

Abstract
Donor/donor carbenes are relatively new in the field of carbene chemistry; although applications in C–H and X–H insertion reactions are few in number, they demonstrate exquisite chemo- and stereo-selectivity. Recent reports have shown that C–H, N–H, B–H, O–H, Si–H, Ge–H, Sn–H and P–H insertion reactions are feasible with a variety of transition metal catalysts, both inter- and intra-molecularly. Furthermore, high degrees of diastereo- and enantioselectivity have been observed in several cases. Methods typically involve the formation of a diazo-based carbene precursor, but procedures using diazo-free metal carbenes have been developed with significant success. This Review covers transition metal catalyzed insertion reactions with donor/donor and donor carbenes, providing context for future developments in this emerging field.
Keywords: C–H Insertion, Donor/Donor, Carbenes, Catalysis
1. Introduction
Metal carbenes are reactive intermediates that are capable of undergoing a wide variety of reactions. Selective insertion into both C–H and X–H bonds by these intermediates forms the basis for many useful transformations. Foundational work in these fields by Taber, Doyle, Davies, and Hashimoto, to name only a few, have brought the chemistry of metal carbenes into common and convenient use.[1–5] Variations in the electronic character of metal carbenes have been found to drastically change the chemo-, regio-, and stereoselective outcome of a given reaction and have been well studied with carbenes appended with electron-withdrawing substituents (Figure 1). This review focuses on metal-catalyzed insertion reactions with carbenes lacking electron-withdrawing substituents. Most donor/donor carbenes have two aromatic rings; the corresponding metal carbenes exhibit unique reactivity and, importantly, much wider functional group tolerance when compared to the more reactive acceptor-substituted carbenes. In some cases, the dihedral angle between one of the aryl rings and the carbene center reduces the donor character, exhibiting slight electron withdrawing properties, wherein the term ‘diaryl’ carbene is more accurate.[6–8] Although the first example of a ‘donor/donor’ carbene was in 1913, their use has been infrequent and underdeveloped;[9] a recent review of donor-substituted carbenes by Zhu and co-workers highlights the appeal of this upcoming field and discusses the subject as a whole.[10]
Figure 1.
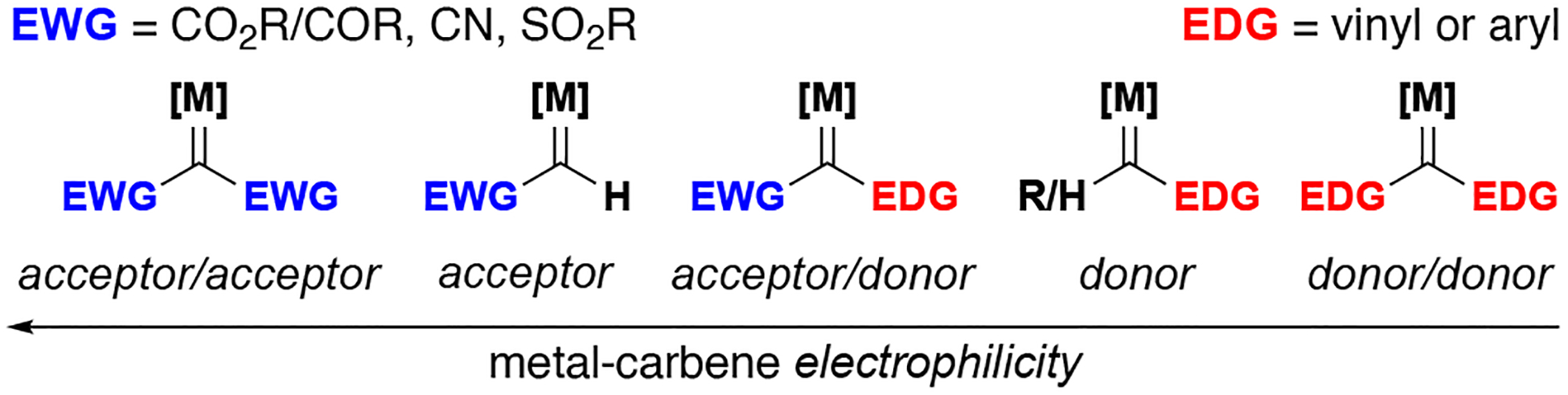
Reactivity of metal carbenes
This review summarizes the catalytic mechanisms and comparitive reactivity of these carbenes toward C–H and X–H insertion reactions.
The majority of donor/donor and donor carbene insertion reactions are catalyzed by rhodium, ruthenium, or copper catalysts; other transition metals have also been found to be proficient, though fewer in number (Figure 2). Similarly, C–H, N–H and Si–H insertion reactions have seen prominent development, while carbene insertions into other X–H bonds remain a nascent field. Many transition metal catalysts have been demonstrated to be effective in C–H insertion reactions, dirhodium paddlewheel complexes have been established as a privileged class of catalysts (Figure 5, vide infra).[11–16]
Figure 2.
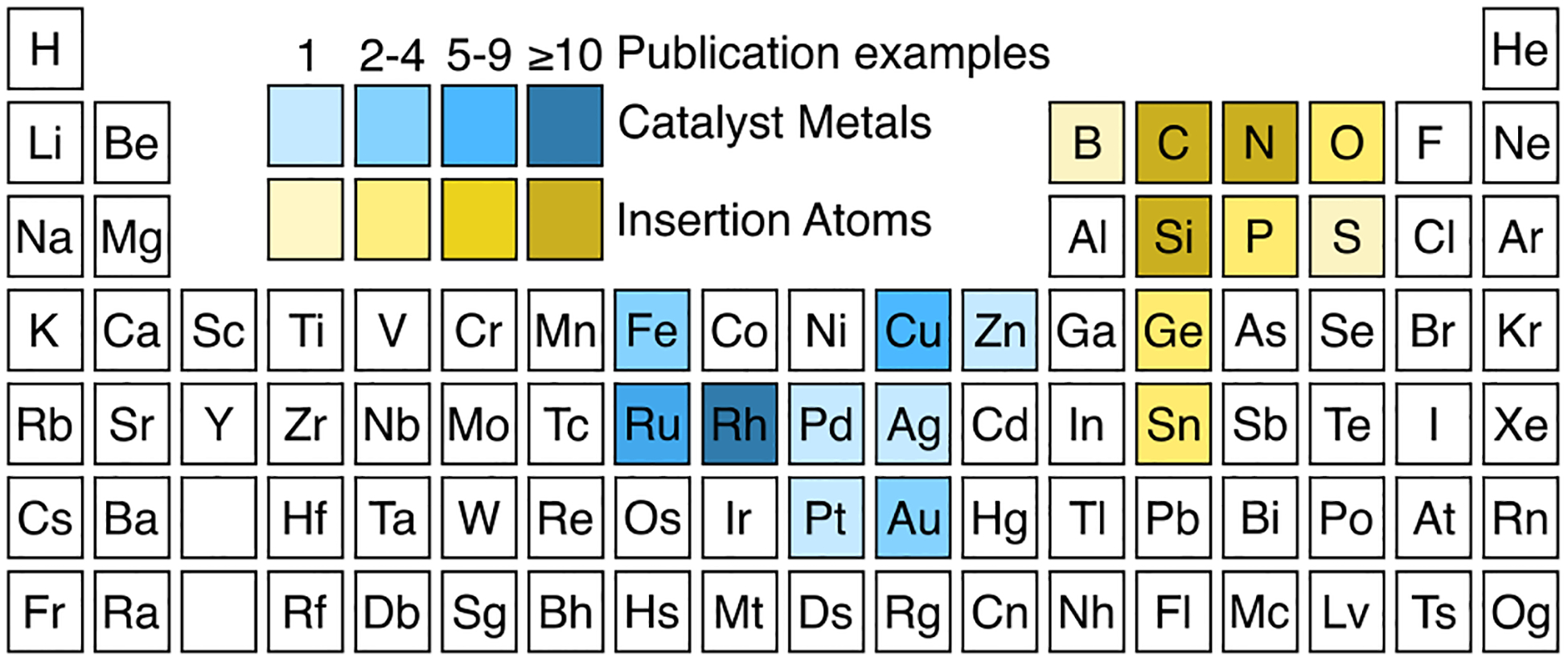
Publication heat map of metals and insertion atoms used in insertion reactions with donor/donor carbenes
Figure 5.
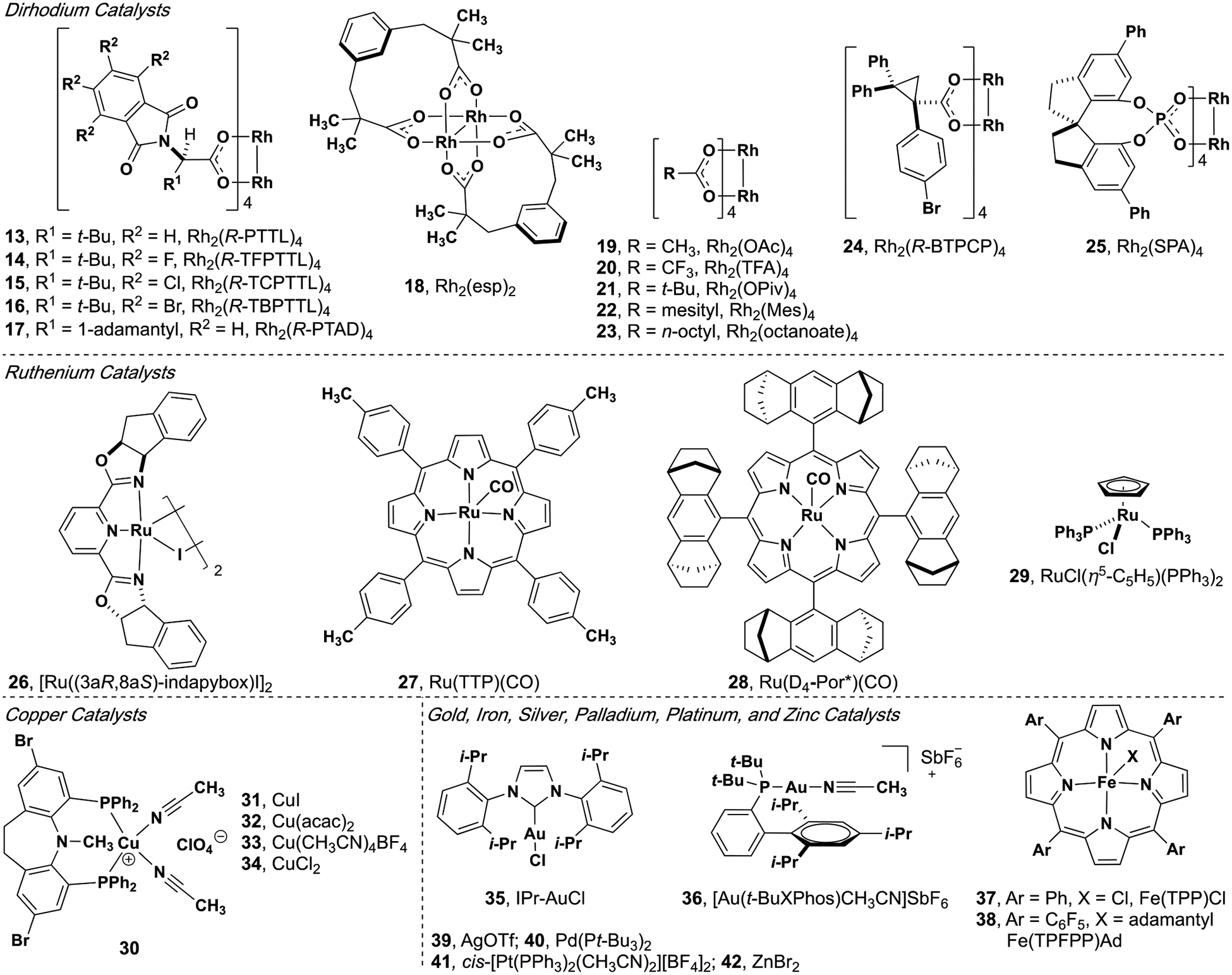
Catalysts used in donor/donor carbene insertion reactions
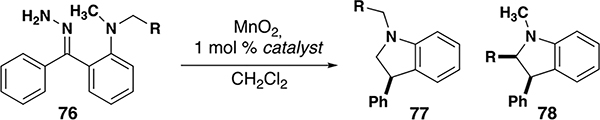
Catalytic reactions via carbene intermediates often employ diazo compounds as precursors to metal carbenes. These intermediates can be isolated in some cases or generated in situ; when exposed to a catalyst, nitrogen gas is extruded and the metal carbene is formed. In most cases of acceptor-substituted carbenes, diazo transfer to a stabilized anion affords the carbene precursor. For donor-substituted carbenes, diazo compounds are typically generated from hydrazone precursors (Figure 3). Tosylhydrazones require base to form diazo compounds, often with heating. This method avoids the use of neat hydrazine, which is toxic and unstable, and can be performed as a one-pot procedure from the requisite ketones. As an alternative to tosylhydrazones, unsubstituted hydrazones can be chemoselectively oxidized with manganese dioxide to form diazo compounds. Although this method requires special handling of hydrazine, this method benefits from the use of a heterogenous and chemoselective oxidant which operates without base or elevated temperature. In some cases, the oxidation can be carried out in the presence of a metal catalyst (e.g. rhodium), enabling one-pot conversion of hydrazones to carbene insertion products.
Figure 3.
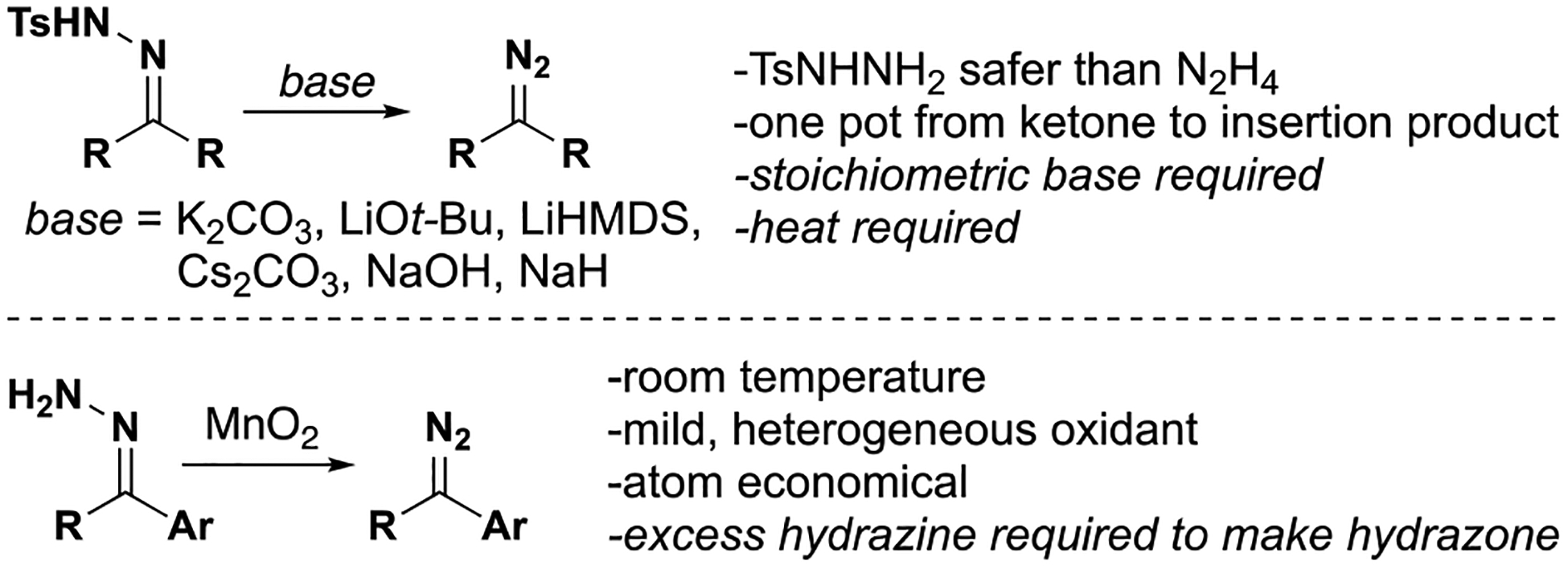
Common methods for diazo formation
Due to the reactive nature of metal carbenes and their requisite precursors, a variety of byproducts may be observed (Figure 4). Tosylhydrazone 2 is known to degrade to sulfone 3 at the elevated temperatures required to form the diazo compound 4. Once formed, diazo intermediates themselves are potentially explosive and are often used on small scale and/or at low concentrations (e.g. dilute solutions and/or inverse addition). Donor/donor diazo compounds readily form metal carbenes; the process is nearly instantaneous at ambient and lower (e.g. −20 °C) temperatures. For hydrazone precursor 1, when oxidation is slower than carbene formation, metal carbene 8 can react with hydrazone 1 to form imine 7. This process can be avoided by filtering off the oxidant after diazo formation is complete, then adding the catalyst. If the insertion reaction is slow, then the diazo 4 can react with the metal carbene to form azine 6. Although most intramolecular insertions are fast enough to avoid this process, azine formation can be suppressed using inverse addition of the diazo compound to a solution of catalyst. Another byproduct of slow insertion reactions is O–H insertion if the reaction is not kept sufficiently anhydrous. If MnO2 is present, benzylic alcohol 11 may be oxidized to ketone 12. When the carbene is substituted with an alkyl group, β-hydride elimination can occur, forming alkene 5 or other degradation products; this byproduct can occasionally be suppressed based on the choice of catalyst/ligand and the character of the β-hydride.[17] Under certain reaction conditions for N–H insertion, imine 10 is observed as the byproduct of β-hydride elimination.[18]. Finally, donor/donor carbenes can occasionally dimerize to form an alkene 9.
Figure 4.
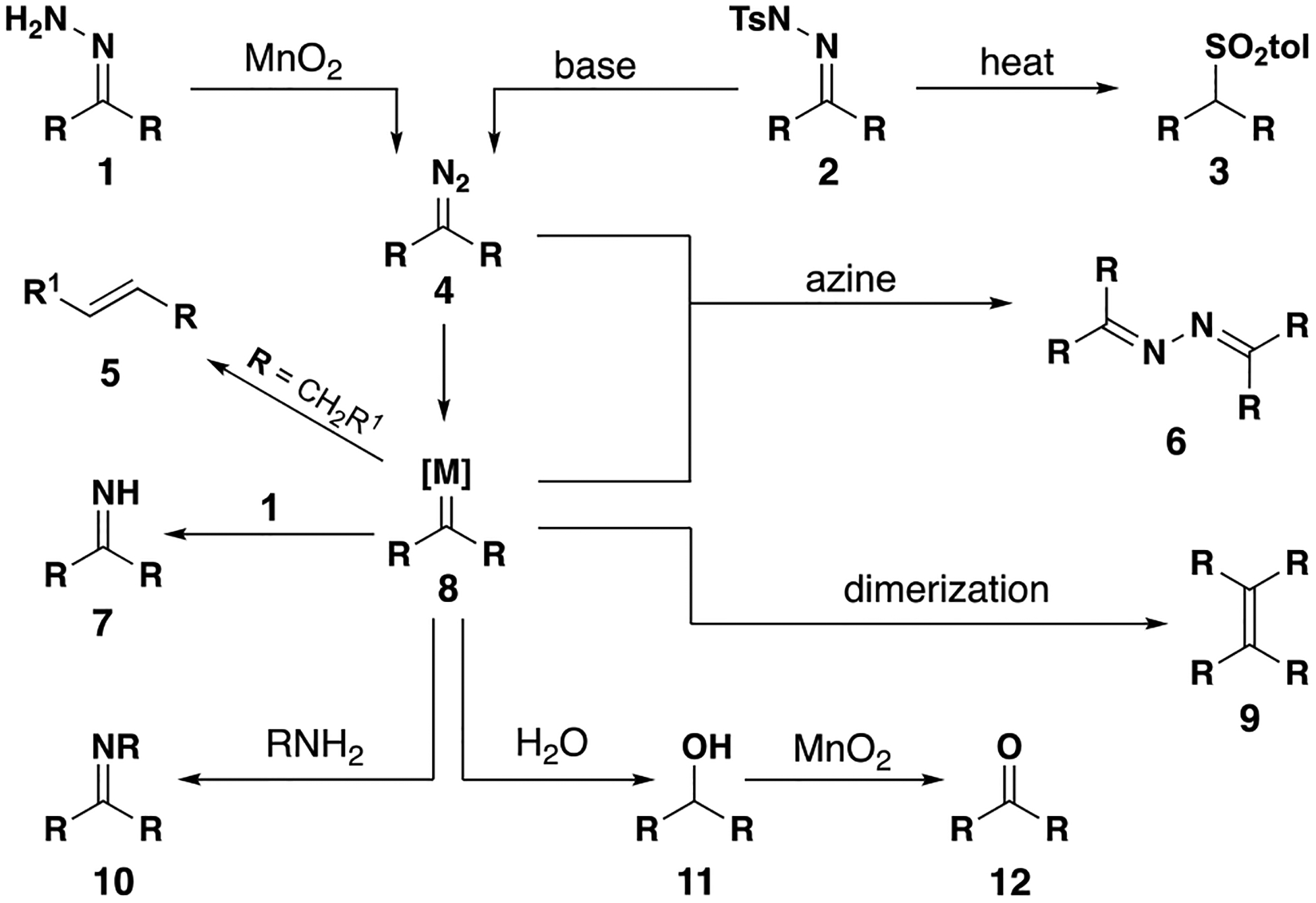
Potential byproducts with carbene precursors and intermediates
2. Mechanism
While relatively little is known about the exact mechanism(s) of insertion reactions with donor/donor carbenes, they can potentially undergo C–H and X–H insertion by several proposed pathways. Although both singlet and triplet carbenes are considered for reactions involving free carbenes, transition metals are known to stabilize the singlet carbene to be the energetic ground state.[19–22] As such, transition metal catalyzed carbene reactions for C–H and X–H are typically considered in the singlet state, supported by experimental selectivity and computational data.[23–25] A notable exception to this notion is in the field of metalloradical catalysis, where triplet-type metal carbenes can undergo a variety of single electron processes. While significant work has been completed in reactions with donor-substituted triplet-type metal carbenes, such reports fall outside of the scope of this review as we are primarily concerned with transition metal- catalyzed insertion reactions that involve singlet carbene, two electron processes.[26–30] Some efforts have also been made to characterize the steric and electronic factors that influence donor/donor carbene stereoselectivity and reactivity, but these examples have primarily been within the context of intermolecular cyclopropanation reactions.[6–8,31]
Metal-catalyzed insertions into atoms other than carbon are generally believed to proceed by a stepwise mechanism.[32] Interestingly, C–H insertion reactions have been postulated to proceed by both concerted and stepwise reactions. A concerted mechanism is proposed in analogy to significant previous computational work supporting this pathway for acceptor-substituted carbenes.[21,33,34] On the other hand, experimental data for reactions with acceptor-substituted carbenes have also suggested a potential step-wise mechanism.[35] DFT computations support a stepwise mechanism for a singular donor/donor carbene system.[36] The potential mechanisms of insertion reactions with donor/donor carbenes can be summarized by four catalytic cycles and a non-catalytic pathway unique to Sn–H substrates.
Mechanistic insight into C–H insertion reactions with donor-substituted metal carbenes is currently limited to intramolecular reactions. For this process, two mechanistic pathways have been proposed (Figure 6). The concerted pathway involves a three-centered transition state 47, while the stepwise pathway produces a zwitterionic intermediate 48. In support of the mechanism first proposed by Doyle,[21] Nakamura and co-workers performed DFT calculations for acceptor-substituted rhodium carbenes, demonstrating a three-center concerted transition state.[34] This mechanism is corroborated implicitly or explicitly in several investigations into various donor-substituted carbene insertions.[4,37–40] In our previous work, the calculated reaction coordinate for a benzodihydrofuran substrate (e.g. Table 1) showed two discrete local maxima: one for hydride transfer and one for C–C bond formation, indicating the brief existence of an ylide, i.e. a stepwise mechanism (Figure 6, 48).[36] Given the breadth of intramolecular insertion substrates to date, it is likely that changes to the substrate (e.g. substituents at the inserting carbon, the steric and electronic character of the carbene, and/or the size of the ring that is formed) can shift the pathway from stepwise to concerted. As donor/donor carbenes continue to be studied, additional experimental and computational mechanistic data will shed light on the factors that favor one pathway or the other.
Figure 6.
C–H insertion reaction mechanism
Table 1.
Synthesis of benzene-fused five-membered rings by C–H insertion
| entry | R1 | R2 | R3 | X | catalyst | # examples | yield (%) | dr (cis.trans) | er | ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ts | CH3 | PMP | O | [Ru], 27 | 1 | 77 | >98:2 | NA | 50 |
| 2 | Ts | CH3, Ph | Ar,[a] alkyl, CO2CH3 | O, NCH2R3, [b] | [Ru], 27 | 8 | 62–89 | 34:66 to 99:1 | 98:2[c] | 51 |
| 3 | H | Ar,[a] alkyl | Ar,[a] alkyl, H, allylic[d] | O, NTs, CH2[e] | [Rh], 17 | 19 | 66–99 | 88:12 to 99:1 | 68:32 to 99:1 | 36,52 |
| 4 | H | Ar[a] | PMP | O | [Rh], 14 | 1 | 87 | >95:5 | 98:2 | 53 |
| 5 | H | Ar[a] | Ar,[a] alkyl | NR5, [f] CH2, S | [Rh], 17 | 22 | 65–99 | 80:20 to >95:5 | 75:25 to 99.5:0.5 | 54 |
Ar = Ph, substituted Ph.
one example of R3 = Ph.
one example with catalyst 28.
includes one example of R3 = propargylic (76%, 95:5 dr, 68:32 er).
one example of X = NTs (98%, 95:5 dr, 87:13 er) and one example of X = CH2 (74%, >97:3 dr, 88:12 er).
R5 = H, Ts, alkyl.
N–H and P–H insertions with donor/donor copper carbenes are reported to proceed by similar mechanisms (Figure 7).[41–43] Tosylhydrazones react with base to generate diazo intermediate 54, which subsequently form metal carbenes in the presence of a copper (I) catalyst. The copper (I) catalyst is produced in situ from reduction by the diazo precursor.[44,45] This metal carbene 57 then undergoes nucleophilic attack by a heteroatom (N or P) to produce zwitterionic intermediate 56. Final proton transfer from the heteroatom to carbon turns over the copper catalyst and yields the X–H insertion product.
Figure 7.
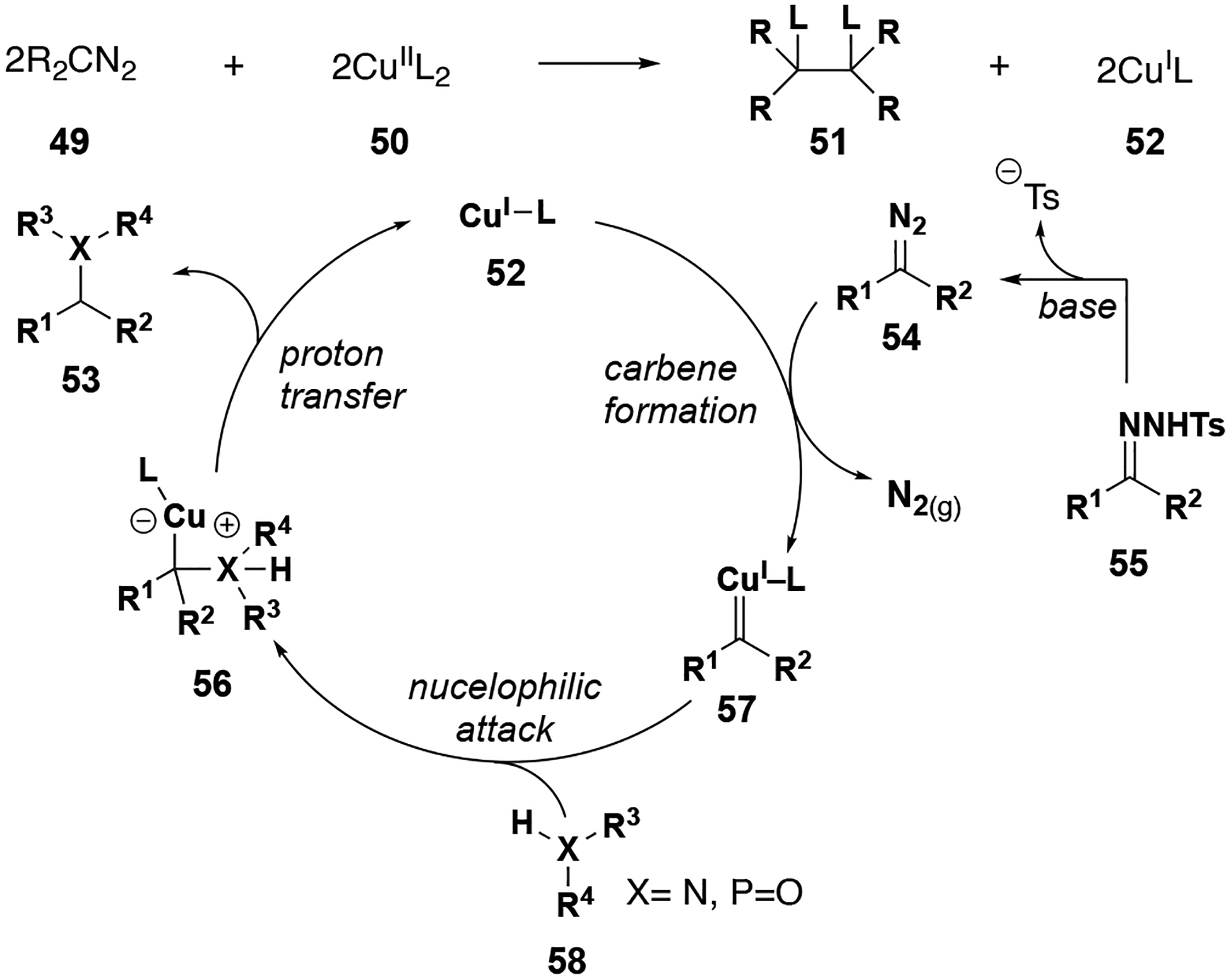
N–H and P–H insertion reaction mechanism
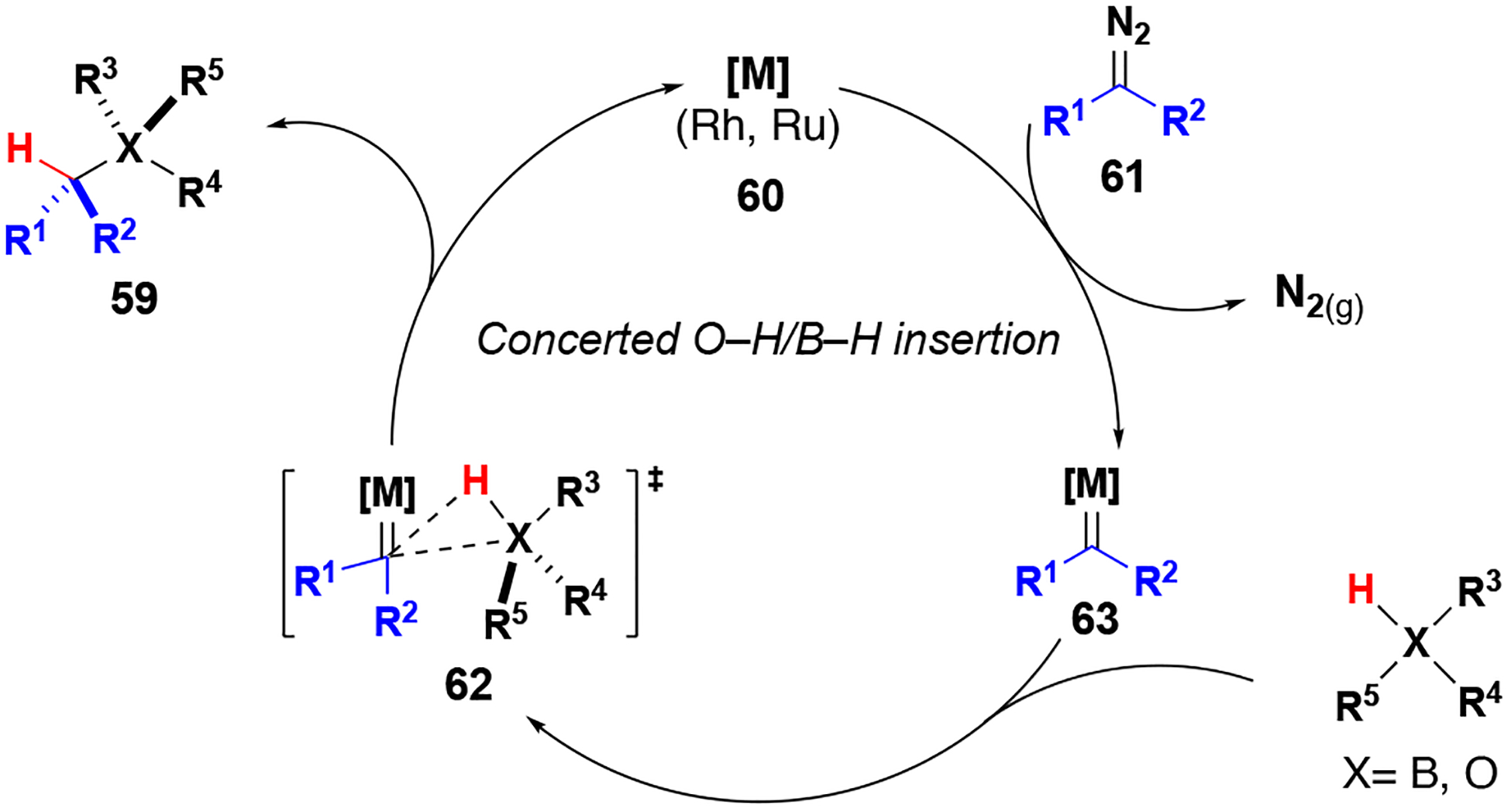
B–H and O–H insertion reactions were suggested to proceed through concerted mechanisms similar to those reported for C–H insertion (Figure 8).[46] Rhodium or ruthenium catalysts produce metal carbenes from diazo compound 61, which then undergo concerted hydride transfer and C–B or C–O bond formation (transition state 62). Due to the strong hydridic character of borane hydrogens when coordinated to Lewis bases, B–H insertion proceeds intramolecularly by a mechanism that resembles proposed transition states for C–H insertion (i.e. explicit concerted asynchronous hydride transfer). It has also been shown that B2Pin2 and BHpin can also decompose aryl/aryl and aryl/alkyl diazo compounds to afford similar products non-catalytically.[47]Although the mechanism for O–H insertion is also proposed to be concerted, the reversed polarity of the insertion partner necessitates nucleophilic attack by oxygen with concomitant proton transfer.
Figure 8.
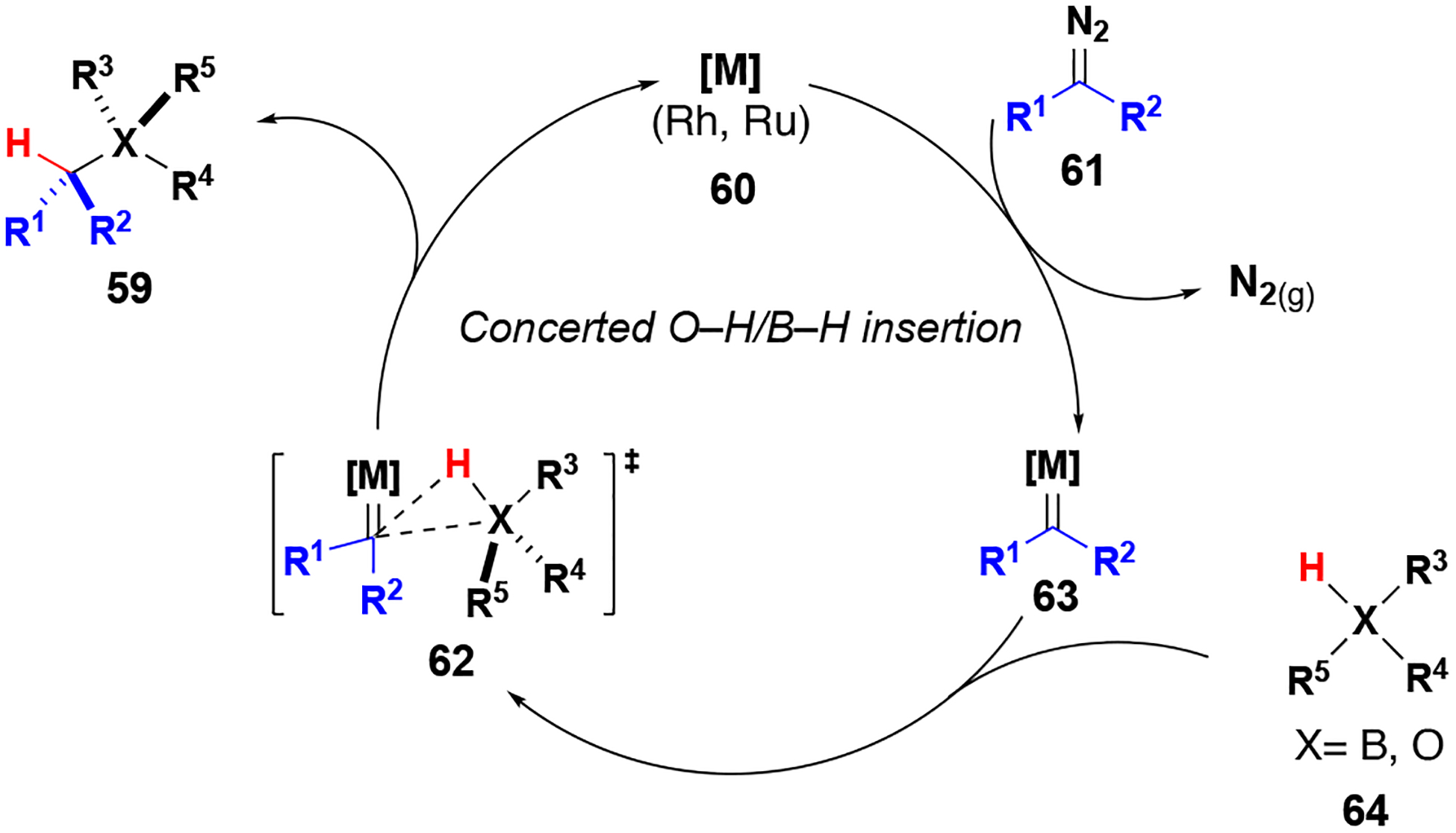
O–H and B–H insertion reaction mechanism
Palladium-catalyzed Si–H insertion reactions with donor/donor carbenes differ from those with rhodium, copper and ruthenium catalysts (Figure 9).[40] Initially, a palladium (0) species undergoes oxidative addition with the silane Si–H bond. This intermediate then decomposes diazo compound 72 to form metal carbene 70, extruding nitrogen gas. This intermediate could then undergo migratory insertion with either Si or H to form either 68 or 69, respectively. Finally, reductive elimination furnishes the Si–H insertion product 65 and regenerates the active catalyst.
Figure 9.
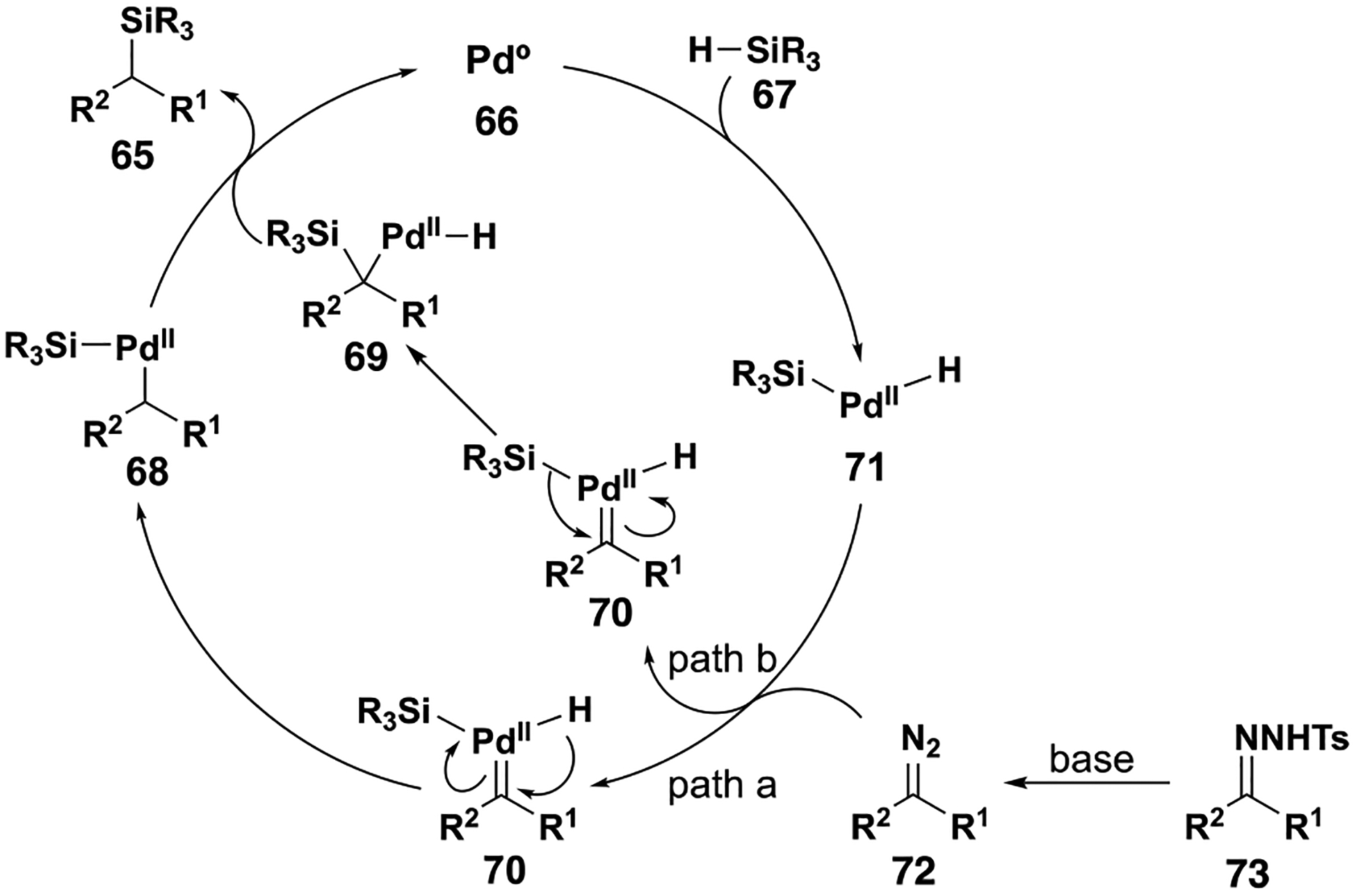
Palladium catalyzed Si–H insertion reaction mechanism
3. C–H Insertion
The functionalization of un-activated C–H bonds, namely Csp3–H bonds, has been an attractive target in synthetic organic chemistry for decades.[48,49] The ubiquity of the moiety in all organic molecules and the potential for dramatic increases in the chemical and stereochemical diversity of simple starting materials have placed C–H activation/insertion methodology at the forefront of transition metal catalysis. While efforts towards intermolecular C–H insertion with acceptor-substituted metal carbenes have seen success, examples with donor/donor carbenes are still few in number. Intramolecular C–H insertion reactions with donor-substituted carbenes, on the other hand, have been proven to proceed with high degrees of selectivity for a broad span of substrate classes.
The most common C–H insertion products are benzene-fused 5-membered ring products, frequently generated with excellent stereoselectivity (Table 1). Che and co-workers first demonstrated this bond-construction using ruthenium porphyrin catalysts 27 and 28.[50,51] Using carbenes generated from tosylhydrazones they achieved the synthesis of benzodihydrofurans and one example of an indoline (Table 1, entries 1–2). Notably, no β-hydride elimination products were reported for acetophenone-derived carbenes, however as a result of the elevated temperatures required, reaction yields and diastereoselectivities were variable. Our lab subsequently showed that carbene formation occurred rapidly at ambient or reduced temperatures in the presence of dirhodium tetracarboxylates (occasionally referred to as paddlewheel complexes).[52] Although initial reactions with achiral rhodium complexes showed nominal reactivity and diastereoselectivity, the use of catalyst 17 improved both the yield and diastereoselectivity while also enabling high degrees of enantioselectivity (Table 1, entry 3). This method was used as the key bond construction for our synthesis of E-δ-viniferin, representing the first enantioselective synthesis of an oligoresveratrol natural product.[52,53] Subsequent studies elucidated the origins of byproducts and computed transition state structures that explain the catalyst control of stereochemistry.[36] Solvent screens for these systems also demonstrated the reduced electrophilicity of these donor/donor carbene systems, showing tolerance of Lewis basic solvents, isopropanol, and even wet acetonitrile.
Intramolecular insertion reactions with donor/donor carbenes have also lead to the stereoselective formation of indanes, benzodihydrothiophenes, and indolines (Table 1, entry 5).[54] Substrates for indane formation lack the activating heteroatom adjacent to the insertion site, reducing reactivity to the point where insertion at a tertiary carbon provided the widest substrate scope. Replacement of oxygen with sulfur resulted in comparable reactivity and, in some cases, higher stereoselectivity. The expansion of the indoline substrate scope from previous studies demonstrated excellent stereocontrol with N–alkyl and N–H substrates. Regioselectivity experiments conducted with indolines showed that steric and electronic effects can contribute to the regioselectivity of insertion reactions (Table 2). With an activated center for insertion (R = Ph) and a catalyst lacking sterically demanding ligands (19), insertion at the more substituted/activated carbon dominates. However, with a more sterically demanding catalyst 22 and a less activated/bulkier substituent (R = i-Pr), insertion at the less activated primary carbon is favored.
Table 2.
Indoline regioselectivity
| entry | R | catalyst | 77:78 |
|---|---|---|---|
| 1 | Ph | [Rh], 19 | 7:93 |
| 2 | i-Pr | [Rh], 22 | 95:5 |
Driver and co-workers reported several examples of polycyclic indanes, indolines, and benzodihydrofurans using Du Bois’s Rh2(esp)2 catalyst 18 from acetophenone-derived tosylhydrazones (Figure 10).[55] This work highlights catalyst control of carbene reactivity: while copper (I)-based aryl/alkyl carbenes were previously found to undergo cyclization/alkyl migrations,[56] rhodium carbenes in this work cleanly furnish allylic insertion products 80 while avoiding cyclopropanation reactions. Interestingly, when the available bond for Csp3–H insertion is at a bridgehead, the preferred pathway is the vinyl Csp2–H insertion reaction to afford 84. It is also notable that in all cases the carbene is appended with a methyl group, which is capable of undergoing a Bamford-Stevens type elimination that is not observed.[57]
Figure 10.
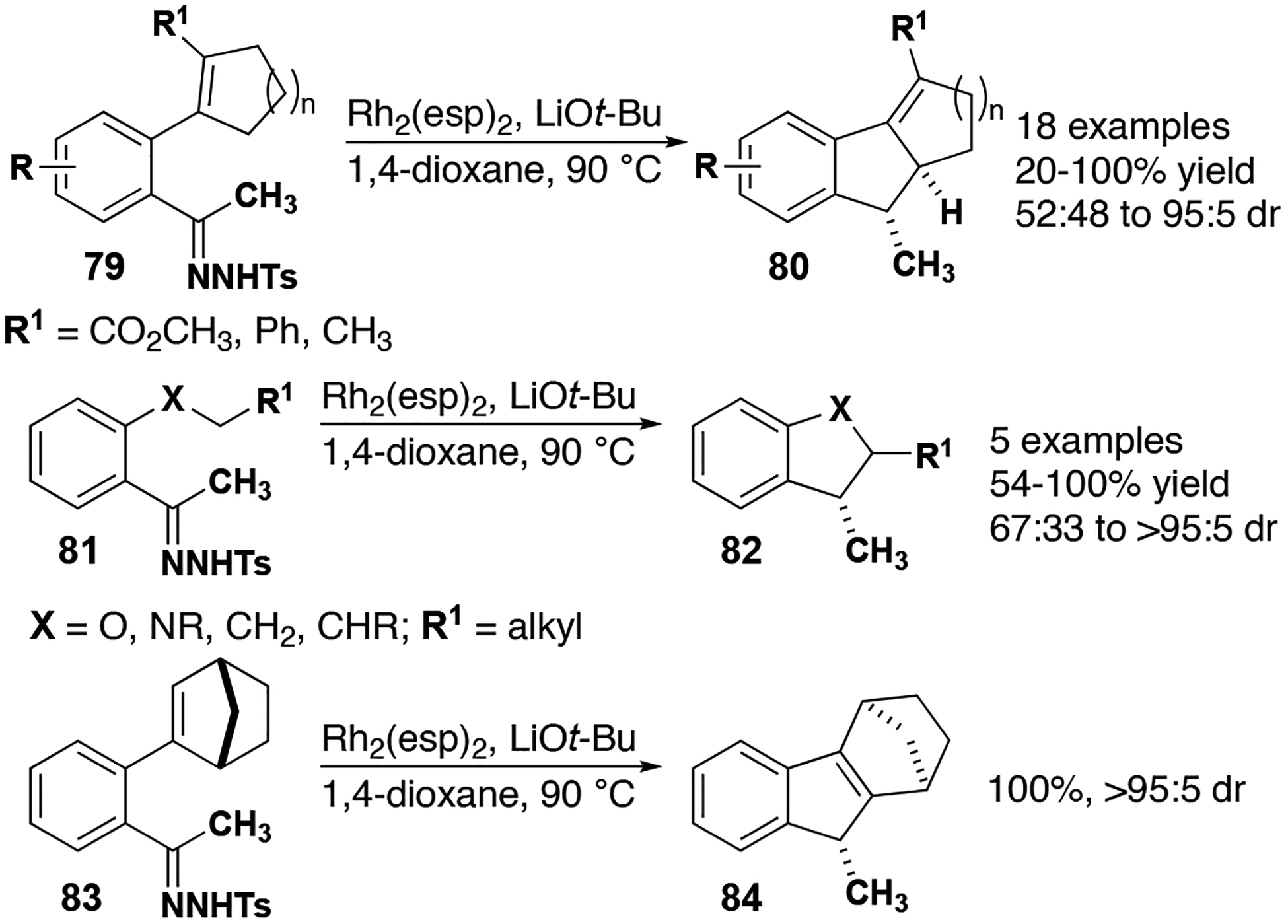
C–H insertion reactions with rhodium carbenes to form indanes
The synthesis of saturated monocyclic 5-membered heterocycles is also enabled using ruthenium porphyrin catalyst 27 (Figure 11). Che and co-workers demonstrated that substituted tetrahydrofurans and pyrrolidines were formed in good yield and, in many cases, high diastereoselectivity.[58] Pyrrolidines with a variety of nitrogen substituents (CH3, Ph, CBz) were also tolerated, demonstrating the robustness of the catalyst and carbene intermediates to Lewis bases. This report further displayed the attenuated reactivity of the system (i.e. catalyst and reaction conditions), given that β-hydride elimination products were not observed despite having two potential elimination sites on the dialkyl carbenes.
Figure 11.
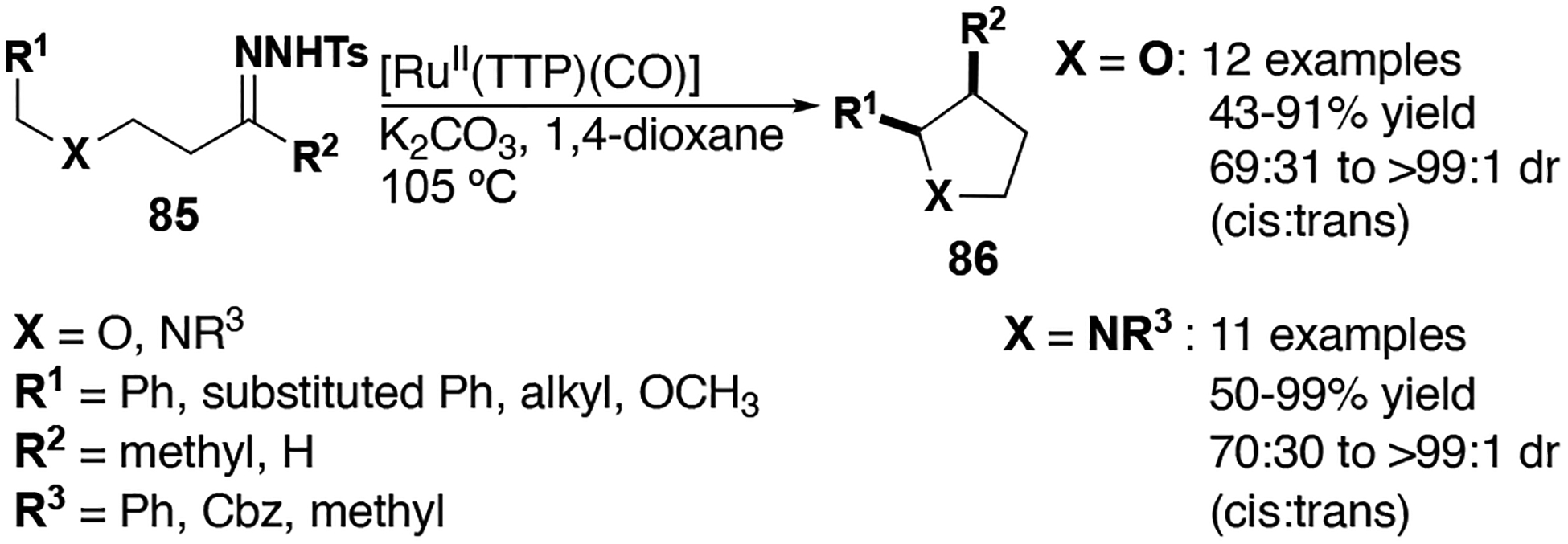
Ruthenium catalyzed C–H insertion reactions with alkyl carbenes
Although the formation of five-membered rings by intramolecular C–H insertion is most common due to kinetic favorability, the formation of six-membered rings is also possible using rhodium catalysts. Hashimoto reported a study of acceptor-substituted carbenes undergoing 1,6-insertion, where a Stevens-type rearrangement was a competing reaction.[59] 1,5-Nucleophilic attack on the metal carbene by a heteroatom (i.e. oxygen in this case) and subsequent [2,3]-sigmatropic shift, reduced the yield of the insertion product in many cases. In contrast, the reduced electrophilicity of donor/donor carbenes enables the formation of isochromans in high yield, as a single diastereomer with excellent enantioselectivity, and without any Stevens-type rearrangement products (Table 3, entry 1).[60] This methodology proved amenable to a broad class of substrates, even tolerating Lewis basic moieties. 1,3-Diaxial interactions were also investigated, demonstrating that the reaction will produce a single diastereomer (with three stereocenters in the cis configuration) from racemic starting materials where R3 is a phenyl or methyl substituent (Table 3, entry 2). Moreover, the first examples of donor/donor C–H insertion to form nitrogen-based six-membered heterocycles were shown to proceed with similarly high levels of stereoselectivity (Table 3, entry 3). Under harsher reaction conditions (i.e. electron deficient catalyst 20, elevated temperatures), a Stevens rearrangement was observed, and the computed transition state demonstrated a polar mechanism involving the dissociation of the rhodium catalyst prior to rearrangement.
Table 3.
Synthesis of isochromans and tethrahydroisoquinolines by enantioselective C–H insertion
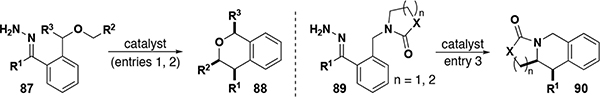 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | X | catalyst | # examples | yield (%) | dr (cis.trans) | er | ref. |
| 1 | Ar,[a] CH3 | Ar,[b] alkyl, alkenyl, propargyl | H | NA | [Rh], 17 | 18 | 54–98 | >95:5 | 63:37 to >99.5:0.5 | 60 |
| 2 | Ph | PMP, alkenyl | Ph, CH3 | NA | [Rh], 22, 15 | 3 | 60–70 | >95:<1 :<1 :<1 | NA | 60 |
| 3 | Ph | NA | H | O, CH2 | [Rh], 17 | 3 | 54–83 | >95:5 | 94:6 to 99:1 | 60 |
Ar = Ph, substituted Ph, 3-pyridyl.
Ar = Ph, substituted Ph
Intermolecular Csp2–H insertion reactions with donor/donor carbenes are possible when oxazoles, thiazoles, benzoxazoles and benzothiazoles are used as the insertion site (Figure 12, A). Functionalized benzoxazoles are widely represented in natural products and drug targets. Wang and co-workers developed a method for the functionalization of these compounds utilizing donor/donor carbenes and copper iodide as a catalyst.[39] The authors propose that an aryl copper intermediate, an adduct between the catalyst and deprotonated azole-derivative, reacts with a diazo species to form an arylcopper (I) carbene that proceeds on to the insertion product. Importantly, substrates containing beta hydrides were tolerated well under these conditions making this method superior to other transition metal cross couplings. In more recent efforts, the Wang lab has expanded this chemistry to intermolecular insertions into oxazole and benzoxazole Csp2–H bonds using bis(trimethylsilyl) diazomethane and copper iodide. Using similar reaction conditions, Zou and co-workers have demonstrated the use of acetophenone-derived tosylhydrazones in insertion reactions with triazolopyridines.[61]
Figure 12.
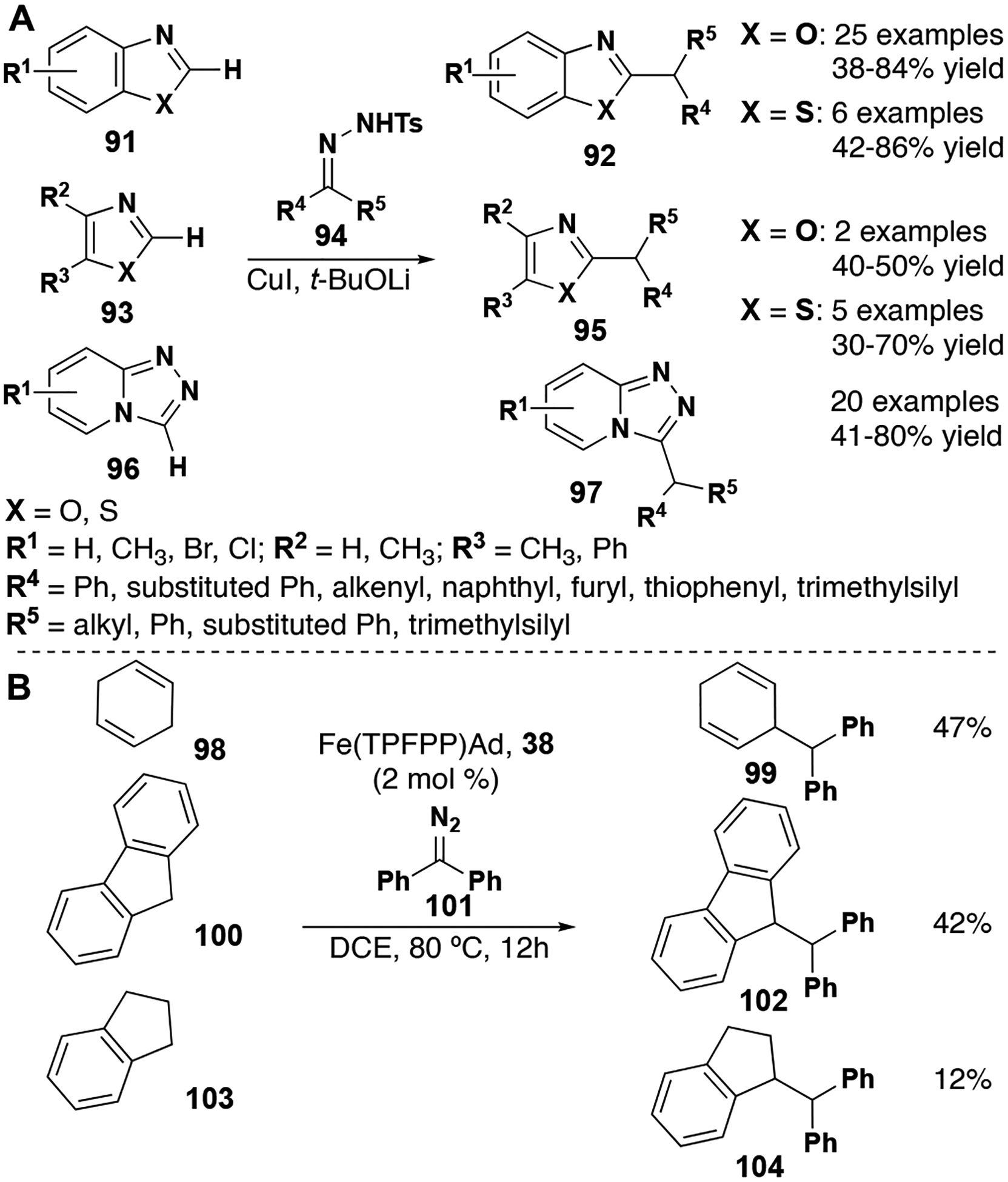
A, Azole Csp2–H insertion reactions. B, Csp3–H insertion reactions.
Early work by Che and co-workers with iron and osmium porphyrin bis-diphenyl carbene adducts showed the potential for these complexes in intermolecular Csp3–H insertion reaction methodology, albeit stoichiometrically.[62,63] Later efforts showed that ruthenium porphyrins were efficient in catalytic stereoselective intramolecular insertion reactions as well.[51] Most recently, Che and co-workers utilized donor/donor carbenes to study the crystallographic and electronic properties of group 8 metal porphyrin-based catalysts.[64] In their studies, the Fe(TPFPP)Cl-2-diazoadamantane decomposition adduct (38) was shown to be particularly stable, suggesting to them that the catalyst may be proficient in typical C–H insertion reaction catalysis. With 2 mol % loading of the iron porphyrin catalyst, they demonstrated that the donor/donor iron-carbenes were able to undergo insertion into allylic and benzylic C–H bonds (Figure 12, B). Although these reactions afforded poor to moderate yields, this work represents the first instance of catalytic intermolecular Csp3–H insertion with donor/donor carbenes. Although this recent publication does not report any diastereo- or enantio-selective variants, it does expose fertile ground for future work.
4. N–H Insertion
With attractive applications in the construction of biologically active compounds and natural products, there have naturally been many reports of donor/donor N–H insertion reactions (Table 3). In the majority of cases, alkyl and aryl tosylhydrazones and copper-based catalysts have seen success inserting into N–H bonds to form secondary and tertiary amine and amide insertion products. The first example of N–H insertion was reported by Del Zotto and co-workers using primary amines in the presence of diaryl diazo compounds and ruthenium catalyst 29 (Table 4, entry 2).[65] In this case, significant quantities of imine byproduct were observed via a β-hydride elimination mechanism. Shortly thereafter, Cu(acac)2 was shown to be effective in insertion reactions with imidazoles (Table 4, entry 4).[66] This development inspired later methods for inserting into aniline and alkyl N–H bonds (Table 4, entries 1 and 3). Impressively, Hamze and co-workers demonstrated that their procedure was highly chemoselective for N–H insertion over O–H moieties present in the substrate;[18,41] Sivasankar and co-workers were able to accomplish a similar result with catalyst 30.[67] Xu and co-workers were able to exploit this chemistry for the synthesis of amides (Table 4, entries 5–6).[42,68] Unfortunately, in most of these examples using copper catalysts, β-hydride elimination is common. Most recently, Che and co-workers were able to demonstrate N–H insertion reactions with iron porphyrin catalyst 38, affording various tertiary and secondary amines in good yields from donor/donor carbenes (Table 4, entries 7–8).[64] While significant progress has been made in the field of N–H insertion reactions, enantioselective variants of these reactions have yet to be successful.
Table 4.
N–H insertion reaction scope with donor/donor metal carbenes
 | |||||||
|---|---|---|---|---|---|---|---|
| entry[b] | R1 | R2 | R3 | catalyst | # examples | yield (%) | ref. |
| 1 | aryl | H, alkyl | H, alkyl, aryl | [Cu], 32 | 40 | 50–93 | 18 |
| 2 | alkyl | H, alkyl | aryl | [Ru], 29 | 10 | 10–48 | 65 |
| 3 | alkyl | H, alkyl | H, alkyl, aryl | [Cu], 30, 32 | 23 | 40–84 | 18,41,67 |
| 4[c] | imidazoles | H, aryl | [Cu], 32 | 10 | 50–80 | 66 | |
| 5[d] | COR | H | H | [Cu], 33 | 38 | 33–99 | 68 |
| 6 | COR | H, alkyl, aryl | alkyl/aryl | [Cu], 31, 33 | 38 | 40–98 | 42,68 |
| 7 | imidazole, carbazole, morpholine | Ph | [Fe], 38 | 3 | 86–94 | 64 | |
| 8 | Ph, Bn | H | Ph | [Fe], 38 | 2 | 51–56 | 64 |
includes Ar = 2-pyridyl napthyl (entry 1); Ar/R3 = fluorenyl (entry 2); Ar = napthyl (entries 5 and 6).
diazo compounds for all entries except 7 and 8 are derived from tosylhydrazones
includes 2 examples with cyclohexanone-derived tosylhydrazones.
includes 3 examples with tosylhydrazones derived from alkyl aldehydes.
5. B–H Insertion
B–H insertion has emerged as a useful method for the construction of organoboron compounds, which have myriad applications in organic synthesis. A variety of donor/donor carbenes undergo B–H insertion reactions to form alkylborane-amine complexes (Figure 13). Zhou and co-workers reported that B–H insertion reactions readily occur between borane-Lewis base complexes 109 and metal carbenes 108 (formed in situ from the decomposition of tosylhydrazones).[46] As with many C–H insertion reactions with donor/donor carbenes, phthalimide and triphenyl cyclopropane-derived rhodium carboxylate catalysts (i.e. 16 and 24, respectively) provided impressive stereoselectivity. In notable contrast to this impressive selectivity, other efforts have shown that B2Pin2 and BHpin can also decompose aryl/aryl and aryl/alkyl diazo compounds to afford similar products as racemates without a metal catalyst or Lewis base additives.[47] Furthermore, this method was observed to be immune to β-hydride elimination despite the presence of β-hydrogen atoms.
Figure 13.
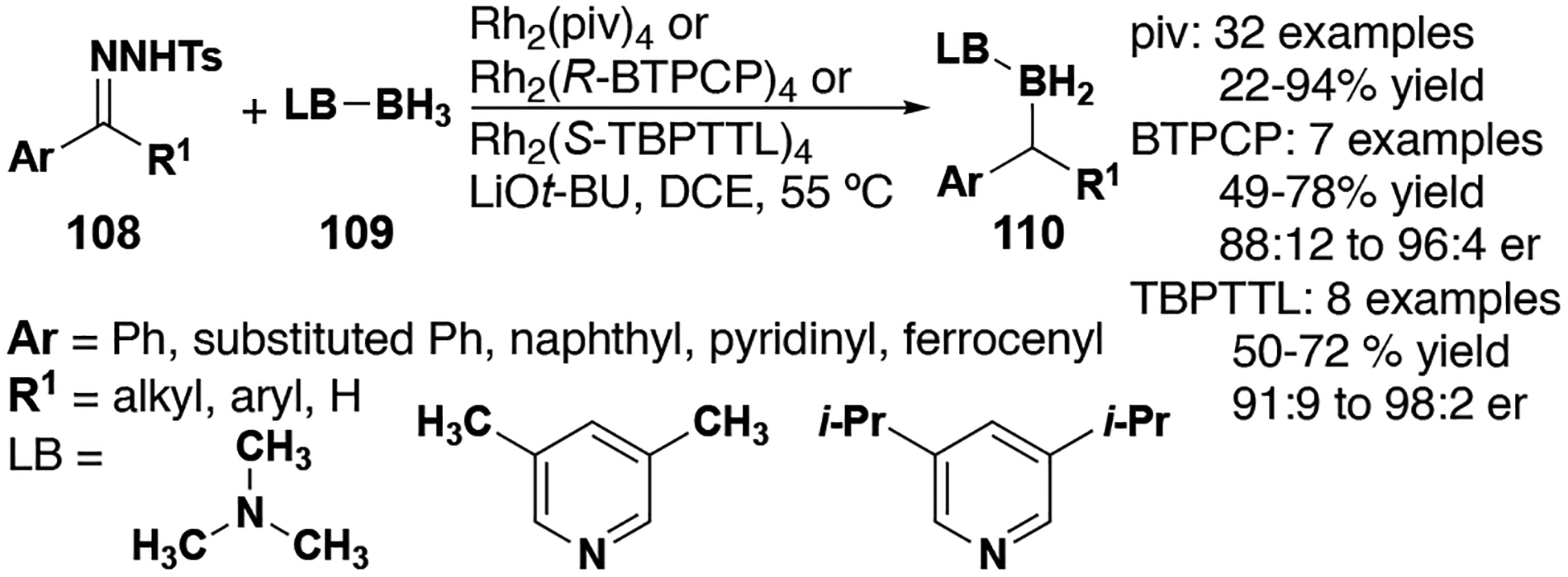
B–H insertion reaction scope
6. O–H/S–H Insertion
Sometimes considered a byproduct in methodologies for C–H insertion reactions, O–H insertion reactions with donor/donor carbenes offer unique routes to functionalized ethers. Bertani et al. detail the decomposition of 9-diazofluorene with neutral and cationic dirhodium carboxylate catalysts, giving poor yields of the O–H insertion product and a distribution of azine, ketone, and dimer byproducts.[69] However, when exposed to a platinum (II) phosphine catalyst 41, quantitative conversion to the O–H insertion product from methanol is observed (Table 5, entry 1). Later work by Barluenga and co-workers demonstrated that these substrates were susceptible to thermal decomposition and formal O–H insertion without a transition metal catalyst, affording a broad variety of ethers.[70] This method used tosylhydrazones derived from acetophenones, alkyl aldehydes, and alkyl ketones without observing any Bamford-Stevens type elimination byproducts, suggesting that O–H insertion is faster than elimination without a metal catalyst. Similar work was recently accomplished by Che and co-workers in O–H insertion reactions using phenyl/phenyl carbenes and iron porphyrin catalyst 38 to insert into phenyl, aryl, and alkyl alcohols (Table 5, entry 3).[64] This methodology proved amenable to S–H insertion reactions as well, affording phenyl and aryl thioethers in excellent yields and one example of an alkoxy thioether in poor yield (Table 5, entry 4). Currently there are no known stereoselective variants for O–H or S–H insertion.
Table 5.
O–H and S–H insertion reaction scope
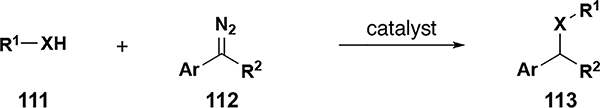 | ||||||
|---|---|---|---|---|---|---|
| entry[a] | X | R1 | Ar, R2 | # examples | yield (%) | ref. |
| 1[b] | O | CH3 | Ar, R2 = fluorenyl | 1 | 100 | 69 |
| 3[e] | O | Ph, Ar, 2-napthyl, alkyl | Ar, R2 = Ph | 5 | 31–92 | 64 |
| 4[e] | S | Ph, Ar, 2-pyridyl, alkyl | Ar, R2 = Ph | 5 | 33–99 | 64 |
diazo compounds derived from tosylhydrazones for entries 1 and 2.
cis-[Pt(PPh3)2(CH3CN)2][BF4]2 41 catalyst
Fe(TPFPP)Ad 38 as catalyst
7. Si/Ge/Sn–H Insertion
Insertion reactions into Si–, Ge–, and Sn–H bonds are newer entries in the list of donor/donor insertion reactions, with the first examples in the literature in 2017. Unlike most insertion reactions that are catalyzed by rhodium or ruthenium complexes, Si–H and related group 14 insertion reactions use several other transition metal catalysts. In the first report of donor/donor carbene insertion into a Sn–H bond, Wang and co-workers discovered that the reaction did not require any transition metal catalyst (Table 4, entry 3).[71] Of great synthetic importance, this method could readily generate reagents to be used in Stille cross-couplings in one-pot from aldehyde or ketone precursors. Without the use of a catalyst, the authors were interested in the mechanistic pathway (Figure 14). The authors posit that diazo compound 115 could be attacked by the alkyl tin hydride, or free carbene 116 forms first and is attacked by the tin hydride. The presence of a free carbene was confirmed by adding a styrene derivative, which formed the corresponding cyclopropanation product 118.
Figure 14.
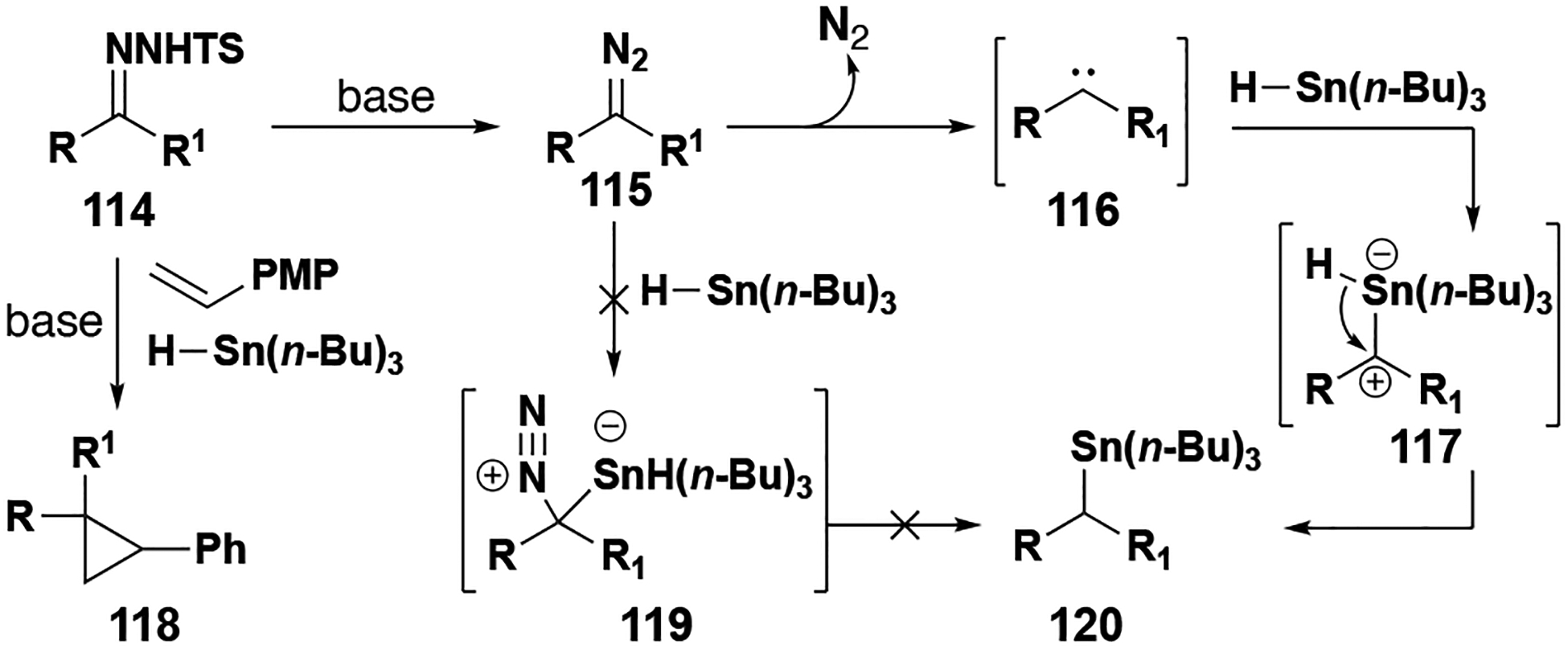
Donor/Donor Sn–H insertion and potential mechanis
Concurrently with Wang, Bi reported silver (I) triflate as a catalyst for Si–, Ge–, and Sn–H insertion reactions using nosylhydrazones derived from aryl aldehydes and aryl ketones (Table 6, entry 4).[72] This process proved to be high yielding and tolerant of a wide range of functional groups albeit at a catalyst loading of 30 mol %. Interestingly, nosylhydrazones derived from aliphatic aldehydes and ketones did not tolerate this method. In 2018, Che demonstrated that iron porphyrins at low catalyst loading (i.e. 2 mol %) could be effective catalysts for Si–, Ge–, and Sn–H insertions (Table 6, entry 1).[73] Unlike previous methods, this procedure allowed for the ability to insert into 3°, 2°, 1° Si–H bonds. Shortly after Che’s publication, Wang established a protocol for Si–H insertion using palladium catalyst 40 (Table 6, entry 2).[40] Though in all previous reports Si–H insertion into tosyl- or nosylhydrazones derived from aliphatic aldehydes or ketones was inaccessible, Wang and co-workers were able to generate several Si–H insertion examples in good yield using these intermediates. Impressively, there were no reported instances of β-hydride elimination in these systems.
Table 6.
Si/Ge/Sn–H insertion reaction scope with donor/donor carbenes
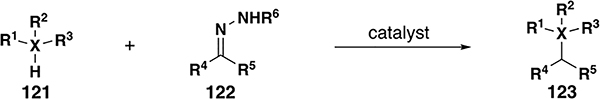 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | X | R1, R2, R3 | R4 | R5 | R6 | catalyst | # examples | yield (%) | Ref. |
| 1 | Si, Ge, Sn | H, alkyl, aryl | Ph, Ar[a] | H, CH3[b] | Ts | [Fe], 37 | 49 | 23–99 | 73 |
| 2 | Si | alkyl, Ph | alkyl, alkynyl, Ph, Ar[a] | H, alkyl, aryl | Ts | [Pd], 40 | 41 | 35–98 | 40 |
| 3 | Sn | alkyl | Ph, Ar[a] | H, alkyl | Ts | NA | 40 | 26–85 | 71 |
| 4 | Si, Ge, Sn | alkyl,aryl | Alkynyl, Ph, Ar[a] | H, alkyl, aryl | Ns | [Ag], 39 | 36 | 37–95 | 72 |
Ar = substituted Ph, napthyl (entry 1); Ar = substituted Ph, ferrocenyl, anthracenyl, napthyl, pyridyl (entry 2); Ar = pyridyl, napthyl (entry 3); Ar = napthyl, furyl, thiophenyl, indolyl (entry 4).
3 examples where X = Sn and Ge (30–44%).
More recently, Franz and co-workers, in collaboration Shaw, developed conditions to undergo the first known stereoselective intermolecular Si–H insertion reaction with donor/donor carbenes to afford chiral-at-silicon products (Table 7, entries 1–2).[74] After substantial optimization, this methodology demonstrated moderate to good enantioselectivity with catalyst 15 for the chiral silane products from symmetrical diazo compounds (Table 7, entry 1). Interestingly, when unsymmetrical, prochiral diazo compounds were used, enantioselectivity was more pronounced, giving up to 95:5 er, in addition to giving good to excellent diastereoselectivity (Table 7, entry 2).
Table 7.
Stereoselective Si–H insertion reactions with donor/donor carbenes
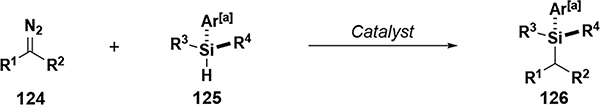 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | R4 | dr | er | catalyst | # examples | yield (%) | Ref. |
| 1 | Ar | R1 | alkyl, siloxy | H | NA | 50:50 to 86:14[b] | [Rh], 15 | 15 | 45–91 | 74 |
| 2 | Ar | Ar ≠ R1 | H, alkyl, aryl | H | 61:39 to 95:5 | 61:3 to 95:5[b,c] | [Rh], 15 | 9 | 55–95 | 74 |
| 3 | Ar | Ar ≠ R1 | CH3, Et, siloxy | R3 | NA | 68:32 to 99.5:0.5[c] | [Rh], 25 | 49 | 36–96 | 75 |
Ar = Ph, substituted Ph, napthyl, indolyl.
er denotes chirality at silicon.
er denotes chirality at carbon.
In another recent publication, Zhou and co-workers show an impressive analysis of electronic parameters for diaryl carbenes undergoing enantioselective Si–H insertion reactions.[75] The authors posit that a difference in the electronic character of the aryl rings of the donor/donor carbene directly impacts the stereochemical outcome of the reaction, enhanced by their new class of chiral spirophosphate-based rhodium paddlewheel catalysts at very low loadings (i.e. Figure 5, 25, 0.1 mol %). A large number of substrates with varied substitution on either aryl ring enabled the extensive Hammett plots which supported their mechanistic claims; a handful of useful silanes were also tolerated (Table 7, entry 3).
Although ‘donor substituents’ on a carbene are typically aryl or alkyl, Micouin developed a unique donor carbene using alkynyl and trimethylsilyl substituents (Figure 15).[76] Using Rh2(esp)2 (18) as catalyst, these carbenes were shown to have excellent selectivity for Si–H insertion, though limited to 3° silanes and moderate yields. It was suggested that these donor carbenes are among the least electrophilic carbenes and could give very high levels of selectivity. To test this, reactions were conducted in phenol, aniline, THF, and dioxane without observing any products from O–H, N–H, and C–H insertions. Cyclopropanation reactions with rhodium or copper catalysts was also unsuccessful, demonstrating the low electrophilicity and concomitant chemoselectivity of these carbenes.
Figure 15.
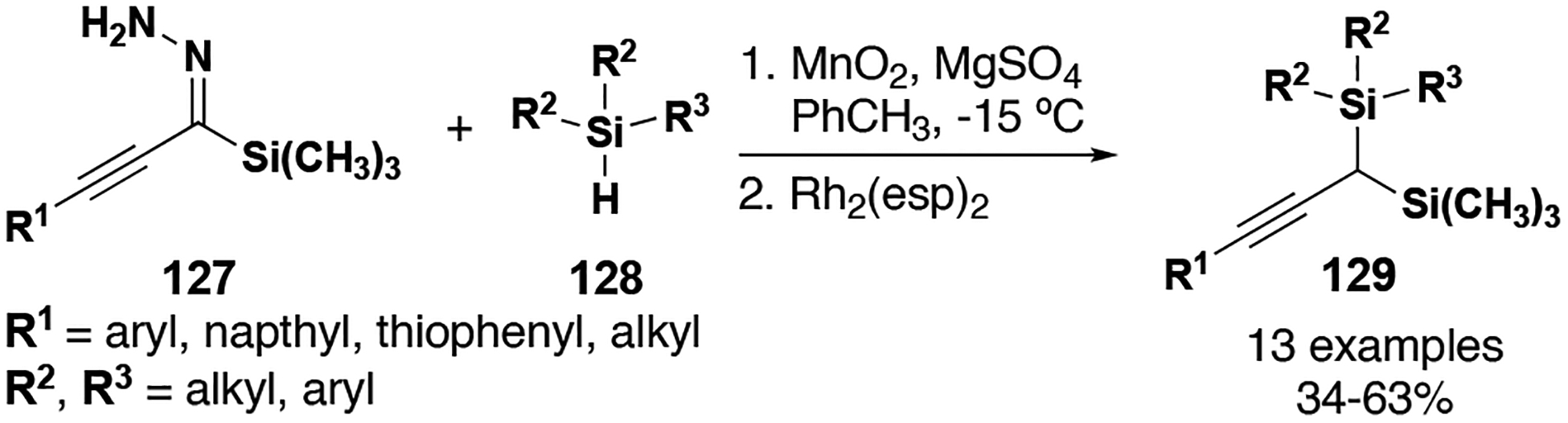
Si–H insertion reactions with alkyl/silyl carbenes
8. P–H Insertion
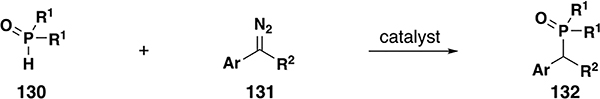
Alkyl phosphonates are useful as reagents in a variety of synthetic procedures and having several methods to generate these reagents increases their accessibility and usefulness. In 1990, Arbuzov and co-workers reported a method to synthesize dialkyl phosphonates by P–H insertion from donor/donor copper carbenes and hydrogen phosphites.[77] In this novel procedure, aryl/aryl diazo species 131 and hydrogen phosphites 130 were added to a refluxing solution of the copper catalyst. This one-pot method afforded 5 examples of dialkyl phosphonates in moderate to good yields (Table 8, entry 1). More recently, Wu and co-workers expanded upon this concept, utilizing copper carbenes derived from the basic decomposition of tosylhydrazones.[43] This work reports the formation of alkylated diaryl phosphine oxides and alkylated dialkyl phosphonates in good yields and without any reported Bamford-Stevens type elimination products (Table 8, entry 2). Currently, no stereoselective variant is known.
Table 8.
P–H insertion scope
| entry | R1 | R2 | catalyst | # examples | yield (%) | ref. |
|---|---|---|---|---|---|---|
| 1 | alkoxy | Ph, fluorenyl | [Cu], 32 | 5 | 52–77 | 77 |
| 2[a], [b] | aryl, alkoxy | H, alkyl | [Cu], 34 | 21 | 50–82 | 43 |
includes Ar = napthyl
diazo compounds derived from tosylhydrazones
9. Diazo-Free Insertion
Access to donor/donor carbenes without the use of diazo precurors offers an alternative method for accessing these ueseful intermediates. Although most donor/donor carbenes are derived from diazo compounds, there are implicit risks involved in the generation and handling of these potentially explosive molecules, especially at larger scales. Several methods have been developed to generate donor-substituted metal carbenes; most prominently, enynones, retro-Büchner ring expansion intermediates, and cyclopropenes can be manipulated in the presence of a transition metal catalyst to form the reactive carbene intermediate (Figure 16). Chelation of the alkyne moiety of an enynone (133) by a suitable rhodium or gold catalyst allows for the rearrangement of the structure to form a furyl-substituted metal carbene (135).[78] Similarly, the alkene moiety of a strained cyclopropene (139) can be chelated by a rhodium or zinc catalyst, opening the ring and forming a vinyl carbene (141). Additionally, when the retro-Büchner ring expansion intermediate (137) is exposed to a rhodium or gold catalyst, the norcaradiene is decomposed to form a metal carbene (138) and benzene. These metal carbene species may then be used in further intra- or intermolecular insertion reactions.
Figure 16.
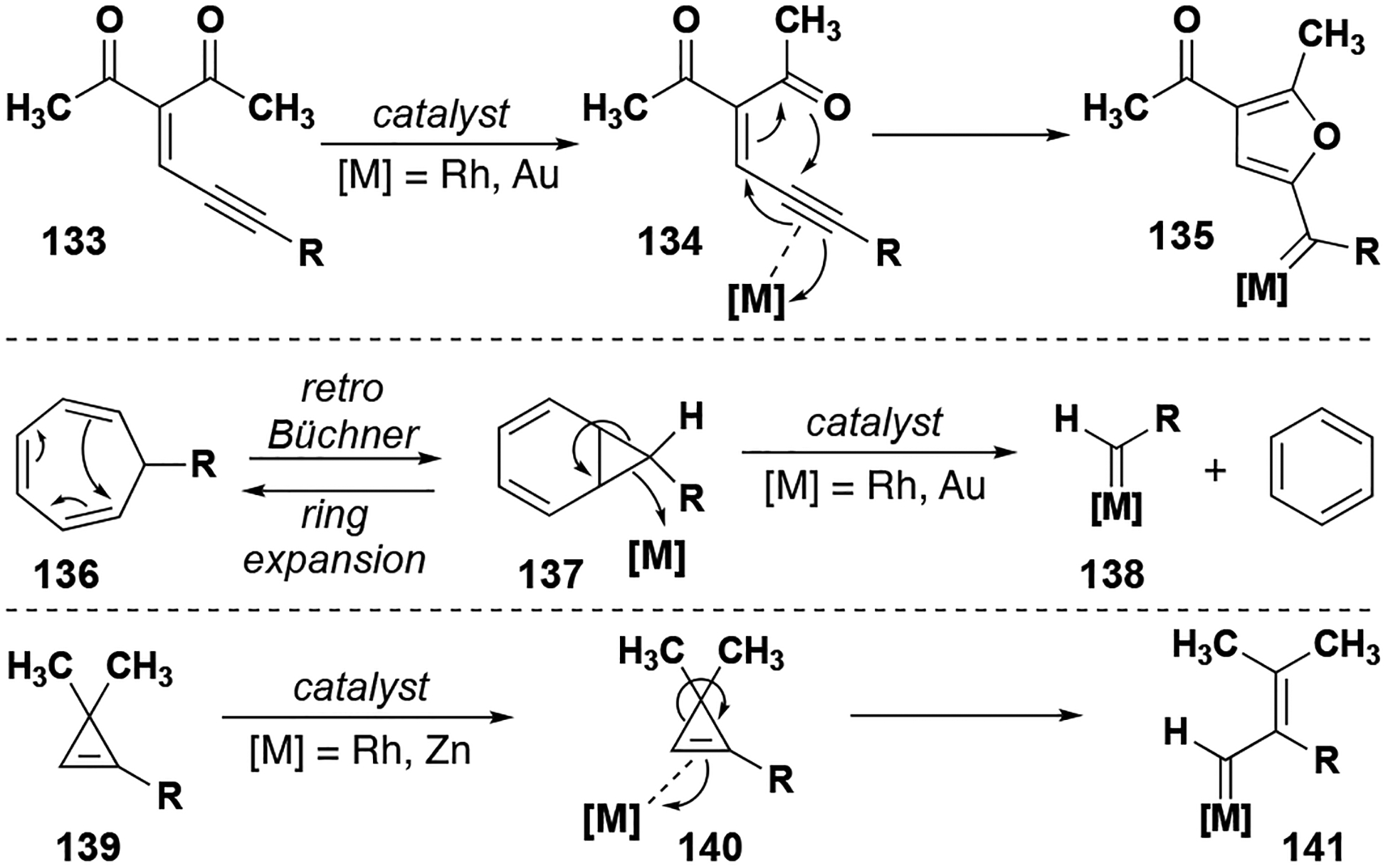
Donor-substituted metal carbene formation from diazo-free substrates
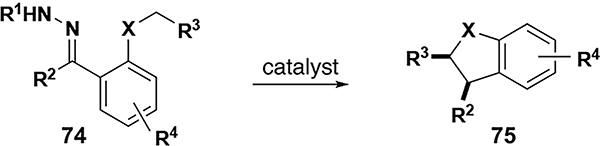
Recent work by Zhu (South China University of Technology) circumvents potential safety issues through a novel diazo-free generation of donor/donor metal carbenes from the cyclization of enynones.[79] These metal carbenes then undergo diastereoselective and enantioselective C–H insertion to form furyl-substituted dihydroindole and dihydrobenzofuran structural cores (Table 9). Entry 1 demonstrates the utility of the methodology with a broad variety of both oxygen and N-acyl substrates; nearly all examples gave excellent diastereo- and enantio-selectivity at low catalyst loadings (1–5 mol %). Entry 3 describes the application of this methodology with a furyl/alkyl carbene, affording excellent yields of the tetrahydrofuran product but with diminished stereoselectivity compared to entry 1.
Table 9.
Diazo-free C–H insertion reactions with donor/donor and donor carbenes from enynones
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| entry | X | R1 | R2 | R3 | catalyst | # examples | yield (%) | dr | er | ref |
| 1[a] | O, NAc | alkyl, alkoxy | alkyl | alkyl, alkenyl, CO2Et, Ar[b] | [Rh], 17 | 29 | 65–99 | 50:50 to >99:1 | 82:18 to 99.5:0.5 | 79 |
| 2[c] | NAc | alkyl,alkoxy, Ph | alkyl, Ph | alkyl, alkenyl, Ar[b] | [Ru], 26 | 34 | 43–97 | 91:9 to >99:1 | 98:2 to >99.5:0.5 | 80 |
| 3[a] | NA | CH3 | CH3 | alkenyl, Ph | [Rh], 17 | 3 | 99 | 77:23 to 95:5 | 80:20 to 87:13 | 79 |
reactions run −20 to −30 °C.
Ar = Ph, substituted Ph, napthyl (entry 1); Ar = Ph, substituted Ph (entry 2).
reactions run 80–120 °C
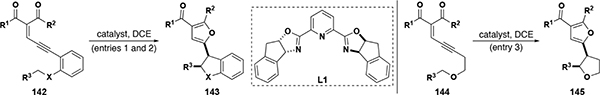
C–H insertion substrates with allylic or propargylic insertion sites often suffer from competing cyclopropanation (and cyclopropenation) reactions. Zhu and co-workers expanded upon their diazo-free methodology to include chemoselective reactions with allylic and alkyl substrates by using an alternative catalyst.[80] The dirhodium carboxylate catalyst used in entries 1 and 3, when applied to allylic insertion sites for N-acyl substrates, was found to give tetrahydroquinoline cyclopropanation products with excellent stereoselectivity. However, when a ruthenium precatalyst is used with indapybox as the ligand (Figure 5, 26, Table 9, L1), the C–H insertion reaction predominates, giving good yields of dihydroindolines with excellent stereoselectivity (Table 9, entry 2). Outside of C–H insertion reactions, Zhu found that these diazo-free donor and donor/donor carbenes generated from enynones are productive in N–H, O–H, and Si–H insertion reactions, in addition to cyclopropanation reactions with gold N-heterocylic carbene catalyst 35 (Figure 17).[81] In a more recent report, Zhu and co-workers (Nankai University) used rhodium paddlewheel complexes with these enynone derived diazo-free carbenes to achieve enantioselective Si–H insertion for a wide selection of substrates (Figure 17, 150).[82]
Figure 17.
Diazo-free N–H, O–H, and Si–H insertion reactions with enynone-derived donor carbenes
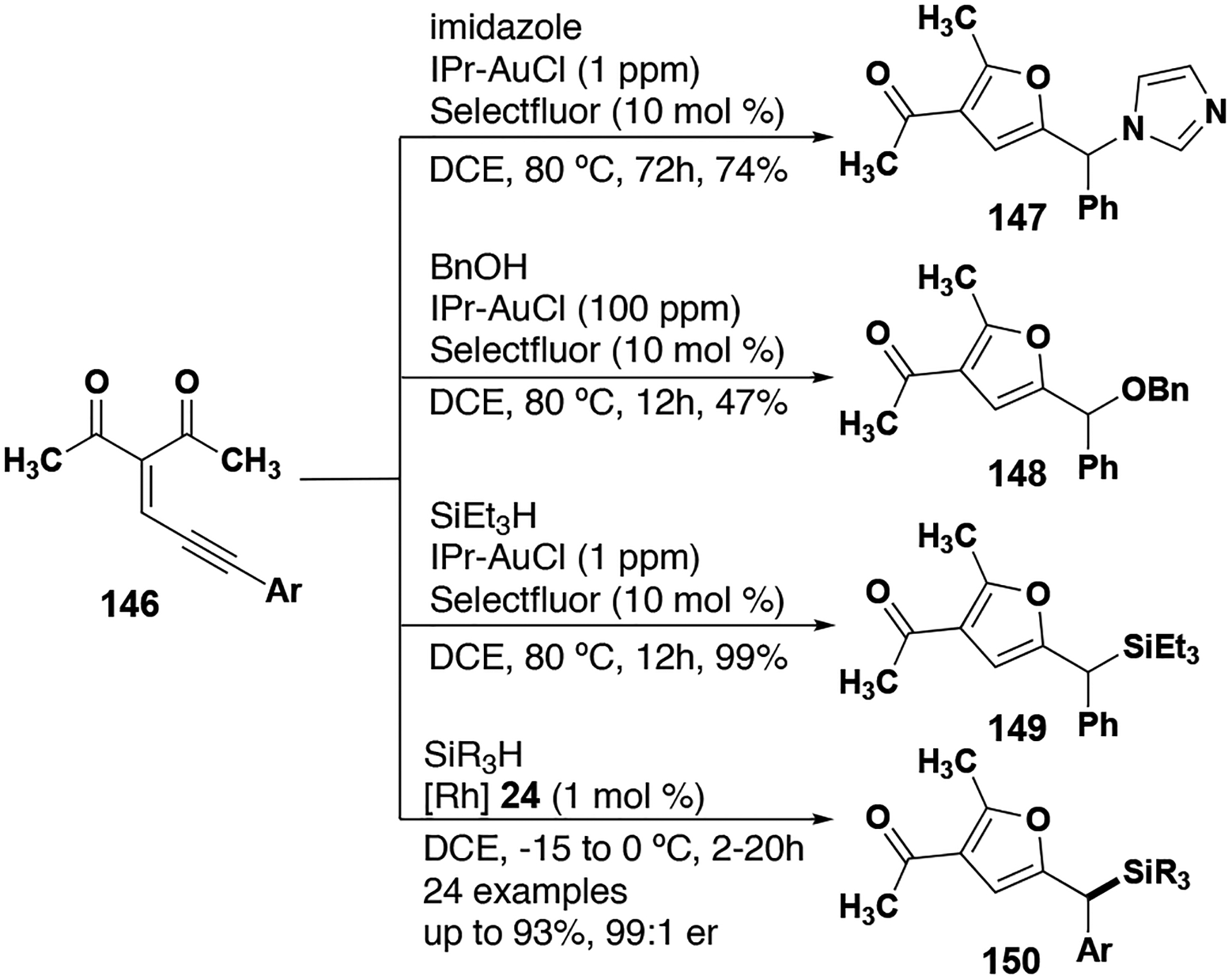
Echavarren and co-workers were able to generate diazo-free donor carbenes through a retro-Büchner ring expansion (Figure 18). With gold phosphine catalyst 36, intramolecular C–H insertion to form an indane was achieved as the minor product, affording mostly cyclopropanation products.[83] In more recent efforts, the Echavarren group used dirhodium paddlewheel complex Rh2(TFA)4 (20) to better effect, generating numerous intermolecular Si–H insertion products (Figure 18).[84]
Figure 18.
Retro-Büchner ring expansion derived donor carbene insertion reactions
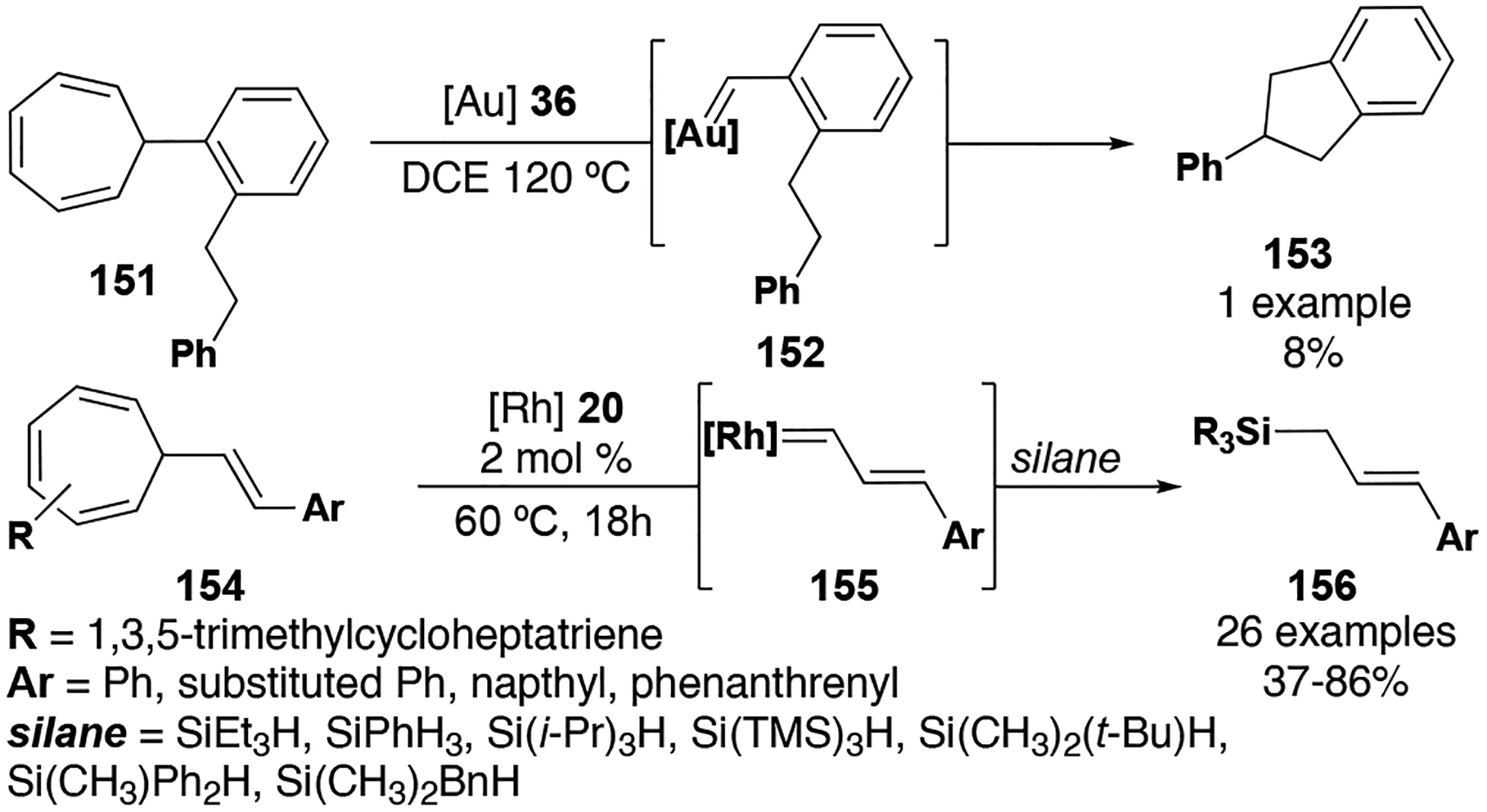
Another robust method for generating diazo-free donor metal carbenes is by the ring opening of substituted cyclopropenes. Early efforts by Cossy and co-workers demonstrated an impressive substrate scope using this method for intramolecular C–H insertion (Figure 19, 157–159). With Rh2(OAc)4 as the catalyst, substituted pyrans (159) were achieved with excellent diastereoselectivity at low catalyst loadings (0.5 mol %); a desymmetrization experiment furthermore demonstrated excellent stereoselectivity, affording 159 (R2 = CH2CH2OR) as a single diastereomer. The authors also offered intriguing stereoselectivity models for these novel substrates and produced convincing evidence for a concerted mechanism for these insertion substrates via stereospecific deuterium labling experiments.[37]
Figure 19.
Cyclopropene derived donor carbene insertion reactions
Vicente and co-workers also used substituted cyclopropenes as diazo-free donor metal carbene precursors with significant success. In a simple and cost-effective system using zinc bromide as the metal catalyst, Vicente was able to generate structurally unique allyl silanes (26 examples) from a variety of substrates and various silanes/siloxanes (Figure 19, 160–162).[85] Notably, this method was applied in Ge–H insertion for the first ever zinc-catalyzed insertions into germanium hydrides. In a later report by Vicente and co-workers, cyclopropene-derived rhodium octanoate (23) carbenes were used to generate a broad scope of substrates (>35 examples) via intermolecular Si–H insertion reactions (Figure 19, 163–165). This method was effective at low catalyst loadings (1 mol %) and tolerated other traditionally reactive moieties on the substrate (i.e. phenols, allyl benzyl groups). For a specialized class of substrates, intramolecular Si–H insertion reactions were also achieved to produce racemic cyclic siloxanes. While these researchers have had considerable success with these diazo-free methodologies, there are still many opportunities to expand substrate scope, further develop enantioselective variants, and introduce new orthogonal diazo-free methods.
10. Summary and Outlook
Donor/donor carbenes exhibit reduced electrophilicity compared to their acceptor-substituted equivalents; in C–H and X–H insertion reactions, this attenuated reactivity provides excellent chemoselectivity, functional group tolerance, and insensitivity to adventitious moisture. Traditionally delicate insertion reactions are accessible without rigorous drying procedures and can even tolerate Lewis basic or other X–H moieties on the substrate without diminished reactivity and/or significant side reactions. In many cases, this difference in reactivity can also produce enhanced stereoselective outcomes. While dirhodium carboxylate catalysts remain a privileged class of catalysts for C–H and some X–H insertion reactions, there are many reports of ruthenium and copper catalyzed reactions (among others) that demonstrate equivalent selectivity and orthogonal reactivity to allow reactions deemed infeasible with rhodium paddlewheel complexes. Mechanistic proposals have suggested that C–H and X–H insertion reactions may proceed by several differing pathways depending on the type of catalyst, the accompanying metal-carbene, and the steric and electronic properties of the insertion site.
Significant work with dirhodium paddlewheel complexes has shown that intramolecular C–H insertion reactions with donor/donor carbenes can achieve high degrees of chemo-, regio-, and stereo-selectivity. While these insertion reactions remain a hot topic, the field of donor-substituted carbene insertion chemistry has been expanded in recent years to include N–H, B–H, O–H, Si–H, Ge–H, Sn–H and P–H moieties. However, whereas C–H insertion reactions have been made stereoselective for many substrate classes, diastereo- and/or enantio-selective X–H insertion reactions remain limited. Zhou and co-workers achieved excellent enantioselectivity with donor/donor carbenes in intermolecular B–H insertions and Franz and Zhu have similarly demonstrated the first catalytic stereoselective intermolecular Si–H insertion reactions, but there remains open methodological territory for creating stereoselective variants of X–H moieties in insertion chemistry. Concerns over the safety of diazo compounds have spawned novel diazo-free procedures that generate unique substrates; these methods have been successfully applied not only in C–H insertion reactions, but also with N–H, O–H, and Si–H bonds. The volume of work in the field of donor-substituted carbene chemistry, while still small compared to acceptor-substituted carbenes, is growing and will continue to offer up unique chemical disconnections, opportunities for stereoselective transformations, and superb chemoselectivity in complex substrates. With these considerations in mind, there is a great outlook for the application of these methods in natural product syntheses, which have remained rare in the literature.
References
- [1].Doyle MP, Duffy R, Ratnikov M, Zhou L, Chem. Rev 2010, 110, 704–724. [DOI] [PubMed] [Google Scholar]
- [2].Davies HML, Morton D, Chem. Soc. Rev 2011, 40, 1857–1869. [DOI] [PubMed] [Google Scholar]
- [3].Davies HML, Beckwith REJ, Chem. Rev 2003, 103, 2861–2904. [DOI] [PubMed] [Google Scholar]
- [4].Wang B, Qiu D, Zhang Y, Wang J, Beilstein J Org. Chem 2016, 12, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Doyle MP, Forbes DC, Chem. Rev 1998, 98, 911–936. [DOI] [PubMed] [Google Scholar]
- [6].Lee M, Ren Z, Musaev DG, Davies HML, ACS Catal 2020, 10, 6240–6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Werlé C, Goddard R, Philipps P, Farès C, Fürstner A, J. Am. Chem. Soc 2016, 138, 3797–3805. [DOI] [PubMed] [Google Scholar]
- [8].Werlé C, Goddard R, Philipps P, Farès C, Fürstner A, Angew. Chem. Int. Ed 2016, 55, 10760–10765. [DOI] [PubMed] [Google Scholar]
- [9].Staudinger H, Endle R, Berichte der Dtsch. Chem. Gesellschaft 1913, 1437–1442. [Google Scholar]
- [10].Zhu D, Chen L, Fan H, Yao Q, Zhu S, Chem. Soc. Rev 2020, 49, 908–950. [DOI] [PubMed] [Google Scholar]
- [11].Hansen J, Davies HML, Coord. Chem. Rev 2008, 252, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Timmons DJ, Doyle MP, J. Organomet. Chem 2001, 617–618, 98–104. [Google Scholar]
- [13].Doyle MP, Chem. Rev 1986, 86, 919–939. [Google Scholar]
- [14].Cheng QQ, Doyle MP, The Selection of Catalysts for Metal Carbene Transformations, Elsevier Inc., 2016. [Google Scholar]
- [15].Deng Y, Qiu H, Srinivas HD, Doyle MP, Curr. Org. Chem 2015, 20, 61–81. [Google Scholar]
- [16].Adly FG, Bollard H, Gardiner MG, Ghanem A, Catalysts 2018, 8, 268. [Google Scholar]
- [17].DeAngelis A, Panish R, Fox JM, Acc. Chem. Res 2016, 49, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aziz J, Brion JD, Hamze A, Alami M, Adv. Synth. Catal 2013, 355, 2417–2429. [Google Scholar]
- [19].Grasse PB, Brauer BE, Zupancic JJ, Kaufmann KJ, Schuster GB, J. Am. Chem. Soc 1983, 105, 6833–6845. [Google Scholar]
- [20].Kirmse W, Özkir IS, J. Am. Chem. Soc 1992, 114, 7590–7591. [Google Scholar]
- [21].Doyle MP, Westrum LJ, Wolthuis WNE, See MM, Boone WP, Bagheri V, Pearson MM, J. Am. Chem. Soc 1993, 115, 958–964. [Google Scholar]
- [22].Collins LR, Van Gastel M, Neese F, Fürstner A, J. Am. Chem. Soc 2018, 140, 13042–13055. [DOI] [PubMed] [Google Scholar]
- [23].Skell PS, Woodworth RC, J. Am. Chem. Soc 1956, 78, 4496–4497. [Google Scholar]
- [24].Davies HML, Bruzinski PR, Lake DH, Kong N, Fall MJ, J. Am. Chem. Soc 1996, 118, 6897–6907. [Google Scholar]
- [25].Hansen J, Autschbach J, Davies HML, J. Org. Chem 2009, 74, 6555–6563. [DOI] [PubMed] [Google Scholar]
- [26].Te Grotenhuis C, Das BG, Kuijpers PF, Hageman W, Trouwborst M, de Bruin B, Chem. Sci 2017, 8, 8221–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Karns AS, Goswami M, de Bruin B, Chem. Eur. J 2018, 24, 5253–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Das BG, Chirila A, Tromp M, Reek JNH, de Bruin B, J. Am. Chem. Soc 2016, 138, 8968–8975. [DOI] [PubMed] [Google Scholar]
- [29].te Grotenhuis C, van den Heuvel N, van der Vlugt JI, de Bruin B, Angew. Chem. Int. Ed 2018, 57, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang Y, Wen X, Cui X, Wojtas L, Zhang XP, J. Am. Chem. Soc 2017, 139, 1049–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Werlé C, Goddard R, Fürstner A, Angew. Chem. Int. Ed 2015, 54, 15452–15456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gillingham D, Fei N, Chem. Soc. Rev 2013, 42, 4918–4931. [DOI] [PubMed] [Google Scholar]
- [33].Taber DF, You KK, Rheingold AL, J. Am. Chem. Soc 1996, 118, 547–556. [Google Scholar]
- [34].Nakamura E, Yoshikai N, Yamanaka M, J. Am. Chem. Soc 2002, 124, 7181–7192. [DOI] [PubMed] [Google Scholar]
- [35].White JD, Hrnciar P, J. Org. Chem 1999, 64, 7271–7273. [Google Scholar]
- [36].Lamb KN, Squitieri RA, Chintala SR, Kwong AJ, Balmond EI, Soldi C, Dmitrenko O, Castiñeira Reis M, Chung R, Addison JB, et al. , Chem. Eur. J 2017, 23, 11843–11855. [DOI] [PubMed] [Google Scholar]
- [37].Archambeau A, Miege F, Meyer C, Cossy J, Angew. Chem. Int. Ed 2012, 124, 11540–11544. [DOI] [PubMed] [Google Scholar]
- [38].Dong K, Fan X, Pei C, Zheng Y, Chang S, Cai J, Qiu L, Yu ZX, Xu X, Nat. Commun 2020, 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhao X, Wu G, Zhang Y, Wang J, J. Am. Chem. Soc 2011, 133, 3296–3299. [DOI] [PubMed] [Google Scholar]
- [40].Liu Z, Huo J, Fu T, Tan H, Ye F, Hossain ML, Wang J, Chem. Commun 2018, 54, 11419–11422. [DOI] [PubMed] [Google Scholar]
- [41].Hamze A, Tréguier B, Brion JD, Alami M, Org. Biomol. Chem 2011, 9, 6200–6204. [DOI] [PubMed] [Google Scholar]
- [42].Xu P, Qi FL, Han FS, Wang YH, Chem. Asian J 2016, 11, 2030–2034. [DOI] [PubMed] [Google Scholar]
- [43].Wu L, Zhang X, Chen QQ, Zhou AK, Org. Biomol. Chem 2012, 10, 7859–7862. [DOI] [PubMed] [Google Scholar]
- [44].Salomon RG, Kochi JK, J. Am. Chem. Soc 1973, 95, 3300–3310. [Google Scholar]
- [45].Shirafuji T, Yamamoto Y, Nozaki H, Tetrahedron 1971, 27, 5353–5358. [Google Scholar]
- [46].Pang Y, He Q, Li Z-Q, Yang J-M, Yu J-H, Zhu S-F, Zhou Q-L, J. Am. Chem. Soc 2018, 140, 10663–10668. [DOI] [PubMed] [Google Scholar]
- [47].Li H, Wang L, Zhang Y, Wang J, Angew. Chem. Int. Ed 2012, 51, 2943–2946. [DOI] [PubMed] [Google Scholar]
- [48].Bard AJ, Whitesides GM, Zare RN, McLafferty FW, Acc. Chem. Res 1995, 28, 91. [Google Scholar]
- [49].Arndtsen BA, Bergman RG, Mobley TA, Peterson TH, Acc. Chem. Res 1995, 28, 154–162. [Google Scholar]
- [50].Zheng S-L, Yu W-Y, Xu M-X, Che C-M, Tetrahedron Lett 2003, 44, 1445–1447. [Google Scholar]
- [51].Cheung W-H, Zheng S-L, Yu W-Y, Zhou G-C, Che C-M, Org. Lett 2003, 5, 2535–2538. [DOI] [PubMed] [Google Scholar]
- [52].Soldi C, Lamb KN, Squitieri RA, González-López M, Di Maso MJ, Shaw JT, J. Am. Chem. Soc 2014, 136, 15142–15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Natori Y, Ito M, Anada M, Nambu H, Hashimoto S, Tetrahedron Lett 2015, 56, 4324–4327. [Google Scholar]
- [54].Souza LW, Squitieri RA, Dimirjian CA, Hodur BM, Nickerson LA, Penrod CN, Cordova J, Fettinger JC, Shaw JT, Angew. Chem. Int. Ed 2018, 57, 15213–15216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Su N, Deng T, Wink DJ, Driver TG, Org. Lett 2017, 19, 3990–3993. [DOI] [PubMed] [Google Scholar]
- [56].Su N, Theorell JA, Wink DJ, Driver TG, Angew. Chem. Int. Ed 2015, 54, 12942–12946. [DOI] [PubMed] [Google Scholar]
- [57].Bamford WR, Stevens TS, J. Chem. Soc 1952, 4735–4740. [Google Scholar]
- [58].Reddy AR, Zhou C-Y, Guo Z, Wei J, Che C-M, Angew. Chem. Int. Ed 2014, 53, 14175–14180. [DOI] [PubMed] [Google Scholar]
- [59].Ito M, Kondo Y, Nambu H, Anada M, Takeda K, Hashimoto S, Tetrahedron Lett 2015, 56, 1397–1400. [Google Scholar]
- [60].Nickerson LA, Bergstrom BD, Gao M, Shiue YS, Laconsay CJ, Culberson MR, Knauss WA, Fettinger JC, Tantillo DJ, Shaw JT, Chem. Sci 2020, 11, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Reddy Lonka M, Zhang J, Gogula T, Zou H, Org. Biomol. Chem 2019, 17, 7455–7460. [DOI] [PubMed] [Google Scholar]
- [62].Li Y, Huang JS, Zhou ZY, Che CM, J. Am. Chem. Soc 2001, 123, 4843–4844. [DOI] [PubMed] [Google Scholar]
- [63].Li Y, Huang J-S, Zhou Z-Y, Che C-M, You X-Z, J. Am. Chem. Soc 2002, 124, 13185–13193. [DOI] [PubMed] [Google Scholar]
- [64].Wang HX, Wan Q, Low KH, Zhou CY, Huang JS, Zhang JL, Che CM, Chem. Sci 2020, 11, 2243–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Del Zotto A, Baratta W, Miani F, Verardo G, Rigo P, Inorg. Chim. Acta 2003, 349, 249–252. [Google Scholar]
- [66].Cuevas-Yañez E, Serrano JM, Huerta G, Muchowski JM, Cruz-Almanza R, Tetrahedron 2004, 60, 9391–9396. [Google Scholar]
- [67].Ramakrishna K, Sivasankar C, J. Org. Chem 2016, 81, 6609–6616. [DOI] [PubMed] [Google Scholar]
- [68].Xu P, Han FS, Wang YH, Adv. Synth. Catal 2015, 357, 3441–3446. [Google Scholar]
- [69].Bertani R, Michelin RA, Mozzon M, Sassi A, Basato M, Biffis A, Martinati G, Zecca M, Inorg. Chem. Commun 2001, 4, 281–284. [Google Scholar]
- [70].Barluenga J, Tomás-Gamasa M, Aznar F, Valdé C, Angew. Chem. Int. Ed 2010, 49, 4993–4996. [DOI] [PubMed] [Google Scholar]
- [71].Qiu D, Wang S, Meng H, Tang S, Zhang Y, Wang J, J. Org. Chem 2017, 82, 624–632. [DOI] [PubMed] [Google Scholar]
- [72].Liu Z, Li Q, Yang Y, Bi X, Chem. Commun 2017, 53, 2503–2506. [DOI] [PubMed] [Google Scholar]
- [73].Wang E-H, Ping Y-J, Li Z-R, Qin H, Xu Z-J, Che C-M, Org. Lett 2018, 20, 4641–4644. [DOI] [PubMed] [Google Scholar]
- [74].Jagannathan JR, Fettinger JC, Shaw JT, Franz AK, J. Am. Chem. Soc 2020, DOI 10.1021/jacs.0c04533. [DOI] [PMC free article] [PubMed]
- [75].Yang L, Evans D, Xu B, Li W, Li M, Zhu S, Houk KN, Zhou Q, 2020, DOI 10.1021/jacs.0c04725. [DOI]
- [76].Courant T, Kumar R, Turcaud S, Micouin L, Org. Lett 2016, 18, 4818–4820. [DOI] [PubMed] [Google Scholar]
- [77].Polozov AM, Polezhaeva NA, Mustaphin AH, Khotinen AV, Arbuzov BA, Synthesis 1990, 515–517. [Google Scholar]
- [78].Ma J, Zhang L, Zhu S, Curr. Org. Chem 2015, 20, 102–118. [Google Scholar]
- [79].Zhu D, Ma J, Luo K, Fu H, Zhang L, Zhu S, Angew. Chem. Int. Ed 2016, 55, 8452–8456. [DOI] [PubMed] [Google Scholar]
- [80].Zhu D, Chen L, Zhang H, Ma Z, Jiang H, Zhu S, Angew. Chem. Int. Ed 2018, 57, 12405–12409. [DOI] [PubMed] [Google Scholar]
- [81].Ma J, Jiang H, Zhu S, Org. Lett 2014, 16, 4472–4475. [DOI] [PubMed] [Google Scholar]
- [82].Huang MY, Yang JM, Zhao YT, Zhu SF, ACS Catal 2019, 2, 5353–5357. [Google Scholar]
- [83].Solorio-Alvarado CR, Wang Y, Echavarren AM, J. Am. Chem. Soc 2011, 133, 11952–11955. [DOI] [PubMed] [Google Scholar]
- [84].Mato M, Echavarren AM, Angew. Chem. Int. Ed 2019, 58, 2088–2092. [DOI] [PubMed] [Google Scholar]
- [85].Mata S, López LA, Vicente R, Angew. Chem. Int. Ed 2017, 56, 7930–7934. [DOI] [PubMed] [Google Scholar]