

Abstract
LKB1-inactivated tumors are vulnerable to the disruption of pyrimidine metabolism, and leflunomide emerges as a therapeutic candidate because its active metabolite, A77–1726, inhibits dihydroorotate dehydrogenase, which is essential for de novo pyrimidine biosynthesis. However, it is unclear whether leflunomide inhibits LKB1-inactivated tumors in vivo, and whether its inhibitory effect on the immune system will promote tumor growth. Here, we carried out a comprehensive analysis of leflunomide treatment in various LKB1-inactivated murine xenograft, PDX, and genetically engineered mouse models. We also generated a mouse-tumor derived cancer cell line, WRJ388, that could metastasize to the lung within a month after subcutaneous implantation in all animals. This model was used to assess the ability of leflunomide to control distant metastasis. Leflunomide treatment shrank a HeLa xenograft and attenuated the growth of an H460 xenograft, a PDX, and lung adenocarcinoma in the immune-competent GEMM. Interestingly, leflunomide suppressed tumor growth through at least three different mechanisms. It caused apoptosis in HeLa cells, induced G1 cell cycle arrest in H460 cells, and promoted S-phase cell cycle arrest in WRJ388 cells. Finally, leflunomide treatment prevented lung metastasis in 78% of the animals in our novel lung cancer metastasis model. In combination, these results demonstrated that leflunomide utilizes different pathways to suppress the growth of LKB1-inactivated tumors, and it also prevents cancer metastasis at distant sites. Therefore, leflunomide should be evaluated as a therapeutic agent for tumors with LKB1-inactivation.
Introduction
LKB1 (liver kinase B1), also known as STK11 (Serine/threonine kinase 11), encodes a Ser/Thr kinase initially discovered as the tumor suppressor gene responsible for the inherited Peutz-Jeghers syndrome (1,2). It is somatically inactivated in several cancer types, such as lung adenocarcinoma, cervical cancer, and melanomas (3–7) with a somatic mutation rate of 14%, 7.7%, and 2.5%, respectively in TCGA PanCancer Atlas Studies (cBioPortal). LKB1/STK11 is one of the most frequently mutated cancer driver genes in lung adenocarcinoma (after p53 and Kras) (8,9). Currently, there are limited therapeutic options for cancers with LKB1-inactivation. For example, LKB1 inactivation mostly occurs in smoking-related lung cancers, which lack EGFR mutation. Thus, they are unlikely to respond to TKI-based therapies (10). Immune checkpoint therapies have shown promise in the treatment of NSCLC, yet recent work has identified distinct molecular features of LKB1/KRAS-mutant tumors, which include suppression or lack of immunoediting (11,12). Furthermore, several clinical trials have demonstrated that lung cancers with LKB1-inactivation are not responsive to immune checkpoint blockade therapy (13,14). Therefore, there is a clinical need for novel treatments for LKB1-inactivated cancers.
Many laboratories have attempted to identify vulnerabilities specifically associated with LKB1-inactivation (15), and 5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide (AICAR) was found to induce apoptosis in LKB1-inactivated cells (16–18). Our previous work revealed that AICAR-induced decreases in intracellular UMP or UMP-related metabolites are responsible for this cell killing mechanism (18). This finding is consistent with a more recent study, indicating that LKB1-inactivated cancers are sensitive to the disruption of pyrimidine metabolism (19). AICAR is not a viable therapeutic agent because of its short half-life (20), but its ability to induce apoptosis in LKB1-inactivated cancer cells prompted the notion to treat this tumor type by inhibiting de novo UMP synthesis (21).
Dihydroorotate dehydrogenase (DHODH) catalyzes the fourth step in de novo pyrimidine biosynthesis, which converts dihydroorotate to orotate. Inhibition of this enzyme is used to treat autoimmune disease because it leads to the depletion of pyrimidine supply for T cells (22). DHODH has previously been evaluated as a target for human cancer, and early studies focused on brequinar, but brequinar was found to be inactive in a phase 2 clinical trial (23). Leflunomide is an immunomodulatory drug that is currently used for the treatment of rheumatoid arthritis and psoriatic arthritis (24). The active metabolite of leflunomide, A77 1726, is an inhibitor of DHODH. It has been shown to share a common binding site with brequinar in a tunnel that leads to the catalytic site but has a different binding region (25). Leflunomide was evaluated in the 1990s as an anti-cancer agent in the form of an EGFR inhibitor (26,27), and several recent publications have renewed interest in its use to treat breast or prostate cancers (28,29). We previously reported that leflunomide is capable of inducing apoptosis in two LKB1-inactivated lung cancer cells in vitro (18), but it is unclear whether leflunomide has the same efficacy as AICAR in promoting apoptosis in all LKB1-inactivated cancer cells, and whether it has in vivo efficacy. Another relevant clinical concern for the use of leflunomide in cancer treatment is that the suppression of the immune system by leflunomide may promote tumor growth in the immunocompetent host. Here, we carried out a comprehensive analysis of the in vivo activity of leflunomide in xenografts, patient-derived xenografts, and immune-competent genetically-engineered mouse model, and we also evaluated leflunomide as an agent to prevent the metastasis of LKB1-inactivated cancers.
Materials and methods
Reagents and antibodies
Leflunomide was purchased from Enzo life sciences (ALX-4300–095-G001). AICAR was purchased from Toronto Research Chemicals Inc (North York, Canada). Uridine was purchased from Sigma (St. Louis, MO). Antibodies against LKB1 (3047s), Caspase-3 (9962s), Cleaved caspase-3 (9664s), PARP (9542s), Kras-G12D (14429), p53 (2524s) were purchased from Cell Signaling Technology (Beverly, MA). Ki67-antibody (ab1667) and TTF-1 antibody (ab76013) were purchased from Abcam (Cambridge, UK). DAB substrate kit was purchased from Vector Laboratories (SK-4100, Burlingame, CA). Mouse monoclonal anti-β-actin antibody (a1978) was purchased from Sigma (St. Louis, MO). Matrigel was purchased from Corning (Cat#356255, Corning, NY).
Cell culture
H460, H1299, and HeLa cells were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA) and were propagated according to the conditions recommended by ATCC. The identities of these two cell lines were validated by STR genotyping service at Emory University. H1299-Cas9 is derived from H1299 with the stable expression of Cas9. T2 and 634 cell lines are generous gifts from Kwok-Kin Wong. WRJ388 cell line was isolated from metastatic lung adenocarcinoma in the lymph node of a KrasG12D/Lkb1null GEMM. It is propagated in DMEM medium (Corning cat#10–013CV, Corning, NY) with EGFR 12.5ng/ml (Invitrogen, Carlsbad, CA, #13247–051) and insulin 10ug/ml (Sigma, #10516) until the majority of fibroblast were eliminated. The genotype of WRJ388 was determined by IDEXX BioAnalystics (Columbia, MO, USA).
Immunoblot analysis
The procedure for the preparation of whole-cell protein lysates and immunoblot was described previously (30). The same blots were used for probing different proteins, and actin was used as a loading control. The immunoblot analyses presented in this study were carried out at least twice, and representative images are shown in the figures.
SRB assay
2000–3000 cells were seeded in 96-well plates, treated with the indicated chemicals and durations followed by SRB assay as described previously (31). Reactions were carried out in quadruplicate, and error bars represent one standard deviation. Dose-response data under each concentration was normalized and fitted with the “inhibitor vs. response—variable slope analysis” to determine the 50% inhibitory concentration (IC50) in GraphPad Prism (version 8.4.1, GraphPad Software Inc., San Diego, CA, USA).
Apoptosis analysis
Cells were seeded in 6-well plates and subjected to indicated treatment. Both floating and attached cells were collected and analyzed by flow cytometry using the Annexin V/7-AAD Apoptosis Detection kit (BD Pharmingen, San Jose, CA) with previously described protocol (32).
Cell cycle analysis
Cell cycle analysis was carried out as previously described (18). Briefly, cells were seeded in 6-well plates and treated with the indicated chemicals for the indicated times. Cells were stained with PI/RNASE staining kit (BD Biosciences, San Jose, CA) and analyzed by FACS analysis (BD Biosciences). A total of 10,000 gated cells were acquired for each analysis. Results were analyzed using FlowJo version 7 software (FlowJo. L.L.C., Ashland, OR).
Leflunomide treatment of xenografts and PDX
All animal studies were approved and conducted according to the Emory University Institutional Animal Care and Use Committee (IACUC) guidelines. 5–6 week old female athymic nude mice (18–20g) were purchased from Harlan Laboratories. Exponentially growing HeLa cells were trypsinized, washed twice with PBS, and diluted to 3×106 cells per 50 μL PBS and 50 μL Matrigel. The cell suspension was injected subcutaneously into the right flank of mice with a 0.5 ml-syringe with a 26½-gauge needle. Mice were randomly allocated into 2 groups (vehicle control and leflunomide treatment), 9 mice per group. A rolling enrollment scheme was used, and treatment began in three different batches when tumors grew to about 91–192 mm3 (at 6, 9, and 15 days after implantation). Leflunomide was prepared in a mixture of 1.5% carboxymethyl-cellulose at a concentration of 3.5 mg/ml and was administered orally at 10 μL/g of body weight (35 mg/kg/day). The control group was administered with 1.5% carboxymethyl-cellulose. All treatments were administered once a day for 21 days. Tumor size and weight were measured every three days, and the tumor volume was calculated with the formula [volume = height × width × width)/2]. Tumors were harvested at the end of the experiment and weighed.
For H460 xenografts, mice were randomly allocated into 2 groups (vehicle control and leflunomide treatment), 10 mice per group. 35 mg/kg/day leflunomide treatment began four days later when tumors grew to about 40 to 80 mm3. All treatments were administered once a day for 23 days. Two mice in the control group were sacrificed on day 21 due to excessive tumor burden, and two paired mice in the treatment group were sacrificed at the same time for proper control.
An LKB1-null patient-derived xenograft (PDX) model (TM00213, Passage 3) was purchased from the JAX laboratory and passaged in NSG mice (JAX laboratory). This PDX was derived by surgical resection from a patient with AJCC (American Joint Committee on Cancer) stage II lung adenocarcinoma. It contains a splice-acceptor mutation in STK11 and a Kras G12S mutation. This PDX was excised and cut into small ~3×3×3 mm fragments and then implanted subcutaneously. Mice were randomly allocated into two groups (vehicle control and leflunomide treatment), 5 mice per group. A rolling enrollment scheme was used, and 35 mg/kg/day leflunomide treatment began when tumors grew to about 200 mm3 (from 6–15 days after implantation).
Leflunomide treatment of KL-GEMM
We used KrasG12DLkb1fl/flRosa-luc GEMM (KL-GEMM) generated previously in our laboratories (33), and animals of both sexes were used in this experiment. We induced lung tumors in anesthetized KL mice by intra-tracheal infection of 1×106 lentiviral-Cre-GFP virus (SL100277, SignaGen Laboratories, Rockville, MD). A cohort of 32 KL-GEMM mice was infected, and mice underwent bioluminescent imaging (BLI) at 6 weeks post-infection. Mice with BLI signals in the lung greater than 3×105 and count over 600 for two consecutive weeks were enrolled in the study with a rolling enrollment design. The mice were treated with 30 mg/kg leflunomide once a day for 44 days using the methods described above. One mouse in the control group was sacrificed on day 40 due to excessive tumor burden.
Leflunomide treatment of WRJ388 in nude mice
WRJ 388 cells were isolated from a lymph node of a female KL-GEMM. 5×106 exponentially growing WRJ-388 cells were implanted using the methods described above. Mice were randomly allocated into two groups (vehicle control and leflunomide treatment, 10 mice per group). 35 mg/kg/day leflunomide treatment began four days later when tumors grew to about 200 mm3. Leflunomide and vehicle control were prepared in the same way above, and all treatments were administered once a day for 24 days except for days 6 and 7. All organs and tumors were harvested and underwent BLI imaging at the end of the experiment.
Statistical methods for xenograft, PDX, and GEMM analysis
The SAS statistical package v9.4 (SAS Institute, Inc., Cary, North Carolina) was used for analyses with a significance level of 0.05. A mixed-effects model was implemented to estimate and compare the growth rate among two experimental groups. The correlation among the repeated measurements in each mouse overtime was accounted for accordingly. The tumor volume was log-transformed to meet the normality and equal variance assumption for the statistical model. The p-value was adjusted for multiple comparisons whenever needed. Kruskal-Wallis p-value was calculated for tumor weight comparison.
Immunohistochemistry analysis
5-μm-thick paraformaldehyde-fixed OCT-embedded mouse lung sections or formalin-fixed paraffin-embedded mouse tumor lung sections were used for IHC analyses as previously described (34). Slides were stained with antibodies against TTF-1, Ki-67 or cleaved caspase-3, and horse anti-rabbit IgG (Vector) was used as the secondary antibody. DAB substrate kit was used to develop immunohistochemistry signals. These samples were blinded and analyzed by a lung cancer pathologist (G.L. Sica).
Results
Leflunomide treatment induces apoptosis in HeLa cells and results in the regression of HeLa xenografts.
We initiated our study with HeLa cells, which we have shown previously to contain a homozygous deletion of LKB1 (5). SRB analysis indicated that the IC50 of leflunomide in vitro against HeLa cells was between 20 to 51 μM at days 3 to 6 (Figure 1A), and 50 μM leflunomide was sufficient to induced detectable caspase-3 and PARP cleavage in 24 hrs (Figure 1B). Leflunomide-induced apoptosis was verified by Annexin-V/7AAD flow cytometry (Figure 1C, 100 μM at 48 and 72 hrs, only shown for 72 hrs). This data led us to evaluate the effect of leflunomide on HeLa xenografts. When HeLa xenografts reached an average size of 137 mm3, mice were treated with vehicle control or 35 mg/kg/day leflunomide through oral gavage (N=9) for 21 days. A rolling enrollment scheme was used, and mice were enrolled in three different batches on days 6 (n=10), 9 (n=4), and 15 (n=4) after initial implantation. As shown in Figure 1D and Supplemental Table 1A, the treatment group had a significantly smaller tumor size compared to the control group on days 3, 6, 12, and 21. We also estimated the increase in log tumor volume per day as growth rate, and found that the treatment group had a negative growth rate compared to the control group (Supplemental Table 1B). There was a significant decrease in tumor weight in the group receiving leflunomide treatment (Figure 1F and Supplemental Table 1C), indicating the regression of HeLa-derived xenografts after this treatment. Leflunomide treatment significantly altered animal weight compared to the control group on day 21, but not on days 3, 6, and 12 before adjustment for p-value for multiple comparisons. After adjusting for multiple comparisons, the animal weight did not significantly differ between the two groups (Supplemental Table 1D). We also compared the animal growth rate between the two groups, and the treatment group had a slower growth rate compared to the control group, but it was not significant at a level of 0.05 (Supplemental Table 1E). Therefore, our analysis indicated that leflunomide treatment led to the regression of HeLa-derived xenografts.
Figure 1. Leflunomide induced apoptosis and tumor regression in HeLa cells.

(A). SRB analysis of leflunomide dose-response in HeLa cells from Day 3 to 6. (B) HeLa cells were treated with the indicated concentration of leflunomide for 24 hrs, and cell lysates were analyzed using indicated antibodies. (C) Annexin-V/7AAD flow cytometry analysis of HeLa cells treated with 0 or 100 μM of leflunomide for 72 hrs. (D) HeLa xenograft growth over the 21-day treatment period (X-axis). Y-axis represents Ln(Tumor Volume in mm3). (E) A rolling enrollment was used for the treatment of HeLa xenografts, and treatment was initiated on day 6 (left), 9 (middle), and 15 (right) after tumor implantation. All mice were treated with vehicle or leflunomide for 21 days. Top panels are photos of nude mice bearing subcutaneous HeLa xenografts 21 days after treatment. The bottom panel is the comparison of isolated HeLa xenografts. (F) Tumor Weight (in grams) comparison in the control and treated group with indicated Kruskal-Wallis p-value.
Leflunomide induces G1 cell cycle arrest but not apoptosis in H460 cells and is capable of attenuating the growth of H460-derived xenografts.
We next evaluated leflunomide treatment against LKB1-inactivated H460 NSCLC cells. While the IC50 of leflunomide in vitro against H460 cells was 80.5 μM at 48hrs and 27 μM at 72 hrs (Figure 2A), we only observed a slight increase in caspase-3 or PARP cleavage with 100 μM leflunomide treatment (Figure 2B). Annexin-V/7AAD flow-cytometry analysis also did not identify substantial increases in apoptotic cells with 100 μM leflunomide treatment at 48 or 72 hrs. We used AICAR as a positive control, and 1 mM of AICAR induced extensive PARP and caspase-3 cleavage in the H460 cells as previously reported (18) (Figure 2B). This result indicated that leflunomide and AICAR had different effects in H460 cells. Cell cycle analysis revealed an increase in G1-phase cells and a decrease in S and G2/M phase cells, suggesting that the dominant mechanism of growth suppression mediated by 100 μM leflunomide treatment was G1 cell cycle arrest (Figure 2C).
Figure 2. Leflunomide induced G1 cell cycle arrest and attenuated the growth of H460 xenograft.

(A) SRB analysis of leflunomide dose-response in H460 cells from Day 1 to 3. (B) H460 cells were treated with 1 mM AICAR, 0 mM, or 100 μM leflunomide for 48 hrs. Cell lysates were analyzed by immunoblot using indicated antibodies. (C) Cell cycle analysis of H460 cells treated with 0 or 100 μM leflunomide for 48 hrs. (D) H460 xenograft growth over the 23-day treatment period (X-axis). Y-axis represents Ln(Tumor Volume in mm3). (E). Tumor Weight (g) comparison in the control and treated group with indicated Kruskal-Wallis p-value. (F) Photos of isolated H460-xenografts at the end of the study. Four xenografts on the left were harvested on Day 21, and the remaining xenografts were collected on Day 23.
We also carried out leflunomide treatment analysis with the H460 xenograft model. Because the H460 xenograft had a fast xenograft growth rate (Supplemental Table 2b), we initiated leflunomide treatment four days after subcutaneous tumor implantation. The treatment group had a significantly smaller tumor size compared to the control group by day 14, 18, and 23 (Supplemental Table 2A). The treatment group also had a slower tumor growth rate compared to the control group (Supplemental Table 2B), but there was no regression of H460-derived xenograft with leflunomide treatment (Figure 2D). Two mice in the control group were sacrificed on day 21 due to excessive tumor burden, and two xenografts from the treatment group were harvested at the same time as a control. There was also a significant decrease in the final tumor weight in the group receiving leflunomide treatment (Figure 2E, Supplemental Table 2C). Comparison of leflunomide-induced animal weight loss between the two groups at different time points did not reach statistical significance, but the comparison of animal weight growth rate did reveal a statistically significant negative growth rate (Supplemental Table 2E). This data indicated that leflunomide does not induce apoptosis in all LKB1-inactivated cancer cells. Nevertheless, the treatment was still capable of attenuating the growth of H460-derived xenografts.
Leflunomide attenuated the growth of LKB1-inactivated patient-derived xenografts.
We obtained an early passage, LKB1-null patient-derived xenograft (PDX) model from the JAX laboratory. The absence of LKB1 expression was confirmed by immunoblot analysis using LKB1 mutant A549 and H460 cell lysates as negative controls and LKB1-wild-type H1299 cell lysate as positive controls (Figure 3A). Because the time required for the establishment of this PDX varied in different NSG mice host, we used a rolling enrollment experimental design and initiated 35 mg/kg/day leflunomide treatment for approximately 30 days after the tumor reached a size above 200 mm3. Leflunomide treatment significantly attenuated the growth of this PDX (Figure 3B and Supplemental Table 3A and 3B), decreasing the daily growth rate from 0.063+/−0.02 in the control group to 0.015+/−0.002 (P<0.001, Supplemental Table 3C). At the end of the treatment period, the tumor weight was significantly lower in the treatment group (0.3 g versus 1.42 g, Kruskal-Wallis p-value =0.009, Figure 3D). The animal weight did not change in the treatment group over time, but mice in the control group grew heavier over time, and there was a statistically significant difference in animal growth rate (Figure 3E and Supplemental Table 3D/3E). Therefore, leflunomide treatment is also capable of suppressing the growth of LKB1-null PDX.
Figure 3. Leflunomide treatment of LKB1-null patient-derived xenograft.
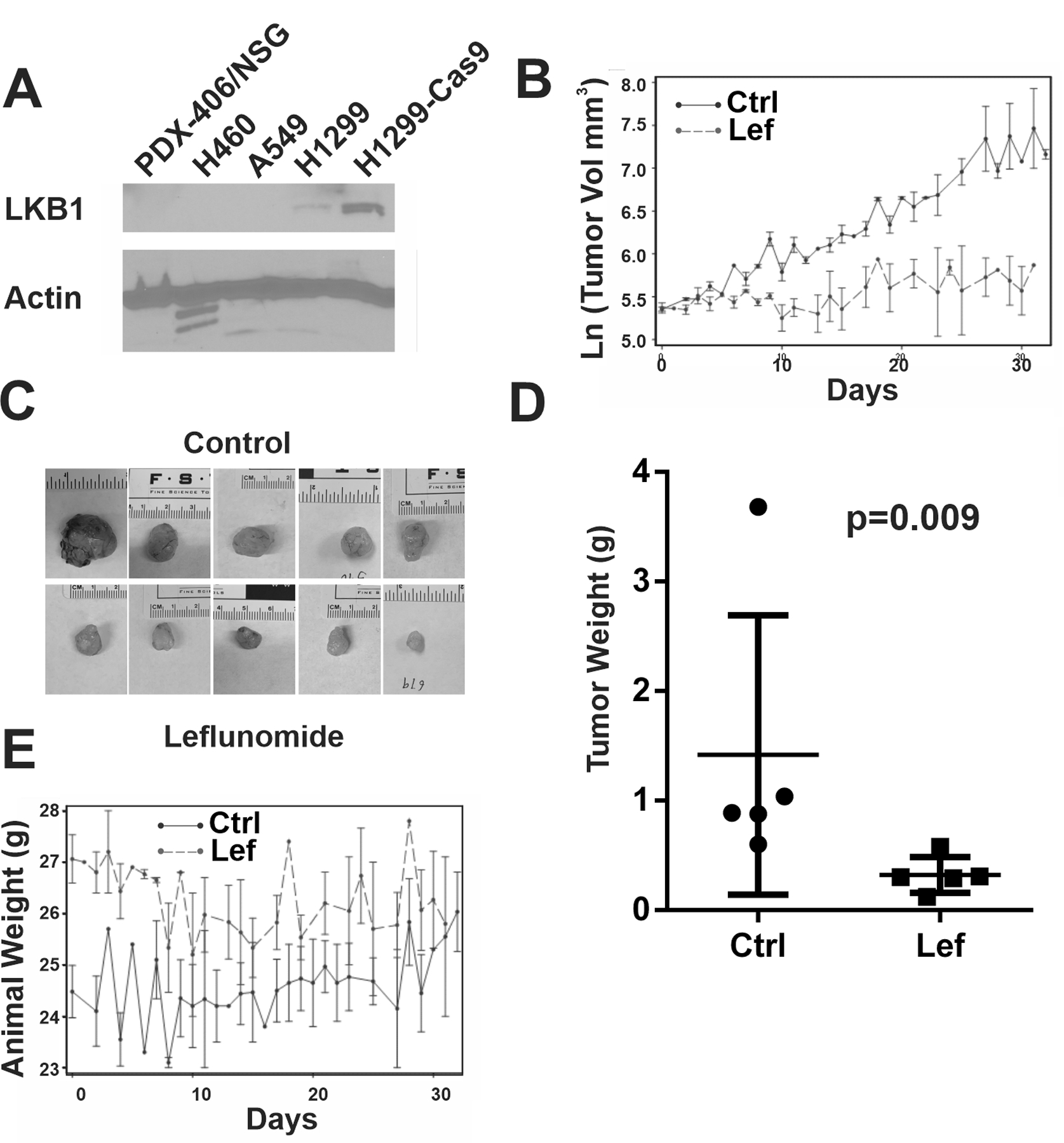
(A) Immunoblot analysis of LKB1 protein expression in indicated PDX or cell lines using actin as a loading control. (B) Estimated PDX growth over the 31-day treatment period (X-axis). Y-axis represents Ln(Tumor Volume in mm3). (C) Photos of isolated PDX at the end of the study. (D). Tumor Weight (g) comparison in the control and treated group with indicated Kruskal-Wallis p-value. (E). Animal weight (g) over the 31-day treatment period (X-axis).
Leflunomide suppresses the progression of lung adenocarcinomas in immune-competent KL-GEMM.
Leflunomide is an immune suppressor, and one reasonable concern is whether leflunomide-mediated immune suppression will promote the growth of LKB1-mutant tumors in an immunocompetent background. To address this issue, we carried out a full-scale pre-clinical trial to test the effects of leflunomide as monotherapy on tumor growth in the KrasG12DLkb1−/−Rosa-luc genetically-engineered mouse model (KL-GEMM). Thirty-two mice were infected with lentivirus via intratracheal delivery, and weekly bioluminescent imaging (BLI) was carried out to monitor tumor development in the lung. We used a rolling enrollment trial design (Figure 4A), and mice with a BLI score over 3×105 and count over 600 in the lung area for two consecutive weeks were enrolled in our study. Twenty-eight mice eventually developed lung adenocarcinoma, and 20 mice were enrolled in this study (control group n=10 and treatment group n=10). Drug toxicity was observed at 35 mg/kg/day dose of leflunomide in our GEMM as several mice lost significant weight or became visually impaired, and 30 mg/kg/day leflunomide was used in this study. The control group had equal sex distribution, and the treatment group had seven males and three females due to a bias against the generation of lung adenocarcinoma in female mice. Enrollment dates ranged from 43 to 113 days after virus infection. Mice were treated with vehicle control or 30 mg/kg/day of leflunomide through oral gavage for 44 days. The BLI score in the control group increased over 100-fold on average in the chest area. At the same time, leflunomide treatment significantly attenuated the increase in BLI signal in the same area (Figure 4B/C and Supplemental Table 4). There was no significant alteration in animal weight with this leflunomide dose (Figure 4D and Supplemental Table 4). Lung weight in control mice was higher than in the treatment group, probably due to the presence of more tumor cells in the lung (Supplemental Table 4). Lungs from the control group contained lung adenocarcinomas similar to the ones described previously (33). All of them stained positive for TTF-1, and we did not observe a significant difference in Ki-67 staining. The overall signal for cleaved caspase-3 was also low in both groups, indicating that leflunomide did not induce caspase-3 cleavage at the end-stage of this treatment. Three mice in the control group had BLI signals above background in the low body area, which may be indicative of distant metastases in these mice (Figure 4B, number 3, 4, and 6 in the control group). No BLI signal outside the chest area was detected in mice treated with leflunomide, suggesting that leflunomide may prevent the formation of distant metastasis.
Figure 4. Leflunomide inhibited the development of lung adenocarcinoma in an immune-competent GEMM.
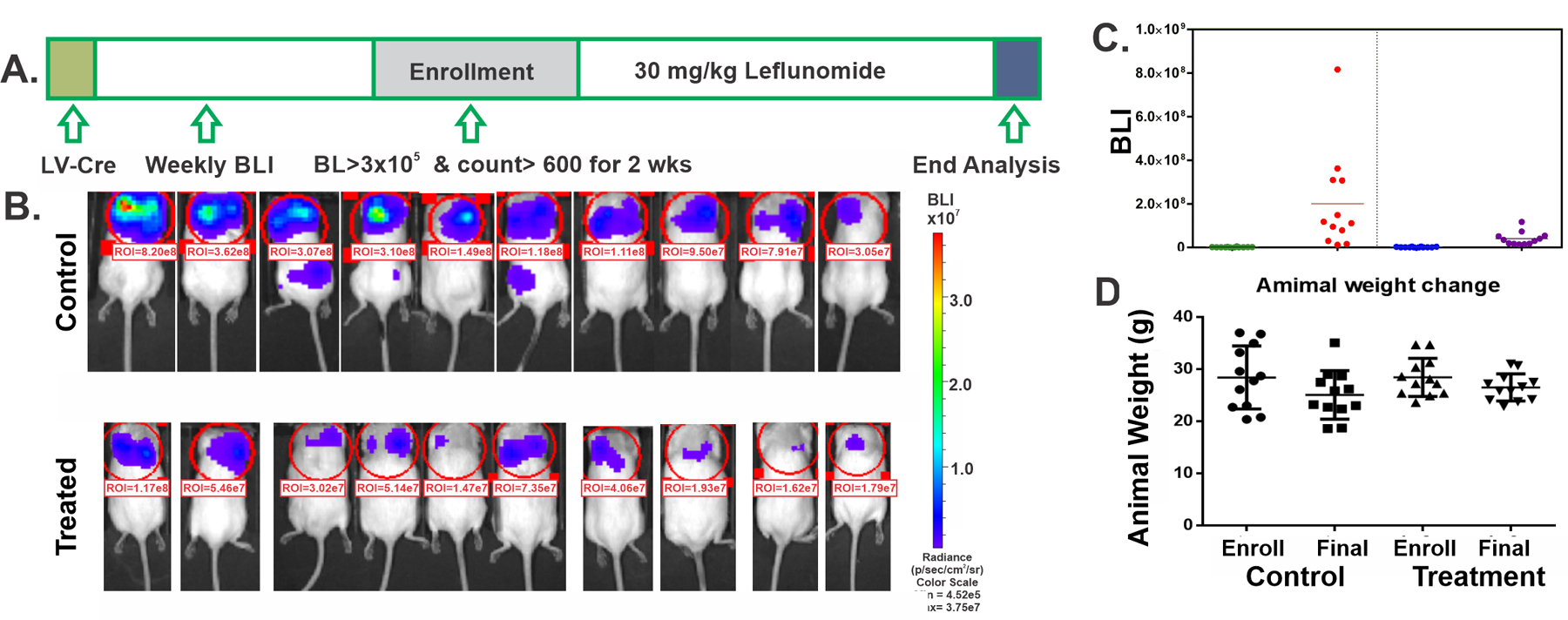
(A) Scheme of the rolling enrollment pre-clinical trial design. LV-Cre: intratracheal delivery of Cre-GFP lentivirus. (B) BLI image of the control and treated mice at the end of the treatment cycle. (C) BLI score for mice at the time of enrollment (Enroll) and the end of the treatment cycle (Final). (D) Animal weight measured at the time of enrollment and the end of the treatment cycle.
Isolation of a novel tumor-derived cancer cell line from lymph node metastasis of the KL-GEMM model
We and others have shown that the loss of LKB1 leads to the aberrant activation of FAK signaling, which significantly enhanced tumor invasion to local tissues and lymph nodes (33,35,36). However, the only distant metastasis reported for the KrasG12D/Lkb1−/− GEMM is to the axillary skeleton (36). In addition, it was not possible to isolate cancer cell lines with only Kras and LKB1 mutations, and the functional studies were done with cell lines containing Kras, LKB1, and TP53 triple mutation (37). We isolated a novel tumor-derived cancer cell line, WRJ388, from a lymph node metastasis of a female KL mouse. The detailed isolation methods will be published elsewhere. Species-specific PCR analysis indicated this cell line was of mouse origin, and a 27-Marker STR strain analysis revealed its profile is novel and consistent with cells derived from the FVB/N mouse strain. Immunoblot analysis indicated that this cell line contained Kras-G12D mutant and full-length p53 protein but completely lacked LKB1 expression (Figure 5A). We also carried out RNAseq analysis in this cell, which verified the expression transcripts containing Kras-G12D mutation, deletion of exons 3–6 of LKB1/STK11, and the lack of mutations in TP53. Therefore, WRJ388 contains Kras and LKB1 mutation but wild-type p53.
Figure 5. Molecular characterization of WRJ388 cells.

(A) Immunoblot analysis of WRJ388, T2 (KrasG12DLkb1−/−p53−/−), and 634 (KrasG12Dp53−/−) cell lysate with indicated antibodies. (B) SRB analysis of leflunomide dose-response in WRJ388 cells at Days 1–4. (C) Uridine rescue of WRJ388 cells treated with 50 μM leflunomide at 72 hrs. (D) Cell cycle analysis of WRJ388 cells treated with 50 μM leflunomide, 2.5 mM uridine, or their combinations at 72 hrs. (E) Annexin-V/7AAD flow cytometry analysis of WRJ388 cells treated with 0, 50, or 200 μM of leflunomide for 72 hrs.
We and others have shown previously that LKB1-inactivated cells are sensitive to the disruption of pyrimidine metabolism (18), and WRJ388 was sensitive to leflunomide treatment with an IC50 of 36 μM on Day 2 and 15 μM on Day 4 (Figure 5B). Therefore, we used 50 μM leflunomide for the remaining analyses for WRJ388 in vitro. Leflunomide-mediated growth suppression was mostly rescued by uridine, which is consistent with the notion that leflunomide inhibits UMP synthesis (Figure 5C). Cell cycle analysis indicated that leflunomide treatment led to a significant decrease in G1 cells and an increase in S-phase cells at 72 hrs, both of which were restored by 2.5 mM uridine rescue (Figure 5D). We also used Annexin V/7-AAD flow cytometry to assess apoptosis. Whereas extensive apoptotic cells were observed with 200 μM leflunomide treatment, we only observed a mild increase in apoptotic cells with 50 μM leflunomide treatment (Figure 5E). In addition, we failed to observe extensive caspase-3 cleavage or PARP-cleavage, which is consistent with the lack of cleaved caspase-3 staining in our GEMM analysis.
Leflunomide inhibits the metastasis of LKB1-inactivated tumors in a novel lung cancer metastasis model.
The subcutaneous implantation of WRJ388 cells in nude mice resulted in a fast-growing tumor (Figure 6A left panel, 6B, and Supplemental Table 5A), and multiple micro-micrometastases were observed in the lung 22 days after the initial tumor implantation (Figure 6A, right panel, and Supplemental Figure 1). The animal weight growth in the control group was not linear, and there was a dramatic decline after day 17 (Figure 6C, and Supplemental Table 5A). Therefore, we used this cell line model to determine whether leflunomide can also be used to prevent the formation of lung metastasis. We initiated daily oral gavage of 35 mg/kg of leflunomide four days after the subcutaneous implantation of WRJ388 cells when the average tumor volume reached 200 mm3. Ten mice were used for each group, and treatment lasted 24 days except for days 6 and 7 when leflunomide was not administered. We excluded one mouse in the treatment group because cells were implanted into the muscle, which prevented the accurate measurement of tumor volume. 35 mg/kg of leflunomide was not toxic to nude mice because animal weight remained stable in the treatment group (Figure 6C and Supplemental Table 5D). Leflunomide treatment significantly attenuated tumor growth for 21 days (Figure 6B), and we observed a significant decrease in tumor size (Figure 6D and Supplemental Table 5B) and final tumor weight (Figure 6E and Supplemental Table 5C) in the treatment group at the end of the study. Tumor growth resumed in the treated mice from day 22–24. However, WRJ388 cells isolated from these tumors were still sensitive to leflunomide treatment in vitro (Supplemental Figure 2). Therefore, this resumed tumor growth is not due to intrinsic changes in WRJ388 cells after leflunomide treatment. We used two methods to assess the micrometastasis of WRJ388 to the lung. First, we measured the BLI signal from the lung and observed a significant decrease in lung BLI signals after leflunomide treatment (Figure 6F and Supplemental Table 5F). We also confirmed this finding by histological analysis of the lung (Supplemental Figure 1). The sample with the highest lung BLI signal in the control group was lost due to poor tissue preservation, but the remaining nine samples all had lung metastasis, and 89% of them were multi-focal (Figure 6G). In contrast, 78% of the lung in the treated group did not have micro-metastasis of WRJ388 cells. Of the two mice that had lung metastases, one of them only had a single focus. This data indicated that leflunomide was also capable of suppressing the metastasis of WRJ388 cells in this mouse model.
Figure 6. Leflunomide treatment attenuated lung metastasis in a WRJ388-based subcutaneous model.

(A) Hematoxylin and eosin (H&E) and TTF1 immunohistochemically stained lung tissue sections from a mouse with subcutaneously implanted WRJ388. (B) Tumor volume analysis with (red) or without (blue) 35mg/kg/day leflunomide treatment. (C) Animal weight analysis during the same period. (D) Photos of isolated subcutaneous WRJ388 tumors on Day 24. (E) Tumor weight comparison in both groups. (F) BLI signal comparison in the lung in control and leflunomide-treated groups. (G) Type of metastasis in the lung by pathological analysis.
Discussion
AICAR is capable of inducing apoptosis in most LKB1-inactivated cell lines (16–18), but the short half-life of this compound limits its clinical utility (20). We previously demonstrated that AICAR-induced apoptosis could be rescued by uridine (18), which is consistent with the notion that LKB1-inactivated cells are sensitive to the disruption of pyrimidine metabolism (19). Here, we chose to target the de novo synthesis of UMP by leflunomide, which is an FDA-approved agent for the treatment of rheumatoid arthritis with well-characterized human clinical pharmacokinetics (38). This agent has a 14-day half-life and 80% bioavailability through oral gavage. Unlike most chemotherapeutic agents that work in the nanomolar range, this agent works in the hundred-micromolar range. The standard daily dosage of 20 mg leflunomide in humans leads to 125–230 μM A77 1726 average steady-state plasma concentration (39,40). Therefore, the concentration of leflunomide used in the in vitro study here is clinically relevant. The pharmacokinetics of leflunomide in animal models are also established (41), and 35 mg/kg/day oral gavage is commonly used in mouse studies (42–44), but we did not determine the corresponding A77 1726 plasma concentration. In the pharmacology review prepared by Hoeschst Marion Roussel Inc for leflunomide (41), at 15 mg/kg oral dose, the average A77 1726 serum levels in mice 24 hours post-dose is 102 μg/ml (377 μM) in males and 71.7 μg/ml (265 μM) in females. They also concluded that “an increase in the dose over 15 mg/kg would have not increased the levels of A 771726 further” based on the exposure data. Therefore, the 30 or 35 mg/kg dose of leflunomide used in this work does result in a higher A77 1726 plasma concentration than the steady-state human plasma concentration.
While leflunomide induced extensive apoptosis in HeLa cells and led to the regression of HeLa xenograft, it did not significantly promote apoptosis in H460, WRJ388, or KrasG12D/LKB1−/− lung adenocarcinoma in our GEMM. Therefore, AICAR and leflunomide had different effects on some LKB1-inactivated cancer cells. AICAR-induced apoptosis in HeLa cells is unlikely to be p53-dependent because HeLa cells express HPV18 E6 protein that leads to p53 degradation. H460 and WRJ388 cells both contain wild-type p53, but the presence of p53 was not sufficient to induce apoptosis in the presence of AICAR. Therefore, p53 is unlikely to play a role in AICAR-induced apoptosis; thus, additional mechanistic studies will be needed to elucidate the difference between AICAR and leflunomide treatment in these tumors. Given the concentration of leflunomide used in this study, multiple targets may be inhibited in our models. We carried out in vitro uridine rescue experiments for WRJ388 (Figure 5C), H460, and HeLa cells, and uridine only reversed approximately 80% of leflunomide-mediated growth suppression, suggesting that other signaling pathways may also be targeted. Nevertheless, the most important finding of our work is that leflunomide treatment significantly attenuates the growth of LKB1-inactivated tumors even in the absence of substantial apoptosis. The ability of leflunomide to suppress the growth of LKB1-inactivated tumors through different mechanisms indicates that this agent may be able to suppress a diverse tumor cell population.
Leflunomide is currently approved for the treatment of autoimmune diseases, such as rheumatoid arthritis or psoriatic arthritis, and it is capable of reducing cycling CD4 T cells and CD8 T cells in peripheral blood (22). Consequently, a reasonable concern with this agent is whether the inhibition of the immune system promotes the growth of KL-mutant lung adenocarcinoma. Our pre-clinical analysis in the immune-competent GEMM refuted that hypothesis, as leflunomide treatment significantly attenuated the growth of adenocarcinoma in the lung. There are two unexpected findings in our GEMM study. The toxicity of leflunomide was well-established in the mouse model (41), and 35 mg/kg of leflunomide was commonly used with xenograft studies (45,46). This dose, however, had a significant toxic effect in the KL-GEMM, as two mice became blind one week after treatment began, and we eventually reduced the dose to 30 mg/kg to avoid the toxicity. The molecular basis of this strain-specific toxicity is not known, but leflunomide was capable of controlling the growth of LKB1-inactivated lung adenocarcinoma at this reduced dose. Another unexpected finding of our GEMM study was female GEMM had reduced frequency of tumor formation in the lung. STK11/LKB1 was found to be more frequently mutated in males than in females in the TCGA dataset (47). The biological basis of the bias in our model is not known, but it will be interesting to determine in the future whether this sex-bias can be studied in the future using our GEMM model.
Most lung cancer patients die of cancer metastasis, and the prevention of micrometastasis after local treatment in early-stage lung cancer patients will significantly improve disease prognosis. In our GEMM study, three mice had distant metastasis in the control group, but none of the mice had such metastasis in the leflunomide treated group. To quantitatively evaluate this phenomenon, we generated a novel subcutaneous tumor model, which formed lung metastasis in 100% of the animals within a month. Leflunomide treatment prevented the formation of lung metastasis in 78% of these animals, indicating that it is a potent agent in preventing the formation of lung metastasis. In combination, our study strongly supports the notion that leflunomide should be considered as a therapeutic option for cancer patients with LKB1-inactivated tumors. The next logical step in this research will be to determine whether leflunomide can be used in combination with existing therapies to promote the elimination of LKB1-inactivated tumors.
Supplementary Material
Acknowledgments
This work was supported in part by the NCI at the NIH [R01 CA203928 (W. Zhou), R01-CA194027 (A.I. Marcus, M. Gilbert-Ross, and W. Zhou), P30CA138292 to Winship Cancer Institute, by Anise McDaniel Brock Scholar fund (W. Zhou) and Winship Invest$ team award (W. Zhou and M. Gilbert-Ross) at the Winship Cancer Institute, and by China Scholarship Council to B. Liu. We would also like to acknowledge Anthea Hammond for editing this manuscript and the technical support from the Shared Resource at Winship for Biostatistics, Bioinformatics and Systems Biology, Cancer Animal Models, Cancer Tissue and Pathology, Integrated Cellular Imaging, and Pediatrics/Winship Flow Cytometry.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare that there is no conflict of interest.
References:
- 1.Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998;391(6663):184–7. [DOI] [PubMed] [Google Scholar]
- 2.Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 1998;18(1):38–43. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res 2002;62(13):3659–62. [PubMed] [Google Scholar]
- 4.Wingo SN, Gallardo TD, Akbay EA, Liang MC, Contreras CM, Boren T, et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS ONE 2009;4(4):e5137 doi 10.1371/journal.pone.0005137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCabe MT, Powell DR, Zhou W, Vertino PM. Homozygous deletion of the STK11/LKB1 locus and the generation of novel fusion transcripts in cervical cancer cells. Cancer Genet Cytogenet 2010;197(2):130–41 doi 10.1016/j.cancergencyto.2009.11.017, S0165–4608(09)00669–4 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guldberg P, thor Straten P, Ahrenkiel V, Seremet T, Kirkin AF, Zeuthen J. Somatic mutation of the Peutz-Jeghers syndrome gene, LKB1/STK11, in malignant melanoma. Oncogene 1999;18(9):1777–80. [DOI] [PubMed] [Google Scholar]
- 7.Rowan A, Bataille V, MacKie R, Healy E, Bicknell D, Bodmer W, et al. Somatic mutations in the Peutz-Jeghers (LKB1/STKII) gene in sporadic malignant melanomas. J Invest Dermatol 1999;112(4):509–11. [DOI] [PubMed] [Google Scholar]
- 8.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 2007;450(7171):893–8 doi 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008;455(7216):1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koivunen JP, Kim J, Lee J, Rogers AM, Park JO, Zhao X, et al. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br J Cancer 2008;99(2):245–52 doi 10.1038/sj.bjc.6604469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015;5(8):860–77 doi 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schabath MB, Welsh EA, Fulp WJ, Chen L, Teer JK, Thompson ZJ, et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2016;35(24):3209–16 doi 10.1038/onc.2015.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov 2018;8(7):822–35 doi 10.1158/2159-8290.CD-18-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer 2019;19(9):495–509 doi 10.1038/s41568-019-0179-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou W, Zhang J, Marcus AI. LKB1 Tumor Suppressor: Therapeutic Opportunities Knock when LKB1 Is Inactivated. Genes & Diseases 2014;1(1):64–74 doi 10.1016/j.gendis.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 2004;101(10):3329–35 doi 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nafz J, De-Castro Arce J, Fleig V, Patzelt A, Mazurek S, Rösl F. Interference with energy metabolism by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside induces HPV suppression in cervical carcinoma cells and apoptosis in the absence of LKB1. Biochem J 2007;403(3):501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu F, Jin R, Liu X, Huang H, Wilkinson SC, Zhong D, et al. LKB1 promotes cell survival by modulating TIF-IA-mediated pre-ribosomal RNA synthesis under uridine downregulated conditions. Oncotarget 2016;7(3):2519–31 doi 10.18632/oncotarget.6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 2017;546(7656):168–72 doi 10.1038/nature22359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosselaar M, Smits P, van Loon LJ, Tack CJ. Intravenous AICAR during hyperinsulinemia induces systemic hemodynamic changes but has no local metabolic effect. J Clin Pharmacol 2011;51(10):1449–58 doi 10.1177/0091270010382912. [DOI] [PubMed] [Google Scholar]
- 21.Jin R, Zhou W. TIF-IA: An oncogenic target of pre-ribosomal RNA synthesis. Biochim Biophys Acta 2016;1866(2):189–96 doi 10.1016/j.bbcan.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Read SW, DeGrezia M, Ciccone EJ, DerSimonian R, Higgins J, Adelsberger JW, et al. The effect of leflunomide on cycling and activation of T-cells in HIV-1-infected participants. PLoS One 2010;5(8):e11937 doi 10.1371/journal.pone.0011937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters GJ. Re-evaluation of Brequinar sodium, a dihydroorotate dehydrogenase inhibitor. Nucleosides Nucleotides Nucleic Acids 2018;37(12):666–78 doi 10.1080/15257770.2018.1508692. [DOI] [PubMed] [Google Scholar]
- 24.Prakash A, Jarvis B. Leflunomide: a review of its use in active rheumatoid arthritis. Drugs 1999;58(6):1137–64 doi 10.2165/00003495-199958060-00010. [DOI] [PubMed] [Google Scholar]
- 25.Liu S, Neidhardt EA, Grossman TH, Ocain T, Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 2000;8(1):25–33. [DOI] [PubMed] [Google Scholar]
- 26.Mattar T, Kochhar K, Bartlett R, Bremer EG, Finnegan A. Inhibition of the epidermal growth factor receptor tyrosine kinase activity by leflunomide. FEBS Lett 1993;334(2):161–4 doi 10.1016/0014-5793(93)81704-4. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh S, Narla RK, Zheng Y, Liu XP, Jun X, Mao C, et al. Structure-based design of potent inhibitors of EGF-receptor tyrosine kinase as anti-cancer agents. Anticancer Drug Des 1999;14(5):403–10. [PubMed] [Google Scholar]
- 28.Brown KK, Spinelli JB, Asara JM, Toker A. Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer. Cancer Discov 2017;7(4):391–9 doi 10.1158/2159-8290.CD-16-0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathur D, Stratikopoulos E, Ozturk S, Steinbach N, Pegno S, Schoenfeld S, et al. PTEN Regulates Glutamine Flux to Pyrimidine Synthesis and Sensitivity to Dihydroorotate Dehydrogenase Inhibition. Cancer Discov 2017;7(4):380–90 doi 10.1158/2159-8290.CD-16-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong D, Guo L, de Aguirre I, Liu X, Lamb N, Sun SY, et al. LKB1 mutation in large cell carcinoma of the lung. Lung Cancer 2006;53(3):285–94. [DOI] [PubMed] [Google Scholar]
- 31.Mao K, Liu F, Liu X, Khuri FR, Marcus AI, Li M, et al. Re-expression of LKB1 in LKB1-mutant EKVX cells leads to resistance to paclitaxel through the up-regulation of MDR1 expression. Lung Cancer 2015;88(2):131–8 doi 10.1016/j.lungcan.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong D, Xiong L, Liu T, Liu X, Liu X, Chen J, et al. The glycolytic inhibitor 2-deoxyglucose activates multiple prosurvival pathways through IGF1R. J Biol Chem 2009;284(35):23225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilbert-Ross M, Konen J, Koo J, Shupe J, Robinson BS, Wiles WGt, et al. Targeting adhesion signaling in KRAS, LKB1 mutant lung adenocarcinoma. JCI Insight 2017;2(5):e90487 doi 10.1172/jci.insight.90487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong D, Morikawa A, Guo L, Colpaert C, Xiong L, Nassar A, et al. Homozygous deletion of SMAD4 in breast cancer cell lines and invasive ductal carcinomas. Cancer Biol Ther 2006;5(6):601–7. [DOI] [PubMed] [Google Scholar]
- 35.Kline ER, Shupe J, Gilbert MM, Zhou W, Marcus AI. LKB1 represses focal adhesion kinase (FAK) signaling via a FAK-LKB1 complex to regulate FAK site maturation and directional persistence. J Biol Chem 2013. 10.1074/jbc.M112.444620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carretero J, Shimamura T, Rikova K, Jackson AL, Wilkerson MD, Borgman CL, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell 2010;17(6):547–59 doi 10.1016/j.ccr.2010.04.026, S1535–6108(10)00199–6 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007;448(7155):807–10. [DOI] [PubMed] [Google Scholar]
- 38.Rozman B. Clinical pharmacokinetics of leflunomide. Clin Pharmacokinet 2002;41(6):421–30 doi 10.2165/00003088-200241060-00003. [DOI] [PubMed] [Google Scholar]
- 39.Grabar PB, Rozman B, Logar D, Praprotnik S, Dolzan V. Dihydroorotate dehydrogenase polymorphism influences the toxicity of leflunomide treatment in patients with rheumatoid arthritis. Ann Rheum Dis 2009;68(8):1367–8 doi 10.1136/ard.2008.099093. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt A, Schwind B, Gillich M, Brune K, Hinz B. Simultaneous determination of leflunomide and its active metabolite, A77 1726, in human plasma by high-performance liquid chromatography. Biomed Chromatogr 2003;17(4):276–81 doi 10.1002/bmc.244. [DOI] [PubMed] [Google Scholar]
- 41.Hoeschst-Marion-Roussel-Inc. Pharmacology Review of Leflunomide (Arava) Application No. NDA:20-905. Volume Application No. NDA:20-9051998. p 1–46. [Google Scholar]
- 42.Glant TT, Mikecz K, Bartlett RR, Deak F, Thonar EJ, Williams JM, et al. Immunomodulation of proteoglycan-induced progressive polyarthritis by leflunomide. Immunopharmacology 1992;23(2):105–16 doi 10.1016/0162-3109(92)90034-a. [DOI] [PubMed] [Google Scholar]
- 43.Stosic-Grujicic S, Dimitrijevic M, Bartlett R. Leflunomide protects mice from multiple low dose streptozotocin (MLD-SZ)-induced insulitis and diabetes. Clin Exp Immunol 1999;117(1):44–50 doi 10.1046/j.1365-2249.1999.00900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, Sun J, Doscas ME, Ye J, Williamson AJ, Li Y, et al. Control of hyperglycemia in male mice by leflunomide: mechanisms of action. J Endocrinol 2018;237(1):43–58 doi 10.1530/JOE-17-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jarman ER, Kuba A, Montermann E, Bartlett RR, Reske-Kunz AB. Inhibition of murine IgE and immediate cutaneous hypersensitivity responses to ovalbumin by the immunomodulatory agent leflunomide. Clin Exp Immunol 1999;115(2):221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu S, Yan X, Xiang Z, Ding HF, Cui H. Leflunomide reduces proliferation and induces apoptosis in neuroblastoma cells in vitro and in vivo. PLoS One 2013;8(8):e71555 doi 10.1371/journal.pone.0071555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan Y, Liu L, Chen H, Wang Y, Xu Y, Mao H, et al. Comprehensive Characterization of Molecular Differences in Cancer between Male and Female Patients. Cancer Cell 2016;29(5):711–22 doi 10.1016/j.ccell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.