

Abstract
RNA-binding proteins (RBPs) play a crucial role in cellular physiology by regulating RNA processing, translation, and turnover. In neoplasms, RBP support of cancer-relevant expression of alternatively spliced, modified, and stabilized mRNA transcripts is essential to self-renewal, proliferation, and adaptation to stress. In this review, we assess the impact of key families of RBPs in leukemogenesis, review progress in targeting those proteins with small molecules, and discuss how multilevel composition of posttranscriptional regulation of gene expression could be used for potential therapies in acute and chronic leukemia.
Introduction
Leukemia is a blood cancer characterized by abnormal proliferation of myeloid or lymphoid progenitors in the bone marrow and their compromised ability to produce fully functional blood cells. Despite the relatively high effectiveness of current conventional and targeted therapeutics, anti-leukemia drugs are facing a number of challenges related to rapidly acquired resistance and intolerable toxicity - critical treatment factors for elderly and physically fragile patients. Mortalities associated with refractory and relapse leukemia indicate a need to optimize risk group stratification and the development of new remedies capable of overcoming resistance to therapeutics.
While alterations in protein-coding genes are considered a driving force of cancer, multiple posttranscriptional events occurring between RNA synthesis and protein production are in control of gene expression and influence cell fate. RNA processing, transport, and translation are orchestrated by various cis- and trans-acting regulatory elements. Cis-acting RNA regulatory elements are the internal RNA motifs recognized by the external trans-acting factors, such as non-coding RNAs and RNA-binding proteins (RBPs). Ribonucleoprotein (RNP) complexes are formed when RNA binds at conventional RBP RNA-binding domains, or through unconventional RNA-protein interactions1. Of the 1,914 RBPs comprising Homo sapiens’ RNA interactome, relatively few have been associated with aberrant development and cancer.
This review provides a snapshot of key families of RBPs involved in leukemogenesis, focusing on their role in messenger RNA (mRNA) fate. We begin with RNA editing and modifying enzymes conferring changes in RNA cis-acting elements. We then discuss the roles of other essential trans-acting factors, such as RNA splicing, export, and translation regulators, as well as several oncofetal RBPs. Last, we look at the current progress and challenges in targeting these proteins with small molecules and discuss their possible applications in leukemia treatment.
RNA editing enzymes
ADAR1
RNA edits are discrete changes in RNA nucleotide sequences introduced after transcription. Hydrolytic deamination of adenine to inosine residues (so-called A-to-I editing) is one of the most prevalent edits on doublestranded mammalian RNA (dsRNA) that is carried out by the adenosine deaminases acting on RNA (ADAR) family of enzymes. ADAR1 is ubiquitously expressed and is the most studied protein of the ADAR family. The ADAR1 gene encodes for two protein isoforms: the constitutively expressed N-terminally truncated p110 isoform, and the full length interferon (IFN)-inducible p150 isoform, both of which shuttle between the nucleus and the cytoplasm2.
One of the adaptive rationales for RNA editing is the ability of eukaryotic cells to discriminate between “self” and “non-self” RNAs. Endogenous RNA editing occurs in transcripts from primate-specific Alu repeats, at the highly conserved regions encoding functional protein domains as well as untranslated coding and non-coding RNAs. Because editing makes the base pairing in RNA duplexes imperfect, the endogenous dsRNAs that are long and entirely aligned are not typically found in the cytoplasm of eukaryotic cells. The perfectly aligned dsRNAs are usually produced during viral replication and trigger pro-apoptotic and pro-inflammatory responses through the activation of melanoma differentiation-associated gene 5 (MDA5), protein kinase R (PKR), and other pathogen-associated molecular patterns receptors. The ADAR1 enzymes balance the immune activation and self-tolerance by attenuating MDA5 and PKR activity3.
ADAR1 role in innate inflammation and apoptosis appears to be critical for embryonic development, especially the hematopoietic lineage, as Adar−/− mice die at E11.4–14 from widespread death of hematopoietic cells in the liver4. Their lethality can be rescued by deleting of genes encoding dsRNA-sensing, pro-inflammatory proteins e.g. Mda55. In addition to embryonic hematopoiesis, ADAR1 is required for the repopulating capacity of hematopoietic stem cells (HSC) in adult mice6.
Elevated mRNA and protein levels of ADAR1 were found in pediatric B-cell acute lymphoblastic leukemia (B-ALL)7, adult acute myeloid leukemia (AML)8, and progressed to blast crisis chronic myeloid leukemia (BC CML)9, Table 1. Several studies indicate that ADAR1 maintains proliferation and self-renewal of myeloid leukemia stem/progenitor cells in cooperation with WNT/β-catenin signaling. Xiao et al. reported that AML samples have significantly higher expression levels of ADAR1 compared to complete AML remission and non-malignant myeloid blood disorders8. ADAR1 knockdown led to decreased expression of WNT signaling effectors (β-catenin, c-MYC, TCF-4, Cyclin D2) and suppressed AML proliferation8.
Table 1.
RNA editing enzyme, ADAR1
| Gene | Protein/RNP function | Target Genes | Biological consequences | Type of cancer | Expression in cancer | Ref |
|---|---|---|---|---|---|---|
| ADAR1 | A-to-I substitutions in dsRNAs, writer | miR-26a, miR-155, let-7 | cell cycle regulation via block of miRNA processing | CML,BC CML | upregulation | 9, 10, 12 |
| GSK-β | mis-spliced mRNA | |||||
| MDM2 | mRNA stabilization through 3’UTR modification | |||||
| INF-ɣ pathway | immune response activation | |||||
| β-catenin, TCF-4, CCND2 | WNT activation | AML | 8 | |||
| not studied | high ADAR1 expressors were in standard-risk groups | pediatric ALL | 7 |
Abbreviations used: Acute myeloid leukemia (AML); acute promyeloblastic leukemia (APL); acute megakaryoblastic leukemia (AMKL); acute lymphoblastic or lymphocytic (ALL); B-cell acute lymphoblastic leukemia (B-ALL); adult T-cell leukemia/lymphoma (ATL); diffuse large B-cell lymphomas (DLBCLs); chronic myeloid leukemia (CML), chronic phase (CP), accelerated phase (AP), blast crisis (BC); chronic lymphoblastic or lymphocytic leukemia (CLL); myelodysplastic syndromes (MDS); multiple myeloma (MM); hepatocellular carcinoma (HCC); leukemia stem cell (LSC); wild type (WT); patient-derived xenograft (PDX); bone marrow (BM); hematopoietic stem/progenitor cells (HSCs, HSPCs).
ADAR1’s p150 isoform was upregulated in BC CML compared to chronic phase (CP) CML and normal cord blood progenitors9. Forced expression of the p150 ADAR1 isoform in CP CML cells increased production of a misspliced form of GSK3 β implicated in leukemia stem cell (LSC) self-renewal, while ADAR1 knockdown impaired self-renewal capacity in BC CML as examined by serial in vivo transplantation9. A comprehensive mechanistic study of ADAR1 functions in LSCs demonstrated JAK2- and BCR-ABL1-dependent activation of ADAR1-mediated RNA editing, which in turn inhibits let-7-mediated differentiation of CML blasts10. Because deregulation of RNA editing is associated with progression and therapeutic resistance of CML, Catriona Jamieson’s group proposed ADAR1 as an important biomarker of CML progression and developed a clinically relevant assay for RNA editing quantification11.
ADAR1-mediated editing influences gene expression by changing both mRNA stability and miRNA expression. Jiang Q et al. showed that A-to-I editing stabilizes MDM2 transcript through modification of miR-155 binding sites within its 3′ UTR region and downregulation of pri-miR-15512, Figure 1 (A, C, I), Table 1. The biological consequences of non-coding RNA editing are likely to be cell type- or context-dependent, contingent on the signaling pathways they target. For example, A-to-I edits inhibiting biogenesis of the tumor suppressor miR-26a enhance proliferation of normal blood progenitors, but slow down the cell cycle transition in BC CML12.
Figure 1, RNA-binding proteins involved in leukemogenesis.
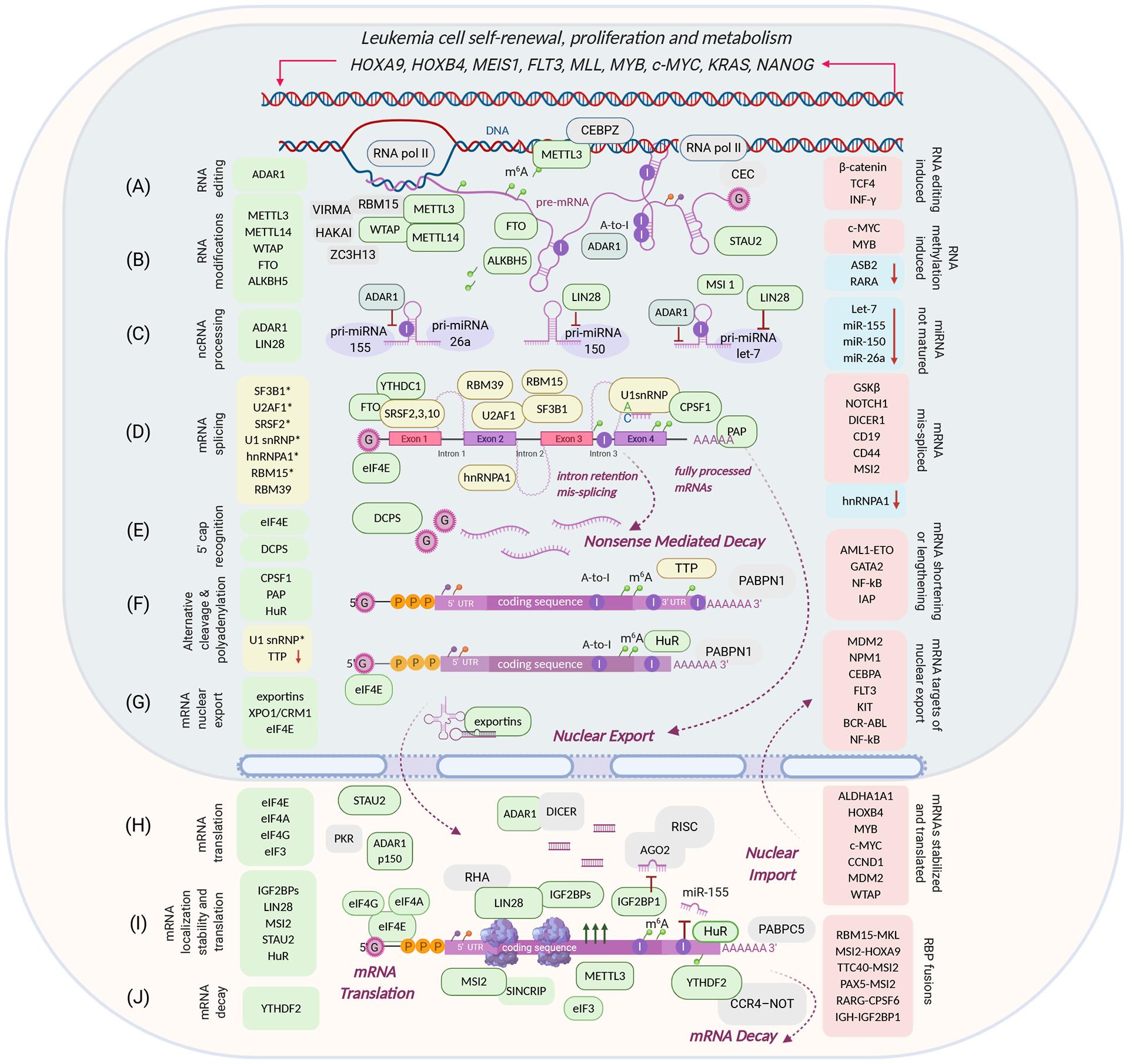
RNA-binding proteins are listed on the left side of the diagram: upregulation, gain-of-function (green), loss- or change-of-function (yellow, arrow down, *mutation). Target genes are listed on the right side of the diagram: upregulated oncogenes (red boxes), tumor suppressor gene inactivation (blue boxes, arrow down). (A) ADAR1 regulates miRNA biogenesis in an A-to-I editing-dependent manner (C), A-to-I editing affects mRNA stability (I); (B) RNA modifying enzymes facilitate m6A methylation (METTL3/14), demethylation (FTO, ALKBH5), substrate recognition (WTAP, RBM15); METTL3 can co-localize with DNA in the nucleus and enhance mRNA translation in the cytoplasm; (C) noncoding RNA processing: ADAR1 and LIN28 suppress maturation of miRNA let-7, miR-155, miR-150, and miR-26a; (D) RNA splicing factors are often mutated in chronic leukemia and/or mis-spliced in acute leukemia producing more mis-spliced pro-oncogenic mRNA isoforms; (E) 5’ cap recognizing enzymes that either promote mRNA nuclear export and translation (eIF4E) or destabilize mRNA (DCPS); (F) alternative cleavage and polyadenylation, occurring during splicing (D), is characterized by mRNA 3’UTRs shortening or lengthening; shorter 3’UTRs increase stability and expression of oncogenic transcripts; downregulation or deactivation of 3’UTR-binding protein TTP increases mRNA abundance; (G) high expression levels of nuclear export regulators (exportins, XPO1/CRM1, eIF4E) increase transport and translation of oncogenic factors; (H) high levels of eIF4E promote nuclear export and translation of selective proto-oncogenic targets; (I) increased mRNA stability and translation of oncogenic transcripts through multiple post-transcriptional events, including reactivation of oncofetal proteins LIN28 and IGF2BPs; (J) m6A reader YTHDF2 targets mRNAs for CCR4-NOT-dependent deadenylation and degradation.
Since a loss of ADAR1 activity induces cell-intrinsic lethality and the induction of cytokines, ADAR1 presents a potentially effective therapeutic target. Gannon et al. suggested possible approaches to disrupt ADAR1 function in cancer cells through inhibition of its adenosine deaminase activity or inactivation of non-enzymatic functions specific for the p150 isoform, such as direct PKR binding13. In accordance with findings describing the immunomodulatory functions of ADAR1, Ishizuka et al. proposed a new strategy for overcoming the resistance to immune checkpoint blockade through ADAR1 inhibition14. Zipeto et al. demonstrated that the previously described inhibitory tool compound 8-azaadenosine (8-aza) reduced ADAR1’s A-to-I editing activity in K562 CML cells10. Multiple studies defining combinatory approaches for ADAR1 inhibition, targeting ADAR-edited transcripts, and immunotherapies suggest a promising future of RNA-editing therapeutics.
RNA modification enzymes
More than 150 types of RNA modifications, ranging from simple methylation or isomerization to more complex multistep chemical transformations, occur co- and post-transcriptionally. Whereas transfer RNA (tRNAs) and ribosomal RNA (rRNA) are the most abundantly modified RNAs in a cell, mRNA is characterized by several modifications including adenosine methylation (N6-methyladenosine (m6A)), which is the most prevalent modification of eukaryotic messenger and long non-coding RNAs15.
m6A’s installation, recognition, and removal are facilitated by protein factors called writers, readers, and erasers, respectively. The main writer is a multicomponent complex that consists of a catalytic methyltransferase-like 3 (METTL3) subunit, a substrate-recognizing subunit METTL14, and other cofactors (WTAP, RBM15/15B, VIRMA, HAKAI, and ZC3H13) that enable adenosine methylation. Another writer installing m6A in RNA sequences in a structure dependent manner is METTL1616. m6A readers (e.g. YTHDCs, YTHDFs, hnRNPs, IGF2BPs) recognize m6A modifications while conveying transcripts’ processing, stability, and translation. The removal of m6A is catalyzed by two erasers: fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5).
RNA modifications influence gene expression by changing RNA secondary structure and folding, consequently affecting functional RNA-RNA and RNA-protein interactions. For example, m6A eraser FTO and nuclear reader YTHDC1 modulate splicing factor activity and exon inclusion17. The levels of m6A RNA modifications have a remarkable effect on cell fate, but this effect is dependent on cellular context16. In fact, METTL3-METTL14 were reported as a tumor suppressor or oncogene in glioblastoma, a tumor suppressor in endometrial cancer, and an oncogene in lung cancer and acute myeloid leukemia18.
METTL3-METTL14 core subunits
Two distinct genetic screens conducted by Barbieri et al. identified METTL3 as an essential gene for AML cell growth. Downregulation of METTL3 resulted in cell cycle arrest, differentiation of leukemic cells, and failure to establish leukemia in immunodeficient mice19. In agreement with these data, Vu et al. demonstrated that shRNA-mediated depletion of METTL3 in human hematopoietic stem/progenitor cells (HSPCs) and AML cell lines promotes cell differentiation, coupled with reduced cell proliferation and induction of apoptosis20. Weng et al. reported that a key component of m6A methyltransferase complex, METTL14, is highly expressed in both normal HSPCs and AML cells carrying t(11q23), t(15;17), or t(8;21)21. METTL14 depletion promoted terminal myeloid differentiation of normal HSPCs and AML cells and inhibited AML tumorigenicity. Therefore, both METTL3 and METTL14 are required for AML sustainability.
Single-nucleotide-resolution mapping of m6A combined with ribosome profiling showed that m6A promotes the translation of c-MYC, BCL2, and PTEN mRNAs in the human AML MOLM-13 cell line, Table 2. Loss of METTL3 led to increased levels of phosphorylated AKT that supported differentiation upon METTL3 depletion20. Similarly, METTL14 exerts its oncogenic role by regulating m6A mRNA modifications and mRNA stability of master regulators of self-renewal and differentiation (e.g., MYB and MYC), whereas its expression levels are negatively regulated by myeloid transcription factor SPI121.
Table 2.
RBPs involved in RNA modification
| Gene | Protein/RNP function | Target Genes | Biological consequences | Type of cancer | Expression in cancer | Ref |
|---|---|---|---|---|---|---|
| METTL3 | RNA hypermethylation, m6A writer | c-MYC, BCL2, PTEN | promotes oncogenes translation | AML MOLM-13 | upregulation | 20 |
| RNA m6A writer, DNA promoter binding through binding CEBPZ | global | methylation of coding regions of mRNAs, m6A-dependent translation, relieving ribosome stalling | AML | 19 | ||
| Attenuate translation, cytoplasmic localization | WTAP | translation, interaction with eIF3, upregulated expression | K562, HeLa | 23 | ||
| METTL14 | RNA recognition | MYB, MYC | enables m6A methylation by METTL3, regulates self-renewal and differentiation | AML (LSCs) | upregulation | 21 |
| WTAP | RNA recognition | global analysis, transcription and RNA processing genes | promotes m6A methylation by METTL3 enables METTL3 nuclear localization | HEK293 cells, HeLa | upregulation | 24 |
| CD4, CD44, CEBPA, CSF1R, MPO, ABCG2, TCL1A, CYP1A1, CYP3A4, FGFR1, PTPRC (CD45), CD83, CD86, CD9 and CCR4. | abnormal proliferation and arrested differentiastion | AML, HL-60, K562 | 25 | |||
| RBM15 | RNA recognition | genes on the X chromosome | transcriptional silencing by lncRNA XIST | n/a | n/a | 26 |
| FTO | RNA hypomethylation, m6A eraser | NANOG, SOX2 WNT signaling | stem cell genes, oncogenes upregulated | AML: MLLr, PML-RARA, FLT3-ITD, NPM1 mut. | upregulation | 28 |
| ASB2, RARA | differentiation factors are downregulated | |||||
| immune checkpoint genes e.g. LILRB4 | immune evasion | AML (LSCs) | 29 | |||
| U1, U2, U6 snRNAs | demethylation, FTO inhibition leads to altered splicing | human TF-1 erythroleukemia cells | tumor suppressor? | 31 | ||
| ALKBH5 | RNA hypomethylation, m6A eraser | TACC3 | functions as an oncogene in AML regardless of TP53 mutation status; significantly associated with shorter overall survival and poor prognosis in AML, similar to solid tumors | NOMO-1 (TP53-mutant), MV4;11 (TP53-WT) and MA9.3-ITDcells (TP53-WT), in vivo | upregulation | 32 |
| promotes LSCs self-renewal through MYC-p21 axis | AML (LSCs) | |||||
| receptor tyrosine kinase AXL | AXL mRNA stability in m6A-dependent manner; MYB, Pol II activity | AML (LSCs) | 33 | |||
| YTHDF2 | m6A reader, cytoplasmic, targets mRNA for degradation | TNFR2 | Inhibition of apoptosis, enhanced self-renewal | AML (LSCs) | upregulation | 34 |
Abbreviations used: Acute myeloid leukemia (AML); acute promyeloblastic leukemia (APL); acute megakaryoblastic leukemia (AMKL); acute lymphoblastic or lymphocytic (ALL); B-cell acute lymphoblastic leukemia (B-ALL); adult T-cell leukemia/lymphoma (ATL); diffuse large B-cell lymphomas (DLBCLs); chronic myeloid leukemia (CML), chronic phase (CP), accelerated phase (AP), blast crisis (BC); chronic lymphoblastic or lymphocytic leukemia (CLL); myelodysplastic syndromes (MDS); multiple myeloma (MM); hepatocellular carcinoma (HCC); leukemia stem cell (LSC); wild type (WT); patient-derived xenograft (PDX); bone marrow (BM); hematopoietic stem/progenitor cells (HSCs, HSPCs).
In addition to previously described methyltransferase (MTase) dimer, Barbieri et al. proposed a METTL14-independent mode of METTL3 function through interaction with chromatin19. The study showed that CAATT-box binding protein CEBPZ recruits METTL3 to the promoters of actively transcribed genes, Figure 1 (B). The promoter bound METTL3 induces m6A modification within the coding region of the associated mRNA transcripts which enhances their translation by relieving ribosome stalling. These observations are relevant to Huang et al. discovery that Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs), known to bind and stabilize coding regions of oncogenic transcripts, are m6A readers22.
METTL3 catalytic activity in the nucleus has a predominant effect on the fate of downstream targets. However, METTL3 can also locate in the cytoplasm and promote the translation of specific mRNAs as a reader. High cytoplasmic levels of METTL3 result in an increase of WTAP protein expression, which might work as a self-regulatory feedback loop necessary for sustaining MTase levels in myeloid leukemia23.
WTAP and RBM15 regulatory subunits
Initially considered as a splicing factor, RNA-binding protein Wilms tumor 1-associated protein (WTAP) has no methyltransferase activity. As a MTase co-factor, WTAP interacts with METTL3 and METTL14, and is required for their recruitment into nuclear speckles. In the absence of WTAP, the RNA-binding capability of METTL3 and m6A levels are strongly reduced, suggesting that WTAP regulates its recruitment to mRNA targets24. Around 30% of AML samples, especially those with FLT3-ITD and NPM1 mutations, show WTAP upregulation, which possesses oncogenic properties in cooperation with functional METTL323, 25.
RNA-binding motif 15 (RBM15) is a multifunctional RBP with an essential role in development and normal and malignant hematopoiesis. As a MTase regulatory subunit, RBM15 binds and recruits the METTL3-METTL14 complex to specific sites of coding and non-coding RNAs26, Table 2. As a splicing factor, RBM15 regulates pre-mRNA splicing of key erythro-megakaryocytic regulators (GATA1, RUNX1, TAL1 etc.) by recruiting SF3B1 splicing complex to intronic regions, Table 3. Perturbations in RBM15 expression are common for infant acute megakaryoblastic leukemia (AMKL), and can potentially be rescued by inhibiting PRMT1 which determines RBM15 protein methylation and stability27.
Table 3.
RBPs involved in mRNA splicing
| Gene | Protein/RNP function | Target Genes | Biological consequences | Type of cancer | Expression in cancer | Ref |
|---|---|---|---|---|---|---|
|
SF3B1 U2AF1(35) SRSF2 ZRSR2 SF3A1 PRPF40B |
splicing factors 3’ splice site recognition in pre-mRNA | 17 genes NMD (SMG1,5,6,7,8 9, DHX34, UPFs, BTZ, Y14, PYM, hNAG, MAGOH, eIF4A3). | apoptosis, G1/M phase arrest, compromise reconstitution capacity | MDS, HeLa | loss or change-of-function due to mutations and mis-splicing | 44 |
| U1 snRNP | splicing factor 5’ splice site recognition thru U1 snRNA:pre-mRNA base pairing | MSI2, POLD1, CD44, ABCD3, global splicing | mis-splicing, intron retention;downregulated genes related to apoptosis, more aggressive CLL | CLL, HCC | mutations in canonical U1 snRNA genes, change-of-function | 58 |
| hnRNPA1 | splicing factor | DICER, NT5C2, global splicing, RNA metabolism | global mis-splicing, BM failure | pediatric B-ALL | loss or change-of-function due to mis-splicing | 49 |
| RBM15 | RNA splicing, erythro-megakaryocytic lineage factors | GATA1, RUNX1, MPL, TAL1 RBM15-MKL1 | altered splicing, abolished megakaryocytic differentiation | AMKL | deletions, fusion (tumor suppressor?) | 27 |
| RBM39 | splicing factor | HOXA9 transcriptional targets | RBM39 inactivation leads to mis-splicing and downregulation of GATA2, BMI-1, MYB | AML, MOLM-13 (MLL-AF9, FLT3ITD), K562 | non-oncogenic “addiction”, upregulated | 50 |
| DCPS | decapping | spliceosomes, transcription, export, nuclear pore complexes | DCPS inactivation causes pre-mRNA mis-splicing, induces a type I interferon response in AML | CALM/AF10 or MLL/AF9 leukemia, AML MOLM-13, AML PDX | AML dependency, upregulated | 51 |
Abbreviations used: Acute myeloid leukemia (AML); acute promyeloblastic leukemia (APL); acute megakaryoblastic leukemia (AMKL); acute lymphoblastic or lymphocytic (ALL); B-cell acute lymphoblastic leukemia (B-ALL); adult T-cell leukemia/lymphoma (ATL); diffuse large B-cell lymphomas (DLBCLs); chronic myeloid leukemia (CML), chronic phase (CP), accelerated phase (AP), blast crisis (BC); chronic lymphoblastic or lymphocytic leukemia (CLL); myelodysplastic syndromes (MDS); multiple myeloma (MM); hepatocellular carcinoma (HCC); leukemia stem cell (LSC); wild type (WT); patient-derived xenograft (PDX); bone marrow (BM); hematopoietic stem/progenitor cells (HSCs, HSPCs).
FTO and ALKBH5 m6A erasers
The m6A eraser FTO is upregulated in AML with the mixed lineage leukemia (MLL) gene rearrangements, PML-RARA, FLT3-ITD, and/or NPM1 mutations28. The molecular analysis of FTO gain-of-function in MLL-rearranged MONOMAC-6 cells identified significantly up- and downregulated hypomethylated mRNAs. The upregulated hypomethylated RNA messengers were enriched in stem cell genes (NANOG, SOX2) and WNT-signaling, while most of the downregulated hypomethylated transcripts belonged to the interferon signaling and genes of the immune system. Ultimately Li et al. showed that FTO enhances leukemogenesis and inhibits all-trans-retinoic acid (ATRA)-induced AML cell differentiation by regulating expression of ASB2 and RARA through reducing m6A levels in these mRNAs28, Figure 1 (B), Table 2.
Subsequently, this research group conducted a massive search for FTO inhibitory compounds, followed by in vitro mRNA target validation and in vivo studies of two highly effective FTO inhibitors, CS1 and CS229. Other inhibitors, namely FB23 and FB23–2, which selectively block FTO m6A demethylase activity, were recently described by Huang et al.30 Similar to genetic depletion, FTO pharmacologic targeting dramatically suppressed proliferation and promoted differentiation of AML cell lines and primary blast cells in vitro and in mouse models. Su et al. demonstrated that in addition to self-renewal and cell cycle control FTO regulates expression of immune checkpoint genes of the LILRB4 family overexpressed in AML. Therefore, FTO inhibition suppressed LILRB4 and sensitized leukemia cells to T cell cytotoxicity29. Given recent findings by Mauer et al. that FTO mediates modifications of small nuclear RNAs (U1, U2, U6 snRNAs) involved in mRNA splicing31, FTO inhibitors may have a broad effect on gene expression.
Two independent studies showed that another RNA demethylase, alkB homologue 5 protein (ALKBH5), is highly expressed in AML32, 33. Wang et al. demonstrated that ALKBH5 transcription is activated by H3K9me3 demethylase KDM4C, and proto-oncogene MYB33. Shen et al. focused on the role of ALKBH5 in mRNA stability and identification of the direct mRNA targets by integrative omics studies of RNA-seq, m6A-seq, and ALKBH5-RNA immunoprecipitant’s sequencing32. Ultimately, both groups illustrated that ALKBH5 selectively supports leukemia stem cell proliferation, metabolism, and self-renewal by regulating essential factors of cell division and kinase signaling such as TACC3 and AXL, Table 2.
YTHDF2 m6A reader
m6A writers and erasers determine the specifics of cis-acting RNA regulatory elements that are recognized and functionally interpreted by m6A readers. Among three cytoplasmic YT521-B homology (YTH) domain family of proteins (YTHDF1, 2, and 3), YTHDF2 targets m6A labeled mRNAs for degradation. Conversely, YTHDF1 and 3 promote translation. Other YTHD readers include nuclear YTHDC1, which regulates splicing and targets some mRNAs for nonsense mediated decay, and cytoplasmic YTHDC2 promoting translation.
Paris et al. reported that YTHDF2 levels are significantly increased in cytogenetically diverse human AML. Importantly from a potential therapeutic standpoint, inactivation of YTHDF2 in AML selectively kills LSCs (most likely by modulating essential regulators of apoptosis) but stimulates expansion of normal HSCs34.
Chemical modulation of m6A RNA methylation
Targeting abnormally overexpressed regulators of RNA methylation has emerged as a promising therapeutic strategy. Within the writer complex, RNA-binding subunit METTL3 is a key m6A methyltransferase containing a targetable S-adenosyl-L-methionine (SAM)-binding pocket. Several biotechnology companies have begun development of METTL3 inhibitors with prospective clinical trials starting in 2021–202235. m6A erasers FTO and ALKBH5 belong to the 2-oxoglutarate and iron-dependent oxygenases respectively, and are sensitive to certain conventional inhibitors, e.g. 2OG competitor succinate and the metal chelator flavonoid15. Because FTO negatively regulates ATRA pathway through ASB2 and RARA, FTO inhibitors can potentially supplement ATRA treatments in myeloid leukemia. Solving crystal structures of these proteins will further aid in the design of selective inhibitors that have high therapeutic potential. However, the physiological consequences of m6A mRNA methylation are context-dependent and may have the opposite effect in different tissues. Another question is why writers and erasers, enzymes with the opposite effects on RNA methylation, both have oncogenic properties. It will be important to understand how cancer cells gain advantage from hundreds of oncogenes and tumor suppressors being simultaneously methylated or demethylated. Elucidating these mechanisms and biological consequences of altering RNA modifications will be critical for the successful clinical implementation of RNA methylation-based therapies.
mRNA splicing
The precursors of eukaryotic mRNA, pre-mRNAs, contain introns that should be excluded from matured RNA messengers. Intron removal happens through splicing, which is carried out by the spliceosomes acting at the regulatory splicing sites in nascent pre-mRNA. Multiple mRNA’s isoforms are usually produced from a single gene by differential exon usage during alternative splicing (AS). Cancer cells often express differentially spliced or aberrant cancer-specific isoforms favoring clonal expansion and survival.
The preferential assembly of the anti-apoptotic long isoform of B-cell lymphoma (BCL-2) gene, and anti-apoptotic short Caspase 9 protein are canonical examples of how acute and chronic myeloid leukemia cells utilize alternative splicing to acquire chemoresistance36. Along with the selective expression of physiologically normal variants, around 30% of differentially expressed transcripts in cancer cells contain products of abnormal splicing. Those events include atypical usage of exons (cassette exon), intron retention, and a disruption of functional open reading frames37. A genome-wide study showed that equal proportions of oncogenes and tumor suppressors are recurrently mis-spliced in AML38. However, distinct sets of splicing-related mutations affect expression of tumor suppressors and oncogenes39. For instance, intron retention, a widespread splicing alteration across various cancers, is a common mechanism for tumor suppressor inactivation40. Although most aberrantly spliced transcripts undergo degradation via nonsense mediated decay, and not all protein products of mis-splicing are equality important for cancer development and progression, clonal enrichments with cancer-specific variants driving chronic myeloid41 and lymphoid42 leukemia as well as the acquired resistance to CAR19 therapies in childhood B-ALL43 were previously described.
The fidelity of canonical splicing hinges on the structural and functional integrity of spliceosomal subunits U1, U2, U4, U5, and U6 snRNPs (five snRNA and around 50 proteins), regulatory RNA sequences in splicing sites flanking introns at 5’-(GT/U) and (AG)-3’, the intronic branch nucleotide adenine (A), exonic or intronic splicing silencers, and enhancers.
In 2011, Kenichi Yoshida et al. were among the first who described the importance of splicing factors (SF) for the pathogenesis of myelodysplasia44. Recurrent mutations in six components of the splicing machinery (SF3B1, U2AF1, SRSF2, ZRSR2, SF3A1, and PRPF40B) were found in about 55% of cases, Figure 1(D), Table 3. Importantly, the heterozygous mutations occurred in a mutually exclusive manner, indicating that the functional splicing factors are required for cell survival.
Among more than 150 proteins involved in splicing, 4 factors (SF3B1, U2AF1, SRSF2, and ZRSR2) are altered most commonly (comprehensively reviewed by Taylor and Lee45). Splicing Factor 3b Subunit 1 (SF3B1, 155 KDa subunit) gene is the most commonly mutated splicing factor in human cancer. It encodes the largest of seven subunits of the SF3B complex, which plays a key role in U2 snRNP positioning to the branchpoint site46. Mutations in the SF3B1 gene are present in about 10% to 20% of acute myeloid and lymphoid leukemia, but are significantly enriched in chronic myeloid malignancies, especially in refractory anemia with ring sideroblasts (RARS)45. Displaying up to 80% frequency for K700E substitution, SF3B1 mutations are likely to be early genetic events in RARS and are associated with favorable prognosis. Conversely, SF3B1 mutations are the subclonal events in chronic lymphoblastic or lymphocytic leukemia (CLL) tumors and linked to poor clinical outcomes. U2 small nuclear RNA auxiliary factor 1 (U2AF1, 35 kDa subunit) is also a core component of the spliceosome that, together with its partner U2AF2, recruits U2 snRNP to the branch site of pre-mRNA. U2AF1 mutations can be found in 10–15% of patients with non-RARS MDS, chronic myelomonocytic leukemia (CMML), and secondary AML (s-AML). Serine and Arginine-(R) Rich Splicing Factor 2 (SRSF2), binds to splicing enhancers and promotes splicing by recruiting a core spliceosome. SRSF2 mutations were found in 50% of CMML cases and in 15–20% of MDS and s-AML cases. The haploid, presumably loss-of-function mutations in the ZRSR2 gene located at Xp22.1, are found in 5–10% patients with MDS, and are more common in males45.
Detailed analysis of SF protein structure showed that the hotspot mutations loosen the strength of the canonical protein-protein and RNA-protein interactions therefore provoking catalytic reactions in otherwise atypical regions. For example, mutations in SF3B1 HEAT domains (HR4-HR7) have a major impact on the formation of the SF3B1 RNA-binding platform. Changes in SF3B1 tertiary structure lead to selection branchpoint sequences with a greater complementarity to U2 snRNA, a shift in the spliceosome position, and usage of cryptic 3’ splicing sites upstream of the canonical site46.
The analysis of SF3B1 mutations in primary human CLL revealed dysregulation of multiple cellular pathways including DNA damage response, telomere maintenance, and Notch signaling47. Although mis-splicing alters multiple mRNAs, dysfunction or inactivation of some factor are critical to disease development. Kim et al. identified a direct connection between SRSF2 P95 mutation, EZH2 mis-splicing and inactivation, and myelodysplasia development. Importantly, restoring EZH2 expression partially rescued hematopoiesis in Srsf2 mutant cells48.
Aberrant splicing can be a feature of leukemic cells without genomic mutations in splicing factors and is likely a result of mutations in cis- and trans-acting RNA elements or the upstream regulators of splicing. Pediatric B-cell malignances lacking genomic mutations of SF display global mRNA mis-splicing, including approximately 100 splicing regulators when compared to normal B-cells49. One of the mis-spliced factors, hnRNPA1, plays an important role in RNA metabolism. The knockdown of hnRNPA1 in B-lymphoblastoid cells initiated a broad change in hnRNPA1-regulated exon usage and production of atypical splice variants of cancer drivers including DICER1 and NT5C249.
Although a number of in vitro and in vivo studies failed to demonstrate a uniform capacity of RBPs to initiate leukemia, three independent genome-wide studies found RBPs indispensable for leukemia sustainability. The CRISPR/Cas9-based library designed by Wang et al. targeted RNA-binding domains of 490 classical RBPs50. The screen identified a network of physically interacting RBPs upregulated in AML, and the RNA splicing factor RBM39 as one of the key factors of AML dependency. RBM39 is required for efficient splicing of many mRNAs, including the HOXA9 transcriptional targets; therefore, genetic or chemical inhibition of this splicing factor caused preferential lethality of cells with spliceosomal mutant AML. The second study by Yamauchi et al. employed a genome-wide CRISPR-Cas9 screening using AML cell lines followed by a second screen in vivo. The screening identified mRNA decapping enzyme scavenger (DCPS) as being essential for AML cell survival, interacting with components of pre-mRNA metabolic pathways including spliceosomes51, Figure 1 (E), Table 3. Finally, a genome-wide in vivo CRISPR/Cas9 screen in BCR-ABL/NUP98-HOXA9-driven CML mouse model showed a significant enrichment with RBPs (~680 genes), suggesting a “disproportionate dependency” on RBPs in myeloid leukemias. In this study, Bajaj et al. identified dsRNA-binding protein Staufen2 (Stau2) as an essential regulator of chromatin modifiers52. The gene expression analysis identified KDM family of H3K4 demethylases being downstream targets of Stau2, Table 6. The biological effects of genetic and pharmacologic inhibition of KDM1A suggest its potential therapeutic value in BC CML.
Alternative cleavage and polyadenylation
Given the important functions of 3’UTR in regulating mRNA fate, mRNAs can be polyadenylated at alternative sites, which, similar to splicing, results in RNA messengers harboring 3’UTRs of different size and content. Notably, global 3’UTR shortening and high expression levels of cleavage and polyadenylation factors, often indicated as alternative polyadenylation (APA), are common for fast proliferative and cancer cells53, 54.
A significant increase in the cleavage and polyadenylation specificity factor 1 (CPSF1) expression was found in t(8;21) AML at diagnosis, and was associated with the short 3’UTR in fusion AML1-ETO transcript. CPSF1 knockdown led to the extension of AML1-ETO 3’UTR, decreased fusion mRNA expression and suppression of leukemia cell growth55. Data analysis of singe cell RNA-seq of 16,843 bone marrow mononuclear cells from healthy donors and AML patients shows that NF-κB, GATA2, and IAP-family genes exhibit APA dynamics specific for altered differentiation and proliferation of leukemic cells56.
The U1 snRNP is an essential component of a spliceosome. Independently from its role in splicing, U1 snRNP plays an important role in controlling premature cleavage and polyadenylation by inhibiting the recognition of proximal and cryptic intronal polyadenylation sites (termed telescripting)57. Because the base pairing between U1 snRNA, a component of U1 snRNP, and pre-mRNA is necessary both for splicing and telescripting, U1 snRNP deficiencies cause global mis-splicing58 and 3’UTR shortening59, Tables 3, 4. The A>C mutation of U1 snRNA was found in eight out of 78 (10.3%) cases of CLL and other types of cancer58, Figure 1 (D, F), Table 3.
Table 4.
RBPs involved in mRNA polyadenylation
| Gene | Protein/RNP function | Target Genes | Biological consequences | Type of cancer | Expression in cancer | Ref |
|---|---|---|---|---|---|---|
| U1 snRNP | telescripting - inhibits premature cleavage and polyadenylation | global transcription elongation | 3’ UTR shortening, truncated mRNA, increased migration and invasion | HeLa | loss or change-of-function |
59 reviewed in57 |
| PAP | Poly(A) Polymerase | n/a (PAP activity in cell extracts) | PAP activity is higher in acute leukemia than in chronic leukemia | AML, ALL, CML | upregulation | reviewed in54 |
| APA (process) | alternative polyadenylation | NF-kB, GATA2, IAP-family of genes | global RNA shortening or lengthening | BM AML | upregulation | 56 |
| CPSF1 | cleavage and polyadenylation, recruits nuclear export | AML1-ETO | oncogenic mRNA stability | AML | upregulation | 55 |
| HuR | AU-rich RNA binding protein, mRNA stability | elF4E, cEBPβ, p21, FOXO3, MEK1, MEK2, DUSP10, ZFP36L1, MYC | mRNA stability | AML, BC CML | upregulation | reviewed in60 |
| TTP | mRNAs degradation via the exosome or via Xrn1 exonuclease | VEGF, cytokines, c-IAP-2 | proapoptotic function in cancers | BC CML, DLBCL, acute phase ATL | loss-of-function, tumor suppressor | reviewed in 60 |
Abbreviations used: Acute myeloid leukemia (AML); acute promyeloblastic leukemia (APL); acute megakaryoblastic leukemia (AMKL); acute lymphoblastic or lymphocytic (ALL); B-cell acute lymphoblastic leukemia (B-ALL); adult T-cell leukemia/lymphoma (ATL); diffuse large B-cell lymphomas (DLBCLs); chronic myeloid leukemia (CML), chronic phase (CP), accelerated phase (AP), blast crisis (BC); chronic lymphoblastic or lymphocytic leukemia (CLL); myelodysplastic syndromes (MDS); multiple myeloma (MM); hepatocellular carcinoma (HCC); leukemia stem cell (LSC); wild type (WT); patient-derived xenograft (PDX); bone marrow (BM); hematopoietic stem/progenitor cells (HSCs, HSPCs).
Ubiquitously expressed human antigen R (HuR, or ELAV-like protein 1), nucleolin, and tristetraprolin protein (TTP) bind to AU-rich elements within 3’UTRs. HuR and nucleolin stabilize mRNAs and are upregulated in a variety of blood cancers, while TTP function as a tumor suppressor by triggering mRNA decay. TTP downregulation or loss of function, reported in several human malignances including leukemia, is associated with poor prognosis60, Figure 1 (F), Table 4.
Chemical modulation of mRNA processing
Given that splicing and RNA processing enzymes are required for cell survival, cancer cells bearing heterozygous SF mutations are dependent on wild-type alleles and are more susceptible to chemical compounds inhibiting spliceosome activity.
The first clinical trials of bacteria-derived chemicals targeting the SF3B complex (spliceostatin A, pladienolide (E7107), and GEX1) did not take into consideration the mutational status of splicing factor genes and presented severe side effects61. Since then, a significant scientific effort has been committed to understanding the spliceosome structure and catalytic activity for the rational design of efficient SF3B inhibitors. Recent work by Michael Seiler and colleagues describes an orally available modulator of the SF3B complex, H3B-8800, which potently and preferentially destroys spliceosome-mutant epithelial and hematologic tumor cells62. The safety, pharmacokinetics and pharmacodynamics of H3B-8800 might be evaluated by the end of 2020, when a phase 1 clinical trial (NCT02841540) in patients with myeloid malignancies carrying spliceosomal mutations is completed.
Whereas the majority of known spliceosome inhibitors target the SF3B complex, sulfonamide-containing compounds were shown to induce the proteasomal degradation of the accessory RNA-splicing factor RBM39. The anti-cancer properties of the molecules indisulam, E7820, and chloroquinoxaline sulfonamide have been known for decades, but the mechanism of their action through inhibiting splicing was only recently discovered63. Another example of possible drug repurposing is the DCPS inhibitor RG3039. A dibasic lipophilic molecule was originally developed to treat spinal muscular atrophy, and its anti-leukemic effect has been recently reported51.
The post-translational modifiers protein arginine methyltransferases PRMT1 and PRMT5, are very promising targets for cancer treatment. These enzymes catalyze arginine methylation on many cellular proteins including histones and cooperate with oncogenic drivers and fusion proteins in promoting cancer. The selective PRMT1 inhibitors (e.g., GSK3368715 and MS023) and PRMT5 inhibitors (e.g., GSK3203591 and GSK3326595) showed a significant synergistic anti-leukemic effect in myeloid malignances64, 65. Mechanistically, a global deficiency of arginine methylation dramatically increased aberrant exon-skipping events64. This suggests that the spliceosomal mutant cancers could be the right category for treatments with PRMTs inhibitors. Indeed, distinct PRMT inhibitors preferentially killed Srsf2-mutant AML compared to the wild type cells65. In addition to spliceosome-mutant cancers, a loss of metabolic regulator MTAP has been shown to increase sensitivity to PRMT1 or a combinatory treatment with PRMT1,5 inhibitors64. The safety, tolerability, and pharmacokinetics of PRMTs inhibitors are under clinical investigation.
mRNA nuclear export and translation
Messenger RNPs are exported to the cytoplasm by a conserved export receptor NXF1-NXT1 (TAP-p15) and various adaptor proteins coupled with mRNA splicing66. The general protein export receptor exportin 1 (XPO1/CRM1), does not have a major role in mRNA export, although mRNAs of some proto-oncogenes and cytokines connect to the XPO1-dependent adaptors through AU-rich sequences in their 3′ UTRs66. Exportins (karyopherin-β proteins) play an important role in cancer including hematologic malignances by exporting ncRNAs and tumor-suppressor proteins (p53, NPM1, NFκB). High expression of XPO1 was reported for AML, ALL, CLL, CML, Non-Hodgkin lymphoma, and multiple myeloma (MM), and was linked to poor survival rates67, Figure 1(G), Table 5. Exportin 1 inhibitor selinexor was tested in various types of cancer and is especially successful against AML and MM68.
Table 5.
RBPs involved in nuclear transport and translation.
| Gene | Protein/RNP function | Target Genes | Biological consequences | Type of cancer | Expression in cancer | Ref |
|---|---|---|---|---|---|---|
| XPO1/CRM1 | Exportin 1, ubiquitous nuclear export | protein export p53, NPM1, NFκB | anti-apoptotic properties | AML, ALL, CML, CLL, lymphoma, MM | upregulation | reviewed in67 |
| eIF4E | cap-dependent mRNA nuclear export and translation | CCND1 | delays granulocytic and monocytic differentiation, promotes leukemogenesis | AML, BC CML, ALL | upregulation | 70 |
| MYC, BCL2, BCL6, B-cell receptor signaling, metabolism, and epigenetic regulation | promotes proliferation, aggressiveness | aggressive double- and triple-hit (DH/TH) DLBCL with active Hsp90 stress pathway | 71 |
Abbreviations used: Acute myeloid leukemia (AML); acute promyeloblastic leukemia (APL); acute megakaryoblastic leukemia (AMKL); acute lymphoblastic or lymphocytic (ALL); B-cell acute lymphoblastic leukemia (B-ALL); adult T-cell leukemia/lymphoma (ATL); diffuse large B-cell lymphomas (DLBCLs); chronic myeloid leukemia (CML), chronic phase (CP), accelerated phase (AP), blast crisis (BC); chronic lymphoblastic or lymphocytic leukemia (CLL); myelodysplastic syndromes (MDS); multiple myeloma (MM); hepatocellular carcinoma (HCC); leukemia stem cell (LSC); wild type (WT); patient-derived xenograft (PDX); bone marrow (BM); hematopoietic stem/progenitor cells (HSCs, HSPCs).
Among several factors of the eIF4F complex required for the initiation of canonical cap-dependent translation, cap-binding protein eIF4E stands as the most powerful oncogene capable of transforming normal cells and inducing cancer in mice69. It is believed that eIF4E’s dual capacity of selectively transporting and initiating translation of cell cycle regulators’ mRNA, e.g. Cyclin D1, initiates tumorigeneses70,71, Figure 1(G, H), Table 5. Inhibition of eIF4E-dependent mRNA export with m7G’-cap competitive inhibitor ribavirin was clinically beneficial, and did not cause significant toxicity in AML patients72. In a subsequent study, however, activation of factor GLI1 led to glucuronidation of ribavirin, loss of the eIF4E-ribavirin interaction, and ultimately drug resistance73. Several ongoing clinical studies assess the possibility of treating AML and lymphomas with ribavirin and monitoring cancer progression by Cyclin D1 levels (NCT03760666, NCT03585725).
Multifunctional oncofetal RBPs
Several multifunctional RBPs expressed in stem and progenitor cells during embryonic development are often upregulated in cancers. Although protein structures of MSI2, LIN28, and IGF2BPs do not match the criteria of well-targeted “druggable” peptides, efforts to develop small molecule inhibitors of those proteins have yielded promising results.
Musashi RNA binding protein 2
Musashi RNA-binding proteins 1 and 2 (MSI1, MSI2) belong to a family of RBPs with a pivotal role in embryonic development of multiple species. Among two homologs, MSI2 plays an essential role in normal and malignant hematopoiesis. Overexpression of MSI2 in human umbilical cord blood-derived HSCs led to a 17-fold increase in short-term repopulating cells and 23-fold ex vivo expansion of long-term HSCs74. MSI2 knockout in mice depleted the HSCs number roughly in half, but even more severely abolished activity of LSCs that are dependent on increased levels of MSI275. By mapping MSI2-mRNA binding in myeloid LSCs and normal HSCs, Nguyen et al. showed that significantly more transcripts were bound to MSI2 in cancer cells than in their normal counterparts75. Interestingly, MSI2 was required for maintaining protein levels of key oncogenes (e.g. MYB, HOXA9) rather than their mRNA abundance. These data are in line with the previous studies demonstrating that MSI2 maintains MLL-leukemia self-renewal programs by retaining efficient translation of HOXA9, MYC, and IKZF2, and where IKZF2 plays a key role in inhibiting myeloid differentiation76, 77. A comparative analysis of myeloid LSCs transcriptomes from Msi-deficient mice identified Tspan3, a transmembrane protein mediating signal transduction, as the important factor for leukemia development, propagation, and AML localization in the bone marrow78.
MSI2 pays an essential role in the development and progression of CML as a translocation partner (e.g., MSI2-HOXA9), or in cooperation with other fusions (e.g., BCR-ABL1, NUP98-HOXA9)79, 80. In NUP98-HOXA9-driven BC CML, MSI2 upregulation was accompanied by the increased expression of self-renewal regulators, HOXA9 and HES1, and downregulation of differentiation factor NUMB79. In addition to control of proliferation and differentiation, MSI2 reprograms the metabolic profile of BC CML by regulating BCAT181, Table 6.
Table 6.
Multifunctional oncofetal RNA-binding proteins
| Gene | Protein/RNP function | Target Genes | Biological consequences | Type of cancer | Expression in cancer | Ref |
|---|---|---|---|---|---|---|
| STAU2 | mRNA transport, localization, translation | KDM1A,1B,5B Kras, Wnt, PTEN, KLF6, VHL | chromatin reorganization, global histone methylation | mouse BCR-ABL, NUP98/HOXA9 CML, human BC CML, AML relapse, AML LSCs | upregulation | 52 |
| MSI2 | HSCs self-renewal, multilineage differentiation and engraftment | Ikzf2, Hoxa9, Myc, Meis1 | promoted self-renewal, LSCs survival | AML, K562 CML | upregulation | 76 |
| Tetraspanin 3 TSPAN3 | CXCR4-mediated chemokine responses | mouse AML, BC CML primary human AML | 78 | |||
| NUMB | Suppressed differentiation in BC CML | BC CML | 79, 80 | |||
| BCAT1 | reprogrammed cellular metabolism | CP CML, BC CML, de novo AML | 81 | |||
|
LIN28 LIN28B |
HSCs self-renewal, differentiation, ncRNA processing, mRNA stability and translation | KRAS, c-MYC, HMGA2, let-7 | promotes proliferation | CP, AP and BC CML | upregulation | 84 |
| miR-150 | suppresses miR-150 maturation, stimulates leukemogenesis | MLL-associated AML, 293T | 85 | |||
| LIN28A | pre-B-cell lineage | preleukemic state of highly invasive myeloid leukemia | miR-125b-driven mouse AML | Lin28A is suppressed by miR-125b | 86 | |
| IGF2BP1 | stemness, proliferation, metabolism, mRNA localization, cell adhesion | ETV6/RUNX1 | survival, proliferation | B-ALL | upregulation | 94 |
| IGF2BP1,3 | HOXB4, MYB, ALDH1A1 | leukemia stem cell properties | K562 CML, HL60 AML, 697 B-ALL | 99 | ||
| IGF2BP2 | n/a | poor prognosis | AML | 98 | ||
| IGF2BP3 | MYC, CDK6 | proliferation, survival, B-cell/myeloid programing | MLL-rearranged B-ALL | 95 |
Abbreviations used: Acute myeloid leukemia (AML); acute promyeloblastic leukemia (APL); acute megakaryoblastic leukemia (AMKL); acute lymphoblastic or lymphocytic (ALL); B-cell acute lymphoblastic leukemia (B-ALL); adult T-cell leukemia/lymphoma (ATL); diffuse large B-cell lymphomas (DLBCLs); chronic myeloid leukemia (CML), chronic phase (CP), accelerated phase (AP), blast crisis (BC); chronic lymphoblastic or lymphocytic leukemia (CLL); myelodysplastic syndromes (MDS); multiple myeloma (MM); hepatocellular carcinoma (HCC); leukemia stem cell (LSC); wild type (WT); patient-derived xenograft (PDX); bone marrow (BM); hematopoietic stem/progenitor cells (HSCs, HSPCs).
The small molecule search identified a selective MSI2 inhibitor that reduced disease burden in a murine MLL-AF9 AML model and suppressed growth of human AML82. Ro 08–2750 specifically diminishes MSI2 mRNA-binding capacity and downregulates MSI2 direct translational targets (SMAD3, c-MYC, HOXA9)82. Because the transcription factor HOXA9 regulates MSI2 expression by binding with the MSI2 promoter79, it is expected that the disruption of RNA-protein interaction between MSI2 and HOXA9 mRNA and similar targets will decrease MSI2 levels and weaken the stem cell program in aggressive leukemia.
LIN28 family of proteins
The LIN28 family consists of the two proteins, LIN28A and LIN28B, which play a central role in regulating pluripotency and differentiation by controlling the fate of coding and non-coding RNA. Fetal hematopoietic progenitors express high levels of Lin28b, which, along with IGF2BP3, is at the center of the fetal-to-adult hematopoietic switch83. Viswanathan et al. reported Lin28/LIN28B upregulation in about 15% of primary human tumors and human cancer cell lines84. LIN28 expression was found to be more common in peripheral blood cells from patients with BC CML or in the accelerated phase than in the chronic phase of CML. Mechanistically, LIN28 blocks maturation of the let-7 family of microRNAs that suppress multiple proliferative factors such as HMGA2, K-RAS, and c-MYC84. Jiang et al. discovered a tumor suppressor miR-150 important for MLL-fusion-mediated leukemogenesis, and showed that pri-miR-150/pre-miR-150 maturation is inhibited by the MLL-fusion/c-MYC/LIN28 axis85. Lin28A, however, is required for cell differentiation and is suppressed in murine miR-125b-driven AML86, Table 6.
The molecular basis of LIN28 and let-7 interaction was thoroughly investigated87, 88. Several groups identified compounds disrupting the antagonistic effect of LIN28 on miR-let-7 biogenesis89, 90. Wang et al. utilized the fluorescence polarization assay to identify small-molecule inhibitors for both domains of LIN28 involved in let-7 interactions. Of 101,017 tested compounds, six inhibited LIN28/let-7 binding and impaired LIN28-mediated let-7 oligouridylation. The selective pharmacologic inhibition of individual domains of LIN28 provides a foundation for their therapeutic targeting in leukemia cells and other LIN28-driven diseases91.
IGF2BPs family of proteins
Insulin-like growth factor 2 RNA-binding proteins (IGF2BPs) comprise another RBP family important for embryonic development. The family consists of three members, IGF2BP1, IGF2BP2, and IGF2BP3, where IGF2BP1 and IGF2BP3 display greater structural similarity and are often co-reactivated in cancer92. IGF2BPs regulate mRNA stability and translation of multiple oncogenes (e.g. IGF2, c-MYC, LIN28B, K-RAS) via binding with 5′UTR, 3’UTR and coding regions of messengers92, 93. Of note, m6A RNA modifications increase the affinity of RNA-IGF2BP binding, therefore, IGF2BPs are considered as m6A readers22. Figure 1(I).
IGF2BP1 and IGF2BP3 are upregulated in ETV-RUNX1 B-ALL and MLL-rearranged leukemia, supporting leukemia proliferation through c-MYC and CDK694, 95, Table 6. Being a downstream target of miR-let-7, IGF2BP1 counteracts let-7 and is often co-expressed with LIN28 enhancing leukemia stem cell properties96, 97. Therefore, upregulation of IGF2BP1 and its paralogs is associated with poor survival rates in subsets of leukemia95, 98, 99.
Given the physiological role of IGF2BP1 in stem cell maintenance and development, we recently investigated the impact of IGF2BP1 expression on LSC properties99. We found that IGF2BP1 supports the LSC phenotype by maintaining levels of HOXB4, MYB, and metabolic factor ALDH1A1. The small molecule inhibitor of IGF2BP1, BTYNB, was assayed in multiple cell lines derived from solid tumors100. In our study, BTYNB sensitized myeloid, B-cell, and T-cell leukemia lines to chemotherapeutics, establishing a proof of principal that IGF2BPs could be successfully targeted by small molecules in leukemia cells.
Concluding Remarks
RBPs are a family of proteins playing a central role in normal cell physiology and are crucial for cancer development and progression. Whereas mutations in functional domains of splicing factors could represent early genetic events predisposing to leukemia, a large body of data depicts abnormal RBP activity as a driving force of leukemia progression and an attribute of aggressive forms of disease. Multiple studies indicate that aberrant activity of RBPs is associated with acquisition of cancer stem cell phenotypes fundamental for resistance to therapies, minimal residual disease, and relapse. Therefore, finding ways of effectively targeting major classes of RBPs, discussed in this review, could potentially improve outcomes of leukemia treatments by lowering rates of refractory and relapsed leukemia. Given the association of RBP deregulation with disease aggressiveness and poor clinical outcomes, constructing a pro-LSCs score by assessing spliceosome mutations or mis-splicing, levels of RNA editing/modifications, and oncofetal proteins expression would be a valuable addition to the existing testing platforms.
Novel molecular-genetic tools and mouse models provided compelling evidence of increased dependency of acute myeloid and blast crisis chronic myeloid leukemia on RBPs. Therefore, a search for chemical modulators of RBP activity is rapidly expanding (summarized in Table 7). The first clinical trials of splicing factor inhibitors highlighted the importance of the deep understanding of RBP functions, which are often context dependent. General toxicity and safety concerns remain a hurdle in targeting proteins that are ubiquitously expressed and are present in normal tissues. In this regard, oncofetal RBPs, which are not widely expressed in normal adult tissues, could have a therapeutic advantage. In addition, the genetic background of leukemic cells should be taken into consideration since a mutational status of splicing factors and other genes can increase the susceptibility to RBP inhibitors. Given a supportive role of RBPs in expression of multiple oncogenes, development of relatively nontoxic compounds would be highly beneficial for combinatorial therapies that would, among other effects, allow lower dosages of conventional cytotoxic drugs in older AML patients.
Table 7.
Small molecule inhibitors of RNA-binding and modifying proteins
| Function | Gene Name | Inhibitor | CAS# | References |
|---|---|---|---|---|
| RNA editing |
ADAR1 JAK2 BCR-ABL |
8-azaadenosine SAR302503 Dasatinib |
10299-44-2 936091-26-8 302962-49-8 |
10 |
| RNA modification |
METTL3 METTL14 |
In development | reviewed in15, 35 | |
| FTO | CS1 (Bisantrene) CS2 (Brequinar) |
78186-34-2 96187-53-0 |
29 | |
| FB23/FB23–2 | 2243736-45-8 | 30 | ||
| RNA splicing | SF3B1 | Spliceostatins A-G Pladienolides A(E7107) Herboxidiene (GEX1A) |
391611-36-2 445493-23-2 142861-00-5 |
reviewed in37, 45, 61 |
| RBM39 | E7820 Indisulam Tasisulam |
289483-69-8 165668-41-7 519055-62-0 |
50, 63 | |
|
PRMT1 (type I PRMTs) |
GSK3368715 MS023 (pan type I PRMTs inh.) |
1629013-22-4 1831110-54-3 |
64, 65 | |
| PRMT5 | GSK3203591 (GSK591) GSK3326595 |
1616391-87-7 1616392-22-3 |
64, 65 | |
| Decapping | DCPS | RG3039 | 1005504-62-0 | 51 |
| Nuclear export & translation | XPO1/CRM1 | Selinexor | 1393477-72-9 | reviewed in68 |
| eIF4E | Ribavirin | 36791-04-5 | reviewed in72 | |
| Oncofetal RBPs | MSI2 | Ro 08-2750 (Ro) | 37854-59-4 | 82 |
| LIN28 | C1632 (C15H15N5O) | 108825-65-6 | 89 | |
| TPEN, LI71 | 91 | |||
| IGF2BP1 | BTYNB | 304456-62-0 | 100 |
Our literature review indicates that leukemia cells may experience a systemic deregulation of RNA network affecting multiple cis- and trans- acting RNA regulatory elements. It is apparent that upregulation of various classes of RBPs are required to meet the anabolic demand of fast proliferating cells. The dynamics and synergistic effect of posttranscriptional aberrations in oncogenic transformation has not been fully investigated and understood. Targeting common pathways and regulatory elements that coordinate abnormal activity of various RBPs might be essential for eradicating the most aggressive forms of leukemia and other cancers.
Acknowledgements
This study was supported in part by the NIH NCI grants CA191550 and CA243167 (V.S.S.). The authors want to thank Alexander Elchev and Rachael Mills for editorial help with the manuscript.
Footnotes
Conflict of interest
The authors declare no conflicts of interest.
References:
- 1.Hentze MW, Castello A, Schwarzl T, Preiss T. A brave new world of RNA-binding proteins. Nat Rev Mol Cell Biol 2018. May; 19(5): 327–341. [DOI] [PubMed] [Google Scholar]
- 2.Nishikura K A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 2016. February; 17(2): 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamers MM, van den Hoogen BG, Haagmans BL. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Frontiers in Immunology 2019. July 25; 10(1763). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Q, Khillan J, Gadue P, Nishikura K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 2000. December 1; 290(5497): 1765–1768. [DOI] [PubMed] [Google Scholar]
- 5.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015. September 4; 349(6252): 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.XuFeng R, Boyer MJ, Shen H, Li Y, Yu H, Gao Y, et al. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc Natl Acad Sci U S A 2009. October 20; 106(42): 17763–17768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma CH, Chong JH, Guo Y, Zeng HM, Liu SY, Xu LL, et al. Abnormal expression of ADAR1 isoforms in Chinese pediatric acute leukemias. Biochem Biophys Res Commun 2011. March 11; 406(2): 245–251. [DOI] [PubMed] [Google Scholar]
- 8.Xiao H, Cheng Q, Wu X, Tang Y, Liu J, Li X. ADAR1 may be involved in the proliferation of acute myeloid leukemia cells via regulation of the Wnt pathway. Cancer Manag Res 2019. September 20; 11: 8547–8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang Q, Crews LA, Barrett CL, Chun H-J, Court AC, Isquith JM, et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A 2013. January 15; 110(3): 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun HJ, et al. ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell 2016. August 4; 19(2): 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crews LA, Jiang Q, Zipeto MA, Lazzari E, Court AC, Ali S, et al. An RNA editing fingerprint of cancer stem cell reprogramming. J Transl Med 2015. February 12; 13: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Q, Isquith J, Zipeto MA, Diep RH, Pham J, Delos Santos N, et al. Hyper-Editing of Cell-Cycle Regulatory and Tumor Suppressor RNA Promotes Malignant Progenitor Propagation. Cancer Cell 2019. January 14; 35(1): 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nature Commun 2018. December 21; 9(1): 5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019. January; 565(7737): 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vu LP, Cheng Y, Kharas MG. The Biology of m(6)A RNA Methylation in Normal and Malignant Hematopoiesis. Cancer Discov 2019. January; 9(1): 25–33. [DOI] [PubMed] [Google Scholar]
- 16.Shi H, Wei J, He C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell 2019. May 16; 74(4): 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez NM, Gilbert WV. Pre-mRNA modifications and their role in nuclear processing. Quant Biol 2018. September; 6(3): 210–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han SH, Choe J. Diverse molecular functions of m(6)A mRNA modification in cancer. Exp Mol Med 2020. May 13; 52(5): 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 2017. December 7; 552(7683): 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 2017. November; 23(11): 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m(6)A Modification. Cell Stem Cell 2018. February 1; 22(2): 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 2018. March; 20(3): 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorci M, Ianniello Z, Cruciani S, Larivera S, Ginistrelli LC, Capuano E, et al. METTL3 regulates WTAP protein homeostasis. Cell Death Dis 2018. July 23; 9(8): 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 2014. February; 24(2): 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 2014. May; 28(5): 1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016. September 15; 537(7620): 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Tran N-T, Su H, Wang R, Lu Y, Tang H, et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife 2015. November 17; 4: e07938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017. January 9; 31(1): 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020. July 13; 38(1): 79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019. April 15; 35(4): 677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat Chem Biol 2019. April; 15(4): 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 2020. July 2; 27(1): 64–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Li Y, Wang P, Han G, Zhang T, Chang J, et al. Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell 2020. July 2; 27(1): 81–97. [DOI] [PubMed] [Google Scholar]
- 34.Paris J, Morgan M, Campos J, Spencer GJ, Shmakova A, Ivanova I, et al. Targeting the RNA m(6)A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019. July 3; 25(1): 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cully M Chemical inhibitors make their RNA epigenetic mark. Nat Rev Drug Discov 2019. November; 18(12): 892–894. [DOI] [PubMed] [Google Scholar]
- 36.El Marabti E, Younis I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front Mol Biosci 2018. September 07; 5: 80–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Obeng EA, Stewart C, Abdel-Wahab O. Altered RNA Processing in Cancer Pathogenesis and Therapy. Cancer Discov 2019. November; 9(11): 1493–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adamia S, Haibe-Kains B, Pilarski PM, Bar-Natan M, Pevzner S, Avet-Loiseau H, et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin Cancer Res 2014. March 1; 20(5): 1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park S, Supek F, Lehner B. Systematic discovery of germline cancer predisposition genes through the identification of somatic second hits. Nature Commun 2018. July 04; 9(1): 2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jung H, Lee D, Lee J, Park D, Kim YJ, Park WY, et al. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet 2015. November; 47(11): 1242–1248. [DOI] [PubMed] [Google Scholar]
- 41.Abrahamsson AE, Geron I, Gotlib J, Dao K-HT, Barroga CF, Newton IG, et al. Glycogen synthase kinase 3β missplicing contributes to leukemia stem cell generation. Proceedings of the National Academy of Sciences 2009. March 10; 106(10): 3925–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puente XS, Beà S, Valdés-Mas R, Villamor N, Gutiérrez-Abril J, Martín-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015. October 22; 526(7574): 519–524. [DOI] [PubMed] [Google Scholar]
- 43.Asnani M, Hayer KE, Naqvi AS, Zheng S, Yang SY, Oldridge D, et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19. Leukemia 2020. April; 34(4): 1202–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011. September 11; 478(7367): 64–69. [DOI] [PubMed] [Google Scholar]
- 45.Taylor J, Lee SC. Mutations in spliceosome genes and therapeutic opportunities in myeloid malignancies. Genes Chromosomes Cancer 2019. December; 58(12): 889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cretu C, Schmitzova J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, et al. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol Cell 2016. October 20; 64(2): 307–319. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Brooks AN, Fan J, Wan Y, Gambe R, Li S, et al. Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell 2016. November 14; 30(5): 750–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015. May 11; 27(5): 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Black KL, Naqvi AS, Asnani M, Hayer KE, Yang SY, Gillespie E, et al. Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res 2018. November 30; 46(21): 11357–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S, et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019. March 18; 35(3): 369–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamauchi T, Masuda T, Canver MC, Seiler M, Semba Y, Shboul M, et al. Genome-wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-mRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018. March 12; 33(3): 386–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bajaj J, Hamilton M, Shima Y, Chambers K, Spinler K, Van Nostrand EL, et al. An in vivo genome-wide CRISPR screen identifies the RNA-binding protein Staufen2 as a key regulator of myeloid leukemia. Nature Cancer 2020. April 20; 1(4): 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol 2017. January; 18(1): 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Danckwardt S, Hentze MW, Kulozik AE. 3’ end mRNA processing: molecular mechanisms and implications for health and disease. EMBO J 2008. February 6; 27(3): 482–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shima T, Davis AG, Miyauchi S, Kochi Y, Johnson DT, Stoner SA, et al. CPSF1 Regulates AML1-ETO Fusion Gene Polyadenylation and Stability in t(8;21) Acute Myelogenous Leukemia. Blood 2017. December 7; 130(Abstract): 2498–2498. [Google Scholar]
- 56.Ye C, Zhou Q, Hong Y, Li QQ. Role of alternative polyadenylation dynamics in acute myeloid leukaemia at single-cell resolution. RNA Biol 2019. March; 16(6): 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Venters CC, Oh JM, Di C, So BR, Dreyfuss G. U1 snRNP Telescripting: Suppression of Premature Transcription Termination in Introns as a New Layer of Gene Regulation. Cold Spring Harb Perspect Biol 2019. February 1; 11(2): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shuai S, Suzuki H, Diaz-Navarro A, Nadeu F, Kumar SA, Gutierrez-Fernandez A, et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 2019. October; 574(7780): 712–716. [DOI] [PubMed] [Google Scholar]
- 59.Oh J-M, Venters CC, Di C, Pinto AM, Wan L, Younis I, et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nature Commun 2020. January 7; 11(1): 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baou M, Norton JD, Murphy JJ. AU-rich RNA binding proteins in hematopoiesis and leukemogenesis. Blood 2011. November 24; 118(22): 5732–5740. [DOI] [PubMed] [Google Scholar]
- 61.Salton M, Misteli T. Small Molecule Modulators of Pre-mRNA Splicing in Cancer Therapy. Trends Mol Med 2016. January; 22(1): 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 2018. May; 24(4): 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017. April 28; 356(6336): 3755. [DOI] [PubMed] [Google Scholar]
- 64.Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, Adams ND, et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019. July 8; 36(1): 100–114. [DOI] [PubMed] [Google Scholar]
- 65.Fong JY, Pignata L, Goy PA, Kawabata KC, Lee SC, Koh CM, et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019. August 12; 36(2): 194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kohler A, Hurt E. Exporting RNA from the nucleus to the cytoplasm. Nat Rev Mol Cell Biol 2007. October; 8(10): 761–773. [DOI] [PubMed] [Google Scholar]
- 67.Mahipal A, Malafa M. Importins and exportins as therapeutic targets in cancer. Pharmacol Ther 2016. August; 164: 135–143. [DOI] [PubMed] [Google Scholar]
- 68.Talati C, Sweet KL. Nuclear transport inhibition in acute myeloid leukemia: recent advances and future perspectives. Int J Hematol Oncol 2018. October; 7(3): IJH04–IJH04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wendel H-G, Silva RLA, Malina A, Mills JR, Zhu H, Ueda T, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev 2007. December 15; 21(24): 3232–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Topisirovic I, Guzman ML, McConnell MJ, Licht JD, Culjkovic B, Neering SJ, et al. Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol Cell Biol 2003. December; 23(24): 8992–9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Culjkovic-Kraljacic B, Fernando TM, Marullo R, Calvo-Vidal N, Verma A, Yang S, et al. Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood 2016. February 18; 127(7): 858–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Borden KL, Culjkovic-Kraljacic B. Ribavirin as an anti-cancer therapy: acute myeloid leukemia and beyond? Leuk Lymphoma 2010. October; 51(10): 1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zahreddine HA, Culjkovic-Kraljacic B, Assouline S, Gendron P, Romeo AA, Morris SJ, et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014. July 3; 511(7507): 90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rentas S, Holzapfel N, Belew MS, Pratt G, Voisin V, Wilhelm BT, et al. Musashi-2 attenuates AHR signalling to expand human haematopoietic stem cells. Nature 2016. April 28; 532(7600): 508–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nguyen DTT, Lu Y, Chu KL, Yang X, Park SM, Choo ZN, et al. HyperTRIBE uncovers increased MUSASHI-2 RNA binding activity and differential regulation in leukemic stem cells. Nat Commun 2020. April 24; 11(1): 2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Park S-M, Gönen M, Vu L, Minuesa G, Tivnan P, Barlowe TS, et al. Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. J Clin Invest 2015. March; 125(3): 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Park SM, Cho H, Thornton AM, Barlowe TS, Chou T, Chhangawala S, et al. IKZF2 Drives Leukemia Stem Cell Self-Renewal and Inhibits Myeloid Differentiation. Cell Stem Cell 2019. January 3; 24(1): 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kwon HY, Bajaj J, Ito T, Blevins A, Konuma T, Weeks J, et al. Tetraspanin 3 Is Required for the Development and Propagation of Acute Myelogenous Leukemia. Cell Stem Cell 2015. August 6; 17(2): 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ito T, Kwon HY, Zimdahl B, Congdon KL, Blum J, Lento WE, et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature 2010. August 5; 466(7307): 765–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kharas MG, Lengner CJ, Al-Shahrour F, Bullinger L, Ball B, Zaidi S, et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med 2010. August; 16(8): 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature 2017. May 25; 545(7655): 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Minuesa G, Albanese SK, Xie W, Kazansky Y, Worroll D, Chow A, et al. Small-molecule targeting of MUSASHI RNA-binding activity in acute myeloid leukemia. Nature Commun 2019. June 19; 10(1): 2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang S, Chim B, Su Y, Khil P, Wong M, Wang X, et al. Enhancement of LIN28B-induced hematopoietic reprogramming by IGF2BP3. Genes Dev 2019. August 1; 33(15–16): 1048–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nature Genet 2009. July; 41(7): 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiang X, Huang H, Li Z, Li Y, Wang X, Gurbuxani S, et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell 2012. October 16; 22(4): 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chaudhuri AA, So AY, Mehta A, Minisandram A, Sinha N, Jonsson VD, et al. Oncomir miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proc Natl Acad Sci U S A 2012. March 13; 109(11): 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ransey E, Björkbom A, Lelyveld VS, Biecek P, Pantano L, Szostak JW, et al. Comparative analysis of LIN28-RNA binding sites identified at single nucleotide resolution. RNA Biol 2017. December 2; 14(12): 1756–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yamashita S, Nagaike T, Tomita K. Crystal structure of the Lin28-interacting module of human terminal uridylyltransferase that regulates let-7 expression. Nat Commun 2019. April 29; 10(1): 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roos M, Pradere U, Ngondo RP, Behera A, Allegrini S, Civenni G, et al. A Small-Molecule Inhibitor of Lin28. ACS Chem Biol 2016. October 21; 11(10): 2773–2781. [DOI] [PubMed] [Google Scholar]
- 90.Yu C, Wang L, Rowe RG, Han A, Ji W, McMahon C, et al. A nanobody targeting the LIN28:let-7 interaction fragment of TUT4 blocks uridylation of let-7. Proc Natl Acad Sci U S A 2020. March 3; 117(9): 4653–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang L, Rowe RG, Jaimes A, Yu C, Nam Y, Pearson DS, et al. Small-Molecule Inhibitors Disrupt let-7 Oligouridylation and Release the Selective Blockade of let-7 Processing by LIN28. Cell Rep 2018. June 5; 23(10): 3091–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Degrauwe N, Suva ML, Janiszewska M, Riggi N, Stamenkovic I. IMPs: an RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev 2016. November 15; 30(22): 2459–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang X, Zhang H, Guo X, Zhu Z, Cai H, Kong X. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in cancer. J Hematol Oncol 2018. June 28; 11(1): 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stoskus M, Vaitkeviciene G, Eidukaite A, Griskevicius L. ETV6/RUNX1 transcript is a target of RNA-binding protein IGF2BP1 in t(12;21)(p13;q22)-positive acute lymphoblastic leukemia. Blood Cells Mol Dis 2016. March; 57: 30–34. [DOI] [PubMed] [Google Scholar]
- 95.Palanichamy JK, Tran TM, Howard JM, Contreras JR, Fernando TR, Sterne-Weiler T, et al. RNA-binding protein IGF2BP3 targeting of oncogenic transcripts promotes hematopoietic progenitor proliferation. J Clin Invest 2016. April 1; 126(4): 1495–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Busch B, Bley N, Müller S, Glaß M, Misiak D, Lederer M, et al. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic acids research 2016. May 5; 44(8): 3845–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou J, Bi C, Ching YQ, Chooi J-Y, Lu X, Quah JY, et al. Inhibition of LIN28B impairs leukemia cell growth and metabolism in acute myeloid leukemia. Journal of hematology & oncology 2017. July 11; 10(1): 138–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.He X, Li W, Liang X, Zhu X, Zhang L, Huang Y, et al. IGF2BP2 Overexpression Indicates Poor Survival in Patients with Acute Myelocytic Leukemia. Cell Physiol Biochem 2018. December; 51(4): 1945–1956. [DOI] [PubMed] [Google Scholar]
- 99.Elcheva IA, Wood T, Chiarolanzio K, Chim B, Wong M, Singh V, et al. RNA-binding protein IGF2BP1 maintains leukemia stem cell properties by regulating HOXB4, MYB, and ALDH1A1. Leukemia 2020. May; 34(5): 1354–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A Novel IMP1 Inhibitor, BTYNB, Targets c-Myc and Inhibits Melanoma and Ovarian Cancer Cell Proliferation. Transl Oncol 2017. October; 10(5): 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]