

Abstract
Ovarian cancer is the most lethal gynecologic malignancy. Poly (ADP-ribose) polymerase inhibitors (PARPi) are effective in treating ovarian cancer. However, cancer cell insensitivity and resistance remain challenges. Determination of the exact chemoresistance mechanisms and potential targeted therapies is urgent. CDCA8 (cell division cycle associated 8) participates in the tumorigenesis of various cancers; however, the exact biological function of CDCA8 in ovarian cancer remains obscure. Here, we found that CDCA8 was overexpressed in ovarian cancer and that high expression of CDCA8 promoted the proliferation of ovarian cancer cells in vitro and in vivo. Moreover, silencing of CDCA8 sensitized ovarian cancer cells to olaparib and cisplatin by inducing G2/M arrest, accelerating apoptosis, increasing DNA damage and interfering with RAD51 accumulation in vitro. In addition, MYBL2 (MYB proto-oncogene-like 2), identified as an upstream transcription factor of CDCA8, was positively correlated with the expression level of CDCA8 in ovarian cancer. Finally, MYBL2 enhanced the aggressive characteristics of ovarian cancer cells by regulating CDCA8. In conclusion, high CDCA8 expression was involved in the tumorigenesis, aggressiveness and chemoresistance of ovarian cancer. CDCA8 silencing combined with olaparib treatment might lead to substantial progress in ovarian cancer targeted therapy.
Keywords: Ovarian cancer, CDCA8, olaparib, G2/M arrest, homologous recombination-mediated repair
Introduction
Ovarian cancer has been the leading cause of gynecologic malignancy-related death. Surgery followed by platinum/paclitaxel-based chemotherapy is still the standard therapy for ovarian cancer patients, although most patients develop resistance to platinum-based chemotherapy [1,2], resulting in recurrence and death.
Recently, poly (ADP-ribose) polymerase inhibitors (PARPi) have emerged as effective ovarian cancer-targeted therapies, especially in patients with deleterious germline BRCA mutations [3-5]. Several PARPi (olaparib, rucaparib and niraparib) have been approved by the FDA as ovarian cancer therapies for maintenance and to prevent recurrence [1,4]. PARPi perform their curative functions through synthetic lethality and PARP trapping [6]. While PARPi have displayed exact antitumor capacity, primary and acquired resistance is still an important clinical problem. Multiple potential resistance mechanisms to platinum-based chemotherapies and PARPi have been explored [7], but the exact mechanism is still undefined.
Human cell division cycle associated 8 (CDCA8), also known as Borealin or Dasar B, is an inseparable part of the vertebrate chromosomal passenger complex (CPC) [8,9]. CPC consists of at least four proteins (AURKB, INCENP, BIRC5/survivin and CDCA8) [10-13] that play an important role in the regulation of the cell mitosis process and tumorigenesis [14-17]. CDCA8 has been shown to be highly expressed, to be related to poor prognosis and to participate in the tumorigenesis of bladder cancer [18], lung cancer [9], breast cancer [19,20] and cutaneous melanoma [13]. CDCA8 had higher expression in chemoresistant papillary serous ovarian cancers than in chemosensitive cancers [21]. However, the exact role of CDCA8 in ovarian cancer remains unexplored.
In this study, we aimed to explore the biological functions of CDCA8 in ovarian cancer. Moreover, we evaluated the preclinical efficacy of silencing CDCA8 in combination with olaparib or cisplatin in ovarian cancer cells. Furthermore, the underlying mechanism was investigated in vitro. Finally, we attempted to elucidate the regulatory mechanism of CDCA8 in ovarian cancer.
Materials and methods
Patients and tissue samples
The high-grade serous ovarian cancer (HGSOC) tissue specimens for Western blot and real-time quantitative PCR (RT-qPCR) were from patients with primary ovarian serous carcinoma without previous surgery or chemotherapy, while the fallopian tube (FT) tissues were from patients who received hysterectomy and bilateral salpingo-oophorectomy due to benign gynecologic tumors. Ethical approval was obtained from the Ethics Committee of Shandong University. All patients provided written informed consent.
Cell lines and cell culture
A2780 (RRID: CVCL_0134) and HEY (RRID: CVCL_0297) cell lines were kind gifts from Jianjun Wei’s Laboratory. HEK293T (RRID: CVCL_0063) cells were purchased from the Chinese Academy of Sciences (Shanghai, China). SKOV3 (RRID: CVCL_0532) cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). A2780 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS); HEK293T, HEY and SKOV3 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS (all from Gibco, Grand Island, NY, USA). All cells were cultured in a humidified incubator at 37°C with 5% CO2. All cell lines were identified within 3 years by short tandem repeat profiling and conformed to be mycoplasma free.
Antibodies and reagents
Antibodies against RAD51 (ab133534), γH2AX (ab26350) and MYBL2 (ab12296) were from Abcam (Cambridge, UK); antibody against CDCA8 (DF6115) was purchased from Affinity (Affinity Biosciences, China); antibody against BrdU-FITC (364104) was from Biolegend, and an antibody against β-actin (A5441) was purchased from Sigma-Aldrich (St. Louis, MO, USA); CDDP (PHR1624) was purchased from Sigma-Aldrich (St. Louis, MO, USA); Olaparib (AZD2281) and BrdU (S7918) were purchased from Selleck Chemicals (Houston, TX, USA).
Bioinformatics analysis
The GEO database was searched to download datasets contain gene expression of ovarian cancer and normal control (GSE14407), of resistance ovarian cancer cells and sensitive ones (GSE15709, GSE98559 and GSE58470). The Cancer Genome Atlas (TCGA), Oncomine (www.oncomine.org) and GEPIA (http://gepia.cancer-pku.cn/) databases were used to analyze the mRNA expression of serous ovarian cancer and normal tissues. The Cancer Cell Line Encyclopedia (CCLE, portals.broadinstitute.org) database was searched to download the expression profile of ovarian cancer cells, then the “limma” package of R software was employed to identify the co-expression genes of TOP2A and CDCA8 with the filter of “correlation > 0.5”, “P < 0.001”. Subsequently, the co-expression genes were analyzed with the clusterProfiler package of R software to performed the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. The cBioPortal website (https://www.cbioportal.org) was used to visualize coexpressed genes with CDCA8. Cistrome (https://cistrome.org) and JASPAR (http://jaspar.binf.ku.dk) websites were used to search for potential transcription factors of CDCA8.
IHC
Immunohistochemical (IHC) staining was performed on 4 μm sections. Tissue sections were deparaffinized in xylene and a graded series of ethanol. After antigen retrieval, the slides were incubated with a primary antibody at 4°C overnight. The I-View 3,3’-diaminobenzidine (DAB; ZSGB-BIO, Beijing, China) detection system was used to detect the staining.
RNA isolation and RT-qPCR
Total RNA was extracted with TRIzol reagent (15596018, Invitrogen). cDNA was synthesized using the PrimeScript RT Reagent Kit (RR037A, TaKaRa, Kyoto, Japan). RT-qPCR was performed with SYBR Premix Ex Taq (RR420A, TaKaRa) on the 7900HT Fast Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). The mRNA levels of specific genes were normalized against those of β-actin using the comparative Ct method (2-ΔΔCt). The primers used are shown in the Table S1.
Protein extraction and western blotting
Cells were harvested and lysed in cell lysis buffer for Western blotting and IP (P0013, Beyotime, Shanghai, China) with PMSF (1%). The protein concentration was quantified with a BCA Protein Assay kit (Merck Millipore, USA). Proteins were separated by SDS-PAGE and transferred to PVDF membranes (Merck Millipore, Burlington, MA, USA) followed by blocking for 1-2 h with 5% skim milk and an incubation with primary antibodies at 4°C overnight. On the second day, the membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies and detected with an enhanced chemiluminescence detection kit (ECL ORT2655, PerkinElmer, Waltham, MA, USA). β-actin was used as an endogenous control. The relative protein level was analyzed using ImageJ 1.47 (US National Institutes of Health).
Plasmid construction, lentivirus production and siRNA transfection
CDCA8 shRNA primers were synthesized by GenePharma (Shanghai, China) and cloned into a PLKO vector. The CDCA8 over-expression plasmid was purchased from GeneChem (Shanghai, China), while the PCMV vector was used as a mock control. psPAX2, pMD2.G, PLKO/shCDCA8 or PCMV/CDCA8 were co-transfected into HEK293T cells to produce lentivirus. Ovarian cancer cells (A2780, SKOV3 and HEY) at appropriate confluence were transfected with lentivirus for 24 h before selection with 2 μg/ml puromycin (Merck Millipore, USA) for 7 days to acquire cells with stable expression.
Specific siRNA and negative control siRNA (NC) synthesized by GenePharma (Shanghai, China) were transfected into cells at appropriate confluence with Lipofectamine 2000 reagent according to the manufacturer’s protocol (11668-019, Invitrogen).
The sequences of specific siRNA and short hair RNA (shRNA) were as follows: siCDCA8, 5’-CAGCAGAAGCUAUUCAGACTT-3’, siMYBL2: 5’-CAGACAAUGCUGUGAAGAATT-3’, siNC: 5’-UUCUCCGAACGUGUCACGUTT-3’; shCDCA8, 5’-AACAGCAGAAGCTATTCAGAC-3’, shMYBL2: 5’-GCCCAAGAGCACACCTGTTAA-3’.
Cell proliferation assay
The 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay was used to detect the cell proliferation ability. A total of 800-1000 cells per well were seeded in 96-well plates and cultured for 5 days. Before detecting the absorbance, 10 μg MTT (Sigma-Aldrich, USA, 0.5 mg/ml) was added to each well, 4 h incubation was continued and the supernatant was replaced by 100 μl DMSO (Sigma-Aldrich, USA). A Varioskan Flash microplate reader (Thermo Scientific) was used to detect the absorbance value at 490 nm.
Colony formation assay
A total of 800-1000 cells were seeded in each well of 6-well plates and cultured for 10-14 days. After fixing with methanol and staining with 0.5% crystal violet, colonies containing more than 50 cells were counted.
Transwell assays
Transwell assays were performed in transwell chambers (8-μm pores, BD Biosciences, USA) inserted in 24-well plates without or with Matrigel (BD Biosciences, USA). The upper chambers were coated with 200 μl serum-free medium containing 1 × 105-2 × 105 cells, while the lower compartment contained 700 μl culture medium supplemented with 20% FBS. After incubating at 37°C for an appropriate time, cells that had migrated to the lower surface of the membrane were fixed with methanol, stained in 0.5% crystal violet, observed and quantified under a light microscope.
Drug treatment
For survival assay, approximately 3 × 103 cells transfected with PLKO.NC or shCDCA8 were seeded in each well of 96-well plates and treated with indicated concentrations of olaparib or cisplatin. MTT assay was performed to detect cell viability.
For clonogenic assays, approximately 1.5 × 103 cells were seeded in each well of 6-well plates. When the clones counted about 50 cells, indicated concentrations olaparib or cisplatin were added and incubated for 48 h.
Flow cytometry assay for cell cycle and apoptosis
Cells were harvested and stained with propidium iodide (PI) according to the manufacturer’s protocol (CCS012, MultiSciences Biotech Co., Ltd.) to analyze the distribution of the cell cycle.
For BrdU incorporation, 10 μM BrdU was added 4 hours before harvesting. Cells were fixed with 75% EtOH overnight at -20°C, then treated with 2 N HCl/0.5% Triton X-100 and 0.1 M Na2B4O7 (A610480, Sangon Biotech, Shanghai, China). Cells were stained with anti-BrdU-FITC (Biolegend, 364104) in the dark for 30 minutes followed by staining with PI for 5 minutes. Stained cells were detected by flow cytometry (FC500, Beckman Coulter, Brea, CA, USA) within one hour.
Apoptosis was quantified with annexin V-FITC and PI. Treated cells were harvested, rinsed twice with PBS and resuspended in 1 × binding buffer (556547, BD Bioscience, Franklin Lakes, NJ, USA). Before detection by flow cytometry, resuspended cells were stained with FITC Annexin V for 25 min and PI for 15 min in the dark at room temperature. The results were analyzed using FlowJo X 10.0.7 R2 software.
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde for 15 min, blocked with normal goat serum at room temperature (RT) for 30 min, and incubated with γH2AX (ab26350; Abcam; dilution 1:100) or RAD51 (ab133534; Abcam; dilution 1:100) antibodies overnight at 4°C. On the second day, cells were incubated with donkey anti-rabbit IgG Alexa Fluor-488 or goat anti-mouse IgG Alexa Fluor-594 (1:150; Invitrogen, Waltham, MA, USA) for 1 h at 37°C in the dark. The nuclei were counterstained with DAPI. Images were captured by Zeiss LSM 780 (Carl Zeiss, Jena, Germany).
Tumor formation assay in nude mice
HEY cells transfected with PLKO.NC and shCDCA8 were harvested, washed and resuspended in PBS. Next, 5 × 106 cells in 150 μl PBS were subcutaneously injected into the left armpit of each mouse. The tumor volumes were measured once every two days using the equation: length × width2 × 0.5. Fourteen days post-injection, the mice were sacrificed and dissected, the tumors were photographed, weighed and the tumor volumes were calculated. Female athymic BALB/c nude mice (4-5 weeks old; NBRI of Nanjing University, Nanjing, China) were maintained in a pathogen-free facility. All animal experiments were performed with the approval of the Shandong University Animal Care and Use Committee.
Luciferase reporter assay
Cells were transiently cotransfected with PCMV or MYBL2, PGL4.26 vector or CDCA8 full-length promoter and pRL-TK using Lipofectamine 2000 for 48 h. Luciferase activity was measured using the Dual-Glo Luciferase Assay System (E2920, Promega, Fitchburg, WI, USA) following the manufacturer’s instructions. The relative luciferase activity was determined by the ratio between firefly luminescence and renilla luminescence.
Chromatin immunoprecipitation (CHIP) assay
CHIP assay was carried out with Beyotime Chromatin immunoprecipitation kit (P2078, Beyotime, Shanghai, China) following the manufacturer’s instructions. Briefly, HEY cells transfected with PCMV-MYBL2 were cross-linked in 1% formaldehyde solution for 10 min at room temperature. Then, 1.1 ml of glycine was added and incubated at room temperature for 5 min. The crosslinked samples were lysed in lysis buffer and sheared by sonification to obtain chromatin fragments that were 200-1000 bp. 10 µl supernatants were retained as input. The DNA-protein complexes were incubated overnight at 4°C with Flag antibody or negative control IgG. After immunoprecipitation, the chromatins were de-crosslinked at 65°C for 4 h and DNA was purified before conducting PCR. The sequences for CDCA8 promoter were as follows: CDCA8 forward, 5’-GGGGAGCGAGGAACGACA-3’; CDCA8 reverse, 5’-GGGTCTTCCGCCCAATGC-3’.
High-throughput differential gene expression analysis
The high-throughput mRNA-Seq experiments were conducted by Biomaker Technologies (Beijing, China). Briefly, A2780 cells were transfected with siCDCA8 or NC (n = 3) for 48 h and total RNAs were extracted using TRIzol reagent. Sequencing libraries were generated using NEBNextR UltraTM Directional RNA Library Prep Kit for IlluminaR (NEB, USA) following manufacturer’s recommendations, then the library preparations were sequenced on an Illumina Hiseq Xten platform and paired-end reads were generated. Subsequently, gene expression levels were estimated by fragments per kilobase of transcript per million fragments mapped (FPKM) and differential expression analysis of two groups was performed using the DESeq R package (1.10.1). Genes with an adjusted P-value < 0.05 and absolute value of log2 (Fold change) > 1 found by DESeq were assigned as differentially expressed genes (DEGs). Finally, gene function was annotated based on GO (Gene Ontology database) and KEGG (The database of Kyoto Encyclopedia of Genes and Genomes) databases. The Gene Ontology (GO) database can be used for functional enrichment analysis from cellular component (CC), molecular function (MF), and biological process (BP) respects. The GO enrichment analysis of the DEGs was implemented by the clusterProfiler R package and the KEGG pathway that are significantly enriched of the DEGs was analyzed by DAVID website (https://david.ncifcrf.gov/tools.jsp).
Statistical analysis
All experiments were repeated at least 3 times independently. The data are expressed as the means ± SEMs. Significance between two and more than two groups was performed using Student’s t test and oneway ANOVA, respectively by SPSS v22.0 (SPSS, Inc., Chicago, IL, USA). Images were processed using GraphPad Prism 8.00 (GraphPad Software, La Jolla, CA, USA) and Adobe Photoshop CC 2019 (Adobe, San Jose, CA, USA). P < 0.05 was considered statistically significant (#P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001).
Results
CDCA8 is key gene high expressed and correlated to chemoresistance in ovarian cancer
In order to identify genes that play a role in progression and chemoresistance in ovarian cancer, we focused on higher expressed genes in ovarian cancer especially cisplatin resistance ovarian cancer. We searched the GEO database and restricted our results to genes with log2 (Fold change) > 1 in GSE14407 and log2 (Fold change) > 0.5 in GSE15709, respectively. Within these restrictions, we found 8 genes that were significantly higher expressed and related to cisplatin resistance in ovarian cancer (Figure 1A). We focused on the top2 genes TOP2A and CDCA8. To confirm our findings, we searched TCGA, GEPIA and Oncomine databases and found that TOP2A and CDCA8 were obviously more highly expressed in serous ovarian cancer samples than in peritoneum or ovarian surface epithelial tissues (Figure 1B-H). Then we verified the expression of these two genes in cisplatin resistance ovarian cancer cells, as shown in Figure 1I-N, CDCA8 was higher expressed in cisplatin resistance A2780, SKOV3 and IGROV1 ovarian cancer cells compared with the sensitive ones, however, the expression of TOP2A was only higher in A2780 cells. We also found that the expression level of these two genes were higher in other cancers such as bladder, lung and breast cancer (Figure 1O, 1P). Furthermore, the mRNA expression data of TOP2A and CDCA8 in ovarian cancer cells were also downloaded from the CCLE and KEGG pathway analysis was performed based on the co-expression genes of TOP2A and CDCA8, respectively. As shown in Figure 1Q and 1R, they both participated in the cell cycle, DNA replication, oocyte meiosis and progesterone-mediated oocyte maturation, while CDCA8 also played a role in base excision repair, nucleotide excision repair and mismatch repair pathways which are important in the DNA damage repair, so we focused on CDCA8. Additionally, the mRNA and protein levels of CDCA8 in HGSOC and FT samples from patients were detected by RT-qPCR and Western blot, and the results showed that CDCA8 had significantly higher expression in HGSOC than in FT tissues (Figures 1S, 1T, S2A). Immunohistochemistry (IHC) was performed to assess the expression profile of CDCA8 in HGSOC. CDCA8-positive staining was located in both the nucleus and cytoplasm, and CDCA8 expression in HGSOC tissues was significantly higher than that in FT tissues (Figure 1U). These results suggest that CDCA8 was distinctively overexpressed in ovarian cancer especially in cisplatin resistance ones, implying a potent role of CDCA8 in the progression and chemoresistance of ovarian cancer.
Figure 1.

CDCA8 is key gene high expressed and correlated to chemoresistance in ovarian cancer. (A) Public data sets were used to identify genes associated with chemoresistance of ovarian cancer. (B-H) Oncomine and GEPIA databases showed the mRNA expression of CDCA8 and TOP2A in serous ovarian carcinoma and normal controls. (I-N) The mRNA expression level of CDCA8 and TOP2A in cisplatin resistance A2780, SKOV3 and TGROV1 cells. (O and P) The mRNA level of CDCA8 and TOP2A in pan cancer. (Q and R) Pathway analysis showed the signal pathways participated in by co-expression genes of CDCA8 and TOP2A. (S and T) The mRNA and protein levels of CDCA8 in HGSOC and FT tissues. (U) Representative IHC staining of CDCA8 in HGSOC and FT tissues (200 ×), Scale bar: 50 µm. (Data are mean ± SEM, #P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, n = 3).
CDCA8 promotes the proliferation and motility of ovarian cancer
To explore the potent role of CDCA8 in the development and progression of ovarian cancer, A2780, SKOV3 and HEY cells were stably transfected with shCDCA8 to knockdown CDCA8 expression (Figures 2A, S2B). Compared to the control, upon CDCA8 silencing, fewer viable cells were observed during cultivation of these three ovarian cancer cells (Figure 2B). Colony formation assays also showed that growth was retarded in the shCDCA8 groups (Figure 2C). To investigate whether CDCA8 could influence the migration and invasion abilities of ovarian cancer cells, we performed transwell assays. The results showed that CDCA8 silencing dramatically decreased cell migration and invasion to the lower surface of the membrane (Figure 2D, 2E). To further explore the effects of CDCA8 on the tumorigenesis of ovarian cancer in vivo, HEY cells transfected with PLKO.NC or shCDCA8 were subcutaneously injected into the left armpit of female nude mice. The results showed that the volumes of the tumors in the shCDCA8 group were evidently decreased compared with those in the control group (Figure 2F, 2G). The expression level of CDCA8 in tumor samples was detected again by IHC. As shown in Figure 2H, the CDCA8 expression level was obviously decreased in the shCDCA8 group. Additionally, the number of Ki-67 positive cells was distinctively decreased in the shCDCA8 group (Figure 2H). To confirm the promotive role of CDCA8 in ovarian cancer, CDCA8-overexpressing lentiviruses (CDCA8) were transfected to SKOV3 and HEY cells to overexpress CDCA8 (Figures 2I, S2C). Conversely, overexpression of CDCA8 obviously accelerated the proliferation and colony formation of SKOV3 and HEY cells (Figure 2J, 2K). The migration and invasion of ovarian cancer cells with CDCA8 overexpression were significantly promoted (Figure 2L). These findings suggest that CDCA8 promoted proliferation, migration and invasion of ovarian cancer.
Figure 2.

CDCA8 promotes the proliferation and motility of ovarian cancer. (A) The protein level of CDCA8 in ovarian cancer cells after transfection with PLKO.NC or shCDCA8. (B) Proliferative curve showed the effect of knocking down CDCA8 on A2780, SKOV3 and HEY cells. (C) The effect of CDCA8 inhibition on colony formation. (D) Transwell assays showed the effects of CDCA8 knockdown on migration and invasion of A2780, SKOV3 and HEY cells (200 ×), Scale bar: 100 µm. (E) Quantification of the number of migration and invasion cells in (D). (F) Tumors photographs were shown. (G) Tumors volumes of each group. (H) Representative IHC staining of Ki-67 and CDCA8 in tumor tissues (400 ×), Scale bar: 25 µm. (I) The protein level of CDCA8 in SKOV3 and HEY cells after transfection with PCMV or CDCA8. (J) MTT assay showed the effect of CDCA8 on SKOV3 and HEY cells. (K) Effect of CDCA8 overexpression on the colony formation of SKOV3 and HEY cells. (L) In SKOV3 and HEY cells, over-expression of CDCA8 affected the migration and invasion ability (200 ×), Scale bar: 100 µm. (Data are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, n = 3; for mouse, n = 6).
CDCA8 knockdown renders ovarian cancer cells more sensitive to olaparib and cisplatin
A2780 and SKOV3 cells were treated with olaparib for 48 h and 72 h respectively, then MTT assay was performed to show the cell survival rate and IC50 (the drug concentration that inhibited cell viability by 50%) of these cells. As shown in Figures 3A and S1, the cell viability decreased in a dose-dependent manner and the IC50 values of olaparib for A2780 and SKOV3 cells were 25.92 and 67.76 μM, respectively. To confirm the role of CDCA8 in the sensitivity of ovarian cancer cells to olaparib, A2780 and SKOV3 cells were treated with 25 μΜ and 100 μM olaparib for 48 h, separately. Western blot assays demonstrated that the CDCA8 level increased after olaparib treatment (Figures 3B, S2D). Cells were treated with the indicated concentrations of olaparib after stable transfection with PLKO.NC or shCDCA8, and MTT assays were performed to measure cell viability 48 h after drug treatment. We observed that the IC50 was lower in the shCDCA8 group than in the control group (Figure 3C). The colony formation assays also indicated that ovarian cancer cells with CDCA8 knockdown were more sensitive to olaparib than control cells (Figure 3D). Due to the similar drug mechanism between olaparib and cisplatin, we also detected the effect of CDCA8 knockdown on the sensitivity to cisplatin. As shown in Figures 3E-G and S2E, cisplatin treatment elevated the expression of CDCA8, and CDCA8 inhibition significantly enhanced the ovarian cancer cells response to cisplatin.
Figure 3.

CDCA8 knockdown renders ovarian cancer cells sensitive to olaparib and cisplatin. (A) The IC50 of olaparib in A2780 and SKOV3 cells. (B) A2780 and SKOV3 cells were treated with 25 μΜ and 100 μΜ olaparib for 48 h, respectively. The expression of CDCA8 protein level was analyzed by western blot. (C and D) A2780 and SKOV3 cells transfected with PLKO.NC or shCDCA8 were treated with olaparib for 48 h at indicated concentrations. MTT assays (C) and colony formation assays (D) were performed to detect cell viability and colony forming capacity. (E) A2780 and SKOV3 cells were treated with 2 μg/ml cisplatin for 48 h. The protein expression level of CDCA8 was analyzed by Western blot. (F and G) A2780 and SKOV3 cells transfected with PLKO.NC or shCDCA8 were treated with cisplatin for 48 h at indicated concentrations. MTT assays (F) and colony formation assays (G) were performed to detect cell viability and colony forming capacity. (Data are mean ± SEM, *P < 0.05, **P < 0.01, n = 3).
Next-generation sequencing (NGS) assay reveals signaling pathways involved in the function of CDCA8
To identify the underlying mechanisms involved in CDCA8-induced biological functions and insensitivity to chemotherapy, Next-generation sequencing (NGS) was performed in CDCA8-knockdown (siCDCA8) and negative control (NC) A2780 cells. Differentially expressed genes (DEGs) were selected according to their fold change (> 2-fold) and statistical significance (P < 0.05). A total of 1263 genes were found, including 760 upregulated and 503 downregulated genes (Figure 4A; Table S2). Biological process (BP) analysis revealed that DEGs mainly participated in the positive regulation of cell proliferation, negative regulation of apoptosis, cell division, negative regulation of cell differentiation, sister chromatid cohesion, DNA damage response and DNA replication initiation (Figure 4B). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was performed to explore the main signaling pathways in which the DEGs participated. The results showed that the downregulated genes mainly participated in the cell cycle, DNA replication, Fanconi anemia pathway, Oocyte meiosis and homologous recombination (Figure 4C). While the top upregulated pathway was the p53 signaling pathway (Figure 4D). We showed partial DEGs and performed RT-qPCR to verify the changes in the representative DEGs involved in the cell cycle, DNA replication, homologous recombination and the p53 signaling pathway (Figure 4E-H). We speculated that CDCA8 mediated the sensitivity of ovarian cancer cells to olaparib and cisplatin through the cell cycle, DNA replication and homologous recombination pathways which were related to the mechanism of these drugs.
Figure 4.

NGS analysis of CDCA8 affected signal pathways. A2780 cells were transfected with siCDCA8 or NC for 48 h and NGS was performed to determine the mRNA expression profile. (A) DEGs between siCDCA8 and NC groups were exhibited in Volcano plot. (B) Biological Process analysis showed the main process participated in by DEGs. (C) The downregulated pathways participated in by the DEGs. (D) The upregulated pathways participated in by the DEGs. (E and F) Heatmap of DEGs in the cell cycle, DNA replication, homologous recombination and P53 signaling pathway between siCDCA8 and NC groups. (G and H) Representative DEGs between siCDCA8 and NC groups were verified by RT-qPCR in A2780 cells. (Data are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, n = 3).
Knockdown of CDCA8 sensitizes ovarian cancer to olaparib and cisplatin by regulating cell cycle, apoptosis and homologous recombination-mediated repair
SKOV3 and HEY cells transfected with PLKO.NC or shCDCA8 were incubated with DMSO or olaparib for 48 h at appropriate concentration. Cell cycle and BrdU incorporation analysis of olaparib-treated cancer cells revealed that combination treatment with knocking down CDCA8 significantly increased the number of cells in G2/M phase (Figure 5A, 5B). We also performed an apoptosis assay to demonstrate whether CDCA8 knockdown affects olaparib sensitivity by increasing the proportion of cells undergoing apoptosis. As shown in Figure 5C, there were more apoptotic cells in the shCDCA8 group than in the control group, and olaparib treatment increased the proportion of apoptotic cells more obviously in the shCDCA8 group. To explore whether depletion of CDCA8 sensitize ovarian cancer cells to olaparib by inducing DNA damage and reducing HR-mediated repair, immunofluorescence analysis was performed, and the number of γH2AX and RAD51 foci inside the cells was counted. CDCA8 knockdown or olaparib monotherapy groups had increased γH2AX expression and reduced RAD51 expression, while these changes were more obvious in the group treated with the combination of CDCA8 silencing and olaparib (Figure 5D, 5E).
Figure 5.

Knockdown CDCA8 increases olaparib sensitivity in SKOV3 and HEY cells by regulating the cell cycle, apoptosis and homologous recombination-mediated repair. HEY and SKOV3 cells transfected with PLKO.NC or shCDCA8 were treated with 40 μM and 100 μM olaparib for 48 h in cell cycle, apoptosis and immunofluorescence assays; for BrdU incorporation the concentration of olaparib was 3 μM and 5 μM, respectively. (A) Cell cycle analysis was performed by flow cytometry. (B) Cell cycle was analyzed via flow cytometry using BrdU incorporation. (C) Flow cytometry assays were performed to analyze the apoptosis. (D and E) The number of γH2AX and RAD51 foci were examined by immunofluorescence (400 ×). Scale bar: 10 µm. (Data are mean ± SEM, *P < 0.05, **P < 0.01, n = 3).
CDCA8 is transcriptionally regulated by MYBL2
To determine the regulatory mechanism of CDCA8 in ovarian cancer, we searched the Cistrome and JASPAR online databases for potential transcription factors that targeted the CDCA8 promoter. Finally, we focused on MYBL2. To determine the direct regulatory role of MYBL2 and CDCA8, we first detected the mRNA and protein levels of CDCA8 after knockdown or overexpression of MYBL2. The results showed that CDCA8 expression decreased with silencing of MYBL2 and that overexpression of MYBL2 increased the CDCA8 level (Figures 6A, 6B, S2F). Then, we performed RT-qPCR to assess the expression profile of MYBL2 in ovarian cancer tissues and FT samples. As shown in Figure 6C, MYBL2 was significantly overexpressed in ovarian cancer tissues. Public databases were also searched to determine the differential expression of MYBL2 between ovarian cancer tissues and normal control tissues. MYBL2 was significantly more highly expressed in ovarian cancer samples than in peritoneum or ovarian surface epithelial tissues (Figure 6D-G). Moreover, a positive correlation between MYBL2 and CDCA8 was confirmed by online databases and RT-qPCR (Figure 6H, 6I). Furthermore, we cloned the promotor of CDCA8 into the pGL4.26 vector to construct the CDCA8 promoter plasmid, and the pGL4.26 vector or CDCA8 promoter was then cotransfected into HEK293T, SKOV3 and HEY cells with PCMV or MYBL2 plasmids, separately. The results showed that MYBL2 overexpression increased the luciferase activity in cells transfected with the CDCA8 promoter (Figure 6J). Finally, ChIP-PCR was performed in HEY cells to confirm that MYBL2 could bind directly to the promoter of CDCA8 at the predicted potentially binding motif (CAGTCAA) about 424 bp upstream of the CDCA8 transcription start site (TSS) (Figure 6K, 6L). Thus, MYBL2 could regulate CDCA8 expression through directly binding to the CDCA8 promoter in ovarian cancer cells.
Figure 6.

CDCA8 is transcriptionally regulated by MYBL2. (A and B) SKOV3 and HEY cells were transfected with NC, siMYBL2, PCMV or MYBL2 for 48 h. The mRNA and protein levels of CDCA8 and MYBL2 were determined by RT-qPCR (A) and western blot (B). (C) The mRNA level of MYBL2 in HGSOC tissues and FT tissues. (D-G) The expression of MYBL2 in ovarian carcinoma and normal controls in Oncomine and GEPIA datasets. (H and I) Correlation between expression levels of MYBL2 and CDCA8 from GEPIA database and in HGSOC tissues. (J) PCMV or MYBL2 plasmids were co-transfected into HEK293T, SKOV3 and HEY cells with CDCA8 promoter or pGL4.26 vector for 48 h and luciferase activity were measured. (K) Schematic illustration of the potential MYBL2-binding sites and sequence on CDCA8 promoter predicted by JASPAR. (L) ChIP-PCR assays were performed to identify that MYBL2 binds directly to CDCA8 promoter. (Data are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, n = 3).
MYBL2 acts as an oncogene in ovarian cancer
To further reveal the definite role of MYBL2 in ovarian cancer, MYBL2 was knocked down by shRNA (shMYBL2). MTT and colony formation assays were performed to assess the growth ability of ovarian cancer cells. After transfection with shMYBL2, the growth rate and colony formation ability of these cells decreased markedly compared to those of cells transfected with PLKO.NC (Figure 7A, 7B). In the transwell assays, migration and invasion of A2780, SKOV3 and HEY cells transfected with shMYBL2 were evidently decreased compared to those of the control cells (Figure 7C, 7D). These findings indicated that inhibition of MYBL2 impaired the growth and metastasis of ovarian cancer cells.
Figure 7.
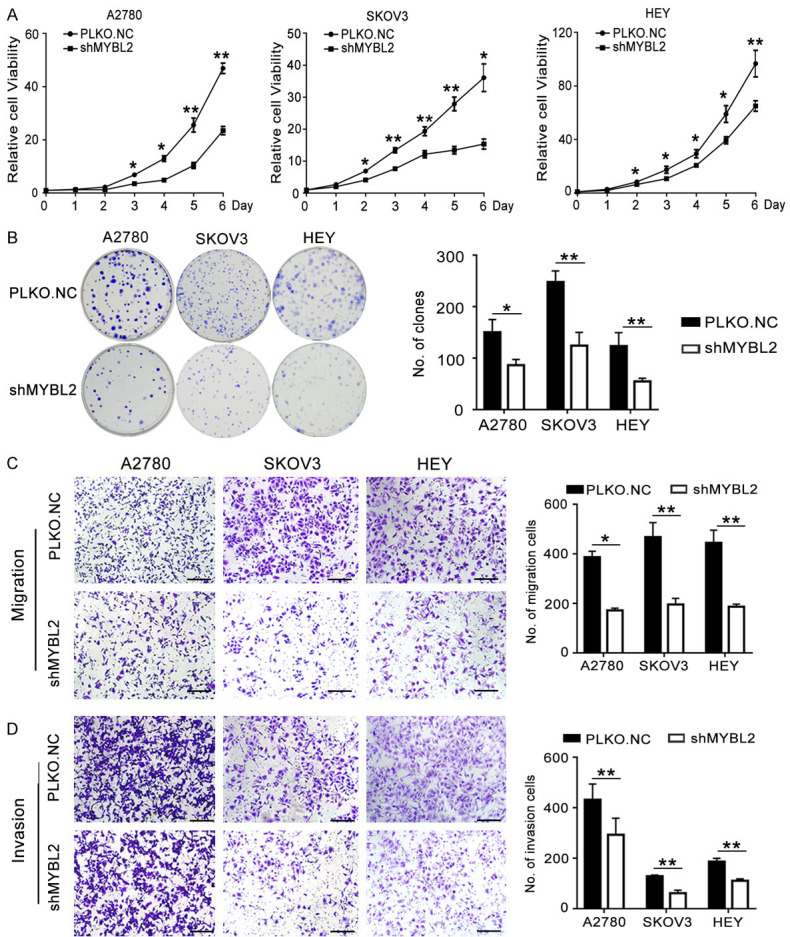
Knockdown of MYBL2 inhibits the proliferation, migration and invasion of ovarian cancer cells. (A) MTT assays were performed to analyze the effect of MYBL2 on cell viability in A2780, SKOV3 and HEY cells. (B) Effect of MYBL2 knocking down on the colony formation of ovarian cancer cells. (C and D) Transwell assays of A2780, SKOV3 and HEY cells after transfection with shMYBL2 or PLKO.NC (200 ×), Scale bar: 50 µm. (Data are mean ± SEM, *P < 0.05, **P < 0.01, n = 3).
Knockdown MYBL2 increases olaparib and cisplatin sensitivity in ovarian cancer cells
To explore the role of MYBL2 in response to olaparib in ovarian cancer cells, HEY and SKOV3 cells were disposed with 40 μΜ and 100 μM olaparib for 48 h, separately. Western blot assays showed an increased protein level of MYBL2 after olaparib treatment (Figures 8A, S2G). Indicated concentrations of olaparib were added to ovarian cancer cells which were stably transfected with PLKO.NC or shMYBL2, and the cell viability was measured by MTT assays 48 h after drug treatment. The shMYBL2 group was more sensitive to olaparib than the control group (Figure 8B). The colony formation assays also indicated that ovarian cancer cells with MYBL2 knockdown were more sensitive to olaparib than control cells (Figure 8C). As shown in Figures 8D-F and S2H, cisplatin treatment elevated the protein level of MYBL2, and knockdown of MYBL2 significantly enhanced the ovarian cancer cells sensitivity to cisplatin.
Figure 8.

Knockdown MYBL2 increases olaparib and cisplatin sensitivity in ovarian cancer cells. (A) Western blot assay was used to show the protein level of MYBL2 in HEY and SKOV3 cells after treating with 40 μΜ and 100 μΜ olaparib. (B and C) The cell viability and colony forming capacity of olaparib treated SKOV3 and HEY cells which were transfected with PLKO.NC or shMYBL2 were detected by MTT assays (B) and colony formation assays (C). (D) SKOV3 and HEY cells were treated with 2 μg/ml cisplatin for 48 h. The protein expression level of MYBL2 was analyzed by Western blot. (E and F) SKOV3 and HEY cells transfected with PLKO.NC or shMYBL2 were treated with cisplatin for 48 h at indicated concentrations. MTT assays (E) and colony formation assays (F) were performed to detect cell viability and colony forming capacity. (Data are mean ± SEM, *P < 0.05, **P < 0.01, n = 3).
MYBL2 promotes aggressive tumor behavior dependent on CDCA8
To determine whether MYBL2 increased the aggression of ovarian cancer cells through CDCA8, we performed a rescue experiment. We introduced CDCA8 or PCMV plasmids into HEY cells that were previously transfected with PLKO.NC or shMYBL2. The results showed that MYBL2 knockdown significantly decreased the proliferation and colony formation of HEY cells, whereas ectopic expression of CDCA8 rescued the inhibitory effect induced by shMYBL2 (Figure 9A, 9B). Similarly, the migration and invasion abilities of HEY cells were impaired in the shMYBL2 group, whereas overexpression of CDCA8 rescued the inhibitory effect of shMYBL2 (Figure 9C). In addition, we introduced siCDCA8 or NC into SKOV3 cells previously transfected with MYBL2 or PCMV. The results showed that CDCA8 knockdown obviously abrogated the proliferation and metastasis induced by MYBL2 overexpression in SKOV3 cells (Figure 9D-F). These results demonstrate that MYBL2 enhanced the aggression of ovarian cancer cells in a CDCA8-dependent manner.
Figure 9.
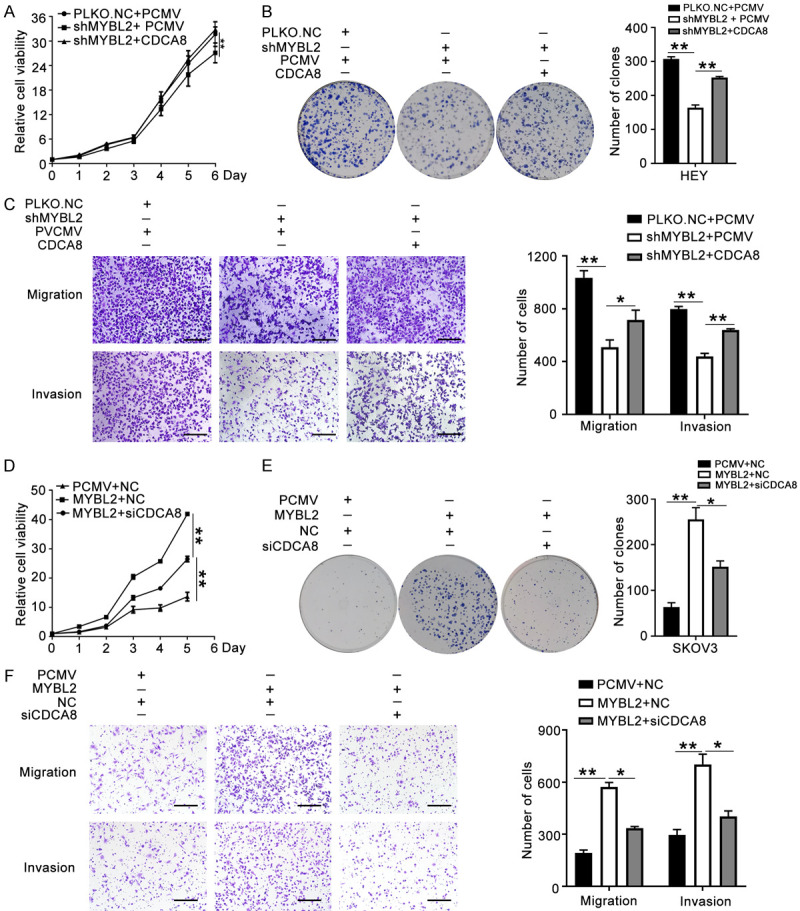
MYBL2 promotes tumor aggressive progression dependent on CDCA8. (A-C) PCMV or CDCA8 plasmids were introduced to HEY cells that transfected with PLKO.NC or shMYBL2. Cell proliferation was detected by MTT assays (A) and colony formation assays (B). Migration and invasion abilities were determined by transwell assays (C) (200 ×), Scale bar: 100 µm. (D-F) MYBL2 over-expressing SKOV3 cells were transfected with siCDCA8 or NC. MTT assays (D), colony formation assays (E) and transwell assays (F) were performed to analyze the effect of CDCA8 depletion on SKOV3 cells stably transfected with MYBL2 (200 ×), Scale bar: 100 µm. (Data are mean ± SEM, *P < 0.05, **P < 0.01, n = 3).
Discussion
CDCA8 has been found to be overexpressed, indicate poor prognosis and participate in the tumorigenesis of several cancers, such as lung cancer [9], breast cancer [8,20], bladder cancer [18] and cutaneous melanoma [13]. In the present study, we found that the expression of CDCA8 was higher in HGSOC tissues than in FT samples. Moreover, overexpression of CDCA8 promoted the proliferation, migration and invasion of ovarian cancer cells in vitro and accelerated the growth of tumors in vivo.
Inherited and acquired resistance to cisplatin impedes the treatment of ovarian cancer [22]; however, the exact reasons leading to cisplatin resistance remain unclear. Recently, PARPi have displayed exciting effects for the current treatment of ovarian cancers. Moreover, future combined treatment strategies that could enhance the antitumor activity of PARPi are underway [23]. Woong Ju et al. found that CDCA8 was upregulated in chemoresistant epithelial ovarian cancer [24]. Therefore, we hypothesized that CDCA8 may be involved in the chemosensitivity of ovarian cancer to cisplatin and olaparib. In this study, we demonstrated that treatment of ovarian cancer cells with cisplatin and olaparib increased CDCA8 expression and that knockdown of CDCA8 sensitized A2780 and SKOV3 ovarian cancer cells to cisplatin and olaparib.
Previous studies have found that olaparib treatment could cause S or G2/M arrest alone or combined with other drugs [5,25-29]. In line with this, we found that treating ovarian cancer cells with olaparib at the IC50 concentration robustly increased the proportion of cells in G2/M, while combination treatment with CDCA8 knockdown enhanced this effect.
PARPi play roles in cancers by trapping PARP at SSB sites and preventing the repair of SSB, which may be converted to DSB [30], finally leading to cancer cell apoptosis [26]. The rH2AX protein is a widely acknowledged indication of DSB damage [31,32], and RAD51 is one of the important proteins in the DNA damage response pathway [33]. Our study revealed that silencing CDCA8 increased the sensitivity to olaparib in ovarian cancer depending on increased accumulation of rH2AX and downregulation of the RAD51 accumulation in the nuclei. However, the exact mechanisms by which CDCA8 influences the rH2AX and RAD51 accumulation and the interactions between CDCA8 and other members of the DSB repair pathway still need to be further investigated.
The interaction and regulatory mechanisms of CDCA8 have been investigated. Junn-Liang Chang [34] revealed that CDCA8 interacted and formed a complex with survivin. Satoshi Hayama et al. [9] demonstrated that CDCA8 was coactivated with and phosphorylated/stabilized by AURKB in lung cancer cells. In addition, CDCA8 promoted the progression of cutaneous melanoma cells via the ROCK pathway [13]. However, few studies have focused on the transcriptional regulation of CDCA8 in ovarian cancer.
The MYBL2 gene belongs to the transcription factor MYB family and has been reported to regulate the cell cycle [35-37], proliferation, survival, differentiation [38] and genome stability maintenance [39-42]. MYBL2, an oncogene, promoted the progression of breast cancer [43], non-small-cell lung cancer [44], gallbladder cancer [45], liver cancer [46] and colorectal cancer [47]. Audra N. Iness et al. reported that the MMB (Myb-MuvB) complex cooperated with FOXM1 to activate G2/M gene expression, and the expression of CDCA8 decreased after the downregulation of MYBL2 in ovarian and breast cancers [35]. Moreover, Rachel Bayley et al. revealed that low MYBL2 levels were associated with the transcriptional deregulation of DNA repair genes [39]. Finally, we hypothesized that CDCA8 might be transcriptionally regulated by MYBL2 in ovarian cancer.
In the current study, we verified that MYBL2 was overexpressed and promoted the progression of ovarian cancer cells. A positive correlation was clarified between MYBL2 and CDCA8 among specimens from ovarian cancer patients. We also illustrated that MYBL2 could bind to the CDCA8 promoter region directly to promote the transcription of CDCA8. Overexpression or knockdown of CDCA8 in MYBL2-depleted or MYBL2-overexpressing cells reversed the effect of MYBL2.
In summary, our study demonstrated that CDCA8 was overexpressed in ovarian cancer tissues. Overexpression of CDCA8 promoted the proliferation, migration and invasion abilities of ovarian cancer cells. Depletion of CDCA8 caused G2/M arrest, increased DNA damage and apoptosis to sensitize ovarian cancer cells to cisplatin and olaparib. In addition, our results illustrated that MYBL2 could promote CDCA8 transcription and act as an oncogene in ovarian cancer dependent on CDCA8. In conclusion, inhibition of CDCA8 combined with cisplatin or olaparib treatment might substantially enhance the therapy of ovarian cancer. We should make efforts to establish methods that can directly and effectively target CDCA8 in the near future.
Acknowledgements
Here, I would like to express my heartfelt thanks to all those who have helped me with this thesis possible and better. This work was financially supported by National Natural Science Foundation of China (81874107, 81902656).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Armstrong DK, Alvarez RD, Bakkum-Gamez JN, Barroilhet L, Behbakht K, Berchuck A, Berek JS, Chen LM, Cristea M, DeRosa M, ElNaggar AC, Gershenson DM, Gray HJ, Hakam A, Jain A, Johnston C, Leath CA III, Liu J, Mahdi H, Matei D, McHale M, McLean K, O’Malley DM, Penson RT, Percac-Lima S, Ratner E, Remmenga SW, Sabbatini P, Werner TL, Zsiros E, Burns JL, Engh AM. NCCN guidelines insights: ovarian cancer, version 1.2019. J Natl Compr Canc Netw. 2019;17:896–909. doi: 10.6004/jnccn.2019.0039. [DOI] [PubMed] [Google Scholar]
- 2.Mukherjee A, Huynh V, Gaines K, Reh WA, Vasquez KM. Targeting the high-mobility group box 3 protein sensitizes chemoresistant ovarian cancer cells to cisplatin. Cancer Res. 2019;79:3185–3191. doi: 10.1158/0008-5472.CAN-19-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhong Q, Peng HL, Zhao X, Zhang L, Hwang WT. Effects of BRCA1- and BRCA2-related mutations on ovarian and breast cancer survival: a meta-analysis. Clin Cancer Res. 2015;21:211–220. doi: 10.1158/1078-0432.CCR-14-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clements KE, Thakar T, Nicolae CM, Liang X, Wang HG, Moldovan GL. Loss of E2F7 confers resistance to poly-ADP-ribose polymerase (PARP) inhibitors in BRCA2-deficient cells. Nucleic Acids Res. 2018;46:8898–8907. doi: 10.1093/nar/gky657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong Q, Hu Z, Li Q, Yi T, Li J, Yang H. Cyclin D1 silencing impairs DNA double strand break repair, sensitizes BRCA1 wildtype ovarian cancer cells to olaparib. Gynecol Oncol. 2019;152:157–165. doi: 10.1016/j.ygyno.2018.10.027. [DOI] [PubMed] [Google Scholar]
- 6.Murai J. Targeting DNA repair and replication stress in the treatment of ovarian cancer. Int J Clin Oncol. 2017;22:619–628. doi: 10.1007/s10147-017-1145-7. [DOI] [PubMed] [Google Scholar]
- 7.Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19:1381–1388. doi: 10.1038/nm.3369. [DOI] [PubMed] [Google Scholar]
- 8.Phan NN, Wang CY, Li KL, Chen CF, Chiao CC, Yu HG, Huang PL, Lin YC. Distinct expression of CDCA3, CDCA5, and CDCA8 leads to shorter relapse free survival in breast cancer patient. Oncotarget. 2018;9:6977–6992. doi: 10.18632/oncotarget.24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayama S, Daigo Y, Yamabuki T, Hirata D, Kato T, Miyamoto M, Ito T, Tsuchiya E, Kondo S, Nakamura Y. Phosphorylation and activation of cell division cycle associated 8 by aurora kinase B plays a significant role in human lung carcinogenesis. Cancer Res. 2007;67:4113–4122. doi: 10.1158/0008-5472.CAN-06-4705. [DOI] [PubMed] [Google Scholar]
- 10.Vagnarelli P, Earnshaw WC. Chromosomal passengers: the four-dimensional regulation of mitotic events. Chromosoma. 2004;113:211–222. doi: 10.1007/s00412-004-0307-3. [DOI] [PubMed] [Google Scholar]
- 11.Gu Y, Lu L, Wu L, Chen H, Zhu W, He Y. Identification of prognostic genes in kidney renal clear cell carcinoma by RNAseq data analysis. Mol Med Rep. 2017;15:1661–1667. doi: 10.3892/mmr.2017.6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan H, Li Z, Shen Q, Wang Q, Tian J, Jiang Q, Gao L. Aberrant expression of cell cycle and material metabolism related genes contributes to hepatocellular carcinoma occurrence. Pathol Res Pract. 2017;213:316–321. doi: 10.1016/j.prp.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 13.Ci C, Tang B, Lyu D, Liu W, Qiang D, Ji X, Qiu X, Chen L, Ding W. Overexpression of CDCA8 promotes the malignant progression of cutaneous melanoma and leads to poor prognosis. Int J Mol Med. 2019;43:404–412. doi: 10.3892/ijmm.2018.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kabisch M, Lorenzo Bermejo J, Dünnebier T, Ying S, Michailidou K, Bolla MK, Wang Q, Dennis J, Shah M, Perkins BJ, Czene K, Darabi H, Eriksson M, Bojesen SE, Nordestgaard BG, Nielsen SF, Flyger H, Lambrechts D, Neven P, Peeters S, Weltens C, Couch FJ, Olson JE, Wang X, Purrington K, Chang-Claude J, Rudolph A, Seibold P, Flesch-Janys D, Peto J, dos-Santos-Silva I, Johnson N, Fletcher O, Nevanlinna H, Muranen TA, Aittomäki K, Blomqvist C, Schmidt MK, Broeks A, Cornelissen S, Hogervorst FB, Li J, Brand JS, Humphreys K, Guénel P, Truong T, Menegaux F, Sanchez M, Burwinkel B, Marmé F, Yang R, Bugert P, González-Neira A, Benitez J, Pilar Zamora M, Arias Perez JI, Cox A, Cross SS, Reed MW, Andrulis IL, Knight JA, Glendon G, Tchatchou S, Sawyer EJ, Tomlinson I, Kerin MJ, Miller N kConFab Investigators; Australian Ovarian Cancer Study Group; Haiman CA, Schumacher F, Henderson BE, Le Marchand L, Lindblom A, Margolin S, Hooning MJ, Hollestelle A, Kriege M, Koppert LB, Hopper JL, Southey MC, Tsimiklis H, Apicella C, Slettedahl S, Toland AE, Vachon C, Yannoukakos D, Giles GG, Milne RL, McLean C, Fasching PA, Ruebner M, Ekici AB, Beckmann MW, Brenner H, Dieffenbach AK, Arndt V, Stegmaier C, Ashworth A, Orr N, Schoemaker MJ, Swerdlow A, García-Closas M, Figueroa J, Chanock SJ, Lissowska J, Goldberg MS, Labrèche F, Dumont M, Winqvist R, Pylkäs K, Jukkola-Vuorinen A, Grip M, Brauch H, Brüning T, Ko YD GENICA Network. Radice P, Peterlongo P, Scuvera G, Fortuzzi S, Bogdanova N, Dörk T, Mannermaa A, Kataja V, Kosma VM, Hartikainen JM, Devilee P, Tollenaar RA, Seynaeve C, Van Asperen CJ, Jakubowska A, Lubinski J, Jaworska-Bieniek K, Durda K, Zheng W, Shrubsole MJ, Cai Q, Torres D, Anton-Culver H, Kristensen V, Bacot F, Tessier DC, Vincent D, Luccarini C, Baynes C, Ahmed S, Maranian M, Simard J, Chenevix-Trench G, Hall P, Pharoah PD, Dunning AM, Easton DF, Hamann U. Inherited variants in the inner centromere protein (INCENP) gene of the chromosomal passenger complex contribute to the susceptibility of ER-negative breast cancer. Carcinogenesis. 2015;36:256–271. doi: 10.1093/carcin/bgu326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen YJ, Chen CM, Twu NF, Yen MS, Lai CR, Wu HH, Wang PH, Yuan CC. Overexpression of Aurora B is associated with poor prognosis in epithelial ovarian cancer patients. Virchows Arch. 2009;455:431–440. doi: 10.1007/s00428-009-0838-3. [DOI] [PubMed] [Google Scholar]
- 16.He X, Yang K, Wang H, Chen X, Wu H, Yao L, Ma S. Expression and clinical significance of survivin in ovarian cancer: a meta-analysis. PLoS One. 2018;13:e0194463. doi: 10.1371/journal.pone.0194463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sui L, Dong Y, Ohno M, Watanabe Y, Sugimoto K, Tokuda M. Survivin expression and its correlation with cell proliferation and prognosis in epithelial ovarian tumors. Int J Oncol. 2002;21:315–20. [PubMed] [Google Scholar]
- 18.Bi Y, Chen S, Jiang J, Yao J, Wang G, Zhou Q, Li S. CDCA8 expression and its clinical relevance in patients with bladder cancer. Medicine (Baltimore) 2018;97:e11899. doi: 10.1097/MD.0000000000011899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bu Y, Shi L, Yu D, Liang Z, Li W. CDCA8 is a key mediator of estrogen-stimulated cell proliferation in breast cancer cells. Gene. 2019;703:1–6. doi: 10.1016/j.gene.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 20.Jiao DC, Lu ZD, Qiao JH, Yan M, Cui SD, Liu ZZ. Expression of CDCA8 correlates closely with FOXM1 in breast cancer: public microarray data analysis and immunohistochemical study. Neoplasma. 2015;62:464–469. doi: 10.4149/neo_2015_055. [DOI] [PubMed] [Google Scholar]
- 21.Ju W, Yoo BC, Kim IJ, Kim JW, Kim SC, Lee HP. Identification of genes with differential expression in chemoresistant epithelial ovarian cancer using high-density oligonucleotide microarrays. Oncol Res. 2009;18:47–56. doi: 10.3727/096504009789954672. [DOI] [PubMed] [Google Scholar]
- 22.Bélanger F, Fortier E, Dubé M, Lemay JF, Buisson R, Masson JY, Elsherbiny A, Costantino S, Carmona E, Mes-Masson AM, Wurtele H, Drobetsky E. Replication protein a availability during DNA replication stress is a major determinant of cisplatin resistance in ovarian cancer cells. Cancer Res. 2018;78:5561–5573. doi: 10.1158/0008-5472.CAN-18-0618. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Matulonis UA. New strategies in ovarian cancer: translating the molecular complexity of ovarian cancer into treatment advances. Clin Cancer Res. 2014;20:5150–5156. doi: 10.1158/1078-0432.CCR-14-1312. [DOI] [PubMed] [Google Scholar]
- 24.Ju W, Yoo BC, Kim IJ, Kim JW, Kim SC, Lee HP. Identification of genes with differential expression in chemoresistant epithelial ovarian cancer using high-density oligonucleotide microarrays. Oncol Res. 2009;18:47–56. doi: 10.3727/096504009789954672. [DOI] [PubMed] [Google Scholar]
- 25.Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, Jeong KJ, Ding Z, Liang J, Wang SW, Dai H, Lee S, Sahni N, Mercado-Uribe I, Kim TB, Chen K, Lin SY, Peng G, Westin SN, Liu J, O’Connor MJ, Yap TA, Mills GB. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. 2019;35:851–867. e857. doi: 10.1016/j.ccell.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng F, Zhang Y, Chen S, Weng X, Rao Y, Fang H. Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed Pharmacother. 2020;123:109661. doi: 10.1016/j.biopha.2019.109661. [DOI] [PubMed] [Google Scholar]
- 27.Caracciolo D, Scionti F, Juli G, Altomare E, Golino G, Todoerti K, Grillone K, Riillo C, Arbitrio M, Iannone M, Morelli E, Amodio N, Di Martino MT, Rossi M, Neri A, Tagliaferri P, Tassone P. Exploiting MYC-induced PARPness to target genomic instability in multiple myeloma. Haematologica. 2021;106:185–195. doi: 10.3324/haematol.2019.240713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mesquita KA, Alabdullah M, Griffin M, Toss MS, Fatah TMAA, Alblihy A, Moseley P, Chan SYT, Rakha EA, Madhusudan S. ERCC1-XPF deficiency is a predictor of olaparib induced synthetic lethality and platinum sensitivity in epithelial ovarian cancers. Gynecol Oncol. 2019;153:416–424. doi: 10.1016/j.ygyno.2019.02.014. [DOI] [PubMed] [Google Scholar]
- 29.Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F. Targeting the ATR/CHK1 axis with PARP inhibition results in tumor regression in BRCA-Mutant ovarian cancer models. Clin Cancer Res. 2017;23:3097–3108. doi: 10.1158/1078-0432.CCR-16-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Helleday T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011;39:3166–3175. doi: 10.1093/nar/gkq1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuo LJ, Yang LX. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo. 2008;22:305–309. [PubMed] [Google Scholar]
- 32.Matsuda Y, Wakai T, Kubota M, Osawa M, Takamura M, Yamagiwa S, Aoyagi Y, Sanpei A, Fujimaki S. DNA damage sensor gamma-H2AX is increased in preneoplastic lesions of hepatocellular carcinoma. ScientificWorldJournal. 2013;2013:597095. doi: 10.1155/2013/597095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, Lu Y, Meric-Bernstam F, Yap TA, Hattersley M, O’Connor MJ, Chen H, Fawell S, Lin SY, Peng G, Mills GB. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33:401–416. e408. doi: 10.1016/j.ccell.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang JL, Chen TH, Wang CF, Chiang YH, Huang YL, Wong FH, Chou CK, Chen CM. Borealin/Dasra B is a cell cycle-regulated chromosomal passenger protein and its nuclear accumulation is linked to poor prognosis for human gastric cancer. Exp Cell Res. 2006;312:962–973. doi: 10.1016/j.yexcr.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Iness AN, Felthousen J, Ananthapadmanabhan V, Sesay F, Saini S, Guiley KZ, Rubin SM, Dozmorov M, Litovchick L. The cell cycle regulatory DREAM complex is disrupted by high expression of oncogenic B-Myb. Oncogene. 2019;38:1080–1092. doi: 10.1038/s41388-018-0490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pilkinton M, Sandoval R, Song J, Ness SA, Colamonici OR. Mip/LIN-9 regulates the expression of B-Myb and the induction of cyclin A, cyclin B, and CDK1. J Biol Chem. 2007;282:168–175. doi: 10.1074/jbc.M609924200. [DOI] [PubMed] [Google Scholar]
- 37.Knight AS, Notaridou M, Watson RJ. A Lin-9 complex is recruited by B-Myb to activate transcription of G2/M genes in undifferentiated embryonal carcinoma cells. Oncogene. 2009;28:1737–1747. doi: 10.1038/onc.2009.22. [DOI] [PubMed] [Google Scholar]
- 38.Musa J, Aynaud MM, Mirabeau O, Delattre O, Grunewald TG. MYBL2 (B-Myb): a central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017;8:e2895. doi: 10.1038/cddis.2017.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bayley R, Blakemore D, Cancian L, Dumon S, Volpe G, Ward C, Almaghrabi R, Gujar J, Reeve N, Raghavan M, Higgs MR, Stewart GS, Petermann E, Garcia P. MYBL2 supports DNA Double strand break repair in hematopoietic stem cells. Cancer Res. 2018;78:5767–5779. doi: 10.1158/0008-5472.CAN-18-0273. [DOI] [PubMed] [Google Scholar]
- 40.Garcia P, Frampton J. The transcription factor B-Myb is essential for S-phase progression and genomic stability in diploid and polyploid megakaryocytes. J Cell Sci. 2006;119:1483–1493. doi: 10.1242/jcs.02870. [DOI] [PubMed] [Google Scholar]
- 41.Lorvellec M, Dumon S, Maya-Mendoza A, Jackson D, Frampton J, Garcia P. B-Myb is critical for proper DNA duplication during an unperturbed S phase in mouse embryonic stem cells. Stem Cells. 2010;28:1751–1759. doi: 10.1002/stem.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shepard JL, Amatruda JF, Stern HM, Subramanian A, Finkelstein D, Ziai J, Finley KR, Pfaff KL, Hersey C, Zhou Y, Barut B, Freedman M, Lee C, Spitsbergen J, Neuberg D, Weber G, Golub TR, Glickman JN, Kutok JL, Aster JC, Zon LI. A zebrafish bmyb mutation causes genome instability and increased cancer susceptibility. Proc Natl Acad Sci U S A. 2005;102:13194–13199. doi: 10.1073/pnas.0506583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen J, Chen X. MYBL2 is targeted by miR-143-3p and regulates breast cancer cell proliferation and apoptosis. Oncol Res. 2018;26:913–922. doi: 10.3727/096504017X15135941182107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiong YC, Chen T, Yang XB, Deng CL, Ning QL, Quan R, Yu XR. Overexpression of MYBL2 promotes proliferation and migration of non-small-cell lung cancer via upregulating NCAPH. Mol Cell Biochem. 2020;468:185–193. doi: 10.1007/s11010-020-03721-x. [DOI] [PubMed] [Google Scholar]
- 45.Liang HB, Cao Y, Ma Q, Shu YJ, Wang Z, Zhang F, Ye YY, Li HF, Xiang SS, Song XL, Xu Y, Zhang YC, Bao RF, Yuan RY, Zhang YJ, Hu YP, Jiang L, Li ML, Wang XA, Wu XS, Wu WG, Zhao S, Fand Y, Cui XP, Lu YS, Zhou J, Zheng L, Gong W, Liu YB. MYBL2 is a potential prognostic marker that promotes cell proliferation in gallbladder cancer. Cell Physiol Biochem. 2017;41:2117–2131. doi: 10.1159/000475454. [DOI] [PubMed] [Google Scholar]
- 46.Wei T, Weiler SME, Tóth M, Sticht C, Lutz T, Thomann S, De La Torre C, Straub B, Merker S, Ruppert T, Marquardt J, Singer S, Gretz N, Schirmacher P, Breuhahn K. YAP-dependent induction of UHMK1 supports nuclear enrichment of the oncogene MYBL2 and proliferation in liver cancer cells. Oncogene. 2019;38:5541–5550. doi: 10.1038/s41388-019-0801-y. [DOI] [PubMed] [Google Scholar]
- 47.Ren F, Wang L, Shen X, Xiao X, Liu Z, Wei P, Wang Y, Qi P, Shen C, Sheng W, Du X. MYBL2 is an independent prognostic marker that has tumor-promoting functions in colorectal cancer. Am J Cancer Res. 2015;5:1542–1552. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.