

Abstract
Bromodomain (BRD) and extra-terminal (BET) proteins are epigenetic readers that regulate gene expression and promote cancer evolution. Pharmacological inactivation of BRD4 has recently been introduced as a promising anti-neoplastic approach that targets MYC oncogene expression. However, resistance against BRD4-targeting drugs has been described. We compared the efficacy of the small-molecule-type BET BRD inhibitor JQ1 with the recently developed BET protein degraders dBET1 and dBET6 in colon, breast, melanoma, ovarian, lung and prostate cancer cell lines. As determined by qPCR, all BRD4 targeting drugs dose-dependently decreased MYC expression, with dBET6 introducing the strongest downregulation of MYC. This correlated with the anti-proliferative activity of these drugs, which was at least one order of magnitude higher for dBET6 (IC50 0.001-0.5 µM) than for dBET1 or JQ1 (IC50 0.5-5 µM). Interestingly, when combined with commonly used cytotoxic therapeutics, dBET6 was found to promote anti-neoplastic effects and to counteract chemoresistance in most cancer cell lines. Moreover, JQ1 and both BET degraders strongly downregulated baseline and interferon-gamma induced expression of the immune checkpoint molecule PD-L1 in all cancer cell lines. Together, our data suggest that dBET6 outperforms first-generation BRD4 targeting drugs like dBET1 and JQ1, and decreases chemoresistance and immune resistance of cancer.
Keywords: BET degrader, dBET6, MYC, PD-L1, solid tumor
Introduction
Epigenetic alterations of the chromatin play an important role in the pathogenesis of cancer. Methylation of DNA and posttranslational alteration of histones such as methylation and acetylation on lysine residues are the most common epigenetic changes. The histone modifications cause loosening of the chromatin and facilitate binding of transcription factors to regulatory sequences of pro-oncogenic genes thereby promoting de-differentiation, cell proliferation and malignant progression. Histone modifying proteins are classified into histone writers, readers and erasers. These proteins add, manage and remove chromatin modifications [1-3]. Epigenetic readers of the bromodomain and extra-terminal domain (BET) protein family use the bromodomain (BRD) to target acetylated lysines in histone proteins and regulate gene expression through interaction with the transcriptional machinery [4,5]. The BET family includes ubiquitously expressed BRD2, BRD3 and BRD4, and testis restricted BRDT [6]. BRD4 has attracted attention as a promising anti-cancer drug target due to its strong effect on expression of the transcription factor MYC, which is a well-known pro-oncogenic master regulator and a major driver of a wide variety of cancers [7-10]. Unfortunately, effective drugs that directly target MYC are still not available. Thus, pharmacological blockade of BRD4 may be a promising indirect strategy to mitigate MYC hyperactivity. Recently, JQ1 has been developed as a potent, selective small molecule inhibitor of BET proteins that displaces the BRD2, BRD3 and BRD4 domains from chromatin through competitive binding to the acetyl-lysine recognition pockets of the domains [11,12].
In hematologic malignancies, JQ1-mediated downregulation of MYC correlated well with reduced cell proliferation, whereas in solid tumors reduction of MYC and of cell growth was less striking after single drug treatment, despite the fact that MYC is a key driver of both blood-borne cancers as well as solid tumors [13-20]. Recently, we characterized several regulatory loops that counteract BET inhibitors and weaken their effects in colon, breast and ovarian cancer [21]. Accordingly, concurrent blockade of these bypass routes improved BET inhibitor mediated growth inhibition significantly indicating that drug combinations can increase the clinical benefit of BET pathway interference in solid tumors. Moreover, we observed that the BET degrader dBET1 outperformed the BET inhibitor JQ1 in colon, breast and ovarian cancer cells [21]. dBET1 belongs to the first generation of bifunctional smallmolecule BET protein degraders. These compounds contain the BRD4 inhibiting JQ1 moiety linked via a carbohydrate bridge to thalidomide that binds cereblon, a component of the ubiquitin ligase complex, and enables degradation of the BET protein by enforcing proximity of the targeted BRD domain with the ubiquitin ligase complex causing ubiquitination and proteasomal disintegration [22]. Subsequent structural modification of dBET1 yielded an optimized compound known as dBET6, which differs from dBET1 by an extended carbohydrate bridge [23]. To our knowledge, this highly potent second generation BET degrader has not yet been used in solid tumors except glioblastoma [24].
The aim of the present study was to compare for the first time the efficacy of this novel BET degrader with the effects of its predecessor dBET1 and the prototypic BET inhibitor JQ1 in a variety of solid tumors including colon, breast, ovarian, lung and prostate cancer, as well as melanoma. Our findings show that dBET6 exerts superior MYC downregulation and anti-proliferation effects, which are further improved, when dBET6 is combined with standard chemotherapy. Moreover, dBET6 potently suppressed interferon gamma (IFN-G)-induced PD-L1 immune checkpoint expression indicating that combining dBET6 with PD-L1 blockade may be a useful therapeutic approach in solid tumors.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), α-modified minimal essential medium (α-MEM) and phosphate buffered saline (PBS) were from Gibco Life Technologies (Gaithersburg, MD), and RPMI 1640 medium and fetal calf serum (FCS) were from PAA Laboratories (Pasching, Austria). The BET inhibitor JQ1 was purchased from Selleckchem (Houston, TX), the BET degraders dBET1 from Chemietek (Indianapolis, IN) and dBET6 from Aobious Inc (Gloucester, MA). 5-Fluorouracil (5-FU), doxorubicin and paclitaxel were obtained from Sigma (St. Louis, MO), and IFN-G was from Merck (Darmstadt, Germany). 3H-thymidine was purchased from Amersham (Buckinghamshire, UK) and Annexin V-FITC from eBiosciences (San Diego, CA).
Cell lines and culture conditions
Colon (HCT15, HCT116, HT29), breast (MCF7, SKBR3, T47D), melanoma (607B, A375, MEL-JUSO), ovarian (A2780, HEY, SKOV3), lung (H1993, H2073) and prostate (DU-145, LNCAP) cancer cell lines were cultured in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA) supplemented with FCS and antibiotics at 37°C in a 5% CO2 atmosphere. Absence of viral/bacterial/fungal/mycoplasma infection in the cell lines was proven by Venor GeM (Minerva Biolabs, Berlin, Germany). Species origins were examined by species-PCR, and cell line identities were tested by fluorescent nonaplex-PCR of short tandem repeat markers (DSMZ, Braunschweig, Germany).
3H-thymidine incorporation
To determine the growth-modulating properties of BET inhibitors and degraders, the cell lines were exposed to JQ1, dBET1 and dBET6 (0.001-5 µM) at 37°C for 48 hours, and the efficacy of dBET6 on chemoresistance was determined by incubating the tumor cells with 5-FU (HCT15, HCT116, HT29, MCF7, SKBR3, T47D), doxorubicin (607B, A375, MEL-JUSO, OPM-2, RPMI-8226, U-266, A2780, HEY, SKOV3) or paclitaxel (H1993, H2073, DU-145, LNCAP) in the presence or absence of dBET6 (at the IC50 of the respective cell line) at 37°C for 48 hours. Then, 1 µCi (0.037 MBq) 3H-thymidine was added (37°C, 16 hours) to determine de novo DNA synthesis. Thereafter, cells were harvested on filter membranes (Packard Bioscience, Meriden, CT) in a Filtermate 196 harvester (Packard Bioscience). Filters were air-dried, and the bound radioactivity was detected in a β-counter (Top-Count NXT, Packard Bioscience).
Quantitative real-time PCR (qPCR)
Cell lines were incubated with JQ1, dBET1 or dBET6 (0.005-5 µM) at 37°C for 16 hours. Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Reverse transcription was performed using Moloney murine leukemia virus reverse transcriptase, random primers, first strand buffer (Invitrogen, Carlsbad, CA), dNTPs (Promega, Mannheim, Germany) and RNasin plus (Promega) according to manufacturer’s instructions (Invitrogen). qPCR was performed using primers (Eurofins MWG Operon, Ebersberg, Germany) for human Actin (forward: 5’-TCGACAACGGCTCCGGCATG-3’; reverse: 5’-CCTCTCTTGCTCTGGGCCTCGTC-3’) and human MYC (forward: 5’-TGCTCCATGAGGAGACACC-3’; reverse: 5’-CCTGCCTCTTTTCCACAGAA-3’). qPCR was performed on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific) using iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA). Expression levels of MYC were normalized to Actin by the standard curve method.
Apoptosis assays
For flow cytometric determination of apoptosis, Annexin V staining was performed. Cell lines were incubated in medium without or with JQ1, dBET1 or dBET6 (0.1-10 µM) at 37°C for 48 hours. Then, cells were washed with PBS and incubated with Annexin V-FITC in binding-buffer containing HEPES (10 mM, pH 7.4), NaCl (140 mM) and CaCl2 (2.5 mM) for 15 minutes. Thereafter, cells were washed and PI (1 mg/mL) was added. Cells were examined in a FACSCanto flow cytometer (BD Biosciences, San Jose, CA).
Immunofluorescence flow cytometry analysis
Baseline expression of immune checkpoint proteins on tumor cells was analyzed by direct immunofluorescence flow cytometry in a FACSCanto (BD Biosciences) using phycoerythrin (PE)-labeled monoclonal antibodies (mAb) against CD28 (clone: CD28.2), CD80 (clone: 2D10), CD83 (clone: HB15e), CD86 (clone: IT2.2), CD273 (PD-L2) (clone: MIH18), CD274 (PD-L1) (clone: 29E.2A3), CD279 (PD-1) (clone: EH12.2H7), CD366 (clone: F38-2E2) (Biolegend, San Diego, CA), CD47 (clone: BH612) and CD243 (clone: 15D3) (BD Biosciences) and PE-conjugated isotype-matched control antibodies. Results were expressed as staining index (SI) defined as ratio between median fluorescence intensity (MFI) obtained with specific mAb and MFI obtained with control mAb (MFIspecific mAb:MFIcontrol mAb). In a separate set of experiments, tumor cells were cultured in the presence or absence of 100 U/ml IFN-G, and without or with various concentrations of JQ1, dBET1 or dBET6 (0.05-2 µM) for 24 hours before PD-L1 expression was analyzed.
Statistical analysis
Data are expressed as mean values plus/minus standard deviation from ≥ 3 independent experiments. Statistical analysis was performed by using ANOVA and post hoc Scheffe test. The correlation r between particular data sets was calculated and its statistical significance was estimated by a correlated t-test.
Results
The baseline expression of BRD4 and MYC varies over a wide range in solid tumor cells
The chromatin reader BRD4 binds to cell-type-specific super-enhancer regions in genes that code for transcription factors and determines cell lineage and cell development. Among them, MYC - a pro-oncogenic master transcription factor of numerous cancer pathways - is the primary effector of BRD4. MYC is essential for cell cycle progression and cell growth, and is a major driver of cancer development. While BRD4 is present in all cell types the super-enhancer regions are associated with variable gene sets, depending on cell type and developmental and environmental signals, enabling cells to rapidly adjust their functional states. Accordingly, MYC, which is under control of a super-enhancer, is expressed at variable levels in cancer cells [25]. Here we demonstrate that the amount of BRD4 and MYC varies considerably in solid tumor cells. While BRD4 and MYC expression significantly differ between individual cell lines even when these cell lines are derived from the same tissue origin (e.g. skin, ovary/fallopian tube, lung, prostate) (Figure 1A), both gene products are expressed at comparable levels (both at either high or low level) in the individual cancer cell lines (r = 0.73, P = 0.0015) (Figure 1B).
Figure 1.
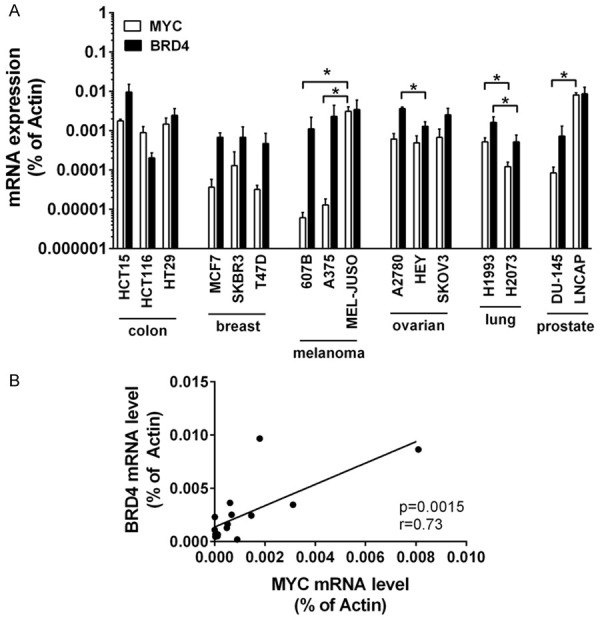
Baseline expression of MYC and BRD4 mRNA in untreated solid tumor cells. A. Baseline expression of MYC and BRD4 mRNA was determined by qPCR analysis. The relative expression levels of MYC and BRD4 mRNA were calculated by the standard curve method and Actin was used as internal control. The figure shows the mean ± SD of 3 independent determinations. Please note that the y-axis is displayed on a logarithmic scale. Statistical analysis was performed using ANOVA and post hoc Scheffe test to compare either MYC mRNA levels between individual cell lines of the same tissue origin or BRD4 mRNA levels between individual cell lines of the same tissue origin. Asterisk (*): P < 0.05. B. Pearson correlation analysis between MYC mRNA level and BRD4 mRNA level across all cell lines revealed a highly significant (P = 0.0015) strong positive linear correlation with an r-value of 0.73.
dBET6 downregulates MYC in solid tumor cells more efficiently than dBET1 and JQ1
MYC is required for growth of most solid tumors and represents an attractive cancer drug target. Unfortunately, however, attempts to develop direct MYC inhibitors have so far been unsuccessful. An indirect approach that interferes with the BET family protein BRD4, the main inducer of MYC expression, would therefore be a promising alternative way to target MYC in solid tumors such as breast, gastric, bladder and prostate cancer [20,26-30]. Here we compared the efficacy of the novel second generation BET protein degrader dBET6 with earlier BET antagonists including dBET1 and JQ1 in 3 colon (HCT15, HCT116, HT29), 3 breast (MCF7, SKBR3, T47D), 3 melanoma (607B, A375, MEL-JUSO), 3 ovarian (A2780, HEY, SKOV3), 2 lung (H1993, H2073) and 2 prostate (DU-145, LNCAP) cancer cell lines. Downregulation of MYC mRNA was used as a well-accepted molecular biomarker to indicate the on-target activity of the inhibitors. Accordingly, qPCR analyses demonstrated that all three BET targeting drugs dose-dependently reduced MYC mRNA levels, with dBET6 clearly being the most effective repressor of MYC (Figure 2).
Figure 2.
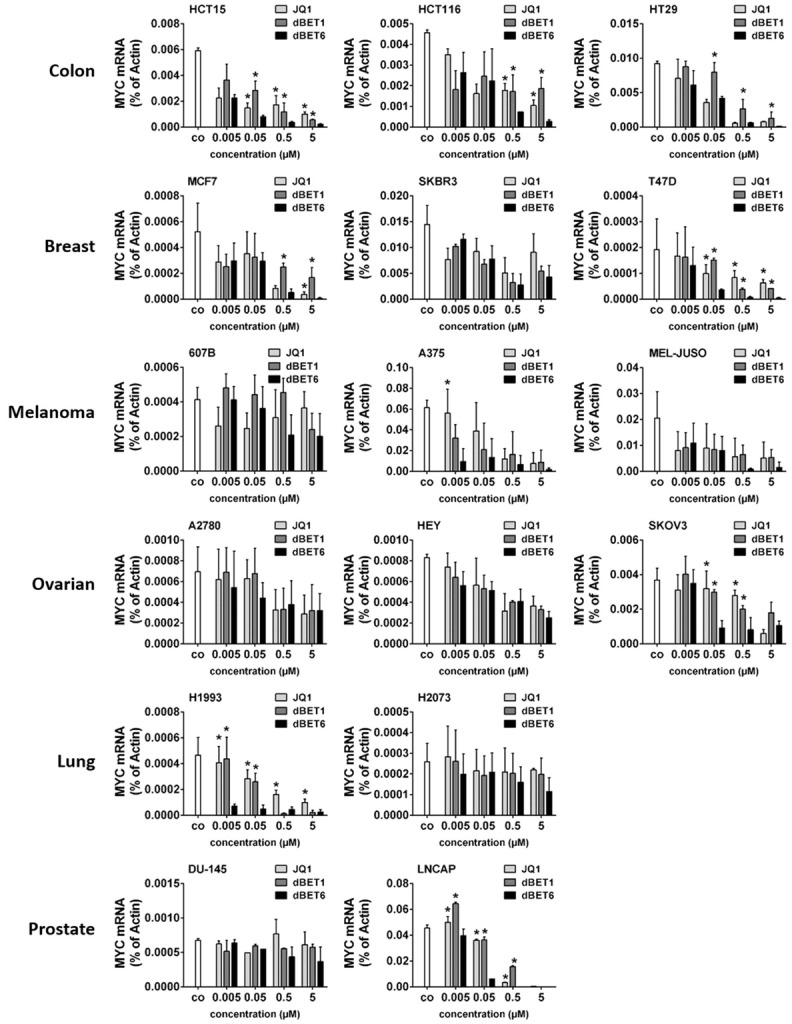
Effects of BRD4 targeting drugs on expression of MYC mRNA in solid tumor cells. Cancer cell lines were incubated in control medium (co) or in medium containing various concentrations of JQ1, dBET1 or dBET6 (0.005-5 µM) at 37°C for 16 hours. Expression of MYC mRNA was determined by qPCR analysis. The relative expression levels of MYC mRNA were calculated by the standard curve method and Actin was used as internal control. The figures show the mean ± SD of 3 independent experiments. Statistical analysis was performed by using ANOVA and post hoc Scheffe test. Asterisk (*): P < 0.05 compared to respective concentration of dBET6 (JQ1 vs. dBET6 and dBET1 vs. dBET6).
dBET6 is a stronger inhibitor of solid tumor cell proliferation than dBET1 and JQ1
Next, we determined the growth-inhibitory effects and potencies of these BET blockers in various tumor cell lines. While JQ1 and dBET1 abrogated DNA synthesis with similar efficacy, dBET6 was a more potent inhibitor of DNA synthesis as demonstrated in dose-response experiments (Figure 3). This became particularly evident when the IC50 values of JQ1 and dBET1 were compared with those of dBET6. As shown in Table 1, the IC50 values of dBET6 were lower than those of dBET1 in each cell line, and were also lower than the IC50 values of JQ1 in most of these lines. Thus, the novel BET degrader dBET6 exerted the strongest anti-proliferative activity. Interestingly, drug-dependent inhibition of cell proliferation correlated weakly with downregulation of MYC (r = 0.44, P < 0.001).
Figure 3.
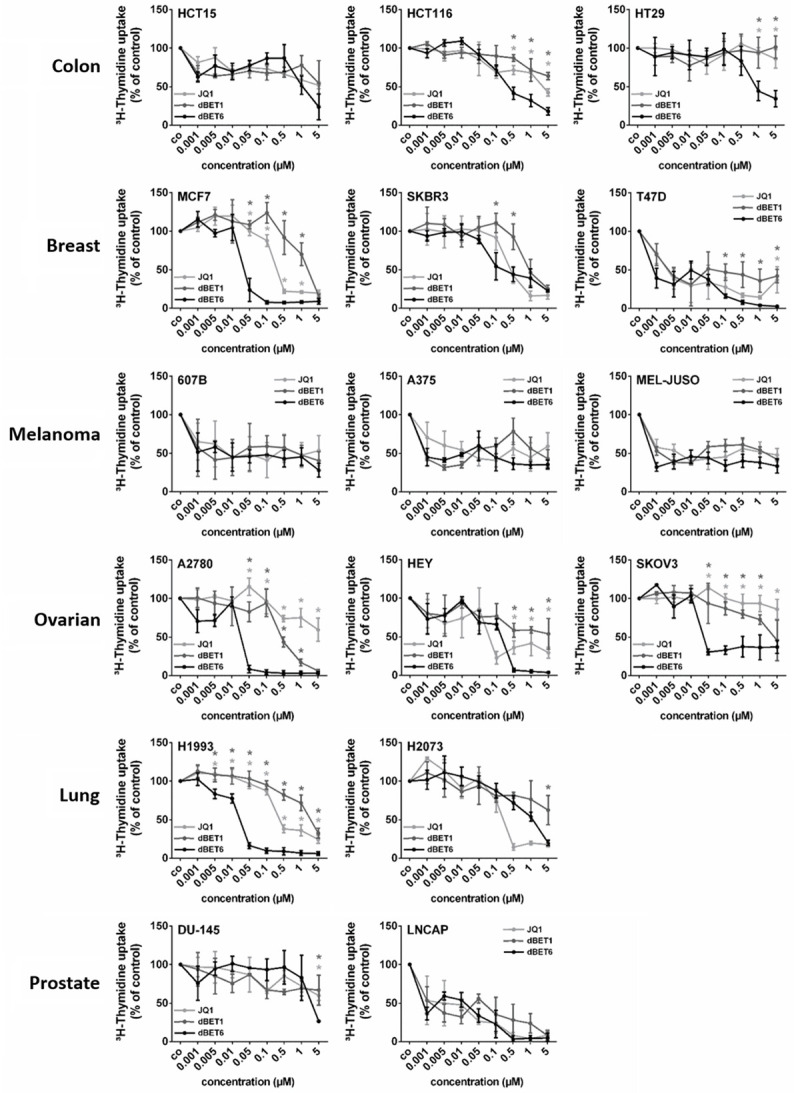
Effects of BRD4 targeting drugs on proliferation of solid tumor cells. Cancer cell lines were incubated in control medium (co) or in medium containing various concentrations of JQ1, dBET1 or dBET6 (0.001-5 µM) at 37°C for 48 hours. Thereafter, 3H-thymidine uptake was measured. Results are expressed as percent of control (co) and represent the mean ± SD of 3 independent experiments. Statistical analysis was performed by using ANOVA and post hoc Scheffe test. Asterisk (*): P < 0.05 compared to respective concentration of dBET6 (light-grey asterisk: JQ1 vs. dBET6; dark-grey asterisk: dBET1 vs. dBET6).
Table 1.
IC50 values for JQ1-, dBET1-, and dBET6-mediated inhibition of 3H-thymidine incorporation in solid tumor cells
| Entity | Cell line | JQ1 IC50 (µM) | dBET1 IC50 (µM) | dBET6 IC50 (µM) |
|---|---|---|---|---|
| Colon cancer | HCT15 | > 5 | > 5 | 1-5 |
| HCT116 | 1-5 | > 5 | 0.1-0.5 | |
| HT29 | > 5 | > 5 | 0.5-1 | |
| Breast cancer | MCF7 | 0.1-0.5 | 1-5 | 0.01-0.05 |
| SKBR3 | 0.1-0.5 | 0.5-1 | 0.1-0.5 | |
| T47D | 0.001-0.005 | 0.001-0.005 | < 0.001 | |
| Melanoma | 607B | 0.5-1 | 0.5-1 | < 0.001 |
| A375 | 1-5 | 1-5 | 0.05-0.1 | |
| MEL-JUSO | 1-5 | 1-5 | < 0.001 | |
| Ovarian cancer | A2780 | > 5 | 0.1-0.5 | 0.01-0.05 |
| HEY | 0.05-0.1 | > 5 | 0.05-0.1 | |
| SKOV3 | > 5 | 1-5 | 0.01-0.05 | |
| Lung cancer | H1993 | 0.1-0.5 | 1-5 | 0.01-0.05 |
| H2073 | 0.1-0.5 | > 5 | 1-5 | |
| Prostate cancer | DU-145 | > 5 | > 5 | 1-5 |
| LNCAP | 0.005-0.01 | 0.05-0.1 | 0.01-0.05 |
Cell lines were cultured in control medium or in medium containing various concentrations of JQ1, dBET1 or dBET6 at 37°C for 48 hours. Thereafter, proliferation was measured by 3H-thymidine incorporation assay. IC50 values (μM) represent the ranges from 3 independent experiments. IC50 values of JQ1 and dBET1: bold numerals: IC50 value higher than IC50 of dBET6; standard numerals: IC50 value equal to IC50 of dBET6; italic numerals: IC50 value lower than IC50 of dBET6.
dBET6 is a stronger inducer of apoptosis in solid tumor cells than dBET1 and JQ1
Next, we examined whether the growth inhibitory effects of JQ1, dBET1 and dBET6 are associated with apoptosis. To this end, Annexin-V labeled control and treated cells were analyzed by flow cytometry. Data shown in Figure 4 reveal that dBET6 increased the apoptotic cell fraction in 14 of the 16 cell lines tested, including HCT15, HCT116, MCF7, SKBR3, T47D, 607B, A375, MEL-JUSO, A2780, HEY, SKOV3, H1993, DU145, and LNCAP. Of these 14 cell lines, 6 (MCF7, T47D, A375, HEY, H1993, and DU145) were sensitive to dBET6 only, but not to dBET1 and JQ1. Two other cell lines (A2780 and SKOV3) were susceptible to both dBET1 and dBET6, but not to JQ1, while the remaining six cell lines (HCT15, HCT116, SKBR3, 607B, MEL-JUSO, and LNCAP) were essentially equally sensitive to the three compounds. Overall, it was found that dBET6 is a major inducer of apoptosis in most cell lines tested (14/16). This was most evident at low drug concentrations. dBET1, on the other hand, was only moderately active, while JQ1 was the least effective apoptotic drug (Figure 4). Further analysis across all cell lines revealed that drug-induced apoptosis correlates with the expression levels of BRD4 and MYC before drug exposure (Table 2).
Figure 4.
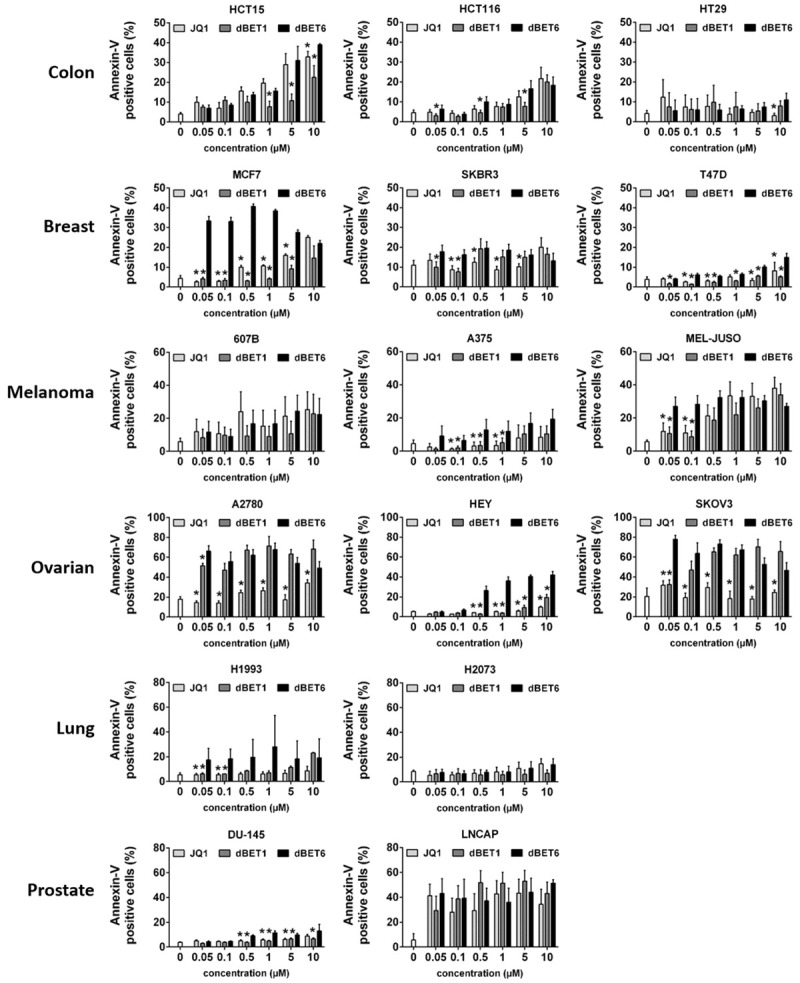
Effects of BRD4 targeting drugs on survival of solid tumor cells. Cancer cell lines were incubated in control medium (co) or in medium containing various concentrations of JQ1, dBET1 or dBET6 (0.1-10 µM) at 37°C for 48 hours. Then, cells were labeled with Annexin-V-FITC and examined by flow cytometry to determine the percentage of apoptotic cells. Statistical analysis was performed by using ANOVA and post hoc Scheffe test. Results represent the mean ± SD of 3 independent experiments. Asterisk (*): P < 0.05 compared to respective concentration of dBET6 (JQ1 vs. dBET6 and dBET1 vs. dBET6).
Table 2.
Pearson correlation between BET drug-induced apoptosis and BRD4 and MYC baseline expression in solid tumor cells
| BRD4 | MYC | |||
|---|---|---|---|---|
|
|
|
|||
| r | P | r | P | |
| JQ1 | 0.81 | 0.0001 | 0.76 | 0.0005 |
| dBET1 | 0.70 | 0.002 | 0.93 | < 0.0001 |
| dBET6 | 0.50 | 0.04 | 0.42 | 0.1 |
Cell lines were cultured in control medium or in medium containing 10 µM of JQ1, dBET1 or dBET6 at 37°C for 48 hours. Thereafter, cells were labeled with Annexin-V-FITC and examined by flow cytometry to determine the percentage of apoptotic cells. Baseline expression of BRD4 and MYC mRNA was determined by qPCR analysis in cells grown in control medium in the absence of drugs.
dBET6 augments the anti-proliferative efficacy of cytotoxic chemotherapy in solid tumor cells
MYC has been identified as a key inducer of cancer stem cell (CSC) survival and expansion. CSCs are typically known to be highly resilient to toxic substances. Moreover, BRD4/MYC signaling has been reported to support cancer chemoresistance [31,32]. Thus, we hypothesized that the anti-proliferative efficacy of clinically proven standard chemotherapy agents may be improved in the presence of a strong inhibitor of the BRD4/MYC pathway such as dBET6. Accordingly, 5-FU (colon and breast cancer), doxorubicin (melanoma and ovarian cancer) or paclitaxel (lung and prostate cancer) were administered at wide dose ranges with or without dBET6 at a concentration ≤ IC50 in each cell line. As shown in Figure 5, the growth inhibitory effects of all cytotoxic drugs applied were significantly increased in the presence of dBET6 in almost all cell lines examined. Notably, dBET6-mediated chemosensitization was most striking at low or moderate concentrations of the therapeutics.
Figure 5.

Effects of dBET6 in combination with chemotherapeutics on proliferation of solid tumor cells. Cancer cell lines were incubated in control medium (co) or in medium containing various concentrations of 5-FU (HCT15, HCT116, HT29, MCF7, SKBR3, T47D), doxorubicin (607B, A375, MEL-JUSO, A2780, HEY, SKOV3) or paclitaxel (H1993, H2073, DU-145, LNCAP) with or without dBET6 (≤ IC50 of the cell lines) at 37°C for 48 hours. Thereafter, 3H-thymidine uptake was measured. Results are expressed as percent of control (co) and represent the mean ± SD of 3 independent experiments.
dBET6 is the most potent repressor of IFN-G-induced PD-L1 expression in solid tumor cells
Hyperexpression of immune checkpoint molecules such as PD-L1, PD-L2, PD-1, CD28, CD47, CD80, CD83, CD86, CD243 and CD366 in neoplastic cells is another mechanism that can cause cancer therapy resistance [33], and IFN-G has been identified as a powerful positive regulator of these checkpoint genes [34]. Here, we set out: 1. To characterize the level of baseline expression of all these checkpoint proteins in the solid tumor cell lines; 2. To determine the impact of IFN-G on checkpoint expression levels in these cells; and 3. To demonstrate the effect of JQ1, dBET1 and dBET6 on IFN-G-induced hyperexpression of these immune checkpoint proteins. To this end, flow cytometry analysis was performed in untreated and treated cancer cell lines. As summarized in Table 3, moderate baseline levels of PD-L1 (CD274) and high levels of CD47 were observed in all cell lines tested, while the remaining checkpoint proteins were not or hardly detectable in any of these lines. Both PD-L1 and CD47 were found to be sensitive to IFN-G, with PD-L1 being induced in all cell lines and CD47 in 50% of them. In contrast, none of the other checkpoint molecules were inducible by IFN-G (Table 3). Next, we asked whether the IFN-G-induced PD-L1 upregulation could be counteracted by co-treatment with the BET targeting drugs. Indeed, we were able to show that JQ1 and both BET degraders diminished the IFN-G-induced upregulation of PD-L1. This effect was most clearly seen for dBET6, which caused a significant dose-dependent downregulation of IFN-G-induced expression of PD-L1 in 14 of 16 cancer cell lines (all except 607B and H1993), while JQ1 reduced PD-L1 in 11 (all except 607B, A375, HEY, H1993 and H2073), and dBET1 only in 10 of them (all except HT29, 607B, HEY, H1993, H2073 and DU145) (Figure 6).
Table 3.
Baseline and IFN-G-induced expression of immune checkpoint proteins in solid tumor cells
| Entity | Cell line | PD-L1 | PD-L2 | PD-1 | CD28 | CD47 | CD80 | CD83 | CD86 | CD243 | CD366 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Colon cancer | HCT15 | +* | - | - | - | ++* | +/- | +/- | - | + | +/- |
| HCT116 | +* | - | - | - | ++* | +/- | +/- | +/- | - | - | |
| HT29 | +* | - | - | - | ++* | +/- | +/- | - | - | - | |
| Breast cancer | MCF7 | +* | - | - | - | ++ | +/- | +/- | - | - | +/- |
| SKBR3 | +* | - | - | - | ++* | - | - | - | - | - | |
| T47D | +* | - | - | +/- | ++* | + | + | - | - | +/- | |
| Melanoma | 607B | +* | - | - | - | +++ | +/- | + | +/- | +/- | +/- |
| A375 | +* | - | - | - | +++* | +/- | + | +/- | - | +/- | |
| MEL-JUSO | ++* | +/- | - | - | +++ | + | + | - | +/- | +/- | |
| Ovarian cancer | A2780 | +/-* | - | - | - | ++* | +/- | +/- | - | - | - |
| HEY | +* | - | - | +/- | ++* | - | +/- | +/- | - | - | |
| SKOV3 | +/-* | - | - | +/- | ++ | +/- | +/- | +/- | - | +/- | |
| Lung cancer | H1993 | +* | +/- | - | - | ++ | + | + | +/- | +/- | + |
| H2073 | +* | +/- | - | - | ++ | +/- | + | +/- | - | +/- | |
| Prostate cancer | DU-145 | +* | +/- | - | + | ++ | +/- | + | +/- | +/- | - |
| LNCAP | +* | - | - | +/- | ++ | +/- | + | +/- | - | - |
Summary of the tested checkpoint protein expression in the cancer cell lines. Scoring system: staining index (SI) was calculated (ratio of median fluorescence intensities obtained with specific antibody and isotype-matched control antibody) and scored according to the following system: -, SI < 1.30; +/-, SI 1.31-3.00; +, SI 3.01-10.00; ++, SI 10.01-100; +++, SI > 100. The asterisk (*) indicates that the expression of the checkpoint marker is increased after 100 U/ml IFN-G treatment for 24 hours. Abbreviations: IFN-G, interferon gamma; PD-L1, programmed cell death-ligand 1; PD-L2, programmed cell death-ligand 2; PD-1, programmed cell death-1; CD, cluster of differentiation.
Figure 6.

Effect of BRD4 targeting drugs on interferon-gamma induced PD-L1 expression in solid tumor cells. Cancer cell lines were incubated in control medium (co) without or with 100 U/ml interferon-gamma alone or in combination with various concentrations (0.05, 0.5, 2 µM) of JQ1, dBET1 or dBET6 at 37°C for 24 hours. Then, PD-L1 expression was determined by flow cytometry. Expression levels are provided as staining index (SI) defined by the ratio of median fluorescence intensities (MFI) obtained with specific PD-L1 mAb and isotype-matched control mAb (SI = MFIPD-L1 mAb:MFIcontrol mAb). Statistical analysis was performed by using ANOVA and post hoc Scheffe test. Results represent the mean ± SD of at least 3 independent experiments. Asterisk (*): P < 0.05 compared to IFN-G treated control (co) cells.
Discussion
In a recent pilot study we observed that first-generation BET-inhibitors and -degraders induce growth inhibition and apoptosis with moderate efficacy in several solid tumors, including cancers of the colon, the breast and the ovary/fallopian tube [21]. This prompted us to analyze the potential of BET-targeting drugs against solid tumors in more detail. Encouraged by the development of novel BET degraders with improved pharmacological features and high activity against hematologic neoplasms, we now aimed at a broader analysis of the efficacy of the advanced second-generation degrader dBET6 on a whole range of different solid tumors and we compared it with the two precursor drugs dBET1 and JQ1. While the main drug target, the chromatin eraser protein BRD4, is known to be ubiquitously expressed, the super-enhancer regions, which are the preferred interaction sites of BRD4 with the DNA, are distributed in the genome in a cell-type specific manner with particularly high densities in the vicinity of genes that determine microenvironmental adaptation and immune defense, and define developmental stage and cell identity. Some of these genes, such as MYC - the major BRD4-target gene-regulate cell cycle and cell growth and become oncogenic during malignant transformation [25]. Thus, sensitivity of solid tumor cells to BRD4-targeting drugs is likely to depend heavily on cell lineage, tissue origin, environmental setting and the actual cell make-up. This prompted us to examine the efficacy of dBET6 on cells from solid tumors of different tissue origins. Apart from one report by Xu et al., who demonstrated growth sensitivity of glioblastoma cells against dBET6 [24], our study is, to the best of our knowledge, the first profound analysis of the anti-cancer effects the BET degrader dBET6. Remarkably, dBET6 efficacy clearly exceeded the anti-cancer effects of dBET1 and JQ1 in our study. The potential reason for the favorable efficacy of dBET6 might be its strong impact on MYC expression, which correlates with its increased cell permeability compared to JQ1 or dBET1 [23]. The MYC oncogene plays a pivotal role in almost all aspects of oncogenesis [35]. Therefore, MYC has been considered an extraordinary attractive molecular cancer drug target. Unfortunately, however, attempts to develop agents that directly neutralize MYC failed repeatedly, rendering it as a yet undruggable oncoprotein. Thus, the BET antagonists JQ1, dBET1 and dBET6 act as indirect MYC inhibitors. JQ1 is an acetyl-lysine mimetic that competes with acetylated histones for binding to the acetyl-lysine binding pocket of both bromodomains located in BRD4 and all other BET proteins. In contrast, the degraders dBET1 and dBET6 represent bifunctional chimeric molecules containing JQ1 for binding and inhibition of the bromodomains of BET proteins and a phthalimide moiety for interaction with the cereblon E3 ubiquitin ligase complex for subsequent proteasomal degradation of the BET proteins. BRD4 preferentially interacts with super-enhancer regions in genes that code for transcription factors such as MYC. Thus, both BET inhibitors as well as BET degraders cause MYC downregulation in cells of solid tumors such as colon and breast cancer [36,37].
The role of MYC in cancer cannot be overestimated, and its functional elimination is of paramount importance for the success of any chemotherapeutic approach, because MYC controls gene programs that promote self-renewal of multiresistant CSC clones and thereby promotes the development of drug resistance in cancer cells [31,32]. With this in mind, we hypothesized that MYC reduction due to BRD4 blockade might sensitize solid tumor cells to cytotoxic drugs. Therefore, we examined the efficacy of dBET6 to counteract resistance of solid tumor cells against clinically established standard chemotherapeutics. Drug combination therapies in cancer cells may work in a synergistic or additive fashion, and as a result, lower therapeutic doses of each individual drug are often required to block cancer cell growth with such combinations [38]. Intriguingly, this was indeed the case, when dBET6 was administered together with various doses of tumor-specific cytotoxic therapeutics. The improved growth inhbition after drug combination is associated with enhanced cooperative downregulation of MYC as previously shown by us for combinations of BET blockers with the multi-kinase inhbitor ponatinib [21].
Cross-regulation between oncogenic transcription programs and the microenvironment is yet another important aspect of MYC function [39,40]. For instance, MYC expression in tumor cells has recently been shown to control transcription of immune regulatory cytokines and to upregulate the immune checkpoint gene products CD47 and PD-L1, which both disable immune effector cells and blunt the host immune response against the neoplastic cells [41,42]. Evidence thus strongly suggests that activated MYC supports resistance against immunotherapy - a very promising form of cancer therapy that has recently been added to the armamentarium [41]. According to Casey et al., MYC appears to modulate PD-L1 expression by binding directly to the promoter of the PD-L1 gene in human and mouse tumor cells [42]. The PD-1/PD-L1 signaling axis plays a key role in the immune escape mechanism of cancer cells. Blocking this pathway is currently the preferred approach in cancer immunotherapy. The goal is to unlock the brake that cancer cells impose on T cells by disrupting the immunosuppressive interaction between cancer borne PD-L1 and its cognate receptor PD-1 expressed on the surface of the T cells [43]. In addition, IFN-G is the key mediator of an autoregulatory negative feedback loop. When expressed by activated cytotoxic tumor-infiltrating T cells, IFN-G stimulates cancer cells to overexpress PD-L1, which then curbs the cytolytic action of the T cells and contributes to immunotherapy resistance [34,44-47]. Here, we demonstrate that exogenous IFN-G upregulates PD-L1 expression in all cancer cell types examined, which underlines its significance as a general mechanism of cancer immune escape. Most intriguingly, by adding BET targeting drugs that interfere with the BRD4/MYC/PD-L1 cascade, we were able to disrupt this resistance loop, with dBET6 being the strongest downregulator of MYC and PD-L1 expression. The fact that degradation of BRD4 generally causes stronger anti-cancer effects than its inhibition has very recently been shown by Bai et al. and Zong et al. in triple-negative breast cancer and in lung cancer [48,49]. Here we demonstrate that advanced new-generation BET degraders with improved membrane permeability further elevate the efficacy of BET degrading drugs.
In summary, we demonstrate that certain BET-degrading drugs show promising effects in solid tumor cells not only as single medication, but even more so in combination with chemotherapy or immunotherapy. This is mainly due to the fact that blockade of BRD4 downregulates oncogenic drivers such as MYC, which regulate pathways that make neoplastic cells resilient to adverse conditions such as toxic microenvironments due to chemicals or immunologic defense or strenuous migration during metastasis [50,51]. Accordingly, the next step will be an in vivo analysis of the anti-cancer efficacy of dBET6 combined with standard chemotherapeutics using human tumor mouse models. Overall, forthcoming studies on the clinical benefit of improved BET degraders such as dBET6 against solid tumors are warranted.
Acknowledgements
This work was supported by a cancer stem cell grant of the Medical University of Vienna and by an Austrian Science Fund (FWF) grant from the Herzfelder’sche Familienstiftung (P30625B28).
Disclosure of conflict of interest
None.
References
- 1.Romanowska J, Joshi A. From genotype to phenotype: through chromatin. Genes. 2019;10:76. doi: 10.3390/genes10020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dawson MA. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science. 2017;355:1147–1152. doi: 10.1126/science.aam7304. [DOI] [PubMed] [Google Scholar]
- 3.Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mio C, Bulotta S, Russo D, Damante G. Reading cancer: chromatin readers as druggable targets for cancer treatment. Cancers (Basel) 2019;11:61. doi: 10.3390/cancers11010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pervaiz M, Mishra P, Gunther S. Bromodomain drug discovery - the past, the present, and the future. Chem Rec. 2018;18:1808–1817. doi: 10.1002/tcr.201800074. [DOI] [PubMed] [Google Scholar]
- 6.Donati B, Lorenzini E, Ciarrocchi A. BRD4 and cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17:164. doi: 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi X, Liu C, Liu B, Chen J, Wu X, Gong W. JQ1: a novel potential therapeutic target. Pharmazie. 2018;73:491–493. doi: 10.1691/ph.2018.8480. [DOI] [PubMed] [Google Scholar]
- 13.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, Vakoc CR. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valent P, Zuber J. BRD4: a BET(ter) target for the treatment of AML? Cell Cycle. 2014;13:689–690. doi: 10.4161/cc.27859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrieu GP, Shafran JS, Deeney JT, Bharadwaj KR, Rangarajan A, Denis GV. BET proteins in abnormal metabolism, inflammation, and the breast cancer microenvironment. J Leukoc Biol. 2018;104:265–274. doi: 10.1002/JLB.5RI0917-380RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J, Curtis J, Piao H, Wong LC, Kung AL, Beroukhim R, Bradner JE, Drapkin R, Hahn WC, Liu JF, Livingston DM. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci U S A. 2015;112:232–237. doi: 10.1073/pnas.1422165112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan Z, Zhang X, Kang T, Zhang L, Chen S. Arsenic sulfide amplifies JQ1 toxicity via mitochondrial pathway in gastric and colon cancer cells. Drug Des Devel Ther. 2018;12:3913–3927. doi: 10.2147/DDDT.S180976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klauber-DeMore N, Schulte BA, Wang GY. Targeting MYC for triple-negative breast cancer treatment. Oncoscience. 2018;5:120–121. doi: 10.18632/oncoscience.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X, Liu D, Tao D, Xiang W, Xiao X, Wang M, Wang L, Luo G, Li Y, Zeng F, Jiang G. BRD4 regulates EZH2 transcription through upregulation of C-MYC and represents a novel therapeutic target in bladder cancer. Mol Cancer Ther. 2016;15:1029–1042. doi: 10.1158/1535-7163.MCT-15-0750. [DOI] [PubMed] [Google Scholar]
- 21.Bauer K, Berger D, Zielinski CC, Valent P, Grunt TW. Hitting two oncogenic machineries in cancer cells: cooperative effects of the multi-kinase inhibitor ponatinib and the BET bromodomain blockers JQ1 or dBET1 on human carcinoma cells. Oncotarget. 2018;9:26491–26506. doi: 10.18632/oncotarget.25474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winter GE, Mayer A, Buckley DL, Erb MA, Roderick JE, Vittori S, Reyes JM, di Iulio J, Souza A, Ott CJ, Roberts JM, Zeid R, Scott TG, Paulk J, Lachance K, Olson CM, Dastjerdi S, Bauer S, Lin CY, Gray NS, Kelliher MA, Churchman LS, Bradner JE. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol Cell. 2017;67:5–18. doi: 10.1016/j.molcel.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu L, Chen Y, Mayakonda A, Koh L, Chong YK, Buckley DL, Sandanaraj E, Lim SW, Lin RY, Ke XY, Huang ML, Chen J, Sun W, Wang LZ, Goh BC, Dinh HQ, Kappei D, Winter GE, Ding LW, Ang BT, Berman BP, Bradner JE, Tang C, Koeffler HP. Targetable BET proteins- and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc Natl Acad Sci U S A. 2018;115:E5086–E5095. doi: 10.1073/pnas.1712363115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.French CA. Small-molecule targeting of BET proteins in cancer. Adv Cancer Res. 2016;131:21–58. doi: 10.1016/bs.acr.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Ocana A, Nieto-Jimenez C, Pandiella A. BET inhibitors as novel therapeutic agents in breast cancer. Oncotarget. 2017;8:71285–71291. doi: 10.18632/oncotarget.19744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahni JM, Keri RA. Targeting bromodomain and extraterminal proteins in breast cancer. Pharmacol Res. 2018;129:156–176. doi: 10.1016/j.phrs.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ba M, Long H, Yan Z, Wang S, Wu Y, Tu Y, Gong Y, Cui S. BRD4 promotes gastric cancer progression through the transcriptional and epigenetic regulation of c-MYC. J Cell Biochem. 2018;119:973–982. doi: 10.1002/jcb.26264. [DOI] [PubMed] [Google Scholar]
- 29.Hao J, Yang Z, Wang L, Zhang Y, Shu Y, Jiang L, Hu Y, Lv W, Dong P, Liu Y. Downregulation of BRD4 inhibits gallbladder cancer proliferation and metastasis and induces apoptosis via PI3K/AKT pathway. Int J Oncol. 2017;51:823–831. doi: 10.3892/ijo.2017.4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen G, Jiang M, Pu J. Dual inhibition of BRD4 and PI3K by SF2523 suppresses human prostate cancer cell growth in vitro and in vivo. Biochem Biophys Res Commun. 2018;495:567–573. doi: 10.1016/j.bbrc.2017.11.062. [DOI] [PubMed] [Google Scholar]
- 31.Schulenburg A, Brämswig K, Herrmann H, Karlic H, Mirkina I, Hubmann R, Laffer S, Marian B, Shehata M, Krepler C, Pehamberger H, Grunt T, Jäger U, Zielinski CC, Valent P. Neoplastic stem cells: current concepts and clinical perspectives. Crit Rev Oncol Hematol. 2010;76:79–98. doi: 10.1016/j.critrevonc.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Elbadawy M, Usui T, Yamawaki H, Sasaki K. Emerging roles of C-Myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: a potential therapeutic target against colorectal cancer. Int J Mol Sci. 2019;20:2340. doi: 10.3390/ijms20092340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kareva I. A combination of immune checkpoint inhibition with metronomic chemotherapy as a way of targeting therapy-resistant cancer cells. Int J Mol Sci. 2017;18:2134. doi: 10.3390/ijms18102134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, Parisi G, Saus CP, Torrejon DY, Graeber TG, Comin-Anduix B, Hu-Lieskovan S, Damoiseaux R, Lo RS, Ribas A. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2019;29:3766. doi: 10.1016/j.celrep.2019.11.113. [DOI] [PubMed] [Google Scholar]
- 35.Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. doi: 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sahai V, Redig AJ, Collier KA, Eckerdt FD, Munshi HG. Targeting BET bromodomain proteins in solid tumors. Oncotarget. 2016;7:53997–54009. doi: 10.18632/oncotarget.9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Posternak V, Cole MD. Strategically targeting MYC in cancer. F1000Res. 2016;5 doi: 10.12688/f1000research.7879.1. F1000 Faculty Rev-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H. Combination therapy in combating cancer. Oncotarget. 2017;8:38022–38043. doi: 10.18632/oncotarget.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, Beavis PA, Darcy PK, Martin BP, Spencer A, Traunbauer AK, Sadovnik I, Bauer K, Valent P, Bradner JE, Zuber J, Shortt J, Johnstone RW. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18:2162–2174. doi: 10.1016/j.celrep.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valent P, Sadovnik I, Eisenwort G, Bauer K, Herrmann H, Gleixner KV, Schulenburg A, Rabitsch W, Sperr WR, Wolf D. Immunotherapy-based targeting and elimination of leukemic stem cells in AML and CML. Int J Mol Sci. 2019;20:4233. doi: 10.3390/ijms20174233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casey SC, Baylot V, Felsher DW. The MYC oncogene is a global regulator of the immune response. Blood. 2018;131:2007–2015. doi: 10.1182/blood-2017-11-742577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M, Felsher DW. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–231. doi: 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi T, Ma Y, Yu L, Jiang J, Shen S, Hou Y, Wang T. Cancer immunotherapy: a focus on the regulation of immune checkpoints. Int J Mol Sci. 2018;19:1389. doi: 10.3390/ijms19051389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan Y, Zhao C, Yang R, Zhou T, Xu N. IFN-gamma induces overexpression of PD-L1 and epithelialmesenchymal transformation of breast cancer cells through activating ERK/Jak2-STAT signaling pathways. Sheng Wu Gong Cheng Xue Bao. 2018;34:2007–2015. doi: 10.13345/j.cjb.180070. [DOI] [PubMed] [Google Scholar]
- 45.Thiem A, Hesbacher S, Kneitz H, di Primio T, Heppt MV, Hermanns HM, Goebeler M, Meierjohann S, Houben R, Schrama D. IFN-gamma-induced PD-L1 expression in melanoma depends on p53 expression. J Exp Clin Cancer Res. 2019;38:397. doi: 10.1186/s13046-019-1403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, Yoshioka Y, Baba T, Konishi I, Mandai M. IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015;112:1501–1509. doi: 10.1038/bjc.2015.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W, Zeng H, Zhang N, Du W, Chen C, Huang JA. PD-L1 induced by IFN-gamma from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. 2017;22:1026–1033. doi: 10.1007/s10147-017-1161-7. [DOI] [PubMed] [Google Scholar]
- 48.Bai L, Zhou B, Yang CY, Ji J, McEachern D, Przybranowski S, Jiang H, Hu J, Xu F, Zhao Y, Liu L, Fernandez-Salas E, Xu J, Dou Y, Wen B, Sun D, Meagher J, Stuckey J, Hayes DF, Li S, Ellis MJ, Wang S. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017;77:2476–2487. doi: 10.1158/0008-5472.CAN-16-2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zong D, Gu J, Cavalcante GC, Yao W, Zhang G, Wang S, Owonikoko TK, He X, Sun SY. BRD4 levels determine the response of human lung cancer cells to BET degraders that potently induce apoptosis through suppression of Mcl-1. Cancer Res. 2020;80:2380–2393. doi: 10.1158/0008-5472.CAN-19-3674. [DOI] [PubMed] [Google Scholar]
- 50.Grunt TW, Somay C, Oeller H, Dittrich E, Dittrich C. Comparative analysis of the effects of dimethyl sulfoxide and retinoic acid on the antigenic pattern of human ovarian adenocarcinoma cells. J Cell Sci. 1992;103:501–509. doi: 10.1242/jcs.103.2.501. [DOI] [PubMed] [Google Scholar]
- 51.Schneider SM, Offterdinger M, Huber H, Grunt TW. Involvement of nuclear steroid/thyroid/retinoid receptors and of protein kinases in the regulation of growth and of c-erbB and retinoic acid receptor expression in MCF-7 breast cancer cells. Breast Cancer Res Treat. 1999;58:171–181. doi: 10.1023/a:1006377006816. [DOI] [PubMed] [Google Scholar]