

Abstract
When the JCVI-syn3.0 genome was designed and implemented in 2016 as the minimal genome of a free-living organism, approximately one-third of the 438 protein-coding genes had no known function. Subsequent refinement into JCVI-syn3A led to inclusion of 16 additional protein-coding genes, including several unknown functions, resulting in an improved growth phenotype. Here, we seek to unveil the biological roles and protein–protein interaction (PPI) networks for these poorly characterized proteins using state-of-the-art deep learning contact-assisted structure prediction, followed by structure-based annotation of functions and PPI predictions. Our pipeline is able to confidently assign functions for many previously unannotated proteins such as putative vitamin transporters, which suggest the importance of nutrient uptake even in a minimized genome. Remarkably, despite the artificial selection of genes in the minimal syn3 genome, our reconstructed PPI network still shows a power law distribution of node degrees typical of naturally evolved bacterial PPI networks. Making use of our framework for combined structure/function/interaction modeling, we are able to identify both fundamental aspects of network biology that are retained in a minimal proteome and additional essential functions not yet recognized among the poorly annotated components of the syn3.0 and syn3A proteomes.
Keywords: structure prediction, computational function annotation, protein–protein interaction, deep learning, essential proteins, JCVI-syn3.0 minimal genome, JCVI-syn3A
Graphical Abstract
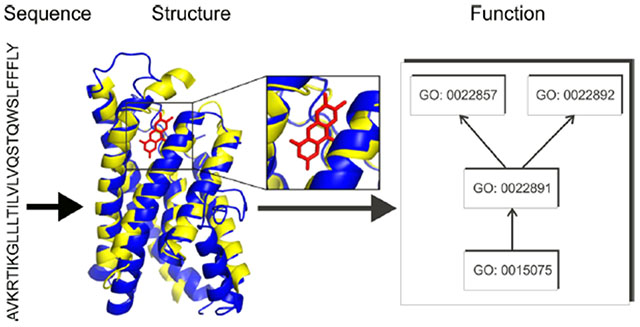
INTRODUCTION
The question of what set of functionalities constitutes the minimal set necessary to enable life is one of the most important unanswered questions of contemporary biology.1–3 While even the question of what constitutes “life” carries a vast range of philosophical difficulties,4,5 for the present purposes, we define a living thing as an entity consisting of one or more membrane-bound cells capable of separating itself from its surroundings, drawing energy from its environment, and using that energy to maintain (and possibly reproduce) itself. As the simplest organisms meeting this definition will be unicellular, and in all known cases such organisms make use of a DNA genome, investigations into the minimal basis for life have almost invariably focused on determining the minimal set of genetic components required to yield a living cell. Studies based on transposon knockout libraries or high-throughput targeted deletions substantially enhanced our ability to rationally design reduced genomes by providing a high-throughput approach for identifying all genes that could not be individually knocked out.6–12 Such knockout libraries cannot, however, provide all needed information for construction of a minimal genome because of the presence of both positive and negative epistatic interactions that cannot be captured in a single pass using such approaches.3,7 More targeted work13 provided a window into the overall reducibility of microbial genomes by deleting all prophages and mobile genetic elements from Escherichia coli MG1655, yielding a genome that was reduced in size by ~15%; the reduced genome strain, MDS42, also showed several useful properties such as increased stability of cloned genes.14,15 A new level of capability in the study of minimal genomes was achieved with the development of JCVI-syn1.0, a completely synthetic Mycoplasma mycoides derivative.16 The subsequent inclusion of repeated cycles of transposon mutagenesis and a “design-build-test” cycle permitted comprehensive mapping of the genes that could not be complemented by any other gene in the original M. mycoides genome, which we refer to as “essential”. The cyclical genome reduction efforts described above yielded a well-defined list of 465 effectively essential genes for a minimal Mycoplasma, 438 of which encode proteins. The resulting organism, syn3.0, has a genome reduced in size by nearly 50%, and shows substantial differences in growth and cellular morphology from the M. mycoides parental strain,17 including a reduced growth rate, reduced colony sizes, and a filamentous and highly heterogeneous cellular morphology.
Simply knowing the identities of all genes needed in a minimal genome, however, does not permit resolution of the fundamental question of what functionalities are needed in a minimal cell. Upon the initial construction of syn3.0, researchers noted that ~1/3 of the protein coding genes in its genome could not be annotated by sequence homologues from characterized protein domain families;17 more recent efforts to enable a complete metabolic reconstruction of syn3.0 still cannot assign a protein to all functions necessary in a minimal metabolic model.18 Initial efforts to determine the functions and biological roles of the remainder of the syn3.0 proteome were based on sequence-based annotations and sequence-profile based protein family assignment,17,19 which have limited sensitivity when there are no close homology templates for annotation transfer. Later, Yang and Tsui attempted to annotate syn3.0 proteins by secondary structure matching,20 which was developed to recognize templates with a similar structure fold but not necessarily of a related function. More recently, Antczak and colleagues applied a multipipeline approach to provide consensus predictions that added functional information for 66 of the proteins of unknown function in syn3.0, demonstrating a particular abundance of putative transporters and other transmembrane proteins.21 The syn3 proteome was also recently expanded through the addition of 19 genes, including 16 protein-coding genes, which while nonessential resulted in an improved growth phenotype; the resulting organism was labeled JCVI-syn3A;18 11 of the new protein-coding genes in JCVI-syn3A are also poorly annotated.
We have recently shown that the inclusion of protein structural information, even from computationally predicted structures, can substantially enhance the accuracy of function predictions for difficult annotation targets.22,23 To this end, we developed an I-TASSER/COFACTOR-based protocol that performs I-TASSER structure prediction followed by COFACTOR structure-based function annotation.24 This pipeline has been shown to accurately assign functions for many proteins in microbes22 and in humans25 and is among the top predictors in the most recent Critical Assessment of Function Annotation round 3 (CAFA3) and CAFA PI competitions.26 Moreover, the recent development of sequence-derived residue–residue contact prediction algorithms based on deep neural networks27,28 has greatly enhanced the accuracy of protein structure assembly, which should in principle enhance the effectiveness of structure-based protein function prediction.
To have a complete understanding of the essential syn3.0 proteome and syn3A expansions, we developed and applied an enhanced C-I-TASSER/COFACTOR pipeline by the combination of contact map-based protein structure simulations with structure-based protein function annotation and protein–protein interaction (PPI) predictions. We found that high-confidence molecular function (MF) and biological process (BP) annotations from gene ontology (GO) can be provided for 86 and 88% of the syn3.0 proteome, respectively, while the utilization of deep neural-network contact-map information shows significant enhancements of both coverage and accuracy of protein structure and functional models. Functions related to nutrient acquisition, microbe–host interactions, and nucleotide metabolism are enriched among the set of previously unannotated genes, likely indicating important and as-yet unresolved portions of syn3.0 physiology. Viewed at the level of the whole-cell PPI network, we further note that the PPI network of syn3.0 follows the scale-free network architecture often noted in natural PPIs but rare in randomly formed networks, suggesting that scale-free layouts persist even when an original, natural PPI network is artificially reduced to a minimal, essential form of itself.
MATERIALS AND METHODS
Protein Structure Prediction
Structure models of all 438 proteins in the syn3.0 genome were predicted by C-I-TASSER,29 our most recent protein structure prediction pipeline based on the I-TASSER structural assembly protocol30 combined with deep learning-based residue–residue contact map predictions.27,28 Briefly, C-I-TASSER first uses DeepMSA31 to search the query protein sequence against three whole-genome and metagenome protein sequence databases, including Uniclust30,32 UniRef90,33 and Metaclust,34 to obtain a multiple sequence alignment (MSA). Next, residue–residue contacts are predicted from the MSA by the deep learning-based algorithms TripletRes/ResTriplet27 and ResPRE28 (see the Supporting Information Text S1 for details). Meanwhile, LOMETS threading35 is performed to search for the query protein sequence against the PDB database to align the query to template structures to extract continuous fragments. These fragments are finally assembled into the full length structures by a replica-exchange Monte Carlo (REMC) simulation under the guidance of a composite force field consisting of the deep learning-predicted contacts, template-derived distance restraints, and knowledge-based energy terms calculated based on statistics of PDB structures. The REMC simulation produces tens of thousands of “decoy” conformations, which are clustered by pairwise structure similarity.36 The centroid of the largest cluster is refined at the atomic level37 to obtain the final C-I-TASSER model.
As a control experiment to study the impact of deep learning-predicted contacts on structure and function prediction, we also performed structure prediction for the same set of 438 proteins using the classical I-TASSER pipeline without contact prediction. Structure-based function annotations were separately performed for the top-ranked models produced by C-I-TASSER and I-TASSER for the same target protein, as detailed below.
Estimation of Structure Model Quality
The global quality of structural models can be assessed by the TM-score38 between modeled and native structures of the target protein
| (1) |
where L is the number of residues in the target, di is the distance between the ith aligned residue pair, and is a length-dependent scaling factor. TM-score ranges between 0 and 1, with TM-score > 0.5 meaning structure models of correct global topology.39
As the native structures of syn3.0 proteins are not available, we estimate the TM-score (eTM) of the C-I-TASSER models using a combination of threading alignment quality, contact satisfaction rate, and convergence of the structure assembly simulations
| (2) |
where the confidence score (C) is defined as
| (3) |
c0 = 0.79, c1 = 0.1077, c2 = 0.00098, w1 = 0.77, w2 = 1.36, and w3 = 0.67 are free parameters obtained by maximizing the correlation between the estimated and actual TM-score on a separate set of 797 training protein domain structures from SCOPe database40 version 2.06. Mtotal is the total number of decoy conformations used for clustering, while M is the number of decoys in the top cluster. ⟨RMSD⟩ is the average rmsd among decoys in the same cluster. Z(m) is the score of the top template by the mth threading method in LOMETS. Z0(m) is a cutoff above which templates are considered reliable. N(CMpred) is the number of contacts predicted by deep learning and used for guiding the REMC simulation, while O(CMnative,CMpred) is the number of common contacts between the final model and the deep learning-predicted contacts. For the (non-contact-based) I-TASSER-predicted structures, the estimated TM-score is calculated similarly, but with c0 = 0.71, c1 = 0.1300, c2 = 0.00060, w1 = w2 = 1, and w3 = 0. The estimated TM-score was shown to highly correlate with the actual TM-score, with a Pearson correlation coefficient (PCC) of 0.91 on 300 test proteins that are nonhomologous to the training proteins of I-TASSER.41
Function Annotation and Enrichment Analysis
Protein functions are predicted from the structure models by COFACTOR,22 which combines models from three complementary submodules based on structure, sequence, and PPI. In the structure-based submodule, the (C-)I-TASSER model is structurally aligned to function templates in the BioLiP database,42 where function annotations are obtained from the function templates identified by global and local structure similarity. In the sequence-based submodule, BLAST and PSI-BLAST43 are used to search for the query sequence against the UniProt Gene Ontology Annotation (UniProt-GOA) database44 to obtain annotations from sequence homologues. Finally, the PPI-based submodule is ported from MetaGO,23 where the query sequence is mapped to the PPI network of STRING,45 with the immediate neighbor (i.e., direct PPI partner) of the query searched against UniProt-GOA for function transfer. Function predictions from these three submodules are combined by weighted averaging to obtain the final prediction. Each predicted function has a confidence score (C-scoreFunc) ranging from 0 to 1, with C-scoreFunc > 0.5 corresponding to a confident function prediction.22,25 While COFACTOR predicts three categories of protein functions, namely, Enzyme Commission (EC) numbers, GO terms, and ligand binding sites (LBSs), we do not separately discuss prediction of EC numbers because they can be mapped to MF GO terms.46
Enrichment of GO terms in previously unannotated syn3.0 proteins (vs proteins with previous UniProt free-text annotation or UniProt-GOA GO term annotations) are quantified by a rate ratio test approach.47 Briefly, for each GO term q, we compute the annotation rate (i.e., the number of proteins annotated with q divided by the total number of proteins) among UniProt-unannotated proteins and that among UniProt-annotated proteins. We then test whether the ratio of the two rates is significantly different from 1. Some GO terms, such as GO:0005515 “protein binding”, are too generic to suggest any specific function. Therefore, similar to our prior study,24 we discard any GO terms associated with >10% of annotated proteins in all steps of our analysis, including the definition of previously unannotated/annotated proteins and the rate ratio test of GO term enrichment.
PPI Prediction
The PPI network of syn3.0 was predicted using the SPRING48 dimer threading program. For a pair of query proteins, SPRING first searches for the sequence of each protein chain to a monomeric template structure database by HHsearch.49 The HHsearch aligned monomeric templates are then structurally aligned to complexes in the PDB dimer template database by TM-align50 to obtain the dimeric complex model. The final score of the dimeric complexes, SPRING-score, is a linear combination of three terms: the Z-score for HHsearch monomeric threading, TM-score of monomer-to-dimer structure alignment by TM-align, and a statistical energy potential for the dimer interface. The two query proteins are considered to interact with each other if there is a good complex hit with SPRING-score >2 and both of the monomer threading Z-scores > −2. The Z-score and SPRING-score cutoffs were trained to optimize the Matthews correlation coefficient (MCC) of classifying interacting versus noninteracting protein pairs on a dataset consisting of 1732 structurally characterized PPI pairs from the SPRING dimer template database and 4117 pairs of noninteracting proteins from the Database of Interacting Proteins (DIP).51 Only heterodimeric interactions are considered in this study.
Data Availability
Protein sequences of syn3.0 were collected from NCBI accession CP014940.1. While the genome consists of 473 genes, this study only considered the 438 protein coding genes, as the other 35 genes encode noncoding RNAs with well-known functions such as tRNAs and rRNAs. The syn3.0 proteins are mapped to the closest UniProt 2019_09 entries from M. mycoides reference proteomes UP000001016 and UP000011126. The GO annotations of these UniProt entries are collected from UniProt-GOA release 2019-09-17. All predicted structure models, functions, and interactions are available at our public Webserver at https://zhanglab.ccmb.med.umich.edu/JCVI-syn3.0/, including a one sentence description of protein function generated using the most specific high confidence predicted GO term.
When we were in the midst of this study, a new version of the minimal genome, JCVI-syn3A (NCBI accession CP016816.2), was published,18 which includes 16 additional protein coding genes not included in the JCVI-syn3.0 genome. Although these new genes are not essential for the survival of the cell, they make the cell less fragile and cause it to have a more stable cellular morphology. For completeness, we have included these 16 new genes in our structure and function prediction as part of our online webserver even though our main analysis focuses on the original JCVI-syn3.0 genome, which still represents the most “minimal” genome achieved in the series. To facilitate comparative study between JCVI-syn3.0 and JCVI-syn3A, the webserver displays the protein names and accessions for both genomes.
RESULTS
Contact-Assisted Protein Structure Prediction and Structure-Based Function Prediction Increase the Coverage of Function Annotation
We began by investigating how many syn3.0 proteins can be assigned specific GO term annotations, which were categorized by the original syn3.0 study17 into five classes (Unknown, Generic, Putative, Probable, and Equivalog) in ascending order of function annotation confidence, based on a protein’s match to the TIGRfam protein family database.52 Specifically, unknown or generic proteins lack functional homologues or do not have homologues with consistent function annotations, while putative, probable, or equivalog proteins can match homologous proteins with related functions in the same family. As shown in Figure 1A–E, for all five classes, the numbers of proteins for which GO terms can be assigned by the structure-based function annotation pipeline C-I-TASSER/COFACTOR are consistently greater than those in UniProt. Here, the UniProt terms in Figure 1A–E refer to the GO annotations from the UniProt-GOA project;44 all UniProt terms for the syn3.0 proteins in our study are from computational approaches such as UniRule and InterProScan53 with evidence codes “Inferred from Electronic Annotation” (IEA) and “Inferred from Sequence or structural Similarity” (ISS). It is therefore fair to compare the coverage (i.e., the percentage of proteins that can be annotated) between UniProt annotations and C-I-TASSER/COFACTOR annotations, as both are computationally predicted GO terms. The broader coverage of C-I-TASSER/COFACTOR is particularly evident for the unknown and generic categories, which are considered uncharacterized in the original syn3.0 study.17 For example, C-I-TASSER/COFACTOR can annotate 49 and 45% of all unknown proteins with specific MF and BP terms, respectively, which are 9 times more than UniProt for the same set of proteins (5% for both MF and BP) (Figure 1A). In both C-I-TASSER/COFACTOR and UniProt GO annotations, the number of proteins with specific cellular component (CC) terms is smaller than those with MF or BP terms. This is partly due to the simple cellular structure of syn3.0 (which has a single cell membrane and no cell wall or membrane-bound organelles), where most proteins localize to the cytoplasm or plasma membrane instead of more specific subcellular locations.
Figure 1.
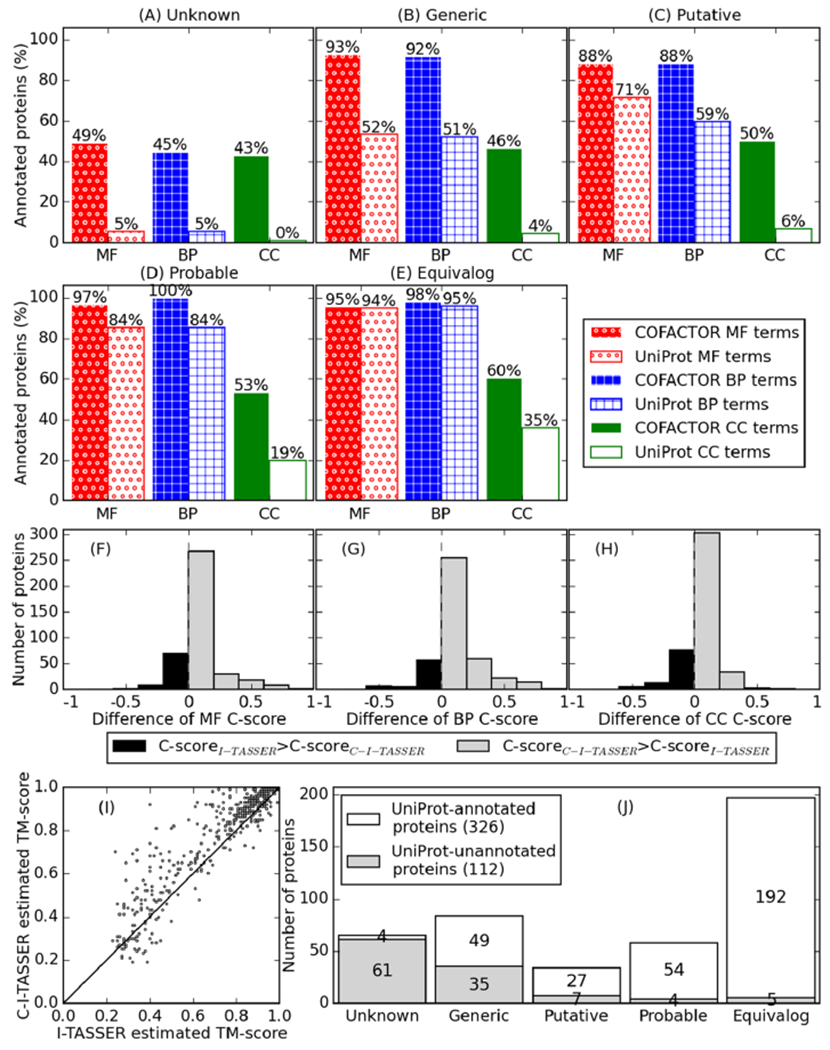
C-I-TASSER/COFACTOR improves coverage of protein function prediction (i.e., percentage of proteins with predicted function) for syn3.0. (A–E) Percentage of proteins that can be annotated with GO terms by C-I-TASSER/COFACTOR and by UniProt for the five categories of syn3.0 proteins classified in the original syn3.0 report, where “unknown” (A) and “generic” (B) proteins were considered unannotated. (F–H) Distribution of difference in confidence scores (C-scores) for COFACTOR GO term prediction using C-I-TASSER models compared to those using I-TASSER models. For each protein, only GO terms predicted with C-score > 0.5 in at least one of C-I-TASSER/COFACTOR and I-TASSER/COFACTOR are considered, and the average C-score difference for using C-I-TASSER compared to using I-TASSER for each protein is shown on the x-axis. The average C-score differences in structure-based GO term prediction using C-I-TASSER versus that using I-TASSER are +0.07, +0.11, and +0.06 for MF (F), BP (G), and CC (H), respectively. (I) Per-target comparison of estimated TM-score between I-TASSER (x-axis) and C-I-TASSER (y-axis). Points on the upper left triangle correspond to targets with better estimated quality in C-I-TASSER than in I-TASSER. (J) Number of proteins with (white) and without (gray) function annotation (GO terms or free-text) in the five categories of syn3.0 proteins.
The high sensitivity of our C-I-TASSER/COFACTOR pipeline can be attributed partly to the use of deep learning-predicted contact maps in the template-based modeling of protein structures by C-I-TASSER. As shown in the recent CASP13 experiment,29 C-I-TASSER is capable of assembling significantly more accurate structure models than traditional threading/homology approaches for the nonhomologous protein sequences, which is particularly important for the proteins from JCVI-syn3.0A. Indeed, the confidence score of COFACTOR GO term prediction is consistently improved by using structure models from contact-assisted C-I-TASSER over the traditional I-TASSER approach for all three aspects of GO terms (Figure 1F–H). Accordingly, the quality of C-I-TASSER structure models in terms of average estimated TM-score (0.76)38 is 8.6% higher than that of I-TASSER (0.70); 328 of the 434 proteins (76%) are estimated to have better structure model quality in C-I-TASSER than in I-TASSER (Figure 1I). Despite the high sensitivity of the C-I-TASSER/COFACTOR pipeline, there are still 14 and 12% of the syn3.0 proteins that cannot be annotated with specific MF and BP terms, respectively, partly because of the high transmembrane contents for the targets (Figure S1), making them more difficult for experimental characterization and computational annotation.
The original method for partitioning syn3.0 protein annotation status into five categories may not be sufficiently specific as a protein not belonging to a characterized TIGRfam protein family can still be individually annotated. Thus, we reclassified annotated versus unannotated proteins based on whether their respective UniProt Gene Ontology Annotation (UniProt-GOA)44 entries in the M. mycoides proteome have specific GO term annotations, excluding overly general GO terms such as “protein binding” (see the Materials and Methods section). As shown in Figure 1J, 112 (26%) of the 438 proteins in syn3.0 are unannotated based on their UniProt entries. This is smaller than the number of proteins with unknown function (149 of 438 proteins) reported in previous studies17,21 as some proteins previously reported to have unknown functions are now annotated as of UniProt release 2019_09. These inconsistencies could have resulted from either the difference in classifying annotated versus unannotated proteins, the recent improvement of the annotation pipeline used in UniProt, or both. For the sake of consistency with contemporary work,54 in later sections we use the term “unannotated proteins” to refer to proteins without UniProt annotation, regardless of their TIGRfam match.
Functions Enriched in Uncharacterized Proteins Highlight the Dependency of syn3.0 on the Environment
To obtain a more nearly complete understanding of the metabolism of syn3.0 and the nature of the required genes that it encodes, we applied a rate-ratio test approach (see the Materials and Methods section for details) to search for the GO terms that were enriched among previously unannotated proteins. Compared to previously annotated proteins, UniProt unannotated proteins are enriched for “transporter activity” and “phosphatase activity” for MF and “response to other organism” and “dephosphorylation” for BP (Figure 2). This is consistent with a previous study that proposed that some of the poorly characterized syn3.0 proteins are transporters.21 Among the newly annotated proteins with “phosphatase activity” annotations, furthermore, at least half appear likely to act on nucleotide substrates, suggesting a particularly important role for these poorly annotated nucleotide phosphatases in syn3.0 for either signal transduction or metabolism. A role in signaling might be possible via second messengers; ppGpp and cyclic-di-AMP, for example, have been shown to be used in various Mycoplasma species,55–57 and syn3.0 does indeed have a probable relA enzyme in MMSYN1_0414 (one should also note that phosphatase and phosphodiesterase activities are sibling nodes in the GO hierarchy and closely related from an enzymatic standpoint). Within the category of metabolism, two appealing explanations exist for the abundance of predicted phosphatases (and particularly nucleotide phosphatases): first, such enzymes might participate in nutrient acquisition and recycling, as has been suggested for enzymes with related activities in Mycoplasma bovis.57 Second, it is possible that these phosphatases are needed to detoxify otherwise harmful products of metabolite damage reactions;58,59 several examples of detoxifying enzymes acting as phosphatases on nucleotide-like substrates have recently been identified.59,60 Further characterization of the enzymes currently flagged in our annotations as phosphatases (GO:0016791) without more detailed current annotations would be particularly useful in investigating these possibilities.
Figure 2.
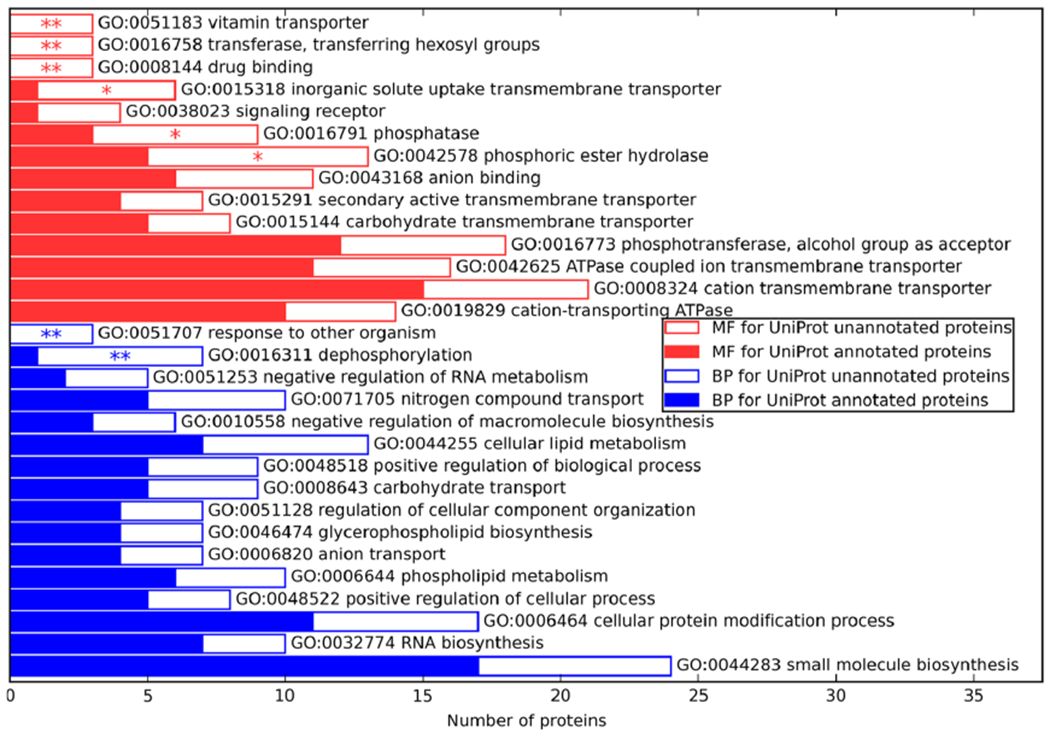
Enrichment of MF (upper half) and BP (lower half) GO terms predicted by C-I-TASSER/COFACTOR in proteins of unknown function (empty bars) compared to proteins of known function (solid bars). One asterisk is shown for significant enrichment of a GO term in the unknown function set (p < 0.05 by rate ratio test) and two asterisks for significant enrichment after adjusting for multiple testing (p < 0.05 with FDR correction). GO terms are ranked in descending order of ratio of annotations rate of a GO term in unannotated proteins vs that in annotated proteins.
As case studies demonstrating the new information provided by the C-I-TASSER/COFACTOR pipeline, we selected MMSYN1_0877 and MMSYN1_0440 (Figure 3) to discuss the derivation and implication of their predicted functions “vitamin transporter” activity and “response to other organism”, respectively, which are the most significantly enriched terms for MF and BP, respectively.
Figure 3.
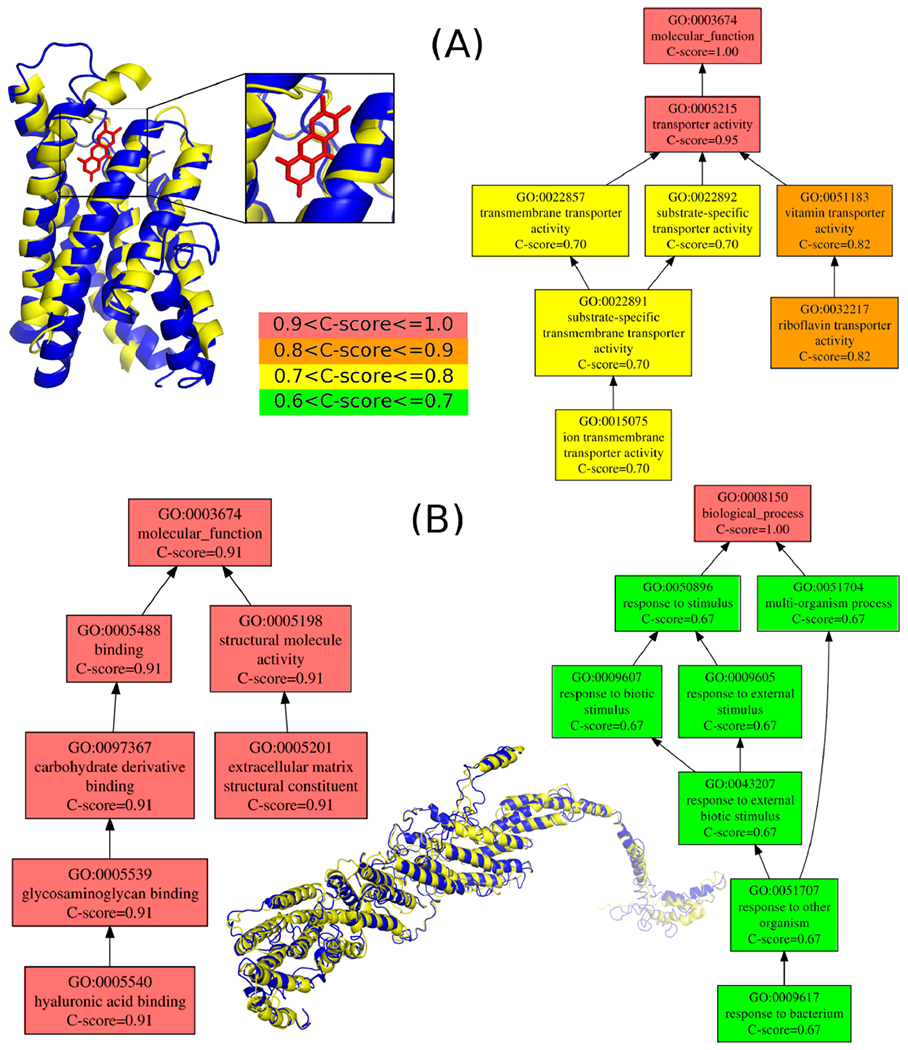
Exemplar proteins corresponding to GO terms that are highly abundant among the newly annotated set. (A) MMSYN1_0877, a protein with predicted “vitamin transporter” activity and (B) MMSYN1_0440, a protein with predicted annotation of the “response to other organisms” GO term. (A) C-I-TASSER structure model (deep blue, estimated TM-score = 0.59) of MMSYN1_0877 (NCBI accession: AMW76711.1) superposed to S. aureus riboflavin transporter RibU (light yellow, PDB ID: 3p5n chain A, TM-score = 0.72) in complex with riboflavin (red stick). Top MF GO term predictions are shown on the right-hand side directed acyclic graph, with different colors representing different ranges of COFACTOR C-scores for the predicted terms (center color map). (B) C-I-TASSER model (deep blue, estimated TM-score = 0.33) of MMSYN1_0440 (NCBI accession: AMW76515.1) superposed to yeast exocyst complex component SEC8 (light yellow, PDB ID: 5yfp chain D with TM-score = 0.84 but sequence identity 0.1). Top predicted MF and BP terms are shown in graphs on the left and right, respectively.
Riboflavin Transporter MMSYN1_0877
MMSYN1_0877 (Figure 3A) is an unannotated essential protein, predicted to have “riboflavin transporter activity” and “vitamin transporter activity” with C-score = 0.82 for MF by the C-I-TASSER/COFACTOR pipeline. The C-I-TASSER structure model exhibits a multipass transmembrane helix bundle with an estimated TM-score of 0.59 (indicating correct topological fold39), with a riboflavin (i.e., vitamin B2) ligand recognized by COFACTOR. The protein is structurally similar to RibU, the riboflavin-binding substrate binding domain of an ECF transporter system from Staphylococcus aureus, with TM-score = 0.72 by TM-align.50 The presence of this putative transporter suggests that syn3.0 relies upon riboflavin uptake from the media for survival. Indeed, we find that M. mycoides have two Riboflavin kinase/FAD synthetase enzymes, ribC (UniProt ID: Q6MUC6) and ribF (UniProt ID: Q6MTQ9), which can make use of riboflavin to synthetize the flavin mononucleotide or the flavin adenine dinucleotide. However, M. mycoides lacks an identifiable pathway for de novo riboflavin biosynthesis and thus presumably relies on uptake from the host or media (presumably via UniProt ID Q6MS70, the homologue of MMSYN1_0877). In the case of syn3.0, the ribC gene is also absent, apparently leaving riboflavin import via MMSYN1_0877 followed by RibF processing as the likely sole path for synthesis of riboflavin-containing compounds. The current lack of annotation of the M. mycoides homologue Q6MS70 is likely because our annotation prediction builds strongly on structural similarity to ECF-type riboflavin uptake proteins from T. maratima61 and S. aureus,62 which have sub-2 Å rmsd’s to the predicted MMSYN1_0877 structure but amino acid sequence identities of less than 22%.
As many ECF systems exist to transport a broad range of target molecules, we performed three additional steps to verify COFACTOR’s assignment of MMSYN1_0877 as a riboflavin transporter. First, to provide an unbiased screen for potential ligands and dock them to MMSYN1_0877, we ran COACHD,63 which identified three potential ligands: riboflavin (RBF), dATP (DTP), and imidazole (IMD). The ligand–protein binding energies for the docked poses, as estimated by X-SCORE,64 were −8.08, −6.73, and −5.17 kcal/mol, respectively, suggesting that riboflavin is the most likely ligand for this protein. In addition, we considered orthogonal evidence by structurally aligning the nine ECF transporters with the highest structural similarity to our predicted MMSYN1_0877 structure, using the STAMP structural alignment program65 via the Multiseq interface66 of VMD 1.9.367 (we note that the structural alignment is essential because the sequence identities of the proteins considered here to MMSYN1_0877 are all 15% or less). We then considered the identity of residues that were within 0.5 nm of the bound riboflavin from a crystal structure of a Staphylococcus riboflavin ECF transporter (PDB ID 3P5N). As shown in Figure S2A, we find that of eight such sites that are shared by the two known riboflavin transporters in our data set (PDB IDs 5KBW and 3P5N) but no other crystal structures in the alignment, the residue in the equivalent position for MMSYN1_0877 is identical for five of them, and one mismatch is glutamate in MMSYN1_0877 but aspartate in the two crystallized riboflavin transporters; MMSYN1_0877 also resembled the riboflavin transporters more closely at several other such substrate-contacting positions, for example, position 71 in the alignment (K in MMSYN1_0877 and 5KBW but T in 3P5N). In addition, MMSYN1_0877 clusters with the two riboflavin transporters in this protein set using both sequences (Figure S2B) and structure (Figure S2C)-based metrics. Finally, to specifically test for specificity among common vitamin targets of ECF transporters, each of four different vitamins (riboflavin, biotin, thiamine, and folate) were docked into the binding pocket of MMSYN1_0877. To this end, a 60 Å × 60 Å × 60 Å cubic searching space was defined around the center of the top 1 binding pocket identified by COACH-D, and AutoDock vina68 was used to generate one docking pose per ligand. The X-SCORE binding affinities calculated using the docking poses are −8.40, −6.77, −7.78, and −8.09 kcal/mol, respectively. Taken together, these data suggest that, while other vitamins could potentially bind to this protein, riboflavin is most likely the main target.
Hyaluronic Acid Binding Protein MMSYN1_0440
Considering that syn3.0 can be cultured in vitro without the need to interact with other organisms, it is initially counterintuitive that we observe several new annotations of the GO term “response to other organism”. However, it must be noted that the ancestral M. mycoides is an obligate parasite of animal hosts, and the culture media used for syn3.0 contains a broad range of animal derivatives (beef heart infusion, peptones, and fetal bovine serum17); it is thus plausible that syn3.0 interacts with animal-derived media components for regulatory or mechanical purposes as well as nutritional purposes. As an example, the essential protein MMSYN1_0440 (Figure 3B) is predicted to be involved in “response to other organism” with C-score = 0.57 for BP. This is substantiated by the predicted MF term “hyaluronic acid binding” with C-score = 0.91, indicating likely interaction with animal-derived hyaluronic acid present in the culture media. The reason for the importance of this particular interaction for the viability of syn3.0 is not immediately clear. One possibility arises from the MMSYN1_0440 structural model, which shows good structural similarity to the yeast membrane tethering protein SEC8; MMSYN1_0440 may play an architectural role in maintaining membrane integrity or cell–cell contacts in syn3.0, likely interacting with hyaluronic acid polymers present in the media. If this inference is correct, one would expect that MMSYN1_0440 would cease to be essential if the cells were grown in chemically defined, rather than biologically derived, media.
Whole-Proteome Dimeric Threading Reveals a Scale-Free PPI Network
Given that many proteins perform their function by interacting with other proteins, we used SPRING, a dimeric threading approach,48 to investigate the organization of pairwise PPIs in the syn3.0 proteome. The interactome predicted by whole-proteome SPRING threading search is relatively sparse, with only 2.6% (2483) of all 95,703 protein pairs being predicted PPI partners (Figure 4A). We initially speculated that because of its simplicity, the syn3.0 network structure might revert to a less ordered state instead of a scale-free layout typical of bacterial networks.69 However, we found that the PPI network is actually scale-free: P(k), the fraction of proteins in the network having k partners, follows a power law distribution
| (4) |
Figure 4.
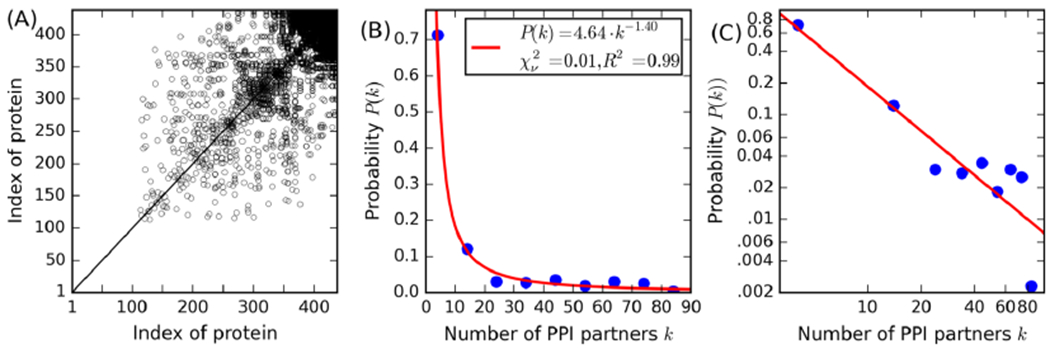
PPI predicted by SPRING. (A) Scatter plot of PPIs for all syn3.0 proteins ranked in ascending number of PPI partners, where a point means the protein pair is predicted to have a PPI. (B,C) Observed distribution (circles) for the number of PPI partners per protein in linear (B) and log (C) scale and the power law fit (lines). Due to the relatively small number of proteins, statistical analysis of the probability distribution of k (the number of PPI partners) is on bins with width of 10. Thus, the first circle from the left in (B) and (C) is for the bin 0 ≤ k < 10, while the second circle for 10 ≤ k < 20, etc. In the inset, χν2 is the reduced χ-squared statistic (lower values are better, with 0 being a perfect fit) and R2 is the coefficient of determination (the higher the better, with 1 being a perfect fit), respectively, to quantify the goodness of fit.
A high goodness-of-fit is achieved with the parameter τ = 1.40, resulting in the reduced χ-squared statistics and the coefficient of determination and approaching 0 and 1, respectively (Figure 4B,C). This is significantly different from a randomly generated PPI network with the same number of positive (2483) and total (95,703) protein pairs (Figure S3), where the number of PPI partners per protein fits poorly to the power law with the reduced χ-squared statistics and the coefficient of determination consistently greater than 1.5 and less than 0, respectively. This suggests that the scale-freeness of the SPRING-predicted PPI network is not coincidental. Scale-free networks were reported previously for naturally evolved biological networks: E. coli, for example, also has a scale-free PPI network69 with τ = 1.3 as estimated by our recent work.70 On the other hand, the present study is the first time that a scale-free PPI network is observed for an artificial proteome, although genes are retained in the syn3.0 genome based solely on their essentiality without explicit consideration for the number of potential PPI. The unintentional retention of a scale-free PPI network in the deeply truncated syn3.0 proteome suggests the universal robustness of the PPI network architecture and the importance of the “hub” proteins (which regulate a large number of proteins with few PPIs) for the overall viability of cells.
DISCUSSION AND CONCLUSIONS
In this study, we extended a unified structure and function prediction pipeline for whole-genome function and PPI modeling of the syn3.0 minimal genome. This pipeline is able to assign function for 9 times more unknown proteins than existing UniProt annotations (Figure 1A) and substantially extends the reach of structure-based function prediction of poorly annotated proteins. These results further demonstrated the usefulness and impact of high-resolution protein structure simulations on large-scale proteome function annotations. In particular, the integration of deep neural network-based contact maps with the structural assembly simulations plays an essential role for not only improving the quality of structure models but also for increasing the coverage and reliability of functional predictions. We expect that the approach employed here will be of substantial utility for providing optimal computational structure/function predictions for other organisms, which are currently progressing in our laboratories.
The annotation efforts detailed here also provide a substantial boost to our ability to understand the biology of the reduced-genome syn3.0 strain, providing confident MF and BP models for 373 and 382 syn3.0 proteins, which represent, respectively, 86 and 88% of the proteome that were previously unannotated. Consistent with the findings of Antczak et al.,21 the spectrum of function annotations for these newly annotated proteins (Figure 2) places a strong emphasis on the importance of nutrient acquisition, demonstrating a broad range of uptake and metabolic pathways that had previously not been appreciated. A substantial number of newly predicted phosphatases (particularly those targeting nucleotides) comprises a substantial additional category of previously unannotated syn3.0 genes and may play roles in nutrient acquisition, removal of toxic metabolic byproducts, or signaling/regulation. The importance of interactions with host tissue and host-derived molecules (including those present in the heavily animal-sourced syn3.0 growth media) is a common thread running throughout the newly identified annotations, ranging from uptake of host-derived nutrients [e.g., the riboflavin transporter shown in (Figure 3A) to architectural proteins binding host-derived glycans (Figure 3B)]. In the ongoing quest to develop a truly minimal genome, it will be intriguing to determine which of the syn3.0 genes represent simple metabolite uptake requirements (e.g., MMSYN1_0877) and which involve detection of host-derived substances that act as growth stimulators (as may be the case for some of the newly annotated proteins bearing the “signaling receptor” and “response to other organism” GO terms); it is likely that the latter class of proteins may be dispensable if the downstream signaling paths can be elucidated, whereas the former likely cannot.
An unexpected discovery of this study is that the artificially reduced minimal syn3.0 genome retains a scale-free PPI network, similar to other naturally occurring PPI networks such as that of E. coli. As the population of proteins with a high number of PPI partners is significantly enhanced in the scale-free networks in comparison with a random network (Figure S3) that follows a Gaussian distribution, the robustness of the scale-free PPI network of the syn3.0 genome likely arises because of the biological importance of network hub proteins, which are unlikely to be removed over the course of genomic pruning and critically contribute to the successful generation of the genome. The scale-free behavior of biological networks should be an important consideration in future synthetic biology experiments.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Eric W. Bell, Zi Liu, Dr. Xiaoqiong Wei, and Qiqige Wuyun for discussion and assistance. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation grant ACI1548562.
Funding
This work was supported in part by National Institutes of Health grants GM083107, GM136422, AI134678, OD026825 (to Y.Z.), P30ES017885 and U24CA210967 (to G.S.O.) and GM128637 (to P.L.F.), and the National Science Foundation grants IIS1901191 (to Y.Z.) and MTM2025426 (to P.L.F. and Y.Z.).
ABBREVIATIONS
- PPIs
protein–protein interactions
- GO
gene ontology
- MF
molecular function
- BP
biological process
- CC
cellular component
- UniProt-GOA
UniProt gene ontology annotation
- MSA
multiple sequence alignment
- PCC
Pearson correlation coefficient
- EC
Enzyme Commission
- LBS
ligand binding site
- MCC
Matthews correlation coefficient
Footnotes
ASSOCIATED CONTENT
Supporting Information
Deep learning-based contact prediction in C-I-TASSER; violin plots for portions of residues predicted by TMHMM2.0 to be within transmembrane helices for JCVI-syn3.0 proteins that are annotated versus unannotated by C-I-TASSER/COFACTOR with C-score > 0.5 for specific GO terms in the MF, BP, and CC aspects; structural alignment of the predicted structure of MMSYN1_0877 with the nine closest structural homologues identified in the Protein Data Bank; substrate binding domains for ECF systems targeting riboflavin; and a random PPI network for syn3.0, where 2483 of all 95,703 protein pairs are randomly selected as the positive PPI pairs (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jproteome.0c00359
The authors declare no competing financial interest.
Contributor Information
Chengxin Zhang, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, United States.
Wei Zheng, Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan 48109, United States.
Micah Cheng, Department of Electrical Engineering and Computer Science, University of Michigan, Ann Arbor, Michigan 48109, United States.
Gilbert S. Omenn, Department of Computational Medicine and Bioinformatics and Departments of Internal Medicine and Human Genetics and School of Public Health, University of Michigan, Ann Arbor, Michigan 48109, United States.
Peter L. Freddolino, Department of Computational Medicine and Bioinformatics and Department of Biological Chemistry, University of Michigan, Ann Arbor, Michigan 48109, United States.
Yang Zhang, Department of Computational Medicine and Bioinformatics and Department of Biological Chemistry, University of Michigan, Ann Arbor, Michigan 48109, United States.
REFERENCES
- (1).Mushegian AR; Koonin EV A Minimal Gene Set for Cellular Life Derived by Comparison of Complete Bacterial Genomes. Proc. Natl. Acad. Sci. U.S.A 1996, 93, 10268–10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Koonin EV Comparative Genomics, Minimal Gene-Sets and the Last Universal Common Ancestor. Nat. Rev. Microbiol 2003, 1, 127–136. [DOI] [PubMed] [Google Scholar]
- (3).Smalley DJ; Whiteley M; Conway T In Search of the Minimal Escherichia Coli Genome. Trends Microbiol. 2003, 11, 6–8. [DOI] [PubMed] [Google Scholar]
- (4).Koshland DE Jr. SPECIAL ESSAY: The Seven Pillars of Life. Science 2002, 295, 2215–2216. [DOI] [PubMed] [Google Scholar]
- (5).Cleland CE; Chyba CF Defining “Life”. Origins Life Evol Biospheres 2002, 32, 387–393. [DOI] [PubMed] [Google Scholar]
- (6).Akerley BJ; Rubin EJ; Camilli A; Lampe DJ; Robertson HM; Mekalanos JJ Systematic Identification of Essential Genes by in Vitro Mariner Mutagenesis. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 8927–8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yu BJ; Sung BH; Koob MD; Lee CH; Lee JH; Lee WS; Kim MS; Kim SC Minimization of the Escherichia Coli Genome Using a Tn5-Targeted Cre/loxP Excision System. Nat. Biotechnol 2002, 20, 1018–1023. [DOI] [PubMed] [Google Scholar]
- (8).Hutchison CA III Global Transposon Mutagenesis and a Minimal Mycoplasma Genome. Science 1999, 2165–2169. [DOI] [PubMed] [Google Scholar]
- (9).Sassetti CM; Boyd DH; Rubin EJ Comprehensive Identification of Conditionally Essential Genes in Mycobacteria. Proc. Natl. Acad. Sci. U.S.A 2001, 98, 12712–12717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Akerley BJ; Rubin EJ; Novick VL; Amaya K; Judson N; Mekalanos JJ A Genome-Scale Analysis for Identification of Genes Required for Growth or Survival of Haemophilus Influenzae. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Judson N; Mekalanos JJ TnAraOut, a Transposon-Based Approach to Identify and Characterize Essential Bacterial Genes. Nat. Biotechnol 2000, 18, 740–745. [DOI] [PubMed] [Google Scholar]
- (12).Giaever G; Chu AM; Ni L; Connelly C; Riles L; Véronneau S; Dow S; Lucau-Danila A; Anderson K; André B; Arkin AP; Astromoff A; El Bakkoury M; Bangham R; Benito R; Brachat S; Campanaro S; Curtiss M; Davis K; Deutschbauer A; Entian K-D; Flaherty P; Foury F; Garfinkel DJ; Gerstein M; Gotte D; Güldener U; Hegemann JH; Hempel S; Herman Z; Jaramillo DF; Kelly DE; Kelly SL; Kötter P; LaBonte D; Lamb DC; Lan N; Liang H; Liao H; Liu L; Luo C; Lussier M; Mao R; Menard P; Ooi SL; Revuelta JL; Roberts CJ; Rose M; Ross-Macdonald P; Scherens B; Schimmack G; Shafer B ; Shoemaker DD; Sookhai-Mahadeo S; Storms RK; Strathern JN; Valle G; Voet M; Volckaert G; Wang C-Y; Ward TR; Wilhelmy J; Winzeler EA; Yang Y; Yen G; Youngman E; Yu K; Bussey H; Boeke JD; Snyder M; Philippsen P; Davis RW; Johnston M. Functional Profiling of the Saccharomyces Cerevisiae Genome. Nature 2002, 418, 387–391. [DOI] [PubMed] [Google Scholar]
- (13).Pósfai G; Plunkett G 3rd; Fehér T; Frisch D; Keil GM; Umenhoffer K; Kolisnychenko V; Stahl B; Sharma SS; de Arruda M; Burland V; Harcum SW; Blattner FR Emergent Properties of Reduced-Genome Escherichia Coli. Science 2006, 312, 1044–1046. [DOI] [PubMed] [Google Scholar]
- (14).Umenhoffer K; Fehér T; Balikó G; Ayaydin F; Pósfai J; Blattner FR; Pósfai G Reduced Evolvability of Escherichia Coli MDS42, an IS-Less Cellular Chassis for Molecular and Synthetic Biology Applications. Microb. Cell Fact. 2010, 9, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Csörgo B; Fehér T; Tímár E; Blattner FR; Pósfai G Low-Mutation-Rate, Reduced-Genome Escherichia Coli: An Improved Host for Faithful Maintenance of Engineered Genetic Constructs. Microb. Cell Fact. 2012, 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gibson DG; Glass JI; Lartigue C; Noskov VN; Chuang R-Y; Algire MA; Benders GA; Montague MG; Ma L; Moodie MM; Merryman C; Vashee S; Krishnakumar R; Assad-Garcia N; Andrews-Pfannkoch C; Denisova EA; Young L; Qi Z-Q; Segall-Shapiro TH; Calvey CH; Parmar PP; Hutchison CA 3rd; Smith HO; Venter JC Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome. Science 2010, 329, 52–56. [DOI] [PubMed] [Google Scholar]
- (17).Hutchison CA 3rd; Chuang R-Y; Noskov VN; Assad-Garcia N; Deerinck TJ; Ellisman MH; Gill J; Kannan K; Karas BJ; Ma L; Pelletier JF; Qi Z-Q; Richter RA; Strychalski EA; Sun L; Suzuki Y; Tsvetanova B; Wise KS; Smith HO; Glass JI; Merryman C; Gibson DG; Venter JC Design and Synthesis of a Minimal Bacterial Genome. Science 2016, 351, aad6253. [DOI] [PubMed] [Google Scholar]
- (18).Breuer M; Earnest TM; Merryman C; Wise KS; Sun L; Lynott MR; Hutchison CA; Smith HO; Lapek JD; Gonzalez DJ; de Crécy-Lagard V; Haas D; Hanson AD; Labhsetwar P; Glass JI; Luthey-Schulten Z Essential Metabolism for a Minimal Cell. Elife 2019, 8, No. e36842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Danchin A; Fang G Unknown Unknowns: Essential Genes in Quest for Function. Microb. Biotechnol 2016, 9, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yang Z; Tsui SK-W Functional Annotation of Proteins Encoded by the Minimal Bacterial Genome Based on Secondary Structure Element Alignment. J. Proteome Res 2018, 17, 2511–2520. [DOI] [PubMed] [Google Scholar]
- (21).Antczak M; Michaelis M; Wass MN Environmental Conditions Shape the Nature of a Minimal Bacterial Genome. Nat. Commun 2019, 10, 3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang C; Freddolino PL; Zhang Y COFACTOR: Improved Protein Function Prediction by Combining Structure, Sequence and Protein-Protein Interaction Information. Nucleic Acids Res. 2017, 45, W291–W299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang C; Zheng W; Freddolino PL; Zhang Y MetaGO: Predicting Gene Ontology of Non-Homologous Proteins Through Low-Resolution Protein Structure Prediction and Protein-Protein Network Mapping. J. Mol. Biol 2018, 430, 2256–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang C; Wei X; Omenn GS; Zhang Y Structure and Protein Interaction-Based Gene Ontology Annotations Reveal Likely Functions of Uncharacterized Proteins on Human Chromosome 17. J. Proteome Res 2018, 17, 4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhang C; Lane L; Omenn GS; Zhang Y Blinded Testing of Function Annotation for uPE1 Proteins by I-TASSER/COFACTOR Pipeline Using the 2018-2019 Additions to neXtProt and the CAFA3 Challenge. J. Proteome Res 2019, 18, 4154–4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhou N; Jiang Y; Bergquist TR; Lee AJ; Kacsoh BZ; Crocker AW; Lewis KA; Georghiou G; Nguyen HN; Hamid MN; Davis L; Dogan T; Atalay V; Rifaioglu AS; Dalklran A; Cetin Atalay R; Zhang C; Hurto RL; Freddolino PL; Zhang Y; Bhat P; Supek F; Fernández JM; Gemovic B; Perovic VR; Davidović RS; Sumonja N; Veljkovic N; Asgari E; Mofrad MRK; Profiti G; Savojardo C; Martelli PL; Casadio R; Boecker F; Schoof H; Kahanda I; Thurlby N; McHardy AC; Renaux A; Saidi R; Gough J; Freitas AA; Antczak M; Fabris F; Wass MN; Hou J; Cheng J; Wang Z; Romero AE; Paccanaro A; Yang H; Goldberg T; Zhao C; Holm L; Törönen P; Medlar AJ; Zosa E; Borukhov I; Novikov I; Wilkins A; Lichtarge O; Chi PH; Tseng W-C; Linial M; Rose PW; Dessimoz C; Vidulin V; Dzeroski S; Sillitoe I; Das S; Lees JG; Jones DT; Wan C; Cozzetto D; Fa R; Torres M; Warwick Vesztrocy A; Rodriguez JM; Tress ML; Frasca M; Notaro M; Grossi G; Petrini A; Re M; Valentini G; Mesiti M; Roche DB; Reeb J; Ritchie DW; Aridhi S; Alborzi SZ; Devignes M-D; Koo DCE; Bonneau R; Gligorijević V; Barot M; Fang H; Toppo S; Lavezzo E; Falda M; Berselli M; Tosatto SCE; Carraro M; Piovesan D; Ur Rehman H; Mao Q; Zhang S; Vucetic S; Black GS; Jo D; Suh E ; Dayton JB; Larsen DJ; Omdahl AR; McGuffin LJ; Brackenridge DA; Babbitt PC; Yunes JM; Fontana P; Zhang F ; Zhu S; You R; Zhang Z; Dai S; Yao S; Tian W; Cao R; Chandler C; Amezola M; Johnson D; Chang J-M; Liao W-H; Liu Y-W; Pascarelli S; Frank Y; Hoehndorf R; Kulmanov M; Boudellioua I; Politano G; Di Carlo S; Benso A; Hakala K; Ginter F; Mehryary F; Kaewphan S; Björne J; Moen H; Tolvanen MEE; Salakoski T; Kihara D; Jain A; Šmuc T; Altenhoff A; Ben-Hur A; Rost B; Brenner SE; Orengo CA; Jeffery CJ; Bosco G; Hogan DA; Martin MJ; O’Donovan C; Mooney SD; Greene CS; Radivojac P; Friedberg I The CAFA Challenge Reports Improved Protein Function Prediction and New Functional Annotations for Hundreds of Genes through Experimental Screens. Genome Biol. 2019, 20, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Li Y; Zhang C; Bell EW; Yu DJ; Zhang Y Ensembling Multiple Raw Coevolutionary Features with Deep Residual Neural Networks for Contact-map Prediction in CASP13. Proteins 2019, 87, 1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Li Y; Hu J; Zhang C; Yu D-J; Zhang Y ResPRE: High-Accuracy Protein Contact Prediction by Coupling Precision Matrix with Deep Residual Neural Networks. Bioinformatics 2019, 35, 4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zheng W; Li Y; Zhang C; Pearce R; Mortuza SM; Zhang Y Deep-Learning Contact-Map Guided Protein Structure Prediction in CASP13. Proteins 2019, 87, 1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zheng W; Zhang C; Bell EW; Zhang Y I-TASSER Gateway: A Protein Structure and Function Prediction Server Powered by XSEDE. Future Generat. Comput. Syst 2019, 99, 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang C; Zheng W; Mortuza SM; Li Y; Zhang Y DeepMSA: Constructing Deep Multiple Sequence Alignment to Improve Contact Prediction and Fold-Recognition for Distant-Homology Proteins. Bioinformatics 2019, 36, 2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mirdita M; von den Driesch L; Galiez C; Martin MJ; Söding J; Steinegger M Uniclust Databases of Clustered and Deeply Annotated Protein Sequences and Alignments. Nucleic Acids Res. 2017, 45, D170–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Suzek BE; Wang Y; Huang H; McGarvey PB; Wu CH; UniProt Consortium. UniRef Clusters: A Comprehensive and Scalable Alternative for Improving Sequence Similarity Searches. Bioinformatics 2015, 31, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Steinegger M; Söding J Clustering Huge Protein Sequence Sets in Linear Time. Nat. Commun 2018, 9, 2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zheng W; Zhang C; Wuyun Q; Pearce R; Li Y; Zhang Y LOMETS2: Improved Meta-Threading Server for Fold-Recognition and Structure-Based Function Annotation for Distant-Homology Proteins. Nucleic Acids Res. 2019, 47, W429–W436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zhang Y; Skolnick J SPICKER: A Clustering Approach to Identify near-Native Protein Folds. J. Comput. Chem 2004, 25, 865–871. [DOI] [PubMed] [Google Scholar]
- (37).Zhang J; Liang Y; Zhang Y Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling. Structure 2011, 19, 1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhang Y; Skolnick J Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins 2004, 57, 702–710. [DOI] [PubMed] [Google Scholar]
- (39).Xu J; Zhang Y How Significant Is a Protein Structure Similarity with TM-Score = 0.5? Bioinformatics 2010, 26, 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Fox NK; Brenner SE; Chandonia J-M SCOPe: Structural Classification of Proteins-extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014, 42, D304–D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhang Y I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinf. 2008, 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Yang J; Roy A; Zhang Y BioLiP: A Semi-Manually Curated Database for Biologically Relevant Ligand–protein Interactions. Nucleic Acids Res. 2012, 41, D1096–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Altschul S; Madden TL; Schäffer AA; Zhang J; Zhang Z; Miller W; Lipman DJ Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Huntley RP; Sawford T; Mutowo-Meullenet P; Shypitsyna A; Bonilla C; Martin MJ; O’Donovan C The GOA Database: Gene Ontology Annotation Updates for 2015. Nucleic Acids Res. 2015, 43, D1057–D1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Szklarczyk D; Gable AL; Lyon D; Junge A; Wyder S; Huerta-Cepas J; Simonovic M; Doncheva NT; Morris JH; Bork P; Jensen LJ; Mering C. v. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hill DP; Davis AP; Richardson JE; Corradi JP; Ringwald M; Eppig JT; Blake JA Program Description. Genomics 2001, 74, 121–128. [DOI] [PubMed] [Google Scholar]
- (47).Wei X; Zhang C; Freddolino PL; Zhang Y Detecting Gene Ontology Misannotations Using Taxon-Specific Rate Ratio Comparisons. Bioinformatics 2020, 36, 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Guerler A; Govindarajoo B; Zhang Y Mapping Monomeric Threading to Protein–Protein Structure Prediction. J. Chem. Inf. Model 2013, 53, 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Söding J Protein Homology Detection by HMM-HMM Comparison. Bioinformatics 2005, 21, 951–960. [DOI] [PubMed] [Google Scholar]
- (50).Zhang Y; Skolnick J TM-Align: A Protein Structure Alignment Algorithm Based on the TM-Score. Nucleic Acids Res. 2005, 33, 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Salwinski L; Miller CS; Smith AJ; Pettit FK; Bowie JU; Eisenberg D The Database of Interacting Proteins: 2004 Update. Nucleic Acids Res. 2004, 32, D449–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Haft DH; Selengut JD; White O The TIGRFAMs Database of Protein Families. Nucleic Acids Res. 2003, 31, 371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Jones P; Binns D; Chang H-Y; Fraser M; Li W; McAnulla C; McWilliam H; Maslen J; Mitchell A; Nuka G; Pesseat S; Quinn AF; Sangrador-Vegas A; Scheremetjew M; Yong S-Y; Lopez R; Hunter S InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Paik Y-K; Lane L; Kawamura T; Chen Y-J; Cho J-Y; LaBaer J; Yoo JS; Domont G; Corrales F; Omenn GS; Archakov A; Encarnación-Guevara S; Lui S; Salekdeh GH; Cho J-Y; Kim C-Y; Overall CM Launching the C-HPP neXt-CP50 Pilot Project for Functional Characterization of Identified Proteins with No Known Function. J. Proteome Res 2018, 17, 4042–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Glaser G; Razin A; Razin S Stable RNA Synthesis and Its Control in Mycoplasma Capricolum. Nucleic Acids Res. 1981, 9, 3641–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Blötz C; Treffon K; Kaever V; Schwede F; Hammer E; Stulke J Identification of the Components Involved in Cyclic DiAMP Signaling in Mycoplasma Pneumoniae. Front. Microbiol 2017, 8, 1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Zhu X; Baranowski E; Dong Y; Li X; Hao Z; Zhao G; Zhang H; Lu D; A. Rasheed M; Chen Y; Hu C; Chen H; Sagne E; Citti C.; Guo A. An Emerging Role for Cyclic Dinucleotide Phosphodiesterase and nanoRNase Activities in Mycoplasma Bovis: Securing Survival in Cell Culture. PLoS Pathog. 2020, 16, No. e1008661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sun J; Jeffryes JG; Henry CS; Bruner SD; Hanson AD Metabolite Damage and Repair in Metabolic Engineering Design. Metab. Eng 2017, 44, 150–159. [DOI] [PubMed] [Google Scholar]
- (59).de Crécy-Lagard V; Haas D; Hanson AD Newly-Discovered Enzymes That Function in Metabolite Damage-Control. Curr. Opin. Chem. Biol 2018, 47, 101–108. [DOI] [PubMed] [Google Scholar]
- (60).Huang L; Khusnutdinova A; Nocek B; Brown G; Xu X; Cui H; Petit P; Flick R; Zallot R; Balmant K; Ziemak MJ; Shanklin J; de Crécy-Lagard V; Fiehn O; Gregory JF 3rd; Joachimiak A; Savchenko A; Yakunin AF; Hanson AD A Family of Metal-Dependent Phosphatases Implicated in Metabolite Damage-Control. Nat. Chem. Biol 2016, 12, 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Karpowich NK; Song J; Wang D-N An Aromatic Cap Seals the Substrate Binding Site in an ECF-Type S Subunit for Riboflavin. J. Mol. Biol 2016, 428, 3118–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhang P; Wang J; Shi Y Structure and Mechanism of the S Component of a Bacterial ECF Transporter. Nature 2010, 468, 717–720. [DOI] [PubMed] [Google Scholar]
- (63).Wu Q; Peng Z; Zhang Y; Yang J COACH-D: improved protein-ligand binding sites prediction with refined ligand-binding poses through molecular docking. Nucleic Acids Res. 2018, 46, W438–W442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wang R; Lai L; Wang S Further Development and Validation of Empirical Scoring Functions for Structure-Based Binding Affinity Prediction. J. Comput.-Aided Mol. Des 2002, 16, 11–26. [DOI] [PubMed] [Google Scholar]
- (65).Russell RB; Barton GJ Multiple Protein Sequence Alignment from Tertiary Structure Comparison: Assignment of Global and Residue Confidence Levels. Proteins 1992, 14, 309–323. [DOI] [PubMed] [Google Scholar]
- (66).Roberts E; Eargle J; Wright D; Luthey-Schulten Z MultiSeq: Unifying Sequence and Structure Data for Evolutionary Analysis. BMC Bioinf. 2006, 7, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Humphrey W; Dalke A; Schulten K VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (68).Trott O; Olson AJ AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Rajagopala SV; Sikorski P; Kumar A; Mosca R; Vlasblom J; Arnold R; Franca-Koh J; Pakala SB; Phanse S; Ceol A; Hauser R; Siszler G; Wuchty S; Emili A; Babu M; Aloy P; Pieper R; Uetz P The Binary Protein-Protein Interaction Landscape of Escherichia Coli. Nat. Biotechnol 2014, 32, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Gong W Guerler A Zhang C Warner E Li C Zhang Y Integrating Multimeric Threading with High-Throughput Experiments for Structural Interactome of Escherichia Coli, bioRxiv, 20202020.10.17.343962 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Protein sequences of syn3.0 were collected from NCBI accession CP014940.1. While the genome consists of 473 genes, this study only considered the 438 protein coding genes, as the other 35 genes encode noncoding RNAs with well-known functions such as tRNAs and rRNAs. The syn3.0 proteins are mapped to the closest UniProt 2019_09 entries from M. mycoides reference proteomes UP000001016 and UP000011126. The GO annotations of these UniProt entries are collected from UniProt-GOA release 2019-09-17. All predicted structure models, functions, and interactions are available at our public Webserver at https://zhanglab.ccmb.med.umich.edu/JCVI-syn3.0/, including a one sentence description of protein function generated using the most specific high confidence predicted GO term.
When we were in the midst of this study, a new version of the minimal genome, JCVI-syn3A (NCBI accession CP016816.2), was published,18 which includes 16 additional protein coding genes not included in the JCVI-syn3.0 genome. Although these new genes are not essential for the survival of the cell, they make the cell less fragile and cause it to have a more stable cellular morphology. For completeness, we have included these 16 new genes in our structure and function prediction as part of our online webserver even though our main analysis focuses on the original JCVI-syn3.0 genome, which still represents the most “minimal” genome achieved in the series. To facilitate comparative study between JCVI-syn3.0 and JCVI-syn3A, the webserver displays the protein names and accessions for both genomes.