

Summary
Stress-induced readthrough transcription results in the synthesis of downstream-of-gene (DoG) containing transcripts. The mechanisms underlying DoG formation during cellular stress remain unknown. Nascent transcription profiles during DoG induction in human cell lines using TT-TimeLapse-seq revealed widespread transcriptional repression upon hyperosmotic stress. Yet, DoGs are produced regardless of the transcriptional level of their upstream genes. ChIP-seq confirmed that stress-induced redistribution of RNA Polymerase (Pol) II correlates with the transcriptional output of genes. Stress-induced alterations in the Pol II interactome are observed by mass spectrometry. While certain cleavage and polyadenylation factors remained Pol II-associated, Integrator complex subunits dissociate from Pol II under stress leading to a genome-wide loss of Integrator on DNA. Depleting the catalytic subunit of the Integrator using siRNAs induces hundreds of readthrough transcripts, whose parental genes partially overlap those of stress-induced DoGs. Our results provide insights into the mechanisms underlying DoG production and how Integrator activity influences DoG transcription.
Graphical Abstract
In brief
Rosa-Mercado et al. report that hyperosmotic stress causes widespread transcriptional repression in human cells, yet downstream-of-gene transcripts (DoGs) arise regardless of the transcriptional response of their upstream genes. Interactions between Pol II and Integrator are disrupted by hypertonicity and knockdown of the Integrator nuclease leads to DoG production.
Introduction
Conditions of cellular stress, such as hyperosmotic stress, heat shock, oxidative stress, viral infection and renal cancer lead to readthrough transcription (Vilborg et al. 2015; Vilborg et al. 2017; Rutkowski et al. 2015; Bauer et al. 2018; Grosso et al. 2015; Vilborg and Steitz 2017). This phenomenon is defined as transcription extending past the annotated termination sites of genes. This stress-induced transcription produces thousands of long noncoding RNAs (lncRNAs), known as downstream-of-gene (DoG) containing transcripts. DoGs arise minutes after stress from approximately 10% of human protein-coding genes and are continuous with their upstream mRNAs. By definition, DoGs extend over 5 kilobases (kb) past the normal termination site of their gene of origin. They can be over 200 kb long, but have an average length of 14 kb. DoGs are retained in the nucleus and largely remain localized close to their site of transcription (Vilborg et al. 2015; Vilborg et al. 2017; Hennig et al. 2018).
Upon influenza and HSV-1 infection, the induction of readthrough transcripts is orchestrated in part by viral proteins NS1 and ICP27, respectively. These proteins interfere with transcription termination by interacting with host cleavage and polyadenylation (CPA) factors (Nemeroff et al. 1998; Bauer et al. 2018; Wang et al. 2020). Accordingly, knockdown of the catalytic subunit of the CPA complex, CPSF73, partially induces DoGs (Vilborg et al. 2015). However, host-dependent mechanisms underlying DoG production upon environmental stress, such as hyperosmotic stress, remain unknown.
Readthrough transcripts have been previously characterized using nuclear RNA sequencing, 4sU-sequencing, mNET-seq and total RNA sequencing (Vilborg et al. 2015; Rutkowski et al. 2015; Hennig et al. 2018; Bauer et al. 2018; Grosso et al. 2015). While these techniques enable the genome-wide identification of DoG transcripts, most do not provide quantitative information regarding the nascent transcription profiles that coincide with DoG induction. Here we use Transient Transcriptome (TT) sequencing coupled with TimeLapse (TL) chemistry (Schwalb et al. 2016; Schofield et al. 2018) to characterize transcriptional profiles that accompany DoG induction upon hyperosmotic stress. This method enables reliable detection of nascent RNAs by using short pulses of metabolic labeling coupled with transcript enrichment and nucleoside recoding chemistry.
The Integrator complex is important for the 3´ end processing of various noncoding RNAs, such as small nuclear RNAs (snRNAs), enhancer RNAs, viral microRNAs, and lncRNAs, as well as replication-dependent histone pre-mRNAs (Albrecht and Wagner 2012; Lai et al. 2015; Xie et al. 2015; Nojima et al. 2018; Skaar et al. 2015). Integrator also regulates promoter-proximal Pol II pausing at protein-coding genes (Stadelmayer et al. 2014; Gardini et al. 2014; Elrod et al. 2019; Beckedorff et al. 2020). Integrator 11 (Int11) is the catalytic subunit of the complex and forms a heterodimer with Int9 (Baillat et al. 2005; Wu et al 2017). These subunits are analogous to CPSF73 and CPSF100 (Dominski et al. 2005; Baillat and Wagner 2015), respectively, which are essential for the 3´ processing of mRNAs (Mandel et al. 2006; Shi et al. 2009).
Here, we report that hyperosmotic stress causes widespread transcriptional repression, from which a minority of human genes escape. Yet, DoGs are induced from genes that experience different transcriptional responses to hyperosmotic stress. ChIP-seq experiments show that Pol II binding profiles correlate with the transcriptional output of genes after hyperosmotic stress. Furthermore, our proteomics results reveal changes in the Pol II interactome after stress. Specifically, we observe a decreased interaction between Pol II and subunits of the Integrator complex, while the interactions between Pol II and factors known to mediate the termination of protein-coding genes are unchanged. ChIP-seq experiments reveal a genome-wide decrease of Int11 and Int3 occupancy on DNA. Correspondingly, siRNA knockdown of Int11 was sufficient to induce readthrough transcription at hundreds of genes. The identity of genes that produce readthrough transcripts upon depletion of functional Int11 partially overlaps the collection of genes that give rise to DoGs after hyperosmotic stress. Together, our results provide mechanistic insights into the biogenesis of a recently characterized class of noncoding RNAs and reveal a novel role for the Integrator complex in transcriptional regulation.
Results
Hyperosmotic stress causes widespread transcriptional repression
We established the nascent transcriptional profiles accompanying DoG induction by performing TT-TL-seq (Schofield et al. 2018) of untreated HEK-293T cells and cells exposed to hyperosmotic stress (Fig. 1A). Specifically, we exposed cells to 80 mM KCl for 60 minutes, but added the nucleoside analog 4-thiouridine (s4U) during the last 5 minutes to label RNAs being actively transcribed (Schwalb et al. 2016). After extracting RNA from HEK-293T cells, RNA from Drosophila S2 cells was added to each sample as a normalization control to ensure accurate differential expression analysis (Lovén et al. 2012; Chen et al. 2015). RNAs containing s4U were then biotinylated using MTS chemistry and enriched on streptavidin beads (Duffy et al. 2015). Finally, U-to-C mutations were induced using TL chemistry to assess the nascent nature of the enriched RNAs (Schofield et al. 2018). TT-TL-seq experiments were performed using conditions previously found to induce DoGs (Vilborg et al. 2017). RT-qPCR results from HEK-293T cells exposed to hyperosmotic stress for different times confirmed that DoGs were robustly induced after 1 hour of treatment (Fig. S1A), consistent with previous results from NIH-3T3 cells (Vilborg et al. 2017).
Figure 1:
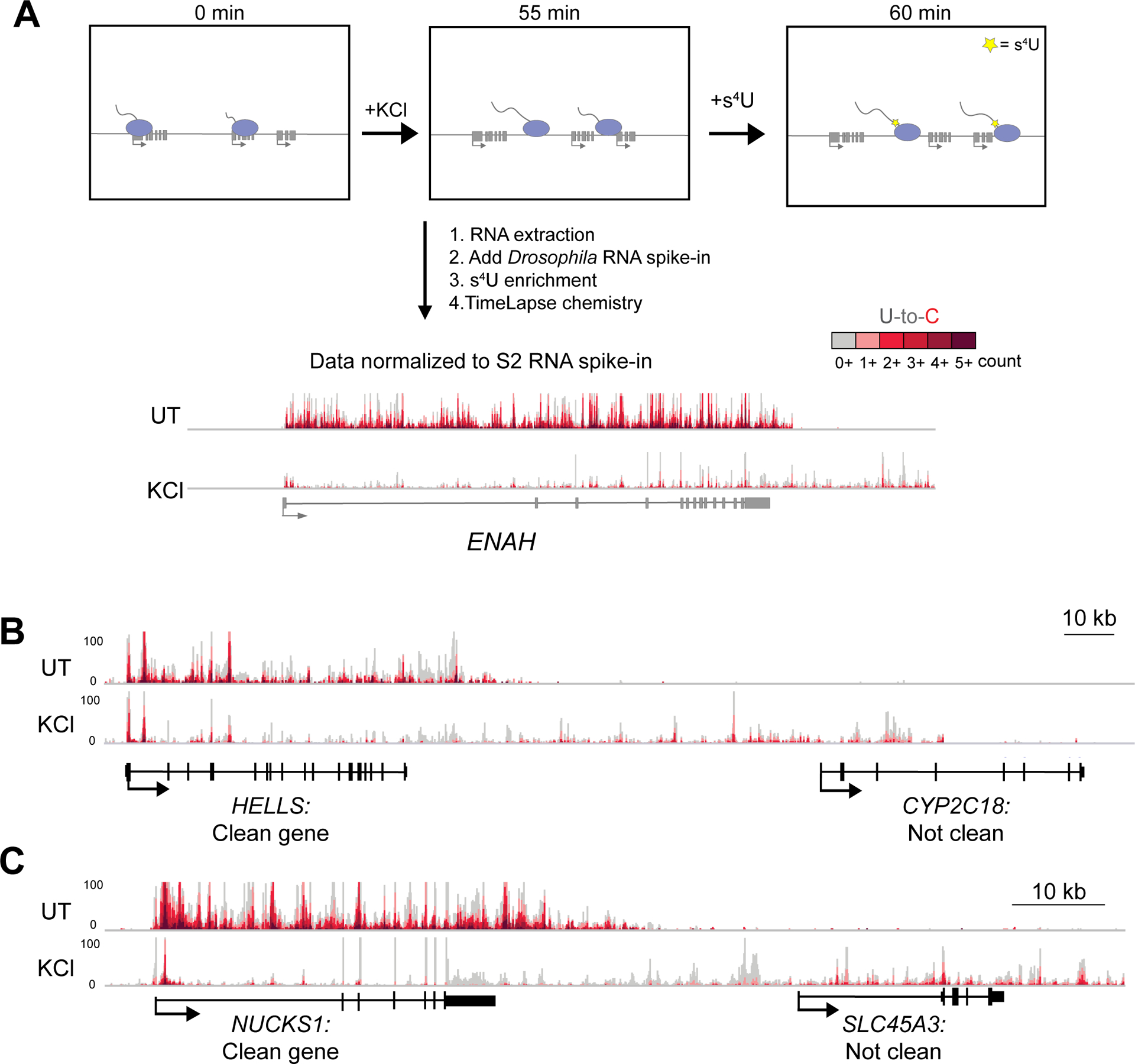
TT-TL-seq reveals transcriptional profiles that accompany DoG induction after hyperosmotic stress. A) Setup for TT-TimeLapse sequencing (TT-TL-seq) experiments in HEK-293T cells. An arrow indicating directionality marks the beginning of each transcription unit. Exons are shown as rectangles and Pol II molecules are light purple ovals with attached nascent RNAs. Genome browser views of TT-TL-seq data for ENAH provide an example of results from untreated (UT) and KCl-treated (KCl) cells after normalization to the spike-in control. B) Browser image of TT-TL-seq data exemplifying a clean gene (HELLS) and a gene that does not meet the criteria for a clean gene (CYP2C18). The DoG produced from HELLS reads into CYP2C18, making the latter appear to be transcriptionally activated by hyperosmotic stress (log2 FC=5.39). C) The DoG produced from NUCKS1 is assigned to SLC45A3 because of extensive read-in transcription, which also complicates accurate differential expression analysis for SLC45A3 (log2 FC=4.13). ~4–5 kb upstream of HELLS and NUCKS1 are shown.
It was previously observed that read-in transcription of DoGs into neighboring genes leads to the mis-characterization of overlapping transcripts as being activated by stress (Rutkowski et al. 2015; Hennig et al. 2018; Cardiello et al. 2018; Roth et al. 2020; Fig. 1B). Moreover, read-in transcription confounds the assignment of DoGs to the corresponding parent gene (Fig. 1C). Our analyses suggest that ~55% of expressed genes experience read-in transcription after hyperosmotic stress (Table S1). Therefore, to ensure accurate differential expression analyses and DoG characterization, we generated a sub-list of genes (referred to as “clean genes” throughout the article). The term “clean genes” describes genes that are expressed, do not overlap with readthrough regions that correspond to neighboring genes on either strand and have higher expression within the gene body than the region 1 kb upstream of the gene’s transcription start site (TSS) (Roth et al. 2020; Figs. 1B & C). We identified 4584 clean genes in HEK-293T cells after hyperosmotic stress and analyzed their transcriptional regulation (Table S1).
Consistent with previous reports analyzing steady-state RNAs in human cells (Amat et al. 2019), we identified changes in transcriptional responses after hyperosmotic stress. Our results reveal predominantly decreases in nascent transcript levels after stress (Figs. 2A–C). Specifically, the number of normalized read counts corresponding to clean genes decreases 3-fold after KCl treatment (Fig. S1D). Yet, we find that a subset of clean genes bypasses this transcriptional repression (Figs. 2B, C & S1E), including GADD45B (Fig. 2D), which is known to be induced by hyperosmotic stress (Mak and Kültz 2004). More than 88% of clean genes were repressed after hyperosmotic stress, while only 3% were upregulated (Figs. 2C & S1F). To validate these observations, we extracted total RNA from untreated and KCl-treated cells and performed RT-qPCR using primers targeting intronic regions of several repressed genes. Results from HEK-293T cells and from SK-N-BE(2)C cells confirm the transcriptional decrease of three representative genes upon hyperosmotic stress (Figs. S1B & C) and demonstrate that mature mRNA levels for these genes remain unaffected, while levels of readthrough transcripts increase after hyperosmotic stress. Together our results reveal that hyperosmotic stress alters the nascent transcription profiles of cells by repressing thousands of genes and activating a small subset of genes.
Figure 2:
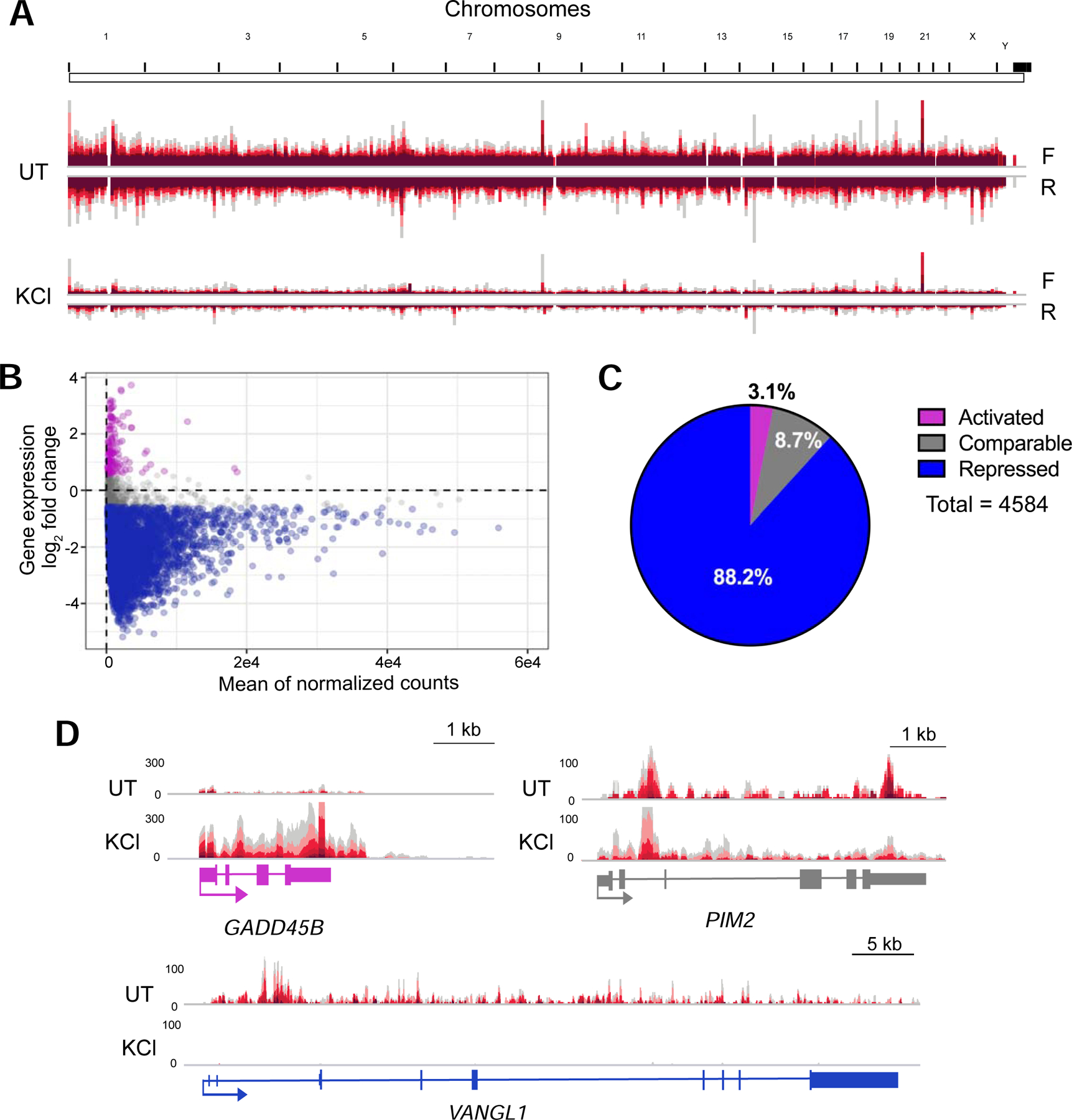
Hyperosmotic stress leads to widespread transcriptional repression. A) Whole-genome view of TT-TL-seq normalized reads for forward (F) and reverse (R) strands in UT and KCl samples. B) Minus average plot showing the log2 fold change for clean genes on the y-axis and the mean of normalized counts on the x-axis. Activated genes are shown in purple, genes retaining comparable expression are gray and repressed genes are blue (n=4584). C) Pie chart illustrating the percentage of clean genes within each of the 3 categories of transcriptional regulation (activated gene, log2 FC >0.58; comparable gene, log2 FC < 0.58 but > −0.58; repressed gene, log2 FC<−0.58). D) Browser shots of TT-TL-seq tracks from HEK-293T cells for VANGL1, which is transcriptionally repressed by hyperosmotic stress (log2 FC= −4.97), PIM2, which retains comparable expression after KCl treatment (log2 FC=0.28), and GADD45B, which is activated by hyperosmotic stress (log2 FC=3.72).
Stress-induced readthrough transcripts arise independent of gene transcription levels
Consistent with widespread transcriptional repression, normalized TT-TL-seq read counts within the bodies of DoG-producing clean genes decreased after hyperosmotic stress, while read counts corresponding to DoG regions increased (Fig. 3A). However, log2 fold changes in nascent RNAs of DoG-producing clean genes show that DoGs are produced from genes that experience all three types of transcriptional responses (Fig. 3B; Table S1). Specifically, 2.9% of DoGs arise from activated clean genes, 87.8% arise from repressed clean genes and 9.3% arise from clean genes that retain comparable expression in stressed and unstressed HEK-293T cells (Fig. 3B). We then asked whether DoGs preferentially arise from genes that are transcriptionally repressed upon hyperosmotic stress. Interestingly, the percentage of DoG-producing genes within each class of transcriptional regulation is consistent, comprising 12–14% (Fig. 3C). We conclude that DoGs are produced regardless of the transcriptional level of their upstream genes (Fig. 3D).
Figure 3:
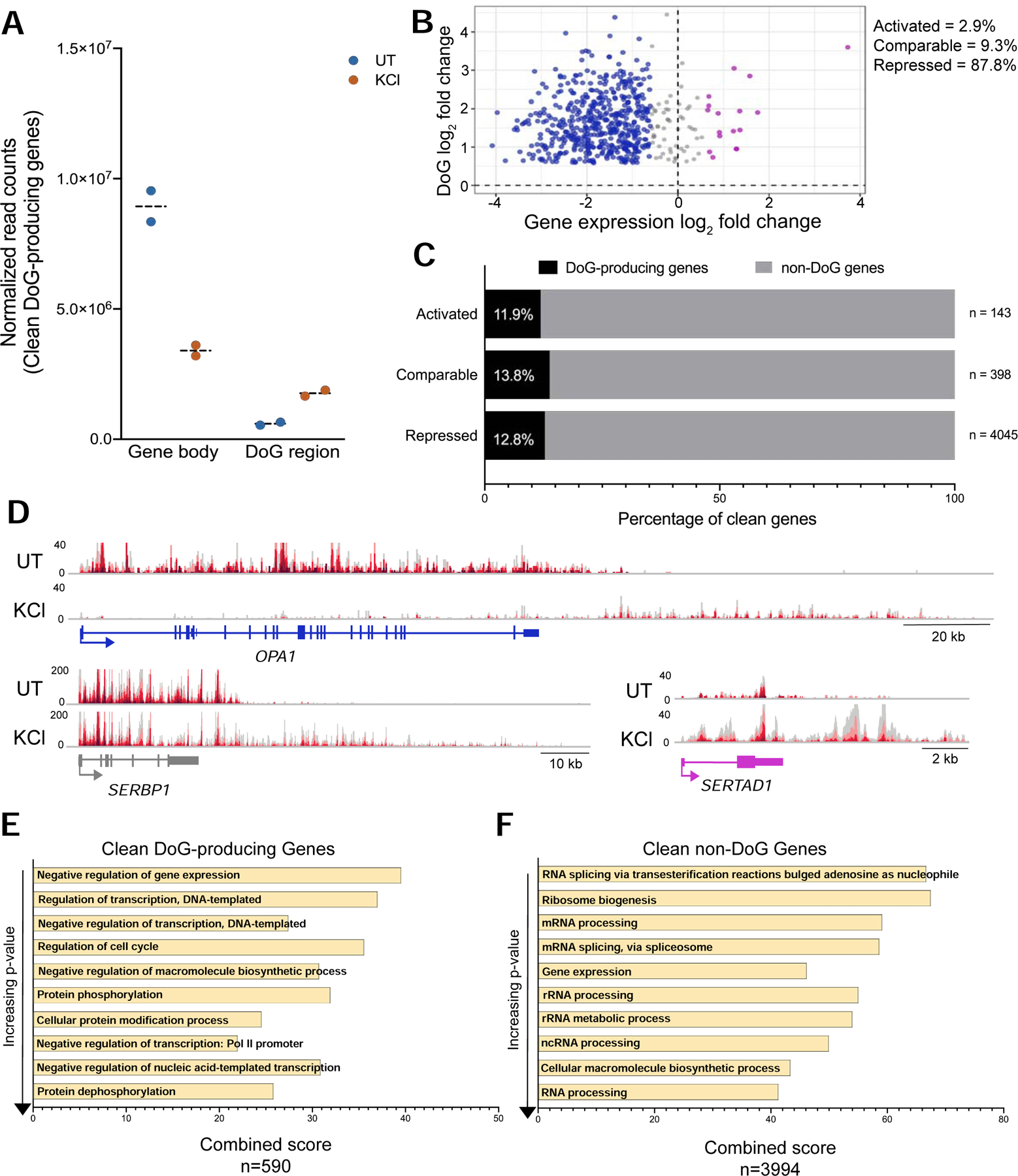
DoGs arise regardless of the transcriptional levels of their upstream genes upon hyperosmotic stress. A) Interleaved scatter plot showing the sum of normalized TT-TL-seq read counts of DoG-producing clean genes and corresponding DoG regions (n=590) in untreated and KCl-treated HEK-293T cells for two biological replicates. B) Scatter plot showing clean gene log2 fold change (FC) for the gene body on the x-axis and the log2 FC for the DoG region on the y-axis. DoG-producing genes that are transcriptionally activated upon stress are represented in purple, genes retaining comparable levels of expression are gray and genes that are repressed are blue. C) Bar graph showing the percentage of DoG-producing clean genes (black) within each category of transcriptional regulation. D) Browser image showing UT and KCl TT-TL-seq reads for OPA1, a transcriptionally repressed DoG-producing clean gene (gene log2 FC= −3.22), for SERBP1, which retains comparable expression after stress (gene log2 FC= −0.45), and for a transcriptionally activated DoG-producing clean gene, SERTAD1 (gene log2 FC= 1.23). E, F) Bar graphs show gene ontology combined scores for the 10 most significantly enriched biological processes in order of increasing p-value for E) DoG-producing clean genes and for F) non-DoG clean genes. Combined scores are the product of the p-value and the z-score as calculated by Enrichr (Chen et al. 2013).
Clean DoG-producing genes are functionally enriched for transcriptional repression
Previous analyses of DoG-producing genes did not reveal any functional enrichment (Vilborg et al. 2017). We suspected that the challenge of assigning DoGs to the correct gene of origin because of their extension into neighboring genes may have complicated previous efforts (Fig. 1C). Therefore, we revisited the question of whether DoG-producing genes are enriched for certain biological processes using only clean genes. We performed gene ontology analysis of DoG-producing clean genes and clean genes that fail to generate DoGs (non-DoG genes) using Enrichr (Chen et al. 2013; Kuleshov et al. 2016; Table S2). Interestingly, 5 out of the 10 enriched terms with the most significant p-values for DoG-producing genes are related to transcriptional repression (Fig. 3E). The remaining 5 terms are related to transcriptional regulation and protein modifications. Non-DoG genes do not show such a striking enrichment for terms related to transcriptional repression compared to other terms (Fig. 3F). Instead, these genes are strongly enriched for general processes related to RNA processing.
Pol II is redistributed along the genome after hyperosmotic stress
We used anti-Pol II ChIP-seq to investigate how Pol II binding correlates with the transcriptional profiles observed through TT-TL-seq in untreated and KCl-treated HEK-293T cells. Meta-analyses of clean DoG-producing genes confirmed a redistribution of Pol II molecules to the downstream regions in stressed cells (Figs. 4A & S2A), consistent with DoG transcription and previous observations (Cardiello et al. 2018; Heinz et al. 2018). As expected, this redistribution of Pol II molecules was not observed downstream of non-DoG genes (Figs. 4A & S2A). Interestingly, our results reveal a decrease in Pol II occupancy near the TES of non-DoG genes after hyperosmotic stress that was not observed for DoG-producing genes.
Figure 4:
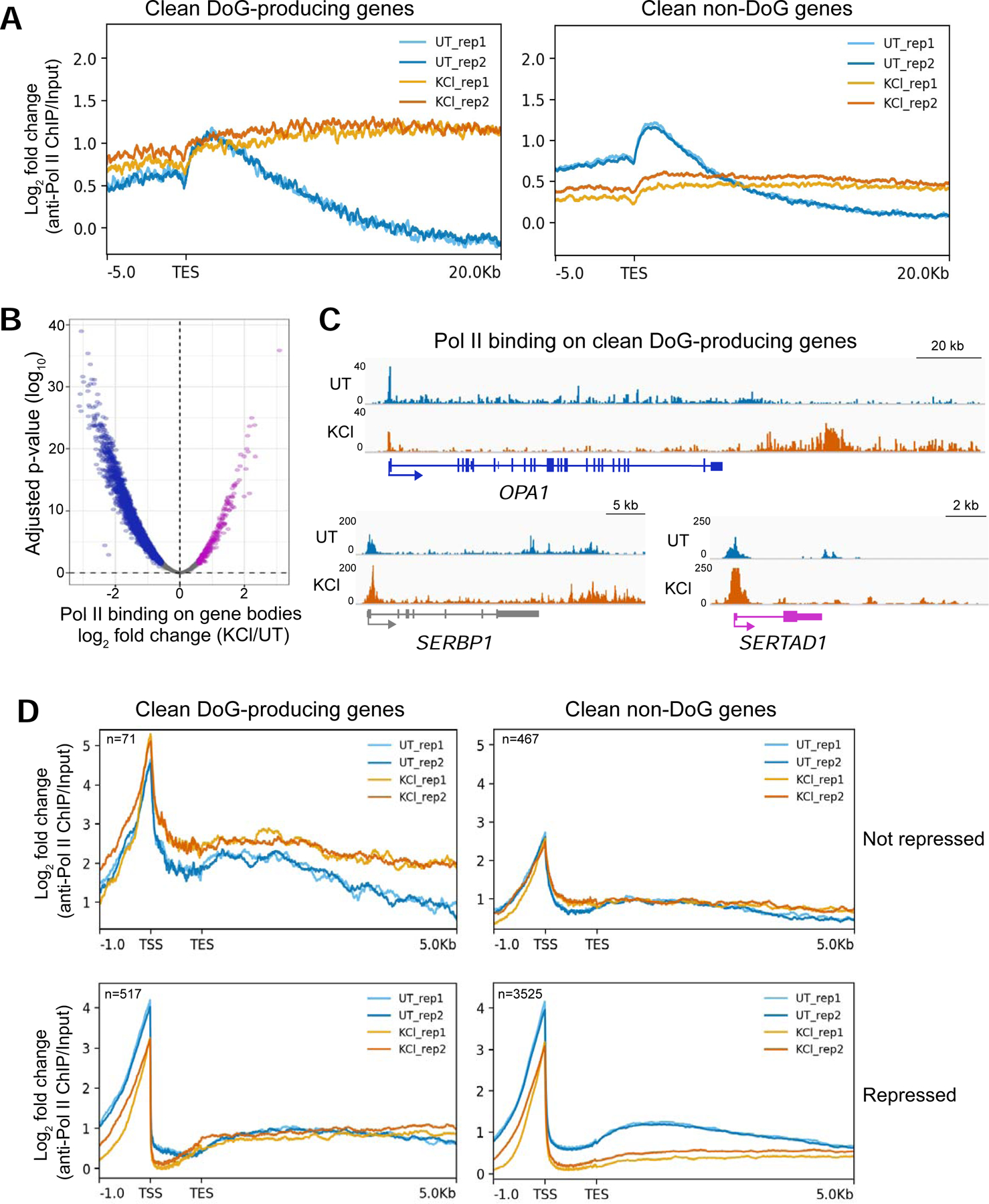
Hyperosmotic stress causes redistribution of Pol II molecules across the genome. A) Meta plots showing the log2 fold change (FC) of anti-Pol II ChIP-seq data normalized to input across the annotated transcription end sites (TES) of DoG-producing clean genes (n=590) and of clean non-DoG genes (n=3994) from UT and KCl-treated HEK-293T cells. B) Volcano plot showing the log2 FC of read counts from anti-Pol II ChIP-seq data normalized to input for clean genes on the x-axis and the corresponding log10 adjusted p-values on the y-axis. Genes with increased Pol II binding across the gene body are shown in purple, genes retaining comparable levels of Pol II binding are gray and those with decreased Pol II binding across the gene body are blue. C) Browser images of DoG-producing clean genes showing anti-Pol II ChIP-seq tracks normalized to input: OPA1 is a repressed gene, SERBP1 retains comparable expression and SERTAD1 is activated by hyperosmotic stress according to TT-TL-seq data. D) Meta plots showing Pol II binding on DoG-producing clean genes and clean non-DoG genes that are not repressed (top) or genes that are repressed (bottom). Two biological replicates are shown in each meta plot for UT and KCl samples.
We asked to what extent the stress-induced redistribution of Pol II molecules into the regions downstream of genes was responsible for the widespread transcriptional repression observed by TT-TL-seq. Alternatively, the decrease in transcription upon stress might be caused by lowered Pol II binding to repressed genes, as was previously reported for hyperosmotic stress induced by NaCl (Amat et al. 2019). Our results show a similar number of Pol II binding peaks after KCl treatment (Fig. S2B): a subset of overlapping peaks show increased Pol II binding after stress, while a majority of the identified peaks either retain comparable levels or exhibit reduced Pol II occupancy (Fig. S2C). Consistently, we found that Pol II binding decreases over most clean gene bodies after KCl treatment (Fig. 4B).
We verified Pol II occupancy patterns for representative genes within each category of transcriptional regulation. OPA1, a DoG-producing gene that is transcriptionally repressed, shows decreased binding close to the TSS and a shift of Pol II peaks into the downstream region in stressed cells (Fig. 4C). In contrast, SERTAD1, a DoG-producing gene that is activated by hyperosmotic stress, and SERBP1, which retains comparable expression according to our TT-TL-seq data, show increased Pol II binding close to each TSS that extends downstream of these genes. Finally, we assessed the distribution of Pol II molecules across repressed clean genes and across clean genes that are either activated or retain comparable expression after hyperosmotic stress (not repressed). Consistent with the individual gene examples discussed above, clean DoG-producing genes that are not repressed by hyperosmotic stress show increased Pol II binding close to their TSSs and across the gene body, while genes that are transcriptionally repressed by hyperosmotic stress show decreased levels of Pol II binding across the gene body. Similar effects were also observed for clean non-DoG genes (Fig. 4D). Our results are, thus, in agreement with the nascent transcription profiles derived from TT-TL-seq (Figs. 2 & 3), supporting a model where hyperosmotic stress induces both redistribution of Pol II molecules along the genome and a decrease in Pol II binding to repressed genes.
The Pol II interactome is altered by hyperosmotic stress
To gain insight into the mechanisms that promote DoG formation upon hyperosmotic stress, we performed immunoprecipitation of chromatin-bound Pol II coupled with mass spectrometry analysis (Harlen et al. 2016) in SK-N-BE(2)C cells (Fig. 5A). This human neuroblastoma cell line, where KCl-induced DoGs were originally described (Vilborg et al. 2015), was used because we observed more robust DoG induction in these cells than in HEK-293T cells (Figs. S1B & C). We confirmed isolation of chromatin through western blots (Fig. S3A). Overall, our results reveal distinct changes in the Pol II interactome (Fig. 5B). Peptides corresponding to several CPA factors, as well as to Xrn2 were detected in both untreated and KCl-treated samples (Table S3) at least once across six experiments. The presence of Xrn2 and CPSF1 in the anti-Pol II immunoprecipitates of untreated and KCl-treated cells was validated through western blots (Figs. S3B–D). In contrast, 11 of the 14 subunits of the Integrator complex were not detected among the Pol II interactors in the KCl-treated samples (Table S3). To validate this observation, we performed western blots to detect Pol II in chromatin-bound anti-Integrator subunit 3 (Int3) immunoprecipitates from SK-N-BE(2)C and HEK-293T cells. Decreased binding between Pol II and Int3 in KCl-treated samples compared to untreated samples was confirmed for both cell lines (Figs. 5C & D). The specificity of the anti-Int3 antibody was verified by performing siRNA-mediated knockdowns of this subunit followed by western blots of HEK-293T whole cell lysates (Fig. S3E). To ensure that the decreased detection of Integrator subunits was not due to protein degradation, we also assessed the levels of Int3 and Int11 in whole cell lysates from SK-N-BE(2)C cells. Western blots confirmed that the levels of these Integrator subunits do not decrease in cells after hyperosmotic stress (Fig. S3F).
Figure 5:
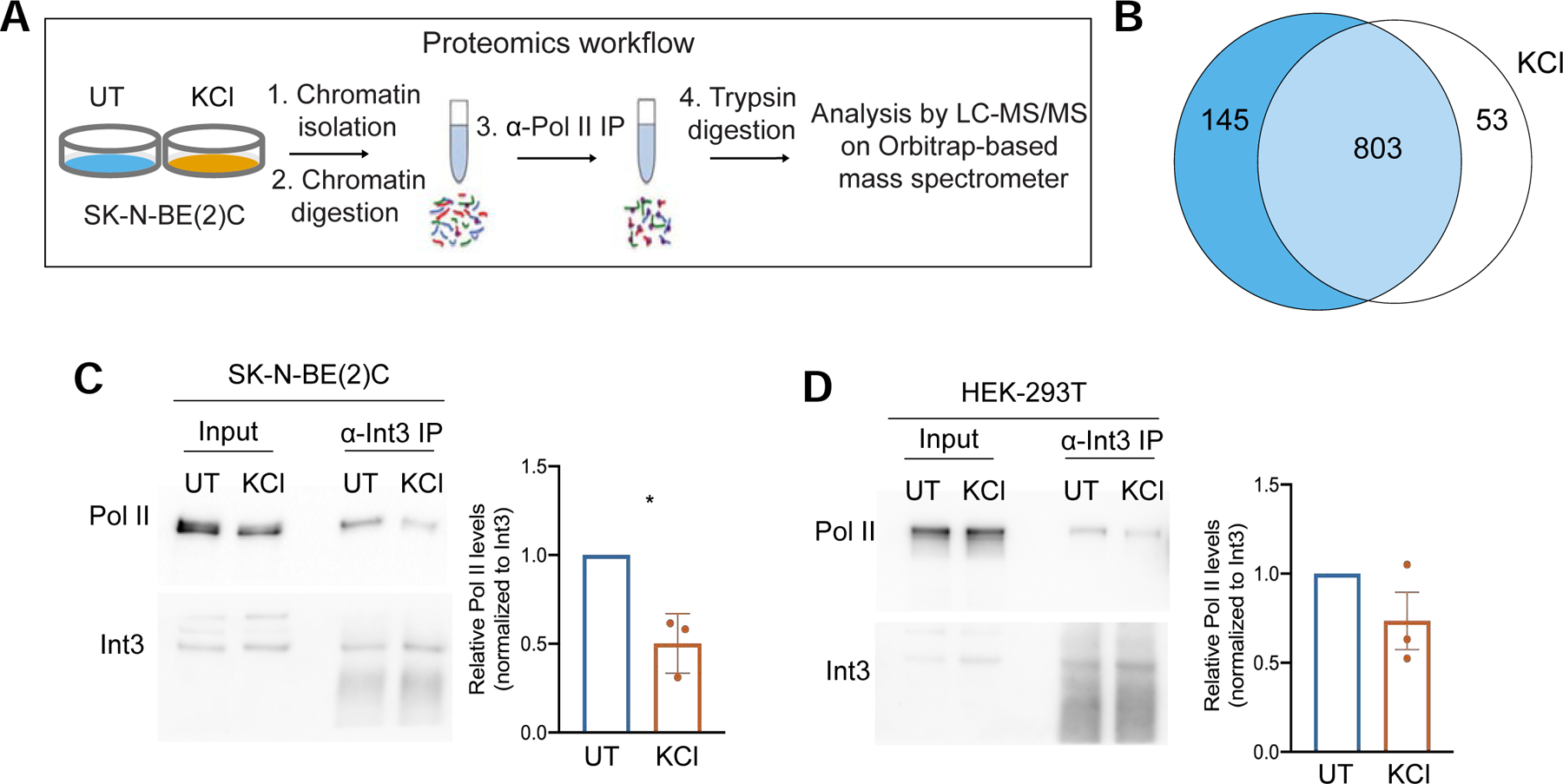
The Pol II interactome changes upon hyperosmotic stress. A) Experimental setup for mass spectrometry of anti-Pol II immunoprecipitates obtained from the chromatin fractions of SK-N-BE(2)C cells. B) Overlap between Pol II-associated proteins found in UT and KCl-treated samples (803). Peptides for 145 proteins were found only in the UT samples (blue) and peptides corresponding to 53 proteins were found only in the KCl-treated samples at least once across six replicates (white). C, D) Western blots of anti-Int3 immunoprecipitates obtained from chromatin fractions reveal decreased levels of associated Pol II in KCl-treated samples compared to untreated samples in C) SK-N-BE(2)C (p-value=0.0357) and in D) HEK-293T cells (p-value=0.2407). Chromatin fractions from these cell lines are shown as input. Around 8% of the total input and 11% of the immunoprecipitates were loaded on the gels.
Together, our proteomic results show that hyperosmotic stress induces changes to the Pol II interactome that could contribute to the transcriptional landscape revealed by high throughput sequencing. Specifically, important termination factors interact with Pol II despite cellular stress, while the interaction between Pol II and the Integrator complex decreases.
Hyperosmotic stress decreases the occupancy of Integrator subunits on DNA
Further insight into the decreased binding of Integrator subunits to Pol II after hyperosmotic stress (Fig. 5) was gained by performing anti-Int11 and anti-Int3 ChIP-seq in untreated and KCl-treated HEK-293T cells. The Integrator complex was previously shown to bind close to the TSS of genes, where it regulates promoter proximal Pol II pause release (Stadelmayer et al. 2014; Elrod et al. 2019; Beckedorff et al. 2020). Consistently, we predominantly observe Int3 and Int11 peaks close to the TSS of clean genes that decrease after hyperosmotic stress (Figs. 6A & S4A). Meta-analysis of DoG-producing clean genes and clean non-DoG genes revealed similar decreases in Int11 and Int3 occupancy close to the TSSs. Additionally, read counts for identified Int11 binding sites confirm decreased occupancy of this protein on DNA after hyperosmotic stress (Fig. 6B).
Figure 6:
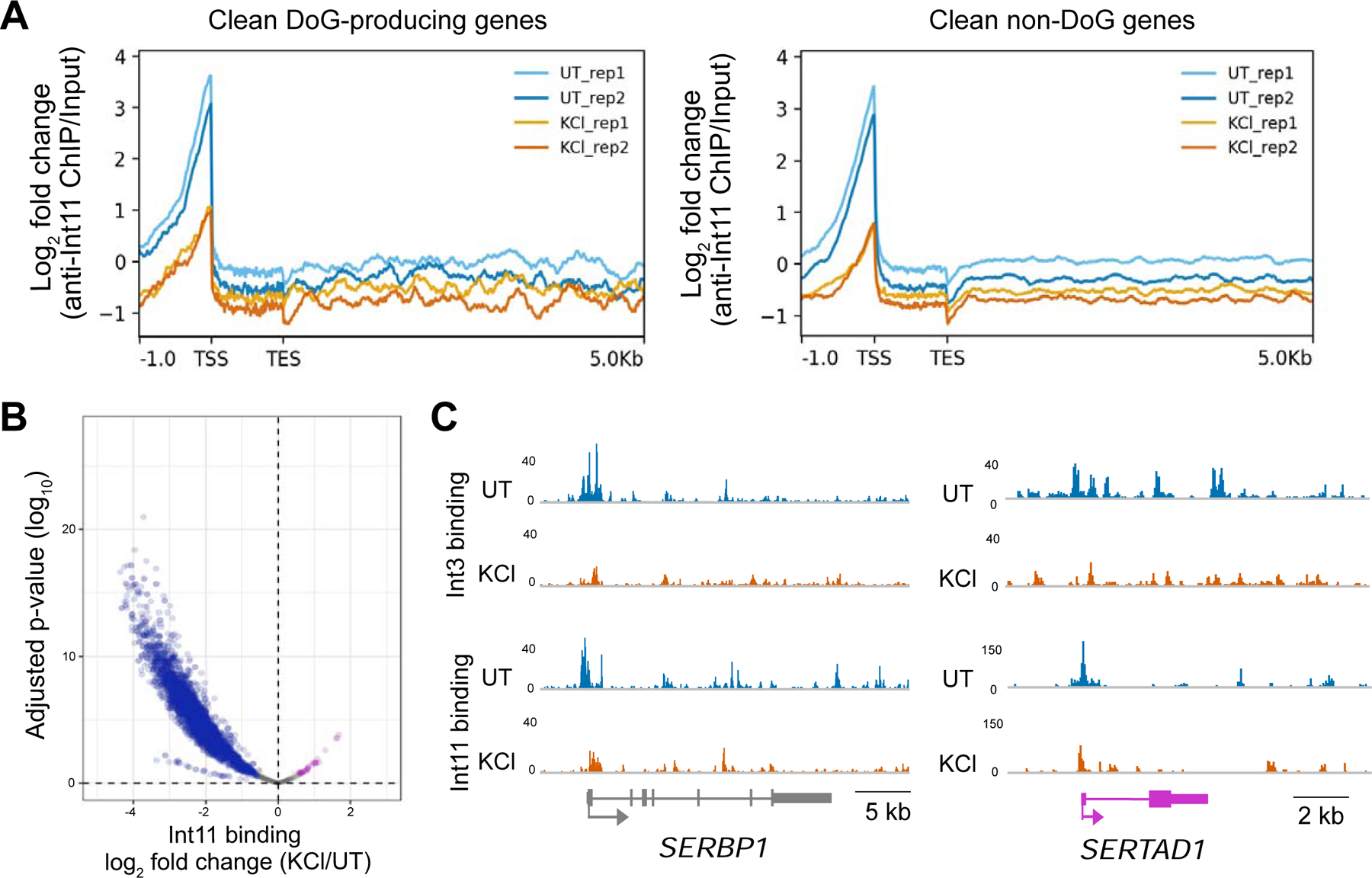
Hyperosmotic stress leads to decreased occupancy of Integrator subunits on DNA. A) Meta-plots showing the log2 fold change (FC) of anti-Int11 ChIP-seq normalized to input reveal decreased occupancy of Int11 near the TSSs of clean DoG-producing genes (n=590) and clean non-DoG genes (n=3994). B) Volcano plot of read counts for Int11 binding sites. Log2 FC of KCl/UT is shown on the x-axis with the corresponding adjusted p-values on the y-axis (n=3881). Sites where Int11 occupancy decreases after hyperosmotic stress are blue, sites where occupancy remains comparable are gray and sites where occupancy increases are purple. C) Browser images of anti-Int3 and anti-Int11 ChIP-seq tracks normalized to input for SERBP1 and SERTAD1, which are DoG-producing clean genes that are not repressed by hyperosmotic stress.
Since our TT-TL-seq and anti-Pol II ChIP-seq experiments reveal that hyperosmotic stress causes transcriptional repression accompanied by a decrease in Pol II binding at downregulated genes, we asked whether the observed decreased occupancy of Integrator subunits on DNA was a consequence of a decrease in Pol II occupancy. The occupancy of Int3 and Int11 decreased near the TSS of genes that are not repressed by hyperosmotic stress, including SERTAD1 and SERBP1 (Figs. 3D, 4C & 6C). Furthermore, we quantified the occupancy of Int11 and Pol II across Int11 binding sites that were identified across two biological replicates and observed that the decrease in Int11 binding is not limited to sites exhibiting a concomitant decrease in Pol II occupancy (Figs. 6C & S4B). We conclude that the decreased binding of Integrator subunits to DNA is not a mere consequence of the transcriptional repression induced by hyperosmotic stress.
Depletion of Integrator endonuclease leads to DoG production
The Integrator complex regulates transcription termination at many noncoding RNA loci and has been shown to bind the 3´ end of certain protein-coding genes (Baillat and Wagner 2015; Gardini et al. 2014). Since the interaction between Pol II and Integrator decreases after hyperosmotic stress (Fig. 5), we investigated whether knocking down the catalytic subunit of the complex, Int11, using siRNAs is sufficient to induce DoGs. We transfected HEK-293T cells with an siRNA against Int11 (siInt11) or with a non-targeting siRNA control (siC). HEK-293T cells stably expressing FLAG-tagged, siRNA-resistant wild-type (WT) or catalytically inactive Int11 (E203Q) were also transfected with siInt11 (Baillat et al. 2005; Xie et al. 2015) for 72 hours. DoG levels were then measured by RT-qPCR, confirming that knockdown of endogenous Int11 is sufficient to induce DoGs. Moreover, expression of exogenous WT Int11 reduces DoG induction compared to samples expressing the E203Q mutant or to samples expressing no rescue (Fig. 7A).
Figure 7:
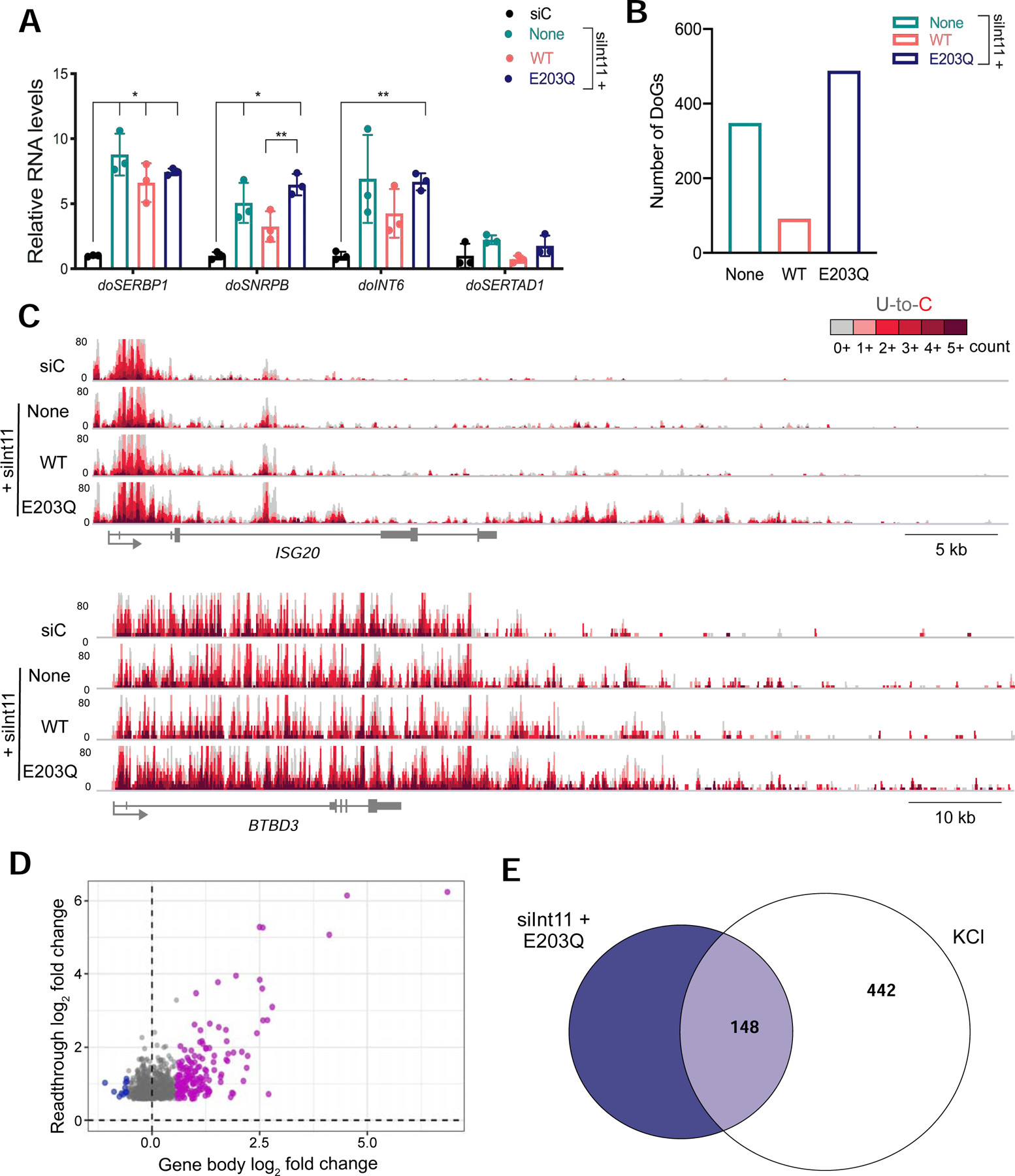
Depletion of Integrator nuclease subunit leads to readthrough transcription. A) RT-qPCR analysis of DoG levels in HEK-293T cells transfected with a scrambled siRNA control (siC) or an siRNA targeting endogenous Int11 (siInt11). The siInt11-transfected cells expressed either exogenous wild-type Int11 (WT), catalytically inactive Int11 (E203Q) or no exogenous Int11 (None). Adjusted p-values compared to siC: None: doSERBP1=0.004; doSNRBP=0.032; doInt6=0.077; doSERTAD1=0.097; WT: doSERBP1=0.011; doSNRBP=0.091; doINT6=0.091; doSERTAD1=0.654; E203Q: doSERBP1=<0.001; doSNRBP=<0.001; doINT6=<0.001; doSERTAD1=0.337. B) Bar graph showing the number of DoGs induced after siRNA knockdown of endogenous Int11. C) Browser image of TT-TL-seq data for two genes that produce DoGs upon depletion of functional Int11. D) Scatter plot showing gene expression log2 fold change (FC) for siInt11+E203Q HEK-293T cells on the x-axis and the log2 FC of the corresponding readthrough transcripts on the y-axis. Genes that are activated in siInt11+E203Q cells compared to siC-transfected cells are purple, unaffected genes are gray and repressed genes are blue. All readthrough sites identified in the siInt11+E203Q sample are represented in this plot (n=840). E) Venn diagram displaying overlap between the identities of clean genes that produce readthrough transcripts in siInt11+E203Q cells (dark blue) and those of DoG-producing clean genes in KCl-treated samples (white).
Endogenous Int11 protein levels were reduced by ~90% in siInt11-transfected samples (Fig. S5A). The efficiencies of exogenous WT and E203Q mutant Int11 expression were verified by measuring the levels of unprocessed U2 through RT-qPCR (Skaar et al. 2015; Fig. S5B): unprocessed U2 accumulates in cells without rescue (None) or expressing the E203Q mutant Int11, but not in cells transfected with siC or cells expressing exogenous WT Int11. Northern blots using probes against snRNA U2 showed that levels of mature U2 do not decrease after knockdown (Fig. S5C), consistent with previous reports (Tatomer et al. 2019). Thus, it is unlikely that the effects of Int11 knockdown on DoG production are because of the role of Integrator in snRNA processing.
We assessed the extent to which knockdown of endogenous Int11 induces DoGs genome-wide. To increase cell viability, we performed TT-TL-seq on HEK-293T cells transfected with an siRNA against Int11 for 48 hours (Fig. S5D). Results obtained from cells lacking functional Int11 reveal hundreds of readthrough sites across the genome that are induced by more than 1.5-fold compared to the siC-transfected sample (Fig. 7B; Table S4). According to TT-TL-seq data, induction of readthrough transcription after depletion of endogenous Int11 was most evident upon expression of the E203Q mutant (Figs. 7B & C). Yet, readthrough transcripts observed in siInt11-transfected cells expressing no rescue and in cells expressing the E203Q mutant Int11 were highly correlated (Fig. S5E). As expected, the most highly induced sites of readthrough transcription (Table S4) corresponded to snRNA genes (Baillat et al. 2005; Albrecht and Wagner 2012). We also detected readthrough downstream of lncRNA and histone genes as previously described (Nojima et al. 2018; Skaar et al. 2015). However, most identified readthrough sites were downstream of protein-coding genes (Table S4). Of the 840 readthrough transcripts induced by knockdown of functional Int11 (E203Q sample), 489 exhibited greater than 80% read coverage in the region 5 kb downstream of the annotated termination site of the gene of origin and, therefore, met all criteria to be classified as DoG RNAs (Figs. 7B & C; Table S4).
Depletion of Integrator subunits has been shown to alter the transcriptional levels of certain genes (Gardini et al. 2014; Elrod et al. 2019; Tatomer et al. 2019; Beckedorff et al. 2020). Consistently, we found that depletion of functional Int11 in HEK-293T cells differentially affects more than a thousand genes (Fig. S5F). Examination of the expression levels of the parent genes revealed that readthrough transcripts predominantly arise from upregulated genes or from genes that retain comparable expression after knockdown, while very few arise from genes that are transcriptionally repressed (Fig. 7D).
Given our observation that the interaction between Integrator subunits and Pol II is disrupted by hyperosmotic stress, we asked how the identities of genes producing readthrough transcripts upon depletion of functional Int11 compare to genes that produce DoGs after hyperosmotic stress. We identified 232 clean genes producing readthrough transcripts in siInt11-transfected cells and 351 clean genes producing readthrough transcripts in the siInt11+E203Q mutant sample. Comparison with the 590 DoG-producing clean genes identified in stressed cells showed that up to 25% of KCl-induced DoGs are detected at loci that also produce readthrough transcripts after depletion of functional Int11 (Figs. 7E & S5G). These readthrough transcripts are generally more robustly induced after KCl treatment than upon depletion of functional Int11 (Fig. S5H), suggesting that, although Int11 knockdown is sufficient to produce readthrough transcription, decreased interactions between Integrator and Pol II are not solely responsible for DoG induction upon hyperosmotic stress.
Discussion
We used TT-TL-seq to evaluate nascent transcription profiles that accompany DoG induction under conditions of hyperosmotic stress (Fig. 1) and observed that hyperosmotic stress triggers transcriptional repression that is widespread and of greater magnitude than anticipated (Robbins et al. 1970; Amat et al. 2019). After an hour-long exposure to mild conditions of hyperosmotic stress, only ~12% of clean genes escaped transcriptional repression (Fig. 2). Interestingly, DoGs are produced independent of the transcriptional levels of their upstream genes, and DoG-producing genes are functionally enriched for transcriptional repression according to gene ontology analyses (Fig. 3). Pol II binding profiles demonstrate a correlation with nascent transcription profiles across DoG-producing clean genes and non-DoG genes (Fig. 4). Proteomic analysis of the Pol II interactome revealed decreased binding between the Integrator complex and Pol II upon exposure to hyperosmotic stress (Fig. 5), accompanied by a genome-wide decrease in the occupancy of Integrator subunits on DNA (Fig. 6). Depletion of the catalytic subunit of the Integrator complex, Int11, is sufficient to induce readthrough transcription downstream of hundreds of genes. Loci that produce readthrough transcripts upon depletion of functional Int11 and those that produce DoGs upon hyperosmotic stress partially overlap (Fig. 7).
Our analysis of clean genes (expressed genes lacking overlap with readthrough regions) has enabled us to accurately characterize the relationship between DoGs and their upstream transcripts (Figs. 1 & 3). Consistent with previous observations in heat-shocked cells (Cardiello et al. 2018), we find that DoGs are produced from genes that experience different transcriptional responses to hyperosmotic stress. Importantly, a similar percentage of clean genes that are activated, repressed or unchanged produce DoGs after hyperosmotic stress, revealing that transcriptional levels and DoG production are not mechanistically linked. Our analyses further suggested that ~55% of genes that are expressed in HEK-293T cells might be affected by read-in transcription upon exposure to hyperosmotic stress (Table S1). Intriguingly, other groups have previously reported that read-in transcription from upstream genes negatively affects the splicing of downstream genes upon HSV-1 infection (Rutkowski et al. 2015). Additionally, there is evidence that heat shock and hyperosmotic stress disrupt splicing (Shalgi et al. 2014; Hennig et al. 2018). These findings suggest that altered splicing patterns may be a consequence of DoG production and highlight the extensive impact of DoG production on the transcriptome of stressed cells. Investigating the diversity of unprocessed transcripts that accumulate upon stress will provide a better understanding of how transcriptional and processing dynamics are impacted by stress.
Previously, we were unable to find any functional enrichment for DoG-producing genes in the context of hyperosmotic stress (Vilborg et al. 2017). Upon revisiting this question using only clean genes, we found that many clean DoG-producing genes encode transcriptional repressors (Fig. 3; Table S2). Transcriptional repression has also been reported under other DoG-inducing conditions, including heat shock and viral infection (Mahat et al. 2016; Rutkowski et al. 2015; Bauer et al. 2018). Thus, it is tempting to speculate that DoGs could serve as a store of unprocessed transcripts to facilitate cell survival upon prolonged stress by providing a source of important mRNAs (requiring processing, but not active transcription), thereby reducing the cell’s energetic costs. Other terms of biological enrichment for DoG-producing genes are related to protein modifications. A repository of transcripts that encode such proteins might facilitate recovery from stress. Nuclear-retained, intron-containing transcripts have been shown to serve as a source of gene regulation in other contexts (Boutz et al. 2015). Therefore, there is precedent for unprocessed transcripts, such as DoGs, functioning to regulate gene expression levels via post-transcriptional processing. This idea is also supported by the fact that DoGs have relatively long half-lives (Vilborg et al. 2015). DoG induction might further promote transcriptional repression during hyperosmotic stress by causing transcriptional interference at neighboring genes (Mazo et al. 2007; Muniz et al. 2017). Alternatively, given the transcriptional changes observed upon stress, DoGs may serve to alter the chromatin landscape surrounding their parent and neighboring genes to facilitate prolonged responses throughout stress and recovery from stress.
In agreement with previous observations in heat shock (Cardiello et al. 2018), anti-Pol II ChIP-seq data from KCl-treated cells demonstrate a decrease in Pol II binding across the bodies of repressed clean genes accompanied by a shift of elongating Pol II molecules into downstream regions (Fig. 4). At loci that are not repressed by stress, we observe an increase in Pol II binding along the gene bodies, extending into their downstream regions. These observations are consistent with our TT-TL-seq results (Figs. 2 & 3). The differing levels of Pol II occupancy observed across parent genes suggest an elegant and tightly regulated mechanism of transcriptional termination at DoG-producing genes. Furthermore, our anti-Pol II ChIP-seq data revealed decreased occupancy near the TES of non-DoG genes after hyperosmotic stress, which was not observed for DoG-producing genes (Figs. 4A & S2A). This observation suggests that one factor distinguishing DoG-producing genes from genes that do not produce DoGs is their dependence on Pol II pausing near their TESs for efficient transcription termination.
Surprisingly, rather than observing decreased interactions between Pol II and CPA factors, our proteomic analyses revealed that the binding between Integrator subunits and Pol II decreases after stress (Fig. 5). This was shown by performing anti-Pol II immunoprecipitations followed by mass spectrometry (Table S3) and anti-Int3 immunoprecipitations followed by western blots using an antibody against Pol II (Fig. 5). Furthermore, we found that hyperosmotic stress decreases the occupancy of Integrator subunits 11 and 3 on DNA. Therefore, we asked whether siRNA depletion of Int11, the catalytic subunit of the complex, is sufficient to induce DoGs. TT-TL-seq of siRNA-treated samples revealed that hundreds of DoGs are induced after functional Int11 is depleted from cells (Fig. 7). Moreover, our results reveal that expression of wild-type Int11 reduces the number of DoGs induced in cells where endogenous Int11 has been knocked down compared to no rescue. Additionally, we observe an increase in the number of DoGs induced upon expression of the E203Q mutant Int11, arguing that the nuclease activity of Int11 is important for repressing DoG production under homeostasis. These results highlight the importance of the Integrator complex in the regulation of mRNA production beyond the promoter proximal pause site (Elrod et al. 2019). Perhaps, cells rely on Integrator to mediate a mechanism of transcription termination at a subset of protein-coding genes that is alternative to the canonical mechanism orchestrated by the CPA complex. This might explain why readthrough transcription is not observed downstream of all protein-coding genes. However, our ChIP-seq experiments did not reveal preferential loss of Integrator subunits on DoG-producing genes (Figs. 6 & S4). Therefore, it is possible that the genome-wide loss of Integrator upon hyperosmotic stress plays an indirect role in regulating DoG biogenesis. More work investigating the role of Integrator binding to mRNA 3énds is imperative.
Our results reveal that as many as 25% of genes that produce DoGs upon KCl-treatment also produce readthrough transcripts upon depletion of functional Int11 (Fig. 7). This partial overlap between Int11-depletion-dependent readthrough transcripts and KCl-induced DoGs suggests that the decreased binding of Integrator subunits to Pol II during the stress response contributes to DoG biogenesis. We asked whether the effects of Int11 knockdown and hyperosmotic stress on DoG induction are additive by exposing HEK-293T cells to KCl after 48 or 72 hours of siInt11 transfection, but did not observe such effects on DoG production upon combined treatment (data not shown). However, our results were inconclusive due to the different extents of DoG induction observed in each context (Fig. S5H) as well as by the temporal differences inherent to the experimental protocols. Because of the changes in the transcriptional landscape upon hyperosmotic stress (Fig. 2), it is unlikely that depletion of a single protein is enough to recapitulate the complexity of this response. Hyperosmotic stress induces widespread transcriptional repression to an extent that is not recapitulated by depletion of functional Int11. Additionally, our results reveal changes in Pol II binding patterns to DNA (Fig. 4) and to other proteins (Fig. 5; Table S3) after hyperosmotic stress. The full extent of DoG induction upon cellular stress might also reflect a stress-induced increase in the processivity of elongating Pol II molecules, which would result in readthrough transcription (Fong et al. 2015).
In agreement with the important roles of Xrn2 and CPSF73 in mediating the termination of protein-coding gene transcripts (Proudfoot. 2016), readthrough transcription is observed upon depletion of these proteins (Fong et al. 2015; Eaton et al. 2018; Eaton et al. 2020). However, unlike stress-induced readthrough transcription, depletion of these proteins was not limited to a subset of genes. Using anti-Pol II immunoprecipitation of chromatin fragments coupled with mass spectrometry and western blots, we observed that the overall binding of Pol II and important termination factors, including Xrn2 and CPA factors, is not affected by hyperosmotic stress. Our data, however, do not exclude the possibility that important termination factors are redistributed along the genome after hyperosmotic stress. Anti-CPSF6 ChIP-seq from cells treated with NaCl to induce hyperosmotic stress revealed decreased binding of this subunit at certain genes (Jalihal et al. 2020); yet it is unclear whether this was accompanied by a decrease in Pol II occupancy at these loci. Moreover, anti-CPSF73 ChIP-qPCR data from heat-shocked cells suggest a decrease of this factor at activated DoG-producing genes (Cardiello et al. 2018). It is possible that at genes activated by stress, disruption of the stoichiometry between Pol II and termination factors contributes to DoG production. However, such a model would not explain readthrough transcription at genes that retain comparable expression levels or are transcriptionally repressed, which comprise the majority of DoG-producing genes upon hyperosmotic stress (Fig. 3B). These observations emphasize the need to further explore the features of DoG-producing genes that might make them susceptible to failed termination despite the presence of important termination factors on chromatin.
Limitations:
Our work provides insight into the transcriptional landscape that accompanies DoG biogenesis after hyperosmotic stress and reveals a role for the Integrator complex in DoG production. However, the Integrator complex does not seem to be the only agent involved in the biogenesis of readthrough transcripts upon stress. Additional work assessing the distribution of other important termination factors on DNA after stress should further elucidate how cells selectively induce DoGs from a subset of genes. Furthermore, our data do not provide information about the exact mechanism by which Integrator regulates DoG biogenesis. More work is needed to determine whether the effect of the Integrator complex on readthrough transcripts produced from protein-coding genes is direct or indirect.
STAR Methods
Lead Contact
Additional information and reagent request should be directed to and will be fulfilled by Joan A. Steitz (joan.steitz@yale.edu).
Material Availability
This study did not generate unique reagents.
Data and Code Availability
All high throughput sequencing datasets generated in this study have been deposited to the Gene Expression Omnibus database and can be found as a super series under the following accession number: GSE152063.
Experimental Model and Subject Details
HEK-293T cells were cultured in DMEM supplemented with 10% fetal bovine serum, 1% penicillin and streptomycin, and 1% L-glutamine. SK-N-BE(2)C cells were cultured in a 1:1 mix of DMEM and F-12K media supplemented with 10% fetal bovine serum, 1% penicillin and streptomycin, and 1% L-glutamine. Stable HEK-293T cell lines expressing WT or E203Q mutant Int11 were supplemented using 2 µg/mL of puromycin, as previously described (Xie et al. 2015). Hyperosmotic stress inductions were performed using 80 mM KCl for 1 hour, as described in Vilborg et al. 2015.
Method Details
RNA preparations and RT-qPCRs:
RNA extractions were performed using TRIzol according to the manufacturer’s instructions. After treating samples with RQ1 DNase (Promega), RNA was recovered through PCA extraction followed by ethanol precipitation. cDNA was made from 2 µg RNA using Super Script III and random primers (Invitrogen). Samples were then diluted 1:10 and 2 µL of diluted cDNA were used for RT-qPCR along with 5 µL of iTaq universal SYBR mix (Bio-Rad), 1.5 µL of primer mix (1.5 µM of forward and 1.5 µM of reverse) and 1.5 µL of water. Plates were run on a Bio-Rad CFX384 machine. Results were analyzed using the comparative CT method (Schmittgen and Livak. 2008). Primers designed for this study were subjected to a primer efficiency test and amplicons were run on agarose gels to ensure that a single band was observed for each pair. Primers used in this study are listed in Table S5.
High throughput sequencing:
TT-TimeLapse:
Samples were fed 1 mM s4U (Sigma) during the final 5 minutes of incubation with either KCl or siRNAs. After extracting RNA from HEK-293T cells, samples were spiked with 4% s4U-labeled RNA from Drosophila S2 cells. TT-TL-seq was performed as described in Schofield et al. 2018. Sequencing was done by the Yale Center for Genomic Analysis (YCGA).
ChIP-seq:
2.5×107 HEK-293T cells were plated in 15 cm dishes (5×106 per dish) and incubated at 37°C overnight. For anti-Int3 ChIP-seq experiments, 3×107 HEK-293T cells were used. Cells were crosslinked during the last 10 minutes of stress induction using 1% formaldehyde and then washed twice with 1x PBS. Chromatin immunoprecipitation was performed as described in Bieberstein et al. 2014 using 30 µL of anti-Pol II antibody (Cell Signaling D8L4Y), 25 µL of anti-Int11 antibody (Abcam ab75276) or 30 µL of anti-Int3 antibody (Bethyl A302–050A) and 180 µL of Protein A beads. The YCGA prepared libraries and sequenced the samples on the NovaSeq platform. We note that the generation of Int11 ChIP-seq datasets was particularly challenging. We suspect that this was due to the biological properties of this protein as well as the reduced interactions between Integrator subunits and Pol II upon hyperosmotic stress. Subsequently, we performed anti-Int3 ChIP-seq experiments and obtained agreeing results.
Bioinformatics:
TT-TL-seq:
TT-TL-seq reads from HEK-293T cells containing spiked-in RNA from Drosophila S2 cells were filtered with FastUniq (Xu et al. 2012) to remove duplicate sequences and trimmed with Cutadapt v1.16 (Martin 2011) to remove adapter sequences. Unfiltered, trimmed reads were mapped to a combined hg38 and dm6 genome using hisat2 version 2.1.0 with default parameters and --mp 4,2 (Kim et al. 2015). Bam files for uniquely mapped reads (SAM flag 83/163 or 99/147) were created, sorted and indexed using samtools version 1.9 (Li et al. 2009). U-to-C mutation calls were performed as previously described (Schofield et al. 2018). To calculate normalization factors, reads aligning to the dm6 genome were filtered and counted with HTseq htseq-count (Anders et al. 2015). Normalization factors for each sample were calculated with edgeR (Robinson et al. 2010) using read counts mapped to the dm6 genome (calcNormFactors with method = ‘upperquartile’).
| Sample | Spike-in normalization factor |
|---|---|
| siC_rep1 | 2.65151515 |
| KCl_rep1 | 0.71138211 |
| siInt11_rep1 | 2.08333333 |
| siC_rep2 | 1.94444444 |
| KCl_rep2 | 0.63405797 |
| siInt11_rep2 | 1.26811594 |
| WT | 2.65151515 |
| mut | 1.82291667 |
Gene counts for reads aligning to the hg38 genome were generated using bedtools multicov version 2.26.0 (Quinlan et al. 2010). Read-in values used to define clean genes were generated using ARTDeco (Roth et al. 2020). DoGs were identified using DoGFinder (Wiesel et al. 2018). Differential expression analysis of normalized read counts in gene bodies and in DoG regions was done using DESeq2 (Love et al. 2014). The list of clean genes is comprised by genes that were expressed by more than 100 read counts after normalization to spike-in, had a read in value ≤ −1 and did not overlap with readthrough regions from neighboring genes on either strand according to bedtools intersect analysis (Quinlan et al. 2010). EnrichR was used for gene ontology analysis of biological enrichments (Chen et al. 2013; Kuleshov et al. 2016). Visualization of the generated data was achieved using ggplot2 (Wickham 2016) and graphPad prism. Eulerr was used to generate proportional Venn diagrams. Normalized tracks were visualized using IGV.
ChIP-seq:
ChIP-seq samples were mapped to the hg38 human genome using bowtie2 (Langmead et al. 2009). Bam files and gene counts were generated as described above. Counts were normalized to input using edgeR normalizeChIPtoInput function (Robinson et al. 2010). MACS2 version 2.1.1 (Zhang et al. 2008) was used to call peaks. Volcano plots were generated using ggplot2 on log2 fold changes and adjusted p-values calculated with DESeq2. Normalized bigwig files and meta plots were created using deepTools version 3.3.0 (Ramírez et al. 2016). Tracks were visualized using IGV.
Cell fractionation and immunoprecipitations:
For cellular fractionation, 4.8×106 cells were plated in three 15 cm dishes and incubated overnight at 37°C. After KCl treatment, cells were washed twice with 1x PBS, scraped on ice and pelleted by centrifugation (1400rpm at 4°C for 5 minutes). Pellets were resuspended using hypotonic lysis buffer for 5 minutes (Nojima et al. 2016). Nuclei were collected by centrifugation (1400rpm at 4°C for 5 minutes) and the cytoplasmic fraction (supernatant) was discarded. Nuclei were incubated in NUN1 and NUN2 (Wuarin and Schibler 1994) on ice for 15 minutes with intermittent vortexing. Samples were then spun down to pellet the chromatin fraction. Chromatin fragments were generated by incubating the chromatin pellets with 2 µL of micrococcal nuclease for 5 minutes at 37°C, at 1400 rpm in a thermomixer (MNase-digested samples) or by incubating the pellets with 25U of benzonase for 45 minutes (Benzonase-digested samples). After digestion, the samples were clarified (16,000 g at 4°C for 5 minutes) and the supernatant was transferred into a new 1.5 mL tube.
For immunoprecipitation, 10 µg of anti-Pol II antibody (MABI0601) or 3 µg of anti-Int3 (Bethyl A302–050A) antibody were incubated with 25 µL of magnetic anti-mouse beads (NEB) or Protein A beads, respectively, overnight. The following day, the antibody-conjugated beads were washed twice with 1 mL of NET-2 buffer and resuspended in 25 µL of NET-2 buffer. Chromatin fragments were pre-cleared by incubating them with the IgG-conjugated beads for 3 hours in a rotator at 4°C. The beads were then collected using a magnetic rack and the supernatant was transferred to a 1.5 mL tube containing the anti-Pol II conjugated beads. The samples were incubated for an additional 2 hours in the rotator at 4°C. After collecting the beads using a magnetic rack, the supernatant was discarded and the beads were washed four times using 1 mL of NET-2 buffer. The final wash was done using 1x PBS and the samples were resuspended using 8 µL of 10 mM Tris-HCL pH 8.5.
Proteomics:
Digestion.
The anti-Pol II immunoprecipitates were solubilized with a 2.5% solution of ALS-110 (Protea) in 50 mM Tris-HCL pH=8.5 with 5 mM EDTA, and 50 mM DTT directly on the beads. Samples were incubated at 95°C for 10 minutes to reduce cystines. Reactions were cooled on ice for 1 minute, 2 µL of 1 M Tris pH 8.5 were added and cysteines were alkylated by adding 4.7 µL of 100 mM iodoacetamide for 30 minutes in the dark. The reactions were then quenched with the addition of 0.7 µL of 200 mM DTT. Afterwards, 108 µL water, 0.67 µL CaCl2, 1.33 µL 1 M Tris-HCL pH 8.5 and 7.8 µL 0.5 mg/mL trypsin (Promega) were added and proteins left to digest for 16 hours at 37°C. Surfactant was cleaved with the addition of 20% trifluoroacetic acid to pH < 3 and samples were incubated at 25°C for 15 minutes. Peptides were then separated from beads using a magnetic rack and desalted using C18 silica MicroSpin Columns (The Nest Group). Column elution was performed in 300 µL 80% acetonitrile 0.1% trifluoroacetic acid and peptides were dried by centrifugal vacuum.
Mass spectrometry:
LC-MS/MS was performed using an ACQUITY UPLC M-Class (Waters) paired with a Q Exactive Plus (Thermo). Peptides were separated on a 65-cm-long, 75-µm-internal-diameter PicoFrit column (New Objective) packed in-house to a length of 50 cm with 1.9 µm ReproSil-Pur 120 Å C18-AQ (Dr. Maisch) using methanol as the packing solvent. Peptide separation was achieved using a non-linear 90-min gradient from 1% ACN 0.1% formic acid to 90% ACN 0.1% formic acid with a flowrate of 250 nl/min. Approximately 3–4 µg of peptides were run for each of the six biological replicates with at least one blank run between samples to avoid peptide carryover.
Bioinformatics:
Mass spectra were searched using Maxquant version 1.5.1.2 (Cox et al. 2008) with Carbamidomethyl (C) as a fixed modification, Acetyl (N-Term), Deamidation (NQ), Oxidation (M) as variable modifications and with up to three missed trypsin cleavages, a 7 amino acid minimum length, and 1% false discovery rate (FDR) against the Uniprot Human database (downloaded Dec-2017).
siRNA knockdowns:
For knockdown experiments, 7.5×104 HEK-293T cells were seeded in six well plates and incubated at 37°C overnight. After 24 hours, the cells were transfected with either 50 nM of scrambled siRNA control (siC), siRNA against Int11 (siInt11) (Xie et al. 2015) or siRNA against Int3 (siInt3) (Skaar et al. 2015) using Lipofectamine RNAimax according to manufacturer’s instructions. Cells were incubated at 37°C for 72 hours after which they were washed with 1x PBS, scraped and pelleted by centrifugation (1400 rpm at 4°C for 5 minutes). After discarding the supernatant, cells were resuspended in 150 µL of NET-2 buffer. 50 µL were aliquoted to verify knockdown efficiency through western blots and the rest of the sample was lysed using TRIzol. siRNA experiments for TT-TL experiments were performed by transfecting 1.5×105 cells for 48 hours instead of 72 hours.
Western blots:
For western blots of whole cell lysates, samples were sonicated using 10 cycles of 30 seconds on and 30 seconds off in a Diagenode Bioruptor Pico sonicator (Withers et al. 2018) and digested with micrococcal nuclease for 45 minutes at 37°C. Samples were run at an increasing voltage (up to 120 V) in NuPAGE 4–12% BisTris gels in 1x MOPS buffer (Invitrogen). Proteins were transferred overnight to a 0.45 µm nitrocellulose membrane at 30 V. Membranes were blocked for 1 hour in 5% milk in 1x PBST. Detection of total Pol II and Tyr1-P containing Pol II molecules was achieved using antibodies from MBL (MABI0601, discontinued) and Active motif (61383) respectively, at a 1:1000 dilution. Antibodies against Ints3 (Bethyl A302–050A), Ints11 (Abcam ab75276), Xrn2 (A301–101) and CPSF160 (A301–580A) were used at a 1:800 dilution. Anti-GAPDH antibody (Cell Signaling 14C10) was used at 1:2000, while the antibody against U1–70K (Kastner et al. 1992; Tarn and Steitz. 1994) was used at a 1:500 dilution. All secondary antibodies were used at a 1:2000 dilution of 1x PBST, 3% milk.
Quantification and Statistical Analysis
One sample t-tests and paired t-tests were performed on the fold changes of western blot band intensities and on RT-qPCR data, respectively, using graphPad prism. Differential expression analysis for TT-TL-seq and ChIP-seq datasets were obtained using DESeq2.
Supplementary Material
Table S1: Differential expression analysis of clean genes and DoG-producing clean genes. Related to Figures 1–3.
Table S2: Gene ontology analysis for DoG-producing clean genes and clean non-DoG genes. Related to Figure 3.
Table S3: Proteins identified by mass spectrometry in untreated and KCl-treated samples. Related to Figure 5.
Table S5: List of oligos used in this study. Related to Figures S1, S3, 7 and S5.
Table S4: Readthrough transcripts induced upon depletion of functional Int11. Related to Figure 7.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CPSF1 | Bethyl Laboratories | A301-580A |
| GAPDH | Cell Signaling | 14C10 |
| H3 | Abcam | ab1791 |
| Int3 | Bethyl Laboratories | A302-050A |
| Int11 | Abcam | ab75276 |
| RNA Pol II CTD | MBL | MABI0601 |
| RNA Pol II (N-terminus) | Cell Signaling | D8L4Y |
| RNA Pol II Tyr1-P CTD | Active Motif | 61383 |
| U1-70K | Kastner et al. 1992; Tarn and Steitz. 1994 | N/A |
| Xrn2 | Bethyl Laboratories | A301-101 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dynabeads Protein A | Invitrogen | 1001D |
| 4-thiouridine | Sigma | T4509 |
| Goat anti-mouse beads | NEB | S1431S |
| Lipofectamine RNAimax | Invitrogen | 13778-150 |
| Micrococcal nuclease | NEB | M0247S |
| RQ1 DNase | Promega | M610A |
| Superscript III | Invitrogen | 56575 |
| Deposited Data | ||
| Raw and analyzed TT-TL-seq data | This paper | GSE152059 |
| Raw and analyzed anti-Pol II ChIP-seq data | This paper | GSE152062 |
| Raw and analyzed anti-Int3 and anti-Int11 data | This paper | GSE159190 |
| Experimental Models: Cell Lines | ||
| HEK-293T | Steitz lab | N/A |
| HEK-293T 2x FLAG-Int11 WT | Xie et al. 2015 | N/A |
| HEK-293T 2x FLAG-Int11 E203Q | Xie et al. 2015 | N/A |
| Drosophila S2 cells | Simon lab | N/A |
| SK-N-BE(2)C | Steitz lab | N/A |
| Oligonucleotides | ||
| RT-qPCR primers | Table S5 | N/A |
| siRNAs | Table S5 | N/A |
| Northern blot probes | Table S5 | N/A |
| Random primers | Invitrogen | 58875 |
| Software and Algorithms | ||
| ARTDeco | Roth et al. 2020 | N/A |
| bedtools | Quinlan et al. 2010 | N/A |
| Bowtie2 | Langmead et al. 2009 | N/A |
| Cutadapt | Martin 2011 | N/A |
| DESeq2 | Love et al. 2014 | N/A |
| DoGFinder | Wiesel et al. 2018 | N/A |
| edger | Robinson et al. 2010 | N/A |
| enrichr | Chen et al. 2013; Kuleshov et al. 2016 | N/A |
| eulerr | Larsson J (2020) | https://github.com/jolars/eulerr |
| FastUniq | Xu et al. 2012 | N/A |
| ggplot2 | Wickham. 2016 | N/A |
| Hisat2 | Kim et al. 2015 | N/A |
| HTSeq | Anders et al. 2015 | N/A |
| MACS2 | Zhang et al. 2008 | N/A |
| Prism | GraphPad | N/A |
| samtools | Li et al. 2009 | N/A |
Highlights.
Hyperosmotic stress triggers transcriptional repression of many genes.
DoG RNAs arise independent of the transcriptional level of their upstream gene.
Interactions between Pol II and Integrator subunits decrease after salt stress.
Depletion of the Int11 nuclease subunit induces the production of hundreds of DoGs.
Acknowledgments
We thank Annsea Park, Kazimierz Tycowski, Salehe Ghasempur and other members of the Steitz lab for critical discussion of the manuscript. We are grateful to Samuel Roth for his assistance in troubleshooting our read-in analysis using ARTDeco. We thank Angela Miccinello for editorial assistance and Mingyi Xie and Mei-Di Shu for cells expressing exogenous Int11. This work was supported by the following NIH grants: CA200147, T32AI055403 and R01 GM137117. N.R. is a Ford Foundation pre-doctoral fellow and J.A.S. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
J.A.S. is a member of the Molecular Cell Advisory Board. The authors have no further competing interest to declare.
References
- Albrecht TR, and Wagner EJ (2012). snRNA 3’ end formation requires heterodimeric association of integrator subunits. Mol Cell Biol 32, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amat R, Bottcher R, Le Dily F, Vidal E, Quilez J, Cuartero Y, Beato M, de Nadal E, and Posas F (2019). Rapid reversible changes in compartments and local chromatin organization revealed by hyperosmotic shock. Genome Res 29, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics, 31(2), 166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Hakimi MA, Naar AM, Shilatifard A, Cooch N, and Shiekhattar R (2005). Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA Polymerase II. Cell 123, 265–276. [DOI] [PubMed] [Google Scholar]
- Baillat D, and Wagner EJ (2015). Integrator: surprisingly diverse functions in gene expression. Trends Biochem Sci 40, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DLV, Tellier M, Martinez-Alonso M, Nojima T, Proudfoot NJ, Murphy S, and Fodor E (2018). Influenza virus mounts a two-pronged attack on host RNA Polymerase II transcription. Cell Rep 23, 2119–2129 e2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckedorff F, Blumenthal E, daSilva LF, Aoi Y, Cingaram PR, Yue J, Zhang A, Dokaneheifard S, Valencia MG, Gaidosh G, et al. (2020). The human Integrator complex facilitates transcriptional elongation by endonucleolytic cleavage of nascent transcripts. Cell Rep 32, 107917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieberstein NI, Straube K, and Neugebauer KM (2014). Chromatin immunoprecipitation approaches to determine co-transcriptional nature of splicing. Methods Mol Biol 1126, 315–323. [DOI] [PubMed] [Google Scholar]
- Boutz PL, Bhutkar A, and Sharp PA (2015). Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev 29, 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardiello JF, Goodrich JA, and Kugel JF (2018). Heat shock causes a reversible increase in RNA Polymerase II occupancy downstream of mRNA genes, consistent with a global loss in transcriptional termination. Mol Cell Biol 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Hu Z, Xia Z, Zhao D, Li W, and Tyler JK (2015). The overlooked fact: fundamental need for spike-in control for virtually all genome-wide analyses. Mol Cell Biol 36, 662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, & Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dominski Z, Yang XC, Purdy M, Wagner EJ, and Marzluff WF (2005). A CPSF-73 homologue is required for cell cycle progression but not cell growth and interacts with a protein having features of CPSF-100. Mol Cell Biol 25, 1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, and Simon MD (2015). Tracking distinct RNA populations using efficient and reversible covalent chemistry. Mol Cell 59, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JD, Davidson L, Bauer DLV, Natsume T, Kanemaki MT, and West S (2018). Xrn2 accelerates termination by RNA Polymerase II, which is underpinned by CPSF73 activity. Genes Dev 32, 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JD, Francis L, Davidson L, and West S (2020). A unified allosteric/torpedo mechanism for transcriptional termination on human protein-coding genes. Genes Dev 34, 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod ND, Henriques T, Huang KL, Tatomer DC, Wilusz JE, Wagner EJ, and Adelman K (2019). The Integrator complex attenuates promoter-proximal transcription at protein-coding genes. Mol Cell 76, 738–752.e737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Brannan K, Erickson B, Kim H, Cortazar MA, Sheridan RM, Nguyen T, Karp S, and Bentley DL (2015). Effects of transcription elongation rate and Xrn2 exonuclease activity on RNA Polymerase II termination suggest widespread kinetic competition. Mol Cell 60, 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini A, Baillat D, Cesaroni M, Hu D, Marinis JM, Wagner EJ, Lazar MA, Shilatifard A, and Shiekhattar R (2014). Integrator regulates transcriptional initiation and pause release following activation. Mol Cell 56, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosso AR, Leite AP, Carvalho S, Matos MR, Martins FB, Vitor AC, Desterro JM, Carmo-Fonseca M, and de Almeida SF (2015). Pervasive transcription read-through promotes aberrant expression of oncogenes and RNA chimeras in renal carcinoma. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, Trotta KL, Smith EE, Mosaheb MM, Fuchs SM, and Churchman LS (2016). Comprehensive RNA Polymerase II interactomes reveal distinct and varied roles for each phospho-CTD residue. Cell Rep 15, 2147–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Texari L, Hayes MGB, Urbanowski M, Chang MW, Givarkes N, Rialdi A, White KM, Albrecht RA, Pache L, et al. (2018). Transcription elongation can affect genome 3D structure. Cell 174, 1522–1536.e1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig T, Michalski M, Rutkowski AJ, Djakovic L, Whisnant AW, Friedl MS, Jha BA, Baptista MAP, L’Hernault A, Erhard F, et al. (2018). HSV-1-induced disruption of transcription termination resembles a cellular stress response but selectively increases chromatin accessibility downstream of genes. PLoS Pathog 14, e1006954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalihal AP, Pitchiaya S, Xiao L, Bawa P, Jiang X, Bedi K, Parolia A, Cieslik M, Ljungman M, Chinnaiyan AM, et al. (2020). Multivalent proteins rapidly and reversibly phase-separate upon osmotic cell volume change. bioRxiv, 748293. [DOI] [PMC free article] [PubMed]
- Kastner B, Kornstadt U, Bach M, and Luhrmann R (1992). Structure of the small nuclear RNP particle U1: identification of the two structural protuberances with RNP-antigens A and 70K. J Cell Biol 116, 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Gardini A, Zhang A, and Shiekhattar R (2015). Integrator mediates the biogenesis of enhancer RNAs. Nature 525, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, and Young RA (2012). Revisiting global gene expression analysis. Cell 151, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Salamanca HH, Duarte FM, Danko CG, and Lis JT (2016). Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol Cell 62, 63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SK, and Kültz D (2004). Gadd45 proteins induce G2/M arrest and modulate apoptosis in kidney cells exposed to hyperosmotic stress. J Biol Chem 279, 39075–39084. [DOI] [PubMed] [Google Scholar]
- Mandel CR, Kaneko S, Zhang H, Gebauer D, Vethantham V, Manley JL, and Tong L (2006). Polyadenylation factor CPSF-73 is the pre-mRNA 3’-end-processing endonuclease. Nature 444, 953–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal, 17, 10–12. [Google Scholar]
- Mazo A, Hodgson JW, Petruk S, Sedkov Y, and Brock HW (2007). Transcriptional interference: an unexpected layer of complexity in gene regulation. J Cell Sci 120, 2755–2761. [DOI] [PubMed] [Google Scholar]
- Muniz L, Deb MK, Aguirrebengoa M, Lazorthes S, Trouche D, and Nicolas E (2017). Control of gene expression in senescence through transcriptional read-through of convergent protein-coding genes. Cell Rep 21, 2433–2446. [DOI] [PubMed] [Google Scholar]
- Nemeroff ME, Barabino SM, Li Y, Keller W, and Krug RM (1998). Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3’end formation of cellular pre-mRNAs. Mol Cell 1, 991–1000. [DOI] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Carmo-Fonseca M, and Proudfoot NJ (2016). Mammalian NET-seq analysis defines nascent RNA profiles and associated RNA processing genome-wide. Nat Protoc 11, 413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Tellier M, Foxwell J, Ribeiro de Almeida C, Tan-Wong SM, Dhir S, Dujardin G, Dhir A, Murphy S, and Proudfoot NJ (2018). Deregulated expression of mammalian lncRNA through loss of SPT6 induces R-Loop formation, replication stress, and cellular senescence. Mol Cell 72, 970–984.e977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot NJ (2016). Transcriptional termination in mammals: Stopping the RNA Polymerase II juggernaut. Science 352, aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins E, Pederson T, and Klein P (1970). Comparison of mitotic phenomena and effects induced by hypertonic solutions in HeLa cells. J Cell Biol 44, 400–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth SJ, Heinz S, and Benner C (2020). ARTDeco: automatic readthrough transcription detection. BMC Bioinformatics 21, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski AJ, Erhard F, L’Hernault A, Bonfert T, Schilhabel M, Crump C, Rosenstiel P, Efstathiou S, Zimmer R, Friedel CC, et al. (2015). Widespread disruption of host transcription termination in HSV-1 infection. Nat Commun 6, 7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schofield JA, Duffy EE, Kiefer L, Sullivan MC, and Simon MD (2018). TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat Methods 15, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Fruhauf K, Demel C, Tresch A, Gagneur J, and Cramer P (2016). TT-seq maps the human transient transcriptome. Science 352, 1225–1228. [DOI] [PubMed] [Google Scholar]
- Shalgi R, Hurt JA, Lindquist S, and Burge CB (2014). Widespread inhibition of posttranscriptional splicing shapes the cellular transcriptome following heat shock. Cell Rep 7, 1362–1370. [DOI] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR 3rd, Frank J, and Manley JL (2009). Molecular architecture of the human pre-mRNA 3’ processing complex. Mol Cell 33, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Ferris AL, Wu X, Saraf A, Khanna KK, Florens L, Washburn MP, Hughes SH, and Pagano M (2015). The Integrator complex controls the termination of transcription at diverse classes of gene targets. Cell Res 25, 288–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmayer B, Micas G, Gamot A, Martin P, Malirat N, Koval S, Raffel R, Sobhian B, Severac D, Rialle S, et al. (2014). Integrator complex regulates NELF-mediated RNA Polymerase II pause/release and processivity at coding genes. Nat Commun 5, 5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn WY, and Steitz JA (1994). SR proteins can compensate for the loss of U1 snRNP functions in vitro. Genes Dev 8, 2704–2717. [DOI] [PubMed] [Google Scholar]
- Tatomer DC, Elrod ND, Liang D, Xiao MS, Jiang JZ, Jonathan M, Huang KL, Wagner EJ, Cherry S, and Wilusz JE (2019). The Integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev 33, 1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, Passarelli MC, Yario TA, Tycowski KT, and Steitz JA (2015). Widespread inducible transcription downstream of human genes. Mol Cell 59, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, Sabath N, Wiesel Y, Nathans J, Levy-Adam F, Yario TA, Steitz JA, and Shalgi R (2017). Comparative analysis reveals genomic features of stress-induced transcriptional readthrough. Proc Natl Acad Sci U S A 114, E8362–E8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, and Steitz JA (2017). Readthrough transcription: How are DoGs made and what do they do? RNA Biol 14, 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hennig T, Whisnant AW, Erhard F, Prusty BK, Friedel CC, Forouzmand E, Hu W, Erber L, Chen Y, et al. (2020). Herpes simplex virus blocks host transcription termination via the bimodal activities of ICP27. Nat Commun 11, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant graphics for data analysis (New York:Springer-Verlag; ). [Google Scholar]
- Wiesel Y, Sabath N, and Shalgi R (2018). DoGFinder: a software for the discovery and quantification of readthrough transcripts from RNA-seq. BMC Genomics 19, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers JB, Li ES, Vallery TK, Yario TA, and Steitz JA (2018). Two herpesviral noncoding PAN RNAs are functionally homologous but do not associate with common chromatin loci. PLoS Pathog 14, e1007389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Albrecht TR, Baillat D, Wagner EJ, and Tong L (2017). Molecular basis for the interaction between Integrator subunits IntS9 and IntS11 and its functional importance. Proc Natl Acad Sci U S A 114, 4394–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin J, and Schibler U (1994). Physical isolation of nascent RNA chains transcribed by RNA Polymerase II: evidence for cotranscriptional splicing. Mol Cell Biol 14, 7219–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Zhang W, Shu MD, Xu A, Lenis DA, DiMaio D, and Steitz JA (2015). The host Integrator complex acts in transcription-independent maturation of herpesvirus microRNA 3’ ends. Genes Dev 29, 1552–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Luo X, Qian J, Pang X, Song J, Qian G, Chen J, and Chen S (2012). FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One. 7, e52249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Differential expression analysis of clean genes and DoG-producing clean genes. Related to Figures 1–3.
Table S2: Gene ontology analysis for DoG-producing clean genes and clean non-DoG genes. Related to Figure 3.
Table S3: Proteins identified by mass spectrometry in untreated and KCl-treated samples. Related to Figure 5.
Table S5: List of oligos used in this study. Related to Figures S1, S3, 7 and S5.
Table S4: Readthrough transcripts induced upon depletion of functional Int11. Related to Figure 7.
Data Availability Statement
All high throughput sequencing datasets generated in this study have been deposited to the Gene Expression Omnibus database and can be found as a super series under the following accession number: GSE152063.