

Abstract
Temporal lobe epilepsy (TLE) is the most frequent type of focal epilepsy in adults, typically resistant to pharmacological treatment, and mostly presents with cognitive impairment and psychiatric comorbidities. The most common neuropathological hallmark in TLE patients is hippocampal sclerosis (HS). However, the underlying molecular mechanisms involved remain poorly characterized. The dentate gyrus (DG), one specific hippocampal subarea, structural and functional changes imply a key involvement of the DG in the development of TLE. In this study, a isobaric tags for relative and absolute quantitation (iTRAQ)-based quantitative proteomic technique was performed for the analysis of hippocampal DG obtained from patients with TLE-HS compared to control samples obtained from autopsy. Our proteomic data identified 5,583 proteins, of which 82 proteins were upregulated and 90 proteins were downregulated. Bioinformatics analysis indicated that differentially expressed proteins were enriched in “synaptic vesicle,” “mitochondrion,” “cell-cell adhesion,” “regulation of synaptic plasticity,” “ATP binding,” and “oxidative phosphorylation.” Protein-protein interaction network analysis found a pivotal module of 10 proteins that were related to “oxidative phosphorylation.” This study has investigated proteomic alterations in the DG region of TLE-HS patients, and paved the way for the better understanding of epileptogenesis mechanisms and future therapeutic intervention.
Keywords: temporal lobe epilepsy, hippocampal sclerosis, dentate gyrus, quantitative proteomics, iTRAQ
Introduction
Temporal lobe epilepsy (TLE) is the most frequent type of focal (partial) epilepsy in adults (1). The most common neuropathological hallmark in TLE patients is hippocampal sclerosis (HS), and seizures of TLE patients with HS (TLE-HS) are typically resistant to pharmacological treatment (2). Moreover, TLE-HS patients mostly present with cognitive impairment and psychiatric comorbidities (3), mandating the research for the better understanding of underlying epileptogenesis and progression mechanisms. Initial precipitating injury events, such as febrile convulsions, inflammation, or trauma may be proposed as a driving force in the development of HS-associated TLE (4, 5). HS is characterized pathologically by the segmental loss of pyramidal neurons, granule cell dispersion (GCD), and reactive gliosis in the hippocampus (2). The structural and functional changes of the dentate gyrus (DG), one specific hippocampal subarea, including GCD, mossy fiber sprouting, aberrant neurogenesis, and increased excitation, imply a key involvement of the DG in the development of TLE (6, 7).
Although the structural alterations in TLE-HS have been widely documented, the underlying molecular mechanisms involved, however, remain poorly characterized. Proteomics has been an important methodology for studying the changes of the protein profiles in physiological and pathological conditions, providing comprehensive knowledge about molecular patterns and functional pathway alterations. To date, several proteomic studies have examined the protein changes associated with TLE in the hippocampus, as opposed to specific subregions of the hippocampus. Identified differentially expressed proteins (DEPs) were related to mitochondria and bioenergetics, neuronal excitability, and cytoskeleton (8–10). Given the complex and heterogeneous architecture of the hippocampus, studying whole hippocampal specimens may hinder the detection of the distinct changes seen in DG that can only be explained by molecular changes occurring within DG. Additionally, to our knowledge, few previous studies have performed quantitative proteomics in the DG of TLE-HS patients. Thus, we aim to investigate the molecular pathogenesis mechanisms that may occur in the DG of TLE-HS.
Isobaric tags for relative and absolute quantitation (iTRAQ) is a robust quantitative proteomic technique that can simultaneously analyze up to 8-plex samples, and have been widely applied in clinical research (11). In the present study, iTRAQ-based tandem mass spectrometry was performed to undertake proteomic analysis of hippocampal DG obtained from patients with TLE-HS compared to the control samples obtained from autopsy. Our proteomic approach may shed light on the molecular changes associated with HS in DG, that could be used for a better understanding of the specific pathomechanisms of HS and identifying potential therapeutic targets.
Materials and Methods
Patients and Tissue Preparation
The study was carried out on seven TLE patients subjected to standard hippocampectomy at the Department of Neurosurgery of Xiangya Hospital. All patients provided their comprehensive medical history, and went through a physical examination, brain magnetic resonance imaging (MRI), and electroencephalograph (EEG). The inclusion criteria of TLE were according to our previous research (12). Preoperative MRI and routine postoperative histopathologic examination of resected tissue confirmed HS. Written informed consent was obtained from all enrolled participants. The study was conducted following the guideline for research involving humans, and approved by the Ethics Committee of Central South University, Xiangya School of Medicine and the affiliated Xiangya Hospital.
Hippocampal samples of the control group were obtained from autopsied patients of Xiangya School of Medicine, and the exclusion criteria were patients with neuropsychiatric disorders and abnormities in the central nervous system (CNS) based on clinical history or histological examination.
The resected brain tissues were snap-frozen in liquid nitrogen and then stored at −80°C. Immunohistochemical staining against NeuN (Merck-Millipore, MAB377, 1:1,000) was performed in the brain slices as described before (13), and for Nissl staining, brain slices were placed under agitation in a cresyl violet staining solution. The staining results were used as localization references, an experienced pathologist therefore separated the DG from the rest of the hippocampus by microdissection (Figure 1).
Figure 1.

Isolation of dentate gyrus and immunohistochemical staining. (A). Schematic drawing depicting the dissected dentate gyrus region that was used for the proteomics study from the human hippocampus. The dashed lines indicate the cut area. (B). Immunohistochemical staining against NeuN and Nissl staining indicate marked neuron loss of dentate gyrus. Calibration bar = 2 mm.
iTRAQ-8plex Labeling and Mass Spectrometry
Quantitative 8-plex iTRAQ proteomic analysis was carried out on eight independent protein samples. Total proteins were extracted with iTRAQ lysis buffer, processed by disulfide bond cleavage and reductive alkylation of proteins with DTT and iodoacetamide, and then the proteins were hydrolyzed by trypsin. The pooled samples were labeled using the iTRAQ kit according to the manufacturer's protocol (Applied Biosystems Sciex).
The iTRAQ-labeled samples were mixed and resuspended in buffer A (2% acetonitrile, pH 10) and eluted with a 2–95% linear gradient of buffer B (90% acetonitrile, pH 10) over 90 min with a flow rate of 1 mL/min.
The samples were separated into 20 fractions on a C18 column with the HPLC system (Thermo Fisher Scientific) and analyzed using a Q Exactive Plus hybrid quadrupole-orbitrap Mass Spectrometer (Thermo Fisher Scientific). The mass spectrometry proteomics data are available via ProteomeXchange with identifier PXD023048.
Proteomic Data Analysis and Differentially Expressed Proteins Identification
Tandem mass spectra were searched for computational analysis in the Homo sapiens protein database using the Proteome Discoverer software. The Sequest algorithm was used to search with a fragment ion mass tolerance of 0.02 Da and a parent ion tolerance of 15 PPM. An FDR of <1% and with at least two unique peptides were used in identifying and quantifying proteins. The analysis of covariance (ANCOVA) model was hired for further analysis adjusting for age. The protein threshold was set to achieve 95% confidence, a P-value and fold-change cut-offs were used to classify an upregulated protein (p < 0.01 and fold-change >2) or a downregulated protein (p < 0.01 and fold-change <0.5). A volcano plot and hierarchical cluster analysis were performed with the software R (Version: 3.5.1).
Bioinformatic Analysis
DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/) was used to conduct enrichment analysis of DEPs and module proteins (14, 15), and the underlying mechanism was identified in the context of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and gene ontology (GO) terms, which consisted of cellular components (CC), molecular functions (MF), and biological processes (BP). p-value <0.05 and gene count ≥2.0 were set as the threshold of significance.
A protein-protein interaction (PPI) network was constructed using STRING (https://string-db.org/). Based on the STRING database, the interaction network was then visualized by Cytoscape software 3.0. Moreover, the Molecular Complex Detection (MCODE) app in Cytoscape was utilized to screen the entire PPI network for hub genes modules, with a degree cutoff ≥2, node score cut-off ≥0.2, K-Core ≥2, and Max Depth = 100.
Results
Demographics and Clinical Characters of Individuals
A total of 14 participants (including seven patients with TLE and seven autopsy controls) were recruited to our study. As is shown in Table 1, the age of the patients with TLE ranged from 9 to 42 years old (25.2 ± 9.2 year) with two female cases and five male cases. The duration of having seizures in the TLE patients was 8.8 ± 5.4 years. Of the sampled autopsy tissue (2 female, 5 male), the age of the subjects at death ranged from 14 to 77 years (50.5 ± 19.9 year). The demographic and clinical data of controls are also shown in Table 1.
Table 1.
Clinical and demographic data of patients with MTLE and autopsied subjects (control samples).
| Data | MTLE patients | Autopsied subjects |
|---|---|---|
| Number of samples | 7 | 7 |
| Gender-female | 2 | 2 |
| Age at surgery or autopsy-year (mean ± SD) | 25.2 ± 9.2 | 50.5 ± 19.9 |
| Seizure duration-year (mean ± SD) | 8.8 ± 5.4 | N/A |
| Presence of febrile seizure | 1 | None |
| Family history of epilepsy | 1 | None |
| Postmortem period | n/a | <10 h |
| Changes in central nervous system | Hippocampal sclerosis | None |
Differentially Expressed Proteins Revealed by iTRAQ Analysis
We identified a total of 5,583 proteins by the iTRAQ experiments. A total of 5,234 proteins were quantified, among which 172 proteins were differentially expressed (p < 0.05, fold-change >2 or <0.5), including 82 upregulated and 90 downregulated proteins (Figure 2, Supplementary Table 1). The 10 most DEPs (upregulated and downregulated) are listed in Table 2. The visualization of hierarchical cluster analysis results revealed a distinction in DG region proteins between TLE-HS patients and control samples (Figure 3).
Figure 2.
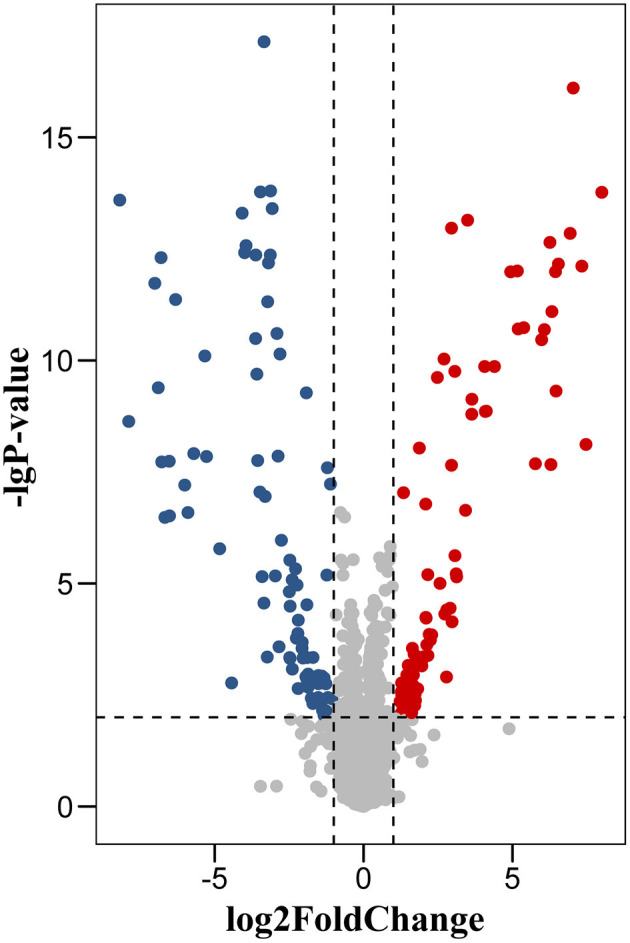
Volcano plot of protein changes in dentate gyrus between TLE-HS patients and control groups. There were 172 proteins with a fold-change of >2 or <0.5 and p-values <0.01 were identified as the significant difference. Red dots represent upregulated proteins, blue dots are downregulated proteins.
Table 2.
Top 10 upregulated and downregulated proteins in MTLE patients compared to controls by iTRAQ.
| Gene | Protein name | Fold-change | P-value | Changes |
|---|---|---|---|---|
| FCHO2 | F-BAR domain only protein 2 isoform X7 | 256.14 | 2.16E-13 | UP |
| STXBP1 | Syntaxin-binding protein 1 isoform b | 177.09 | 3.08E-08 | UP |
| TATDN1 | Putative deoxyribonuclease TATDN1 isoform d | 160.63 | 4.23E-12 | UP |
| MPZL1 | Myelin protein zero-like protein 1 isoform b precursor | 131.58 | 1.53E-15 | UP |
| NDUFA5 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 5 isoform 1 | 122.48 | 1.08E-12 | UP |
| SVOP | Synaptic vesicle 2-related protein | 93.36 | 4.27E-12 | UP |
| ATP5ME | ATP synthase subunit e, mitochondrial | 88.21 | 2.01E-09 | UP |
| NUP50 | Nuclear pore complex protein Nup50 isoform a | 87.22 | 8.44E-12 | UP |
| MAP4K2 | Mitogen-activated protein kinase kinase kinase kinase 2 isoform X7 | 80.07 | 5.16E-11 | UP |
| RIN1 | Ras and Rab interactor 1 isoform 3 | 78.40 | 4.75E-08 | UP |
| RBBP4 | Histone-binding protein RBBP4 isoform a | 0.003 | 1.83E-13 | DOWN |
| UQCR10 | Cytochrome b-c1 complex subunit 9 isoform a | 0.004 | 1.1E-08 | DOWN |
| EGLN1 | Egl nine homolog 1 | 0.008 | 1.52E-11 | DOWN |
| GSTT2 | Glutathione S-transferase theta-2 isoform b | 0.008 | 1.92E-09 | DOWN |
| SNAPIN | SNARE-associated protein Snapin | 0.009 | 3.27E-12 | DOWN |
| NDUFS5 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 5 | 0.009 | 6.33E-08 | DOWN |
| LRRTM1 | Leucine-rich repeat transmembrane neuronal protein 1 precursor | 0.010 | 1.05E-06 | DOWN |
| RPS17 | 40S ribosomal protein S17 | 0.011 | 6.58E-08 | DOWN |
| MTFR1L | Mitochondrial fission regulator 1-like isoform a | 0.011 | 1.38E-08 | DOWN |
| ABCF2 | ATP-binding cassette sub-family F member 2 isoform a | 0.013 | 3.05E-11 | DOWN |
Figure 3.

Hierarchical cluster analysis of 172 differentially expressed proteins in TLE-HS and control groups. Each protein expression level from different samples is presented horizontally, and the expression levels of all proteins in each sample are listed vertically. The log2-transformed expression levels are shown in a red-blue color scale (red is upregulated, blue is downregulated). The pink color in the heatmaps indicates TLE-HS patients, the blue color indicates control samples.
Gene Ontology, KEGG Pathway Analysis of Differentially Expressed Proteins
GO terms were further assigned to DEPs according to their CC, MF, and BP (Figure 4). From the perspective of CC, it can be noticed that the top DEPs were enriched in “cytoplasm,” “extracellular exosome,” “synaptic vesicle,” and “mitochondrion.” From the BP perspective, the most enriched terms included “cell-cell adhesion,” “regulation of protein stability,” “synaptic vesicle exocytosis,” and “regulation of synaptic plasticity.” As for MF, “protein binding,” “ATP binding,” and “syntaxin binding” were the most enriched terms. “Parkinson's disease” and “oxidative phosphorylation” were the two significantly enriched KEGG pathways (Figure 4). Functional enrichment analysis of up and downregulated proteins are presented in Supplementary Table 2.
Figure 4.

Enriched KEGG pathway and GO analysis of 172 differentially expressed proteins using DAVID. The identified proteins were annotated with DAVID into molecular function, biological process, cellular component, and KEGG pathway.
PPI Network Construction and Analysis of Modules
A total of 119 nodes and 144 edges were selected to plot the PPI network by STRING, which consisted of 82 upregulated proteins and 90 downregulated proteins (Figure 5A). We counted up the degree of the protein to highlight specific proteins with top linkage (Figure 5B).
Figure 5.

PPI network and module analysis. (A): Protein-protein interaction network of differentially expressed genes by the STRING database. We identified 144 PPI pairs among 119 differentially expressed genes. (B): Degree distribution histogram of hub genes. (C): A significant module selected from the entire PPI network using the Cytoscape MCODE app with the default parameters (Degree cutoff ≥2, Node score cut-off ≥0.2, K-Core ≥2 and Max Depth = 100). Red nodes represent upregulated proteins and blue nodes represent downregulated proteins.
Subsequently, in the entire PPI network, a pivotal module of 10 proteins (RPS14, RPS17, NDUFS5, RPL23A, ATP5I, NDUFA5, MT-ND4, UQCR10, KCNA1, C18orf32) was identified with the default parameters using MCODE (Figure 5C). Moreover, it is worth noting that, functional and KEGG pathway enrichment analysis revealed that genes in this module were found to relate to “oxidative phosphorylation,” “Parkinson's disease,” “metabolic pathways,” and “mitochondrial respiratory chain complex” (Table 3).
Table 3.
Functional enrichment analysis of differentially expressed genes in the module.
| Category | Term | P-value | Genes |
|---|---|---|---|
| KEGG_PATHWAY | Oxidative phosphorylation | 4.47E-06 | NDUFA5, UQCR10, NDUFS5, ND4, ATP5I |
| KEGG_PATHWAY | Parkinson's disease | 2.84E-04 | NDUFA5, UQCR10, NDUFS5, ND4 |
| KEGG_PATHWAY | Ribosome | 0.007635 | RPS17, RPS14, RPL23A |
| KEGG_PATHWAY | Non-alcoholic fatty liver disease (NAFLD) | 0.00935 | NDUFA5, UQCR10, NDUFS5 |
| KEGG_PATHWAY | Alzheimer's disease | 0.011486 | NDUFA5, UQCR10, NDUFS5 |
| KEGG_PATHWAY | Huntington's disease | 0.014838 | NDUFA5, UQCR10, NDUFS5 |
| KEGG_PATHWAY | Metabolic pathways | 0.021831 | NDUFA5, UQCR10, NDUFS5, ND4, ATP5I |
| GOTERM_BP | Mitochondrial electron transport, NADH to ubiquinone | 2.96E-04 | NDUFA5, NDUFS5, ND4 |
| GOTERM_BP | Mitochondrial respiratory chain complex I assembly | 4.90E-04 | NDUFA5, NDUFS5, ND4 |
| GOTERM_BP | SRP-dependent co-translational protein targeting to membrane | 0.001088 | RPS17, RPS14, RPL23A |
| GOTERM_BP | Viral transcription | 0.001539 | RPS17, RPS14, RPL23A |
| GOTERM_BP | Nuclear-transcribed mRNA catabolic process, nonsense-mediated decay | 0.001735 | RPS17, RPS14, RPL23A |
| GOTERM_BP | Translational initiation | 0.002291 | RPS17, RPS14, RPL23A |
| GOTERM_BP | rRNA processing | 0.005487 | RPS17, RPS14, RPL23A |
| GOTERM_BP | Translation | 0.007591 | RPS17, RPS14, RPL23A |
| GOTERM_BP | Ribosomal small subunit assembly | 0.01014 | RPS17, RPS14 |
| GOTERM_CC | Mitochondrial inner membrane | 2.19E-05 | NDUFA5, UQCR10, NDUFS5, ND4, ATP5I |
| GOTERM_CC | Mitochondrial respiratory chain complex I | 1.96E-04 | NDUFA5, NDUFS5, ND4 |
| GOTERM_CC | Ribosome | 0.002228 | RPS17, RPS14, RPL23A |
| GOTERM_CC | Mitochondrion | 0.016474 | NDUFS5, ND4, RPS14, ATP5I |
| GOTERM_CC | Cytosolic small ribosomal subunit | 0.020882 | RPS17, RPS14 |
| GOTERM_MF | NADH dehydrogenase (ubiquinone) activity | 2.81E-04 | NDUFA5, NDUFS5, ND4 |
| GOTERM_MF | Structural constituent of ribosome | 0.005832 | RPS17, RPS14, RPL23A |
Discussion
For patients with refractory epilepsy, surgical therapy is an important alternative treatment, and surgical resection of the epileptogenic zone offers valuable materials for investigating the molecular mechanisms underlying epilepsy. Studies in surgical specimens revealed the critical role of the hippocampus for TLE and, particularly, of the DG in several pathological processes that were related to the epileptogenesis and development of the disease (16). However, the proteomics alterations of DG in TLE-HS patients have not been well-studied. In this regard, we utilized iTRAQ-based tandem mass spectrometry to resolve and compare the protein expression of DG to characterize the molecular changes associated with HS pathology. Differential expression analysis identified 82 upregulated proteins and 90 downregulated proteins in the samples with TLE-HS compared to the autopsy samples. Subsequent functional analysis and PPI network analysis suggested that these proteins might be involved in biological changes that relate to epilepsy.
Our results showed that oxidative phosphorylation and metabolic pathways were significantly enriched KEGG pathways. The proteins that participate in those two pathways included NDUFA5, UQCR10, NDUFS5, ND4, and ATP5I. Among that, NDUFA5, UQCR10, NDUFS5, and ATP5I were also in the top 10 dysregulated proteins list, and NDUFA5, UQCR10, NDUFS5, ND4, and ATP5I were components of the significant module in the PPI network. ATP5I and NDUFA5 have been reported to be dysregulated in the HS of TLE patients and animal models (17–19). Oxidative phosphorylation takes place in the mitochondria of eukaryotic cells, and couples the process with the oxidation of substrates and ATP generation (20). Since mitochondrial oxidative phosphorylation meets the energy demands in neurons and mitochondria participate in cellular Ca2+ homeostasis, mitochondria malfunction has a strong impact on synaptic transmission and neuronal excitability, which has been proposed to play a pivotal role in epileptogenesis (21). Moreover, human and experimental TLE with a variety of impaired mitochondrial functions have been reported (22). Additionally, mitochondrial dysfunction is highly relevant for neuronal loss, which is a distinct pathological feature in TLE-HS patients (23).
There was also significant enrichment of several KEGG pathways associated with neurodegeneration: Parkinson's disease, Alzheimer's disease, and Huntington's disease. The overlap proteins of these pathways included NDUFA5, NDUFS5, ND4, which are subunits of complex I (CI; NADH-ubiquinone oxidoreductase) (24). CI is the first complex of the mitochondrial oxidative phosphorylation system, deficiencies in which have been associated with TLE, Huntington's disease, and Parkinson's disease (25–27). Humans and animals exposed to the CI inhibitor develop Parkinsonism, which strengthened CI deficiencies in Parkinson's disease (28, 29). Ketone bodies metabolism actively produces energy in periods of energy shortage in the brain, which may also be part of the compensatory mechanism of CI deficiencies (30). Ketogenic diet has been shown to be involved in energy metabolism, and to stimulate mitochondrial biogenesis (31). Moreover, ketogenic diet induced a coordinated upregulation of mitochondrial genes, exhibited neuroprotective effects in the hippocampus of a TLE model (32, 33), and reduced the amyloid pathology in an experimental model of Alzheimer's disease (34). Although these neurodegenerative diseases differ in their underlying etiology, they share a common feature, in that each disorder implicates a certain mitochondrial dysfunction through a specific pathway. Above all, this perspective may raise the possibility for the development of novel neuroprotective agents in addition to the conventional anticonvulsant therapy of TLE.
While 172 DEPs were identified, we believe that the following selection may be of interest for further investigation. Three proteins related to neurotransmitter release, STXBP1, SNAPIN, and SVOP, also appeared in the top 10 dysregulated proteins list. Synaptic transmission is critical for normal brain functioning and homeostasis, and abnormal regulation could break the delicate balance between excitatory and inhibitory synapses, which causes seizures (35). STXBP1 encodes a syntaxin1-binding protein, and helps regulate the release of neurotransmitters through the regulation of transmembrane attachment protein receptors (syntaxin1) (36). STXBP1 gene mutations have been reported to cause STXBP1 encephalopathy or early infantile epileptic encephalopathy-4 (EIEE4) (37). The synaptic vesicle glycoprotein 2 (SV2) family and SV2-related proteins (SVOP) are located in synaptic neurotransmitter-containing vesicles, and regulate the secretion of neurotransmitters from presynaptic neurons, possibly via synaptic vesicle recycling (38, 39). SV2A is the target site of levetiracetam (LEV) and related compounds, which are approved for use in patients with multiple types of seizures and epilepsies (40, 41). Although SVOP is structurally similar to SV2, little is known about its function (39). Bando et al. noted that the expression of SVOP was significantly upregulated in the hippocampus of refractory mesial TLE patients (42). KCNA1, which appeared in the significant module of the PPI network, encoded a voltage-gated potassium channel that mediates primarily potassium transport in excitable membranes of the CNS, and contributes to the regulation of neuronal excitability especially in mossy fibers of the hippocampus (43, 44). KCNA1−/− mice display early seizures and premature unexpected death, and can be used as a valuable sudden unexpected death (SUDEP) model in epilepsy (45). Notably, KCNA1 is the target of Isoflurane, a commonly used inhalation agent, which has anticonvulsant properties and can terminate refractory status epilepticus in patients (46).
Previous proteomic research analyzing samples of patients with epilepsy have identified different differential protein profiles. This is because of (i) different types of epilepsy; (ii) diverse sample sources or brain regions; and (iii) different controls. Two previous studies have performed proteomics on the DG region of epilepsy patients: Liu et al. (47) compared the proteomes of basal and dispersed granule cells in the hippocampus of MTLE patients with GCD (not including surgical or post-mortem healthy controls), and found that upregulated proteins in dispersed samples were involved in developmental cellular migratory processes. Pire et al. (48) performed proteomic analysis of the hippocampal CA1-3 region, frontal cortex, and DG samples from epilepsy (without specific classification) and control cases, the pathway analysis of altered proteins in the DG involved in mitochondrial dysfunction showed the most enrichment. Interestingly, by analyzing the gene expression profiles of dentate granule cells from surgically resected hippocampal specimens from patients with MTLE with and without HS, Griffin et al. (19) demonstrated that differentially expressed genes were associated with the oxidative phosphorylation pathway, which is generally consistent with our findings. Our proteomic study further expanded the biomedical resource for molecular mechanism research on epilepsy. Together, more detailed research including different brain subregions, specific cell components, and even single-cell sequencing would offer more promising options to bridge the understanding of epileptogenesis from a different perspective.
However, several points should be taken into account when analyzing and interpreting the results. Firstly, subjects enrolled in our study appear to be refractory patients as they still have seizures even after combined antiepileptic drugs (AEDs) treatment; AEDs may have an impact on the proteome of the brain tissue, however, no published literature has explored this. Second, the age of patients with TLE was younger than that of control subjects, which may still be a tentatively confounding factor after an adjustment. Third, it is not possible to use samples with identical post-mortem times for ethical and practical reasons, but we have to collect samples from autopsies within a maximum post-mortem time of 10 h. Fourth, tissue samples from distinct sample acquisition methodologies for TLE and control groups (hippocampectomy and autopsy) may induce possible effects in protein content integrity. Therefore, control samples with a smaller post-mortem time were selected as much as possible, and protein samples were subjected to strict quality control. Last, as a limited quantity of DG brain tissue was obtained from each subject, proteins profiled by iTRAQ-based tandem mass spectrometry were not verified by immunohistochemistry assays or western blot, which should warrant further validation in larger independent samples.
Conclusion
For the first time we applied an iTRAQ-based proteomic study to investigate the molecular alterations in the DG region of TLE-HS patients and autopsy controls. Our proteomic work revealed a number of dysregulated proteins and involved critical biological pathways associated with HS pathological mechanisms. The gained information will pave the way to a better understanding of epileptogenesis and future therapeutic intervention.
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD023048.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Central South University, Xiangya School of Medicine and the affiliated Xiangya Hospital. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
WX, HL, and YZ contributed to the study conception and design. WX, ZY, XY, TT, HL, LL, and LF collected the patients' data. WX, HL, ND, and WC analyzed and interpreted the data. WX, YZ, WC, and BX funded the study. All authors contributed to writing the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Foremost, we thank the patients, autopsy controls, and their families for their cooperation and for donating tissue samples for this study.
Footnotes
Funding. This study was financially supported by the National Natural Science Foundation of China (Nos. 81974206, 81641039, and 81801279), the Project from Hunan Provincial Science and Technology Department (No. 2019SK1012), and the Fundamental Research Funds for the Central Universities of Central South University (No. 2018zzts248).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.626013/full#supplementary-material
References
- 1.Tellez-Zenteno JF, Hernandez-Ronquillo L. A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res Treat. (2012) 2012:630853. 10.1155/2012/630853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blumcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A, et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE Commission on diagnostic methods. Epilepsia. (2013) 54:1315–29. 10.1111/epi.12220 [DOI] [PubMed] [Google Scholar]
- 3.Kanner AM. Management of psychiatric and neurological comorbidities in epilepsy. Nat Rev Neurol. (2016) 12:106–16. 10.1038/nrneurol.2015.243 [DOI] [PubMed] [Google Scholar]
- 4.Kobow K, Blumcke I. The emerging role of DNA methylation in epileptogenesis. Epilepsia. (2012) 53(Suppl. 9):11–20. 10.1111/epi.12031 [DOI] [PubMed] [Google Scholar]
- 5.Thom M. Review: hippocampal sclerosis in epilepsy: a neuropathology review. Neuropathol Appl Neurobiol. (2014) 40:520–43. 10.1111/nan.12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng Z, Houser CR. Temporal patterns of FOS expression in the dentate gyrus after spontaneous seizures in a mouse model of temporal lobe epilepsy. J Neurosci. (2005) 25:7210–20. 10.1523/JNEUROSCI.0838-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pitkanen A, Lukasiuk K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. (2011) 10:173–86. 10.1016/S1474-4422(10)70310-0 [DOI] [PubMed] [Google Scholar]
- 8.Persike DS, Marques-Carneiro JE, Stein M, Yacubian E, Centeno R, Canzian M, et al. Altered proteins in the hippocampus of patients with mesial temporal lobe epilepsy. Pharmaceuticals. (2018) 11:95. 10.3390/ph11040095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang JW, Czech T, Felizardo M, Baumgartner C, Lubec G. Aberrant expression of cytoskeleton proteins in hippocampus from patients with mesial temporal lobe epilepsy. Amino Acids. (2006) 30:477–93. 10.1007/s00726-005-0281-y [DOI] [PubMed] [Google Scholar]
- 10.Yang JW, Czech T, Yamada J, Csaszar E, Baumgartner C, Slavc I, et al. Aberrant cytosolic acyl-CoA thioester hydrolase in hippocampus of patients with mesial temporal lobe epilepsy. Amino Acids. (2004) 27:269–75. 10.1007/s00726-004-0138-9 [DOI] [PubMed] [Google Scholar]
- 11.Treumann A, Thiede B. Isobaric protein and peptide quantification: perspectives and issues. Expert Rev Proteomics. (2010) 7:647–53. 10.1586/epr.10.29 [DOI] [PubMed] [Google Scholar]
- 12.Long HY, Feng L, Kang J, Luo ZH, Xiao WB, Long LL, et al. Blood DNA methylation pattern is altered in mesial temporal lobe epilepsy. Sci Rep. (2017) 7:43810. 10.1038/srep43810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu SY, Zhang QL, Zhang Q, Wan L, Jiang J, Tu T, et al. Regional and cellular mapping of sortilin immunoreactivity in adult human brain. Front Neuroanat. (2019) 13:31. 10.3389/fnana.2019.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. (2009) 4:44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 15.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. (2009) 37:1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siebzehnrubl FA, Blumcke I. Neurogenesis in the human hippocampus and its relevance to temporal lobe epilepsies. Epilepsia. (2008) 49(Suppl. 5):55–65. 10.1111/j.1528-1167.2008.01638.x [DOI] [PubMed] [Google Scholar]
- 17.Bosco DB, Zheng J, Xu Z, Peng J, Eyo UB, Tang K, et al. RNAseq analysis of hippocampal microglia after kainic acid-induced seizures. Mol Brain. (2018) 11:34. 10.1186/s13041-018-0376-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu XY, Yang JL, Chen LJ, Zhang Y, Yang ML, Wu YY, et al. Comparative proteomics and correlated signaling network of rat hippocampus in the pilocarpine model of temporal lobe epilepsy. Proteomics. (2008) 8:582–603. 10.1002/pmic.200700514 [DOI] [PubMed] [Google Scholar]
- 19.Griffin NG, Wang Y, Hulette CM, Halvorsen M, Cronin KD, Walley NM, et al. Differential gene expression in dentate granule cells in mesial temporal lobe epilepsy with and without hippocampal sclerosis. Epilepsia. (2016) 57:376–85. 10.1111/epi.13305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson DF. Oxidative phosphorylation: regulation and role in cellular and tissue metabolism. J Physiol. (2017) 595:7023–38. 10.1113/JP273839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Folbergrova J, Kunz WS. Mitochondrial dysfunction in epilepsy. Mitochondrion. (2012) 12:35–40. 10.1016/j.mito.2011.04.004 [DOI] [PubMed] [Google Scholar]
- 22.Kudin AP, Zsurka G, Elger CE, Kunz WS. Mitochondrial involvement in temporal lobe epilepsy. Exp Neurol. (2009) 218:326–32. 10.1016/j.expneurol.2009.02.014 [DOI] [PubMed] [Google Scholar]
- 23.Rowley S, Patel M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Radic Biol Med. (2013) 62:121–31. 10.1016/j.freeradbiomed.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuperfain AB, Zhang ZL, Kennedy JL, Goncalves VF. The complex interaction of mitochondrial genetics and mitochondrial pathways in psychiatric disease. Mol Neuropsychiatry. (2018) 4:52–69. 10.1159/000488031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parker WJ, Boyson SJ, Luder AS, Parks JK. Evidence for a defect in NADH: ubiquinone oxidoreductase (complex I) in Huntington's disease. Neurology. (1990) 40:1231–4. 10.1212/WNL.40.8.1231 [DOI] [PubMed] [Google Scholar]
- 26.Kunz WS, Kudin AP, Vielhaber S, Blumcke I, Zuschratter W, Schramm J, et al. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann Neurol. (2000) 48:766–73. [DOI] [PubMed] [Google Scholar]
- 27.Smigrodzki R, Parks J, Parker WD. High frequency of mitochondrial complex I mutations in Parkinson's disease and aging. Neurobiol Aging. (2004) 25:1273–81. 10.1016/j.neurobiolaging.2004.02.020 [DOI] [PubMed] [Google Scholar]
- 28.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. (2000) 3:1301–6. 10.1038/81834 [DOI] [PubMed] [Google Scholar]
- 29.Cannon JR, Greenamyre JT. Neurotoxic in vivo models of Parkinson's disease recent advances. Prog Brain Res. (2010) 184:17–33. 10.1016/S0079-6123(10)84002-6 [DOI] [PubMed] [Google Scholar]
- 30.Peralta S, Torraco A, Wenz T, Garcia S, Diaz F, Moraes CT. Partial complex I deficiency due to the CNS conditional ablation of Ndufa5 results in a mild chronic encephalopathy but no increase in oxidative damage. Hum Mol Genet. (2014) 23:1399–412. 10.1093/hmg/ddt526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wenz T, Luca C, Torraco A, Moraes CT. mTERF2 regulates oxidative phosphorylation by modulating mtDNA transcription. Cell Metab. (2009) 9:499–511. 10.1016/j.cmet.2009.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Linard B, Ferrandon A, Koning E, Nehlig A, Raffo E. Ketogenic diet exhibits neuroprotective effects in hippocampus but fails to prevent epileptogenesis in the lithium-pilocarpine model of mesial temporal lobe epilepsy in adult rats. Epilepsia. (2010) 51:1829–36. 10.1111/j.1528-1167.2010.02667.x [DOI] [PubMed] [Google Scholar]
- 33.Kobow K, Kaspi A, Harikrishnan KN, Kiese K, Ziemann M, Khurana I, et al. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. (2013) 126:741–56. 10.1007/s00401-013-1168-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao J, Chen S, Mao Z, Cadenas E, Brinton RD. 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer's disease. PLoS ONE. (2011) 6:e21788. 10.1371/journal.pone.0021788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casillas-Espinosa PM, Powell KL, O'Brien TJ. Regulators of synaptic transmission: roles in the pathogenesis and treatment of epilepsy. Epilepsia. (2012) 53(Suppl. 9):41–58. 10.1111/epi.12034 [DOI] [PubMed] [Google Scholar]
- 36.Stamberger H, Weckhuysen S, De Jonghe P. STXBP1 as a therapeutic target for epileptic encephalopathy. Expert Opin Ther Targets. (2017) 21:1027–36. 10.1080/14728222.2017.1386175 [DOI] [PubMed] [Google Scholar]
- 37.Lanoue V, Chai YJ, Brouillet JZ, Weckhuysen S, Palmer EE, Collins BM, et al. STXBP1 encephalopathy: connecting neurodevelopmental disorders with alpha-synucleinopathies? Neurology. (2019) 93:114–23. 10.1212/WNL.0000000000007786 [DOI] [PubMed] [Google Scholar]
- 38.Nowack A, Yao J, Custer KL, Bajjalieh SM. SV2 regulates neurotransmitter release via multiple mechanisms. Am J Physiol Cell Physiol. (2010) 299:C960–7. 10.1152/ajpcell.00259.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao J, Bajjalieh SM. SVOP is a nucleotide binding protein. PLoS ONE. (2009) 4:e5315. 10.1371/journal.pone.0005315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. (2004) 101:9861–6. 10.1073/pnas.0308208101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H. Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A. Br J Pharmacol. (2008) 154:1662–71. 10.1038/bjp.2008.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bando SY, Alegro MC, Amaro EJ, Silva AV, Castro LH, Wen HT, et al. Hippocampal CA3 transcriptome signature correlates with initial precipitating injury in refractory mesial temporal lobe epilepsy. PLoS ONE. (2011) 6:e26268. 10.1371/journal.pone.0026268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Wijst J, Glaudemans B, Venselaar H, Nair AV, Forst AL, Hoenderop JG, et al. Functional analysis of the Kv1.1 N255D mutation associated with autosomal dominant hypomagnesemia. J Biol Chem. (2010) 285:171–8. 10.1074/jbc.M109.041517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Imbrici P, D'Adamo MC, Kullmann DM, Pessia M. Episodic ataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvbeta1.1 and Kv1.4-1.1/Kvbeta1.2. Eur J Neurosci. (2006) 24:3073–83. 10.1111/j.1460-9568.2006.05186.x [DOI] [PubMed] [Google Scholar]
- 45.Ren Y, Chang J, Li C, Jia C, Li P, Wang Y, et al. The effects of ketogenic diet treatment in KCNA1-null mouse, a model of sudden unexpected death in epilepsy. Front Neurol. (2019) 10:744. 10.3389/fneur.2019.00744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mirsattari SM, Sharpe MD, Young GB. Treatment of refractory status epilepticus with inhalational anesthetic agents isoflurane and desflurane. Arch Neurol. (2004) 61:1254–9. 10.1001/archneur.61.8.1254 [DOI] [PubMed] [Google Scholar]
- 47.Liu J, Dzurova N, Al-Kaaby B, Mills K, Sisodiya SM, Thom M. Granule cell dispersion in human temporal lobe epilepsy: proteomics investigation of neurodevelopmental migratory pathways. Front Cell Neurosci. (2020) 14:53. 10.3389/fncel.2020.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pires G, Leitner D, Drummond E, Kanshin E, Nayak S, Askenazi M, et al. Proteomic differences in the hippocampus and cortex of epilepsy brain tissue. bioRxiv [Preprint]. 10.1101/2020.07.21.209163 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD023048.