparts.(1–3, 9, 11–13) Moreover, our ex vivo transwell experiments demonstrated that undiluted mucus prevented adequate delivery of OXAL to LS174T cells, while serial dilution of the mucus correlated with increased drug delivery and cytotoxic effect. These data provide convincing evidence for the direct cytoprotective effects of extracellular mucus and a solid rationale for targeting mucus as a therapeutic strategy for MCP. Extracellular mucus can impact drug delivery through a variety of mechanisms both at the nano/microscale as well as at the macroscale and this effect can be quite heterogeneous.(14, 16, 30) At the microscale level, factors like pore size, charge, solubility, and pH can affect drug diffusion. At the macroscale, factors including density/extent of extracellular matrix and microvasculature density/permeability are major determinants of drug diffusion, especially via their influence on interstitial fluid pressure (IFP). The presence of bulky extracellular mucus in MCP, with small pore size between mucin fibers (10-500 nm), hydrophilic and highly charged nature of mucus, high interstitial pressure and microvascular compression imposed by the bulky tumors, has a significant negative impact on drug transport. Therefore, destruction (mucolysis) of this complex lattice-like structure, composed of amide, glycosidic, and disulfide bonds, would likely reduce the direct bulk-effect of extracellular mucus (compressive organ dysfunction) and improve chemotherapeutic effect (improved drug delivery).
We utilized a combination of BRO + NAC to lyse extracellular mucus since BRO hydrolyzes esters, amides and glycosidic linkages, while NAC reduces disulfide bonds. This combination destroyed the complex lattice structure of mucus, evident by the conversion of MCP-derived mucus gel into solution in ex vivo experiments. Following mucolysis we did not detect significant changes in the viscosity of the drug solution and were unable to detect MUC2 protein in the solution, suggesting that mucus/MUC2 protein structure was completely degraded by the mucolytic combination. From a clinical perspective, complete dissolution of the abundant extracellular mucus would potentially allow for minimally invasive techniques to sequentially remove mucinous ascites following chronic IP mucolytic therapy, decrease bulky mucinous tumor related symptoms and avoid the need for aggressive open operations to remove bulky gelatinous tumors.
Mucolysis prior to chemotherapeutic drug exposure in our ex vivo and in vitro experiments allowed for improved drug delivery and increased cytotoxic effects. In addition to mucolytic properties, BRO has previously been shown to have direct cytotoxic effects against cancer cell lines as well. BRO has been shown to inhibit cyclooxygenase-2 (COX-2), fatty acid synthase (FASN), mitogen-activated protein kinase (MAPK), phosphatidylinositol-3-kinase (PI3K)/AKT, and NF-κB signaling pathways and induce cell death via apoptosis, autophagy, and ferroptosis.(31–35) Similarly, NAC has mucolytic and anticancer effects, including reduced MAPK signaling, induction of cyclin dependent kinase (CDK)-inhibitor-mediated cell cycle arrest, and activation of apoptosis.(36, 37) Increased cytotoxicity from OXAL following pretreatment with low-dose BRO (20 μg/ml) + NAC (10 mM) in our ex vivo experiments was independent of any direct anticancer activity related to low-dose BRO and NAC, as demonstrated by the significantly higher apoptosis with combination therapy (BRO + NAC + MMC) compared to BRO + NAC alone or MMC alone. On the other hand, additional independent anticancer effects of high-dose BRO (300 μg/ml) + NAC (250 mM) would be beneficial in a clinical setting to treat MCP.
We successfully reduced mucinous tumor growth in the mouse PDX model of early MCP. Treatment was started when the implanted tumors were still small and not clinically appreciable. Such a treatment strategy may have a clinical role as adjuvant therapy for resected MCP patients, given that recurrence rates remain high despite aggressive surgical resection. Placement of a subcutaneous IP-port with adjuvant BRO + NAC therapy may prevent mucinous tumor growth via the mucolytic and direct anticancer effects of these drugs. At the same time, adjuvant long-term IP-chemotherapy (currently used at some centers for peritoneal metastases from ovarian and colorectal cancers) may be more effect in combination with IP-BRO + NAC to improve drug delivery. We found that the mouse PDX model of late (advanced) MCP was inadequate to assess mucolytic benefits of BRO + NAC since the animals are unable to tolerate drug volumes (200 μl) once the tumor burden was severe. In our ex vivo studies drug volume to tumor weight ratio of > 10:1 was required to achieve effective mucolysis, and this ratio of mucolytic solution to tumor could not be achieved in the mouse model due to intolerance to this large volume. Therefore, we generated rat PDX models of late (advanced) MCP so that larger drug volumes could be given IP, despite larger tumor burden. In addition, this larger animal model allowed for IP-catheter placement and aspiration following mucolytic therapy. Following the administration of adequate drug volumes in the larger animal model, efficient mucloysis and improved chemosensitivity was achieved. Following mucolysis, we utilized a larger-bore IP catheter to drain mucinous ascites at a single time-point (week 5). More frequent removal of mucinous ascites was not feasible since the animals did not tolerate long-term placement of the catheter and also suffered infectious complications. From a clinical perspective, long-term large-bore IP-catheters (e.g. 9.5F Tenckoff dialysis catheters) are likely to be feasible for this therapeutic strategy. Although the combinatorial therapy was well tolerated, we did find that mucinous ascites in the mucolytic combination treated animals was often hemorrhagic at the time of drainage. BRO is known to have anti-platelet and thrombolytic properties which may lead to adverse consequences from prolonged therapy. We did not find this to be the case in our toxicity studies in normal mice and rats and we suspect that the bulky friable mucinous tumors are likely more susceptible to hemorrhage following mucolysis. These data provide a strong rationale for an early phase clinical trial to assess the feasibility, safety and efficacy of IP-BRO + NAC for appendiceal MCP. Valle and colleagues recently published results from a phase 1 study showing mucolytic activity against mucinous tumors in patients with unresectable appendiceal MCP following IP administration of BRO + NAC, however this was a small trial, with a majority of patients undergoing local BRO + NAC injection into discrete tumors.(38)
In summary, we found that a combination of BRO + NAC efficiently lysed extracellular mucus in clinically relevant models of MCP. Conversion of solid mucinous tumors into mucinous ascites decreases tumor bulk and allows for minimally invasive drainage of liquified tumors. Lysis of extracellular mucus removes the protective mucinous coating surrounding cancer cells and improves chemotherapeutic drug delivery/efficacy in cancer cells.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
AT A GLANCE COMMENTRARY.
Background
Abundant extracellular mucus may act as a protective barrier for cancer cells against external insults like chemotherapy. Clinical data demonstrate that mucinous appendix, colorectal, breast, and ovarian cancers are less responsive to palliative systemic chemotherapy than their non-mucinous counterparts.
Translational Significance
Using patient-derived ex vivo and in vivo models of mucinous appendix cancer, we demonstrate that mucolytic therapy (bromelain + N-acetylcysteine) removes the protective mucus coating around cancer cells to improve chemotherapeutic drug delivery/efficacy and reduces mucinous tumor growth. Our research supports the targeting of extracellular mucus as a therapeutic strategy against mucinous tumors and provides a rationale for combining mucolytic therapy with standard cytotoxic drugs.
ACKNOWLEDGMENTS
We thank Foley Lesley May and Hitchens T Kevin for MRI image acquisition and technical assistance.
Funding: This is work is supported by NIH R21CA3241004-02. This work was supported by the Pseudomyxoma Peritonei Philanthropic Research Fund and the David C. Koch Regional Therapy Cancer Center, University of Pittsburgh. Grant R50 CA211241. This project used the resource of the UPMC Hillman Cancer Center Cancer Pharmacokinetics and Pharmacodynamics Facility (CPPF), In Vivo Imaging Facility, and Hillman Animal Facility and Tissue and Research Pathology/Pitt Biospecimen Core shared resource that is supported in part by NIH award P30-CA-047904.
Abbreviations:
- BRO
bromelain
- CDK
cyclin dependent kinase
- COX-2
cyclooxygenase-2
- DMEM
Dulbecco’s modified eagle’s medium
- FBS
fetal bovine serum
- H&E
hematoxylin and eosin
- HG
high-grade
- IF
immunofluorescence
- IP
intraperitoneal
- IFP
interstitial fluid pressure
- LG
low-grade
- MAPK
mitogen activated protein kinase
- MCP
mucinous carcinoma peritonei
- MMC
mitomycin C
- MRI
magnestic resonance imaging
- MTS
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)
- MUC2
mucin 2
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PDX
patient-derived xenograft
- NAC
N-acetylcysteine
- OXAL
oxaliplatin
- PBS
phosphate buffered saline
- PI3K
phosphoinositide 3-kinase
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
- AAS
atomic absorption spectrophotometry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: All authors have read the journals policy on disclosure of potential conflicts, and have no competing interests to declare. All authors have read the journal’s authorship agreement, and approved the final version of the manuscript.
REFERENCES
- 1.Hugen N, Brown G, Glynne-Jones R, de Wilt JH, Nagtegaal ID. Advances in the care of patients with mucinous colorectal cancer. Nat Rev Clin Oncol. 2016;13(6):361–9. [DOI] [PubMed] [Google Scholar]
- 2.Khan M, Loree JM, Advani SM, Ning J, Li W, Pereira AAL, et al. Prognostic Implications of Mucinous Differentiation in Metastatic Colorectal Carcinoma Can Be Explained by Distinct Molecular and Clinicopathologic Characteristics. Clin Colorectal Cancer. 2018;17(4):e699–e709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo C, Cen S, Ding G, Wu W. Mucinous colorectal adenocarcinoma: clinical pathology and treatment options. Cancer Commun (Lond). 2019;39(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Connell JT, Hacker CM, Barsky SH. MUC2 is a molecular marker for pseudomyxoma peritonei. Mod Pathol. 2002;15(9):958–72. [DOI] [PubMed] [Google Scholar]
- 5.O’Connell JT, Tomlinson JS, Roberts AA, McGonigle KF, Barsky SH. Pseudomyxoma peritonei is a disease of MUC2-expressing goblet cells. Am J Pathol. 2002;161(2):551–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugarbaker PH. New standard of care for appendiceal epithelial neoplasms and pseudomyxoma peritonei syndrome? Lancet Oncol. 2006;7(1):69–76. [DOI] [PubMed] [Google Scholar]
- 7.Carr NJ, Cecil TD, Mohamed F, Sobin LH, Sugarbaker PH, Gonzalez-Moreno S, et al. A Consensus for Classification and Pathologic Reporting of Pseudomyxoma Peritonei and Associated Appendiceal Neoplasia: The Results of the Peritoneal Surface Oncology Group International (PSOGI) Modified Delphi Process. Am J Surg Pathol. 2016;40(1):14–26. [DOI] [PubMed] [Google Scholar]
- 8.Davison JM, Choudry HA, Pingpank JF, Ahrendt SA, Holtzman MP, Zureikat AH, et al. Clinicopathologic and molecular analysis of disseminated appendiceal mucinous neoplasms: identification of factors predicting survival and proposed criteria for a three-tiered assessment of tumor grade. Mod Pathol. 2014;27(11):1521–39. [DOI] [PubMed] [Google Scholar]
- 9.Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4(1):45–60. [DOI] [PubMed] [Google Scholar]
- 10.Choudry HA, O’Malley ME, Guo ZS, Zeh HJ, Bartlett DL. Mucin as a therapeutic target in pseudomyxoma peritonei. J Surg Oncol. 2012;106(7):911–7. [DOI] [PubMed] [Google Scholar]
- 11.Lupinacci RM, Mello ES, Coelho FF, Kruger JA, Perini MV, Pinheiro RS, et al. Prognostic implication of mucinous histology in resected colorectal cancer liver metastases. Surgery. 2014;155(6):1062–8. [DOI] [PubMed] [Google Scholar]
- 12.Vigano L, Russolillo N, Ferrero A, De Rosa G, Ferreri E, Forchino F, et al. Resection of liver metastases from colorectal mucinous adenocarcinoma: is this a different disease? Results of a case-control study. Ann Surg. 2014;260(5):878–84; discussion 84–5. [DOI] [PubMed] [Google Scholar]
- 13.Jonckheere N, Skrypek N, Van Seuningen I. Mucins and tumor resistance to chemotherapeutic drugs. Biochim Biophys Acta. 2014;1846(1):142–51. [DOI] [PubMed] [Google Scholar]
- 14.Lai SK, Wang YY, Wirtz D, Hanes J. Micro- and macrorheology of mucus. Adv Drug Deliv Rev. 2009;61(2):86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menzel C, Bernkop-Schnurch A. Enzyme decorated drug carriers: Targeted swords to cleave and overcome the mucus barrier. Adv Drug Deliv Rev. 2018;124:164–74. [DOI] [PubMed] [Google Scholar]
- 16.Murgia X, Loretz B, Hartwig O, Hittinger M, Lehr CM. The role of mucus on drug transport and its potential to affect therapeutic outcomes. Adv Drug Deliv Rev. 2018;124:82–97. [DOI] [PubMed] [Google Scholar]
- 17.Henke MO, Ratjen F. Mucolytics in cystic fibrosis. Paediatr Respir Rev. 2007;8(1):24–9. [DOI] [PubMed] [Google Scholar]
- 18.Poole P, Sathananthan K, Fortescue R. Mucolytic agents versus placebo for chronic bronchitis or chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2019;5:CD001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sadowska AM. N-Acetylcysteine mucolysis in the management of chronic obstructive pulmonary disease. Ther Adv Respir Dis. 2012;6(3):127–35. [DOI] [PubMed] [Google Scholar]
- 20.Amini A, Masoumi-Moghaddam S, Ehteda A, Liauw W, Morris DL. Depletion of mucin in mucin-producing human gastrointestinal carcinoma: Results from in vitro and in vivo studies with bromelain and N-acetylcysteine. Oncotarget. 2015;6(32):33329–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amini A, Masoumi-Moghaddam S, Ehteda A, Morris DL. Bromelain and N-acetylcysteine inhibit proliferation and survival of gastrointestinal cancer cells in vitro: significance of combination therapy. J Exp Clin Cancer Res. 2014;33:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akhter J, Pillai K, Chua TC, Alzarin N, Morris DL. Efficacy of a novel mucolytic agent on pseudomyxoma peritonei mucin, with potential for treatment through peritoneal catheters. Am J Cancer Res. 2014;4(5):495–507. [PMC free article] [PubMed] [Google Scholar]
- 23.Pillai K, Akhter J, Chua TC, Morris DL. A formulation for in situ lysis of mucin secreted in pseudomyxoma peritonei. Int J Cancer. 2014;134(2):478–86. [DOI] [PubMed] [Google Scholar]
- 24.Dilly A, Honick BD, Lee YJ, Bartlett DL, Choudry HA. Rational application of targeted therapeutics in mucinous colon/appendix cancers with positive predictive factors. Cancer Med. 2020;9(5):1753–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dilly AK, Honick BD, Lee YJ, Guo ZS, Zeh HJ, Bartlett DL, et al. Targeting G-protein coupled receptor-related signaling pathway in a murine xenograft model of appendiceal pseudomyxoma peritonei. Oncotarget. 2017;8(63):106888–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liedert B, Pluim D, Schellens J, Thomale J. Adduct-specific monoclonal antibodies for the measurement of cisplatin-induced DNA lesions in individual cell nuclei. Nucleic Acids Res. 2006;34(6):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mavanur AA, Parimi V, O’Malley M, Nikiforova M, Bartlett DL, Davison JM. Establishment and characterization of a murine xenograft model of appendiceal mucinous adenocarcinoma. Int J Exp Pathol. 2010;91(4):357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choudry MHA, Shuai Y, Jones HL, Pai RK, Pingpank JF, Ahrendt SS, et al. Postoperative Complications Independently Predict Cancer-Related Survival in Peritoneal Malignancies. Ann Surg Oncol. 2018;25(13):3950–9. [DOI] [PubMed] [Google Scholar]
- 29.Blackham AU, Swett K, Eng C, Sirintrapun J, Bergman S, Geisinger KR, et al. Perioperative systemic chemotherapy for appendiceal mucinous carcinoma peritonei treated with cytoreductive surgery and hyperthermic intraperitoneal chemotherapy. J Surg Oncol. 2014;109(7):740–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dewhirst MW, Secomb TW. Transport of drugs from blood vessels to tumour tissue. Nat Rev Cancer. 2017;17(12):738–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang TC, Wei PL, Makondi PT, Chen WT, Huang CY, Chang YJ. Bromelain inhibits the ability of colorectal cancer cells to proliferate via activation of ROS production and autophagy. PLoS One. 2019;14(1):e0210274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chobotova K, Vernallis AB, Majid FA. Bromelain’s activity and potential as an anticancer agent: Current evidence and perspectives. Cancer Lett. 2010;290(2):148–56. [DOI] [PubMed] [Google Scholar]
- 33.Dave S, Kaur NJ, Nanduri R, Dkhar HK, Kumar A, Gupta P. Inhibition of adipogenesis and induction of apoptosis and lipolysis by stem bromelain in 3T3-L1 adipocytes. PLoS One. 2012;7(1):e30831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park S, Oh J, Kim M, Jin EJ. Bromelain effectively suppresses Kras-mutant colorectal cancer by stimulating ferroptosis. Anim Cells Syst (Seoul). 2018;22(5):334–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sobolewski C, Cerella C, Dicato M, Ghibelli L, Diederich M. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol. 2010;2010:215158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aldini G, Altomare A, Baron G, Vistoli G, Carini M, Borsani L, et al. N-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why. Free Radic Res. 2018;52(7):751–62. [DOI] [PubMed] [Google Scholar]
- 37.Salamon S, Kramar B, Marolt TP, Poljsak B, Milisav I. Medical and Dietary Uses of N-Acetylcysteine. Antioxidants (Basel). 2019;8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valle SJ, Akhter J, Mekkawy AH, Lodh S, Pillai K, Badar S, et al. A novel treatment of bromelain and acetylcysteine (BromAc) in patients with peritoneal mucinous tumours: A phase I first in man study. Eur J Surg Oncol. 2019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.