

Abstract
6-phosphofructo-2-kinase (PFKFB3) is a crucial regulator of glycolysis that has been implicated in angiogenesis and the development of diverse diseases. However, the functional role and regulatory mechanism of PFKFB3 in early-onset preeclampsia (EOPE) remain to be elucidated. According to previous studies, noncoding RNAs play crucial roles in EOPE pathogenesis. The goal of this study was to investigate the functional roles and co-regulatory mechanisms of the metastasis-associated lung adenocarcinoma transcript-1 (MALAT1)/microRNA (miR)-26/PFKFB3 axis in EOPE. In our study, decreased MALAT1 and PFKFB3 expression in EOPE tissues correlates with endothelial cell (EC) dysfunction. The results of in vitro assays revealed that PFKFB3 regulates the proliferation, migration, and tube formation of ECs by modulating glycolysis. Furthermore, MALAT1 regulates PFKFB3 expression by sponging miR-26a/26b. Finally, MALAT1 knockout reduces EC angiogenesis by inhibiting PFKFB3-mediated glycolysis flux, which is ameliorated by PFKFB3 overexpression. In conclusion, decreased MALAT1 expression in EOPE tissues reduces the glycolysis of ECs in a PFKFB3-dependent manner by sponging miR-26a/26b and inhibits EC proliferation, migration, and tube formation, which may contribute to abnormal angiogenesis in EOPE. Thus, strategies targeting PFKFB3-driven glycolysis may be a promising approach for the treatment of EOPE.
Keywords: PFKFB3, endothelial cells, angiogenesis, glycolysis, early-onset preeclampsia
Graphical Abstract
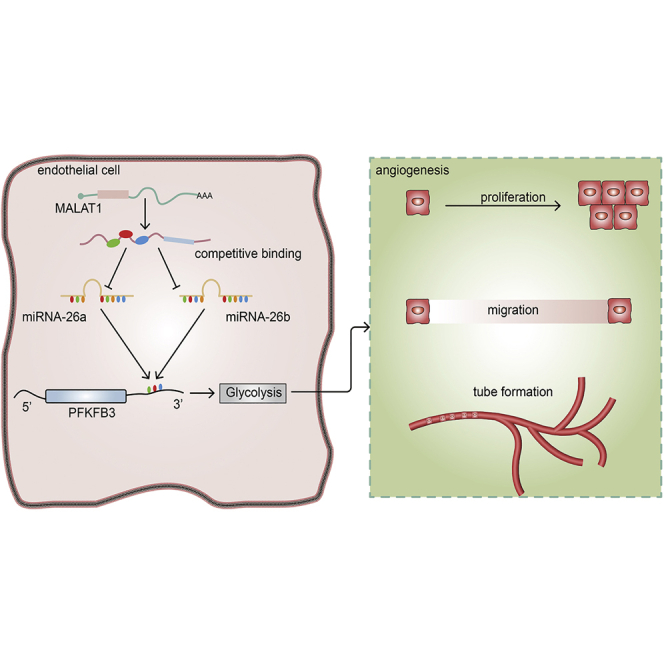
This study showed that decreased MALAT1 modulates PFKFB3-driven glycolysis through sponge miR-26a/26b in ECs, thus contributing to the abnormal angiogenesis observed in patients with early-onset preeclampsia (EOPE). These findings have improved our understanding of the mechanism of action of the MALAT1/miR-26/PFKFB3 axis in EOPE and provide PFKFB3 as a promising therapeutic target.
Introduction
Preeclampsia (PE) is a heterogeneous disease that affects 3%–5% of all pregnant women.1 According to the time of onset, PE is commonly classified into two major subtypes: early-onset PE (EOPE; ≤34 weeks) and late-onset PE (LOPE; >34 weeks). Based on accumulating evidence, EOPE and LOPE are two distinct disorders,2,3 with EOPE exhibiting a greater placental pathology than LOPE.4,5 The appropriate formation of placental blood vessels ensures an appropriate supply of blood containing oxygen and nutrients to the fetus, which is a prerequisite for the uneventful progression of pregnancy.6 Abnormal placental vascular development leads to the occurrence of placental vascular diseases and can affect fetal intrauterine development, severe cases of which endanger the safety of the mother and the fetus.7 Endothelial cells (ECs) play active roles in angiogenesis in both healthy and disease states.8 Vascular EC dysfunction reduces placental angiogenesis and promotes the onset of preeclampsia,9,10 but its exact pathophysiology remains to be elucidated.
Before 2009, vascular ECs were believed to be regulated only by pro-angiogenic growth factor signals. Recent studies have shown that ECs rely on specific metabolic pathways to undergo angiogenesis.11 The different metabolic pathways of ECs, such as glycolysis, fatty acid oxidation (FAO), and the pentose phosphate pathway (PPP), play important roles in vessel formation.12,13 Among these pathways, glycolysis is the primary mechanism by which ECs generate energy, with over 85% of ATP in ECs produced via glycolysis.12,13 Despite the immediate availability of oxygen in the blood, vascular ECs primarily consume glucose to generate energy. The 6-phosphofructo-2-kinase (PFKFB3) isozyme belongs to the PFKFB family. PFKFB enzymes synthesize fructose-2,6-bisphosphate (F-2,6-BP), a strong allosteric activator of PFK-1 that is involved in the rate-limiting step of glycolysis.14,15 Of all PFKFB isoenzymes, PFKFB3 exhibits much higher kinase activity than phosphatases (700 times), allowing it to function as an effective glycolytic activator that increases the glycolysis rate.16, 17, 18 PFKFB3 is widely expressed in different organs and cells, such as breast, ovary, and thyroid cells, and plays corresponding roles in cell proliferation, migration, and tumor angiogenesis.17 However, few reports have described the roles of PFKFB3 and glycolysis in PE. Therefore, additional studies are needed to determine the mechanisms by which PFKFB3-driven glycolysis exerts its pathological effects on PE.
Recently, a large number of noncoding RNAs, including microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), have attracted increased attention due to their biological effects on PE initiation and progression.19,20 As shown in previous studies, microRNA (miR)-26a and miR-26b expression is upregulated in the placenta and plasma of women with PE.21, 22, 23 Notably, miR-26a and miR-26b are members of the miR-26 family.24 Interestingly, both of these miRNAs bind the 3′ untranslated region (3′ UTR) of PFKFB3 to regulate its expression, allowing them to regulate tumor cell migration and invasion by targeting glycolysis.25,26 Other studies of miR-26a/26b have also revealed their involvement in EC angiogenesis.27,28
With the consideration of the expression of miR-26a/26b in PE tissues and potential regulatory effects on PFKFB3, we conducted a bioinformatics analysis that predicted a binding interaction between miR-26a/26b and the lncRNA metastasis-associated lung adenocarcinoma transcript-1 (MALAT1). MALAT1 was previously shown to be downregulated in PE tissues29,30 and has been reported to regulate the function of trophoblast and mesenchymal stem cells in the pathogenesis of PE.30, 31, 32 However, no study has explored whether MALAT1 affects EC behavior to exert its function in PE. Additionally, recent studies have highlighted the potential role of MALAT1 as a competing endogenous RNA (ceRNA), a class of lncRNAs that is capable of serving as “molecular sponges” for miRNAs, thereby modulating their downstream functions.33,34 Therefore, we hypothesized that MALAT1 may function as a ceRNA of miR-26a/26b to regulate the expression of PFKFB3.
In this study, we observed decreased MALAT1 and PFKFB3 expression in EOPE tissues. Further experiments revealed that PFKFB3 overexpression promotes EC proliferation, migration, and tube formation by activating glycolysis, whereas PFKFB3 silencing exerts the opposite effect. Then, we showed that MALAT1 functions as a ceRNA of miR-26a and miR-26b and regulates the expression of PFKFB3 in ECs. Furthermore, we indicated that MALAT1 knockdown reduced the angiogenesis of ECs by inhibiting PFKFB3-mediated glycolysis flux, whereas overexpression of PFKFB3 reversed this effect. These data provide novel insights into the roles of the MALAT1/miR-26/PFKFB3 axis in EOPE pathogenesis.
Results
Decreased expression of MALAT1 and PFKFB3 in patients with EOPE
We first measured the relative levels of MALAT1 and the PFKFB3 mRNA in 16 placental tissues from patients with EOPE and 16 normal control tissue samples using quantitative real-time PCR to determine whether MALAT1 and PFKFB3 are involved in EOPE pathogenesis. The MALAT1 and PFKFB3 mRNA levels were significantly decreased in placental tissues from patients with EOPE compared with normal placental tissues (p < 0.001; Figures 1A and 1B). Then, we performed a fluorescence in situ hybridization (FISH) assay to detect the expression and localization of MALAT1 in the tissues. MALAT1 was predominately localized in the cytoplasm and was significantly downregulated in placental tissues from patients with EOPE compared with normal tissues (Figure 1C). We also examined the expression of the PFKFB3 protein in these patients using western blotting (WB) and immunohistochemistry (IHC) analyses. The expression of the PFKFB3 protein was decreased in placental tissues from patients with EOPE compared with normal tissues. Representative images of WB and IHC analyses are shown in Figures 1D and 1E.
Figure 1.

MALAT1 and PFKFB3 expression in tissues from patients with EOPE
(A and B) The relative expression levels of the MALAT1 mRNA (A) and PFKFB3 mRNA (B) in 16 tissues from patients with EOPE and 16 tissues from normal controls were analyzed using quantitative real-time PCR. (C) Representative FISH images of MALAT1 expression in normal tissues and tissues from patients with EOPE. (D) Representative WB images of PFKFB3 levels in normal tissues and tissues from patients with EOPE. (E) Representative IHC images of PFKFB3 expression in normal tissues and tissues from patients with EOPE. All images were obtained at 200× magnification. The data are presented as the mean ± SD of three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 using Student’s t test.
PFKFB3 regulates glycolytic activity and EC angiogenesis
We expanded the overexpression and knockout study to human umbilical vein ECs (HUVECs), which are primary cells derived from the normal human umbilical vein, to study the role of PFKFB3 in more detail. We transfected HUVECs with overexpression plasmids and small interfering RNAs (siRNAs), which effectively upregulated or downregulated PFKFB3 mRNA and protein expression, respectively (p < 0.001; Figures S1A−S1E). After assessing the knockdown efficacy of siRNAs targeting PFKFB3, siRNA-3 was identified as exhibiting the best PFKFB3 knockdown efficacy and was used in subsequent experiments (p < 0.0001; Figure S1C).
Since enhanced glycolysis flux is essential for EC activity during angiogenesis, we first confirmed that PFKFB3 regulates glucose metabolism in ECs. We measured the cellular levels of ATP, nicotinamide adenine dinucleotide phosphate (NADPH), reactive oxygen species (ROS), and extracellular lactate production to represent glycolytic activity in ECs. ECs produce lactate to generate ATP during glycolysis, and an increase in glycolysis reduces ROS production and protects ECs from hyperoxic microenvironments.14 NADPH is produced by a side pathway of glycolysis and is used in fatty acid synthesis to promote cell proliferation.35 PFKFB3 overexpression increased the cellular levels of ATP, NADPH, and extracellular lactate while decreasing the ROS levels, whereas the silencing of PFKFB3 produced the exact opposite effects (p < 0.05; Figures 2A and 2B).
Figure 2.

PFKFB3 regulates glycolytic activity and EC angiogenesis
Cells were transfected with the vector, PFKFB3 plasmid, si-NC, or si-PFKFB3. (A and B) The extracellular lactate production and the cellular levels of ATP, NADPH, and ROS were measured in ECs. (C and D) The proliferation of ECs was determined using CCK-8 assays. (E and F) The cell cycle phases of ECs were determined using flow cytometry. (G and H) The migration of ECs was determined using Transwell assays. (I and J) Tube formation of ECs was determined using tube-formation assays. (K and L) Filopodia (arrowheads) and lamellipodia (arrows) were measured in ECs stained with Alexa Fluor 488-labeled phalloidin. All images in (G) and (H) were obtained at 200× magnification, whereas images in (I) and (J) were obtained at 100× magnification, and images in (K) and (L) were obtained at 1,000× magnification. The data are presented as the mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 using Student’s t test.
The proliferation, migration, and tube-formation abilities of ECs are crucial for angiogenesis. Therefore, we performed Cell Counting Kit-8 (CCK-8), Transwell, and tubule-formation experiments to study the effects of PFKFB3 on cell proliferation, migration, and tube formation. CCK-8 assays showed that overexpression of PFKFB3 increased cell proliferation, and PFKFB3 silencing significantly reduced the cell proliferation rate (p < 0.05; Figures 2C and 2D). Next, a flow cytometry analysis was performed to examine whether PFKFB3 affected the proliferation of ECs by altering cell cycle progression. Overexpression of PFKFB3 increased the percentage of cells in the S phase, and PFKFB3 silencing induced cell cycle arrest at the G0/G1 phase (p < 0.05; Figures 2E and 2F). Cell migration and tube-formation abilities were further measured using Transwell and tubule-formation experiments. As shown in Figures 2G−2J, PFKFB3 overexpression increased the migration and tubule formation of ECs, whereas the silencing of PFKFB3 exerted the opposite effect (p < 0.05). Cellular protrusions have an important role in cell migration.36 We then explored whether PFKFB3 regulated the motility of ECs by altering cell protrusion formation. The protrusive structures at the leading edge of a motile cell consist of lamellipodia and filopodia. Alexa Fluor 488-labeled phalloidin staining indicated that PFKFB3 overexpression increased the length of filopodia and the area of lamellipodia in ECs, whereas PFKFB3 silencing led to shorter filopodia and smaller lamellipodia (p < 0.01; Figures 2K and 2L).
In summary, PFKFB3 overexpression increased glycolytic activity and the proliferation, migration, and tubule formation of ECs. PFKFB3 overexpression increased the proliferation of ECs by accelerating the cell cycle and increased the motility of ECs by promoting the formation of cell protrusions. The silencing of PFKFB3 exerted the opposite effects.
MALAT1 functions as a ceRNA of miR-26a and miR-26b and regulates the expression of PFKFB3 in ECs
According to previous studies, the expression of MALAT1 is decreased, and miR-26a/26b expression is increased in PE.21, 22, 23,29,30 Therefore, we transfected short hairpin (sh)-MALAT1 and miR-26a/26b mimic plasmids into ECs and detected the expression of PFKFB3. shRNAs targeting MALAT1 were designed and assessed for knockdown efficacy; shRNA-5277 displayed the best MALAT1 knockdown efficacy and was used in subsequent experiments (p < 0.0001; Figure S1F). MALAT1 knockdown in ECs significantly downregulated PFKFB3 expression at the mRNA and protein levels, while regulating the miR-26a/26b mRNA levels (p < 0.01; Figures 3A and 3B). Moreover, the miR-26a/26b mimic reduced the levels of the PFKFB3 mRNA and protein (p < 0.001; Figure 3C), consistent with the effect of sh-MALAT1 on the expression of PFKFB3.
Figure 3.

MALAT1 regulates PFKFB3 expression via miR-26a and miR-26b in ECs
(A and B) After transfection with sh-NC and sh-MALAT1, the mRNA expression levels of MALAT1, miR-26a, miR-26b, and PFKFB3 (A) and the protein expression level of PFKFB3 (B) were measured in ECs. (C) PFKFB3 mRNA and protein expression in ECs transfected with the mimic-NC, miR-26a mimic, and miR-26b mimic. (D) Schematic representation of the putative miR-26a and miR-26b binding sites in MALAT1. (E) Dual-luciferase reporter assay in cells co-transfected with the MALAT1 WT or MUT reporter plasmid and miR-26a/26b. (F) RNA pull-down assays were performed using biotin-labeled sense or antisense MALAT1, and quantitative real-time PCR analyses for MALAT1 and miR-26a/26b expression were performed using the pull-down products. (G) Schematic representation of the putative miR-26a and miR-26b binding sites in PFKFB3. (H) Dual-luciferase reporter assay in cells co-transfected with the PFKFB3 WT or MUT reporter plasmid and miR-26a/26b. (I and J) After transfection with sh-NC, sh-MALAT1, the sh-NC + miR-26a inhibitor, and the sh-MALAT1 + miR-26a inhibitor, the mRNA expression levels of MALAT1, PFKFB3, and miR-26a (I) and the protein expression level of PFKFB3 (J) were measured in ECs. (K and L) After transfection with sh-NC, sh-MALAT1, the sh-NC + miR-26b inhibitor, and the sh-MALAT1 + miR-26b inhibitor, the mRNA expression levels of MALAT1, PFKFB3, and miR-26b (K) and the protein expression level of PFKFB3 (L) were measured in ECs. The data are presented as the mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 using one-way ANOVA and Student’s t test. n.s., not significant.
Based on these findings, we next speculated that MALAT1 might regulate PFKFB3 expression by sponging miR-26a/26b. We used RNA hybrid software to predict the possible binding sites for miR-26a/26b on MALAT1 and PFKFB3 and verified them by conducting dual-luciferase experiments (Figures 3D, 3E, 3G, and 3H). The miR-26a/26b mimic reduced the luciferase activity of wild-type (WT) but not mutant (MUT) MALAT1 (p < 0.05; Figure 3E). Thus, miR-26a/26b directly binds to MALAT1. We performed RNA pull-down assays followed by quantitative real-time PCR to further verify the correlation between MALAT1 and miR-26a/26b expression. Compared with the antisense group, the expression of miR-26a/26b in the sense group was significantly upregulated, and miR-26a/26b was pulled down by sense MALAT1 (p < 0.001; Figure 3F). These data validated the interaction between MALAT1 and miR-26a/26b in ECs. Another assay revealed that the miR-26a/26b mimic reduced the luciferase activity of the WT PFKFB3 construct but not the MUT PFKFB3 construct (p < 0.001; Figure 3H). Based on these results, miR-26a/26b directly targets the 3′ UTR of PFKFB3. We co-transfected sh-MALAT1 and miR-26a/26b inhibitors into cells and then assessed PFKFB3 expression to elucidate the associated mechanism. As expected, the inhibition of miR-26a/26b in MALAT1-silenced cells reversed the decrease in PFKFB3 mRNA and protein expression (p < 0.05; Figures 3I−3L). Therefore, MALAT1 might regulate PFKFB3 expression by sponging miR-26a/26b.
MALAT1 regulates glycolytic activity and EC angiogenesis through PFKFB3
Cells were divided into four groups and transfected with the sh-negative control (NC), sh-MALAT1, sh-NC + PFKFB3 plasmid, or sh-MALAT1 + PFKFB3 plasmid, which effectively upregulated or downregulated the expression of MALAT1 and PFKFB3, respectively, to further explore the interaction between MALAT1 and PFKFB3 in the regulation of glycolytic activity and angiogenesis in ECs (p < 0.001; Figures 4A and 4B).
Figure 4.

MALAT1 regulates glycolytic activity and EC angiogenesis through PFKFB3
Cells were transfected with sh-NC, sh-MALAT1, the sh-NC + PFKFB3 plasmid, or the sh-MALAT1 + PFKFB3 plasmid. (A) MALAT1 and PFKFB3 mRNA expression in ECs. (B) PFKFB3 protein expression in ECs. (C) Extracellular lactate production and the cellular levels of ATP, NADPH, and ROS were measured in ECs. (D) The proliferation of ECs was determined using CCK-8 assays. (E) The cell cycle phase of ECs was determined using flow cytometry. (F) The migration of ECs was determined using Transwell assays. (G) Tube formation of ECs was determined by performing tube-formation assays. (H) Filopodia (arrowheads) and lamellipodia (arrows) were measured in ECs stained with Alexa Fluor 488-labeled phalloidin. All images in (F) were obtained at 200× magnification, whereas images in (G) were obtained at 100× magnification, and images in (H) were obtained at 1,000× magnification. The data are presented as the mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 using one-way ANOVA.
Knockdown of MALAT1 significantly decreased the cellular ATP, NADPH, and extracellular lactate levels but increased the ROS levels, whereas PFKFB3 overexpression in MALAT1-silenced cells reversed these effects (p < 0.05; Figure 4C). Similarly, overexpression of PFKFB3 reversed the inhibitory effects on proliferation, cell cycle progression, migration, tube formation, and filopodia and lamellipodia formation induced by MALAT1 knockdown (p < 0.05; Figures 4D−4H). Taken together, MALAT1 knockdown reduces glycolytic activity and contributes to inhibit the angiogenesis of ECs through the glycolytic rate-limiting enzyme PFKFB3, whereas overexpression of PFKFB3 reverses these changes.
Discussion
PFKFB3 is a recently discovered regulator of cell metabolism, and it triggers robust glycolysis in ECs by producing F-2,6-BP. Furthermore, PFKFB3 is involved in the development of many diseases by controlling angiogenesis, apoptosis, drug resistance, and the tumor microenvironment.12 However, the pathophysiological mechanism of PFKFB3 in EOPE is poorly characterized. In this study, we documented a decrease in PFKFB3 and MALAT1 expression in placenta tissues from patients with EOPE. Further in vitro studies revealed that decreased MALAT1 expression downregulated PFKFB3 expression by functioning as a ceRNA of miR-26a/26b in ECs, which reduced glycolytic activity and contributed to abnormal angiogenesis in EOPE. MALAT1 knockdown inhibited the proliferation of ECs by inducing cell cycle arrest at the G0/G1 phase and decreased the motility of ECs by inhibiting cell protrusion formation in a PFKFB3-dependent manner. Overexpression of PFKFB3 improved the effects on angiogenesis caused by decreased MALAT1 expression in ECs. Based on these findings, the MALAT1/miR-26/PFKFB3 axis regulates EC angiogenesis by modulating glycolysis and plays a key role in the pathogenesis of EOPE.
The ceRNA hypothesis was first proposed by Salmena et al.37 in 2011 and describes how lncRNAs function as molecular sponges of miRNAs to regulate mRNA expression levels, which may have important functions in pathophysiological processes. Many studies have shown that ceRNAs are an important mechanism underlying the regulatory functions of MALAT1.38 Although the relevant miRNAs were shown to be able to regulate or be regulated by MALAT1, they are rarely reported in PE.31 In the present study, PFKFB3 expression was decreased when MALAT1 was knocked out, and a negative regulatory relationship was observed between MALAT1 and miR-26a/26b and between miR-26a/26b and PFKFB3. The binding sites for MALAT1 in miR-26a/26b and for miR-26a/26b in PFKFB3 were confirmed by the results of dual-luciferase experiments. RNA pull-down assays were performed to further verify the correlation between MALAT1 and miR-26a/26b expression. Finally, we co-transfected cells with sh-MALAT1 and miR-26a/26b inhibitors. The expression of PFKFB3 in the inhibitor-transfected group was increased compared with its expression in the control group transfected with only sh-MALAT1. These outcomes confirmed that MALAT1 regulated PFKFB3 expression by sponging miR-26a/26b via a ceRNA mechanism.
Decreased MALAT1 expression in tissues from patients with EOPE reduces glycolysis in ECs in a PFKFB3-dependent manner and inhibits EC proliferation, migration, and tube formation. Thus, we suggest that PFKFB3-driven glycolysis plays a role in EOPE pathogenesis. Yalcin et al.39,40 reported that F-2,6-BP, the product of PFKFB3, promotes cell cycle progression and controls cell proliferation by activating cyclin-dependent kinase 1 (Cdk1). Additionally, Jia et al.41 identified the interaction of PFKFB3 with Cdk4, which controls the transition from the G1 phase to S phase of the cell cycle. Compared to these studies, our research also supported the participation of PFKFB3 in the regulation of the cell cycle of ECs. MALAT1 knockdown caused cell cycle arrest at the G1/S phase, whereas overexpression of PFKFB3 reversed the effect.
On the other hand, we verified that PFKFB3 promotes protrusion formation, as PFKFB3 overexpression reversed the inhibitory effects of MALAT1 knockdown on filopodia and lamellipodia formation. The effect of PFKFB3 on cell motility is consistent with the findings reported by De Bock et al.14 and Schoors et al.,42 who revealed that PFKFB3 is important for EC movement and that PFKFB3-driven glycolysis regulates vessel sprouting. The study by De Bock and colleagues14 confirmed that glycolysis occurs in filopodia and lamellipodia during cell migration, whereas PFKFB3 binds to the actin cytoskeleton to promote high local ATP production required for cytoskeletal remodeling. Currently, the mechanisms underlying the effect of PFKFB3-driven glycolysis on EC angiogenesis have not been completely elucidated. According to recent studies, up- or downregulation of PFKFB3 expression does not alter the relative expression levels of genes involved in tip or stalk cell behavior, suggesting that glycolysis directly modulates angiogenesis without any genetic modulation.12,14,43 Thus, EC metabolism, in parallel with genetic signals, mediates angiogenesis. However, researchers have not clearly determined how ECs change their metabolic state, for example, from a static state to an active state, and the specific regulatory mechanism requires further study.
In addition to PFKFB3-mediated regulation of cell cycle progression and cell motility, many other glycolysis-related mechanisms are involved in angiogenesis and the development of diseases. For instance, lactate levels are increased as an end result of the glycolytic process, and lactate functioning as a signaling molecule stimulates angiogenesis by activating hypoxia-inducible factor 1-alpha (HIF-1α) and increasing the vascular endothelial growth factor (VEGF) levels44 or by inducing nuclear factor κB (NF-κB) activation and interleukin-8 (IL-8) expression.45 Additionally, the metabolite crosstalk between ECs and other cell types is also an interesting, yet outstanding, question. Based on accumulating evidence, communicating cells regulate metabolic pathways in opposing cells and may promote disease development via this mechanism.46 For example, in the heart, cardiomyocyte-derived exosomes modulate glycolysis in ECs.47 In addition to abundant vascular ECs, a large number of trophoblasts are also present in the placenta. However, little is known about the mechanism of metabolic regulation between ECs and trophoblasts. Further studies are needed to determine whether trophoblast metabolites affect the glycolysis of ECs and play a role in EOPE.
Although our study documented a role for PFKFB3-mediated glycolysis in angiogenesis, other metabolic pathways, such as the PPP, polyol pathway, and FAO pathway, may also influence this process.48, 49, 50 For example, FAO plays an important role in EC function. Carnitine palmitoyl transferase 1a (CPT1a) is a rate-controlling enzyme in the fatty acid metabolic pathway and is abundant in ECs. Decreased CPT1a levels result in vascular sprouting defects due to impaired cell proliferation, and glucose and glutamine metabolism are unable to compensate for this effect.50 Thus, different metabolic changes or differences in the activity levels of specific pathways may drive ECs through different mechanisms. However, different metabolic pathways are not completely independent, and they often exert synergistic effects. For example, the PPP is a side pathway of glycolysis that uses glucose-6-phosphate (G6P), an intermediate product of glycolysis, to generate NADPH for redox balance.51 In addition, NADPH also plays an important role in the synthesis of lipids, nucleotides, and amino acids and promotes the angiogenic activity of ECs.52 Many questions remain as to how ECs integrate the regulation of multiple metabolic pathways, although a few regulators have been identified. Therefore, a good practice is to study transcript and protein and metabolite levels using multiomics technologies to clarify the mechanism of EC metabolism. In addition to the reported metabolic pathway, these techniques may also recognize new metabolic pathways related to EC function.
Angiogenesis plays an important role in the development of EOPE. Therefore, many studies have been conducted to target this process by regulating angiogenic signals, such as VEGF. However, this effect may be lost by compensating for other angiogenic factors. Emerging evidence highlights that genetic and metabolic signals regulate angiogenesis. Therefore, we suggest that the regulation of angiogenesis by targeting PFKFB3-mediated glycolysis of ECs should be further investigated as a promising therapeutic strategy in EOPE treatment.
Conclusions
In summary, decreased MALAT1 and PFKFB3 expression in tissues from patients with EOPE correlated with EC dysfunction (Figure 5). By performing in vitro experiments, we showed that PFKFB3 regulates EC proliferation, migration, and tube formation by modulating glycolysis. Furthermore, we described a novel regulatory mechanism between MALAT1 and PFKFB3, where MALAT1 functions as a ceRNA of miR-26a/26b to regulate PFKFB3 expression in ECs. Finally, MALAT1 regulates EC angiogenesis via PFKFB3-driven glycolysis. Taken together, our results suggest that the MALAT1/miR-26a and miR-26b/PFKFB3 axes participate in regulating EC angiogenesis through effects on glycolysis. These findings have improved our understanding of the specific mechanism of action of the MALAT1/miR-26/PFKFB3 axis in EOPE and provide PFKFB3 as a potential therapeutic target for EOPE.
Figure 5.

A schematic diagram depicting how MALAT1 competitively binds miR-26a and miR-26b to regulate EC angiogenesis by modulating PFKFB3-driven glycolysis in ECs
Materials and methods
Tissue collection
Placental and umbilical cord tissues from the healthy control group (n = 16) and placental tissues from patients with EOPE (n = 16) were collected during cesarean section. These patients were aged 26–37 years. PE was diagnosed according to the criteria of the American College of Obstetricians and Gynecologists (ACOG).53 EOPE was defined as PE that developed prior to 34 weeks of gestation. Patients who smoked or had multiple gestations, fetal structural or genetic anomalies, maternal infection, and any other confounding pathology (diabetes mellitus, renal disease, chronic hypertension, hyperthyroidism, and hypothyroidism) were excluded.
After delivery of the placenta, specimens, approximately 3 to 5 cm from the umbilical cord attachment site (on the chorionic side, full thickness), were clipped and then washed with sterile phosphate-buffered saline (PBS). Each specimen was divided into two parts: one was fixed with 4% formaldehyde and then embedded in paraffin for IHC, and the other was frozen in liquid nitrogen to extract RNA and protein. HUVECs were extracted from normal cord samples for subsequent experiments. Informed consent was obtained with approval from the Local Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology. All patients signed an informed consent form before entering the study.
Cell culture
In this study, we used primary HUVECs to build a model of vascular ECs in vitro.
HUVECs were isolated from freshly obtained human umbilical cords of normal patients without EOPE. The tissues were digested enzymatically with collagenase type I (0.1%; Sigma-Aldrich, St. Louis, MO, USA) for 15 min at 37°C. After the incubation, the collagenase solution containing HUVECs was flushed from the cord by perfusion with EC medium (ECM) to stop digestion. Then, the filtrate was centrifuged at 1,000 rpm for 5 min to separate HUVECs from the collagenase, and HUVECs were resuspended in ECM containing 10% fetal bovine serum (FBS). The HUVECs were placed in a culture flask and incubated in an atmosphere with 5% CO2 at 37°C.
quantitative real-time PCR
Total RNA, including miRNAs, was extracted from the tissue samples and cultured cells using TRIzol Reagent (Vazyme Biotech, Nanjing, China). The cDNA templates were synthesized with a PrimeScript RT Reagent Kit (Takara, Tokyo, Japan), and quantitative real-time PCR was performed using a StepOnePlus Real-Time PCR system (Applied Biosystems, CA, USA) with a preset PCR program. β-Actin was used as an internal control to quantify mRNA expression, and U6 was used as an internal control to quantify miRNA expression. The quantitative real-time PCR primer sequences are shown in Table S1. The PCR cycling conditions were as follows: 95°C for 120 s, followed by 95°C for 10 s, 60°C for 30 s, and 72°C for 30 s, with a dissociation program consisting of 95°C for 60 s, 55°C for 60 s, and 95°C for 10 s. The relative expression levels of the target genes were calculated using the 2−ΔΔCt method.
FISH assay
The FISH assay was performed in paraffin-embedded placental tissues. Carboxyfluorescein (FAM)-labeled, MALAT1-specific probes were designed and synthesized by Siwega (Wuhan, China), and the probe sequence was 5′-GATTCTGTGTTATGCCTGGTTAGGTATGAGC-3′. Briefly, paraffin-embedded tissues were de-waxed and rehydrated. After prehybridization in PBS, the tissues were hybridized overnight at 37°C in hybridization solution. Then, cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; C1002; Beyotime, Jiangsu, China). Images were obtained using a fluorescence microscope (200× magnification; Nikon, Tokyo, Japan).
WB analysis
The total proteins extracted from cells and tissues were quantified with a bicinchoninic acid (BCA) protein assay kit (P0010S; Beyotime). The protein samples were incubated for 10 min at 95°C; then, proteins (30 mg) were subjected to 10% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (0.45 μm pore size; Millipore, MA, USA). The blots were incubated with 5% skim milk in Tris-buffered saline containing 0.05% Tween 20 (TBST) at room temperature for 1 h and then incubated overnight at 4°C with the rabbit polyclonal anti-PFKFB3 (1:1,000; 13763-1-AP; Proteintech, Chicago, IL, USA) or rabbit monoclonal anti-beta-actin (1:2,000; 20536-1-AP; Proteintech) primary antibodies. Subsequently, the membranes were washed with TBST and incubated with a secondary anti-rabbit antibody (1:4,000; Affinity Biosciences, OH, USA) for 1 h at room temperature. The proteins were visualized using the enhanced chemiluminescence method (WBKLS0500; Millipore), according to the manufacturer’s recommendations.
IHC analysis
The placental tissues were embedded in paraffin for the IHC analysis. The procedures for IHC detection are described below. First, for antigen retrieval, rehydrated paraffin sections were heated in citrate buffer (pH 6.0) for 15 min. Then, endogenous peroxidase activity was quenched by incubating the sections with 3% H2O2 for 20 min and blocking the sections with 10% normal goat serum for 1 h. After washing, the sections were incubated with the primary antibody against PFKFB3 (1:500; 13763-1-AP; Proteintech) at room temperature for 1 h. Finally, the slides were incubated with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG; 1:1,000; Santa Cruz, CA, USA) for 30 min. Diaminobenzidine tetrahydrochloride was used as a substrate, and sections were lightly counterstained with hematoxylin, dehydrated, and mounted.
Cell transfection
The pEX-1 vector (GenePharma, Shanghai, China) was used for PFKFB3 overexpression, and an empty plasmid vector was used as a control. PFKFB3 was silenced by siRNAs, and si-NC, serving as a NC, was purchased from RiboBio (Guangzhou, China). Three siRNAs targeting the PFKFB3 gene were designed and synthesized, and the most effective siRNA (si-3) identified by quantitative real-time PCR was utilized for further experiments. Four shRNAs targeting the MALAT1 gene were designed and synthesized by GenePharma, and the most effective shRNA (sh-5277) identified by quantitative real-time PCR was used for further experiments. The overexpression and inhibition of miR-26a/26b were achieved using miR-26a/26b mimics and miR-26a/26b inhibitors, respectively, with NC mimics and NC inhibitors serving as controls (GenePharma). These plasmids were transfected into cells using Lipofectamine 3000 Reagent (Thermo Fisher Scientific, Waltham, MA, USA) as required. The cells were transfected for 48 h and then collected for subsequent analyses.
Measurements of total lactate, ATP, and ROS levels and the NADPH/NADP+ ratio
Extracellular lactate, cellular ATP, ROS, and intracellular NADPH levels were measured using the following assay kits, according to the manufacturers’ instructions: lactate (K607-100; BioVision, Milpitas, CA, USA), ATP (S0027; Beyotime), ROS (S0033; Beyotime), and NADPH/NADP+ ratio (ab65349; Abcam, Cambridge, MA, USA).
Luciferase reporter assay
The 3′ UTR of PFKFB3 or MALAT1 containing miR-26a/26b putative binding sites was amplified and cloned into a pmirGlo vector (GenePharma). 1 day before transfection, cells were plated in 24-well plates at a density of 5 × 105 cells per well. The cells were co-transfected with the WT or MUT luciferase vectors and miR-26a, miR-26b, or the controls using Lipofectamine 3000 Reagent (Thermo Fisher Scientific). After a 48-h incubation, the relative luciferase activity was measured with a Dual-Luciferase Reporter System (Promega, Madison, WI, USA) using a Multimode Reader (Infinite M1000; Tecan, Switzerland).
RNA pull-down assay
Biotin-labeled sense and antisense MALAT1 sequences were synthesized by Genecreate (Wuhan, China). RNA pull-down assays were performed with a Magnetic RNA-Protein Pull-Down Kit (20164; Pierce, Rockford, IL, USA). According to the manufacturer’s instructions, biotinylated RNA was captured with streptavidin magnetic beads and then incubated with cell lysates. TRIzol was used to purify the bound RNAs, and quantitative real-time PCR was employed to measure the levels of MALAT1 and miR-26a/26b.
Cell proliferation assay
The cell proliferation assay was performed using a CCK-8 assay (C0038; Beyotime). Transfected cells were seeded on 96-well plates at a density of 2 × 103/100 μL cells per well. The proliferation level was determined at 0, 24, 48, and 72 h after transfection. The CCK-8 solution (10 μL) was added to each well, followed by 1 h of incubation in the dark at 37°C. The absorbance at 450 nm was measured using a Multimode Reader (Infinite F50; Tecan).
Cell cycle analysis using flow cytometry
Cells were fixed overnight at 4°C with 70% ethanol. Afterward, the cells were washed with PBS and stained with propidium iodide (PI). The cell cycle was analyzed using a flow cytometer (LSRFortessa; BD Biosciences, San Jose, CA, USA), and percentages of cells in the G0/G1, S, and G2/M phases were calculated using ModFit software.
Cell migration assay
Transwell units (24-well plates, insert membranes with 8 μm pores; Corning Costar, NY, USA) were not coated with Matrigel to investigate the cell migration capacities. In each well, transfected HUVECs (5 × 104 cells/well) were resuspended in 200 μL of serum-free ECM and placed in the upper chamber, and 500 μL of complete medium was added to the lower chamber. The Transwell units were incubated under an atmosphere with 5% CO2 at 37°C for 24 h; the cells and Matrigel were removed from the upper-membrane surface and stained with 0.1% crystal violet for 20 min at 37°C. The number of cells on the underside of the membrane was counted under a light microscope (200× magnification; Olympus, Tokyo, Japan). Five randomly selected fields were counted per insert.
Tube-formation assay
HUVECs (3 × 104 cells/well) were resuspended in 100 μL of conditioned medium and then seeded in 96-well plates coated with Matrigel (356234; BD Biosciences). After an incubation for 3−6 h at 37°C, tube-like structures formed and were imaged using a microscope (100× magnification; Olympus). Photomicrographs from each well were captured, and the total length of tubes was analyzed using ImageJ software (NIH, Bethesda, MD, USA).
Filopodia and lamellipodia formation assay
The protrusive structures at the leading edge of a motile cell are called lamellipodia and filopodia. Filopodia were defined as thin, finger-like structures that extend from the cell surface. Lamellipodia were defined as broad and flat sheet-like cellular protrusions.36
Cells were fixed with 4% formaldehyde, incubated with Alexa Fluor 488-labeled phalloidin, and the nuclei were stained with mounting medium containing DAPI. All images were captured using a confocal microscope (1,000× magnification; Olympus). The length of filopodia and the area of lamellipodia in cells were analyzed using ImageJ software.
Statistical analysis
All data are presented as the mean ± SD. Statistical analyses were performed using GraphPad Prism 5.01 software (GraphPad Software) and SPSS 19.0 statistical software (SPSS). For variables with a normal distribution, unpaired Student’s t test was used to determine the significance of differences between two groups, whereas one-way ANOVA was used for comparisons among three or more groups. p <0.05 was considered significant. All data were obtained from ≥3 independent experiments.
Acknowledgments
This project was supported by the National Natural Science Foundation of China (grant number 81873844).
Author Contributions
Q.L., Y.Zhao., and L.Z. conceived the study and wrote the paper. X.L., W.L., Y. Zhang, M.W., and Z.C. performed the experiments and analyzed the data. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2021.01.005.
Supplemental information
References
- 1.Mol B.W.J., Roberts C.T., Thangaratinam S., Magee L.A., de Groot C.J.M., Hofmeyr G.J. Pre-eclampsia. Lancet. 2016;387:999–1011. doi: 10.1016/S0140-6736(15)00070-7. [DOI] [PubMed] [Google Scholar]
- 2.Huppertz B. Placental origins of preeclampsia: challenging the current hypothesis. Hypertension. 2008;51:970–975. doi: 10.1161/HYPERTENSIONAHA.107.107607. [DOI] [PubMed] [Google Scholar]
- 3.Raymond D., Peterson E. A critical review of early-onset and late-onset preeclampsia. Obstet. Gynecol. Surv. 2011;66:497–506. doi: 10.1097/OGX.0b013e3182331028. [DOI] [PubMed] [Google Scholar]
- 4.Egbor M., Ansari T., Morris N., Green C.J., Sibbons P.D. Morphometric placental villous and vascular abnormalities in early- and late-onset pre-eclampsia with and without fetal growth restriction. BJOG. 2006;113:580–589. doi: 10.1111/j.1471-0528.2006.00882.x. [DOI] [PubMed] [Google Scholar]
- 5.Long W., Rui C., Song X., Dai X., Xue X., Lu Y., Shen R., Li J., Li J., Ding H. Distinct expression profiles of lncRNAs between early-onset preeclampsia and preterm controls. Clin. Chim. Acta. 2016;463:193–199. doi: 10.1016/j.cca.2016.10.036. [DOI] [PubMed] [Google Scholar]
- 6.Harris L.K., Benagiano M., D’Elios M.M., Brosens I., Benagiano G. Placental bed research: II. Functional and immunological investigations of the placental bed. Am. J. Obstet. Gynecol. 2019;221:457–469. doi: 10.1016/j.ajog.2019.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Burton G.J., Jauniaux E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 2018;218(2S):S745–S761. doi: 10.1016/j.ajog.2017.11.577. [DOI] [PubMed] [Google Scholar]
- 8.Eelen G., de Zeeuw P., Treps L., Harjes U., Wong B.W., Carmeliet P. Endothelial cell metabolism. Physiol. Rev. 2018;98:3–58. doi: 10.1152/physrev.00001.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts J.M., Taylor R.N., Musci T.J., Rodgers G.M., Hubel C.A., McLaughlin M.K. Preeclampsia: an endothelial cell disorder. Am. J. Obstet. Gynecol. 1989;161:1200–1204. doi: 10.1016/0002-9378(89)90665-0. [DOI] [PubMed] [Google Scholar]
- 10.Chaiworapongsa T., Chaemsaithong P., Yeo L., Romero R. Pre-eclampsia part 1: current understanding of its pathophysiology. Nat. Rev. Nephrol. 2014;10:466–480. doi: 10.1038/nrneph.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X., Sun X., Carmeliet P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. 2019;30:414–433. doi: 10.1016/j.cmet.2019.08.011. [DOI] [PubMed] [Google Scholar]
- 12.Stapor P., Wang X., Goveia J., Moens S., Carmeliet P. Angiogenesis revisited - role and therapeutic potential of targeting endothelial metabolism. J. Cell Sci. 2014;127:4331–4341. doi: 10.1242/jcs.153908. [DOI] [PubMed] [Google Scholar]
- 13.Rohlenova K., Veys K., Miranda-Santos I., De Bock K., Carmeliet P. Endothelial cell metabolism in health and disease. Trends Cell Biol. 2018;28:224–236. doi: 10.1016/j.tcb.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 14.De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B.W., Cantelmo A.R., Quaegebeur A., Ghesquière B., Cauwenberghs S., Eelen G. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 15.Bando H., Atsumi T., Nishio T., Niwa H., Mishima S., Shimizu C., Yoshioka N., Bucala R., Koike T. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin. Cancer Res. 2005;11:5784–5792. doi: 10.1158/1078-0432.CCR-05-0149. [DOI] [PubMed] [Google Scholar]
- 16.Bartrons R., Rodríguez-García A., Simon-Molas H., Castaño E., Manzano A., Navarro-Sabaté À. The potential utility of PFKFB3 as a therapeutic target. Expert Opin. Ther. Targets. 2018;22:659–674. doi: 10.1080/14728222.2018.1498082. [DOI] [PubMed] [Google Scholar]
- 17.Shi L., Pan H., Liu Z., Xie J., Han W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017;2:17044. doi: 10.1038/sigtrans.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eelen G., de Zeeuw P., Simons M., Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ. Res. 2015;116:1231–1244. doi: 10.1161/CIRCRESAHA.116.302855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lv Y., Lu C., Ji X., Miao Z., Long W., Ding H., Lv M. Roles of microRNAs in preeclampsia. J. Cell. Physiol. 2019;234:1052–1061. doi: 10.1002/jcp.27291. [DOI] [PubMed] [Google Scholar]
- 20.Moradi M.T., Rahimi Z., Vaisi-Raygani A. New insight into the role of long non-coding RNAs in the pathogenesis of preeclampsia. Hypertens. Pregnancy. 2019;38:41–51. doi: 10.1080/10641955.2019.1573252. [DOI] [PubMed] [Google Scholar]
- 21.Choi S.Y., Yun J., Lee O.J., Han H.S., Yeo M.K., Lee M.A., Suh K.S. MicroRNA expression profiles in placenta with severe preeclampsia using a PNA-based microarray. Placenta. 2013;34:799–804. doi: 10.1016/j.placenta.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Hu Y., Li P., Hao S., Liu L., Zhao J., Hou Y. Differential expression of microRNAs in the placentae of Chinese patients with severe pre-eclampsia. Clin. Chem. Lab. Med. 2009;47:923–929. doi: 10.1515/CCLM.2009.228. [DOI] [PubMed] [Google Scholar]
- 23.Wu L., Zhou H., Lin H., Qi J., Zhu C., Gao Z., Wang H. Circulating microRNAs are elevated in plasma from severe preeclamptic pregnancies. Reproduction. 2012;143:389–397. doi: 10.1530/REP-11-0304. [DOI] [PubMed] [Google Scholar]
- 24.Zhu Y., Lu Y., Zhang Q., Liu J.J., Li T.J., Yang J.R., Zeng C., Zhuang S.M. MicroRNA-26a/b and their host genes cooperate to inhibit the G1/S transition by activating the pRb protein. Nucleic Acids Res. 2012;40:4615–4625. doi: 10.1093/nar/gkr1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du J.Y., Wang L.F., Wang Q., Yu L.D. miR-26b inhibits proliferation, migration, invasion and apoptosis induction via the downregulation of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 driven glycolysis in osteosarcoma cells. Oncol. Rep. 2015;33:1890–1898. doi: 10.3892/or.2015.3797. [DOI] [PubMed] [Google Scholar]
- 26.Wei Z., Chang K., Fan C., Zhang Y. MiR-26a/miR-26b represses tongue squamous cell carcinoma progression by targeting PAK1. Cancer Cell Int. 2020;20:82. doi: 10.1186/s12935-020-1166-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Icli B., Wara A.K., Moslehi J., Sun X., Plovie E., Cahill M., Marchini J.F., Schissler A., Padera R.F., Shi J. MicroRNA-26a regulates pathological and physiological angiogenesis by targeting BMP/SMAD1 signaling. Circ. Res. 2013;113:1231–1241. doi: 10.1161/CIRCRESAHA.113.301780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng J., Hu L., Cheng J., Xu J., Zhong Z., Yang Y., Yuan Z. lncRNA PVT1 promotes the angiogenesis of vascular endothelial cell by targeting miR-26b to activate CTGF/ANGPT2. Int. J. Mol. Med. 2018;42:489–496. doi: 10.3892/ijmm.2018.3595. [DOI] [PubMed] [Google Scholar]
- 29.Song X., Luo X., Gao Q., Wang Y., Gao Q., Long W. Dysregulation of LncRNAs in placenta and pathogenesis of preeclampsia. Curr. Drug Targets. 2017;18:1165–1170. doi: 10.2174/1389450118666170404160000. [DOI] [PubMed] [Google Scholar]
- 30.Li X., Song Y., Liu F., Liu D., Miao H., Ren J., Xu J., Ding L., Hu Y., Wang Z. Long non-coding RNA MALAT1 promotes proliferation, angiogenesis, and immunosuppressive properties of mesenchymal stem cells by inducing VEGF and IDO. J. Cell. Biochem. 2017;118:2780–2791. doi: 10.1002/jcb.25927. [DOI] [PubMed] [Google Scholar]
- 31.Wu H.Y., Wang X.H., Liu K., Zhang J.L. LncRNA MALAT1 regulates trophoblast cells migration and invasion via miR-206/IGF-1 axis. Cell Cycle. 2020;19:39–52. doi: 10.1080/15384101.2019.1691787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen H., Meng T., Liu X., Sun M., Tong C., Liu J., Wang H., Du J. Long non-coding RNA MALAT-1 is downregulated in preeclampsia and regulates proliferation, apoptosis, migration and invasion of JEG-3 trophoblast cells. Int. J. Clin. Exp. Pathol. 2015;8:12718–12727. [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J., Liu X., Xu Y., Zhang K., Huang J., Pan B., Chen D., Cui S., Song H., Wang R. TFAP2C-activated MALAT1 modulates the chemoresistance of docetaxel-resistant lung adenocarcinoma cells. Mol. Ther. Nucleic Acids. 2019;14:567–582. doi: 10.1016/j.omtn.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Q., Meng W.Y., Jie Y., Zhao H. LncRNA MALAT1 induces colon cancer development by regulating miR-129-5p/HMGB1 axis. J. Cell. Physiol. 2018;233:6750–6757. doi: 10.1002/jcp.26383. [DOI] [PubMed] [Google Scholar]
- 35.Méndez I., Díaz-Muñoz M. Circadian and metabolic perspectives in the role played by NADPH in cancer. Front. Endocrinol. (Lausanne) 2018;9:93. doi: 10.3389/fendo.2018.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattila P.K., Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008;9:446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- 37.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng N., Shi L., Zhang Q., Hu Y., Wang N., Ye H. Microarray profiling of circular RNAs in human papillary thyroid carcinoma. PLoS ONE. 2017;12:e0170287. doi: 10.1371/journal.pone.0170287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yalcin A., Clem B.F., Imbert-Fernandez Y., Ozcan S.C., Peker S., O’Neal J., Klarer A.C., Clem A.L., Telang S., Chesney J. 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27. Cell Death Dis. 2014;5:e1337. doi: 10.1038/cddis.2014.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yalcin A., Clem B.F., Simmons A., Lane A., Nelson K., Clem A.L., Brock E., Siow D., Wattenberg B., Telang S., Chesney J. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J. Biol. Chem. 2009;284:24223–24232. doi: 10.1074/jbc.M109.016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jia W., Zhao X., Zhao L., Yan H., Li J., Yang H., Huang G., Liu J. Non-canonical roles of PFKFB3 in regulation of cell cycle through binding to CDK4. Oncogene. 2018;37:1685–1698. doi: 10.1038/s41388-017-0072-4. [DOI] [PubMed] [Google Scholar]
- 42.Schoors S., De Bock K., Cantelmo A.R., Georgiadou M., Ghesquière B., Cauwenberghs S., Kuchnio A., Wong B.W., Quaegebeur A., Goveia J. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19:37–48. doi: 10.1016/j.cmet.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 43.Bousseau S., Vergori L., Soleti R., Lenaers G., Martinez M.C., Andriantsitohaina R. Glycosylation as new pharmacological strategies for diseases associated with excessive angiogenesis. Pharmacol. Ther. 2018;191:92–122. doi: 10.1016/j.pharmthera.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 44.Hunt T.K., Aslam R.S., Beckert S., Wagner S., Ghani Q.P., Hussain M.Z., Roy S., Sen C.K. Aerobically derived lactate stimulates revascularization and tissue repair via redox mechanisms. Antioxid. Redox Signal. 2007;9:1115–1124. doi: 10.1089/ars.2007.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Végran F., Boidot R., Michiels C., Sonveaux P., Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71:2550–2560. doi: 10.1158/0008-5472.CAN-10-2828. [DOI] [PubMed] [Google Scholar]
- 46.Teuwen L.A., Draoui N., Dubois C., Carmeliet P. Endothelial cell metabolism: an update anno 2017. Curr. Opin. Hematol. 2017;24:240–247. doi: 10.1097/MOH.0000000000000335. [DOI] [PubMed] [Google Scholar]
- 47.Garcia N.A., Moncayo-Arlandi J., Sepulveda P., Diez-Juan A. Cardiomyocyte exosomes regulate glycolytic flux in endothelium by direct transfer of GLUT transporters and glycolytic enzymes. Cardiovasc. Res. 2016;109:397–408. doi: 10.1093/cvr/cvv260. [DOI] [PubMed] [Google Scholar]
- 48.D’Souza D.R., Salib M.M., Bennett J., Mochin-Peters M., Asrani K., Goldblum S.E., Renoud K.J., Shapiro P., Passaniti A. Hyperglycemia regulates RUNX2 activation and cellular wound healing through the aldose reductase polyol pathway. J. Biol. Chem. 2009;284:17947–17955. doi: 10.1074/jbc.M109.002378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riganti C., Gazzano E., Polimeni M., Aldieri E., Ghigo D. The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012;53:421–436. doi: 10.1016/j.freeradbiomed.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 50.Schoors S., Bruning U., Missiaen R., Queiroz K.C., Borgers G., Elia I., Zecchin A., Cantelmo A.R., Christen S., Goveia J. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. 2015;520:192–197. doi: 10.1038/nature14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stincone A., Prigione A., Cramer T., Wamelink M.M., Campbell K., Cheung E., Olin-Sandoval V., Grüning N.M., Krüger A., Tauqeer Alam M. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015;90:927–963. doi: 10.1111/brv.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu J.Z., Yang H.K., Zhang W.G. NADPH metabolism: a survey of its theoretical characteristics and manipulation strategies in amino acid biosynthesis. Crit. Rev. Biotechnol. 2018;38:1061–1076. doi: 10.1080/07388551.2018.1437387. [DOI] [PubMed] [Google Scholar]
- 53.American College of Obstetricians and Gynecologists. Task Force on Hypertension in Pregnancy Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013;122:1122–1131. doi: 10.1097/01.AOG.0000437382.03963.88. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.