

Abstract
Subclonal reconstruction from bulk tumor DNA sequencing has become a pillar of cancer evolution studies, providing insight into the clonality and relative ordering of mutations and mutational processes. We provide an outline of the complex computational approaches used for subclonal reconstruction from single and multiple tumor samples. We identify the underlying assumptions and uncertainties in each step, and suggest best practices for analysis and quality assessment. This guide provides a pragmatic resource for the growing user community of subclonal reconstruction methods.
Introduction
Cancers evolve from a single cell through the sequential acquisition of somatic mutations, some of which enable the hallmark traits of cancer1,2. The descendants of this cell, which share its genotype, form the initial cancer clone. Selection, mutation, drift and spatial separation of clonal populations may then give rise to related but genetically distinguishable descendant subpopulations within a single tumor. These subclones can be evaluated from DNA sequencing studies, which have started to quantify key aspects of tumor development, such as metastatic seeding patterns3,4 and mutations present in all tumor cells (i.e. clonal mutations) that may be targets for treatment and early intervention5,6. Tumor heterogeneity has important clinical consequences: tumors with complex subclonal structures can be more aggressive7,8 and are more likely to develop drug resistance and metastases9.
The process of subclonal reconstruction involves three key aspects. First, it characterizes the major populations of cells in a given tumor by identifying the somatic mutations present in each one. Second, it quantifies the proportion of cells from each clone in the tumor (its cellular prevalence; Lexicon). Third, it reconstructs the phylogenetic path by which the different clones evolved from their common ancestor, and ultimately from a normal host cell. Subclonal reconstruction can be performed from DNA sequencing data of a single tumor sample, or from multiple samples collected over time and/or space. The DNA sequencing data itself can be generated via a variety of sequencing strategies10. Thus, the accuracy and resolution of each feature of the subclonal reconstruction is shaped by the experimental design and the mutational characteristics of the specific tumor being reconstructed.
Box 1: Lexicon.
Branching clones: a non-linear set of clones descending from a common ancestor (e.g., sibling or cousin clones).
Cancer cell fraction (CCF): the fraction of cancer cells from the sequenced sample carrying a set of SNVs, i.e. CCF = CP / purity. It can be inferred from the VAF (f), given a sample purity (ρ), local copy-number (NT) and the inferred multiplicity of the mutations
Cellular prevalence (CP): the fraction of all cells (both tumor and admixed normal cells) from the sequenced tissue carrying a set of SNVs.
Clonal mutation: mutation present in all the tumor cells of a tumor sample or biopsy.
Clone: a lineage of cells descended from a common ancestor that inherited its genotype. Clones can be characterized by 1) the genotype of the MRCA of that lineage, which is the set of initial SNVs that will be carried by the descendant cells, i.e. detected at the same CP or CCF and 2) the fraction of (cancer) cells carrying these SNVs, i.e. the CP or CCF of the initial SNVs.
Crossing rule: when performing multi-sample or multi-region sequencing, when clone A and B are descendant of clone C and the CCF of clone A is higher than the CCF of clone B in one sample but the opposite is true in another sample, then clone A and B must be branching subclones. It stems from the more general rule that the shared subclones across samples arose from the same phylogeny, which further constrains the possible phylogenetic relationships between subclones.
Illusion of clonality: a mutation that is clonal in the sequenced tumor sample but is not clonal in the whole tumor.
Infinite sites hypothesis: hypothesis that the size of the genome tends to infinity. Consequently, mutated positions are only mutated once and never revert to wild type. This approximation results from the observation that, given the large size of the genome, a set of mutations is unlikely to have happened twice during tumor evolution. The infinite sites hypothesis is likely occasionally violated for single nucleotide variants94, but their frequencies remain very low when considering larger sets of SNVs spread along the genome such as those making up the genotype of large subclones (see pigeonhole principle).
logR: total copy number log ratio, which can be estimated from local normalized tumor to normal read depth , where the logR at position i is the log-ratio of two normalised depths, the total depth in the tumor or normal at that position (Ti or Ni, respectively) divided by the average depth across positions in the tumor or normal ( or , respectively).
Linear clones: a set of clones where one or more clones is an ancestor of another clone in the set (e.g., parent-child clones).
Most recent common ancestor (MRCA): the most recent common ancestor is the most recent cell that spawned a set of cells. By extension, the MRCA also refers to the genotype of that ancestor cell. The MRCA of a given tumor is sometimes used to implicitly refer to the MRCA of all cells in a set of sequenced samples. Note that the MRCA of a tumor sample (or set of samples) is not necessary the MRCA of the whole tumor, due to the illusion of clonality.
Multiplicity of a mutation: the number of DNA copies bearing a mutation m, which can be estimated from the VAF f, sample purity ρ and total copy number of the region in the tumor cells (NT) as . In regions of clonal copy number, the multiplicity of a mutation is a strictly positive integer, so the most likely value can be obtained by rounding to the nearest non-zero integer: , where round is a function that returns the nearest integer or by performing probabilistic assignment to integer values. In genomic regions with subclonal copy number alterations, subclonal cell populations may have differing multiplicities. Further, subclonal copy number losses may cause mutations to be lost from some subclones, resulting in multiplicities of zero for these subclones.
Pigeonhole principle: in the context of subclonal reconstruction, the sum of CCFs of branching subclones should be less than the CCF of their parent clone. Indeed, if it was greater, this would mean that mutations have occurred independently in branching lineages. However, according to the infinite sites hypothesis, the same set of random mutations is unlikely to have happened twice independently. Therefore, the smaller subclone must be a descendant of the bigger subclone, i.e. they are linear subclones, which is compatible with the infinite sites hypothesis.
Purity, sample purity or tumor purity (ρ): the purity is the fraction of cancer cells in the tumor sample. Thus, the cellular prevalence of clonal mutations is the purity. Consequently, the fraction of non-cancer cells in the tissue sample is 1-ρ.
Subclonal mutation: mutation that is present in a subset of tumor cells in a tumor sample or biopsy.
Subclone: a clone that is a descendant of the MRCA of the tumor sample, i.e. with associated CCF<1 in at least one region.
Superclonal cluster: an apparent clone with CCF > 1, usually indicative of germline contamination or purity estimation errors.
Subclonal reconstruction: the exercise of reconstituting the subclonal structure from sequencing data, i.e. number of (sub)clones, size of subclones in terms of fraction of cancer cells, and genotype of the subclones, as well as their phylogenetic relationships.
Sufficiency of subclonality: a mutation that is subclonal in the sequenced tumor sample will be subclonal in the whole tumor.
Sum rule: see Pigeonhole Principle.
Variant allele fraction or frequency (VAF): the fraction of mutated reads for a given variant, which is a readout for the proportion of DNA mutated in the sequenced tissue.
Weak parsimony: the vast majority of the SNVs with detectable VAFs are associated with a small number of subclonal lineages.
We focus here on computational methods for subclonal reconstruction using bulk DNA sequencing data, which remains the most widely-used approach, although single-cell techniques continue to rapidly improve in quality and cost. We first outline the fundamental principles of subclonal reconstruction from a single heterogeneous tumor sample, then extend them to subclonal reconstruction from multiple samples. We next review the key approaches used for subclonal reconstruction, along with their limitations. Finally, we close with some perspectives on how the field may move and summarize our recommendations for subclonal reconstruction in practice (Table 1), providing a step-by-step practical guide with an example case (Supplementary Information).
Table 1:
Checklist of recommended best practices
| Recommendation | Rationale |
|---|---|
| High-depth sequencing (>60x) of biopsy samples with the highest pathological purity possible, ideally complemented with deep targeted sequencing of SNVs | Increasing read depth increases the limit of detection for minor subclones and the resolution of CCF estimation22,23,54. High purity ensures most of the reads come from the tumor cells, increasing NRPCC. |
| Ensure the number of SNVs called is sufficient for subclonal reconstruction | A low coding substitution rate can lead to insufficient data for accurate subclonal reconstruction in exome-based studies23,90. |
| Sequence multiple regions from a single tumor | Single-region bulk sequencing systematically underestimates the number of subclones and locally dominant subclones can be mistaken as clonal13,21,91. Multi-region sequencing also provides better subclone resolution and allows phylogeny inference. |
Minimize germline variant contamination:
|
Germline contamination can lead to false-positive SNVs with high VAF that can be mislabeled as a cluster. Using a consensus call set can improve sensitivity and specificity of variant detection23. |
| Call somatic variants with a highly sensitive algorithm | Increased algorithm sensitivity facilitates low VAF SNV detection, improves clustering accuracy and better captures the level of tumor heterogeneity. Highly sensitive detection algorithms can also improve the chances of detecting clinically relevant minor subclones22,23,92. However, users should be cautious of false-positive SNVs which are often seen at low VAF and may form a low VAF cluster. |
| For CNA reconstructions, review solutions for incorrect CP and WGD estimation and adjust accordingly. Optimally, perform experimental ploidy validation. | CNA reconstructions must decide between multiple equally likely ploidy and purity solutions. Ideally, inform CNA calling with experimental ploidy estimates using FACS, image cytometry, or FISH. |
| Carry out orthogonal copy number estimation | Multiple copy number solutions are usually possible; estimating copy number from WES data can be especially challenging90. |
| Perform CNA + SNV based reconstruction using a method that incorporates a Binomial or Beta-binomial noise model | Binomial and Beta-binomial noise models better capture the noise in read sampling for a given read depth and CP, improving SNV clustering accuracy. |
| If possible, use phasing or single-cell sequencing data to support inferred mutation ordering. Ideally, perform multi-sample sequencing. | An unambiguous phylogeny is not always possible based only on the crossing and sum rules. Phasing or single-cell sequencing information can support or refute a proposed phylogeny14,16,73. Our preferred setup is multi-sample sequencing, intelligent designs combine high and low depth sequencing to minimize cost93. |
| Validate subclonal SNVs of interest using high-depth targeted sequencing | SNVs detected in one sample may occur at very low VAFs in another, and high-depth targeted sequencing can detect these rare subclonal populations17,23,92. |
Overview of subclonal reconstruction
Mutations belonging to the initiating cell of the most recent clonal sweep are expected to occur in every cell in the tumor. We refer to these as clonal, and distinguish them from subclonal mutations which arose in descendant subpopulations. We assume familiarity with some key technical terms in cancer genomics (see Lexicon).
Figure 1 outlines the standard workflow for single sample subclonal reconstruction. Most subclonal reconstruction methods consider single nucleotide variants (SNVs), small indels, and larger copy number alterations (CNAs; Figure 1a). They use the variant allele frequency (VAF) of SNVs to infer the proportion of sampled cells bearing the SNV (cellular prevalence; CP, see Lexicon). They do so by using the read structure (Figure 1b) to first reconstruct copy number state, learning regions of clonal and subclonal copy number change (Figures 1b–c). Algorithms then group SNVs with similar cellular prevalences, assuming that these occurred within a single distinct clone. Knowing the proportion of the sampled cells that are cancerous (the sample’s purity), SNVs can be clustered by the proportion of tumor cells bearing the mutation, called the cancer cell fraction (CCF; Figure 1d). Some algorithms may then attempt to infer the evolutionary relationships between clones (their phylogeny) based on cluster CCF and mutation co-occurrence, although we believe this is usually only advisable for multi-sample data.
Figure 1 |. Standard Workflow and Input Data for Subclonal Reconstruction.
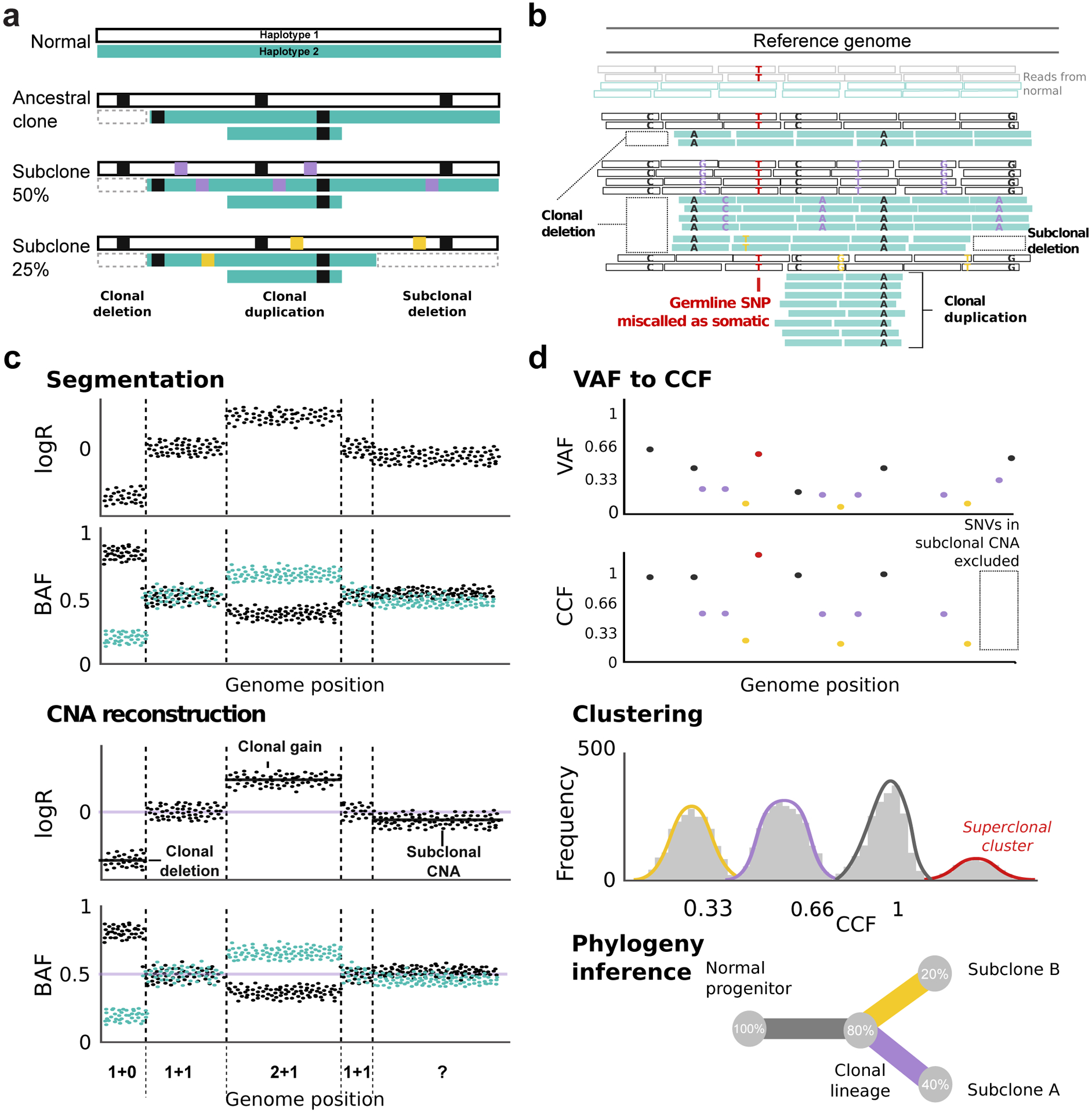
(a) A simplified example of tumor clonal genotypes. We illustrate a tumor containing two subclones at 50% (purple) and 25% (yellow) CCF, both descended from a common ancestral clone (100% CCF, black). The remaining 25% of tumor cells are indistinguishable from the ancestor. (b) First, somatic mutations are called from aligned reads. Read depth must be much higher (coverage >60x) than illustrated for mutation calling and subclonal reconstruction. Similarly, an elevated local mutation burden is illustrated. A somatic variant caller identifies somatic SNVs by comparing to a matched normal, although germline SNP contamination may occur (see main text). (c) Second, CNA reconstruction is performed. It typically uses read depth and B-allele frequency (BAF) data for heterozygous SNPs. (d) Third, CNAs are used to translate the measured SNV VAF to a CCF/CP estimate. This procedure relies on an accurate SNV multiplicity estimates (see Lexicon) which are typically inaccurate in subclonal CNAs so we exclude these regions from the analysis. SNV CCFs are then clustered to identify (sub)clonal lineages in the sample. False positive SNVs or inaccurate CNAs can cause spurious superclonal clusters (i.e. with CCF>1. Finally, phylogenetic reconstruction infers the ancestral relationships among lineages.
Inferring CP or CCF from VAFs requires estimating allele-specific copy number as CNAs drastically impact VAF interpretation (Figure 1c–d; Lexicon). CNAs can be inferred from sequencing data by comparing local read depth in tumor and reference samples (quantified as the log2 of the tumor and normal depth ratio; logR) as well as the allele counts of heterozygous single nucleotide polymorphisms (SNPs), altering the allelic ratio in the tumor relative to the normal sample (quantified as the B-allele frequency; BAF) (Figure 1c). Using these metrics, CNA reconstruction algorithms estimate the purity and ploidy of the sample, identify genomic segments with copy number changes, infer allele-specific copy number, and attempt to distinguish between clonal and subclonal CNAs.
Each of these steps involves uncertainty and can introduce errors into subclonal reconstruction. Indeed, many other sources of error exist upstream of subclonal reconstruction (e.g. sequencing, alignment, variant detection). For example, low tumor purity, errors in sequence alignment, and low sequence coverage in specific regions of either the tumor or matched reference normal genomes can all lead to germline variants being misclassified as somatic11,12 (Figure 1b). These errors can propagate uncertainty into the subclonal reconstruction results.
Study design and data collection for subclonal reconstruction
The resolution and accuracy of subclonal reconstruction are strongly influenced by how the input data are sampled. Single-sample sequencing, even at high depth, can underestimate the number of subclones, and subclonal populations or mutations can appear clonal (Figure 2). This is called the illusion of clonality (Lexicon)9,13. Multi-region sequencing can improve separation of subclones based on CCF differences across samples, and thereby facilitate phylogenetic inference13,14 (Figure 2a). Increasing sequencing depth improves the precision of CCF estimates and ability to distinguish subclones with similar CCFs. Given these trade-offs, in general, sequencing more samples will improve subclonal reconstruction more than higher-depth sequencing, given that depth is sufficient to accurately identify variants and resolve peaks in CCF space.
Figure 2 |. Subclonal Reconstruction Using Multiple Samples.
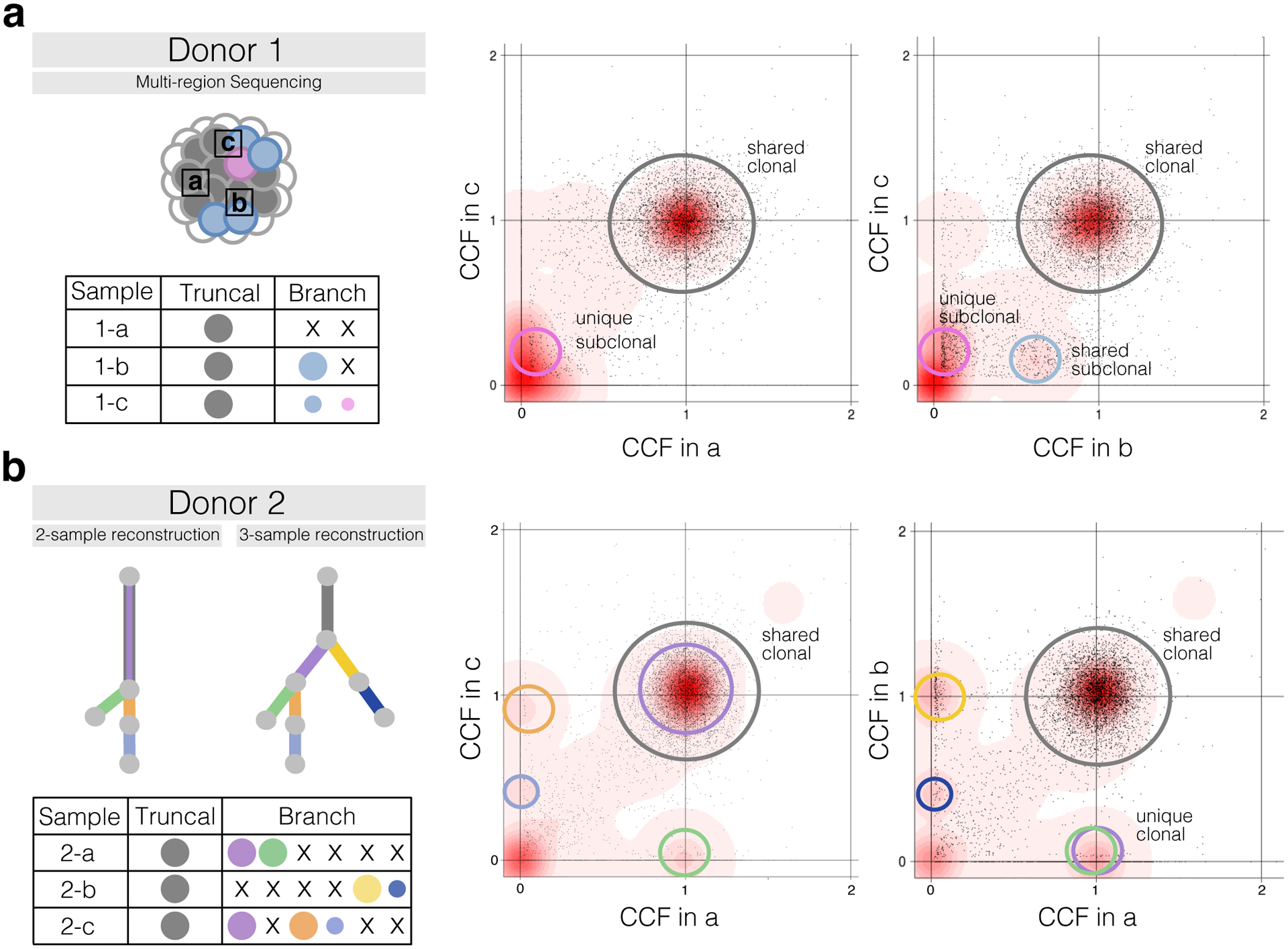
(a) Multiple samples can reveal additional subclones. Left: a tumor with three sequenced samples (a,b,c). The table, shows clones in each sample with color-coded circles proportional to their CCF in size. Truncal is defined as CCF = 1 in all samples and branch as CCF < 1 in at least one sample. Right: two sample density plots for the tumor. SNV CCFs from each sample are plotted along the axes. Circles indicate clone clusters, while the red background shows SNV density. SNVs clustered around (1,1) occur in all tumour cells in both samples; subclones on the axes are sample-specific, and clusters off the axes appear subclonal in both samples. For example, a subclonal cluster occurs in ~15% of cells in c but is absent in a. However, region (b) shows that this cluster was a mixture of two subclones: one unique to c and one shared by b and c. (b) Sequencing multiple samples clarifies clonal relationships. Left: phylogenetic trees for 2- and 3-sample subclonal reconstruction from multi-region sequencing (a, b, c). Subclones are represented by color-coded circles, as in (a). Right: density plots, as in (a). Looking only at samples a and c, mutations from the purple cluster appear clonal. However, it is absent in sample (b) and thus subclonal.
However, optimizing subclonal reconstruction is not typically the only or primary goal of a study. As a result, we recommend that study design match the biological questions being investigated given technical and financial limitations. For example, if the patient number is large (e.g. a clinical trial) single sample analysis may be appropriate to maximize statistical power for clinical inference. Single-sample studies can put lower bounds on subclonal heterogeneity, as a mutation found subclonally in a single sample is sufficient to be deemed subclonal in the whole tumor. Thus, mutations subclonal in single sample studies are potentially poor clinical targets (the sufficiency of subclonality, see Lexicon). Single sample subclonal reconstruction in large cohorts can be useful for showing coarse trends in mutation timing, as clonality errors due to spatial heterogeneity are likely random, but detailed phylogenetic inference will likely be imprecise in these studies8,15,16.
In other studies, evaluating clonal evolution over time may be critical, for example in understanding metastatic processes. Samples taken at different times (e.g. at diagnosis and relapse) permit inference of temporal features of tumor evolution. Samples taken from different spatial points (e.g. different regions of the primary site or metastases) permit inference on lineage frequency changes that can hint at subclone fitness17. For example, an intelligent design to look at evolution across many metastatic sites captures variants through high-depth sequencing and follows them spatially through shallow-coverage sequencing18. Because of the illusion of clonality, multi-sample designs are superior when searching for clonal targets, e.g. clonal neo-antigens19. In general, sequencing even one additional sample per tumor can help resolve additional subclones (Figure 2a) and clarify phylogenetic relationships (Figure 2b), and additional samples would further improve accuracy20,21. However, in practice there are many logistical and physical limitations to multi-sample sequencing: tumor size, tissue quality, availability of material for research, cost, feasibility of tissue collection in the given clinical setting, and the attainable sequencing depth for each sample. As a result, while multi-sample subclonal reconstruction may be technologically superior, clinical and financial considerations can strongly constrain the possible sample number and space combinations.
Optimizing sequencing depth
When balancing sequencing depth and sample number, an important consideration is that overall sequencing coverage, as well as the specific sequencing technology used, which influences local read depth distribution, determines the sensitivity and specificity of clonal and subclonal SNV detection22,23. Both tumor ploidy and purity impact the depth of sequencing coverage needed to detect low-CCF SNVs22,23.
One useful metric for evaluating sequencing depth for any given tumor is the number of reads per tumor chromosomal copy (NRPCC, Box 2). As NRPCC increases, the signal of true CCF peaks becomes clearer relative to read-sampling noise, and clones are easier to distinguish. In a large single-sample pan-cancer study, most samples with NRPCC>10 exhibited at least one subclone15. An NRPCC of 10 represents a read depth of ~40x in a diploid 50% purity tumor. Some studies have successfully used large numbers of samples sequenced to moderate depth (30–50x), but generally deeper sequencing improves subclonal reconstruction accuracy and resolution22,24. Sequencing depth of the reference normal sample is also important to limit false-positive identification of germline variants as somatic ones. This is particularly true in studies sequencing multiple samples for each tumor and using a single reference sample as a control for all, as these false positives can enlarge the apparent number of clonal mutations. Current copy-number detection algorithms appear to be more robust to lower sequencing depths than current SNV detection methods22,25. Indeed, some CNA methods are already amenable to single-cell resolution26, although few methods reconstruct phylogenies from CNAs27–29.
Box 2: NRPCC.
The number of reads per tumor chromosomal copy (NRPCC) can be defined as:
where d is the depth of sequencing, ρ is the purity and ψ is the average tumor sample ploidy, i.e. Ψ = ρΨT + (1−ρ) ΨN , where ΨT is tumor ploidy and ΨN = 2 is normal ploidy.
Consider a diploid tumor sample (ΨN = 2) with purity ρ = 0.5. In this diploid context, because 50% of the cells are non-tumor cells, 50% of the reads would derive from non-tumor cells. If a mutation occurs on one of the two tumor copies, then ~25% of reads will carry it . Next consider a ρ = 0.5 tumor that undergoes a whole genome duplication and becomes tetraploid (ΨT = 4). In this case, two thirds of the reads derive from tumor cells. Note that mutations that have happened after the whole genome duplication will only be present on one of the four tumor copies, and therefore only ~16.7% of reads will carry these clonal mutations . Thus, the fraction of mutated reads of subclonal mutations in a tetraploid tumor is lower than in a diploid tumor.
One rule of thumb is that most variant detection algorithms will not identify a somatic SNV without at least three variant reads95.
Let us imagine a tetraploid tumor ΨT = 4, with purity ρ = 0.7, i.e. Ψ = 0.7 × 4 + 0.3 × 2 = 3.4. An SNV at CCF = 0.33 (present in a third of cancer cells), will be present in f fraction of the reads as quantified by:
And the expected number of mutated reads will depend on the depth and is:
Or in terms of NRPCC:
This equation illustrates why the NRPCC is a relevant measure. It defines the expected number of mutated reads at given CCF values, and given that (as a rule of thumb) mutation with Nmut < 3 are not being called, it defines the detection threshold, or sensitivity threshold, i.e. the power to detect these subclonal mutations.
We model the number of mutated reads as following a Binomial distribution:
If we want to select a minimum depth that allows to detect most of these mutations, the probability to miss or to call them must be low or high, respectively. For example, not to miss more than 5% mutations in that subclone, i.e. to have Nmut < 3 with probability P < 0.05 (Nmut ≥ 3 with probability P ≥ 0.95), we have:
The depth of sequencing must be greater than 91x, which corresponds to NRPCC=18.7. For clonal mutations, i.e. CCF=1, the depth should be greater than 29x.
Sequencing breadth
While in general, higher-depth sequencing will improve subclonal reconstruction, it is also important to consider the portion of the genome directly measured – sequencing breadth. Subclonal reconstruction can be applied to whole-genome sequencing (WGS) or to targeted sequencing of genes, either the whole exome (WES)30, or subsets of selected genes31. The major differences between these approaches are the number of SNVs/indels detected, local depth and variability of coverage, and resolution and accuracy of CNA reconstruction. Moderate depth (30–50x) WGS supports high-quality CNA calls and allows estimation of copy number CCFs for the characterization of subclonal CNAs. WES detects ~50-fold fewer SNVs and indels, and decreases CNA reconstruction resolution and quality, thus hindering subclonal lineage detection25,32. Conversely, higher depth of sequencing usually allows for better accuracy in CCF estimates, and WES may yield higher resolution subclonal reconstruction than WGS in samples with many SNVs and indels and few CNAs, particularly if it permits use of multi-sample data by lowering costs per sample9,33. Targeted sequencing using smaller gene panels rarely supports meaningful subclonal reconstruction, unless an initial round of WGS or WES is performed to define SNVs representative of individual subpopulations (cells with nearly identical genotypes)34. If there are few genes in the panel, allele-specific CNA reconstruction will be unreliable unless additional data (e.g. from a SNP chip) is available.
We recommend only attempting subclonal reconstruction with fresh frozen tissue. Standard WGS with formalin fixed paraffin embedded (FFPE) samples has variable DNA quality35, and FFPE derived artefacts can introduce CNA errors36. If FFPE samples must be used, protocols that optimize library preparation and sequencing for accurate SNV and CNA detection from FFPE samples are an active research area37, but downstream subclonal reconstruction results will need to be interpreted with significant caution.
The computational workflow for subclonal reconstruction
CNA reconstructions
Overview
The majority of CNA reconstructions use germline single nucleotide polymorphisms (SNPs; Figure 1c), and evaluate their read depth and allele frequencies. For each SNP, copy number state is inferred from the changes in the relative depth, i.e. the logR, and an imbalance in the number of maternal and paternal alleles, i.e. the BAF, in the corresponding region. Larger allelic imbalances show up as horizontal bands when the SNP BAFs are plotted against genomic position. More subtle differences (typically subclonal CNAs) can be detected by phasing SNPs within a haplotype block using a large phased genome panel38. CNA reconstruction algorithms use the presence or absence of BAF and logR shifts to segment the genome into regions with constant copy number states. The logR is typically noisier than BAF because it is influenced by local effects such as GC content and replication timing, whereas BAF is relatively unaffected (Figure 3a). Many algorithms correct logR for these types of covariates39.
Figure 3 |. CNA reconstructions and Uncertainty from Whole Genome Duplications.
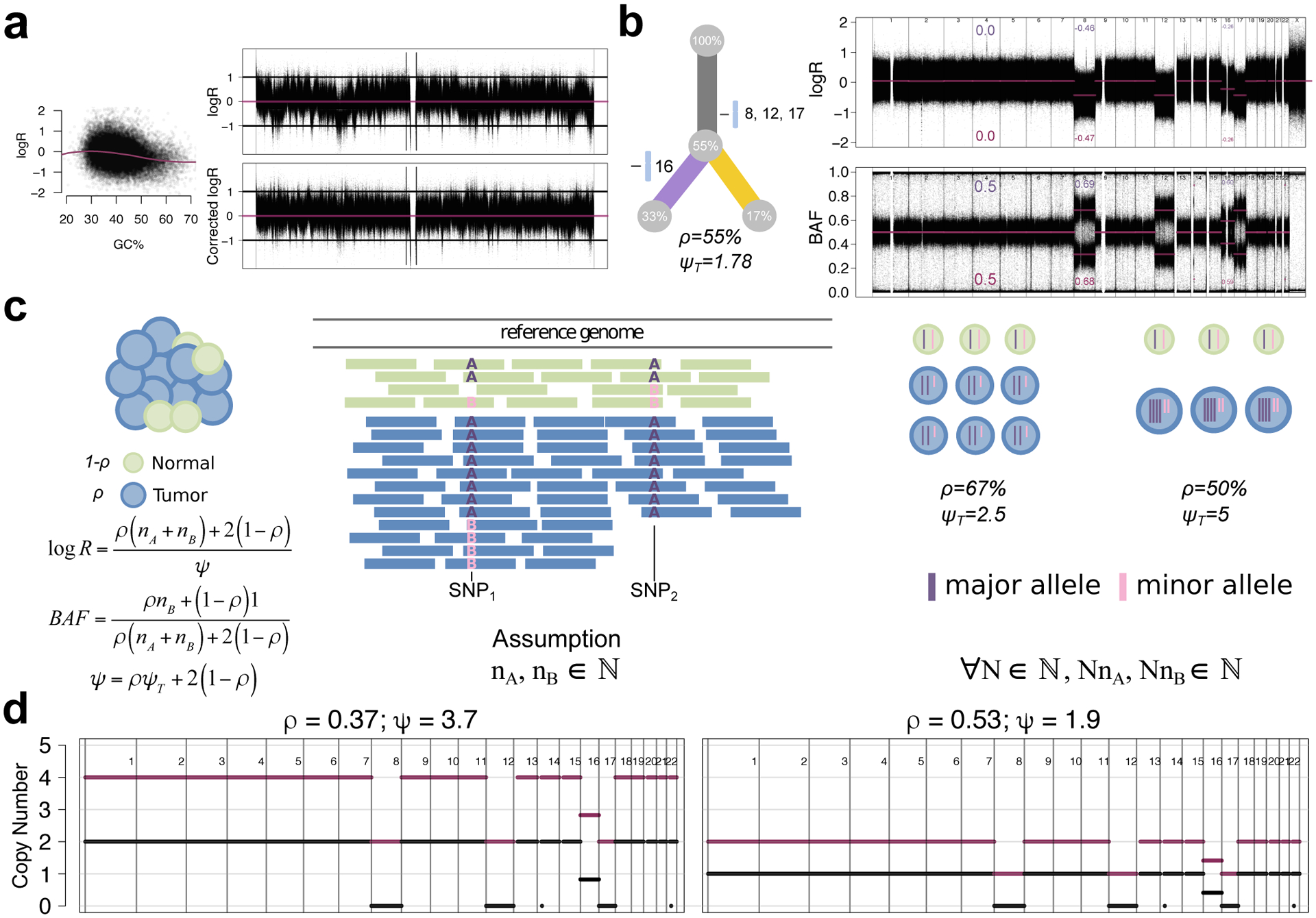
(a) Effect of GC-content on logR. Left: the GC content (% in 500 kbp bins) around SNPs vs. logR for a PCAWG tumour16 with a loess fit (purple). Right: chromosome 22 logR before (top) and after GC and replication timing correction (bottom). (b) logR and BAF reflect relative allele-specific DNA content. Left: the subclonal structure for a tumour with clonal and subclonal chromosomal CNAs. Right: genome-wide logR and BAF with expected (violet) and measured (purple) values for CNAs22. (c) Schematic illustration of ploidy ambiguity. The bulk sample contains tumor (blue) and non-tumor (green) cells. The number of reads from each allele from normal and the tumor cells depends on the number of allelic copies. We show a toy example with two heterozygous SNP positions (A and B alleles). logR and BAF can be expressed as a function of purity ρ, tumor ploidy ψT and the number of major and minor allele copies (nA and nB) in the tumor, which clonally should be integers. Combinations of purity and ploidy values that best align nA and nB to integers are often used to derive copy number profiles. However, multiple combinations can explain the observed data -- multiples of 2ΨT (i.e. a whole genome duplication; WGD) apart. In this example, ΨT = 2.5 and ΨT = 2 × 2.5 = 5 both explain the data. (d) Copy number profiles inferred by Battenberg. Left: along the genome (x-axis) copy number of the major (violet) and minor (grey) allele (default fit, which favored a WGD solution because it fit the subclonal event on chromosome 16 near integers). Right: same as left, after manual refitting.
Copy-number calling algorithms make the relatively strong biological assumption that most copy number events should be clonal, i.e. allow to interpret the logR and BAF as being generated by integer copy number values (Figure 3b). From the literature, experimental ploidy validation has suggested that this assumption was satisfied in most cases (e.g. breast cancers, ovarian cancers, cancer cell lines40) despite most reconstructions bearing at least one subclonal CNA16. Concordantly, emerging DNA single-cell sequencing datasets show that CNA-defined subclones present at a single time point only differ by a few genomic segments26, showing that this assumption is reasonable in many cases.
Segmentation is a critical step in CNA reconstruction because it defines the boundaries of each region of constant copy number state. Segmentation can be done by identifying breakpoints where there is a change in the average read depth and/or BAF. Differences in segmentation lead to many reconstruction differences between methods41. Some methods iteratively join segments of fixed size42; others use changepoint detection algorithms, commonly including circular binary segmentation43, piecewise constant fitting44 or Hidden Markov Models28,38,45,46. Structural variant breakpoints can also help inform segmentation15,47,48.
Once segments are defined, the average major and minor allele copy number for each segment in the cancer’s genome can be estimated from its logR and BAF, and purity and ploidy are estimated from clonal CNAs40 (Figure 3c). CNA segments with integer or near integer values are assumed to be clonal, i.e. all cancer cells in a sample have the same copy number state in that region. Average copy number values that significantly differ from integers (fractional values) typically indicate subclonal CNAs38.
CNA reconstruction then requires interpreting fractional average copy numbers as a mixture of whole number states and ascertaining the proportion of cells in each one. Solutions to this problem are intrinsically ambiguous: for any segment, it is always possible to posit a larger copy number and smaller CP that explain the BAF and logR equivalently well (Figure 3c). Purity and ploidy estimates based on CNA reconstructions are ambiguous for the same reason. CNA detection algorithms usually assess reconstructions based on multiple purity and ploidy estimates. Nevertheless, the final estimates may be incorrect and users should carefully evaluate solutions (Figure 3d).
For clonal CNAs, ambiguity is partially resolved by requiring that the CP of all clonal CNAs must be the same (i.e. equal to the purity). However, in moderate sequencing depth experiments, estimates of exact copy number remain uncertain, as many solutions may be equally likely at any given purity. This can be particularly problematic for estimating copy number in highly amplified regions. There are three main ways to resolve this ambiguity for subclonal CNAs. Genome-wide methods attempt to group subclonal CNAs into subpopulations with the same CP28,48. They can correct for errors in individual segments, but are vulnerable to large-scale errors if they group subclonal CNAs into lineages incorrectly. Event-based methods, such as Battenberg38, apply a set of parsimony rules separately to each copy number segment. They make reconstruction errors in segments where their heuristic is wrong, but those errors are restricted to individual segments. Neither of these approaches can robustly infer more than two subclonal copy number states within a single region41, since they depend on exactly two informative inputs (BAF and logR). The third approach assigns subclonal CNAs to subclonal lineages with defined CP, e.g. via SNV clustering46,49. None of these methods fully resolve the subclonal copy number ambiguity, so we recommend usersconsider only using SNVs in regions of normal copy number or clonal copy number change for subclonal reconstruction.
Troubleshooting CNA Reconstructions
Detecting CNA breakpoints can be challenging, especially in tumors with low effective depth (i.e. low NRPCC). Missing a CNA breakpoint can lead to a series of segments with different clonal copy number states being called as a single segment, which may be miscalled as normal diploid or subclonal. Noise in sequencing data (e.g. from library preparation artifacts) can lead to over-segmentation. Miscalled copy number states can also produce spurious clusters in the final subclonal reconstruction by distorting local CCF estimates. Methods to avoid overfitting include prioritizing BAF over logR38, correcting for GC content38,40 and replication timing effects50, using structural variants as breakpoints15, and automatic adjustment of segmentation parameters38,40.
An intrinsic ambiguity in CNA reconstruction arises from whole-genome duplication (WGD). Any given CNA reconstruction is equivalent to another with each copy number doubled and purity lowered (Figure 3c,d). To resolve this, CNA reconstruction methods often return multiple solutions or select tetraploid solutions only when there is positive evidence of them, e.g. from odd (1, 3, 5, …) values for major or minor allele copy number states. Because some WGD uncertainty usually persists, we recommend using CNA reconstruction methods that allow the user to set the ploidy and re-derive copy numbers and purity to facilitate user-driven assessment of different CNA reconstructions.
Several features in the data can help diagnose purity/ploidy errors41. If the majority of CNAs appear subclonal or known early drivers in the tumor type being studied appear subclonal, the correct purity may be lower than inferred. Missed clonal WGD can sometimes be diagnosed by the presence of subclones with ~50% CCF which contain clonal SNVs acquired after WGD (mutations occurring on one of the four copies), while those occurring at 100% CCF really represent mutations that occur before WGD and are hence present on two of the four copies. Observing a 50% CCF subclone in multiple samples would further support WGD, as subclones are unlikely to occur at the same CCF across samples otherwise. In the case of multi-sample reconstruction, purity can be estimated from CNA-adjusted VAFs of SNVs present in all regions9. Comparing purity estimates from CNA-only, SNV-only and SNV+CNA subclonal reconstructions can further guide purity inference and is especially useful in tumors with few clonal CNAs (e.g. papillary thyroid carcinomas and some leukemias)15.
Experimental purity and ploidy estimates can also support CNA fitting. CNA-detection methods can fit copy number solutions informed by experimental ploidy estimates, as in silico estimates of tumor ploidy have been repeatedly shown to match experimental values40,51,52. Experimental purity and ploidy estimates can be obtained through FISH, image cytometry, FACS, or single-cell sequencing53.
SNV clustering
Overview
Before SNVs can be clustered, their measured variant allele frequencies must be transformed into cellular frequencies (CCF or CP) using purity and copy number. It is essential to adjust for CNAs when converting VAFs to cellular frequencies, or to only cluster SNVs in normal diploid regions, as copy number gains and losses will alter the fraction of reads bearing a SNV. Neglecting to adjust for these effects can lead to incorrect clustering (Figure 1b,d)54. Cellular prevalence estimates for SNVs in normal diploid regions can be clustered first to identify the major subclonal lineages. In normal diploid regions, CP is precisely twice VAF, so clustering by VAF and implied CPs is equivalent. The cluster with the highest CP can be deemed clonal, and the remaining clusters can be assigned CPs and associated with a subclonal lineage. Errors can arise from incorrect cluster number estimation or SNV-lineage mis-assignment, potentially shifting the clonal peak, from which purity can be estimated. In general, the clustering principles outlined in this section apply equivalently to indels.
The assumed noise distribution in VAF estimates will influence subclonal reconstruction accuracy. Using an inappropriate noise model can lead to over- or under-estimating cluster number. Binomial noise models can capture the influence of the read depth, copy number state and CP on the accuracy of the assessed SNV VAF, while in general fixed variance Gaussian noise models cannot. Over-dispersed binomial models (e.g. beta-binomial31 or negative binomial, which are often used for bisulfite, exome or single-cell RNA sequencing data) assume greater variance than standard binomial models, but it is unclear whether they are better-suited for subclonal reconstruction from DNA data. Like Binomial models, Beta models are also suited for subclonal reconstruction, as they model VAF directly24,55.
Care should be taken when translating SNV VAFs to CCF space prior to clustering, as this requires estimating the multiplicity of each SNV, i.e. the number of tumor DNA copies harboring it. SNV multiplicity estimates depend on the accuracy of copy number calls, as before assignment, it is necessary to enumerate the space of possible multiplicities. This requires allele-specific copy number estimates, as total copy number is insufficient to set boundaries of possible multiplicities. For example, in the case of copy-neutral loss-of-heterozygosity, the total copy number is two, and individual SNVs can have a multiplicity of one (for that mutations that occur after the duplication) or two (for those mutations that happen before it). By contrast, in balanced diploid regions, the total copy number is again two, but each individual SNV must have a multiplicity of one.
If the CCF values of clonal SNVs are computed assuming a multiplicity of one, then the resulting estimated CCFs will be approximately equal to the (real) multiplicity of the SNV in the clonal lineage (see Supplementary Information). However, subclonal changes in SNV multiplicity equate to subclonal copy number changes, and because subclonal copy number states are ambiguous, the affected SNVs may generate spurious clusters. Nonetheless, CCF clustering can sometimes detect, but not correct, errors in CNA reconstruction. For example, large SNV clusters with CCFs > 1 (super-clonal clusters, see Lexicon) are theoretically impossible for somatic mutations. When they are detected in subclonal reconstruction, this can be diagnostic of failure in detecting the clonal lineage during CNA reconstruction, or of large segments with wrong copy number calls. In the former case, purity will be incorrect (i.e. underestimated), and the clonal peak will be shifted upwards in CCF space. In the latter case, SNV multiplicity and thus CCF will be incorrect. These errors often occur in tumors without CNAs or without any clonal CNAs, or as a result of contamination by germline variants (discussed below). In these cases, the CNA-based purity will be underestimated, leading to CCFs > 1 for SNVs in these super-clonal lineages.
To address these concerns, a number of methods use generative models of VAFs that incorporate CNA reconstructions. These methods assess the impact of clonal and subclonal CNAs on SNV multiplicity (and their associated changes in VAF) using maximum likelihood25,31,38,46,56. Of these, PhyloWGS25 and LICHEe56 attempt full phylogenetic reconstructions. CloneHD46 does not explicitly enforce consistent tree structure, PyClone31 assumes a single CNA change per segment without reconstructing a clonal tree, and DPClust38 assigns SNV multiplicities to the most likely value, given the CNA reconstruction.
Assumptions underlying SNV clustering
Methods based on SNV clustering rely on several assumptions about cancer evolution. The first assumption is that the majority of SNVs with detectable VAFs are associated with a small number of subclonal lineages (weak parsimony)25. Given the large number of cells in a bulk tumor and the positive mutation rate per cell division, the existence of a very large number of low-prevalence SNVs is uncontroversial. Their low VAF typically precludes detection by typical somatic mutation calling algorithms, as detectable subclonal lineages are primarily established through selective sweeps and early drift57,58. This assumption is somewhat controversial however, as the lowest-VAF cluster may contain a mixture of SNVs coming from numerous parallel lineages growing neutrally59,60. Therefore, the lowest VAF cluster might be a mix of subclones, and efforts are ongoing to characterize this to capture non-tail subclones24.
A second assumption implicitly made by many clustering-based algorithms is that a given genomic position is subject to an SNV only once during the development of an individual tumor, and never reverts to the germline state (the infinite sites assumption, Lexicon). As a result, each SNV can be uniquely assigned to a specific subclonal lineage31,56,61. It is known that the infinite sites assumption can be violated, as exemplified by the existence of parallel acquisition of the same driver SNVs and by tri-allelic loci. As a result, some methods do not make this assumption62. Infinite sites violations occur rarely enough that they are not expected to impact clustering based on hundreds to thousands of SNVs. As a result, this approximation remains widely used for subclonal reconstruction through bulk whole-genome and whole-exome sequencing, and it may even be reasonable for targeted sequencing studies of driver genes.
Subclonal losses of chromosomal segments may also lead to somatic variants disappearing in these lineages and thereby variants may appear to revert to the germline state, leading to apparent violations of the infinite sites assumption. This situation is relatively frequent, particularly in multi-sample reconstruction, and may lead to the appearance of spurious clusters. Apart from disregarding mutations in regions showing subclonal copy number events, another elegant way to account for this in subclonal reconstruction is by modelling mutations through a Dollo process, which assumes mutations can occur only once, but can subsequently disappear again (only once)14.
Many methods also make implicit “hidden” assumptions about the number of clusters or their density. For example many clustering methods rely on some form of Dirichlet Process clustering14,25,31,38,56,63. This technique has an important hyperparameter called the concentration parameter, which can be inferred from the data or be constrained with strong priors. We recommend testing different values for these parameters to quantify their impact on the subclonal reconstruction for any given tumor (see Supplementary Information).
Troubleshooting SNV clustering
When two subclonal lineages show similar CPs, their VAF distributions may overlap. As a result, clustering algorithms will merge these lineages without further information (e.g. using additional samples). Nonetheless, it is possible to be highly confident about the presence and CP of a lineage while being uncertain about the assignment of many of its SNVs.
Variant calling accuracy will impact clustering accuracy22,23. Germline SNPs wrongly called as somatic SNVs have high VAFs (~0.5) (Figure 1b,d), and may be clustered into their own high CCF lineage (>100% CCF). These clusters tend to have few SNVs, which have a specific mutational signature (primarily C>T and T>C), and contain few or no CNAs. Filtering against a database of germline variants (e.g. dbSNP) and higher-depth normal sample sequencing can reduce the risk of germline contamination, although filtering against a database like dbSNP can lead to increased numbers of false-negative somatic SNVs, particularly as these databases grow. False-positive SNVs due to sequencing errors have low VAFs and mostly will be assigned to a low-frequency lineage, which can help identify them. They may also have a distinct mutational signature64. False-negatives in somatic SNV calling can lead to overestimating the CPs of low frequency subclones because only the higher part of the VAF distribution is observed. CPs can be adjusted for this bias15, and using a highly sensitive SNV detection algorithm can also help mitigate this effect.
We have observed that algorithms that assume a diploid copy neutral state can struggle with mutations on the male sex chromosomes. An easy fix is to exclude SNVs on these chromosomes. In theory, these SNVs could be reassigned post-hoc with appropriate treatment, although many CNA-reconstruction methods do not report calls on chromosome Y. Tetraploid tumors will likely be annotated as diploid if they have few other CNAs, leading to spurious clusters (see the trouble-shooting section on CNA reconstructions, above). Artefact clusters also sometimes result from incorrect copy number calls, which can lead to incorrect adjustment of allele frequencies to CPs. Such clusters are easily identified, as all the associated SNVs are located in a single chromosomal region.
Phylogenetic reconstruction
Given the weak parsimony assumption, SNV clusters represent groups of mutations that occurred in one (or a few) subclones at one point in the tumor’s evolution, inherited by all of their descendants, i.e. members of their subclonal lineages. These clusters represent the ancestors of the subpopulations present in the tumor at the time of sample acquisition. Thus, SNVs clustered in CCF space are assigned to the same lineage. Assigning these mutations to existing subpopulations in the sample requires additional information, namely the ancestor relationships between these lineages or the tumor phylogeny, often represented as a clone tree (Figure 1d). We believe tree inference should generally be restricted to multi-sample studies: while subclones can be identified in single samples, they are only weakly informative of the underlying tree (Figure 2b).
The weak parsimony and infinite sites assumptions restrict the phylogenies consistent with a set of inferred CPs. Assuming SNVs do not revert to their germline state implies that descendent subpopulations inherit all the mutations in their ancestors. The CP of an ancestral lineage must be at least as large as the sum of the CPs of its direct descendants38 (this concept is known as the pigeonhole principle, see Lexicon). Consider a common ancestor A with two descendants (lineages B & C), whose relationship is unknown. If CP(B) > CP(C), and CP(B) + CP(C) > CP(A), then B must be an ancestor of C63. Tumor phylogenetic methods employ some version of these rules either explicitly or implicitly63.
When there are multiple subclonal lineages, and few samples, multiple phylogenies are usually consistent with a given set of lineage CPs. Because any set of CPs from a single sample are often consistent with a linear phylogeny, it is typically difficult to unambiguously call a branching phylogeny from single sample CPs. Branching can be inferred in some cases when the CP of an SNV cluster is incompatible with being either an ancestor or a descendant of a CNA, which can sometimes be detected automatically in a single sample14,25. Branching can also be detected in a single sample when nearby or overlapping SNVs on the same chromosome copy are identified as mutually exclusive through read phasing41 (Figure 1b). However, with short-read data, it is often difficult to phase enough somatic SNVs for a high-quality reconstruction, though some methods automatically apply this approach65. As some phylogenetic ambiguity usually persists, and choice of a single phylogeny is often arbitrary or depends on weakly validated assumptions61,66, phylogenetic methods should report uncertainty in the reported phylogeny.
In single sample studies15, phasing of SNVs can inform branching subclones and the pigeonhole principle helps identify linear subclones, although most phylogenies remain unresolved. Multiple samples can greatly clarify phylogenetic relationships amongst subclones (Figure 2b). For example, if CP(B) > CP(C) in one sample but CP(C) > CP(B) in another sample, then these subclones must be cousins or siblings. Thus, though branching is usually impossible to establish using a single sample, it is often possible with just two samples and becomes increasingly easy to identify with additional samples. Subclonal reconstruction methods generalize trivially to clustering SNVs in multiple CCF dimensions and the same principles and assumptions apply. Notably, chromosomal losses leading to variants being lost in subclonal lineages and thereby apparent infinite sites violations are quite common and, if overlooked, can lead to errors in both multi- and single sample phylogeny reconstruction.
Evolution of the Field
DNA sequencing technologies continue to improve, with major ongoing advances in long-read and single-cell sequencing. We anticipate these technologies will be useful for reducing subclonal reconstruction uncertainty and improving accuracy. Long-read sequencing allows more accurate breakpoint detection, copy number state characterization and longer-range phasing relative to short read sequencing67–69. These will all improve CNA reconstruction, reducing downstream errors, while long-range phasing will facilitate phylogeny inference through mutual exclusivity. Single-cell WGS will become increasingly useful for phylogenetic inference as its CNA and SNV detection accuracy improves70,71. Both these technologies can be combined with short-read sequencing to reduce costs and leverage their strengths but retain the resolution of high-depth sequencing72,73. Subclonal reconstruction algorithms will need to accommodate the data, biases, and errors these and other new technologies generate. Carefully characterizing error profiles at each step is critical for interpretation as errors are likely to propagate and impact the final reconstruction.
Both WGS and WES have limited sensitivity for studying intra-tumor heterogeneity in single samples74,75. Bulk sequencing of multiple tumor regions still misses many subclones, particularly when subclones are not evenly distributed across the tumor mass. While cell turnover facilitates subclone mixing, spatial subclone segregation can arise through local differences in the microenvironment and subclone competition. As subclone distributions remain difficult to predict, and cannot be exhaustively sampled, emerging liquid biopsy76 and homogenized tissue sampling technologies77 can provide a more complete evolutionary profile than even dense tissue sampling, by providing a complementary sampling of the tumor with different biases. These technologies are especially useful when detecting minor subclones is a priority (e.g. to detect evolving post-treatment resistance). They typically employ targeted gene panels to allow sufficient depth for accurate SNV calling and subclonal reconstruction. However, their sensitivity may depend on tumor features and their efficacy in diverse clinical contexts is not yet clear17.
Single-cell sequencing can resolve phylogenies in greater detail and with less uncertainty than bulk sequencing10,14,78,79 and the technology is rapidly developing80. Due to artefacts left during amplification of the genetic material, and limited genome coverage, calling SNVs in single-cell data remains an important challenge, with active development70. Many alternatives have been explored, such as using the cell’s own high-fidelity replicative machinery by expanding colonies from single cells, on which to perform bulk whole-genome sequencing81; using direct library preparation of thousands of cells82, which simply skip the error-prone amplification; or using single-cell targeted sequencing approaches to maximize local depth of coverage, also allowing allele-specific copy number inference in single-cells26.
Although inferring the genotypes of each subclone may be more difficult using bulk than single-cell sequencing, bulk-sequencing tissue samples from multiple tumor regions can mitigate some of these limitations13. We thus recommend combining bulk and single-cell when possible for phylogeny reconstruction, which has shown great potential to resolve ambiguities in the phylogenetic tree reconstruction due to the single cell resolution, while maintaining high-quality somatic mutation calls from the bulk14,81. However, if cells are sampled from a small number of regional biopsies and there is limited clonal mixing, spatial biases may still impact single-cell phylogenetic reconstruction83.
Subclonal reconstruction is a fast-evolving field. Further work is underway to integrate additional data types into subclonal reconstructions (e.g. structural variants which, given the higher noise in their VAF, are assigned to subclones post hoc84,85, or epigenetic marks56) but these will require careful validation given the uncertainty already present in subclonal reconstruction. Similarly, mechanistic models and approaches have recently been proposed13,59,60, but remain in their infancy59,60,86–89. In the future, combining the phenomenological models discussed with mechanistic models is likely to prove invaluable24. A thorough assessment of new and existing methods, particularly for subclonal CNA detection, is acutely needed. Precise metrics of reconstruction accuracy, increasingly realistic synthetic tumor genomes and joint single-cell and bulk tumor sequencing datasets22,73 are all required to establish community-accepted benchmarks that drive algorithm development and application.
Supplementary Material
Acknowledgments
A.S. was supported by an NSERC CGS. M.T. and P.V.L. are supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001202), the UK Medical Research Council (FC001202), and the Wellcome Trust (FC001202). M.T. is a postdoctoral fellow supported by the European Union’s Horizon 2020 research and innovation program (Marie Skłodowska-Curie Grant Agreement No. 747852-SIOMICS). P.V.L. is a Winton Group Leader in recognition of the Winton Charitable Foundation’s support towards the establishment of The Francis Crick Institute. M.N.L. was supported by a Junior Research Fellowship (Trinity College, University of Oxford). P.C.B. was supported by a Terry Fox Research Institute New Investigator Award and a CIHR New Investigator Award. Q.M. is supported by an Associate Investigator Award from the Ontario Institute of Cancer Research, and holds a Canada CIFAR AI chair. This work was supported by the NIH/NCI under award number P30CA016042 (P.C.B), P30-CA008748 (Q.M) and through support from the ITCR (1U24CA248265–01) to PCB.
Footnotes
Competing interests
P.C.B is a member of the Scientific Advisory Boards of BioSymetrics Inc. and Intersect Diagnostics Inc. M.T., A.S., A.G.D., M.N.L., J.W., D.C.W., Q.D.M., and P.V.L. declare no competing interests.
Ethics Declaration
This article does not involve animals nor human participants.
References
- 1.Hanahan D & Weinberg RA Hallmarks of Cancer : The Next Generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Nowell PC The clonal evolution of tumor cell populations. Science 194, 23–28 (1976). [DOI] [PubMed] [Google Scholar]
- 3.Gundem G et al. The Evolutionary History of Lethal Metastatic Prostate Cancer. Nature 520, 353–357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong MKH et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nature Communications 6, 1–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell TJ et al. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell 173, 611–623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turajlic S et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell 173, 581–594 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andor N et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nature medicine 22, 105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Espiritu SMG et al. The Evolutionary Landscape of Localized Prostate Cancers Drives Clinical Aggression. Cell (2018). [DOI] [PubMed] [Google Scholar]
- 9.Jamal-Hanjani M et al. Tracking the evolution of non--small-cell lung cancer. New England Journal of Medicine 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Fittall MW & Van Loo P Translating insights into tumor evolution to clinical practice: promises and challenges. Genome medicine 11, 20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sendorek DH et al. Germline contamination and leakage in whole genome somatic single nucleotide variant detection. BMC Bioinformatics 19, 28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alioto TS et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nature Communications 6, 1–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun R et al. Between-region genetic divergence reflects the mode and tempo of tumor evolution. Nature genetics 49, 1015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salehi S et al. ddClone: joint statistical inference of clonal populations from single cell and bulk tumour sequencing data. Genome biology 18, 44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dentro SC et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. bioRxiv 312041 (2020) doi: 10.1101/312041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerstung M et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abbosh C et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 545, 446 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noorani A et al. Genomic evidence supports a clonal diaspora model for metastases of esophageal adenocarcinoma. Nature Genetics 52, 74–83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGranahan N et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez K et al. Somatic evolutionary timings of driver mutations. BMC cancer 18, 85 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Opasic L, Zhou D, Werner B, Dingli D & Traulsen A How many samples are needed to infer truly clonal mutations from heterogenous tumours? BMC cancer 19, 403 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salcedo A et al. A community effort to create standards for evaluating tumor subclonal reconstruction. Nature Biotechnology 38, 97–107 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffith M et al. Optimizing cancer genome sequencing and analysis. Cell systems 1, 210–223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caravagna G et al. Subclonal reconstruction of tumors by using machine learning and population genetics. Nature Genetics 52, 898–907 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deshwar AG et al. PhyloWGS: reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome biology 16, 35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laks E et al. Clonal decomposition and DNA replication states defined by scaled single-cell genome sequencing. Cell 179, 1207–1221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwarz RF et al. Phylogenetic quantification of intra-tumour heterogeneity. PLoS computational biology 10, e1003535 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ha G et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome research 24, 1881–1893 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El-Kebir M SPhyR: tumor phylogeny estimation from single-cell sequencing data under loss and error. Bioinformatics 34, i671--i679 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roth A et al. PyClone: statistical inference of clonal population structure in cancer. Nature methods 11, 396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi W et al. Reliability of whole-exome sequencing for assessing intratumor genetic heterogeneity. Cell reports 25, 1446–1457 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yates LR, Gerstung M, Knappskog S & Desmedt C Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nature Medicine 21, 751–759 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuh A et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood 120, 4191–4196 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Boutros PC et al. Spatial genomic heterogeneity within localized , multifocal prostate cancer. Nature Publishing Group 47, 736–745 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Robbe P et al. Clinical whole-genome sequencing from routine formalin-fixed, paraffin-embedded specimens: pilot study for the 100,000 Genomes Project. Genetics in Medicine 20, 1196–1205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chin S-F et al. Shallow whole genome sequencing for robust copy number profiling of formalin-fixed paraffin-embedded breast cancers. Experimental and molecular pathology 104, 161–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nik-zainal S et al. The Life History of 21 Breast Cancers. Cell 149, 994–1007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deshpande A, Walradt T, Hu Y, Koren A & Imielinski M Robust foreground detection in somatic copy number data. bioRxiv 847681 (2019). [Google Scholar]
- 40.Van Loo P et al. Allele-specific copy number analysis of tumors. Proceedings of the National Academy of Sciences 107, 16910–16915 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dentro SC, Wedge DC & Van Loo P Principles of reconstructing the subclonal architecture of cancers. Cold Spring Harbor perspectives in medicine 7, a026625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiang DY et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nature methods 6, 99 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olshen AB, Venkatraman ES, Lucito R & Wigler M Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Nilsen G et al. Copynumber: efficient algorithms for single-and multi-track copy number segmentation. BMC genomics 13, 591 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lai D & Shah S HMMcopy: copy number prediction with correction for GC and mappability bias for HTS data. R package version 1, (2012). [Google Scholar]
- 46.Fischer A, Vázquez-Garc’ia I, Illingworth CJR & Mustonen V High-definition reconstruction of clonal composition in cancer. Cell reports 7, 1740–1752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McPherson AW et al. ReMixT: clone-specific genomic structure estimation in cancer. Genome biology 18, 140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oesper L, Mahmoody A & Raphael BJ THetA: inferring intra-tumor heterogeneity from high-throughput DNA sequencing data. Genome biology 14, R80 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang Y, Qiu Y, Minn AJ & Zhang NR Assessing intratumor heterogeneity and tracking longitudinal and spatial clonal evolutionary history by next-generation sequencing. Proceedings of the National Academy of Sciences 113, E5528--E5537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Müller CA et al. The dynamics of genome replication using deep sequencing. Nucleic Acids Research 42, e3--e3 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carter SL et al. Absolute quantification of somatic DNA alterations in human cancer. Nature biotechnology 30, 413 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steele CD et al. Undifferentiated sarcomas develop through distinct evolutionary pathways. Cancer Cell 35, 441–456 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Almendro V et al. Genetic and phenotypic diversity in breast tumor metastases. Cancer research 74, 1338–1348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farahani H et al. Engineered in-vitro cell line mixtures and robust evaluation of computational methods for clonal decomposition and longitudinal dynamics in cancer. Scientific Reports 7, 13467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller CA et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS computational biology 10, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Popic V et al. Fast and scalable inference of multi-sample cancer lineages. Genome biology 16, 91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nowell PC The clonal evolution of tumor cell populations. Science 194, 23–28 (1976). [DOI] [PubMed] [Google Scholar]
- 58.Sottoriva A et al. A Big Bang model of human colorectal tumor growth. Nature genetics 47, 209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williams MJ, Werner B, Barnes CP, Graham TA & Sottoriva A Identification of neutral tumor evolution across cancer types. Nature genetics 48, 238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams MJ et al. Quantification of subclonal selection in cancer from bulk sequencing data. Nature genetics 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Strino F, Parisi F, Micsinai M & Kluger Y TrAp: a tree approach for fingerprinting subclonal tumor composition. Nucleic acids research 41, e165--e165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marass F et al. A phylogenetic latent feature model for clonal deconvolution. The Annals of Applied Statistics 10, 2377–2404 (2016). [Google Scholar]
- 63.Jiao W, Vembu S, Deshwar AG, Stein L & Morris Q Inferring clonal evolution of tumors from single nucleotide somatic mutations. BMC bioinformatics 15, 35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ewing AD et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nature Methods 12, 623–630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou T, Müller P, Sengupta S & Ji Y PairClone: a Bayesian subclone caller based on mutation pairs. Journal of the Royal Statistical Society: Series C (Applied Statistics) 68, 705–725 (2019). [Google Scholar]
- 66.El-Kebir M, Satas G & Raphael BJ Inferring parsimonious migration histories for metastatic cancers. cancer 2, 5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Esteki MZ et al. Concurrent whole-genome haplotyping and copy-number profiling of single cells. The American Journal of Human Genetics 96, 894–912 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mantere T, Kersten S & Hoischen A Long-read sequencing emerging in medical genetics. Frontiers in genetics 10, 426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dong X et al. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nature methods 14, 491–493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martelotto LG et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nature medicine 23, 376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huddleston J et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome research 27, 677–685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malikic S, Jahn K, Kuipers J, Sahinalp SC & Beerenwinkel N Integrative inference of subclonal tumour evolution from single-cell and bulk sequencing data. Nature communications 10, 2750 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abécassis J et al. Assessing reliability of intra-tumor heterogeneity estimates from single sample whole exome sequencing data. PloS one 14, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu LY et al. Quantifying the Influence of Mutation Detection on Tumour Subclonal Reconstruction. bioRxiv 418780 (2020) doi: 10.1101/418780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parikh AR et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nature medicine 25, 1415–1421 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Litchfield DK et al. Representative Sequencing: Unbiased Sampling of Solid Tumor Tissue. (2019). [DOI] [PubMed]
- 78.Eirew P et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 518, 422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim C et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 173, 879–893 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gawad C, Koh W & Quake SR Single-cell genome sequencing: current state of the science. Nature Reviews Genetics 17, 175 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Yoshida K et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578, 266–272 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zahn H et al. Scalable whole-genome single-cell library preparation without preamplification. Nature methods 14, 167 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Chkhaidze K et al. Spatially constrained tumour growth affects the patterns of clonal selection and neutral drift in cancer genomic data. PLoS computational biology 15, e1007243 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eaton J, Wang J & Schwartz R Deconvolution and phylogeny inference of structural variations in tumor genomic samples. Bioinformatics 34, i357--i365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cmero M et al. Inferring structural variant cancer cell fraction. Nature communications 11, 1–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Noorbakhsh J & Chuang JH Uncertainties in tumor allele frequencies limit power to infer evolutionary pressures. Nature genetics 49, 1288 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tarabichi M et al. Neutral tumor evolution? Nature genetics 50, 1630 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heide T et al. Reply to ‘Neutral tumor evolution?’ Nature Genetics 50, 1633–1637 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Williams MJ, Werner B, Barnes CP, Graham TA & Sottoriva A Reply: Uncertainties in tumor allele frequencies limit power to infer evolutionary pressures. Nature Genetics 49, 1289–1291 (2017). [DOI] [PubMed] [Google Scholar]
- 90.Zare F, Dow M, Monteleone N, Hosny A & Nabavi S An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC bioinformatics 18, 286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gerlinger M, Horswell S, Larkin J, Rowan AJ & Salm MP Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nature Communications 46, 225–233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vinci M et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nature medicine 24, 1204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Noorani A et al. Genomic evidence supports a clonal diaspora model for metastases of esophageal adenocarcinoma. Nature Genetics 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuipers J, Jahn K, Raphael BJ & Beerenwinkel N Single-cell sequencing data reveal widespread recurrence and loss of mutational hits in the life histories of tumors. Genome research 27, 1885–1894 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rieber N et al. Coverage Bias and Sensitivity of Variant Calling for Four Whole-genome Sequencing Technologies. PLOS ONE 8, e66621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.