

Abstract
Circular RNAs (circRNAs) are highly stable RNA molecules that are attractive templates for expression of therapeutic proteins and non-coding RNAs. In eukaryotes, circRNAs are primarily generated by the spliceosome through backsplicing. Here, we interrogate different molecular elements including intron type and length, Alu repeats, internal ribosome entry sites (IRESs), and exon length essential for circRNA formation and exploit this information to engineer robust backsplicing and circRNA expression. Specifically, we leverage the finding that the downstream intron can tolerate large inserts without affecting splicing to achieve tandem expression of backspliced circRNAs and tRNA intronic circRNAs from the same template. Further, truncation of selected intronic regions markedly increased circRNA formation in different cell types in vitro as well as AAV-mediated circRNA expression in cardiac and skeletal muscle tissue in vivo. We also observed that different IRES elements and exon length influenced circRNA expression and translation, revealing an exonic contribution to splicing, as evidenced by different RNA species produced. Taken together, these data provide new insight into improving the design and expression of synthetic circRNAs. When combined with AAV capsid and promoter technologies, the backsplicing introns and IRES elements constituting this modular platform significantly expand the gene expression toolkit.
Graphical Abstract
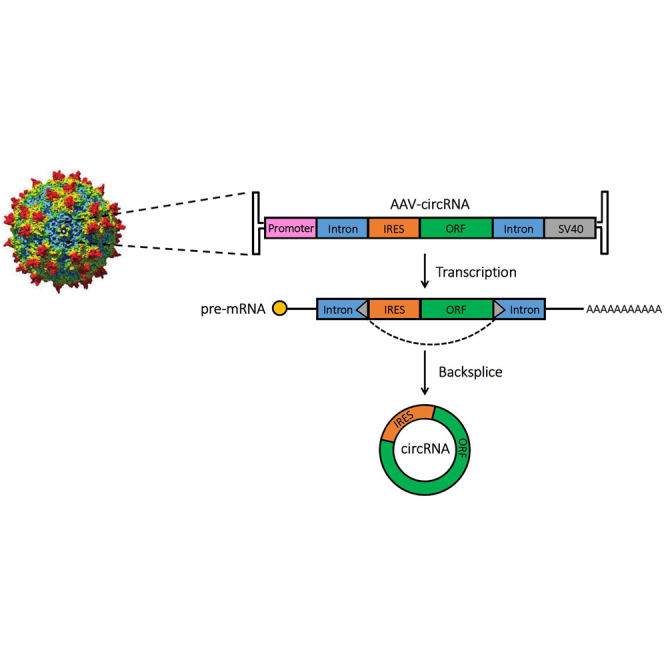
In this study, Asokan and colleagues interrogate a battery of molecular elements including introns, Alu repeats, and internal ribosome entry sites and exploit this information to engineer robust RNA backsplicing. The approach yields a modular circular RNA expression platform for gene therapy applications.
Introduction
Circular RNAs (circRNAs) are a class of covalently closed RNA molecules characterized by their highly stable nature.1 CircRNAs are both widely and highly expressed; they can be formed from introns (e.g., self-splicing introns,2 tRNA introns,3 or retained lariats4,5) or, most commonly in metazoans, by the backsplicing of pre-mRNA exons. Exonic circRNAs are formed through a backsplicing process, in which the spliceosome joins a downstream donor site to an upstream acceptor site, creating a covalently closed circularized exon.6,7 Sequencing experiments have revealed thousands of circRNAs expressed from different cell types, tissue types, and organisms.1 However, the function of the majority of circRNAs is unknown, with the exception of a few well-characterized circRNAs, which act as miRNA or protein sponges or are translated to produce protein products.7, 8, 9
Recent work has shown that overexpression of both endogenous and synthetic circRNAs can enhance outcomes in multiple disease states. For instance, restoration of the endogenous circRNA circFndc3b, which is downregulated following cardiac injury such as myocardial infarct, has been shown to enhance cardiac function and repair in mice by regulating vascular endothelial growth factor (VEGF) signaling.10 Another recent study demonstrated that synthetic circRNAs can be engineered into artificial miRNA sponges to decrease increased miRNA levels associated with cardiac dysfunction in a hypertrophic mouse model of disease.11 Similarly, overexpression of circRNA ZNF532, an endogenous circular miRNA sponge, has been shown to mitigate retinal degeneration in a diabetic mouse model.12 Thus, circRNAs present a promising new class of therapeutic RNA molecules.
The aforementioned studies were enabled by recombinant adeno-associated virus (AAV) vectors that allow for the expression of therapeutic circRNAs from target tissues/cell types.13, 14, 15 In humans, a major cis element for circRNA formation is the presence of complementary Alu elements in the flanking introns.1,16 Inclusion of these backsplicing elements within the AAV genomic cassette allows for the production of circRNA products in transduced cells with significantly greater stability than linear mRNAs.17 Further, addition of an internal ribosome entry site (IRES) element preceding an open reading frame (ORF) in the backsplicing cassette can enable circRNA translation.17 These elements work in tandem to regulate both the levels of circRNAs generated and the rate of protein synthesis. While AAV-circRNAs show promise for several therapeutic applications, work to improve circRNA expression using AAV vectors has not been explored.
In the current work, we interrogate molecular elements that influence circRNA backsplicing and translation in vitro and in vivo. We find that intronic backsplicing introns can tolerate insertions, in an asymmetric manner that allows for the expression of dual circRNA products. Further, we show that targeted intronic deletions result in significantly enhanced circRNA generation. Additionally, changes to exonic sequences affect multiple aspects of the splicing process, leading to changes in the type and quantity of RNAs produced. We also assess the impact of these newly optimized elements on circRNA expression in mice and find that both circRNA formation and translation can be tissue specific. Our findings highlight how the nature of different intronic and exonic elements can significantly impact synthetic circRNA backsplicing and expression. Further, these studies provide modular design principles for expression of synthetic circRNA-based tools and a potential new sub-class of gene-based therapeutics.
Results
Backsplicing introns tolerate insertions, allowing for dual circRNA expression
A common feature of introns that mediate backsplicing is the presence of inverted Alu elements or other complementary sequences.1 To interrogate how the distance between these complementary regions and splice donor/acceptor sites affects backsplicing of a circRNA, we utilized a previously reported system as a template, wherein introns from the HIPK3 gene drive circularization of a split GFP exon.17 Briefly, upon transcription, the introns drive formation of a circRNA that unites the GFP ORF. Translation of the GFP ORF is then initiated by an IRES within the circGFP RNA (Figure 1A; sequences in Figure S1).
Figure 1.

Distance requirements between Alu element and splice site differ in upstream and downstream introns
(A) A reporter was constructed by extracting portions of introns from the HIPK3 gene and placing them around a split GFP reporter exon. Inverted Alu repeats in the introns interact, allowing for backsplicing to occur, forming a circRNA. The presence of an IRES sequence drives translation, leading to GFP protein expression. See Figure S1 for intron sequences. (B) Sequence ranging from 100 nt to 1,500 nt was inserted into the left HIPK3 intron at the indicated position. See also Figure S2. (C and D) Constructs were transfected into HEK293 cells and expression assayed at 4 days post-transfection by (C) western blot analysis, with actin as a loading control (quantification below), and (D) northern blot analysis, probing for GFP sequences (quantification below). (E) Sequence ranging from 100 nt to 1,500 nt was inserted into the right HIPK3 intron at the indicated position. (F and G) Constructs were transfected into HEK293 cells and expression assayed at 4 days post-transfection by (F) western blot analysis, with actin as a loading control (quantification below), and (G) northern blot analysis, probing for GFP sequences (quantification below). (H) Sequences driving formation of a tricRNA containing Broccoli (tricY-Broccoli) were inserted into the same location in the right HIPK3 intron. (I) tricY-Broccoli expression was verified by gel electrophoresis followed by DFHBI-1T staining. (J and K) circRNA formation was assayed by (J) western blot analysis, with actin as a loading control (quantification below left), and (K) northern blot analysis, probing for GFP sequences (quantification left). On northern blots, the asterisk refers to an additional circular band. Western and northern blots were quantified as detailed in Materials and methods and graphed relative to the unchanged HIPK3 intron construct. Student’s t test was performed to test for statistical significance. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.0005; ∗∗∗∗p < 0.00005.
To probe the impact of spacing in the introns, we first artificially increased the distance between each Alu element and the splice site. Random inserts ranging from 100 to 1,500 nt were designed with nucleotide content similar to the HIPK3 introns but devoid of any strong predicted secondary structure. We first inserted these into the “left” intron (upstream of the splice acceptor site) at a site ∼100 nt from the splice acceptor (Figure 1B). The insertion of additional distance was highly detrimental, as seen from western blots of GFP protein and northern blots of the circRNA (Figures 1C and 1D). Insertion of only 100 nt lowered the circRNA formation by ∼5-fold, whereas greater distances effectively ablated circRNA formation. Given that the HIPK3 splice acceptor has a large poly-pyrimidine tract (Figure S1A) and to control for a possible impact on the branch point environment, we created a second series of constructs in which the additional sequences were inserted distal to the splice junction and proximal to the Alu element (Figure S2A). This second set behaved virtually identically, with insertion of 100 nt or more greatly reducing circRNA formation and GFP expression (Figures S2B and S2C).
We also created insertions in the “right” intron (downstream of the splice donor site) (Figure 1E). Surprisingly, the effect of right insertions was distinct from the left intron insertions, with sequences up to 1.5 kb in length showing no effect on GFP expression or circRNA formation (Figures 1F and 1G). Different RNA concatemers formed in the case of the original HIPK3 intron were also observed in these constructs with inserts in the right intron. Notably, these additional bands/molecular entities were later resolved and found to be dependent on other elements such as the IRES, as demonstrated later in this report. We then replaced the randomized insert with a sequence encoding a Drosophila tRNA:TyrGUA gene carrying an intron containing the noncoding RNA-based fluorescent aptamer Broccoli.3,18 In theory, this construct is expected to independently produce both a GFP circRNA and a Broccoli tricRNA (tricY-Broccoli) (Figure 1H). We observed robust tricY-Broccoli expression using an in-gel assay (Figure 1I). Further, western and northern blot analyses confirmed that tricRNA formation did not affect GFP protein or circRNA levels (Figures 1J and 1K). Thus, not only can the distance between the splice donor and the Alu element be increased without effect, but an additional functional RNA element can be inserted in the intronic region.
Synthetic intronic sequences increase circRNA expression
In counterpoint to the above experiments, we investigated the effect of decreasing the distance between the Alu element and splice site by deleting intronic sequences. As mentioned earlier, the HIPK3 splice acceptor has a large poly-pyrimidine tract; therefore, we preserved ∼125 nt proximal to the splice junction to create the LΔ42–267 construct. In the right intron, we designed a full deletion (RΔ10–654) and two smaller deletions (RΔ10–497 and RΔ161–654), both of which preserve ∼150 nt distance (Figure 2A, deletions numbered from the start of their respective introns). The ability of these modified introns to support circRNA formation and translation was then analyzed by GFP fluorescence, western blot, and northern blot. With the LΔ42–267 construct we observed ∼2-fold higher GFP and circRNA expression in comparison to the original construct (Figures 2B–2D). The large RΔ10–654 deletion, which places the Alu element within 20 nt of the splice junction, reduced circularization efficiency drastically. However, the smaller deletions were tolerated without impacting circRNA biogenesis or GFP expression. Note that both smaller deletions splice equally well, indicating a sequence-independent effect. We then combined the upstream LΔ42–267 and downstream RΔ10–497 deletions to create the minimal LΔRΔ intronic elements. The combination of deletions led to a synergistic effect, increasing GFP expression and circRNA levels by ∼5-fold (Figures 2B–2D). Deletion of either the left or right Alu regions (Figure 2E) abrogated GFP expression and circRNA formation, confirming the necessity of Alu elements in this system (Figures 2F and 2G). Similar results were observed in both the U87 glioblastoma and Huh7 hepatocarcinoma cell lines (Figure S3), suggesting that regulation of backsplicing is conserved across diverse cell types.
Figure 2.

Partial truncation of intronic sequences increases circRNA expression
(A) Portions of the HIPK3 left and right introns were deleted; deletions are numbered from the start of their respective intron. (B–D) Constructs were transfected into HEK293 cells and expression assayed at 4 days post-transfection by (B) GFP fluorescence, (C) western blot analysis, with actin as a loading control (quantification below), and (D) northern blot analysis, probing for GFP sequences (quantification below). (E) Either the left or right partial Alu element was deleted from the HIPK3 introns. (F and G) Constructs were transfected into HEK293 cells and expression assayed at 4 days post-transfection by (F) western blot analysis, with actin as a loading control, and (G) northern blot analysis, probing for GFP sequences. On northern blots, the asterisk refers to an additional circular band. Western and northern blots were quantified as detailed in Materials and methods and graphed relative to the unchanged HIPK3 intron construct. Student’s t test was performed to test for statistical significance. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.0005; ∗∗∗∗p < 0.00005.
Intronic spacing effects on circRNA formation are conserved
To ascertain if the conclusions generated from the HIPK3-derived introns apply more generally, we conducted experiments with reporters driven by two different intron pairs derived from the human ZKSCAN1 gene or the Drosophila melanogaster Laccase2 gene (Figures S1B and S1C). We created three insertions in each intron pair (L100, L500, and R1500) (Figures S4A and S4D). In both cases, insertions in the left intron decreased circRNA expression. Insertion into the right Laccase2 intron had no effect, while insertion into the right ZKSCAN1 intron slightly decreased expression (Figures S4B, S4C, S4E, and S4F). We also created deletion constructs for both Laccase2 and ZKSCAN1, designed to remove as much sequence as possible based on our results in the HIPK3 intron pair. For ZKSCAN1, it was not possible to design a deletion in the left intron because of space constraints between the Alu element and splice site (Figure S4G). For Laccase2, LΔ403–507 and RΔ15–118 constructs and a combined deletion were created (Figure S4J). The ZKSCAN1 and Laccase2 deletions were all tolerated and expressed at equivalent levels to the original (Figures S4H, S4I, S4K, and S4L). Therefore, the results from additional intron pairs confirm that intronic spacing effects on splicing efficiency appear to be conserved in general.
IRES elements regulate circRNA levels and translation
In our system, GFP expression is a product of both circRNA levels and IRES activity. As circRNAs do not contain a 5′ end, canonical cap-dependent translation cannot occur, and thus must rely on IRES elements to initiate cap-independent translation and protein synthesis. We created versions of the HIPK3 split GFP construct containing the encephelomyocarditis virus (EMCV) IRES, poliovirus IRES, Kaposi sarcoma-associated herpesvirus (KSHV) vFLIP IRES, or hepatitis C virus (HCV) IRES.19 circRNAs containing these different IRES elements displayed variable levels of GFP expression, with the poliovirus IRES-containing circRNA construct yielding ∼5-fold higher expression compared to EMCV and KSHV IRES elements. In contrast, no GFP expression was observed with the HCV IRES (Figures 3A and 3B). To directly assess translation efficiency, we isolated polyribosome fractions and visualized the bound RNAs by northern blot using a probe specific for GFP (Figure 3C). RNAs associated with multiple ribosomes (polyribosomes) are translated more efficiently, while those associated with two (disome) or a single (monosome) ribosome are translated less efficiently. The circGFP RNA containing the EMCV IRES was present on mono- and disomes, although much of the circRNA was untranslated, confirming previous results.20 The KSHV IRES-containing circRNA was also present in fractions corresponding to monosomes and disomes, although, like the EMCV circRNA, a large portion of the circRNA was untranslated, showing that low GFP protein levels are due to low translation efficiency of these circRNAs. The poliovirus IRES-containing circGFP RNA, in contrast, was detected in higher fractions corresponding to multiple bound ribosomes, correlating with the observed higher levels of GFP expression (Figure 3C). Confirming the lack of protein expression with the HCV IRES, a large majority of the circRNA was in an unbound fraction; in contrast, a control linear GFP mRNA was detected in high-weight polysome fractions (Figures S5A and S5B). Overall, levels of RNA and protein of these constructs in U87 and Huh7 cell lines were similar to those observed in 293T cells (Figure S6), suggesting that IRES-mediated translation efficiency of these circRNA vectors is similar in vitro.
Figure 3.

IRES elements affect translation efficiency and circRNA expression levels
The HIPK3 split GFP construct was created with either the EMCV, Polio, KSHV vFLIP, or HCV IRES elements. (A and B) Constructs were transfected into HEK293 cells and expression assayed at 4 days post-transfection by (A) GFP fluorescence and (B) western blot analysis, with actin as a loading control (quantification below). (C) To assay translation efficiency, the indicated constructs were transfected into HEK293 cells and harvested in cycloheximide followed by a sucrose gradient and fractionation. Top: OD trace of the gradient, with fractions marked by lines. Middle: RNA was extracted from gradients, separated by gel electrophoresis, transferred to a membrane, and stained with methylene blue to visualize the ribosomal RNA. Bottom: the same membrane was probed for GFP sequences. The arrowhead marks the last fraction in which the circRNA is detected. (D) RNA from transfected cells was assayed by northern blot and probed for GFP sequences (quantification left). (E) RNA was digested by RNase R (RnR) followed by northern blotting, probing for GFP sequences. On northern blots, the asterisk refers to an additional circular band. Western and northern blots were quantified as detailed in Materials and methods and graphed relative to the unchanged HIPK3 intron construct. Student’s t test was performed to test for statistical significance. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.0005; ∗∗∗∗p < 0.00005.
When visualizing the total RNA produced from these constructs through northern blot, we noticed some differences. First, the total amount of circRNA produced varied among the four constructs. The HCV IRES construct expressed the highest levels of circRNA, followed by KSHV, poliovirus, and finally EMCV IRES (Figure 4D). Although the four IRES elements varied in size, there was not a strong correlation between IRES (and therefore circRNA) size and circRNA levels. Second, there were dramatic differences in the type of RNA species produced, other than the circRNA. Notably, we observed an RNA species smaller than the circRNA with both the poliovirus and KSHV constructs. Additionally, the KSHV construct produced several larger RNA species (Figure 4D). Interestingly, these higher bands were detected in polyribosome fractions, while the small RNA species was not (Figure 4C). Both the small RNA species and the larger KSHV RNA species were RNase R resistant, suggesting that they may also be circular (Figure 4E).
Figure 4.

circRNA size can be increased without loss of expression
(A) A self-cleaving P2A peptide followed by the dsRed ORF was added to the exon at the end of the GFP fragment either in the original HIPK3 construct or in the LΔRΔ intron pair. (B and C) Constructs were transfected into HEK293 cells and expression assayed at 4 days post-transfection by (B) GFP and dsRed fluorescence and (C) western blot analysis, with actin as a loading control (quantification below). (D) To assay translation efficiency, the construct was transfected into HEK293 cells and harvested in cycloheximide followed by a sucrose gradient and fractionation. Top: OD trace of the gradient, with fractions marked by lines. Middle: RNA was extracted from gradients, separated by gel electrophoresis, transferred to a membrane, and stained with methylene blue to visualize the ribosomal RNA. Bottom: the same membrane was probed for GFP sequences. The arrowhead marks the last fraction in which the circRNA is detected. (E) RNA from transfected cells was analyzed by northern blot analysis, probing for GFP sequences (quantification above right). (F) RNA was treated with RNase R, then analyzed by northern blot, probing for GFP sequences. (G) RNase H (RnH) digestion was performed with an oligonucleotide targeting the backsplice junction. Samples were analyzed by northern blot and probed against GFP sequences. (H) RNA was analyzed by northern blot and probed with an oligonucleotide spanning the backsplice junction. On northern blots, the asterisk refers to an additional circular band. Western and northern blots were quantified as detailed in Materials and methods and graphed relative to the unchanged HIPK3 intron construct. Student’s t test was performed to test for statistical significance. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.0005; ∗∗∗∗p < 0.00005.
IRES elements significantly affect circRNA splicing patterns
To further interrogate the identity of the additional RNA species observed, we first carried out RNase H digestion using an oligonucleotide that binds the backsplice junction, confirming the circular nature of these RNA species. As expected, the bands corresponding to the main circRNAs remained. Intriguingly, many of the higher KSHV bands disappeared altogether or decreased in intensity, while the bands corresponding to the main circRNA and small circRNA increased in intensity, suggesting that these larger species may be concatemers (Figure S7A). We then probed total RNA with a probe specific to the backsplice junction. The RNA banding pattern was similar to that observed with a probe to the GFP exon, suggesting that these RNA species represent spliced products (Figure S7B).
To elucidate the identity of the additional spliced products produced by poliovirus and KSHV circRNAs, we probed total RNA against the construct’s respective IRES elements. This probe detected most of the RNA species but was unable to detect the small circRNA, indicating that this species lacks the IRES, suggesting that the IRES element was spliced out of the RNA product (Figure S7C). We then performed a virtual northern blot and extracted RNA corresponding to the small circRNA.21 Following cDNA synthesis, PCR products were obtained using primers amplifying across the IRES and sequenced, confirming that the IRES was indeed spliced out of these circRNA products. In both cases, a weak splice donor in the last two codons of GFP was spliced to a weak acceptor at the end of the IRES (Figure S7D). Having identified the splice sites involved, we abolished the alternative donor site by making silent mutations in the GFP coding sequence, which consequently eliminated formation of the small circRNA (Figure S7E). These results highlight the importance of identifying weak donor/acceptor splice sites in the design of circRNA vectors.
Larger circRNA generation does not affect expression
The size of endogenous circRNA exons can range from ∼100 nt to >1 kb, averaging ∼700 nt.1,6 Our circGFP RNAs range in size from 1.1 to 1.5 kb in length, fairly large compared to endogenous circRNAs, but express at high levels. To investigate whether exon length could be further increased, a P2A self-cleaving sequence followed by the dsRed ORF downstream of the end of the GFP ORF fragment (-FP) was added, increasing the exon length by ∼750 nt to a total of 2.2 kb. This larger exon was paired with both the original and LΔRΔ HIPK3 intron pairs and yielded GFP expression comparable to previous results (Figure 2), in addition to expressing dsRed (Figures 4A–4C). Polyribosome analysis revealed that the GFP-dsRed circRNA was present in fractions containing multiple translating ribosomes, showing that it is efficiently translated (Figure 4D). When the P2A sequence was added, removal of the GFP stop codon mutated the weak splice donor site identified earlier. Accordingly, when the total RNA species was analyzed by northern blot, only two species were detected: the circGFP RNA and a larger RNA species (Figure 4E). RNase R and RNase H analyses confirmed the circular nature of the putative circRNA band and revealed that the larger species was linear (Figures 4F and 4G). Probing for the backsplice junction revealed that both bands contained the backsplice junction, indicating that the larger linear species has undergone a splicing event (Figure 4H). These results show that both the endogenous and synthetic HIPK3 backsplicing introns can mediate splicing of large circRNA products.
circRNAs can be highly expressed using AAV vectors in cardiac and muscle tissue
To gather further insight into circRNA backsplicing and IRES-mediated translation efficiency in vivo, we packaged DNA constructs containing a cytomegalovirus (CMV) promoter driving (1) the original HIPK3 intron and poliovirus IRES or (2) the LΔRΔ intron and poliovirus IRES with the split GFP exon and flanked by inverted terminal repeats into AAV vectors (Figure 5A).17 Mice in each cohort were injected intravenously with 3 × 1011 vector genomes per animal, and tissues were harvested at 4 weeks post-injection. The AAV vector genome copy numbers were statistically indistinguishable between cohorts, confirming equivalent dosing (Figure 5B). We then assayed circRNA expression in cardiac, liver, and skeletal muscle tissues. Quantitative RT-PCR was performed using a primer pair across the circRNA backsplice junction and revealed that the LΔRΔ intron pair led to significantly higher circRNA expression: ∼4-fold higher expression in heart and liver and ∼50-fold higher expression in muscle compared to the original HIPK3 introns. In particular, circRNA expression in skeletal muscle was detected at very low levels with the original HIPK3 introns, while expression dramatically increased with the LΔRΔ intron pair to levels comparable to the heart and liver (Figure 4C). Immunofluorescent staining of sectioned tissue for GFP confirmed that the LΔRΔ construct demonstrated significantly higher GFP expression in the heart (Figure 5D) as well as skeletal muscle (Figure 5E). GFP expression in the liver was low with both constructs, despite the difference seen in circRNA expression, indicating that the poliovirus IRES does not translate efficiently in liver (Figure S8). These results demonstrate that the altering intronic distances can be exploited to regulate synthetic circRNA levels in different tissues in vivo.
Figure 5.

The LΔRΔ intron pair afford increased circRNA expression in multiple murine tissues in vivo
(A) The indicated constructs were packaged into recombinant AAV9 vectors and injected intravenously into C57BL/6 mice (n = 5) at a dose of 3 × 1011 vector genomes per animal, then harvested 4 weeks post-injection. (B) AAV vector genomes per cell were quantified in each tissue by quantitative PCR (qPCR) for the CMV promoter, normalized to the mouse lamin B2 locus. (C) Quantitative RT-PCR performed with primers amplifying GFP across the backsplice junction revealed increased circRNA expression with the LΔRΔ intron pair. circRNA expression is graphed relative to GAPDH (left) or normalized to HIPK3 Polio expression in each tissue (right). (D) Immunofluorescent staining of sectioned cardiac tissue for GFP expression. The level of GFP expression was quantified by the corrected total cell fluorescence (CTCF) method (right). (E) Immunofluorescent staining of sectioned skeletal muscle tissue showing GFP expression. The level of GFP expression was quantified by the CTCF method (right). Student’s t test was performed to test for statistical significance. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.0005; ∗∗∗∗p < 0.00005.
Discussion
To date, the best-characterized cis elements associated with backspliced circRNAs are inverted repeat sequences such as Alu elements.1,16 Current models postulate that these inverted repeat sequences base pair, bringing the splice sites in physical proximity. Our studies provide further insight into the role of intronic and exonic elements that can influence backsplicing efficiency. First, the physical spacing between the base pairing and splice sites does not appear to follow a simple relationship. Instead, we discovered an asymmetric effect, wherein the upstream inverted repeat must be within a few hundred nucleotides of the splice acceptor site and the downstream inverted repeat can be separated from the splice donor site by >2 kb. This spacing effect appears to be conserved for three different intron pairs derived from two distinct organisms, suggesting that these principles could be applied to many or all complementarity-driven circRNA intron pairs in designing synthetic circRNAs.
Given that it is detrimental to move the upstream repeat element far from the splice site, we reasoned that moving the Alu element closer may increase expression. In the HIPK3 intron pair, we removed ∼700 nt, which synergistically and significantly increased circRNA expression. This effect was observed in multiple transformed cell lines as well as various mouse tissues. Interestingly, the deletions “activated” circRNA expression in skeletal muscle tissue, which otherwise expressed extremely low levels. While the mechanism behind this observation remains to be determined, our findings imply that tissue-specific circRNA expression can be achieved in a promoter-independent manner.
Second, when generating a circRNA for protein expression, the IRES is a critical element controlling translation.19,22,23 In our simplest interpretation, we postulated that different IRES elements (and exonic elements in general) could affect the strength of protein expression as well as the cell/tissue specificity. While this was indeed the case with different IRES elements, we also observed unanticipated effects on splicing. It should be noted that in all cases the intended circRNA was formed, and assays that directly detect only the desired species would not report findings of additional RNA species. The ability of a 3′ splice site within an IRES to activate weak upstream 5′ splice sites has been previously shown in reporters of translation.24 Indeed, in three out of four tested IRES elements, weak splice sites led to the removal of the entire IRES. This linear splice is consistent with known multi-exon circRNAs, in which intervening intron sequences are not contained in the mature circRNA. We also discovered that inclusion of the KSHV vFLIP IRES led to creation of multiple large circRNAs, which may represent concatemer formation. We therefore propose that when designing a synthetic circRNA, splicing analyses performed on sequences to be circularized could reveal additional species. In corroboration, we also demonstrate that removal or decrease in level of these aberrant splice products by mutation of weak splice sites is readily feasible. Lastly, we also demonstrate that the size of a backspliced circRNA can be increased significantly without decreasing expression. This observation is consistent with earlier reports that circularized exons tend to be longer than control exons1 and supports the notion that small exons would be sterically suboptimal for backsplicing to occur.
Interest in circRNA-based therapeutics has been growing,10,11,25 largely in part due to their long life span and resistance to RNA decay mechanisms. Although the current study investigated molecular aspects of circRNA formation, our findings provide a road map for rational design of synthetic circRNA cassettes. For instance, our discovery that additional inserts are tolerated within the right intron can be exploited to create bi-functional RNA molecules from a single template. Specifically, we demonstrate that a tRNA intron-derived circRNA can be incorporated within the intronic sequence without affecting backspliced circRNA formation. This insert could also be replaced by other non-coding RNAs such as miRNA precursors, shRNAs, tRNAs, or RNA aptamers. Such a design allows for both RNA species to be produced from the same RNA transcript, enabling co-regulated expression. Additionally, the LΔRΔ introns described in this work are a minimal intronic pair that increases circRNA expression significantly (both in cell culture and in vivo), implying that synthetic introns can be further engineered to improve backsplicing efficiency. Further, this approach also helped reduce the length of the expression cassette by two-thirds from the original intron, enabling packaging into recombinant AAV vectors.
It is also noteworthy to compare the platform described here to other systems for expressing synthetic circRNAs such as the Tornado system, which relies on ribozyme cleavage and tRNA ligation machinery,26 and the use of permutated group I self-splicing introns.27 Although each system has benefits and drawbacks, a backspliced circRNA platform provides several distinct advantages, including (1) creation of seamless circRNAs with no “scars” remaining in the mature product; (2) ability to generate large (>1 kb) circRNA with high efficiency; (3) control by RNA polymerase II (RNA Pol II) promoters, allowing for cell- and tissue-specific expression or controlled expression;28,29 (4) lack of immune response arising from exogenous linear RNAs;30,31 and (5) compatibility with AAV-based delivery systems that allow long-term gene expression in a wide range of tissues compared to non-viral delivery platforms.14,15 Overall, the molecular determinants regulating circRNA expression examined in this work not only provide insight into circRNA biogenesis but also provide design principles for engineering synthetic circRNAs for gene therapy applications.
Materials and methods
Plasmids
Plasmids containing portions of the HIPK3, ZKSCAN1, and Laccase2 genes were previously described.16,32 New vector cassettes were constructed using these naturally occurring intron sequences and cloning them into a plasmid backbone separated by a multiple cloning site. A cassette containing an IRES (EMCV, Polio, KSHV, or HCV) and split GFP was cloned between these intron sequences. The split GFP cassette used was derived from the circGFP plasmid.33 All plasmids were constructed in the pcDNA3.1 backbone. Random sequences were designed to match the G/C base content of the HIPK3 introns but controlled for lack of strong secondary structure. 1.5 kb randomized sequences for the left and right introns were then synthesized. Sequences were inserted into the introns or exons using PCR and restriction cloning. When necessary, site-directed mutagenesis was used to make 1–2 nt changes to create a restriction enzyme site.
Cell culture
HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Gaithersburg, MD, USA) supplemented with 10% FBS (Millipore-Sigma, Burlington, MA, USA) and 1% penicillin-streptomycin and maintained at 37°C and 5% CO2. Cells were seeded into 6-well plates and transfected at ∼70% confluency. Transfections were performed using 2.5 μg of plasmid DNA (test constructs were equimolar, with mass equalized using empty vector) and 12.5 μL of 1 mg/mL PEI-MAX 40,000 (Polysciences, Warrington, PA, USA).
U87 cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Gaithersburg, MD, USA) supplemented with 5% FBS (Millipore-Sigma, Burlington, MA, USA) and 1% penicillin-streptomycin and maintained at 37°C and 5% CO2. Cells were seeded into 6-well plates and transfected at ∼70% confluency. Transfections were performed using 2.5 μg of plasmid DNA (test constructs were equimolar, with mass equalized using empty vector) and Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s protocol.
Huh7 cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Gaithersburg, MD, USA) supplemented with 5% FBS (Millipore-Sigma, Burlington, MA, USA) and 1% penicillin-streptomycin and maintained at 37°C and 5% CO2. Cells were seeded into 6-well plates and transfected at ∼70% confluency. Transfections were performed using 2 μg of plasmid DNA (test constructs were equimolar, with mass equalized using empty vector) and Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) using equal amounts of P3000 and Lipofectamine 3000 reagents, with the transfection mixture removed and replaced with fresh medium after 24 h.
For all cell types, cells were imaged 4 days post-transfection using an EVOS FL epifluorescence cell imaging system (AMC/Life Technologies, Carlsbad, CA, USA) equipped with the GFP light cube (excitation 470 nm, emission 510 nm) or the Texas Red light cube (excitation 585 nm, emission 624 nm). Cells were harvested from the plate by pipetting with cold 1× PBS (HEK293, U87) or trypsinization (Huh7). The cell suspension was separated into two tubes and centrifuged at 300 × g for 4 min at 4°C. Cell pellets were suspended in either 1× Passive Lysis Buffer (Promega, Madison, WI, USA) or TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), placed on a rocker for 10 min at 4°C, and then stored at −80°C prior to use. Lysates in Passive Lysis Buffer were centrifuged for 5 min at 16,000 × g at 4°C to remove cell debris prior to freezing.
Western blotting
Lysates were recovered using 1× Passive Lysis Buffer (Promega, Madison, WI, USA) and stored at −80°C. Equal volumes of samples were denatured in sample buffer (200 mM DTT, 100 mM Tris pH 8, 2.5% SDS, 20% glycerol) and heated to 100°C for 5 min before separation on a 10% Tris-glycine gel and transfer to a nitrocellulose membrane. Membranes were blocked overnight at 4°C in 5% non-fat milk in 1× TBST. Membranes were blotted with primary antibody against GFP (SC9996 1:1,000, Santa Cruz Biotechnology, Dallas, TX, USA) or Actin (GT5512 1:10,000, GeneTex, Irvine, CA, USA). Stabilized peroxidase-conjugated anti-mouse antibody was used as a secondary antibody (NA931V 1:3,000, GE Healthcare, Chicago, IL, USA). Blots were developed using SuperSignal West Femto substrate (Thermo Scientific, Waltham, MA, USA) and visualized by the Chemiluminescence High Sensitivity protocol on a ChemiDoc XRS+ (Bio-Rad Laboratories, Hercules, CA, USA). Quantifications were performed in ImageJ using the gel analysis method.34
RNA extraction
RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s protocol. RNA was resuspended in nuclease-free water and stored at −80°C until use.
Northern blotting
5 μg of RNA was resuspended in denaturing buffer (67% deionized formamide, 6.7% formaldehyde, 1× morpholino propanesulfonate [MOPS] running buffer), incubated at 65°C for 7 min, and cooled on ice. Samples were separated on a 1.2% denaturing formaldehyde-agarose gel in 1× MOPS running buffer and subsequently transferred to Hybond N+ membrane (GE Healthcare, Chicago, IL, USA). Radiolabeled probe was generated using the Prime-It II Random Primer Labeling Kit (Agilent Technologies, Santa Clara, CA, USA) according to manufacturer’s instructions. DNA templates for probe labeling were generated by PCR or restriction enzyme digest from a plasmid template. Radiolabeled probe was purified using illustra MicroSpin G-50 columns (GE Healthcare, Chicago, IL, USA), following the manufacturer’s protocol. Probe was then hybridized to the membrane in Rapid-Hyb buffer (GE Healthcare, Chicago, IL, USA) at 55°C for 3 h. After washing, blots were visualized by exposure to film and radiolabel signal was quantified by exposure to a PhosphorImager screen followed by scanning and quantification using an Amersham Typhoon (GE Healthcare, Chicago, IL, USA). ImageQuant TL software was used to quantify blots using the 1D gel analysis tool; background was subtracted using the rolling circle method. For oligonucleotide probing, the oligo was labeled with radioactive ATP using T4 Polynucleotide Kinase (NEB, Ipswich, MA, USA), following the manufacturer’s protocol. Radiolabeled probe was purified as above and hybridized to membrane in Rapid-Hyb buffer (GE Healthcare, Chicago, IL, USA) at 42°C for 3 h. After washing, blots were visualized and quantified as described above.
In-gel tricRNA visualization
5 μg of RNA was resuspended in denaturing buffer (67% deionized formamide, 6.7% formaldehyde, 1× MOPS running buffer), incubated at 65°C for 7 min, and cooled on ice. Denatured samples were separated on a 10% Trisborate/EDTA (TBE)-urea gel run at 300 V. The gel was washed three times in water and then stained for 30 min in DFHBI-1T fluorescent probe staining solution (40 mM HEPES pH 7.4, 100 mM KCl, 1 mM MgCl2, 10 μM DFHBI-1T). The stained gel was imaged with an Amersham Typhoon (GE Healthcare, Chicago, IL, USA), using the 488 excitation/526 emission setting.
RNase R treatment
5 μg of RNA was treated with 5 units of RNaseR (Lucigen, Middleton, WI, USA) at 37°C for 10 min. Enzyme was inactivated at 95°C for 5 min. The resulting RNA was visualized by northern blotting. RNase R minus samples were treated in the same manner in the absence of enzyme.
Polyribosome isolation
HEK293 cells were seeded overnight into 10 cm plates and were transfected at ∼70% confluency using PEI Max with 8 μg of the indicated plasmids. 24 h post-transfection, cells were split and seeded 1:2 into 10 cm plates. Cells were grown to 60–70% confluency, then incubated in media containing cycloheximide (CHX, 100 μg/mL) for 10 min at 37°C, followed by two washes with ice-cold PBS containing CHX. Cells were lysed (20 mM Tris-HCl pH 7.4, 140 mM KCl, 5 mM MgCl2, 1 mM DTT, 1% Triton X-100) and passed through a 27 1/2-gauge needle five times to disrupt cell membranes. Lysates were spun to remove nuclei and spun again to remove remaining cellular debris. Clarified lysate was loaded on top of a linear 10–50% sucrose gradient prepared in polysome gradient buffer (20 mM Tris-HCl pH 7.4, 140 mM KCl, 5 mM MgCl2) and spun for 2 h at 32,000 rpm in a SW41 swinging bucket rotor with no break. Gradients were fractionated into 750 μL fractions with continuous monitoring of absorbance at 254 nm using a Brandel gradient fractionator system. RNA was extracted from each gradient fraction using TRIzol reagent according to the manufacturer’s directions and visualized by northern blot as described above.
Virtual northern procedure
5 μg of RNA was separated on a denaturing agarose as for a northern blot (see above). The gel was stained with ethidium bromide to visualize the ribosomal RNA. Pieces of the gel corresponding to the larger and smaller circRNAs were cut based on distance from the 18S rRNA (distances measured from a northern blot). RNA was extracted from the gel with a QIAquick Gel Extraction Kit (QIAGEN, Germantown, MD, USA), using a modified protocol (J. Knobloch, Heinrich Heine University, Düsseldorf, Germany). RNA was DNase treated using the Turbo DNA-free kit (Ambion, Austin TX, USA). Equal nanogram amounts of DNase-treated RNA were converted to cDNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA, USA). Products of this reverse transcription reaction were utilized as template for PCR using primers specific to the backsplice (5′-CTGCTTGTCGGCCATGATATAGACGTTGTGGC-3′, 5′-CAAGCTGACCCTGAAGTTCATCTGCACCACC-3′) or primers spanning the IRES element (5′-GGCCGACAAGCAGAAGAACGGCATCAAG-3′, 5′-GGTGGTGCAGATGAACTTCAGGGTCAGCTTG-3′), followed by sequencing of the purified PCR products.
Recombinant AAV vector production
A triple plasmid transfection protocol with modifications was used to generate recombinant AAV vectors. Briefly, the transfection mixture contained (1) the pXR helper plasmid; (2) the adenoviral helper plasmid pXX6-80; and (3) the indicated transgene, driven by a CMV promoter and a SV40 polyA, flanked by AAV2 inverted terminal repeats (ITRs). Vector purification was carried out following polyethylene glycol (PEG) precipitation (8% w/v) from media supernatant using iodixanol gradient ultracentrifugation and desalting with ZebaSpin desalting columns (40K MWCO, Thermo Scientific, Waltham, MA, USA). Vector genome (vg) titers were obtained by quantitative PCR (Lightcycler 480, Roche Applied Sciences, Pleasanton, CA, USA) using SYBR Green (Roche Applied Sciences, Pleasanton, CA, USA) and primers designed to selectively bind AAV2 inverted terminal repeats (forward, 5′-AACATGCTACGCAGAGAGGGAGTGG-3′; reverse, 5′-CATGAGACAAGGAACCCCTAGTGATGGAG-3′).
Intravenous administration of AAV vectors
Animal experiments reported in this study were conducted with C57BL/6 mice bred and maintained in accordance with NIH guidelines as approved by the Duke Institutional Animal Care and Use Committee (IACUC). Animals were injected intravenously through the tail vein with 3 × 1011 vg/animal. At 4 weeks post-injection, mice were overdosed with tribromoethanol (Avertin) (0.2 mL/10 g of 1.25% solution) via the intraperitoneal route. This was followed by transcardial perfusions of phosphate-buffered saline. Portions of the harvested organs (heart, liver, skeletal muscle) were cut and stored in RNAlater solution (Invitrogen, Waltham, MA, USA); the remainder was postfixed in 4% paraformaldehyde. For this study, n = 5 mice were injected for each construct.
Quantification of vector genome copy number in tissues
Genomic DNA was extracted from sections of fixed tissue using the QiaAmp DNA FFPE Tissue Kit (QIAGEN, Germantown, MD, USA). To calculate viral genome copy numbers, quantitative PCR was performed with primers specific to the CMV promoter (5′-CAAGTACGCCCCCTATTGAC-3′ and 5′-AAGTCCCGTTGATTTTGGTG-3′). The vector genome copy numbers were normalized to the mouse lamin B2 locus as the housekeeping gene (primers 5′-GGACCCAAGGACTACCTCAAGGG-3′ and 5′-AGGGCACCTCCATCTCGGAAAC-3′).
RT-PCR
5 μg of RNA was DNase treated using the Turbo DNA-free kit (Ambion, Austin, TX, USA). Equal nanogram amounts of DNase-treated RNA were converted to cDNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA, USA). Products of this reverse transcription reaction were utilized as template for PCR (or quantitative PCR) using gene-specific primers for GFP (5′-CTGCTTGTCGGCCATGATATAGACGTTGTGGC-3′, 5′-CAAGCTGACCCTGAAGTTCATCTGCACCACC-3′) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (5′-CCACTCCTCCACCTTTGAC-3′, 5′-ACCCTGTTGCTGTAGCC-3′). Quantitative PCR was carried out using a Roche Light-Cycler 480 and SYBR Green Mastermix (Roche Applied Sciences, Pleasanton, CA, USA).
Tissue processing and immunohistochemistry
For heart and liver, 50-μm-thick sections were obtained from fixed tissue using a Leica VT 1200 S vibrating blade microtome (Leica Biosystems, Buffalo Grove, IL, USA). Immunohistochemical analyses of GFP expression were conducted using an antibody against GFP (G10632, Invitrogen, Waltham, MA, USA) and Alexa Fluor goat anti-rabbit 488 (1:500, Invitrogen A-11008) secondary antibody. Sections were mounted on slides in ProLong Gold Antifade Mountant with DAPI (P36935, Invitrogen, Waltham, MA, USA). Imaging was performed on an Aperio ScanScope XT (brightfield) or an Aperio ScanScope FL (fluorescence) at 20× magnification (Translational Pathology Lab, UNC). Fluorescence was quantified in ImageJ by the corrected total fluorescence method.34 Skeletal muscle samples were processed by the UNC Translational Pathology Lab for mounting and sectioning, followed by immunofluorescence (GFP ab13970, Abcam, Cambridge, UK) and slide scanning. Fluorescence was then quantified in ImageJ by the corrected total fluorescence method.34
Statistical analysis
Statistical analysis was carried out using an unpaired, one-tailed Student’s t test. Significance symbols are defined as follows: n.s., not significant; ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.0005; ∗∗∗∗p < 0.00005. Data are graphed as mean ± standard deviation.
Acknowledgments
We would like to thank Noah Legall and Dr. Alain Laederach at UNC-Chapel Hill for their help in designing the random insertion sequences. We would also like to thank Dr. A. Gregory Matera for providing the tricY-Broccoli construct. We thank the UNC Translational Pathology Laboratory (TPL) for expert technical assistance. This work was supported by the National Institutes of Health (R01NS-099371 to A.A., W.F.M., and J.E.W.; R01HL-089221, R01GM-127708, and UG3AR-075336 to A.A.; and 5T32GM-007092 to R.M.M.).
Author contributions
R.M.M., W.F.M., and A.A. conceived and designed the project. R.M.M., and J.L. designed and performed experiments. R.M.M., K.E.M. and M.M.F. performed mouse experiments. R.M.M., A.E.H. and N.J.M. performed polyribosome fractionation experiments. J.E.W. provided expertise and feedback. R.M.M., W.F.M., H.V. and A.A. wrote the paper with input from the authors.
Declaration of interests
A.A., W.F.M, and R.M.M. are inventors on patent applications filed on the subject matter of this manuscript. A.A. is a co-founder at TorqueBio, LLC.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2021.01.003.
Supplemental information
References
- 1.Jeck W.R., Sorrentino J.A., Wang K., Slevin M.K., Burd C.E., Liu J., Marzluff W.F., Sharpless N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. doi: 10.1261/rna.035667.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pyle A.M. Group II Intron Self-Splicing. Annu. Rev. Biophys. 2016;45:183–205. doi: 10.1146/annurev-biophys-062215-011149. [DOI] [PubMed] [Google Scholar]
- 3.Lu Z., Filonov G.S., Noto J.J., Schmidt C.A., Hatkevich T.L., Wen Y., Jaffrey S.R., Matera A.G. Metazoan tRNA introns generate stable circular RNAs in vivo. RNA. 2015;21:1554–1565. doi: 10.1261/rna.052944.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Panda A.C., De S., Grammatikakis I., Munk R., Yang X., Piao Y., Dudekula D.B., Abdelmohsen K., Gorospe M. High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res. 2017;45:e116. doi: 10.1093/nar/gkx297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y., Zhang X.O., Chen T., Xiang J.F., Yin Q.F., Xing Y.H., Zhu S., Yang L., Chen L.L. Circular intronic long noncoding RNAs. Mol. Cell. 2013;51:792–806. doi: 10.1016/j.molcel.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 6.Zhang X.O., Wang H.B., Zhang Y., Lu X., Chen L.L., Yang L. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–147. doi: 10.1016/j.cell.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Ashwal-Fluss R., Meyer M., Pamudurti N.R., Ivanov A., Bartok O., Hanan M., Evantal N., Memczak S., Rajewsky N., Kadener S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell. 2014;56:55–66. doi: 10.1016/j.molcel.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 8.Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 9.Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 10.Garikipati V.N.S., Verma S.K., Cheng Z., Liang D., Truongcao M.M., Cimini M., Yue Y., Huang G., Wang C., Benedict C. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat. Commun. 2019;10:4317. doi: 10.1038/s41467-019-11777-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavenniah A., Luu T.D.A., Li Y.P., Lim T.B., Jiang J., Ackers-Johnson M., Foo R.S. Engineered Circular RNA Sponges Act as miRNA Inhibitors to Attenuate Pressure Overload-Induced Cardiac Hypertrophy. Mol. Ther. 2020;28:1506–1517. doi: 10.1016/j.ymthe.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Q., Liu C., Li C.P., Xu S.S., Yao M.D., Ge H.M., Sun Y.N., Li X.M., Zhang S.J., Shan K. Circular RNA-ZNF532 regulates diabetes-induced retinal pericyte degeneration and vascular dysfunction. J. Clin. Invest. 2020;130:3833–3847. doi: 10.1172/JCI123353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madigan V.J., Asokan A. Engineering AAV receptor footprints for gene therapy. Curr. Opin. Virol. 2016;18:89–96. doi: 10.1016/j.coviro.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C., Samulski R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020;21:255–272. doi: 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- 15.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang D., Wilusz J.E. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014;28:2233–2247. doi: 10.1101/gad.251926.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meganck R.M., Borchardt E.K., Castellanos Rivera R.M., Scalabrino M.L., Wilusz J.E., Marzluff W.F., Asokan A. Tissue-Dependent Expression and Translation of Circular RNAs with Recombinant AAV Vectors In Vivo. Mol. Ther. Nucleic Acids. 2018;13:89–98. doi: 10.1016/j.omtn.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filonov G.S., Moon J.D., Svensen N., Jaffrey S.R. Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 2014;136:16299–16308. doi: 10.1021/ja508478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terenin I.M., Smirnova V.V., Andreev D.E., Dmitriev S.E., Shatsky I.N. A researcher’s guide to the galaxy of IRESs. Cell. Mol. Life Sci. 2017;74:1431–1455. doi: 10.1007/s00018-016-2409-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borchardt E.K., Meganck R.M., Vincent H.A., Ball C.B., Ramos S.B.V., Moorman N.J., Marzluff W.F., Asokan A. Inducing circular RNA formation using the CRISPR endoribonuclease Csy4. RNA. 2017;23:619–627. doi: 10.1261/rna.056838.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hurowitz E.H., Brown P.O. Genome-wide analysis of mRNA lengths in Saccharomyces cerevisiae. Genome Biol. 2003;5:R2. doi: 10.1186/gb-2003-5-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Douin V., Bornes S., Creancier L., Rochaix P., Favre G., Prats A.C., Couderc B. Use and comparison of different internal ribosomal entry sites (IRES) in tricistronic retroviral vectors. BMC Biotechnol. 2004;4:16. doi: 10.1186/1472-6750-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Othman Z., Sulaiman M.K., Willcocks M.M., Ulryck N., Blackbourn D.J., Sargueil B., Roberts L.O., Locker N. Functional analysis of Kaposi’s sarcoma-associated herpesvirus vFLIP expression reveals a new mode of IRES-mediated translation. RNA. 2014;20:1803–1814. doi: 10.1261/rna.045328.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baranick B.T., Lemp N.A., Nagashima J., Hiraoka K., Kasahara N., Logg C.R. Splicing mediates the activity of four putative cellular internal ribosome entry sites. Proc. Natl. Acad. Sci. USA. 2008;105:4733–4738. doi: 10.1073/pnas.0710650105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L., Han B., Zhang Z., Wang S., Bai Y., Zhang Y., Tang Y., Du L., Xu L., Wu F. Extracellular Vesicle-Mediated Delivery of Circular RNA SCMH1 Promotes Functional Recovery in Rodent and Nonhuman Primate Ischemic Stroke Models. Circulation. 2020;142:556–574. doi: 10.1161/CIRCULATIONAHA.120.045765. [DOI] [PubMed] [Google Scholar]
- 26.Litke J.L., Jaffrey S.R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 2019;37:667–675. doi: 10.1038/s41587-019-0090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wesselhoeft R.A., Kowalski P.S., Anderson D.G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. 2018;9:2629. doi: 10.1038/s41467-018-05096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng C., Baum B.J. Evaluation of promoters for use in tissue-specific gene delivery. Methods Mol. Biol. 2008;434:205–219. doi: 10.1007/978-1-60327-248-3_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X., Wu J.M., Hornischer K., Kel A., Wingender E. TiProD: the Tissue-specific Promoter Database. Nucleic Acids Res. 2006;34:D104–D107. doi: 10.1093/nar/gkj113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y.G., Kim M.V., Chen X., Batista P.J., Aoyama S., Wilusz J.E., Iwasaki A., Chang H.Y. Sensing Self and Foreign Circular RNAs by Intron Identity. Mol. Cell. 2017;67:228–238.e5. doi: 10.1016/j.molcel.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wesselhoeft R.A., Kowalski P.S., Parker-Hale F.C., Huang Y., Bisaria N., Anderson D.G. RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In Vivo. Mol. Cell. 2019;74:508–520.e4. doi: 10.1016/j.molcel.2019.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kramer M.C., Liang D., Tatomer D.C., Gold B., March Z.M., Cherry S., Wilusz J.E. Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins. Genes Dev. 2015;29:2168–2182. doi: 10.1101/gad.270421.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Wang Z. Efficient backsplicing produces translatable circular mRNAs. RNA. 2015;21:172–179. doi: 10.1261/rna.048272.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.