

Abstract
TGF-β is a pleiotropic cytokine with immunosuppressive activity. In pre-clinical models blockade of TGF-β enhances the activity of radiation and invokes T cell anti-tumor immunity. Here, we combined galunisertib, an oral TGF-β inhibitor, with stereotactic body radiotherapy (SBRT) in patients with advanced hepatocellular carcinoma (HCC) and assessed safety, efficacy and immunological correlatives. Patients (n=15) with advanced HCC who progressed on, were intolerant of, or refused sorafenib were treated with galunisertib (150mg PO BID) on days 1–14 of each 28-day cycle. A single dose of SBRT (18-Gy) was delivered between days 15–28 of Cycle 1. Site of index lesions treated with SBRT included liver (9), lymph node (4) and lung (2). Blood for high-dimensional single cell profiling was collected. The most common treatment-related adverse events (AEs) were fatigue (53%), abdominal pain (46.6%), nausea (40%) and increased alkaline phosphatase (40%). There were two instances of grade 2 alkaline phosphatase increase and two instances of grade 2 bilirubin increase. One patient developed grade 3 achalasia possibly-related to treatment. Two patients achieved a partial response. Treatment with galunisertib was associated with a decrease in the frequency of activated T regulatory cells in the blood. Distinct peripheral blood leukocyte populations detected at baseline distinguished progressors from non-progressors. Non-progressors also had increased CD8+PD-1+TIGIT+ T cells in the blood after treatment. We found galunisertib combined with SBRT to be well-tolerated and associated with anti-tumor activity in patients with HCC. Pre- and post-treatment immune profiling of the blood was able to distinguish patients with progression versus non-progression.
Keywords: TGF-β, HCC, liver cancer, hepatocellular carcinoma, radiation, SBRT, T cells, immunotherapy
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common cause of primary liver cancer worldwide (1, 2). Recently, immune checkpoint inhibitors targeting the programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) pathways have demonstrated a role for immunotherapy in HCC (3, 4). However, response rates remain low and only a minority of patients have responses that are durable (5). This observation supports the existence of additional immune escape mechanisms. Specifically, immunosuppression within tumors and restricted infiltration by effector T cells in late stage disease are key determinants that may impair the efficacy of immunotherapy in HCC (6).
Transforming growth factor beta (TGF-β) is a pleiotropic cytokine with immunosuppressive properties and a central role in HCC pathogenesis. TGF-β promotes tumor growth, migration and metastasis (7). TGF-β also coordinates immunosuppression by enhancing T regulatory cell (Treg) generation and survival, inhibiting T cell differentiation and proliferation, and suppressing antigen-presenting cell functions (6, 8–10). In animal models, neutralizing TGF-β antibodies combine with radiation to trigger T cell immunity against both irradiated and non-irradiated lesions (11). Similarly, in patients with metastatic cancer, treatment with a TGF-β blocking antibody in combination with hypo-fractionated radiation associates with increases in central memory CD8+ T cells detected in the blood (12). These findings suggest a role for TGF-β blockade in overcoming resistance to radiotherapy and as a strategy to augment anti-tumor T cell immunity.
Galunisertib (LY2157299) is an oral small molecule inhibitor of TGF-β receptor I (13, 14). Based on clinical and preclinical data, we conducted a pilot study using galunisertib in combination with stereotactic body radiotherapy (SBRT) for the treatment of patients with advanced HCC. The primary objective was to define safety and tolerability of galunisertib when given in combination with SBRT. Secondary objectives were to evaluate clinical activity and immunological correlatives.
MATERIALS AND METHODS
Patients and study design
This was a single institution pilot study. Eligible patients had inoperable HCC and had progressed on, been intolerant of, or refused sorafenib, which was the only US Food and Drug Association (FDA) approved systemic therapy for advanced HCC at the time of study initiation. Additional eligibility criteria are detailed in the Supplementary Materials. The protocol was approved by the institutional review board of the University of Pennsylvania. Written informed consent was obtained from all patients prior to study-related procedures in accordance with federal and institutional guidelines.
The primary objective was assessment of safety of combining galunisertib with SBRT in patients with advanced HCC. Clinical secondary objectives included progression free survival (PFS), overall response rate (ORR) and overall survival (OS). Scientific secondary endpoints included evaluation of peripheral immune subsets at baseline and after treatment.
Patients were treated with galunisertib at a dose of 150mg PO BID on days 1–14 of each 28-day cycle (Fig. 1A). SBRT was delivered in one fraction of 18-Gy between days 15–28 of the first treatment cycle. Lesions measuring between 1–10 cm were chosen at the discretion of the treating radiation oncologist for treatment with SBRT. The index lesion could not have been previously treated with external beam radiation therapy or transarterial radioembolization and was required to have evidence of viable tumor by MRI. Galunisertib was supplied by Lilly Oncology (Indianapolis, IN). The chemical structure of galunisertib has been previously reported (15). Clinical evaluation, and safety assessment are described in the Supplementary Materials. The DLT evaluable period was defined as start of study treatment to day 28. The toxicity evaluable population were all patients who received at least one dose of study treatment.
Figure 1. Trial design and clinical outcomes.
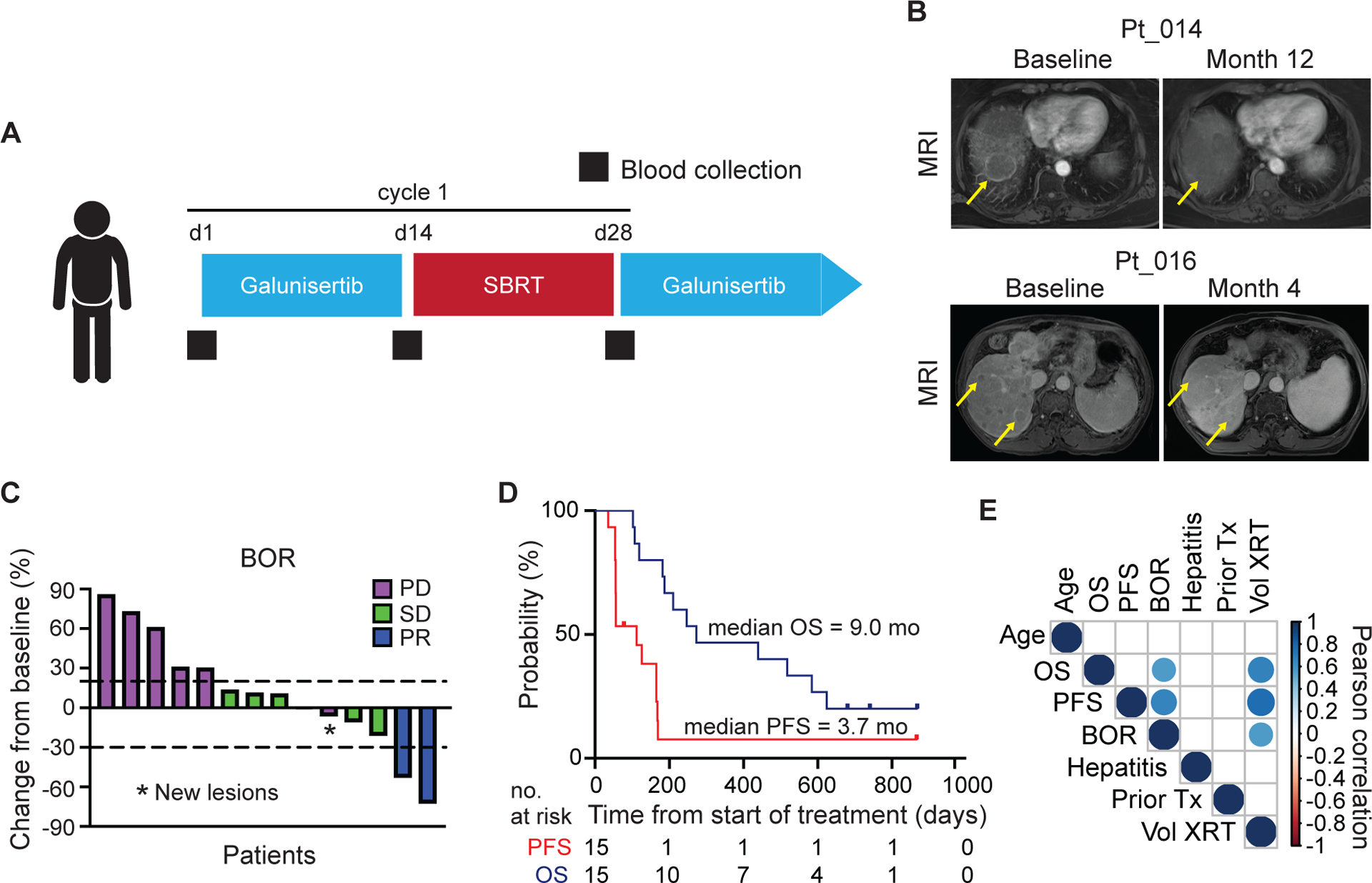
(A) Study schema. Patients received galunisertib 150 mg PO twice daily for days 1–14 of each 28-day cycle. Radiation (SBRT 18-Gy in one fraction) was delivered during cycle one only between days 15–28. Blood for isolation of PBMCs was collected at baseline, prior to SBRT and prior to start of cycle 2 (black squares). (B) Sequential contrast enhanced MRI for patients 014 and 016. Yellow arrows mark representative non-irradiated lesions. (C) Best overall response showing percent change in target lesions from baseline measured by RECIST 1.1. One patient withdrew consent and was not evaluable for radiologic response. One patient had non-target lesion progression (*). (D) Kaplan Meier plots showing progression free survival and overall survival. (E) Correlation matrix of clinical variables. Colored circles represent Pearson’s correlations with a significance of p < 0.05. BOR, best overall response; OS, overall survival; PFS, progression free survival.
Mass Cytometry
Blood was collected at baseline, on the day of SBRT administration between days 15 – 28, and on day 1 of Cycle 2. Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll centrifugation and cryopreserved in liquid nitrogen. For analysis, PBMCs were thawed and washed with FACS buffer (metal free PBS + 2% FBS + 1 mM EDTA). 2×106 or fewer cells per sample were incubated at room temperature with 1 μM 198Pt monoisotopic cisplatin (Fluidigm) for one minute and then washed twice with FACS buffer. Cells were incubated in Cytofix fixation buffer (BD) for 25 minutes on ice. Each sample was barcoded using unique palladium metal barcodes as per the manufacturer’s protocol (Fluidigm). After barcoding, samples were combined and blocked with Human TruStain FcX (Biolegend) for ten minutes at room temperature. Subsequently, cells were stained with a master mix of metal-tagged antibodies for 30 minutes at room temperature (antibody information is available in Supplementary Table 1). Cells were washed and fixed a second time with 2.4% formaldehyde in PBS containing 125 nM Iridium DNA intercalator (Fluidigm). Data was acquired on a Helios mass cytometer (Fluidigm) at a speed below 600 events/sec.
Mass cytometry data were normalized and sample barcodes were resolved using Fluidigm’s CyTOF Software (ver. 6.7.1014). Individual samples were manually gated in FlowJo (BD) to exclude normalization beads, debris, dead cells and doublets to identify expression markers of interest (e.g., CD45, CD3, CD4, CD8, CD25, Foxp3, CD19, CD14, HLA-DR, CCR7, CD45RO, PD-1, TIGIT, CTLA-4, Ki67). CD45+ cisplatin negative events and CD8+ cisplatin negative events were exported for downstream processing. Data were transformed using cytofAsinh and downsampled to 5000 events per sample. For dimensionality reduction, t-Distributed Stochastic Neighbor Embedding (tSNE) with perplexity of 30 and Max iterations of 1000 and Phenograph clustering analysis with K of 30 were performed in R using the cytofkit package. All markers not used for manual gating were included in the dimensionality reduction analysis. Marker expression level plots and density plots were generated using ggplot2 and color pallets from the viridis package.
Statistical Analysis
Statistical analyses were performed using Prism 8.0 software (GraphPad) and R. PFS and OS were estimated by the Kaplan-Meier method. Unless otherwise indicated, mass cytometry analysis were conducted first by unsupervised analysis with correction for multiple comparisons and findings were validated by manual gating. Student’s t-test using the two-stage linear step up procedure of Benjamini, Krieger and Yekutieli with q = 5% and ANOVA with Tukey’s correction were used to compare the frequencies of unsupervised clusters generated from Phenograph. Mann-Whitney and Wilcoxon tests were used to compare manually gated immune cell populations.
RESULTS
Patients Characteristics and Treatment
Fifteen patients with advanced HCC were enrolled and treated between May 2, 2017 and May 23, 2018. The median age of patients was 65 (range: 48–77). Patients were predominantly male (80%) and most were white (73.3%). Nine patients (60%) had extrahepatic spread of disease and five patients (33%) had macroscopic vascular invasion. Two patients (13%) had both extrahepatic disease and macroscopic vascular invasion and three patients (20%) had no extrahepatic disease or macroscopic vascular invasion. The Barcelona Clinic liver cancer stage was B in three patients (20%) and C in twelve patients (80%). Two patients had active hepatitis C and one patient had active hepatitis B. Six patients (40%) had received prior sorafenib and two of these patients had received additional systemic therapy. Refusal of sorafenib was the primary reason for not receiving systemic therapy prior to study enrollment. Baseline characteristics are summarized in Table 1.
Table 1.
Demographic and baseline characteristics
| Characteristic | Value |
|---|---|
| Age at diagnosis - yr | |
| Median | 65 |
| Range | 48 – 77 |
| Sex - no. (%) | |
| Male | 12 (80) |
| Female | 3 (20) |
| Race or ethnic group - no. (%)a | |
| White | 11 (73) |
| Black | 3 (20) |
| Other | 1 (7) |
| ECOG - no. (%) | |
| 0 | 10 (66) |
| 1 | 5 (33) |
| Extrahepatic spread - no. (%) | |
| Present | 9 (60) |
| Absent | 6 (40) |
| Macroscopic vascular invasion - no (%) | |
| Present | 5 (33) |
| Absent | 10 (66) |
| Childs Pugh Score - no. (%) | |
| A | 15 (100) |
| B7 | 0 (0) |
| Barcelona Clinic liver cancer stage - no. (%) | |
| B | 3 (20) |
| C | 12 (80) |
| Prior systemic therapy - no. (%) | |
| 0 | 9 (60) |
| 1–2 | 4 (27) |
| >2 | 2 (13) |
| Prior liver directed therapy - no. (%) | |
| Yes | 13 (87) |
| No | 2 (13) |
| History of infectious hepatitis - no. (%) | |
| Hepatitis C | 7 (47) |
| Hepatitis B | 2 (13) |
| No history of infectious hepatitis | 6 (40) |
| Biochemical analysis | |
| Albumin - g/dL | |
| Median | 3.8 |
| Range | 2.9 – 4.4 |
| Total bilirubin - mg/dL | |
| Median | 0.6 |
| Range | 0.3 – 1.6 |
| Alpha-fetoprotein - ng/mL | |
| Median | 26.6 |
| Range | 3.6 – 13×104 |
Race was reported by the investigator
Safety results
Fifteen patients were evaluable for toxicity (Table 2). The most common treatment-related AEs included fatigue (53%), abdominal pain (46.6%), nausea (40%), increased alkaline phosphatase (40%) and constipation (26.6%). In regard to changes in liver chemistries, two instances of grade 2 and four instances of grade 1 alkaline phosphatase increase were observed. Grade 2 bilirubin increase occurred in two patients and grade 1 bilirubin increased occurred in one patient. Grade 1 increases in ALT and AST occurred in two patients. Additionally, no treatment-related decline in liver function by synthetic function parameters (albumin and INR) was observed. All treatment-related toxicities were grade 1 or 2, with the exception of one patient who developed grade 3 achalasia in the setting of disease progression after 2.5 months of therapy. SBRT was not thought to contribute given the index lesion was distant from the esophagus. After exclusion of alternative etiologies, achalasia was considered to be at least possibly-related to study drug. There was no evidence of cardiac toxicity and no dose-limiting toxicities or treatment delays.
Table 2.
Adverse events at least possibly related to therapy
| Event | Any Grade | Grade 1 | Grade 2 | Grade 3 |
|---|---|---|---|---|
| number of patients (%) | ||||
| Gastrointestinal | ||||
| Abdominal pain | 7 (46.6) | 6 (40) | 1 (6.6) | 0 |
| Nausea | 6 (40) | 6 (40) | 0 | 0 |
| Constipation | 4 (26.6) | 3 (20) | 1 (6.6) | 0 |
| Vomiting | 3 (20) | 2 (13.3) | 1 (6.6) | 0 |
| Abdominal distention | 2 (13.3) | 2 (13.3) | 0 | 0 |
| Flatulence | 2 (13.3) | 2 (13.3) | 0 | 0 |
| Diarrhea | 1 (6.6) | 1 (6.6) | 0 | 0 |
| Dysgeusia | 1 (6.6) | 1 (6.6) | 0 | 0 |
| Achalasia | 1 (6.6) | 0 | 0 | 1 (6.6) |
| Hepatic | ||||
| Alkaline phosphatase increased | 6 (40) | 4 (26.6) | 2 (13.3) | 0 |
| Bilirubin increased | 3 (20) | 1 (6.6) | 2 (13.3) | 0 |
| ALT increased | 2 (13.3) | 2 (13.3) | 0 | 0 |
| AST increased | 2 (13.3) | 2 (13.3) | 0 | 0 |
| Hematologic | ||||
| Platelet count decreased | 2 (13.3) | 1 (6.6) | 1 (6.6) | 0 |
| Neutrophil count decreased | 2 (13.3) | 0 | 2 (13.3) | 0 |
| White blood cells decreased | 2 (13.3) | 2 (13.3) | 0 | 0 |
| Lymphocyte count decreased | 1 (6.6) | 0 | 1 (6.6) | 0 |
| Pulmonary | ||||
| Cough | 1 (6.6) | 1 (6.6) | 0 | 0 |
| Constitutional | ||||
| Fatigue | 8 (53.3) | 7 (46.6) | 1 (6.6) | 0 |
| Rash | 2 (13.3) | 2 (13.3) | 0 | 0 |
| Pruritis | 2 (13.3) | 1 (6.6) | 1 (6.6) | 0 |
| Dizziness | 1 (6.6) | 1 (6.6) | 0 | 0 |
| Dry mouth | 1 (6.6) | 1 (6.6) | 0 | 0 |
| Dry skin | 1 (6.6) | 1 (6.6) | 0 | 0 |
| Edema | 1 (6.6) | 1 (6.6) | 0 | 0 |
| Weight loss | 1 (6.6) | 1 (6.6) | 0 | 0 |
| Neurological | ||||
| Headache | 2 (13.3) | 2 (13.3) | 0 | 0 |
| Renal | ||||
| Hypomagnesemia | 1 (6.6) | 1 (6.6) | 0 | 0 |
AEs were graded using the NIC Common Terminology for Adverse Events (Version 4.1)
Clinical Outcomes
Fourteen patients were evaluable for disease response. Two partial responses (PR) were observed. One PR lasted five months and the other is ongoing with a duration of 28 months at time of data cut-off. For both of the patients who achieved PR, non-irradiated lesions reduced in size (Fig. 1B). Six patients had stable disease (SD). The ORR was 14.2% with a disease control rate (DCR) of 57% (Fig. 1C). We performed a second response analysis excluding irradiated lesions to provide insight into the systemic impact of combination galunisertib and SBRT. One patient met criteria for PR when the irradiated lesion was excluded and a second patient had tumor reduction of 28%. One patient who was initially defined as having SD was reclassified as having PD when the irradiated lesion was excluded. Additionally, one patient had a single site of disease which was irradiated, however, this patient developed progression with new lesions identified at first restaging. The median PFS was 3.7-months and median OS was 9.0-months (Fig. 1D). SBRT in one-fraction was delivered between days 15 and 28 of Cycle 1. Three patients received SBRT on day 15; five patients received SBRT between days 16 and 17; and seven patients received SBRT between days 20 and 27.
Nine patients underwent SBRT to the liver; two patients received SBRT to the lung; and four patients received SBRT to involved lymph nodes. Median irradiated tumor volume was 8.1cm3 (range: 1.4cm3-352.4cm3). Radiation doses delivered to organs-at-risk are described in Supplementary Table 2. Volume of radiation delivered was associated with PFS, OS and best overall response (Fig. 1E). For the two patients with PRs, SBRT was delivered to the liver with a volume of 352.4cm3 and 3cm3, respectively.
Discontinuation of study therapy occurred for disease progression (10 patients), death (1 patient) and withdrawal of consent (2 patients).
Relationship of Baseline Immune Correlates with Treatment Outcome
To identify specific immune populations, we conducted high-dimensional single cell phenotyping using mass cytometry and a 37-marker metal-tagged antibody panel. We analyzed blood samples collected from patients prior to treatment, after 14-days of galunisertib treatment and after radiation just prior to the start of Cycle 2. One patient was not evaluable for radiologic response and was excluded from the analysis. Patients were dichotomized as being either progressors (n = 6) or non-progressors (n = 8) based on best overall response (Fig. 2A). Frequencies of immune cell subsets detected in the peripheral blood were compared between the two groups.
Figure 2. Pre-treatment immune subsets in the peripheral blood associate with outcomes to galunisertib and radiation.
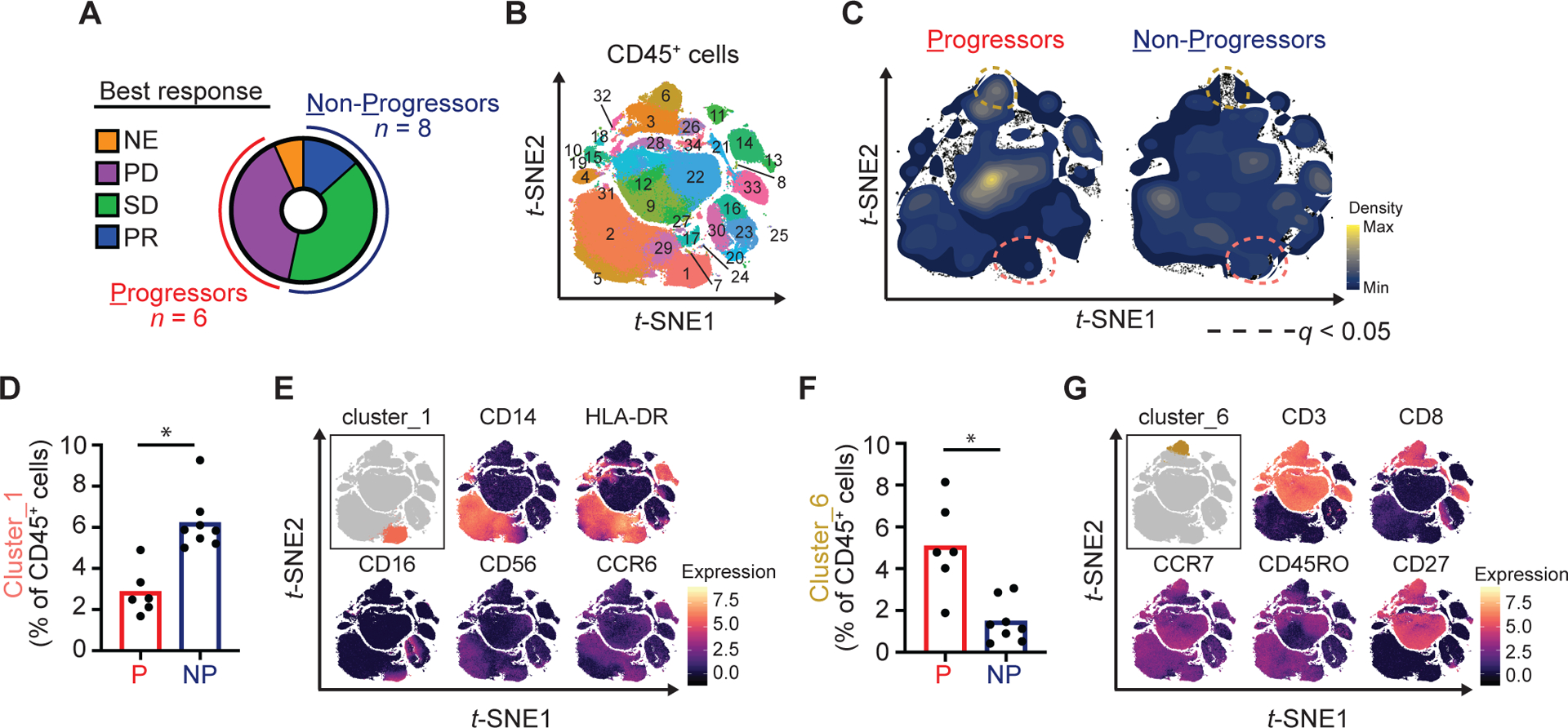
(A) Donut chart showing classification of patients as Progressors (P) or Non-Progressors (NP) based on best overall response measured with RECIST 1.1. One patient was not evaluable for radiologic response. (B) PBMC collected at baseline were analyzed by high-dimensional single cell mass cytometry. Shown is phenograph clustering analysis performed after exclusion of doublets and dead cells and positive selection for CD45. (C) Density plots showing clusters significantly altered between Progressors as compared to Non-Progressors. (D) Quantification of cluster_1 (percentage of CD45+ cells). (E) Marker expression level plots. (F) Quantification of cluster_6 (percentage of CD45+ cells). (G) Marker expression level plots. Multiple t-tests were performed with Benjamini, Krieger and Yekutieli correction. *, q < 0.05. NE, not evaluable; PD, progressive disease; SD, stable disease; PR, partial response.
We first focused our analysis on immune cell subsets present at baseline in progressors and non-progressors. Using an unsupervised clustering algorithm (16), we identified 34 unique clusters (Cl_x) (Fig. 2B, Supplementary Fig. S1A). In non-progressors, Cl_1, consistent with a non-classical monocyte (CD14+CD16highHLA-DR+CCR6+), was increased (6.1% vs. 2.8% of CD45+ cells; q=0.01) (Fig. 2C, Supplementary Fig. S1B, Fig. 2D–E). In progressors, Cl_6, consistent with a naïve-like CD8+ T cell (CD3+CD8+CCR7+CD27+CD45ROneg), was increased (5.1% vs. 1.5% of CD45+ cells; q=0.02) (Fig. 2F–G). Similarly, Cl_9, consistent with a naïve-like CD4+ cell, was increased in progressors (14.3% vs. 5.2% of total CD45+ cells; p=0.015, q=0.17).
Next, we examined the differentiation status of CD8+ T cells detected at baseline in the blood. We identified 21 distinct clusters, representing subsets of naïve (CCR7+CD45ROneg), effector (CCR7negCD45ROneg), effector memory (CCR7negCD45RO+) and central memory (CCR7+CD45RO+) CD8+ T cells (Fig. 3A). We performed principle component analysis and found that CD8+ T cell profiles segregated progressors from non-progressors (Fig. 3B). Manual gating of CD8+ T cell subsets (Fig. 3C) revealed that non-progressors had significant skewing of T cell differentiation towards an effector-like phenotype (effector:naïve ratio, 6.0 vs. 1.4; p=0.01) (Fig. 3D).
Figure 3. Pre-treatment CD8+ T cell profiles in the peripheral blood associate with outcomes to galunisertib and radiation.
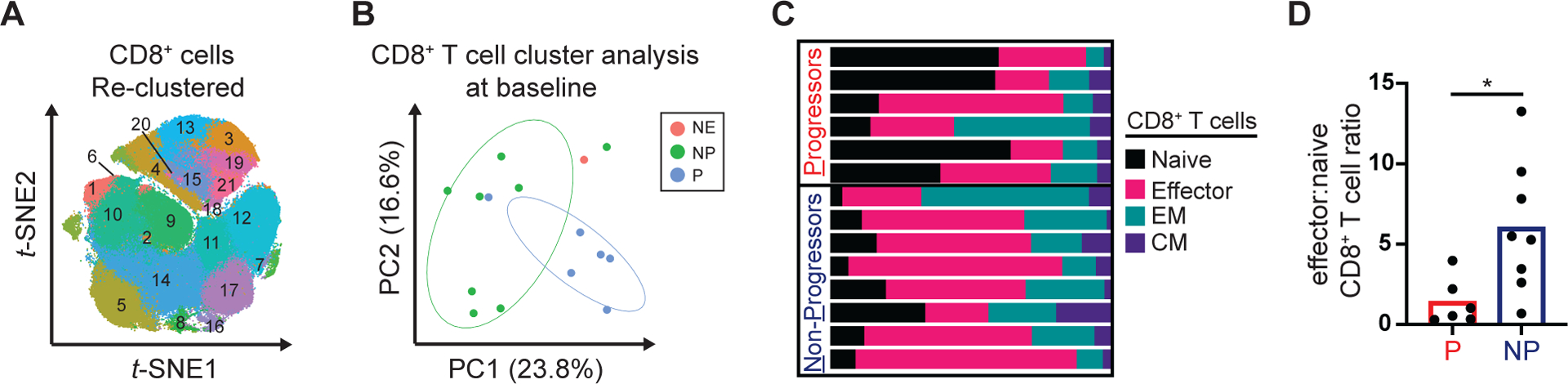
Baseline PBMC samples were analyzed by high-dimensional single cell profiling using mass cytometry. (A) Shown is phenograph clustering analysis performed after the exclusion of doublets, dead cells and CD19 cells and positive selection for CD45, CD3 and CD8. (B) PCA analysis of phenograph defined CD8+ clusters. (C) Stacked bar graph showing manually gated CD8+ naïve (CCR7+CD45ROneg), effector (CCR7negCD45ROneg), effector memory (CCR7negCD45RO+) and central memory (CCR7+CD45RO+) cell frequencies in individual patients. (D) Effector (CCR7negCD45ROneg) to naïve (CCR7+CD45ROneg) CD8+ T cell ratio at baseline among Progressors and Non-Progressors. Mann-Whitney tests were performed. *, p < 0.05. NE, not evaluable. NP, Non-Progressor. P, Progressor. EM, effector memory. CM, central memory.
Treatment-induced Immune Correlates
We found no change in CD4+ or CD8+ T cells, NK cells, Treg cells, monocytes, or dendritic cells after treatment with galunisertib or SBRT (Supplementary Fig. S2A). In contrast, treatment with SBRT produced a significant decrease in B cells, consistent with known lymphotoxic effects of radiation (Supplementary Fig. S2B–C) (17). We compared the frequency of CD45+ cell clusters identified among progressors and non-progressors during treatment. Progressors showed a significant increase in Cl_2, consistent with a monocyte-like population (CD14+HLA-DR+CD56+), after treatment with galunisertib (16.6% vs. 20.4% of CD45+ cells; corrected p=0.014) (Supplementary Fig. S3A–B).
We next examined the effect of treatment on CD8+ T cell phenotype. We found one distinct cell population that increased after combination therapy in non-progressors but not progressors (Fig. 4A). This population expressed CD45RO, PD-1 and T cell immunoreceptor with Ig and ITIM domains (TIGIT) and lacked expression for CCR7, LAG3 and Tim3 (Fig. 4B). Manual gating also showed a significant increase in CD8+PD-1+TIGIT+ T cells after combination therapy compared to baseline in non-progressors (8.1% vs. 4.6% of CD8+ T cells; p = 0.04), without change in progressors (2.8% vs. 2.7% of CD8+ T cells; p=0.6) (Fig. 4C). An unsupervised analysis of CD8+ T cells identified Cl_17, which resembled the manually gated PD-1+TIGIT+CD8+ T cell subset (Supplementary Fig. S3C). Cl_17 showed a similar increase in non-progressors after combination therapy compared to baseline (11.1% vs. 5.3% of CD8+ T cells; corrected p=0.079) (Supplementary Fig. S3D–E).
Figure 4. Galunisertib and radiation is associated with changes in T cell subsets in the peripheral blood after treatment.
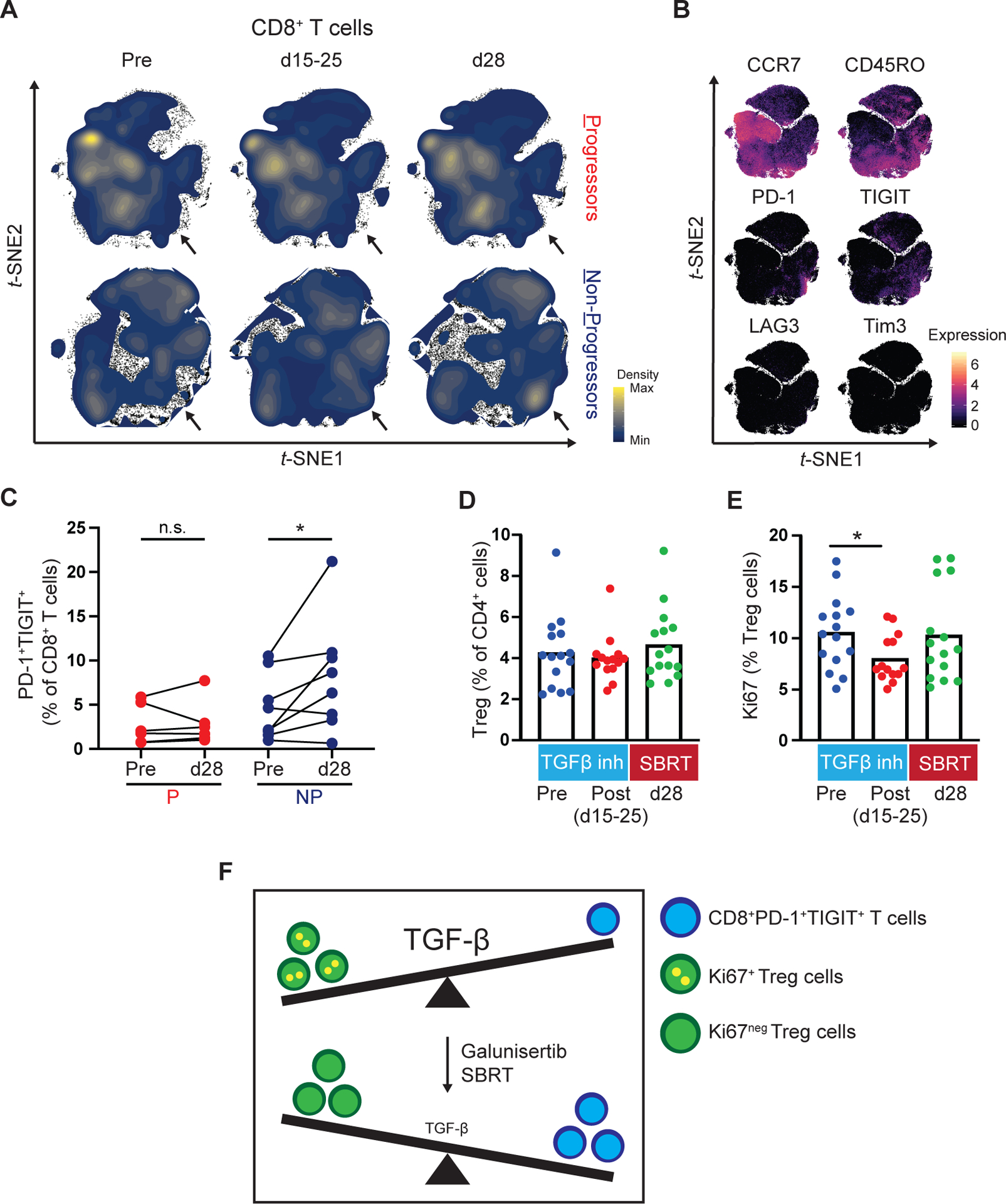
PBMC from Progressors (P) and Non-Progressors (NP) were analyzed at the indicated timepoints by high-dimensional single cell mass cytometry. Shown are (A) Density plots and (B) Marker expression level plots. (C) Paired comparison of manually gated CD8+PD-1+TIGIT+ cells at baseline (pre) and after combination treatment (d28) in Progressors and Non-Progressors. (D) Quantification of Treg cell (CD25+CD127lowFoxp3+) frequency (percentage of CD3+CD4+) pre-treatment (pre) and post-treatment with galunisertib (d15–25) and SBRT (d28). Cell subsets were gated after the exclusion of doublets and dead cells and positive selection for CD45, CD3 and CD4. (E) Quantification of Ki67+Treg cells (percentage of Treg cells) in samples pre-treatment and post-treatment with galunisertib and SBRT. (F) Schematic detailing the proposed association between TGF-β signaling and relevant peripheral blood leukocytes. At baseline Ki67+Tregs and low frequencies of CD8+PD-1+TIGIT+ T cells are found in the peripheral blood. After treatment, in non-progressors, the peripheral blood leukocyte composition is shifted towards higher frequencies of CD8+PD-1+TIGIT+ T cells and lower frequencies of Ki67+Tregs. The size of TGF-β lettering indicates higher (large) or lower (small) TGF-β signaling activity. Wilcoxon matched-pairs tests were performed (C). Mann-Whitney tests were performed (D-E). *, p < 0.05. SBRT, stereotactic body radiation therapy.
TGF-β inhibition and T Regulatory Cell Frequency and Phenotype
We next studied the impact of galunisertib on Treg frequency and phenotype. After combination treatment there was no change in frequency of Tregs (CD4+CD25+CD127lowFoxp3+) in the peripheral blood or expression of CTLA-4 or HLA-DR (Fig. 4D, Supplementary Fig. S4A–B, Supplementary Fig. S5A–B). In contrast, there was a significant decrease in Ki67+ Tregs after treatment with galunisertib compared to baseline (8.0% vs. 10.6% of Treg cells; p=0.036) (Fig. 4E). Notably, the frequency of Ki67+ Tregs increased between days 15–25 and d28 (8.0% vs. 10.3% of Treg cells; p=0.27) when galunisertib was not administered.
DISCUSSION
We evaluated the safety, efficacy and immunological impact of combining a TGF-β receptor I inhibitor (galunisertib) with SBRT for the treatment of patients with advanced HCC. Combination treatment was well tolerated and potential anti-tumor activity was seen. In addition, an exploratory analysis showed that pre- and post-treatment immune profiles in the blood were distinct among patients with progressive and non-progressive disease as best response.
Given the known role for TGF-β in the pathogenesis of HCC, early phase clinical trials have studied TGF-β blockade as monotherapy and in combination with sorafenib (13, 14). These studies have shown safety and potential efficacy. We sought to use galunisertib to alleviate immunosuppression and radioresistance to SBRT, which is commonly used for the treatment of HCC (18). The combination of galunisertib and SBRT was chosen based on the observation that radiation, especially in combination with immunotherapy, can liberate tumor-associated antigens generating an in situ vaccine (i.e. abscopal effect) (19). In our study, AEs compared favorably to those seen with galunisertib monotherapy. In regards to efficacy, prior studies of patients with advanced HCC showed an ORR of 2% to galunisertib monotherapy and 4.5% to galunisertib in combination with sorafenib (13, 14). In contrast, in our study we found an ORR of 14%, suggesting potential synergy of galunisertib and SBRT.
There are several important limitations to our study. First, our sample size is small and the secondary endpoints are hypothesis generating only. Second, the optimal dose and sequencing of SBRT in combination with galunisertib was unknown at the start of our study. The trial was designed to provide a run in of immunomodulation with TGF-β inhibition followed by administration of radiation. A radiation dose of 18-Gy was chosen based on prior experience and data showing safety of SBRT for the treatment of spinal metastases (20). Third, there was heterogeneity in the tumor volume irradiated, the site of irradiation and timing of SBRT in relation to galunisertib. The index lesion for SBRT was chosen based on safety considerations and size. Fourth, variability in the timing of blood collection respective to treatment and a limited number of time points assessed may have restricted our ability to identify additional treatment associated immune changes. Fifth, a lack of on-treatment biopsies limits our ability compare peripheral blood immune changes with intra-tumoral immune composition. While our study suggests the combination of galunisertib and SBRT is well tolerated, further study is needed to define the optimal dosing and sequencing of treatment.
At the start of our study, sorafenib was the only systemic therapy approved by the FDA for the treatment of advanced HCC. Since April 2017, a number of FDA approved therapies have drastically altered the therapeutic landscape of HCC. Recently, the combination of atezolizumab (anti-PD-L1) and bevacizumab (anti-VEGF) showed improved OS in comparison to sorafenib for the treatment of patients with newly diagnosed advanced HCC, setting a new standard of care (21). Additionally, multikinase inhibitors, checkpoint inhibitors and VEGF targeted agents, are now also approved for HCC in the first and second-line setting (22). As most patients will now be treated with atezolizumab/bevacizumab in the first-line, it becomes especially important to design novel immunotherapy strategies for those patients that progress on or are refractory to checkpoint inhibition. Additionally, although atezolizumab/bevacizumab is effective, the vast majority of patients with advanced HCC will still ultimately die from their disease. Thus, in regard to future directions for the combination of TGF-β inhibition and SBRT, we suggest that additional immunomodulatory agents could be incorporated into this treatment strategy in order to boost efficacy. Galunisertib is currently being studied in combination with nivolumab (anti-PD-1) for the treatment of patients with advanced solid tumors including HCC (NCT02423343). Incorporating radiation into a treatment regimen including anti-PD-1 therapy and TGF-β blockade would be a reasonable next step. Additionally, our study supports the continued development of novel TGF-β and radiation combinations as is actively being studied. Of particular interest, M7824 (bintrafusp alpha), which is a first-in-class bifunctional fusion protein containing the extracellular domain of the TGF-βRII (acting as a TGF-β trap) fused to a PD-L1 blocking antibody (23), is currently being evaluated in combination with SBRT for the treatment of head and neck cancer (NCT04220775).
A pre-existing immune reaction is a key determinant of outcomes to immunotherapy (24). We found distinct differences in baseline CD8+ T cell differentiation among patients with progression and non-progression as best response. Both cancer and chronic viral hepatitis are associated with changes in CD8+ T cell differentiation (25). However, we saw no clear association between history of viral hepatitis and immune composition. Additionally, our findings are consistent with observations in other malignancies where increased frequency of CD8+ memory T cell subsets at baseline are associated with improved outcomes (26–28). Together, these data support profiling of pre-treatment immune subsets in the blood of patients with HCC as a non-invasive method for identifying potential responders to therapy.
We also incorporated a longitudinal analysis of peripheral blood and found that non-progressor patients had increased CD8+PD-1+TIGIT+ T cells after combination treatment (Fig. 4F). PD-1 and TIGIT are upregulated early during T cell activation and can mark tumor-specific T cells (29). However, CD8+PD-1+TIGIT+ T cells may also play a detrimental role in HCC biology. Elevated baseline frequencies of CD8+PD-1+TIGIT+ T cells in patients with hepatitis B-associated HCC correlate with reduced survival and higher disease burden (30). Thus, it is possible that CD8+PD-1+TIGIT+ T cells observed with galunisertib and SBRT treatment may initially enact effector functions but ultimately assume an exhausted state with limited effector potential. Given our findings, inclusion of longitudinal profiling of CD8+ T cells in future prospective studies may be useful to identify biomarkers of response.
Treg cells are key mediators of immune homeostasis (31–34). The immunosuppressive activity of Tregs is well-suited for preventing autoimmunity. However, this same biology impedes the development and productivity of anti-tumor immunity. For these reasons, strategies to modulate or inhibit Tregs have garnered interest as an approach to improve the efficacy of cancer immunotherapy. Ki67 is a clinically relevant marker of activated Tregs (35). For example, an increase in intra-tumoral Ki67+ Tregs detected at the time of surgery is associated with poor disease free survival in patients with melanoma treated with neo-adjuvant anti-PD-1 therapy (36). Our data suggest that inhibition of TGF-β signaling may impair Treg activation, as demonstrated by a decreased frequency of Ki67+ Tregs in the blood after treatment with galunisertib. However, we did not observe evidence for immune-related adverse events to indicate increased risk for autoimmunity, consistent with prior studies using galunisertib (13, 14).
In conclusion, results from this pilot study demonstrate safety of combined galunisertib and SBRT in patients with advanced HCC. Despite a small sample size, we observed early evidence of clinical benefit. Further investigation will be necessary to determine a role for TGF-β inhibition in the treatment of HCC. Additionally, our study suggests the value of incorporating immune profiling of peripheral blood to identify potential benefit of treatment in patients with HCC.
Supplementary Material
Acknowledgements
We thank the Human Immunology Core (P30-CA016520) at the University of Pennsylvania for assistance in processing and storing patient samples. The authors would like to thank Michael Galantino and Carla Wright for their critical assistance with this trial and their dedication to patient care.
The clinical trial reported in this manuscript was supported by Lilly Oncology. Additional support was provided by ASCO Conquer Cancer Foundation Career Development Award to K.A.R.; The National Institutes of Health (grant number T32-HL007439-41 to M.M.W. and P30-CA016520 to R.H.V.); and the Stand Up to Cancer (SU2C) Innovative Research Grant (grant number SU2C-AACR-IRG 13-17 to G.L.B).
Footnotes
Conflict of interest:
K.A.R reports receiving research support from Lilly Oncology, Bristol-Myers Squibb, Tesaro and Clovis Oncology. G.L.B. reports prior or active roles as a consultant/advisory board member for Seattle Genetics, Boehinger Ingelheim, Cour Pharmaceuticals, Aduro Biotech, AstraZeneca, Bristol-Myers Squibb, Genmab, Incyte, Janssen, Opsona, Merck, and BiolineRx; reports receiving commercial research grants from Incyte, Bristol-Myers Squibb, Verastem, Halozyme, Biothera, Newlink, Novartis, Arcus, and Janssen. G.L.B. is an inventor of intellectual property and recipient of royalties related to CAR T cells that is licensed by the University of Pennsylvania to Novartis. E.P.D. is a speaker for Taiho Oncology. E.L.C. reports receiving honoraria from Imedex and AstraZeneca, as well as commercial research grants from Janssen, Merck, and Becton Dickinson. E.L.C. also received research funding from the Parker Institute for Cancer Immunotherapy and the Penn Pancreatic Cancer Research Center. R.H.V. reports having received consulting fees or honoraria in the past 5 years from Celgene, Celldex, Janssen, Lilly, Medimmune, and Verastem; and research funding from Apexigen, Fibrogen, Inovio, Janssen, and Lilly. He is an inventor on a licensed patent relating to cancer cellular immunotherapy and receives royalties from Children’s Hospital Boston for a licensed research-only monoclonal antibody.
References
- 1.Villanueva A Hepatocellular Carcinoma. N Engl J Med. 2019;380(15):1450–62. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Stat Facts: Liver and Intrahepatic Bile Duct Cancer SEER: National Cancer Institute, DCCPS, Surveillance Research Program; 2018. [https://seer.cancer.gov/statfacts/html/livibd.html].
- 3.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yau T, Kang Y-K, Kim T-Y, El-Khoueiry AB, Santoro A, Sangro B, et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. Journal of Clinical Oncology. 2019;37(15_suppl):4012-. [Google Scholar]
- 5.El Dika I, Khalil DN, Abou-Alfa GK. Immune checkpoint inhibitors for hepatocellular carcinoma. Cancer. 2019;125(19):3312–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12(12):681–700. [DOI] [PubMed] [Google Scholar]
- 7.Fransvea E, Angelotti U, Antonaci S, Giannelli G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 2008;47(5):1557–66. [DOI] [PubMed] [Google Scholar]
- 8.Wan YY, Flavell RA. ‘Yin-Yang’ functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol Rev. 2007;220:199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134(3):392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Gingold JA, Su X. Immunomodulatory TGF-β Signaling in Hepatocellular Carcinoma. Trends Mol Med. 2019;25(11):1010–23. [DOI] [PubMed] [Google Scholar]
- 11.Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, et al. TGFβ Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015;75(11):2232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, et al. Focal Irradiation and Systemic TGFβ Blockade in Metastatic Breast Cancer. Clin Cancer Res. 2018;24(11):2493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faivre S, Santoro A, Kelley RK, Gane E, Costentin CE, Gueorguieva I, et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019;39(8):1468–77. [DOI] [PubMed] [Google Scholar]
- 14.Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, Merle P, et al. A Phase 2 Study of Galunisertib (TGF-β1 Receptor Type I Inhibitor) and Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Clin Transl Gastroenterol. 2019;10(7):e00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yingling JM, McMillen WT, Yan L, Huang H, Sawyer JS, Graff J, et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget. 2018;9(6):6659–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levine JH, Simonds EF, Bendall SC, Davis KL, Amir e-A, Tadmor MD, et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162(1):184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhuang Y, Yuan BY, Chen GW, Zhao XM, Hu Y, Zhu WC, et al. Association Between Circulating Lymphocyte Populations and Outcome After Stereotactic Body Radiation Therapy in Patients With Hepatocellular Carcinoma. Front Oncol. 2019;9:896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wahl DR, Stenmark MH, Tao Y, Pollom EL, Caoili EM, Lawrence TS, et al. Outcomes After Stereotactic Body Radiotherapy or Radiofrequency Ablation for Hepatocellular Carcinoma. J Clin Oncol. 2016;34(5):452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520(7547):373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rwigema JC, Parikh SD, Heron DE, Howell M, Zeh H, Moser AJ, et al. Stereotactic body radiotherapy in the treatment of advanced adenocarcinoma of the pancreas. Am J Clin Oncol. 2011;34(1):63–9. [DOI] [PubMed] [Google Scholar]
- 21.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–905. [DOI] [PubMed] [Google Scholar]
- 22.Finn RS, Zhu AX. Evolution of Systemic Therapy for Hepatocellular Carcinoma. Hepatology. 2020. [DOI] [PubMed] [Google Scholar]
- 23.Spira A, Awada A, Isambert N, Estellés DL, Nemunaitis J, Penel N, et al. Abstract P3-09-06: Bintrafusp alfa (M7824), a bifunctional fusion protein targeting transforming growth factor-β and programmed death ligand 1, in advanced triple-negative breast cancer: Preliminary results from a phase 1 cohort. Cancer Research. 2020;80(4 Supplement):P3-09-6-P3–6. [Google Scholar]
- 24.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol. 2019;37:457–95. [DOI] [PubMed] [Google Scholar]
- 26.Wistuba-Hamprecht K, Martens A, Heubach F, Romano E, Geukes Foppen M, Yuan J, et al. Peripheral CD8 effector-memory type 1 T-cells correlate with outcome in ipilimumab-treated stage IV melanoma patients. Eur J Cancer. 2017;73:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tada K, Kitano S, Shoji H, Nishimura T, Shimada Y, Nagashima K, et al. Pretreatment Immune Status Correlates with Progression-Free Survival in Chemotherapy-Treated Metastatic Colorectal Cancer Patients. Cancer Immunol Res. 2016;4(7):592–9. [DOI] [PubMed] [Google Scholar]
- 28.Manjarrez-Orduño N, Menard LC, Kansal S, Fischer P, Kakrecha B, Jiang C, et al. Circulating T Cell Subpopulations Correlate With Immune Responses at the Tumor Site and Clinical Response to PD1 Inhibition in Non-Small Cell Lung Cancer. Front Immunol. 2018;9:1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8⁺ T cells in melanoma patients. J Clin Invest. 2015;125(5):2046–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Li M, Wang X, Dang Z, Jiang Y, Kong Y, et al. PD-1 + TIGIT + CD8 + T cells are associated with pathogenesis and progression of patients with hepatitis B virus-related hepatocellular carcinoma. Cancer Immunol Immunother. 2019;68(12):2041–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–6. [DOI] [PubMed] [Google Scholar]
- 32.Konkel JE, Zhang D, Zanvit P, Chia C, Zangarle-Murray T, Jin W, et al. Transforming Growth Factor-β Signaling in Regulatory T Cells Controls T Helper-17 Cells and Tissue-Specific Immune Responses. Immunity. 2017;46(4):660–74. [DOI] [PubMed] [Google Scholar]
- 33.Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25(3):441–54. [DOI] [PubMed] [Google Scholar]
- 34.Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25(3):455–71. [DOI] [PubMed] [Google Scholar]
- 35.Santegoets SJ, Dijkgraaf EM, Battaglia A, Beckhove P, Britten CM, Gallimore A, et al. Monitoring regulatory T cells in clinical samples: consensus on an essential marker set and gating strategy for regulatory T cell analysis by flow cytometry. Cancer Immunol Immunother. 2015;64(10):1271–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med. 2019;25(3):454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.