

The spinal cord appears especially vulnerable in CLN1 disease, a fatal inherited lysosomal storage disorder. Our data reveal a much earlier onset of spinal cord disease and accompanying behavioural changes in CLN1 mice, while spinal maturation is still ongoing. Our multi‐disciplinary data provide new insights into the spatio‐temporal staging of CLN1 pathogenesis during ongoing postnatal maturation, and highlight the need to deliver therapies during the presymptomatic period.

Keywords: batten disease, gait, neurodegeneration, neuronal ceroid lipofuscinosis, postnatal development, spinal cord
Abstract
Aim
To understand the progression of CLN1 disease and develop effective therapies we need to characterize early sites of pathology. Therefore, we performed a comprehensive evaluation of the nature and timing of early CLN1 disease pathology in the spinal cord, which appears especially vulnerable, and how this may affect behaviour.
Methods
We measured the spinal volume and neuronal number, and quantified glial activation, lymphocyte infiltration and oligodendrocyte maturation, as well as cytokine profile analysis during the early stages of pathology in Ppt1‐deficient (Ppt1−/−) mouse spinal cords. We then performed quantitative gait analysis and open‐field behaviour tests to investigate the behavioural correlates during this period.
Results
We detected significant microglial activation in Ppt1−/− spinal cords at 1 month. This was followed by astrocytosis, selective interneuron loss, altered spinal volumes and oligodendrocyte maturation at 2 months, before significant storage material accumulation and lymphocyte infiltration at 3 months. The same time course was apparent for inflammatory cytokine expression that was altered as early as one month. There was a transient early period at 2 months when Ppt1−/− mice had a significantly altered gait that resembles the presentation in children with CLN1 disease. This occurred before an anticipated decline in overall locomotor performance across all ages.
Conclusion
These data reveal disease onset 2 months (25% of life‐span) earlier than expected, while spinal maturation is still ongoing. Our multi‐disciplinary data provide new insights into the spatio‐temporal staging of CLN1 pathogenesis during ongoing postnatal maturation, and highlight the need to deliver therapies during the presymptomatic period.
Abbreviations
- AFSM
Autofluorescent Storage Material
- ALS
Amyotrophic Lateral Sclerosis
- ANOVA
Analysis of variance
- CRP
C‐ Reactive Protein
- CB
Calbindin
- CGRP
Calcitonin Gene‐Related Peptide
- ChAT
Choline Acetyl Transferase
- CLN1
CLN1 disease or infantile neuronal ceroid lipofuscinosis
- CLN5
CLN5 disease or variant late infantile neuronal ceroid lipofuscinosis
- CNS
Central Nervous System
- CR
Calretinin
- DAPI
4′,6‐diamidino‐2‐phenylindole
- GABA
Gamma aminobutyric acid
- GFAP
Glial Fibrillary Acidic Protein
- IFNg
Interferon Gamma
- IL‐1A
−1B, −6, −10, −33, Interleukins −1A, −1B, −6, −10, −33
- IP‐10
Interferon gamma‐induced protein 1
- LSD
Lysosomal Storage Disorder
- MBP
Myelin Basic Protein
- MCP‐1
Monocyte chemoattractant protein‐1
- MCP‐3
Monocyte chemoattractant protein‐3
- MIP‐1 alpha
Macrophage Inflammatory Protein‐1 alpha
- MIP‐1 beta
Macrophage Inflammatory Protein‐1 beta
- MIP‐2
Macrophage Inflammatory Protein‐2
- NCL
Neuronal Ceroid Lipofuscinosis
- PPT1
Palmitoyl Protein Thioesterase‐1
- PV
Parvalbumin
- RANTES
Regulated on Activation, Normal T‐cell Expressed and Secreted
- SEM
Standard Error of the Mean
- SMA
Spinal Muscular Atrophy
- SubP
Substance‐P
- TBS
Tris‐Buffered Saline
- TBS‐T
Tris‐Buffered Saline with Triton‐X 100
- TGF‐Beta
Transforming Growth Factor Beta
- VEGF‐A
Vascular endothelial growth factor A
- WT
Wild‐type
Introduction
The neuronal ceroid lipofuscinoses (NCLs) are fatal neurodegenerative lysosomal storage disorders that affect children and young adults [1]. The NCLs are classified together based on broadly shared clinical symptoms including a loss of vision, seizures of increasing severity and progressive loss of cognitive and motor function [2, 3]. CLN1 disease or infantile NCL is an early onset, rapidly progressing form of NCL, caused by mutations in the CLN1 gene that encodes the lysosomal enzyme Palmitoyl Protein Thioesterase‐1 (PPT1) [3, 4]. There is currently no effective therapy for CLN1 disease, and all cases are fatal [5, 6, 7, 8].
The Ppt1‐deficient (Ppt1−/−) mouse recapitulates most human phenotypes including a shortened lifespan (typically 7.5‐8 months), visual defects, epileptic seizures and gait defects later in disease progression [9, 10]. Ppt1−/− mice also display pathological changes characteristic of the human disorder [11, 12] including pronounced accumulation of autofluorescent storage material (AFSM), and glial activation that precedes the pronounced neuronalloss that occurs in the brain, cerebellum and retina [13, 14, 15, 16]. A preliminary analysis showed significant pathology in the spinal cords of human CLN1 patients at autopsy and in Ppt1−/− mice, and that directing gene therapy neonatally to both brain and spinal cord is important for disease outcome [17].
In many later onset neurodegenerative disorders, pathological changes only occur once the CNS has fully developed and matured, but this is unlikely in a disorder like CLN1 disease with an onset in childhood.Reasoning that PPT1 deficiency will impact the CNS during its development, we investigated the extent to which pathological changes occur during the early postnatal period, when Ppt1−/− mice display no overt disease phenotypes. The events that occur in the CNS during the early stages of CLN1 disease are poorly understood, and unlikely to be treated effectively if therapy is delivered after the onset of symptoms and subsequent diagnosis. As such, a more comprehensive analysis of the regional neurological, pathological and molecular consequences of PPT1 deficiency in its initial stages will be important for designing therapies that are more effectively targeted to vulnerable cell types and regions of the CNS.
Here, we have characterized an onset of spinal pathology at least 2 months earlier (a quarter of their normal lifespan) than any previously documented CNS pathology in these Ppt1−/− mice. This is accompanied by progressive changes in cytokine expression, and a transient early period when Ppt1−/− mice have a significantly altered gait compared to control wild‐type mice, before subsequently declining in performance. These data reveal that the pathological and behavioural consequences of PPT1 deficiency occur in the context of ongoing maturation of the CNS and suggest an interplay between neurodegenerative and developmental processes. Collectively our findings define novel and unexpectedly early behavioural and spinal pathological phenotypes that will help inform timing and targeting of therapy for CLN1 disease.
Methods
Patients and clinical assessment
Written informed consent was received from the participant’s guardians for data, pictures or videos appearing in the manuscript. This study was approved by the institutional ethics committee at Centre for Rare Diseases, Department of Paediatrics and Adolescent Medicine, Aarhus University Hospital (Data S1).
Mice
Ppt1‐deficient (Ppt1−/−) mice were first generated by Gupta et al [10]. Both congenic Ppt1−/− and wild‐type (WT) mice were maintained separately on a C57Bl/6J background through homozygous mating. All animals were housed in an animal facility at Washington University School of Medicine (St. Louis, MO) under a 12hr light/dark cycle, and provided food and water ad libitum. All procedures were performed in accordance with NIH guidelines under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University School of Medicine. Unless otherwise stated, n = 5 mice of each genotype were used, except for gait analysis (n = 12; 6 males,6 females), locomotor activity and open‐field behaviour (n = 12; 8 males, 4 females).
Tissue processing and cresyl fast violet staining
Tissue processing and analysis for tinctorial and immunohistochemical staining was performed as previously described [13, 18] (Data S1).
Immunostaining
A one in forty‐eight series of 40µm coronal spinal cord sections from each mouse were stained on slides using a modified immunofluorescence protocol for the following antibodies: rabbit anti‐calbindin, 1:500, rabbit anti‐calretinin, 1:500 and rabbit anti‐parvalbumin 1:500, Swant, goat anti‐choline acetyl transferase (ChAT), 1:50, Chemicon, rabbit anti‐calcitonin gene‐related peptide (CGRP), 1:200, Enzo life sciences, rabbit anti‐substance‐P (SubP), 1:200, Chemicon, rat anti‐CD4,1:100, Bio‐Rad, rat anti‐CD8, 1:100, Bio‐Rad, rabbit anti‐GFAP, 1:1000, DAKO, rat anti‐mouse CD68, 1:400, Bio‐Rad, rabbit anti‐Olig2, 1:200, GeneTex, rabbit anti‐NG2 chondroitin sulphate proteoglycan (NG2), 1:200, Chemicon and rat anti‐MBP, 1:500, Merck Millipore (Data S1).
Measurements of regional volume and area
Area measurements and Cavalieri estimates of regional volume were obtained as previously described [18], using StereoInvestigator software (MBF Bioscience Inc, Williston, VT) (Data S1).
Cytokine profile analysis
Cytokine profiling was performed using an Affymetrix multiplex assay through the Center for Human Immunology and Immunotherapy Programs (CHiiPS) Immunomonitoring Lab (IML) at Washington University School of Medicine. A standard 96‐well Luminex assay plate was used to run the samples in duplicate. Procartaplex Luminex Assay mouse 9 plex (ThermoFisher) EPX090‐20821‐901 (GRO alpha; IP‐10; MCP‐1; MCP‐3; MIP‐1 alpha; MIP‐1 beta; MIP‐2; RANTES; Eotaxin) plus CRP, ENA‐78, IFNg, IL‐1A, IL‐1B, IL‐6, IL‐10, IL‐33, TGF‐Beta 1 and VEGFa (ThermoFisher) assays were run to create an 19plex (Data S1).
Neuron counts
Counts of neurons in the dorsal and ventral horns of each hemisection of cord were performed by a design‐based optical fractionator method in a one in forty‐eight series of Cresyl fast violet‐stained sections [13, 19] using StereoInvestigator (MBF Bioscience). Cells were sampled with counting frames (70 × 40 μm) distributed over a sampling grid of 150 × 150 μm superimposed over the region of interest at 100 × magnification. Counts for interneurons (calbindin, calretinin and parvalbumin) in laminae IV‐X of each spinal hemisection, motor neurons (ChAT) in the ventral horn and counts of CD4‐ and CD8‐positive cells across entire hemisections of cord, were carried out by manual stereological estimations in a one in forty‐eight series of sections using Image‐Pro Premier software (Media Cybernetics, Chicago, IL) according to anatomical landmarks [20].
Thresholding image analysis and image densitometry
To analyse AFSM accumulation and glial activation in the grey matter (GFAP‐positive astrocytes + CD68‐positive microglia), markers of oligodendrocyte precursors (Olig2 and NG2) in the white matter and CGRP‐positive fibres in laminae III‐IV of the dorsal horn of the spinal cord, semiautomated thresholding image analysis was performed using Image‐Pro Premier (Media Cybernetics) [17]. This involved collecting slide‐scanned images at 10× magnification from each animal followed by demarcation of regions of interest. Images were subsequently analysed using Image‐Pro Premier (Media Cybernetics) using an appropriate threshold that selected the foreground immunoreactivity above background. Due to the density of interneurons in laminae I‐III of the dorsal horn [20, 21, 22], image densitometry for the average pixel luminance data was gathered for those antigens that showed a higher density of staining (calbindin, calretinin and parvalbumin) and for other antigens with higher staining densities (CGRP, Substance P). Slide‐scanned images at 10x magnification were collected, followed by collecting the mean luminance data across all pixels from regions of interest delineated using StereoInvestigator (MBF Bioscience).
Quantitative gait analysis
The CatWalk XT gait analysis system (Noldus Information Technology bv, Wagenigen, Netherlands) was used to study gait abnormalities, as previously described [23, 24, 25]. To evaluate general activity levels in Ppt1−/− and WT mice, mice were evaluated over a 1‐hour period in transparent polystyrene enclosures at monthly intervals, as previously described [26, 27] (Data S1).
Statistical analysis
All measurements were performed blind to genotype. All statistical analyses were performed using GraphPad Prism version 8.0.0 for MacOS (GraphPad Software, San Diego, CA, www.graphpad.com). All data were analysed using a two‐way ANOVA with a post hoc Bonferroni correction, except stereological counts of Nissl‐stained neurons at 2 months, which used a two‐tailed, unpaired, parametric t‐test. A p‐value of ≤ 0.05 was considered significant.
Results
Interneurons are the earliest affected neuron populations in the Ppt1−/− mouse spinal cord
Extensive neuron loss in the CNS is a crucial hallmark of CLN1 disease, with cortical and hippocampal interneuron populations particularly vulnerable [13, 14, 15, 16]. We have previously performed stereological counts of Nissl‐stained dorsal and ventral horn neurons in the lumbar spinal cord of WT and Ppt1−/− mice at 1, 3, 5 and 7 months [17],which revealed that significant neuron loss first became apparent in the ventral horn at 3 months and the dorsal horn at 5 months. To obtain information about spinal interneuron loss, we examined populations of interneurons immunostained for parvalbumin (PV), calbindin (CB) or calretinin (CR), which are affected in the brains of Ppt1−/− mice [13, 14]. There was a significant reduction in pixel luminance of PV, CB and CR immunoreactivity in laminae I‐III as early as 2 months that worsened with age, and a significant decrease in the number of PV‐ and CR‐positive neurons in laminae IV‐IX at 2 months, and of CB‐positive neurons at 3 months (Figure 1 A‐C). In light of these findings of early interneuron loss, we also performed stereological counts on Nissl‐stained sections at 2 months to assess overall neuron survival. However, there was no significant loss of Nissl‐stained neurons in either the ventral or dorsal horns of Ppt1−/− mice at 2 months (Figure 1 D), indicating that while interneuron numbers are reduced at this early time point, overall neuron numbers only begin to decrease at 3 months, as our previous analyses had suggested [17].There was also no significant difference in the number of choline acetyltransferase (ChAT) positive motor neurons in the ventral horns of Ppt1−/− and WT spinal cord even at 3 months when interneuron loss is already underway (Figure 1 E). Collectively, these data demonstrate the early and selective loss of spinal interneurons in the Ppt1−/− spinal cord starting at 2 months of age, when no other populations of spinal neurons appear to be affected [14, 15].
Figure 1.
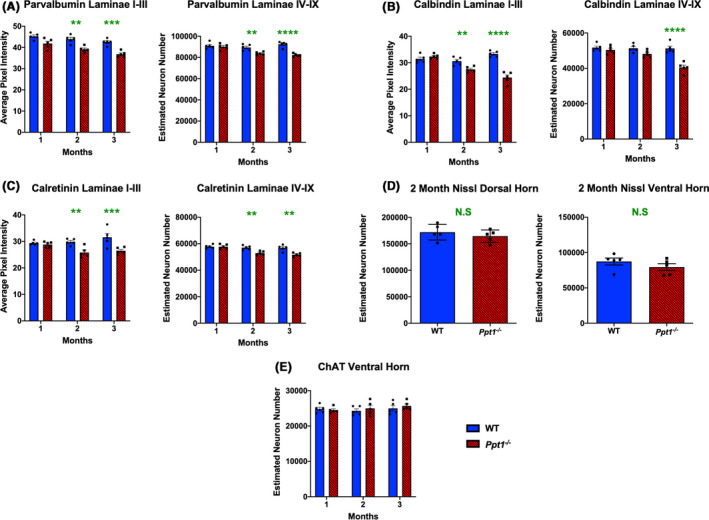
Early interneuron loss in the spinal cords of Ppt1−/− mice. (A‐C) Analysis of lumbosacral cords immunostained for interneuron markers – parvalbumin, calbindin and calretinin respectively. Image densitometry (average pixel luminance) revealed a significant decrease in intensity of immunostaining for these markers in laminae I‐III as early as 2 months for all antibodies stained, progressing at 3 months. Manual stereological counts of neurons positively immunostained for these markers in laminae IV‐IX [98] revealed a significant loss of parvalbumin‐ and calretinin‐positive interneurons as early as 2 months, and calbindin‐positive interneurons at 3 months. (D) Unbiased stereological estimates of neuron numbers from Nissl‐stained spinal cord sections showed no significant difference between WT (wild‐type) and Ppt1−/− mice at 2 months in either the dorsal or ventral horns of the lumbosacral spinal cord. Two‐tailed, unpaired, parametric t‐test (n = 5 mice/group). (E) Manual stereological estimates of choline acetyltransferase (ChAT) positive motor neurons showed no significant loss at any time point measured. P‐values ‐ **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
Ppt1−/− mice show early increases in pain‐associated neuropeptides
The significant early loss of interneurons (Figure 1) raised the possibility of accompanying alterations in sensory signal processing in the dorsal horns of Ppt1−/− mice. Substance P (SubP) and calcitonin gene‐related peptide (CGRP) are neuropeptides which have been shown to have altered expression in conditions such as persistent hyperalgesia [28, 29, 30]. Analysing immunoreactivity for these antigens between 1 and 3 months in Ppt1−/− mice spinal dorsal horns revealed a significant increase in the intensity of immunofluorescence (average pixel luminance) of SubP at 2 months, but no change in SubP intensity at either 1 or 3 months. Similarly, the area of SubP immunoreactivity in laminae I‐II did not significantly differ between genotypes at any time point (Figure 2 A). We also observed an increase in the intensity of CGRP immunoreactivity in laminae I‐II of Ppt1−/− mice at 2 and 3 months, and a larger area of CGRP immunoreactivity in these laminae of Ppt1−/− mice at one month. We also saw an increase in CGRP‐positive fibres in laminae III and IV of Ppt1−/− mice across all time points (Figure 2 B). Our data demonstrate significant early alterations in neuropeptide expression in Ppt1−/− mouse dorsal horns that parallel the onset of interneuron loss.
Figure 2.
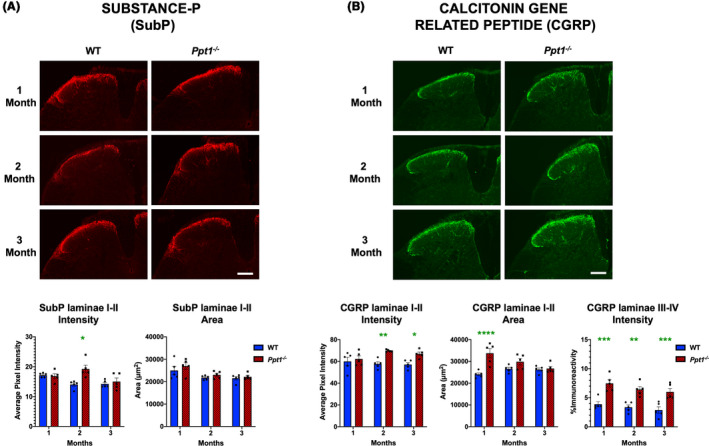
Early alterations in nociceptive markers in Ppt1−/− mouse spinal cords. Immunostaining for (A) Substance‐P (SubP) and (B) calcitonin gene‐related peptide (CGRP) reveal age‐related changes in these markers in the dorsal horn of Ppt1−/− mice. Representative images of dorsal horns of spinal cords reveal an increase in SubP immunoreactivity in laminae I‐II at 2 months and increased CGRP immunoreactivity in laminae I‐II beginning at 2 months as measured by image densitometry (average pixel luminance). Measures of area of immunoreactivity (µm2) confirmed a greater area of CGRP immunoreactivity at 1 month in Ppt1−/− mice across all time points as compared to WT (wild‐type) mice. Thresholding image analysis of positively stained fibres in laminae III‐IV of CGRP‐stained tissue showed a consistent increased in values in Ppt1−/− mice across all time points as compared to WT mice. Scale bars = 200µm. P‐values ‐ *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
Early onset innate immune responses in the Ppt1−/− spinal cord precede storage material accumulation and lymphocyte infiltration
Given the early onset of interneuron loss in Ppt1−/− mouse spinal cord the spinal cord (Figure 1), we next characterized the progression of AFSM accumulation, a characteristic hallmark of CLN1 disease [13, 14, 15, 16]. Thresholding image analysis of AFSM at these early stages of disease revealed that compared to WT mice, significantly elevated levels of AFSM were not apparent in either the dorsal or ventral horns of Ppt1−/− spinal cords until 3 months (Figure 3 A), after significant interneuron loss was already underway (Figure 1).
Figure 3.
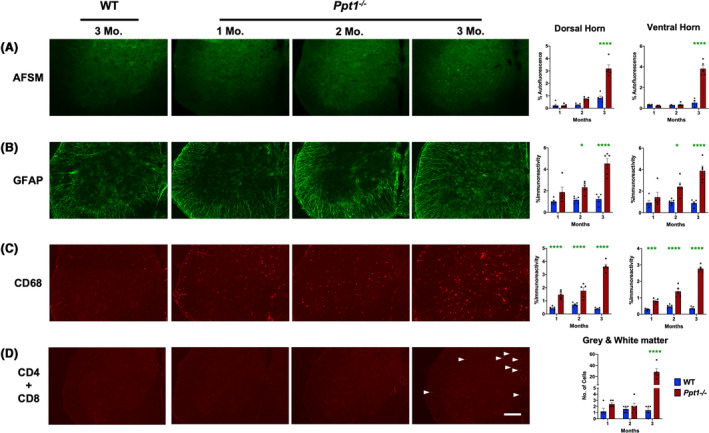
Early accumulation of storage material, glial activation and peripheral lymphocyte infiltration in Ppt1−/− mouse spinal cords. Representative images of ventral horns of spinal cords and thresholding image analysis of dorsal and ventral horns reveal (A) a significant increase in autofluorescent storage material (AFSM) accumulation at 3 months (Mo.), (B) significant increase in glial fibrillary acidic protein (GFAP, astrocytosis) immunoreactivity beginning at 2 months and (C) significant increase in CD68 (microglial activation) immunoreactivity as early as 1 month in Ppt1−/− mice as compared to WT mice. These data are indicative of a significant early glial immune response and subsequent accumulation of storage material. (D) Manual stereological estimates of CD4‐ and CD8‐positive lymphocytes reveal a significant increase of these cells in the spinal cord of Ppt1−/− mice at 3 months (white arrowheads), compared to WT cords, indicative of an early humoral immune response. Scale bars = 200µm. P‐values ‐ P‐values ‐ *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001; two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
Both microglial activation and astrocytosis are also closely associated with neuron loss in the forebrains of Ppt1−/− mice, typically preceding neurodegeneration [14, 15]. Staining spinal cord tissue for glial fibrillary acidic protein (GFAP, astrogliosis) showed significantly elevated GFAP immunoreactivity within the spinal grey matter starting at 2 months, in both the dorsal and ventral horns with accompanying changes in astrocyte morphology and staining intensity already apparent at this age (Figure 3 B). In contrast, staining for CD68 (microglial activation) revealed intense CD68‐positive hypertrophied brain macrophages within the spinal grey matter in both the dorsal and ventral horns of Ppt1−/− mouse spinal cords, as early as 1 month of age (Figure 3 C). These data demonstrate that astrocytes and microglia are activated during very early stages of disease in the Ppt1−/− cord, with microglial activation evident at least 2 months before the onset of any previously documented pathology in the CNS of these mice [13, 14, 15, 16, 17], also preceding the onset of significant interneuron loss (Figure 1).
The forebrain of Ppt1−/− mice is also progressively infiltrated by both helper and cytotoxic T lymphocytes, with the numbers of these cells increasing with age [16, 31]. Therefore, we immunostained lumbosacral spinal sections at these early time points simultaneously for CD4 and CD8 to gauge the overall impact of peripheral lymphocyte infiltration into the spinal cord, rather than to reveal any subtype specificity as we have done previously in the CNS [16, 31]. There were no significant differences between genotypes in staining for these lymphocyte markers at 1 and 2 months. However, there was a dramatic increase in CD4‐ and CD8‐positive lymphocytes in Ppt1−/− spinal cords at 3 months (Figure 3 D). This lymphocyte infiltration of the spinal cord parallels similar events in the optic nerve but precedes their occurrence in the brain [16].
Cytokine profile analysis reveals specific early alterations in inflammation‐related pathways
We further investigated the progression of neuroimmune responses in the in Ppt1−/− spinal cord by measuring the expression level of various cytokines and chemokines (Figure 4, Figure S1). There was already a significant and persistent increase of IL‐33 and VEGF‐A expression in Ppt1−/− spinal cords at 1 month, and significant increases in IP‐10 and MIP‐1α expression at 2 months as compared to WT spinal cords. There was also a significant decrease in IL‐10, Interferon‐γ and CXCL5 expression in Ppt1−/− spinal cords at 2 months (Figure 4 Ppt1−/− spinal). Other cytokines including MIP‐1β, MIP‐2 and MCP‐1 were significantly increased at 3 months, similar to the brain [15]and MCP‐3, Eotaxin (CCL11), RANTES and GRO‐α (Figure S1). In contrast, cytokines including IL‐1α, IL‐1β, IL‐6, TGF‐β and C‐reactive protein (CRP), were not significantly altered at these early time points in Ppt1−/− spinal cords (Figure S1). These findings demonstrate the presence of significant, but specific neuroinflammatory changes in Ppt1−/− spinal cords as early as 1 month, correlating with the onset of microglial activation at this time point, subsequently involving progressively more chemokines and cytokines.
Figure 4.
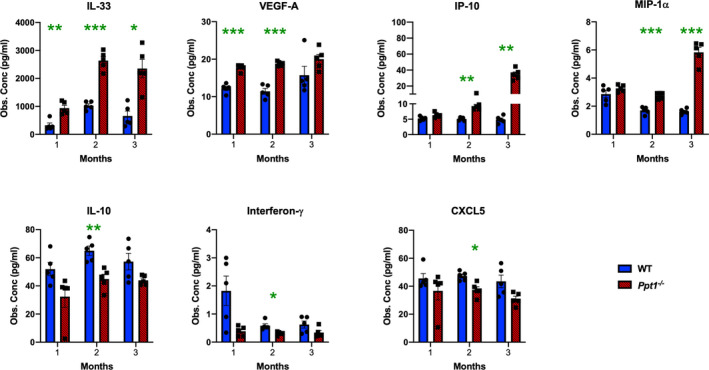
Early cytokine profile alterations in Ppt1−/− mouse spinal cords reveal specific inflammatory changes. Observed concentrations (Obs. Conc) in pg/µl of various cytokines measured in spinal cord homogenates from WT and Ppt1−/− mice revealing significant early upregulation of IL‐33 and VEGF‐A as early as 1 month, and of IP‐10 and MIP‐1α at 2 months in Ppt1−/− cords and significant downregulation of IL‐10, Interferon‐γ and CXCL5 were also seen at 2 months in Ppt1−/− cords as compared to WT cords. P‐values ‐ P‐values ‐ *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
Early volumetric changes and oligodendrocyte maturation defects in Ppt1−/− mouse spinal cords
The mammalian spinal cord continues to develop during the postnatal period, and this process continues from birth to 3 months in mice [21, 32]. While the number of neurons remains largely the same, there are dramatic increases in nerve fibre density, dendritic arbour and white matter volumes during this postnatal period [21, 32].Significant cerebral atrophy occurs both in CLN1 patients [33, 34] and in Ppt1−/− mice [18]. Given the early onset pathological changes in Ppt1−/− cords, we next investigated spinal cord volume. Cavalieri regional volumetric estimates of lumbosacral spinal cord volumes revealed that while WT and Ppt1−/− mice did not differ in regional volume at 1 month, WT mice subsequently showed a progressive increase across all volume measures until 3 months. Ppt1−/− mice showed significantly lower total spinal, grey matter and white matter volumes from 2 months of age onwards (Figure 5). The decreased spinal cord volumes observed in Ppt1−/− mice may be due to the onset of neurodegeneration described here and accompanying axon loss, altered postnatal maturation of the cord or a combination of these factors.
Figure 5.
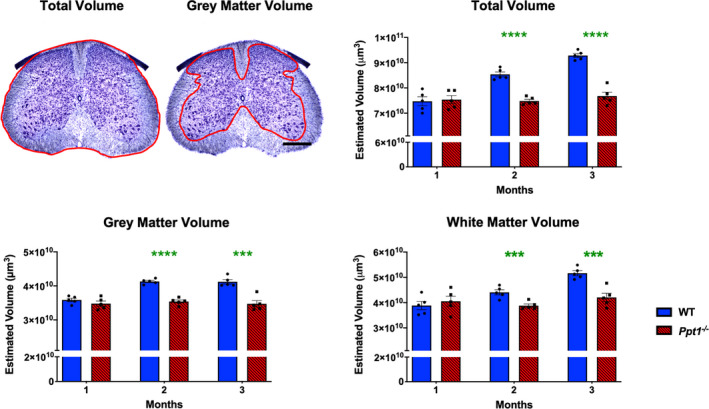
Progressive changes in Ppt1−/− mouse spinal cord volumes. Representative images of a Nissl‐stained mouse lumbar spinal cord section demonstrating the delineated boundaries (red) used to determine total and grey matter volumes. Scale bar = 400µm. Stereological Cavalieri estimates show decreased volumes of the lumbosacral spinal cord in Ppt1−/− mice as compared to age‐matched wild‐type controls (WT) as early as 2 months of age in total, grey matter and white matter volumes indicating a possible postnatal maturation deficit. Scale bars = 400µm. P‐values ‐ ***p ≤ 0.001, ****p ≤ 0.0001; two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
We next immunostained for oligodendrocyte transcription factor 2 (Olig2) and NG2 chondroitin sulphate proteoglycan (NG2), two established markers of oligodendrocyte lineage cells [35, 36] to ascertain whether oligodendrocyte maturation is altered in the postnatal Ppt1−/− cord, contributing to the altered white matter volumes observed. There was a significant increase in Olig2‐positive cells in both the grey and white matter of Ppt1−/− mice at 1 month, which subsequently decreased over time (Figure 6 A), compared to WT mouse cords. In WT mouse spinal cords, there was an increase in NG2‐positive cells at 2 months, especially in the lateral and dorsal funiculi of the lumbosacral spinal cords (Figure 6 B). In contrast, in Ppt1−/− mice, there was a persistently higher level of NG2‐positive cells in all white matter regions from 1 month onwards, with significantly higher values in the lateral and dorsal funiculi at these ages (Figure 6 B). We next stained lumbosacral spinal sections for myelin basic protein (MBP), a marker of myelinating oligodendrocytes [37]. Ppt1−/− mice showed increased levels of MBP across the dorsal, lateral and ventral funiculi at 1 month, and within the ventral horn grey matter. These white matter increases in MBP staining persisted in the ventral funiculus at 2 months, but MBP staining intensity subsequently decreased across all regions measured for Ppt1−/− mice compared to WT cords at 3 months (Figure S2). These data demonstrate that in addition to an early onset of neuron loss and a marked early neuroimmune response, the Ppt1−/− cord also shows evidence of altered maturation of oligodendrocytes, which may contribute to the volumetric changes seen in their spinal white and grey matter.
Figure 6.
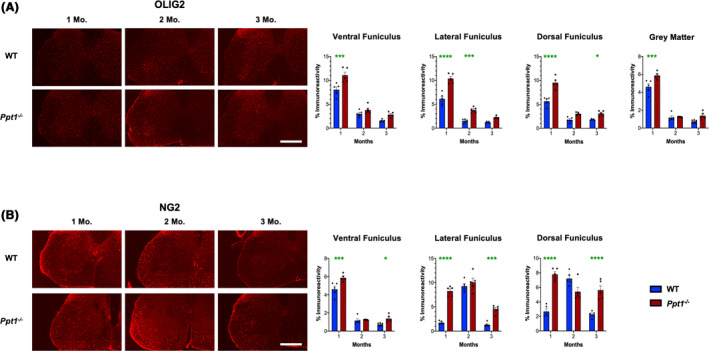
Dysregulated oligodendrocyte maturation in Ppt1−/− mouse spinal cords. (A) Representative images and thresholding image analysis data for lumbosacral spinal cord sections immunostained for the oligodendrocyte precursor marker Olig2 reveal significant increase in immunoreactivity for this marker in the ventral, lateral and dorsa funiculi of the spinal white matter, as well as the total grey matter area at 1 month (Mo.), as well as significant increases in the lateral funiculus at 2 months, and dorsal funiculus at 3 months in Ppt1−/− mice as compared to WT mice. (B) Representative images and thresholding image analysis data for lumbosacral spinal cord sections stained for the oligodendrocyte precursor marker NG2 chondroitin sulphate proteoglycan (NG2) reveal significant increases in NG2 immunoreactivity in the ventral, lateral and dorsal funiculi of the white matter at 1 month and 3 months in Ppt1−/− mice as compared to WT mice. P‐values ‐ *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001; two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
Early gait and behavioural abnormalities in Ppt1−/− mice
Decreased rotarod performance and other behavioural changes [9, 15] have previously been reported in Ppt1−/− mice and altered gait has been described late in disease progression, but not quantified [10]. In light of our findings of significant early spinal pathology, we sought to quantitatively determine the onset of gait abnormalities, and how their timing relates to pathology in the spinal cord and elsewhere in the CNS of these mice. CatWalk XT measures [23, 24, 25]revealed no significant differences in any gait parameter between genotypes at 1 month. However, at 2 months of age Ppt1−/− mice of both sexes significantly outperformed WT mice in many gait parameters, before these values normalized at 3 months. Subsequently, we quantified a significant and progressive decline across gait parameters in Ppt1−/− mice, that worsened and became pronounced with disease progression. This pattern was apparent for average speed (significantly higher in Ppt1−/− mice than WT mice at 2 months) (Figure 7 B), average stride length (significantly longer in Ppt1−/− mice at 2 months) (Figure 7 C), and cadence or frequency of steps (elevated at 2 months) (Figure 7 D). Conversely, body speed variation in each run showed an opposite pattern to overall body speed, with Ppt1−/− mice showing significantly less variation at 2 months, but significantly increased variation from 4 months onwards, compared to their WT counterparts (Figure 7 E). Collectively these data reveal for the first time an early period of supra‐normal gait of Ppt1−/− mice at 2 months of age, moving more quickly and taking larger steps than WT mice, before a subsequent progressive and dramatic decline in their mobility with disease progression.
Figure 7.
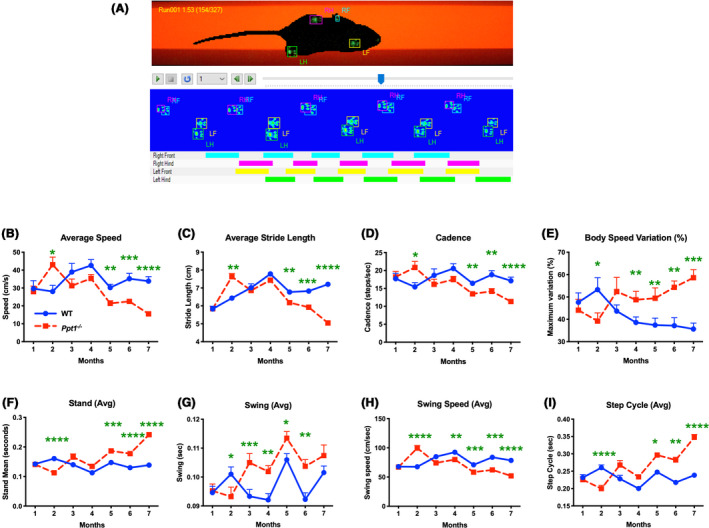
CatWalk XT data acquisition and early gait abnormalities in Ppt1−/− mice. (A) Representative frame of the captured video from a single run on the Catwalk XT gait analysis system showing each paw print as a fluorescent green mark. These are then classified as Right Fore (RF), Left Fore(LF), Right Hind (RH) or Left Hind (LH). The schematics below show placement of these classified paw prints over the duration of the run, with coloured bars indicating the periods when each foot was in contact with the glass floor of the apparatus. CatWalk XT measures showing Ppt1‐deficient (Ppt1−/−) mice having a higher average speed (cm/sec) (B), stride length (cm) (C) and cadence (steps/sec) (D) at 2 months compared to wild‐type (WT) mice, followed by a subsequent progressive decline across all these parameters with increased age. Variation in body speed (E) over a run (%)mirrored this pattern, with markedly less variation at 2 months which increases with age in Ppt1−/− mice as compared to WT mice. The measures of stand (sec) (F), swing (G), swing speed (cm/sec) (H) and step cycle (sec) (I) were also indicative of a supra‐normal gait at 2 months. P‐values ‐ *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; two‐way repeated measures ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 12 mice/group).
To reveal more about the nature of these early gait abnormalities, we determined the average time spent on each paw either in contact with the ground (Stand) or in motion between successive placements (Swing) (Figure 7 F,G). Ppt1−/− mice spent significantly less time during both the stand and swing phase at 2 months, compared to WT mice, with both measures increasing with age, reaching significance at 5 months for stand and 4 months for swing. Swing speed showed a predictable inverse relation to swing, indicating a faster limb motion at 2 months followed by a progressive decline (Figure 7 H). The Step cycle which is a combined measure of stand and swing times between successive placements of an individual paw, showed a similar pattern with significantly shorter step cycle times for Ppt1−/− mice at 2 months, which then progressively increased to significantly greater times at 5 months (Figure 7 I). At 2 months, Ppt1−/− mice also showed a significantly increased average paw print area measures in their fore paws, as compared to hind paws. These fore paw measures subsequently normalized to WT mouse values at 3 months and continued to decrease with age (Figure S3).
We then investigated whether the early gait alterations Ppt1−/− mice display were sustained over longer periods of time. We therefore assessed the one‐hour locomotor activity and open‐field behaviour of WT and Ppt1−/− mice (Figure 8), focusing at early (1‐, 2‐ and 3‐month) time point. Consistent with previous observations of decreased overall motility early in disease [9], the open‐field locomotor activity revealed that Ppt1−/− mice covered significantly shorter distance, made a lower number of ambulations, and an inversely greater number of rests over a 1‐hour period vs. WT mice at 1, 2 and 3 months. Ppt1−/− mice also showed a significant decrease in the number of rearing motions at 1 month as compared to WT mice (Figure 8). Rearing is not only used as a measure of locomotor activity but also a measure of exploration and anxiety [38].We have not seen altered fear conditioning in Ppt1−/− mice at this age [9], and in the current study Ppt1−/− mice showed no significant difference in time spent in the centre versus the edge of the chamber during this test. Nevertheless, these data show that Ppt1−/− mice travel shorter distances and transit between the centre and the edge fewer times vs. WT mice (Figure S4). Therefore, Ppt1−/− mice show an overall lower mobility as early as 1 month, which is earlier than any previously established behavioural phenotype for this model.
Figure 8.
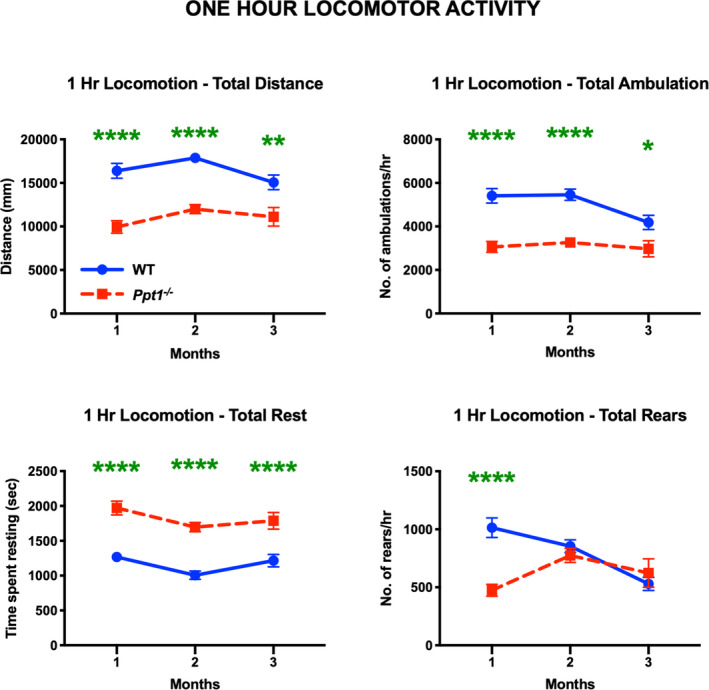
Decreased overall locomotion in Ppt1−/− mice. One‐hour locomotor activity and open field test reveal that Ppt1−/− mice moved over a significantly shorter distance, showed fewer ambulations and increased number of rests at all time points. Ppt1−/− mice also show a significant decrease in number of rearing motions on their hind limbs at 1 month, as compared to WT (wild‐type) mice. P‐values ‐ *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001; two‐way repeated measures ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 12 mice/group).
The relentless decline in motor performance by CLN1 disease children is well‐recognized [39, 40], but this is preceded by clumsiness and an early onset hyperkinesia that precedes a dramatic and progressive loss of mobility [3, 8]. Given our findings in Ppt1−/− mice, we undertook clinical observations of gait in a case of CLN1 disease who is homozygous for a well‐known CLN1 mutation; c.541G> A (p.Val181Met). The walking abilities of the patient seen on the video (Video S1), recorded early in his motor deterioration at 2 years and 4 months of age, clearly demonstrate a nonrhythmic gait, with periods of pausing followed by walking that is faster than usual and proceeds in an uncoordinated manner. This applies to both hip and knee movements, and when initiating a movement from a standing posture. During the same period his family reported that he was irritable, had restless sleep and often cried without obvious reason. When the parents tried to support his gait by keeping hold of his arms or body, he pushed them away, and generally he did not like to be touched. These observations indicate a significantly altered gait early in disease progression, consistent with our observations in Ppt1−/− mice. Although, the basis for this unexpected early gait presentation remains unclear, early spinal pathology may play a contributory role along with the progressive involvement of cerebral, cerebellar and possibly peripheral nervous system pathology.
Discussion
Very little is known about the events that occur early in CLN1 disease progression before children or mice become symptomatic. However, CNS damage is likely to occur during this critical period and this will influence therapeutic efficacy. Our findings provide insights into these early events in Ppt1−/− mice, identifying that PPT1 deficiency has both pathological and behavioural impacts upon the CNS during a period when these mice otherwise appear overtly unaffected. Our data place the onset of selective neuron loss, a neuroinflammatory cascade and gait abnormalities all starting during the period when the spinal cord is still developing. Our data suggest that postnatal maturation appears impaired and there may be an interplay between neurodegeneration and developmental processes that has so far been underappreciated.
The loss of spinal interneurons in Ppt1−/− mice at 2 months, reveals them to be the most vulnerable and earliest affected CNS neuron population, being lost significantly earlier than motor neurons (Figure 1). Given that neuron loss may be preceded by a period of neuronal dysfunction, it is likely that this vulnerable neuron population will be functionally impaired even earlier [41, 42]. This early dysfunction may impact the spinal dorsal horn, a site critical for processing sensory inputs before their transmission to the brain [43, 44], and we documented an early increased expression of Substance‐P and CGRP (Figure 2), neuropeptides that are associated with the propagation and maintenance of hyperalgesia [30]. This may help explain the restlessness, irritability and sleep disturbances that are reported in ~90% of CLN1 cases [5, 8], and it will be important to assess sensory and nociceptive function in Ppt1−/− mice.
Pain, hypersensitivity, irritability and altered mobility are all associated with glial activation [45, 46], which we demonstrate starts within the Ppt1−/− spinal cord as early as one month of age. This spinal microglial activation is the earliest histologically identified pathology anywhere in the CNS of these mice (Figure 3). This is placed at the beginning of a chronologically staged series of pathological events that subsequently includes astrocytosis and selective interneuron loss (2 months). These all occur far earlier than similar events within the cerebrum or cerebellum of Ppt1−/− mice and are accompanied by a progressively more complex spinal chemokine and cytokine response. Subsequently, lymphocyte infiltration occurs in the spinal cord, paralleling its appearance in the optic nerve, but preceding its onset in the rest of CNS [16, 31]. This is also the time when significant storage material accumulation is detected, but precedes motor neuron loss by several months. These data further emphasize the early onset and rapid progression of spinal disease while this structure is still maturing, and a full quarter lifespan before we thought had previously thought it to be affected. A developmental component to CLN5 disease has recently been suggested [47], but so far very little attention has been paid to these aspects of CLN1 disease. Given the similar expression profiles of CLN1 and CLN5 during development [48] it will be important to extend our analysis of Ppt1−/− mice into the prenatal period.
Our data for early changes in the expression of a restricted subset of cytokines is in stark contrast to the generalized inflammatory responses that subsequently occur in the brain in CLN1 disease [16, 17, 49]. Cytokine profile analysis of the early inflammatory response revealed a specific upregulation of IL‐33 and VEGF‐A in Ppt1−/− mice as early as 1 month (Figure 4). While VEGF‐A is known to largely play an anti‐inflammatory role [50, 51], IL‐33 is of particular interest as an ‘alarmin’ that mediates a downstream microglia‐mediated inflammatory response and is also secreted by damaged or dying cells [52], and by dysregulated oligodendrocytes [53], implicated in mediating neuropathic pain. As such, dysfunctional neurons, glia or oligodendrocyte precursors in Ppt1−/− mice may secrete IL‐33 to co‐ordinate the early onset of the glial immune response and recruitment of vascular macrophages. We found further evidence suggestive of such a sequence of events, with the subsequent increase in IP‐10 and MIP‐1α at 2 months and RANTES at 3 months, all of which are secreted by activated microglia [54, 55]. As such, our data potentially reveal the VEGF‐A and IL‐33 signalling cascade as a novel early immunomodulatory target for CLN1 therapy.
The early onset of pathology in the Ppt1−/− mouse spinal cord, was mirrored by altered locomotion when these mice otherwise appear unaffected. Most behavioural analyses in Ppt1−/− mice [9], have concentrated on motor performance with altered gait only qualitatively reported at later stages of the disease [9, 15]. Collectively our data suggest an overall lower mobility of Ppt1−/− mice, but that at 2 months of age, upon initiating locomotion, they do so more rapidly than wild‐type mice.
This mirrors the hyperkinetic gait of CLN1 children, which also includes periods of immobility interspersed by rapid walking movements (Video S1). The subsequent severe decline in gait has always considered to be sequelae of the devastating neurodegeneration that occurs in the brain and cerebellum [14, 15]. However, our description of behavioural changes in young Ppt1−/− mice (Figures 7,8) before onset of detectable brain or cerebellar pathology, suggests that spinal pathology likely also contributes to these early gait phenotypes. Interneurons in the ventral part of the spinal cord, which we found were lost significantly earlier than motor neurons, are critical for precisely controlling the speed and coordination of locomotion [56, 57]. Other previously described pathologies in the cerebrum and cerebellum may also contribute to these gait alterations. Nevertheless, there is consistency between what is known about pathology in Ppt1−/− mice and the altered gait seen both in these mice and also in our CLN1 child. The characteristically altered gait and mobility of Ppt1−/− mice at 2 months (Figure 7), and the increased expression of Substance‐P and CGRP (Figure 2) in the dorsal horn, also hint at the involvement of peripheral nerve pathology in this disease. It will be important to characterize the onset and progression of pathology within both peripheral nerve and skeleto‐muscular components, and how these may influence behavioural changes. Clumsiness and a lack of motor development are typically first presenting symptoms of CLN1 disease [3, 8], with an early onset hyperkinesia and subsequent progressive loss of mobility. Our clinical observations of a CLN1 child also demonstrated altered gait and sensory perception at early stages of disease, similar to our observations in Ppt1−/− mice. While the pathological basis of such behavioural changes is likely to be complex and multisystemic, the timing of their onset implicates the spinal cord to play at least a contributory role in their presentation in mice.
Our data reveal vulnerable neuronal populations, altered oligodendrocyte maturation and early neuroinflammatory changes that can potentially serve as new therapeutic targets. We also demonstrate early behavioural consequences that may involve the spinal cord and potentially other components of the central and peripheral nervous systems and how these may relate to human CLN1 disease. Our findings highlight the need for further ultrastructural investigation of this regional pathology across both mouse and human tissue to further understand the basis of this vulnerability. Significantly, the disease process starts in the spinal cord of Ppt1−/− mice during a period when they previously did not display signs of being affected, but while development is still ongoing. These data highlight the need to initiate therapy as soon as possible, and while this is feasible in a mouse model, this has important implications for identifying and treating CLN1 children while they are presymptomatic. Finally, our data also raise the important question of whether the spinal cord is similarly affected in other forms of NCL.
Funding
This work was supported by National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke (NINDS) Grant 043205 to MSS and JDC, institutional support from Department of Pediatrics, Washington University in St. Louis Medical School to JDC and a King’s College London Graduate School International Studentship award to HRN.
Conflict of interests
JDC has received research support from BioMarin Pharmaceutical Inc. in addition to Abeona Therapeutics Inc., Regenexbio Inc. and CereSpir Inc. The remaining authors declare no conflicts of interest.
Authors' contributions
JDC, JRO and MSS conceived and designed the study. JRO performed the clinical observations and obtained consent for these and for their publication, HRN carried out the pathology experiments, gait analysis and western blotting; JTD performed the open‐field behavioural testing. As acknowledged below, chemokine and cytokine analyses were performed by the core facility in the Bursky Center for Human Immunology and Immunotherapy Programs, Washington University School of Medicine and Barnes‐Jewish Hospital in St. Louis, MO. HRN, and JDC wrote the manuscript with input from all the authors. All authors contributed to the data analysis, interpretation. All authors read and approved the final manuscript.
Ethics approval and consent to publication
Our clinical observation study was approved by the institutional ethics committee and was conducted at Centre for Rare Diseases, Department of Paediatrics and Adolescent Medicine, Aarhus University Hospital. Written informed consent was received from the participant’s guardians for data as well as any pictures or videos appearing in this manuscript (Data S1). All animal procedures were performed in accordance with National Institutes of Health (NIH) guidelines under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University School of Medicine in St. Louis, MO.
Supporting information
Figure S1. Cytokine profiles showing alterations at 3 months or no significant changes in Ppt1‐/‐ mice. The levels of various cytokines were measured in spinal cord homogenates from WT and Ppt1‐/‐ mice revealing GRO‐α, RANTES, Eotaxin (CCL11), MIP‐1β, MIP‐2 and MCP‐1 and MCP‐3 were significantly increased at 3 months indicative of an overall progressive increase in inflammatory response with age. However, other cytokines including IL‐1α, IL‐1β , TGF‐β, C‐reactive protein (CRP) and IL‐6, which are also associated with general inflammatory responses, were not significantly altered at these early time points in Ppt1‐/‐spinal cords. P‐values ‐ P‐values ‐ *p≤0.05, **p≤0.01, two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
Figure S2. Dysregulated MBP expression in Ppt1‐/‐ mouse spinal cords. Representative images and image densitometry (average pixel luminance) measures of lumbosacral spinal cord sections stained for Myelin Basic Protein (MBP) show significantly higher immunoreactivity in the ventral funiculus at 1 and 2 months (arrowheads are representative of these areas) and the lateral and dorsal funiculi at 1 month followed by a significant decrease in the dorsal and lateral funiculi at 3 months. Scale bars = 400µm. P‐values ‐ *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
Figure S3. Progressive changes in Ppt1‐/‐ mouse paw print areas. CatWalk XT (Noldus) measures of print area (cm2) show an early increase in fore paw areas at 2 months, coinciding with an increased speed and stride length in Ppt1‐/‐ mice, but not in the hind paws. There was also a significant increase of fore paw measures in Ppt1‐/‐ mice at 5 months, followed by a significant decrease in both fore and hind paw measures in these mutant mice at 6 months, that recovers in the fore paws but progresses in the hind paws. Overall, this is indicative of an increased reliance on the fore‐paws for locomotion in Ppt1‐/‐ mice with disease progression. P‐values ‐ *p≤0.05, **p≤0.01, ****p≤0.0001; two‐way repeated measures ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 12 mice/group).
Figure S4. Decreased locomotion in Ppt1‐/‐ mice. Edge versus centre measures of one‐hour open‐field locomotor activity reveal that Ppt1‐/‐ mice spend similar amounts of time in the centre vs. edge of the open‐field locomotion apparatus as compared to WT mice, but travelled a significantly lower distance in the edge across all time points and at 2 months in the centre, as compared to WT mice. Ppt1‐/‐ mice also consistently crossed fewer times between the centre and edge zones across all time points as compared to WT mice. P‐values ‐ *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; two‐way repeated measures ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 12 mice/group).
Methods S1.
Video S1.
Acknowledgements
We thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes‐Jewish Hospital in St. Louis, MO., for the use of the Bursky Center for Human Immunology and Immunotherapy Programs, which provided the Luminex multiplex service. The authors acknowledge Dr Frances Platt, University of Oxford, for her advice on analysing the gait analysis data and Dr Diane Bender, Washington University in St. Louis for assistance with cytokine profile analysis. We also thank DrsThomas Gillingwater, Marco Sardiello, Marion Bonneau and Alison Barnwell for their advice and comments on the manuscript, and Samuel Cooper for skilled assistance with video processing.
Nelvagal H. R., Dearborn J. T., Ostergaard J. R., Sands M. S. and Cooper J. D. (2021) Neuropathology and Applied Neurobiology 47, 251–267 Spinal manifestations of CLN1 disease start during the early postnatal period
Data availability statement
The data sets analysed in this study are available from the corresponding author upon reasonable request.
References
- 1. Mole SE, Anderson G, Band HA, Berkovic SF, Cooper JD, Kleine Holthaus S‐M, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol 2019; 18: 107–16 [DOI] [PubMed] [Google Scholar]
- 2. Kohlschütter A, Schulz A, Bartsch U, Storch S. Current and emerging treatment strategies for neuronal ceroid lipofuscinoses. CNS Drugs 2019; 33: 315–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases — clinical perspectives. Biochimica et Biophysica Acta (BBA). Molecular Basis of Disease 2013; 1832: 1801–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Warrier V, Vieira M, Mole SE. Genetic basis and phenotypic correlations of the neuronal ceroid lipofusinoses. Biochim Biophys Acta 2013; 1832: 1827–30 [DOI] [PubMed] [Google Scholar]
- 5. Dolisca S‐B, Mehta M, Pearce DA, Mink JW, Maria BL. Batten Disease. J Child Neurol 2013; 28: 1074–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haltia M, Rapola J, Santavuori P. Infantile type of so‐called neuronal ceroid‐lipofuscinosis. Acta Neuropathol 1973; 26: 157–70 [DOI] [PubMed] [Google Scholar]
- 7. Santavuori P, Haltia M, Rapola J, Raitta C. Infantile type of so‐called neuronal ceroid‐lipofuscinosis. 1. A clinical study of 15 patients. J Neurol Sci 1973; 18: 257–67 [DOI] [PubMed] [Google Scholar]
- 8. Williams RE, Aberg L, Autti T, Goebel HH, Kohlschütter A, Lönnqvist T. Diagnosis of the neuronal ceroid lipofuscinoses: an update. Biochim Biophys Acta 2006; 1762: 865–72 [DOI] [PubMed] [Google Scholar]
- 9. Dearborn JT, Harmon SK, Fowler SC, O’Malley KL, Taylor GT, Sands MS, et al. Comprehensive functional characterization of murine infantile Batten disease including Parkinson‐like behavior and dopaminergic markers. Sci Rep 2015; 5: 12752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, et al. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci U S A 2001; 98: 13566–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim Biophys Acta 2013; 1832: 1807–26 [DOI] [PubMed] [Google Scholar]
- 12. Tyynelä J, Cooper JD, Khan MN, Shemilts SJA, Haltia M. Hippocampal pathology in the human neuronal ceroid‐lipofuscinoses: distinct patterns of storage deposition, neurodegeneration and glial activation. Brain Pathol 2004; 14: 349–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bible E, Gupta P, Hofmann SL, Cooper JD. Regional and cellular neuropathology in the palmitoyl protein thioesterase‐1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiology of Disease 2004; 16: 346–59 [DOI] [PubMed] [Google Scholar]
- 14. Kielar C, Maddox L, Bible E, Pontikis CC, Macauley SL, Griffey MA, et al. Successive neuron loss in the thalamus and cortex in a mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis 2007; 25: 150–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Macauley SL, Wozniak DF, Kielar C, Tan Y, Cooper JD, Sands MS. Cerebellar pathology and motor deficits in the palmitoyl protein thioesterase 1‐deficient mouse. Exp Neurol 2009; 217: 124–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Groh J, Kühl TG, Ip CW, Nelvagal HR, Sri S, Duckett S, et al. Immune cells perturb axons and impair neuronal survival in a mouse model of infantile neuronal ceroid lipofuscinosis. Brain 2013; 136: 1083–101 [DOI] [PubMed] [Google Scholar]
- 17. Shyng C, Nelvagal HR, Dearborn JT, Tyynelä J, Schmidt RE, Sands MS, et al. Synergistic effects of treating the spinal cord and brain in CLN1 disease. Proc Natl Acad Sci U S A 2017; 114: E5920–E9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kühl TG, Dihanich S, Wong AMS, Cooper JD. Regional brain atrophy in mouse models of neuronal ceroid lipofuscinosis. J Child Neurol 2013; 28: 1117–22 [DOI] [PubMed] [Google Scholar]
- 19. West MJ. Design‐based stereological methods for counting neurons. Prog Brain Res 2002; 135: 43–51 [DOI] [PubMed] [Google Scholar]
- 20. Sengul G, Watson C, Tanaka I, Paxinos G. Atlas of the spinal cord of the rat, mouse, marmoset, rhesus, and human. London, Waltham, MA: Academic Press, 2013. [Google Scholar]
- 21. Fu Y, Rusznák Z, Herculano‐Houzel S, Watson C, Paxinos G. Cellular composition characterizing postnatal development and maturation of the mouse brain and spinal cord. Brain Struct Funct 2013; 218: 1337–54 [DOI] [PubMed] [Google Scholar]
- 22. Etlin A, Bráz JM, Kuhn JA, Wang X, Hamel KA, Llewellyn‐Smith IJ, et al. Functional synaptic integration of forebrain GABAergic precursors into the adult spinal cord. J Neurosci 2016; 36: 11634–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hamers FPT, Koopmans GC, Joosten EAJ. CatWalk‐Assisted gait analysis in the assessment of spinal cord injury. J Neurotrauma 2006; 23: 537–48 [DOI] [PubMed] [Google Scholar]
- 24. Vrinten DH, Hamers FFT. 'CatWalk' automated quantitative gait analysis as a novel method to assess mechanical allodynia in the rat; a comparison with von Frey testing. Pain 2003; 102: 203–9 [DOI] [PubMed] [Google Scholar]
- 25. Coulthard P, Simjee SU, Pleuvry BJ. Gait analysis as a correlate of pain induced by carrageenan intraplantar injection. J Neurosci Methods 2003; 128: 95–102 [DOI] [PubMed] [Google Scholar]
- 26. Schaefer ML, Wong ST, Wozniak DF, Muglia LM, Liauw JA, Zhuo M, et al. Altered stress‐induced anxiety in adenylyl cyclase type VIII‐deficient mice. J Neurosci 2000; 20: 4809–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, et al. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis 2004; 17: 403–14 [DOI] [PubMed] [Google Scholar]
- 28. Hokfelt T. Neuropeptides in perspective: the last ten years. Neuron 1991; 7: 867–79 [DOI] [PubMed] [Google Scholar]
- 29. Shi X, Guo T‐Z, Wei T, Li W‐W, Clark DJ, Kingery WS. Facilitated spinal neuropeptide signaling and upregulated inflammatory mediator expression contribute to postfracture nociceptive sensitization. Pain 2015; 156: 1852–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Greco R, Tassorelli C, Sandrini G, Di Bella P, Buscone S, Nappi G. Role of calcitonin gene‐related peptide and substance P in different models of pain. Cephalalgia 2008; 28: 114–26 [DOI] [PubMed] [Google Scholar]
- 31. Groh J, Ribechini E, Stadler D, Schilling T, Lutz MB, Martini R. Sialoadhesin promotes neuroinflammation‐related disease progression in two mouse models of CLN disease. Glia 2016; 64: 792–809 [DOI] [PubMed] [Google Scholar]
- 32. Sakla FB. Quantitative studies on the postnatal growth of the spinal cord and the vertebral column of the albino mouse. J Comp Neurol 1969; 136: 237–47 [DOI] [PubMed] [Google Scholar]
- 33. Vanhanen SL, Puranen J, Autti T, Raininko R, Liewendahl K, Nikkinen P, et al. Neuroradiological findings (MRS, MRI, SPECT) in infantile neuronal ceroid‐lipofuscinosis (infantile CLN1) at different stages of the disease. Neuropediatrics 2004; 35: 27–35 [DOI] [PubMed] [Google Scholar]
- 34. Baker EH, Levin SW, Zhang Z, Mukherjee AB. MRI brain volume measurements in infantile neuronal ceroid lipofuscinosis. Am J Neuroradiol 2017; 38: 376–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu X, Zuo H, Maher BJ, Serwanski DR, LoTurco JJ, Lu QR, et al. Olig2‐dependent developmental fate switch of NG2 cells. Development 2012; 139: 2299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dawson MR, Levine JM, Reynolds R. NG2‐expressing cells in the central nervous system: are they oligodendroglial progenitors? J Neurosci Res. 2000; 61: 471–9 [DOI] [PubMed] [Google Scholar]
- 37. Bergles DE, Richardson WD. Oligodendrocyte development and plasticity. Cold Spring Harb Perspect Biol 2015; 8: a020453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zimcikova E, Simko J, Karesova I, Kremlacek J, Malakova J. Behavioral effects of antiepileptic drugs in rats: Are the effects on mood and behavior detectable in open‐field test? Seizure 2017; 52: 35–40 [DOI] [PubMed] [Google Scholar]
- 39. Santavuori P. Neuronal ceroid‐lipofuscinoses in childhood. Brain Dev 1988; 10: 80–3 [DOI] [PubMed] [Google Scholar]
- 40. Santavuori P, Lauronen L, Kirveskari K, Aberg L, Sainio K. Neuronal ceroid lipofuscinoses in childhood. Suppl Clin Neurophysiol 2000; 53: 443–51 [DOI] [PubMed] [Google Scholar]
- 41. Kielar C, Wishart TM, Palmer A, Dihanich S, Wong AM, Macauley SL, et al. Molecular correlates of axonal and synaptic pathology in mouse models of Batten disease. Hum Mol Genet 2009; 18: 4066–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koster KP, Francesconi W, Berton F, Alahmadi S, Srinivas R, Yoshii A. Developmental NMDA receptor dysregulation in the infantile neuronal ceroid lipofuscinosis mouse model. eLife 2019; 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Petitjean H, Bourojeni FB, Tsao D, Davidova A, Sotocinal SG, Mogil JS, et al. Recruitment of spinoparabrachial neurons by dorsal horn calretinin neurons. Cell Rep 2019; 28(1429–38): e4 [DOI] [PubMed] [Google Scholar]
- 44. Smith KM, Boyle KA, Madden JF, Dickinson SA, Jobling P, Callister RJ, et al. Functional heterogeneity of calretinin‐expressing neurons in the mouse superficial dorsal horn: implications for spinal pain processing. J Physiol 2015; 593: 4319–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Beh SC, Greenberg BM, Frohman T, Frohman EM. Transverse myelitis. Neurol Clin 2013; 31: 79–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hausmann ON. Post‐traumatic inflammation following spinal cord injury. Spinal Cord 2003; 41: 369–78 [DOI] [PubMed] [Google Scholar]
- 47. Singh Y, Leinonen H, Fazaludeen F, Jaronen M, Guest D, Buckley N, et al. Loss of Cln5 leads to altered Gad1 expression and deficits in interneuron development in mice. Hum Mol Genet 2019; 28: 3309–22 [DOI] [PubMed] [Google Scholar]
- 48. Heinonen O, Kyttälä A, Lehmus E, Paunio T, Peltonen L, Jalanko A. Expression of palmitoyl protein thioesterase in neurons. Mol Genet and Metab. 2000; 69: 123–9 [DOI] [PubMed] [Google Scholar]
- 49. Macauley SL, Wong AMS, Shyng C, Augner DP, Dearborn JT, Pearse Y, et al. An anti‐neuroinflammatory that targets dysregulated gliaenhances the efficacy of CNS‐directed gene therapy in murine infantile neuronal ceroid lipofuscinosis. J Neurosci 2014; 34: 13077–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li J, Chen S, Zhao Z, Luo Y, Hou Y, Li H, et al. Effect of VEGF on inflammatory regulation, neural survival, and functional improvement in rats following a complete spinal cord transection. Front Cell Neurosci 2017; 11: 381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang H, Wang Y, Li D, Liu Z, Zhao Z, Han D, et al. VEGF inhibits the inflammation in spinal cord injury through activation of autophagy. Biochem Biophys Res Commun 2015; 464: 453–8 [DOI] [PubMed] [Google Scholar]
- 52. Cao K, Liao X, Lu J, Yao S, Wu F, Zhu X, et al. IL‐33/ST2 plays a critical role in endothelial cell activation and microglia‐mediated neuroinflammation modulation. J Neuroinflammation 2018; 15: 136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zarpelon AC, Rodrigues FC, Lopes AH, Souza GR, Carvalho TT, Pinto LG, et al. Spinal cord oligodendrocyte‐derived alarmin IL‐33 mediates neuropathic pain. FASEB J 2016; 30: 54–65 [DOI] [PubMed] [Google Scholar]
- 54. Kettenmann H, Hanisch U‐K, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011; 91: 461–553 [DOI] [PubMed] [Google Scholar]
- 55. Kremlev SG, Roberts RL, Palmer C. Differential expression of chemokines and chemokine receptors during microglial activation and inhibition. J Neuroimmunol 2004; 149: 1–9 [DOI] [PubMed] [Google Scholar]
- 56. Cote MP, Murray LM, Knikou M. Spinal control of locomotion: individual neurons, their circuits and functions. Front Physiol 2018; 9: 784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Takakusaki K. Neurophysiology of gait: from the spinal cord to the frontal lobe. Mov Disord 2013; 28: 1483–91 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Cytokine profiles showing alterations at 3 months or no significant changes in Ppt1‐/‐ mice. The levels of various cytokines were measured in spinal cord homogenates from WT and Ppt1‐/‐ mice revealing GRO‐α, RANTES, Eotaxin (CCL11), MIP‐1β, MIP‐2 and MCP‐1 and MCP‐3 were significantly increased at 3 months indicative of an overall progressive increase in inflammatory response with age. However, other cytokines including IL‐1α, IL‐1β , TGF‐β, C‐reactive protein (CRP) and IL‐6, which are also associated with general inflammatory responses, were not significantly altered at these early time points in Ppt1‐/‐spinal cords. P‐values ‐ P‐values ‐ *p≤0.05, **p≤0.01, two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
Figure S2. Dysregulated MBP expression in Ppt1‐/‐ mouse spinal cords. Representative images and image densitometry (average pixel luminance) measures of lumbosacral spinal cord sections stained for Myelin Basic Protein (MBP) show significantly higher immunoreactivity in the ventral funiculus at 1 and 2 months (arrowheads are representative of these areas) and the lateral and dorsal funiculi at 1 month followed by a significant decrease in the dorsal and lateral funiculi at 3 months. Scale bars = 400µm. P‐values ‐ *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; two‐way ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 5 mice/group).
Figure S3. Progressive changes in Ppt1‐/‐ mouse paw print areas. CatWalk XT (Noldus) measures of print area (cm2) show an early increase in fore paw areas at 2 months, coinciding with an increased speed and stride length in Ppt1‐/‐ mice, but not in the hind paws. There was also a significant increase of fore paw measures in Ppt1‐/‐ mice at 5 months, followed by a significant decrease in both fore and hind paw measures in these mutant mice at 6 months, that recovers in the fore paws but progresses in the hind paws. Overall, this is indicative of an increased reliance on the fore‐paws for locomotion in Ppt1‐/‐ mice with disease progression. P‐values ‐ *p≤0.05, **p≤0.01, ****p≤0.0001; two‐way repeated measures ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 12 mice/group).
Figure S4. Decreased locomotion in Ppt1‐/‐ mice. Edge versus centre measures of one‐hour open‐field locomotor activity reveal that Ppt1‐/‐ mice spend similar amounts of time in the centre vs. edge of the open‐field locomotion apparatus as compared to WT mice, but travelled a significantly lower distance in the edge across all time points and at 2 months in the centre, as compared to WT mice. Ppt1‐/‐ mice also consistently crossed fewer times between the centre and edge zones across all time points as compared to WT mice. P‐values ‐ *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001; two‐way repeated measures ANOVA with post hoc Bonferroni correction. Values shown are mean ± SEM. (n = 12 mice/group).
Methods S1.
Video S1.
Data Availability Statement
The data sets analysed in this study are available from the corresponding author upon reasonable request.